
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe chemical assessment of surfaces and air (CASA) study: using chemical and physical perturbations in a test house to investigate indoor processes†
Delphine K.
Farmer
 *a,
Marina E.
Vance
*b,
Dustin
Poppendieck
c,
Jon
Abbatt
d,
Michael R.
Alves
e,
Karen C.
Dannemiller
fg,
Cholaphan
Deeleepojananan
h,
Jenna
Ditto‡
d,
Brian
Dougherty
c,
Olivia R.
Farinas
f,
Allen H.
Goldstein
e,
Vicki H.
Grassian
h,
Han
Huynh§
d,
Deborah
Kim
h,
Jon C.
King
f,
Jesse
Kroll
i,
Jienan
Li
a,
Michael F.
Link
c,
Liora
Mael¶
b,
Kathryn
Mayer||
a,
Andrew B.
Martin**
b,
Glenn
Morrison
j,
Rachel
O'Brien
k,
Shubhrangshu
Pandit
h,
Barbara J.
Turpin
j,
Marc
Webb
j,
Jie
Yu
d and
Stephen M.
Zimmerman
c
*a,
Marina E.
Vance
*b,
Dustin
Poppendieck
c,
Jon
Abbatt
d,
Michael R.
Alves
e,
Karen C.
Dannemiller
fg,
Cholaphan
Deeleepojananan
h,
Jenna
Ditto‡
d,
Brian
Dougherty
c,
Olivia R.
Farinas
f,
Allen H.
Goldstein
e,
Vicki H.
Grassian
h,
Han
Huynh§
d,
Deborah
Kim
h,
Jon C.
King
f,
Jesse
Kroll
i,
Jienan
Li
a,
Michael F.
Link
c,
Liora
Mael¶
b,
Kathryn
Mayer||
a,
Andrew B.
Martin**
b,
Glenn
Morrison
j,
Rachel
O'Brien
k,
Shubhrangshu
Pandit
h,
Barbara J.
Turpin
j,
Marc
Webb
j,
Jie
Yu
d and
Stephen M.
Zimmerman
c
aDepartment of Chemistry, Colorado State University, Fort Collins, CO, USA. E-mail: Delphine.Farmer@colostate.edu
bDepartment of Mechanical Engineering, University of Colorado, Boulder, CO, USA. E-mail: Marina.Vance@colorado.edu
cNational Institute of Standards and Technology, Gaithersburg, MD, USA
dDepartment of Chemistry, University of Toronto, Toronto, ON, Canada
eDepartment of Environmental Science, Policy and Management, University of California Berkeley, Berkeley, CA, USA
fDepartment of Civil, Environmental, and Geodetic Engineering, Division of Environmental Health Sciences, The Ohio State University, Columbus, OH, USA
gSustainability Institute, The Ohio State University, Columbus, OH, USA
hDepartment of Chemistry and Biochemistry, University of California San Diego, La Jolla, CA, USA
iMassachusetts Institute of Technology, Cambridge, MA, USA
jDepartment of Environmental Sciences and Engineering, Gillings School of Global Public Health, University of North Carolina, Chapel Hill, NC, USA
kDepartment of Civil and Environmental Engineering, University of Michigan, Ann Arbor, MI, USA
First published on 21st June 2024
Abstract
The Chemical Assessment of Surfaces and Air (CASA) study aimed to understand how chemicals transform in the indoor environment using perturbations (e.g., cooking, cleaning) or additions of indoor and outdoor pollutants in a well-controlled test house. Chemical additions ranged from individual compounds (e.g., gaseous ammonia or ozone) to more complex mixtures (e.g., a wildfire smoke proxy and a commercial pesticide). Physical perturbations included varying temperature, ventilation rates, and relative humidity. The objectives for CASA included understanding (i) how outdoor air pollution impacts indoor air chemistry, (ii) how wildfire smoke transports and transforms indoors, (iii) how gases and particles interact with building surfaces, and (iv) how indoor environmental conditions impact indoor chemistry. Further, the combined measurements under unperturbed and experimental conditions enable investigation of mitigation strategies following outdoor and indoor air pollution events. A comprehensive suite of instruments measured different chemical components in the gas, particle, and surface phases throughout the study. We provide an overview of the test house, instrumentation, experimental design, and initial observations – including the role of humidity in controlling the air concentrations of many semi-volatile organic compounds, the potential for ozone to generate indoor nitrogen pentoxide (N2O5), the differences in microbial composition between the test house and other occupied buildings, and the complexity of deposited particles and gases on different indoor surfaces.
Environmental significancePeople in the United States spend most of their time in indoor environments, which can have high concentrations of reactive compounds. These compounds may originate from outdoor air, indoor activities, indoor chemistry, or even the building itself. The chemical and physical fate of indoor chemicals impacts their air and surface concentrations, and thus the potential for human exposure. The Chemical Assessment of Surfaces and Air (CASA) study aimed to understand the transformations of indoor and outdoor air pollutants in the built environment, demonstrating a ‘lab-in-field’ approach to indoor chemistry. This work provides insight on transformations of indoor chemicals by the addition of urban smog and wood smoke and investigates potential mitigation strategies for common indoor air pollutants. |
1 Introduction
According to the National Human Activity Pattern Survey,1 people in the United States spend around 90% of their lives indoors, meaning that indoor environments can be significant contributors to human exposure to chemical contaminants.2,3 While there is an emerging body of literature on indoor chemistry,4–9 comprehensive field measurements in the built environment remain rare. Several recent field studies conducted in real buildings have investigated the indoor chemical composition, demonstrating the persistent presence of elevated levels of reactive organic carbon relative to outdoor air,2,10–13 oxidation products despite low levels of oxidants,14 large surface reservoirs of organic compounds,15 and a strong role of surfaces in mediating indoor chemistry and exposure.16–20 Laboratory studies have proven essential for investigating the fundamental chemical reactions controlling indoor chemistry, with models providing new insights on indoor chemistry occurring on temporal and spatial scales spanning several orders of magnitude.4,21,22 Despite this acceleration in indoor chemistry measurements, questions persist about how air pollutants transport and transform in buildings and the subsequent spatial gradients,21 how well different air quality mitigation approaches work,23 how outdoor air pollutants interact with indoor air,24 how indoor air impacts the outdoor environment,25 and the extent to which this chemistry impacts human exposure and health outcomes.2,3Indoor chemistry includes surfaces and air; air contains both gaseous and particulate matter, requiring a wide suite of measurements to measure the relevant processes. Spatial and temporal variability of chemical contaminants indoors is poorly constrained by observations21 due, in part, to the reactive nature of many indoor compounds, which creates challenges for analytical measurement techniques.26 Numerous compounds exist on the surfaces and in the air of the indoor environment, including reactive organic and inorganic species, some of which are present at much higher concentrations indoors than outdoors.7
While much of the reactive organic carbon in indoor air originates from the built environment and human activities indoors (e.g., cooking, cleaning),2,10,27,28 outdoor air pollutants can also enter the built environment. Urban smog includes ozone (O3), particulate matter, and other known air toxics. Wildfire smoke is an increasing concern in the western United States,29 and is a well-established source of indoor particulate matter.30,31 However, while the secondary chemistry of outdoor air pollution is well-studied, its behavior may be very different indoors due to high surface area to volume ratios, low light levels, and large concentrations of reactive material. The increasing frequency of wildfire events and the recent coronavirus disease pandemic (COVID-19) have raised public interest in indoor air quality, including identifying strategies to reduce indoor sources of pollutants and quantifying the effectiveness of mitigation strategies.
Upon entering indoor air, the potential fate of compounds includes (1) transport throughout the building via mechanical ventilation systems and other mechanisms of indoor air movement; (2) transformation via reaction with oxidants or other reactive species in the gas, surface, or particle phase; and (3) interaction with indoor surfaces via physical or chemical sorption, and thus either temporary or permanent removal from air. These different fates impact not only indoor air quality and human exposure, but also outdoor air quality following exfiltration through wall leakage, opening of windows or doors, and the exhaust of mechanical airflows. The migration of indoor reactive species to the outdoors means that buildings not only act as primary sources of outdoor air pollutants, but also as reactors that chemically process outdoor air and thus release secondary air pollutants.
The Chemical Assessment of Surfaces and Air (CASA) study investigated the underlying chemical transformations occurring inside a house, including the fate and chemical transformations associated with wildfire smoke and other air pollutants after entering buildings. Specific questions addressed by this project include:
• How does outdoor air pollution impact a home's indoor chemistry?
• How is wildfire smoke transported and transformed once inside a house?
• How do gases and particles in the air interact with the surfaces of a home?
• How do the home's environmental conditions (e.g., relative humidity (RH), temperature, ventilation conditions) impact indoor chemistry?
This study aimed to further develop our understanding of indoor chemistry and to produce evidence-based actionable information that can be communicated to the public and other stakeholders. This paper provides an overview of the CASA study's experimental design and instrumentation; selected results that exemplify the aims of the project are also included.
2 Methods
Acknowledging the heavy use of acronyms in the field of indoor chemistry, we point the reader to the ESI Section S1† for a list of acronyms, excluding chemical names.2.1 House description
The CASA study took place from 22 February to 14 April 2022 at the Net Zero Energy Residential Test Facility32 (NZERTF) at the National Institute of Standards and Technology (NIST) campus in Gaithersburg, Maryland (Fig. 1 and 2). Constructed in 2012, the south-facing 1100 m3 NZERTF is a three story, unoccupied, unfurnished test house with painted wallboard and hardwood flooring throughout the first (330 m3) and second floors (294 m3). The basement (380 m3) has painted wallboard, an unfinished ceiling, and a concrete floor. For this study, computer-controlled operations created activities and loads within the NZERTF that mimic those of a four-member family.33 These activities and loads included operating the dishwasher and clothes washer, lighting the in-room on a set schedule, and releasing CO2 to mimic the same four family members. A daily tracer gas decay test using sulfur hexafluoride (SF6) provided the total air change rate (average 0.24 h−1) of the NZERTF via mechanical ventilation and infiltration. A mechanical heat recovery ventilation (HRV) and exhaust system exchanged indoor air from the bathrooms with outdoor air and then supplied pre-conditioned outdoor air to the bedrooms and kitchen at a fixed, continuous flow rate of 250 ± 20 m3 h−1 (150 ± 10 cfm), leading to a ≈ 0.2 h−1 mechanical air change rate. To enhance mixing, the indoor blower of the NZERTF's primary heat pump system operated continuously at its maximum setting, returning air to the unit from the first and second floors and supplying air to all three levels. When heating, this max-setting flow rate was 1150 ± 30 m3 h−1 (705 ± 16 cfm). When cooling, the max-setting flow was 820 ± 20 m3 h−1 (480 ± 10 cfm). Whole-house humidifiers, whose operation was restricted to times when the heat pump was heating, were used during certain tests to raise the RH throughout the test house. | ||
| Fig. 1 Photos of (a) the NZERTF test house, with the garage holding instruments on left, joined by a breezeway to the main house. (b) The dining area contained surface samples, trace gas analyzers, low-cost sensors, and several particle sizing instruments. Inlets (marked with a red arrow) carried sample air to (c) instruments in the garage. (d) The smoke chamber in the breezeway enabled injection of aged smoke into the house. Photos (a), (b), and (d) were taken by John Eisele; (c) was taken by Dustin Poppendieck. | ||
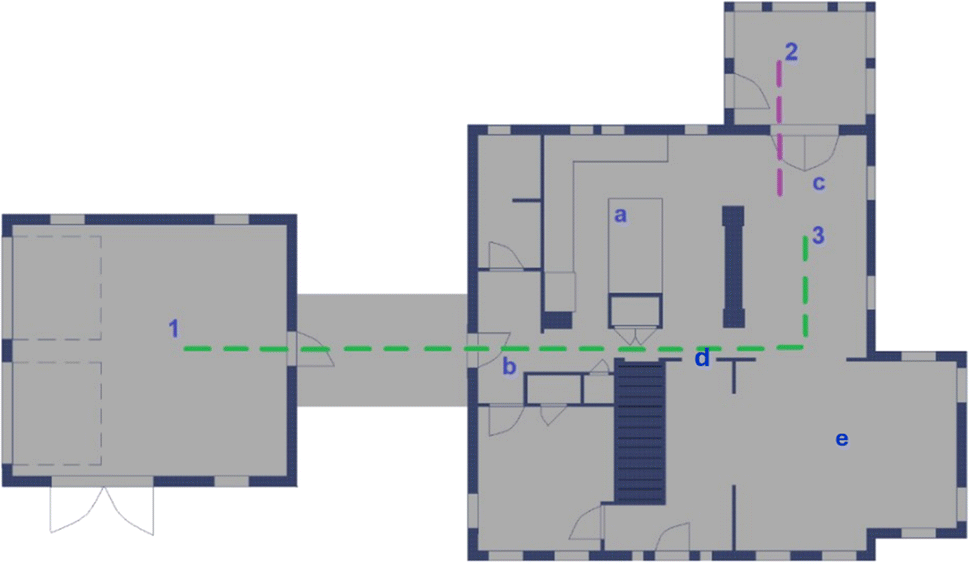 | ||
| Fig. 2 NZERTF diagram of the first floor. Instruments were located in (1) the garage, (2) the enclosed porch, or (3) the dining room. Locations for (a) cooking, (b) aged smoke injection, (c) chemical cocktail, and (d) fresh smoke. The pesticide was sprayed in (e) the living room. Other chemicals, including ozone (O3), carbon dioxide (CO2), and ammonia (NH3), were injected directly into the supply air flow of the ventilation system (not shown). Dashed lines show sample lines from the garage (green) and porch (purple). Image created by Rileigh Robertson. | ||
2.2 Study description
The experimental design of the CASA study focused on a series of physical and chemical perturbations to the test house, which was monitored by a suite of gas, aerosol, and surface measurements. Fig. 3 summarizes the experimental plan, which included physical environmental perturbations (changing temperature, RH, ventilation rate) and chemical perturbations (addition of wood smoke, commercial products, O3, cooking emissions, etc.) to the house. Fig. 4 shows the schedule of experiments. Specific experiments are detailed in Sections 3.1–3.10 below. Table 1 shows average conditions for some of the indoor and outdoor measurements during CASA. Fig. S1† shows PM2.5 concentrations measured by low-cost sensors in 12 locations inside the house and one outdoors. We express air levels of some chemical components as mixing ratios in parts-per notation defined as the number of moles of an analyte per mole of air, where parts per million (ppm) is μmol mol−1, parts per billion (ppb) is nmol mol−1, and parts per trillion (ppt) is pmol mol−1.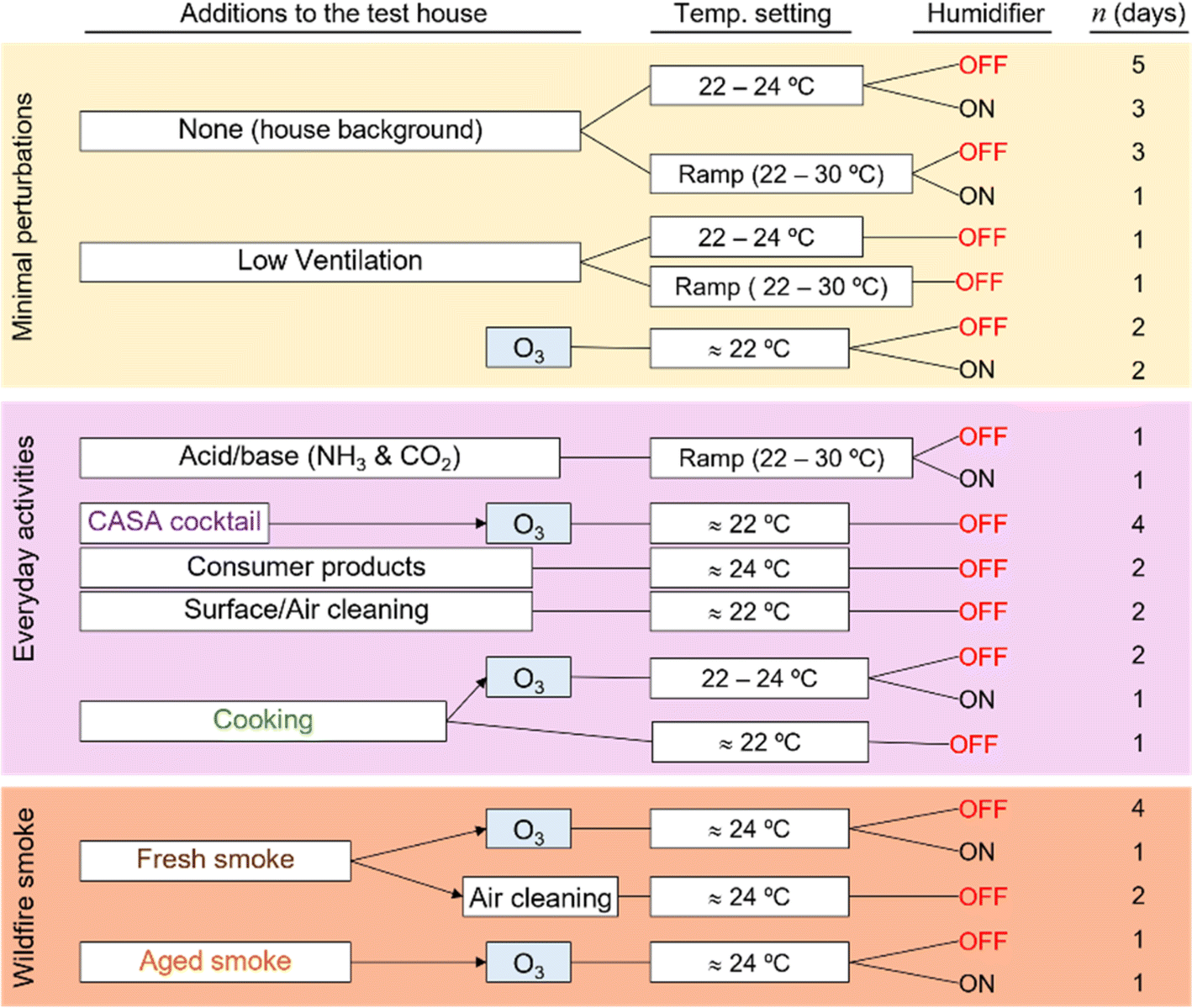 | ||
| Fig. 3 The experimental matrix for CASA included perturbations to investigate the house during background conditions (yellow), additions of chemicals to represent both everyday indoor activities and the dynamic range of chemicals present indoors (pink), and additions of wildfire smoke (brown). Most experiments included replicates, and many were conducted under varying environmental or O3 conditions. Stated temperatures represent typical ranges. | ||
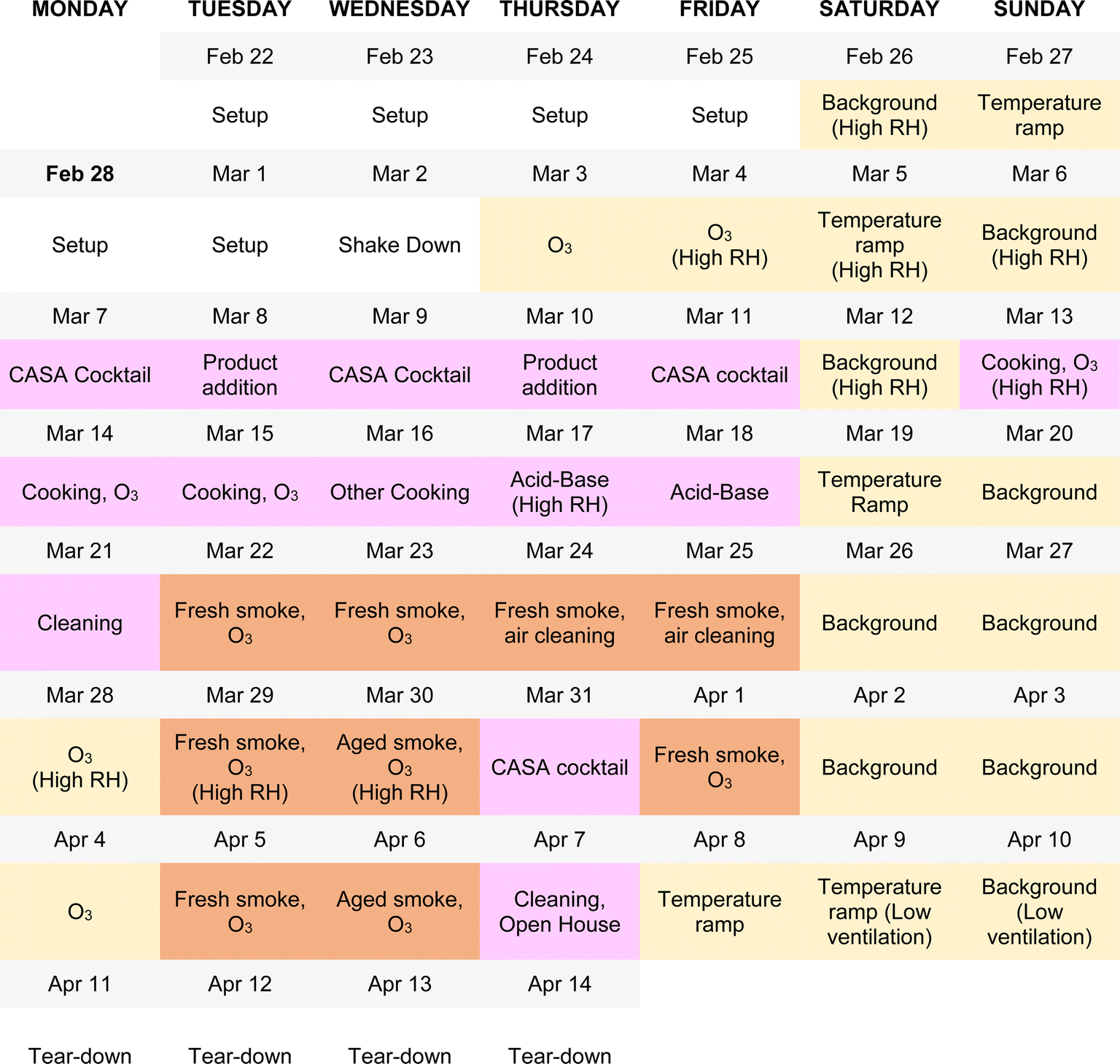 | ||
| Fig. 4 The experimental schedule for CASA. Colors indicate the experimental categories described in Fig. 3: background days, changes in environmental conditions, and O3 additions under background conditions are shown in yellow; everyday products and activities are shown in pink; and smoke additions are shown in brown. Ozone was injected in combination with almost all experimental conditions. The schedule reflects the dominant activity or condition for each day; several additional tests took place beyond those noted on this schedule (Section 3.10 for details). | ||
| Indoor (average ± σ) | Outdoor (average ± σ) | |
|---|---|---|
| a Average from first floor. Excludes temperature ramp and high RH tests. b Average excludes CO2 injections, average from basement, first and second floor. c Average excludes O3 injections and when there were high NO concentrations indoors. d Average from basement, first and second floor; attic average was 36 ± 5.2 ppb. e Average from basement, first and second floor; attic average was 161 ± 27.7 ppb. f Average from basement, first and second floor. g Average from dining area, from corrected PurpleAir measurements. h Average background measurements, excluding perturbations. i Average from outdoors on 11 April. | ||
| Temperature (°C) | 24 ± 1 a | 8 ± 6 |
| Relative humiditya (%) | 32 ± 6 a | 50 ± 20 |
| Mechanical ventilation (m3 h−1) | 250 ± 20 | N/A |
| Air change rate (h−1) | 0.24 ± 0.04 | N/A |
| CO2 (ppm) | 500 ± 100 b | 450 ± 30 |
| O3 (ppb) | 8 ± 1 c | 31 ± 4 |
| HCHO (ppb) | 13 ± 5 d | 1.2 ± 0.7 |
| HCOOH (ppb) | 30 ± 10 e | 0.7 ± 0.3 e |
| NO (ppb) | 1 ± 3 f | 2 ± 7 |
| NO2 (ppb) | 4 ± 3 f | 5 ± 6 |
| PM2.5 (μg m−3) | 4 ± 20g | 3 ± 6 |
| WSOCg (μg C m−3) | 220 ± 30h | 5 ± 1i |
| WSOCp (μg C m−3) | 6 ± 3 | Below LOD |
2.3 Meteorological and trace gas data
Wind and temperature sensors, along with a pyranometer, collected meteorological data on the roof of the NZERTF.34 Indoor house measurements included temperature in 20 locations, RH in seven locations, and air change rates from daily injections of SF6 (Fig. S2†). Pitot tubes in the outdoor air, return, and exhaust ducts of the HRV provided mechanical ventilation air flow rates. A photoacoustic gas monitor provided CO2 mixing ratios in five locations throughout the house and one location outdoors. Several trace gas monitoring instruments were placed in the passively-conditioned attic and shared an automated inlet switching system to sample air from the basement, first floor, second floor, attic and outdoors. These measurements included nitrogen oxides (NOx), O3, formaldehyde (HCHO), and formic acid (HCOOH). Total airborne water soluble organic carbon was measured on the first floor throughout the campaign, with one day of outdoor sampling. Additional reactive trace gas measurements included an array of organic and inorganic compounds, with inlet and instrument details in Section 2.4.2.4 Instruments
Instruments were located in the house, attic, garage, and covered porch (Tables 2–4). Instruments in the garage and porch used inlet lines to sample from the dining room in the house; instruments in the house had short inlet lines, with most interior instruments located in the dining room. Several low-cost sensors were placed throughout the house. All gas-phase instruments had inlets made of 0.3625 cm (1/4′′) o.d. perfluoroalkoxy (PFA) tubing unless otherwise noted.| Institution | Measurement | Instrument | Sample line/Instrument inlet details | Limit of detection (LOD) and time resolution | Comments |
|---|---|---|---|---|---|
| a Lpm = Liters per minute, PTFE = polytetrafluoroethylene, PFA = perfluoroalkoxy. | |||||
| House trace gas system | |||||
| National Institute of Standards and Technology (NIST) | CO2 | Photoacoustic gas monitor | Sample line:3.2 mm (1/8′′) o.d. PTFE, <50 m | LOD: 1.5 ppm | Manifold system switching between six locations on a 4 min interval: First floor, second floor, basement, attic and outside |
| Flowrate: 0.9 Lpm | Time resolution: 40 s | ||||
| NIST | O3 | Dual-cell, UV photometric | Sample line: 6.4 mm (¼′′) o.d. PFA, 9 m | LOD: 0.5 ppb | 8-Port valve switching between five locations on 5 min intervals: outside, first floor, second floor, basement, attic |
| Flowrate: 10 Lpm | Time resolution: 1 min | ||||
| NIST | Formaldehyde (HCHO), formic acid (HCOOH) | Laser absorption spectrometer | Sample line: 6.4 mm (¼′′) o.d. PFA, 9 m | LOD:0.1 ppbv HCHO | Same as NIST O3 inlet |
| Flowrate: 10 Lpm | Time resolution: 1 s | ||||
| Flowrate: 0.75 Lpm | |||||
| Colorado State University (CSU) | NOx | Chemiluminescent detector with molybdenum oxide NO2 converter (Thermo Fisher model 42i TL) | Sample line: 6.4 mm (¼′′) o.d. PFA, 9 m | LOD: 2.3 ppb | Same as NIST O3 inlet |
| Flowrate: 10 Lpm | |||||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||
| Other trace gases | |||||
| NIST | CO, CH4 | Cavity ring down spectroscopy | No sample line | LOD: 5 ppb CO; 0.3 ppb CH4 | Instrument was located in the dining room for a subset of experiments |
| Flowrate: 0.07 Lpm | Time resolution: 3 s | ||||
| CSU | SO2 | Teledyne laser induced phosphorescence detector | No sample line | LOD: 0.5 ppb | Instrument was located in the dining room |
| University of California (UC) Berkeley | NH3, CO2, H2O, select VOCs | Cavity ring down spectroscopy, Picarro model SI9110 | Sample line: 6.4 mm (¼′′) o.d. PTFE, 0.5 m | LOD: Ranges from 2 – 60 ppb | Instrument was located in the dining room |
| Massachusetts Institute of Technology (MIT) | VOCs | Low-cost VOC sensors (6 metal oxide, 3 electrochemical, 3 photo-ionization detectors) | Flowrate: 0.3 Lpm | LOD: Variable (sensor dependent) | Two units placed in kitchen, one with Nafion dryer inlet and one without; one unit placed in upstairs bedroom 3 |
| Sample line: 6.4 mm (¼”) o.d. PTFE, < 0.5 m | Time resolution: 1 s | ||||
| MIT | CO | Infrared absorption gas monitor (Teledyne T300) | No inlet | LOD: 0.04 ppm | One unit placed in kitchen |
| Time Resolution: 70 s | |||||
| MIT | PM, CO | QuantAQ Modulair (low-cost sensor Node with Alphasense OPC N-3, Plantower PMS, Alphasense CO–B4 sensors) | No inlet | LOD: Variable | One unit placed in kitchen |
| Time resolution: 5 s | |||||
| University of Washington Bothell | Various | Low-cost indoor air quality monitor (AirAssure, TSI, Inc – O3, NO2, SO2, CO, CO2, VOC, PM) | No inlet | LOD: Variable | One unit place in kitchen |
| University of Toronto | VOCs | Proton transfer reaction mass spectrometer (PTR-MS) with gas chromatography | Heated inlet line: 6.4 mm (¼”) i.d. PFA tubing held at 50 °C, 30.5 m long | LOD: Variable | Calibrations and zeros performed every 3–6 h using a VOC mixed standard cylinder diluted in VOC-free air |
| Flow rate: 4 Lpm through PTFE filter to scrub out particles | Time resolution: 4 s, averaged to 1 min | Operated largely in real-time mode, with periodic use of GC-PTR-MS mode | |||
| NIST | VOCs | PTR-MS | Heated inlet line: 6.4 mm (½′′) o.d. PFA tubing held at 50 °C, 30.5 m | LOD: Variable | Hourly zeros and calibrations using a VOC mixed standard cylinder diluted in VOC-free air. Operated only from 1 April to 11 April |
| Flowrate: 5 Lpm through PTFE filter to scrub out particles | Time resolution: 1 s, averaged to 10 s | Operated exclusively in real-time mode | |||
| CSU | Oxidized organic and inorganic compounds | Time-of-flight chemical ionization mass spectrometer with iodide ionization (iodide-CIMS)10,35 | Same heated line as NIST PTR-MS | LOD: Variable | Hourly zeros and calibrations; mass resolution ≈4000 (m/Δm); 5 min outdoor sampling bihourly |
| Time resolution: 1 s | |||||
|
|||||
| Gas + particle instruments | |||||
| University of North Carolina | Water soluble organic carbon in gas (WSOCg) and particle-phase (PM2.5; WSOCp) | Gas-phase with mist chamber (MC) (alternating with particle-phase by particle-into-liquid sampler, PILS36), both analyzed with Sievers M9 total organic carbon analyzer | MC inlet: 9.5 mm (3/8′′) i.d. PTFE, 7.6 m | WSOCg & WSOCp alternate every 6 min | Sampling in dining room. Instrument in attached enclosed porch |
| PILS inlet: 9.5 mm (3/8′′) o.d. Refrigeration-grade copper, 12 m | MC LOD: 0.5 μg-C m−3 | Daily blanks; MC collection efficiency measured for indoor WSOCg (data corrected) plus glyoxal, formic acid, acetic acid, acetone; PILS collection efficiency 95%37 | |||
| PILS flowrate: 13 Lpm | PILS LOD: 2.0 μg-C m−3 | Precision: WSOCg 16%, WSOCp 10%38 |
|||
| Time resolution: 4 s analysis time, 3.25 min response time | LOD limited by water background | ||||
| UC Berkeley | Semi-volatile organic compounds (SVOC), typically organic compounds with vapor pressures C14–C30+ alkanes | Semi-volatile thermal Desorption gas Chromatography (SVTAG)39 | Sample line: 19.0 mm (¾′′) stainless steel, 1 m | LOD: Variable from high ppq to low ppt | Gas and/or particle measurements. Daily internal standard correction. Sampling through 2.5 μm cyclones and a denuder (for particle-only measurements) |
| Flowrate: 10 Lpm | Time resolution: 1 h | ||||
| Institution | Measurement | Instrument | Sample LineInstrument inlet details | LOD and time resolution | Comments |
|---|---|---|---|---|---|
| CSU | Aerosol composition | High resolution time-of-flight Aerosol Mass Spectrometer40 (AMS) | Two identical 15.9 mm (5/8′′) o.d. copper sample lines – one with an inlet in the dining room, the other with an inlet outside the garage; house sampling line temperature was maintained at 21 °C with heating tape | Time resolution: 5 s | Inlet alternated indoors (51 min) and outdoors (9 min) |
| CSU | Size distributions (27.4–723.4 nm) | Scanning mobility particle Sizer (SMPS, EC TSI 3080, DMA – TSI 3081, CPC – TSI 3010) | Same heated line as AMS | Time resolution: 3 min | Inlet passed through a silica bead diffusion dryer |
| University of Colorado (CU) Boulder | Size distributions (27.4–723.4 nm) | Scanning mobility particle Sizer (SMPS, EC – TSI 3082, DMA – TSI 3081, CPC – TSI 3750) | Same heated line as CSU | Time resolution: 3 min | Undried samples |
| NIST | Size distributions | Scanning mobility particle Sizer | No sample line | ||
| CU Boulder | Low-cost sensors | PurpleAir PA-II, 12 units distributed inside the house and 1 outdoors | No sample line | LOD: ≈1 μg m−3 PM2.5 | Each unit contains two Plantower PMS-5003 particle detectors |
| 50% at 0.3 μm, 98% at ≥0.5 μm | |||||
| Time resolution: 2 min | |||||
| CU Boulder | Light absorbance | 5-Wavelength aethalometer (MicroAeth MA 200) | Dining room | LOD: 30 ng m−3 | |
| Time resolution: 1 min |
| Institution | Sample type/collection approach | Sampling location | Measurement/instrument | Sampling notes |
|---|---|---|---|---|
| University of Michigan | Aerosol sample collection with Micro-Orifice Uniform Deposition Impactor (MOUDI) | Kitchen | Offline-AMS and Fourier Transform Ion Cyclotron Resonance Mass Spectrometry (FT-ICR) | Samples collected daily during activities |
| University of Michigan | Surface samples collected for off-line analysis | Kitchen, upstairs, living room | Offline-AMS and FT-ICR | Samples collected after activities over varying lengths (3 days to the full campaign) |
| University of California San Diego | Sample collection for off-line analysis | Kitchen | nanoIR2 (AFM-IR spectroscopy) with 30 nm spatial resolution | Samples collected daily |
| In situ surface sampling | Kitchen | Quartz Crystal Microbalance (QCM) | Data collected every 6 s | |
| Sample collection for off-line analysis | Kitchen | ATR-FTIR (Model Nicolet iS50), Thermo Fisher Scientific | Samples were collected after cocktail, smoke, and other events | |
| Sample collection for off-line analysis | Kitchen | GC-MS (Thermo trace 1300/TSQ 8000 Evo Triple Quadrupole GC-MS) | Samples were collected before and after cocktail event | |
| Sample collection for off-line analysis | Kitchen | High-resolution mass spectrometer (Thermo Orbitrap Elite Hybrid MS) | Samples were collected before and after smoke event | |
| Yale University | Sample collection for off-line analysis | Kitchen | Passive polydimethylsiloxane samplers for offline analysis of volatile and semi-volatile organic compounds via gas chromatography orbitrap high resolution mass spectrometry | Most samples were deployed before experimental days and collected prior to the subsequent experiment. Multiple samples were collected during cooking and smoke days |
| Data can be used for targeted and non-targeted analysis | ||||
| University of Toronto flux chamber | Surface fluxes of volatile organic compounds within an enclosed stainless steel chamber clamped to a painted windowsill | Windowsill on east-facing wall of house | PTR-MS measurements | 30 minutes, spaced periodically throughout campaign |
| Multiple Universities | Vacuum floor dust samples for offline analysis | Entire house (first and second floor) | Various | Samples collected before and after smoke events |
2.5 Safety considerations
All experiments and instrument deployments underwent review to determine potential for hazards or exposure concerns. Except for cleaning, cooking, and a subset of direct smoke and commercial product spray injections, no researchers were in the NZERTF during the experiments. The NZERTF was accessed daily to collect surface samples and check instrumentation located within the building envelope, but with at least 30 minutes between ending indoor activities and the start of the next perturbation.3 Experimental details and selected findings
The CASA experiments can be divided into nine categories: house background, varying environmental parameters, ozone additions, smoke additions, air and surface cleaning, the CASA cocktail, product additions, acid/base additions, and cooking. An array of additional brief experiments were conducted throughout the project. Each group of experiments was designed to answer a specific set of questions. These questions, the subsequent experimental plan, and key data are presented below to demonstrate the chemical perturbation approach of the CASA study. Most perturbation experiments were compared to the house ‘background’ days described below. Finally, Section 3.11 summarizes the dust sample collection and subsequent microbial analysis that took place at the beginning and end of the project.3.1 House background
The unperturbed house background includes measurements taken while no people were in the house and in the absence of intentional activities or perturbations. All exterior doors and windows remained closed. The balanced ventilation and exhaust system was set to 250 m3 h−1. The indoor blower of the two-stage heat pump operated continuously while its compressor cycled on, as needed, to maintain the first floor temperature at the 24 °C setpoint. The whole house humidifiers were disabled for most background days, but operated for several consecutive hours on three background days. The house background thus includes influences from emissions of building materials, infiltration of outdoor air, HVAC system operation, and other automated activities including the dishwasher, clothes washer, clothes dryer and bathroom water lines that were automatically turned on and off on a predetermined schedule. CASA included five full days of ‘house background’, spread across the entire project, plus three days with elevated humidity and four days of ramped temperature setpoints at either high or low humidity. On two additional days (April 9 and 10), the fixed, continuous mechanical ventilation rate was decreased from the typical rate of 250 m3 h−1 (150 cfm) to 50 m3 h−1 (30 cfm); the first low-ventilation day included a temperature ramp. On some background days, individuals were present in the house for short periods of time for maintenance activities.Specific science questions investigated during the house background conditions included:
• What are the influences from building material emissions and outdoor air infiltration into indoor air?
• How does the cycling of HVAC and other indoor automated activities (e.g., dishwasher and clothes washer) affect the composition of indoor air?
• How do changes in ventilation conditions, indoor temperature, and humidity change indoor chemical composition of gases, particles, and surfaces?
These background periods enabled analysis of how outdoor pollution impacted indoor air. For example, Link et al.41 used the CASA data and a chemical box model to show that nitric oxide (NO) of outdoor origin could titrate the low levels of indoor O3 and inhibit the production of indoor NO3.
3.2 Environmental parameters
These perturbation experiments included days in which RH, temperature, or ventilation rates were varied from the background house conditions. These three parameters can be mechanically controlled in residences and affect human comfort and health. However, these environmental parameters may also affect chemical processes in a house. For example, temperature influences the partitioning of semi-volatile species between surfaces or particles and air, as well as chemical reaction rates. Air concentrations of phthalates correlate with indoor temperature, demonstrating a link between thermal comfort and indoor air composition.42 Increased ventilation reduces indoor air pollutant concentrations if the outdoor air is lower in concentration than indoor air, or if the air is treated in a mechanical ventilation system to reduce, for example, particulate matter or O3.During the COVID-19 pandemic, recommendations for increased ventilation aimed to reduce respiratory aerosol concentrations. However, when ventilation dilutes indoor air with outdoor air, shifts in surface-air partitioning can drive semi-volatile compounds from surface reservoirs into the indoor air. Wang et al. provide an example of this dilution-driven partitioning from the HOMEChem study, in which concentrations of semi-volatile organic compounds decreased during enhanced ventilation events, but increased once the enhanced ventilation was stopped and the house returned to normal operation.15
Relative humidity also impacts indoor concentrations, causing increased liquid water content of surfaces and aerosol, thus enhancing uptake of water-soluble compounds43,44 and potentially enabling aqueous chemical reactions. Water vapor may also react with compounds in the air, including carbonyl oxide intermediates created from ozonolysis of alkenes.45 Additionally, water vapor may displace other molecules on surfaces and shift surface-air partitioning. Water vapor leads to hygroscopic aerosol growth, increasing aerosol mass and size, and thus influencing deposition rates – both within a building and within the human respiratory system. However, the extent of these transformations in the indoor environment, and their potential impacts on air toxics and other chemical contaminants, surface chemical reservoirs, or human health are poorly constrained.
The CASA study included multiple manipulations of the house systems to investigate how environmental parameters impact chemical processes in the indoor environment. Specific questions of interest included:
• What is the pool of semi-volatile organic and inorganic components in indoor surface reservoirs?
• To what extent can temperature changes influence indoor chemical composition of gases, particles, and surfaces?
• How do surfaces and particles take up water?
• How do the concentrations of water soluble gases change with humidity?
Under background “low RH” conditions, humidifiers were kept off and the average RH was 32%, but ranged between 30 and 50%. On high RH days, the whole house in-duct humidifiers were turned on and set to their maximum speed. On these high RH days, the temperature typically increased from the 24 °C setpoint to about 27 °C and RH increased to about 70% RH. Each high RH day was paired with an identically scripted low RH day (i.e., humidifiers off) day. These paired RH perturbation days included:
• Background conditions (one high RH versus nine low RH days).
• O3 additions (two high RH versus two low RH days).
• Cooking (one high RH versus two low RH days).
• Temperature ramps (one high RH versus one low RH day).
• Acid-base additions (one high RH versus one low RH day).
• Fresh smoke additions (one high RH versus three low RH days).
• Aged smoke additions (one high RH versus one low RH day).
Because of the auxiliary resistive heating coupled with the refrigerant-based heat pump heating, the NZERTF can be heated much more quickly than it can be cooled. Moreover, a target humidity level, especially a higher one like that used for CASA high RH days, can be sufficiently controlled when increasing the NZERTF's dry bulb temperature. Reducing the indoor temperature while maintaining a target humidity level was beyond the capabilities of this house. Consequently, all temperature ramps conducted during CASA progressed in one direction, from coolest to warmest. Prior to starting a temperature ramp, the house was cooled to between 18 and 20 °C. The system was then switched to heating while maintaining continuous indoor blower operation. The house was heated by 2 °C increments from 20 to 30 °C; each temperature step was maintained for approximately two hours. Overall, the entire six-step temperature ramp took approximately eighteen hours to complete. A few hours were needed before temperatures returned to the background conditions of 24 °C. Temperature ramps were conducted at normal low RH (5 March), high RH (19 March), and low ventilation (9 April) conditions. This low ventilation temperature ramp was the sole experiment conducted during CASA at different ventilation conditions. Additional experiments were conducted with a limited subset of instrumentation after the end of the CASA experiment.
We observed that environmental parameters influenced indoor SVOC concentration. During a temperature ramp with RH held at 40%, each 1 °C rise in temperature corresponded to an increase in SVOC concentration by about 25% with respect to the house background signal (Fig. S3†). In comparison, during the high RH day (80% RH), each 1 °C rise in temperature corresponded to an increase in the SVOC concentration by about 50% of the background sample signal. Thus, RH plays a significant role in modulating SVOC concentrations in indoor air and may even enhance the temperature-dependent partitioning for certain compounds.
Further evidence that multiple processes control surface-gas exchange comes from temperature ramps on low RH versus high RH days. We observed that many gas-phase SVOCs were consistently higher in concentration under higher humidity conditions (80% RH) compared to background (≈32% RH). While temperature varied on high RH days, the SVOCs are more clearly linked to changes in RH. Fig. 5 shows that during a high RH event, the more volatile SVOCs, such as the C8–10 carboxylic acids, increase in gas-phase concentration. This response is weaker, but still apparent, for alkanes with similar volatility (C12–16) indicating that volatility is not the only factor driving this humidity effect. We hypothesize that water molecules compete with active sorption sites on surfaces when the humidity is increased, thus liberating some of these SVOC molecules. Some SVOC species, such as levoglucosan and vanillic acid/vanillin (not shown here), show no dependence on RH.
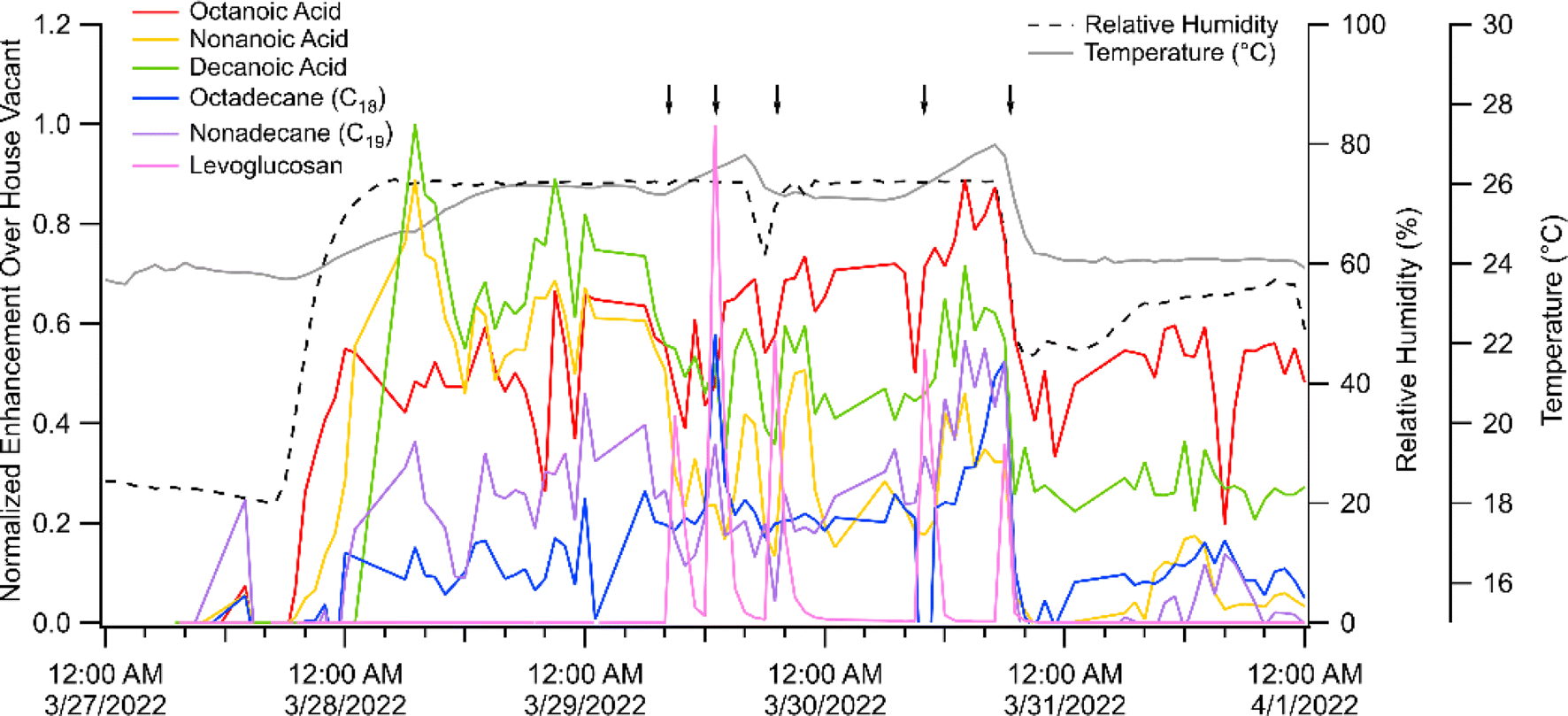 | ||
| Fig. 5 Signal from the SV-TAG during smoke additions conducted under changing RH. The signal has been normalized to the greatest signal intensity and is shown as an enhancement to the house background sample (27 March 1:00 AM). Relative humidity and temperature are shown in the dashed black and solid grey lines respectively. Black arrows correspond to fresh or aged smoke injections. Various cleaning, filtering, and ozone additions occurred on 28 March, which results in some variability in the signal. | ||
3.3 Ozone additions
Ozone is a well-studied oxidant in the indoor environment that can react with unsaturated organic compounds on surfaces and in air. During CASA, indoor O3 averaged 8 ± 1 ppb with an average indoor to outdoor concentration ratio of 0.26, in line with other North American residential environments.9,14,46,47 Indoor levels are typically much lower than outdoor levels due to high indoor reactivity. One recent study indicated that ozonolysis products from squalene, a reactive component of human skin oil, can be persistent indoors.14 The addition of ozone to indoor environments can also contribute to secondary organic aerosol.48 However, the extent to which indoor ozonolysis contributes to secondary air pollutants formed indoors is poorly quantified. To this end, two types of O3 perturbations were conducted during the CASA study: extended O3 additions (longer releases of O3 of 60–180 minutes peaking 80 ppb on the first floor) and punctuated O3 additions (short bursts in O3 release with indoor concentrations rising to 60 ppb within 30 minutes). The mass of O3 injected was not equal during these two experiment types. However, these two contrasting approaches enable studies of both multigenerational oxidation chemistry from longer exposures, and first order decay time analyses from shorter exposures.Ozone additions were designed to mimic the enhancement of ozone, though not the levels of other pollutants, upon opening a window on a high-O3 day. The O3 injection system was comprised of a cylinder of high-purity oxygen (O2) and a remotely-controlled two-way solenoid valve from the regulator to a mass flow controller on the porch of the test house. The mass flow controller, also located in the enclosed porch, delivered the O2 to an O3 generator at a flowrate of ≈1 Lpm. The power supply for the high-voltage O3 generator was also remotely controlled. Ozone from the generator transferred through a 3.2 mm (1/8′′) PTFE line to the HRV supply duct in the basement of the test house. The HRV supply duct distributed air to the dining room on the first floor and to each of the second floor bedrooms. The HRV flows between the first and second floors were not perfectly balanced, and the mass of O3 delivered to these spaces may have been slightly different.
Ozone addition experiments were performed periodically throughout the CASA study to investigate the following questions:
• What is the fate of O3 in the built environment, and to what extent can outdoor ozone impact indoor air quality?
• Does the house's reactivity to O3 change with time, or after O3 exposure?
• What are the main O3 products and their kinetics following both chemical perturbations and changes in environmental parameters?
Four punctuated O3 injections caused increased concentrations in the main spaces of the test house (first and second floors), while only changing slightly in the basement, and not changing at all in the attic or outdoors (Fig. 6). A key finding from O3 addition experiments was that adding ozone enabled formation of NO3 radicals following oxidation of ambient NO2; these NO3 radicals could then initiate oxidation reactions that impacted VOCs and generated organic nitrate products – highlighting the problems with adding ozone to buildings.41
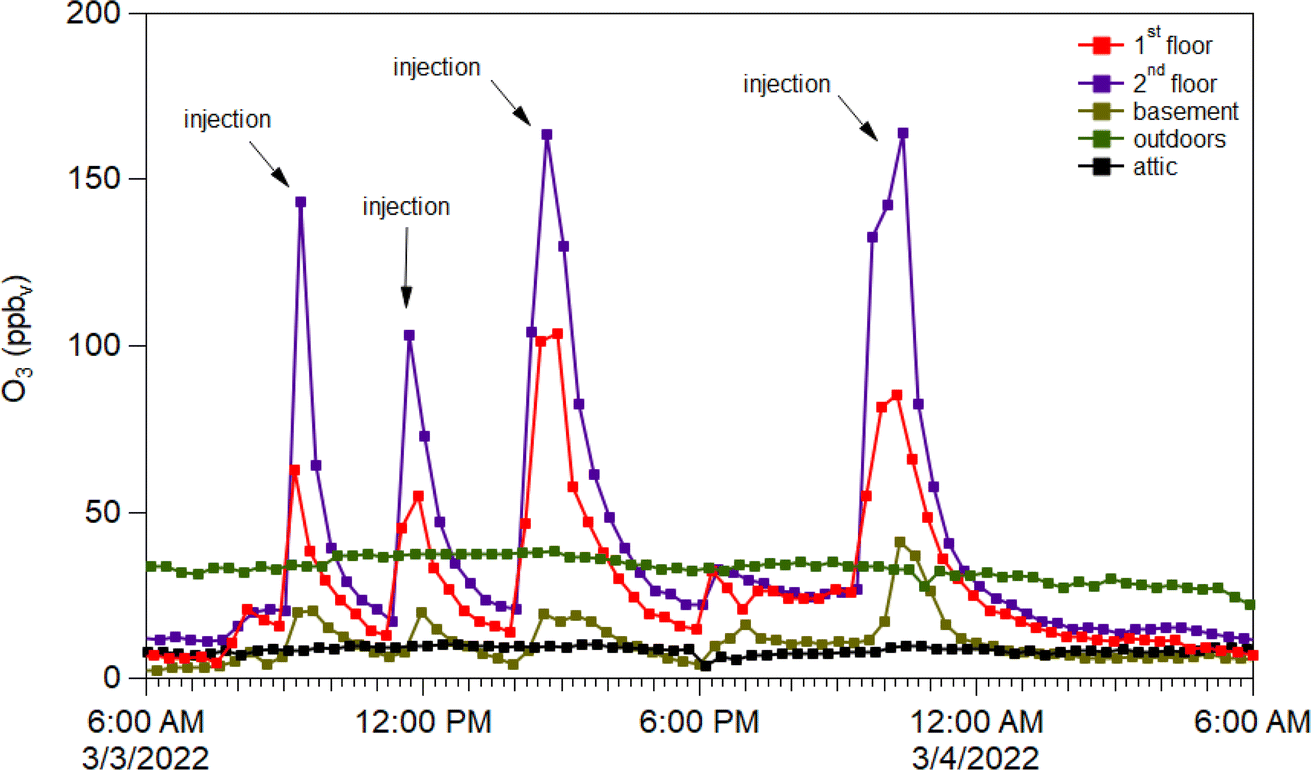 | ||
| Fig. 6 Ozone concentrations (ppb) on 3 March 2022 as detected from inlet lines located on the 1st (red), 2nd (purple), and basement (brown) floors in contrast to the outdoor (green) concentrations. | ||
In one subset of ozone addition experiments, we diluted the inflow to the O3 generator with ≈10% of 18O-labeled O2. 18O is a rare stable isotope, and its addition was observed in secondary reaction products observed by the iodide CIMS, with an 18O enhancement on the order of the same ratio as the injection ozone (11%, Fig. 7a). The presence of 18O-labeled N2O5 (Fig. 7b) directly demonstrates that O3 reacts with NO2 indoors to form N2O5. N2O5 is well established to react with aerosol surfaces to form HNO3, providing a potential mechanism for infiltration or addition of O3 pollution to acidify indoor surfaces.
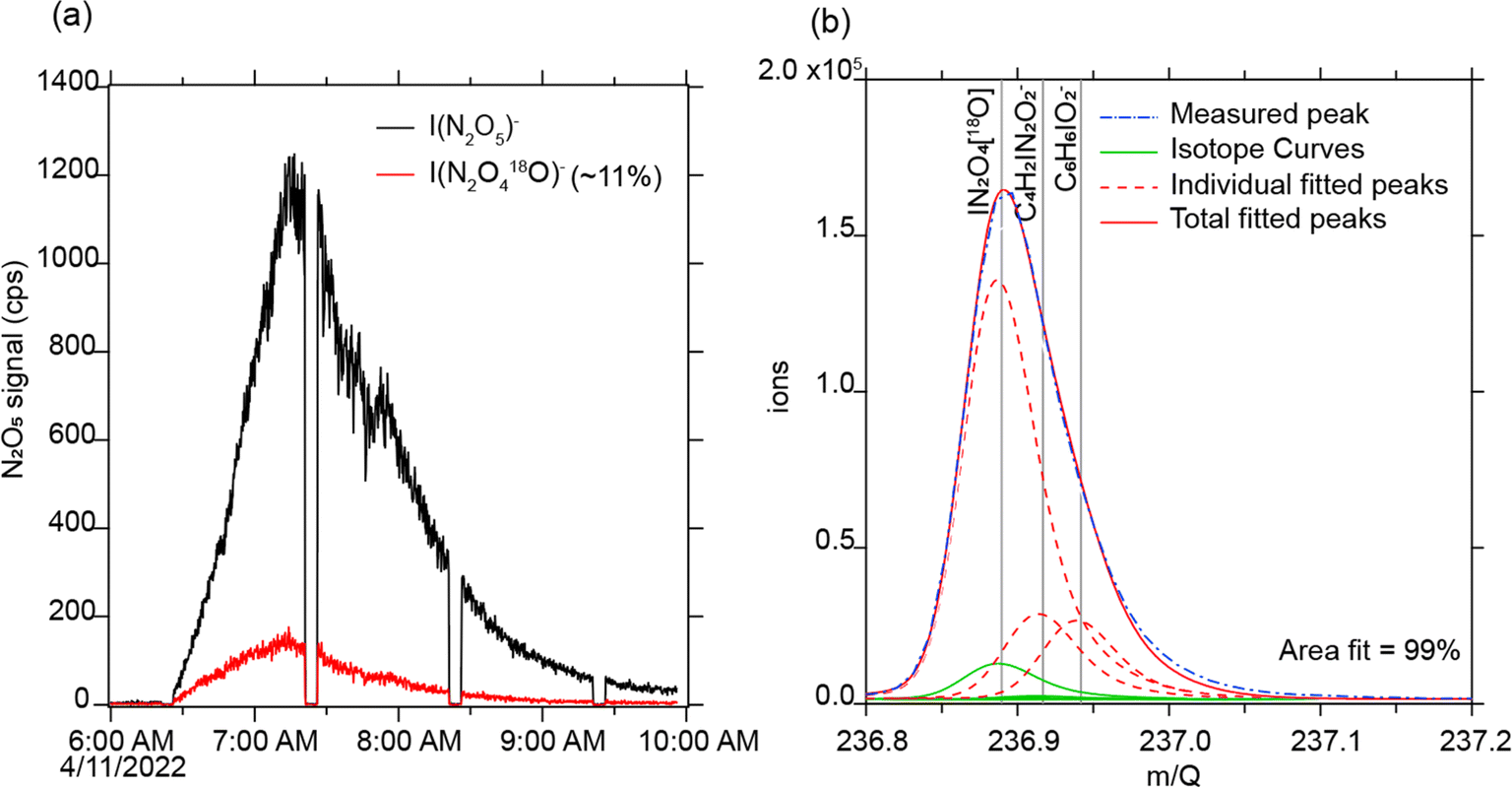 | ||
| Fig. 7 (a) Adding labeled O3 to the house enhanced 18O–N2O5. (b) The I-CIMS separates 18O-substituted N2O5 from adjacent ion peaks in the mass spectrum. Note that the timeseries includes periodic zero air measurements; signals decrease during this time. | ||
3.4 Smoke additions
Over the last few decades, wildfires have increased in frequency across the western United States, raising concerns over their influence on outdoor air quality and public health.29,49 Smoke is a complex mixture including both particulate matter and trace gases. While concentrations of particulate matter are typically lower indoors than outdoors during poor air quality events, indoor concentrations can still reach levels of concern.30,31 Further, the influx of outdoor air pollution to the built environment, either through door or window opening, mechanical ventilation, or leaks in the building envelope, may add new chemical compounds to the indoor chemical system and induce secondary chemical transformations. Residential wood burning also introduces smoke compounds in the gas or particle phase into the indoor environment. The extent to which wood smoke in buildings can impact human exposure to various chemical contaminants on timescales beyond the outdoor pollution event is poorly understood. For example, recent wildfires at the wildland–urban interface have raised concerns about smoke damage to indoor spaces and the efficacy of different mitigation approaches.50 Wildfire events are not the only sources of wood smoke to indoor environments: wood stoves and fireplaces can directly release smoke to the indoor environment,51 while simultaneously releasing smoke to the local area, potentially impacting other homes.The CASA study provided an opportunity to investigate how wood smoke impacts indoor chemistry. Specific questions of interest included:
• Where does smoke go in a house (i.e., deposition versus ventilation versus reactive chemistry), and how does it chemically transform on the timescales of air change rates?
• Do deposited smoke compounds undergo chemical reactions, and on what timescales?
• How does smoke interact with ozone? How does humidity impact smoke and resulting surface soiling?
Introducing fresh wood smoke to a house involves two challenges: first, safely and consistently making smoke that is chemically comparable to wood smoke, and second, introducing the smoke into the house in a reproducible manner. At CASA, we used a portable cocktail smoker (Breville, “the Smoking Gun”, BSM600SILUSC), loaded with Ponderosa pine woodchips to generate wood smoke. This smoke was then added either directly to the house as bursts of fresh smoke, or to an outdoor chamber where it was oxidized by ozone and then injected to the house as simulated aged smoke.
For fresh smoke, a fixed amount of wood chips (about 0.5 g per event) was added to the cocktail smoker inside the house, which then directly released smoke into the house near the kitchen. To simulate aged smoke, we first added large levels of O3 to an approximately 1 m3 Teflon chamber located outside of the house between the garage and the house (chamber mixing ratios on order of 20 ppm O3). Smoke was then added to the chamber by processing about 6 g of woodchips. The O3 and smoke-filled chamber was then left undisturbed with no air flow in or out for 45 minutes. To inject the aged smoke into the house, a second valved port on the chamber was connected to a short length of copper tubing that originated from just inside the house while being routed through a fixed partition which, during CASA, had replaced the house's west-side exterior door. Having connected a zero air (Environics, Series 7000) source to the chamber via the tubing previously used for O3, valves within the inlet and outlet ports were opened while flowing air into the chamber at a rate of about 20 Lpm for 40 minutes. A fan located inside the house, next to the inlet from the smoke chamber, was remotely turned on during the addition to increase mixing, and concentrations of particulate matter and other smoke markers immediately increased. After 30–40 minutes, particulate matter concentrations in the house leveled off, indicating that the bulk of the aged smoke had entered the house. Despite the high levels of ozone used in the chamber, ozone did not increase in the house during injections of aged smoke. Smoke experiments took place in the latter part of the CASA study to ensure that any changes to the background did not influence other experiments. As discussed by Li et al.,52 we found that smoke VOCs partitioned to surfaces of the house and were released back into the house over hours to months timescales, which are longer than the house's air change rate timescale.
Peak indoor PM2.5 concentrations were 120 ± 40 μg m−3 for fresh smoke additions and 150 ± 50 μg m−3 for simulated aged smoke additions. The fresh smoke PM2.5 was dominated by submicron particles, while the aged smoke PM2.5 was primarily composed of supermicron particles, likely due to the aging process in the ozone chamber. On low RH days, fresh smoke injections led to higher concentrations of brown carbon compared to simulated aged smoke, while concentrations of black carbon were similar between fresh and aged smoke additions (Fig. 8).
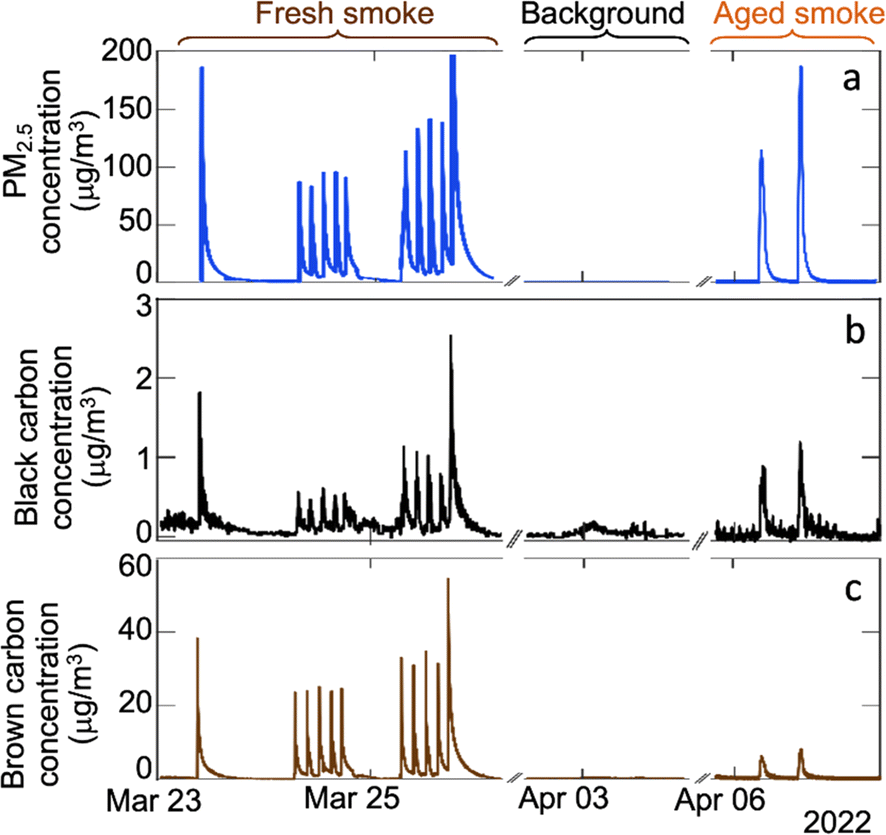 | ||
| Fig. 8 (a) Indoor PM2.5 concentrations obtained from combined SMPS (electrical mobility diameter = 24.4–723.4 nm) and APS (aerodynamic diameter = 0.7–2.5 μm) measurements assuming a density of 1.2 g cm−3, (b) black carbon, and (c) brown carbon concentrations from aethalometer measurements from fresh smoke additions (23–25 March), house background (2–3 April) and simulated aged smoke additions (6–7 April). Note that the y axes have different scales between panels. The EPA's optimized noise-reduction algorithm was applied; MAC values for the analysis were 24.1, 19.1, 17.0, 14.1, and 10.1 at wavelengths of 375, 470, 528, 625, and 880 nm, respectively.53 Aethalometer data was not analyzed during high RH days because humidity causes significant interference in those measurements. | ||
The bulk organic chemical composition of the aerosol produced from the cocktail smoker is similar to that observed in western wildfires. Li et al.52 compared the mass spectrum of the submicron non-refractory smoke aerosol directly output from the cocktail smoker to wildfire smoke measured during the WE-CAN 2018 study, in which we previously collected ambient aerosol data on an aircraft flying through wildfire plumes in the Western United States.54 The aerosol mass spectra are very similar (r2 = 0.90),52 with the key exceptions being in particulate nitrate. The relatively low temperature of the cocktail smoker is designed to maximize smoke generation and minimize flaming, which results in negligible NOx and HONO, and hence a lower generation of particulate nitrate. However, submicron aerosol in wildfire smoke is dominantly (85–96%) organic, and the cocktail smoker appears to be an adequate generation device for simulated wildfire smoke. In terms of gas-phase emissions, the emissions ratios for key VOCs (formaldehyde, benzene, toluene, formic acid, acetic acid, acetone, furan) relative to CO agreed to within an order of magnitude of the western wildfire smoke plumes sampled during WE-CAN (Fig. S4†).55 However, emissions ratios for pyrrole were an order of magnitude larger, and emissions of acetonitrile were an order of magnitude lower, for CASA than at WE-CAN. We thus take the smoke generated by the cocktail smoker to be a useful simulation of wildfire smoke for this project.
Smoke components deposit on indoor surfaces, with the deposited particles showing surprisingly complex morphology and composition. We investigated this deposition and subsequent chemistry through off-line microspectroscopic analysis of window glass samples that were placed both vertically and horizontally in the kitchen and living room area for 24 hours before, during, and after a smoke injection experiment. Upon collection, samples were sealed and subsequently analyzed using atomic force microscopy-infrared (AFM-IR) spectroscopy (Fig. 9). Two different types of particle morphologies were identified on exposed glass surfaces: (1) aggregate-like (Fig. 9a) and (2) rounded (Fig. 9b). The aggregate-like smoke particles were much larger, taller, and rougher than the rounded particles – similar to ash. Aggregate-like particles were up to 2.6 μm in height, while rounded particles were <100 nm in height.
 | ||
| Fig. 9 AFM 2D height images of smoke particle deposition on glass in two different regions on the surface of two different particle morphologies – (a) aggregate-like particles and (b) rounded particles. The left side represents a height image for a larger region, while the right side is a zoomed-in height image. (c) and (d) are the corresponding deflection images along with AFM-IR point spectra. Each spectrum was obtained by co-averaging seven spectra on the same point as shown by the colored dots in the deflection image and the color-coded spectra. All infrared spectra were collected at 100% power in tapping mode. AFM images were processed using the Gwyddion software; AFM-IR spectra were plotted in Origin. | ||
The two particle types (aggregate-like vs. rounded particles) are not only morphologically distinct, but also chemically distinct. As shown in the deflection images for aggregate-like and rounded particles, point spectra were collected in different regions of each of the particle types. For example, the blue and orange spectra shown in Fig. 9 have broad peaks around 1166 cm−1, which can be associated with C–O stretching found in esters, C–O–C stretching, or potentially sulfate derivatives.56 Spectra shown in blue and green also have a distinct peak at 1420 cm−1, consistent with aliphatic carbon chains from sugars in the wood. The spectrum shown in orange also has a very distinctive peak centered around 1784 cm−1 associated with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching motion of esters. The spectrum shown in green also has a peak above 1700 cm−1 associated with protonated CO in protonated carboxylic acids, whereas the two peaks at 1500 and 1450 cm−1 are indicative of carboxylate stretching motions. The three different observed spectra on a single aggregate thus clearly depict the complexity of these samples and provide evidence for the presence of multiple functional groups within a single aggregate-like particle. In contrast, rounded smoke particles exhibit more consistent spectra with broad bands ranging from 1000 cm−1 to 1300 cm−1, indicative of C–C and O–H functional groups that are commonly present in biomass burning aerosol, including levoglucosan or catechol.57,58 Peaks at 1604 cm−1, 1620 cm−1 and 1674 cm−1 suggest the presence of aromatic, unsaturated CC, and aldehyde functional groups, respectively.56,58–60 Although more analysis is necessary to fully characterize these smoke-deposited aerosols, these data provide microscopic evidence for the deposition of smoke particles on window glass surfaces that vary in not only size and morphology, but also in composition.
O stretching motion of esters. The spectrum shown in green also has a peak above 1700 cm−1 associated with protonated CO in protonated carboxylic acids, whereas the two peaks at 1500 and 1450 cm−1 are indicative of carboxylate stretching motions. The three different observed spectra on a single aggregate thus clearly depict the complexity of these samples and provide evidence for the presence of multiple functional groups within a single aggregate-like particle. In contrast, rounded smoke particles exhibit more consistent spectra with broad bands ranging from 1000 cm−1 to 1300 cm−1, indicative of C–C and O–H functional groups that are commonly present in biomass burning aerosol, including levoglucosan or catechol.57,58 Peaks at 1604 cm−1, 1620 cm−1 and 1674 cm−1 suggest the presence of aromatic, unsaturated CC, and aldehyde functional groups, respectively.56,58–60 Although more analysis is necessary to fully characterize these smoke-deposited aerosols, these data provide microscopic evidence for the deposition of smoke particles on window glass surfaces that vary in not only size and morphology, but also in composition.
3.5 Air and surface cleaning
CASA included both surface cleaning (vacuuming, dusting, mopping) and use of portable air cleaners. Cleaning includes both the addition of commercial or homemade products with the aim of removing soluble or mobile contaminants, and the direct removal of dust and other potentially harmful components present in an indoor space. Cleaning products are well-established to release inorganic and organic compounds into indoor air,27,35,64 leave residue on indoor surfaces,65 and have unintended effects on indoor chemistry. For example, the addition of bleach solutions can induce multiphase chemistry,35,64,66 while the addition of hydrogen peroxide can induce formation of hydroxyl radicals and other oxidants.67,68 Persistent organic pollutants and several other chemical contaminants are well-established to adsorb and accumulate on dust surfaces,69 meaning that dust removal is a potentially effective tool to reduce exposure to harmful contaminants – not to mention allergens, bioparticles, and other indoor solid-phase pollutants. While most cleaning is focused on surfaces, air cleaning is growing in interest, particularly in light of the broad recognition of airborne pathogens during the COVID-19 pandemic. Air cleaners generally use physical or chemical techniques to remove air pollutants. Physical approaches include filtration or removal by other adsorptive substances to physically extract particulate matter or volatile organic compounds from air. Chemical approaches to air cleaning include the use of ultraviolet light, photocatalytic oxidation, ions, or oxidants such as hydrogen peroxide or hypochlorous acid to destroy pathogens, induce deposition of particulate matter, or chemically transform other chemical air contaminants. While use of filters is well-established in the scientific literature, chemical air cleaning techniques are less well understood and potentially subject to unintended chemical consequences.23 Surface cleaning at CASA was limited to two specific events – one just prior to the first smoke additions on 21 March, and one at the end of the smoke additions on 7 April. These two surface cleaning events included manually vacuuming the floors and dusting all horizontal surfaces on the first floor of the house, shortly followed by mopping and surface cleaning with an aqueous cleaning mixture. The aqueous mixture was based on the Red Cross recommendation for cleaning after wildfire smoke events, and included a scented cleaning product mixed to manufacturer instructions, along with trisodium phosphate (4 tbsp of trisodium phosphate, 1 cup of cleaner, 1 gallon of water). We collected dust and other residue from the vacuum cleaner (116 g prior to smoke events; 35 g after smoke events), which were stored in a refrigerator or freezer for off-line analyses.To assess the efficacy of various air cleaners, four consumer-grade portable air cleaners were procured from different manufacturers with prices ranging between $500 and $1000. These air cleaners were chosen to cover a broad spectrum of physical and cleaning technologies, including dual polarity ions, photocatalytic oxidation (PCO), UV, electrostatic charge, HEPA filter, and activated carbon filter. Additionally, two custom-built Corsi-Rosenthal boxes70 were tested, one with MERV-13 filters and the other with combined activated carbon and MERV-8 filters. All air cleaners were situated in the dining and kitchen area during operation.
These cleaning events allowed us to investigate:
• Can surface cleaning affect indoor air before and after simulated wildfire smoke additions?
• How effective are surface versus air cleaning approaches to cleaning indoor air?
• Does the indoor dust and microbial composition change before and after wildfire smoke events?
• How effective are common portable air cleaners at removing particulate matter and volatile organic compounds?
• Do portable air cleaners induce secondary chemistry with observable effects on indoor air composition?
Results from these experiments will be discussed in future manuscripts
3.6 CASA cocktail
To investigate how the fate of organic compounds varies with molecular properties, we injected a gaseous mixture of compounds covering a range of volatilities and organic functional groups into the house. The injection represented a rapid pulse (e.g., from sudden infiltration of outdoor air or short-term emission from indoor sources). This mixture – the ‘CASA cocktail’ – comprised 1-hexene, 1-octene, isoprene, α-pinene, toluene-d8, acetone, 2-pentanone, chlorobenzene, o-xylene, 2-heptanone, and furfural; these compounds were selected to represent specific chemicals or structural features of interest in the indoor environment. The experiment was performed five times: during the first four trials, 0.34–0.55 g of each species was injected to the house, and during the fifth trial, this was diluted by a factor of 10 such that 0.034–0.055 g of each was added. To ensure the mixture was injected in the gas-phase, we heated the mixture in a water bath on a hot plate at 90 °C, passing zero air through the mixture in a bubbler at 10 Lpm and into a port on the porch door. The mixture took, on average, eight minutes to evaporate, during which time a box fan near the inlet in the dining room was remotely turned on. Following addition of the CASA cocktail to the house, no personnel entered the house, nor were other activities conducted for at least five hours. For half of the CASA cocktail experiments, ozone was injected at the six-hour mark, while for the other half of the experiments, the CASA cocktail was left to decay over twelve uninterrupted hours with no ozone addition.The CASA cocktail experiment allowed us to investigate the following science questions, among others:
• How does the temporal behavior and fate of indoor pollutants vary with their molecular properties?
• How do differences in volatility and the presence of functional groups affect indoor VOC sorption on different building materials?
During the CASA cocktail experiment, we deployed thin, porous films of varying building materials on a horizontal glass slide in the dining room collection area. These materials included clay (kaolinite), commercial zeolite, cement (CaO + CaCO3 mixture) and rutile (TiO2). These porous films were exposed to the CASA cocktail and then sealed and stored refrigerated prior to shipping to UC San Diego. Any organic compounds that remained adsorbed on or within the thin film were extracted with acetone and analyzed by GC-MS and compared to a glass slide with no porous film coatings (Fig. 10a). The instrument was operated using a TG-WAXMS column (30 m × 0.25 mm i.d. × 0.25 μm film thickness, Thermo Scientific) with the injection temperature of 230 °C and the following oven temperature program: 40 °C for 3 min, then gradually increased to 230 °C at a rate of 15 °C min−1, and the final temperature was held for 10 min. The identification of adsorbed surface products was based on the NIST electron ionization mass spectral library. Most interestingly, 1-octene adsorbed strongly on several – but not all – of the surfaces. Glass showed little to no surface product, whereas kaolinite showed the highest adsorption of 1-octene and the formation of oxidized products of the cocktail gases including oxidized α-pinene. Other reactive surfaces included cement and titanium dioxide. This outcome suggests that CASA cocktail precursor compounds did transform on indoor-relevant surfaces as some of the surface products were unique from the gas-phase precursors and varied according to surface type.
 | ||
| Fig. 10 (a) Extracted organic compounds from different surfaces deployed during the CASA cocktail experiment as detected by GC-MS. (b) Attenuated Total Reflection Fourier Transform Infrared spectroscopic data of organic compounds on a TiO2 surface collected during the CASA cocktail experiment. | ||
In addition, a TiO2 thin film deposited on an Attenuated Total Reflection element (ATR) was analyzed with FTIR spectroscopy (Fig. 10b). The ATR data show vibrational frequencies consistent with alkenes, aromatics, and carbonyls. Taken together, the ATR and GC-MS data demonstrate that different building materials retain and potentially react differently with gas-phase species. For some materials, there is no adsorption or product formation, while in others, adsorption and product formation are evident.
3.7 Product addition
Where the CASA cocktail experiment focused on a curated suite of VOCs, commercial products include complex mixtures that can be released in the indoor environment through spray cans. The fate of such commercial products and the mechanisms by which they can contribute to human exposure are poorly understood. Spray cans are typically expected to release large particles that rapidly deposit to surfaces. Insecticides are lower-volatility compounds; however, these lower-volatility components could enter the gas-phase and partition either to surfaces or to preexisting indoor aerosol particles. We sprayed a commercially available flying insect pesticide containing permethrin and tetramethrin as active ingredients in the living room of the house for 25 seconds in accordance with manufacturer instructions. This spray time corresponded to a product mass loss of about 30 g from the bottle. A box fan in the living room that directed air towards the living/dining room area was turned on two minutes prior to the start of the spray, and turned off four minutes after the event. According to the manufacturer, the insecticide contained 0.15% permethrin, 0.15% tetramethrin, and 99.70% ‘other ingredients’ including isobutane, propane, hydrotreated light petroleum distillates, and light aromatic solvent naptha from petroleum.This insecticide addition allowed us to investigate:
• What is the chemical behavior of low-volatility consumer product sprays in indoor air and surfaces?
• What is the timescale of the different components in the insecticide spray in the indoor air?
Results from this experiment will be discussed in detail in a forthcoming manuscript.
3.8 Acid/base addition
Surfaces play a major role in indoor chemistry, in part because sorption to abundant indoor surfaces prolongs the indoor residence time of compounds, providing more time for reactions to take place. There is evidence that sufficient surface-bound water can exist on soiled indoor surfaces to enable the uptake of water-soluble gases71 and studies of surface-bound tobacco smoke suggest that changes in surface pH can alter the gas-surface partitioning of indoor contaminants.72,73 However, the degree to which variations in pH in surface water reservoirs affects most indoor species is poorly understood. To approach this topic, we asked:• How does the intentional introduction of a weak acid gas (CO2) or base gas (NH3) influence the dynamic concentrations of water-soluble organic compounds, target organic acids and bases and other target species?
• How do the effects of acid-base additions vary with RH?
In these experiments, CO2 or NH3 was injected into the supply air flow of the mechanical ventilation system for about 30 minutes (500 g min−1 CO2; about 40 mg min−1 NH3) with the goal of reaching mixing ratios that were high but realistic (on the order of 5000 ppm CO2 and 500 ppb NH3). They were then allowed to decay for at least 3 h. The process was repeated three to four more times. The first day involved injections at high RH, while the second day involved injections at background RH.
NH3 decayed faster than the air change rate due to surface uptake. We measured about 20% lower mixing ratios of NH3 at high RH, suggesting enhanced surface removal into sorbed water. We observed various gas-phase bases or acids increase following injection of CO2 or NH3, likely due to shifts in the pH of surface water reservoirs. High RH likely increased the surface reservoir of water, thereby enhancing the impact on more water-soluble aids and bases. Results will be presented in detail in forthcoming manuscripts.
3.9 Cooking
Cooking experiments investigated the following questions:• How are particles emitted from cooking in the kitchen transported throughout the house and how does the transport timescale compare to the air change rate?
• How do cooking emissions and resulting surface/particle/air contributions change between high and low RH conditions?
• Is the water uptake of cooking emissions different from that of smoke?
• How do cooking emissions change as particles and gases get transported throughout the house?
• How do cooking emissions interact with ozone?
The NZERTF used for CASA lacks any combustion-based appliances (e.g., natural gas). We used a two-burner electric cooktop at maximum setting and an electric air fryer for cooking experiments, which took place over a 3 day period from March 13 to 15. The first day of experiments was performed at high RH (60–80% on the first floor, slightly lower upstairs) and the other two days at low RH (30–50%). Eight cooking experiments were performed in these three days involving the same ingredients and preparation. Six of these were done on a frying pan while the other two were conducted with an air fryer. The cooking process consisted of heating up the pan or air fryer with canola oil (10 g for pan fryer), then sequentially adding vegetables, frozen potato products, and bacon; each food item was cooked for ten minutes. Specific details for all cooking events are in Table S1.† At the conclusion of each cooking event, the cook removed all food from the test house. Fig. 11 shows the average particle number size distributions measured during air frying and pan cooking, demonstrating that air frying released a larger amount of smaller aerosol particles compared to pan cooking for similar meal preparations and same ingredients.
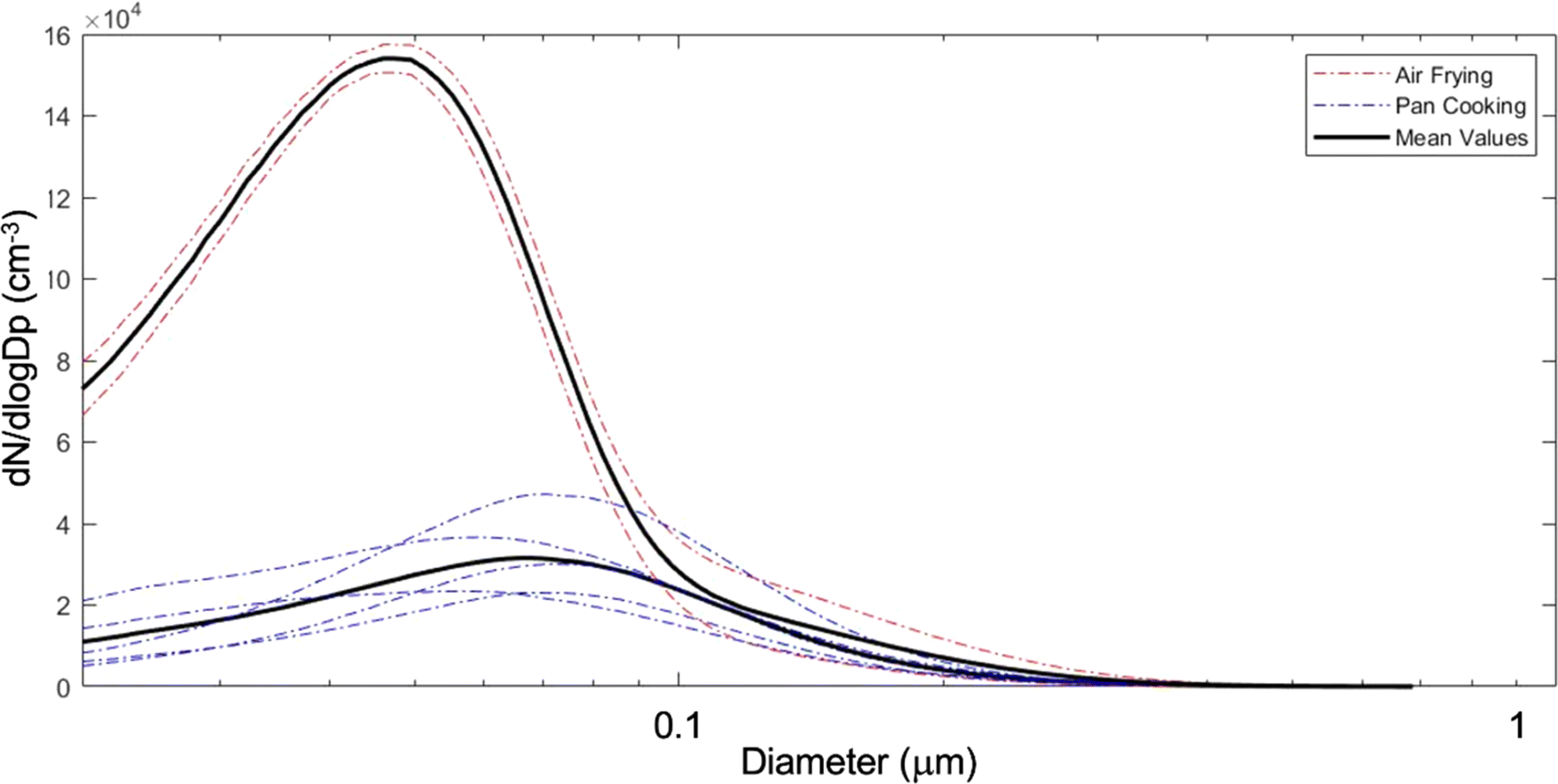 | ||
| Fig. 11 Average particle size distributions sampled in the dining room during all air frying and pan cooking events. Each dashed curve represents one average size distribution for one cooking event (air frying in red and pan frying in blue). Averages for air frying and pan cooking are represented by the dark lines. | ||
3.10 Additional experiments
CASA provided the opportunity for additional experiments to probe different aspects of indoor chemistry and indoor–outdoor interactions, including:• Microwaving popcorn, which may emit volatile organic compounds74,75
• Instrument testing and intercomparisons: For example, several instruments measured the same compounds with different approaches (e.g., formic acid was measured by a laser absorption system, iodide-CIMS, and PTR-MS), providing useful tests for instrument validation.
• Low-cost sensors for measuring key components of indoor air (e.g., PM, VOCs) were continually sampling throughout the campaign, providing insight into how such techniques can be used to characterize indoor air quality and chemistry.
• Residential laundry effects on air quality. As the NZERTF is used for long-term residential water quality studies, the dishwasher and laundry machines were automated to continue running (typically once per night). During CASA, we noticed elevated levels of chlorine-containing organic compounds that coincided with runs of the laundry or dishwasher, indicating an effect of residential water quality on indoor air quality (Fig. S5†).
3.11 Microbial analysis of dust samples
We collected floor dust samples from a vacuum cleaner twice during CASA, as described in Section 3.5. Microbial analysis of those samples enabled us to investigate:• What is the microbial composition in the test house during background periods?
• Can an intense smoke and ozone injection affect the indoor microbial composition in a house?
The collected dust allowed us to investigate microbial composition in the NZERTF. Bacteria in NZERTF dust (n = 3) were measured using next-generation DNA sequencing (details in methods in ESI Section S34†). Between 86064 and 97141 sequences per sample and 1059 to 1102 amplicon sequencing variants (ASVs) per sample were detected. The dominant phyla (i.e., 92% of reads) were Proteobacteria (36%), Cyanobacteria (25%), Actinobacteria (17%), Bacteroidetes (9%), and Firmicutes (4%) (ESI file†). The most abundant families were Oxalobacteraceae (7%), Sphingomonadaceae (5%), Cytophagaceae (4%), Microbacteriaceae (4%), and Xenococcaceae (3%). The genera Corynebacterium, Lactobacillus, Staphylococcus, and Streptococcus – which are human commensals commonly found in buildings76,77 – collectively made up approximately 2% of detected bacteria.
Fungi in dust samples were measured with qPCR and next-generation DNA sequencing, then compared to fungi in dust samples from occupied homes across the United States (n = 9).78 The study on occupied homes was approved by The Ohio State University Institutional Review Board (2019B0457). Sequencing of NZERTF dust yielded between 240025 and 276854 fungal reads per sample. Concentrations of fungi in test house dust were lower compared to occupied homes (Welch's t-test; p < 0.001) (Fig. 12). However, NZERTF dust samples were vacuumed from hardwood floor, whereas all samples from occupied homes with complete metadata (n = 8 of 9) had carpeted and/or mixed flooring. Between 203 and 217 ASVs were detected per NZERTF dust sample. Fungal Shannon diversity in NZERTF dust was lower than in occupied homes (Welch's t-test, p < 0.001) and community composition differed significantly (PERMANOVA; R2 = 0.093, p < 0.001)79 (Fig. 12). NZERTF dust was dominated by just four genera – Epicoccum (71%), Neoascochyta (5%), Cladosporium (3%), and Vishniacozyma (3%) (ESI file†). Both Epicoccum and Neoascochyta are plant pathogens.
 | ||
| Fig. 12 Comparison of fungi in floor dust samples of the NZERTF during CASA (n = 3) versus those from occupied homes across the United States (n = 9). Boxplots display (a) fungal concentration in spore equivalents mg−1 dust, and (b) Shannon diversity with significance of Welch's t-test indicated by asterisks as follows: *p < 0.05, **p < 0.01, ***p < 0.001. (c) Comparisons of fungal composition using principal coordinates analysis plot of Bray–Curtis dissimilarity with result of PERMANOVA displayed on plot. | ||
The microbial community present in the NZERTF dust was compositionally similar to reports in literature from occupied buildings.76,80,81 However, there were certain differences. Human-associated bacteria such as Corynbacterium, Lactobacillus, Streptococcus, and Staphylococcus comprised approximately 2% of reads in the NZERTF. In contrast, a previous study showed that dust from classroom floors contained at least 10% human-associated taxa.77 While these differences between the microbial community in the NZERTF compared to literature for occupied buildings are most likely driven by differences in human occupancy, it is also possible that HEPA air filtration82 and a higher cleaning-to-occupancy ratio contributed.
One NZERTF dust sample was analyzed following ozone/smoke exposure. In comparison with two pre-exposure dust samples, bacterial and fungal measures were similar albeit slightly lower, which suggests that short term ozone/smoke exposure does not substantially alter the microbial community of floor dust. However, next-generation DNA sequencing cannot distinguish between living and non-viable microorganisms; thus, additional testing would be necessary to fully understand impacts from ozone and smoke.
5 Conclusions
The CASA study represents a ‘lab-in-field’ approach to indoor chemistry, using a chemically comprehensive suite of measurements to capture gas, particle, and surface composition of a test house. While this approach shares similarities to the HOMEChem project, CASA emphasized chemical additions over everyday activities with the goal of improving fundamental understanding of the chemistry of the built environment. While initial studies are published41,52 and an array of papers are forthcoming, several themes have already emerged:(1) Introduction of both outdoor pollutants (ozone, NOx, smoke) and indoor pollutants (pesticides, cooking) induce changes in indoor surface and air chemistry.
(2) Environmental parameters, including RH, impact indoor air composition in significant ways.
(3) Seemingly small indoor changes through the use of commercial products or everyday activities can change the chemical composition of indoor air.
(4) The complex interplay of mechanical ventilation, human activities, movement of air through buildings, and the diversity of surfaces means that test houses can be useful for studying indoor chemistry, albeit with limitations in their representativeness in terms of both microbial and human occupation.
The CASA campaign, involving perturbations conducted in a highly instrumented and well-characterized test house, is designed to test and refine our foundational understanding of processes that control indoor chemistry, improving the ability to understand future challenges. Reducing exposures to chemical air contaminants requires an improved understanding of how outdoor and indoor air are chemically linked, and the fate of chemicals in the built environment.
Disclaimer
Certain equipment, instruments, software, or materials are identified in this paper in order to specify the experimental procedure adequately. Such identification is not intended to imply recommendation or endorsement of any product or service by NIST, nor is it intended to imply that the materials or equipment identified are necessarily the best available for the purpose. The policy of NIST is to use the International System of Units in all publications. In this document, however, some units are presented in the system prevalent in the relevant discipline.Data availability
Substantial CASA data are available at https://osf.io/hymek/. Additional data are available upon request, and through emerging publications.Conflicts of interest
The authors declare no conflict of interest in this work.Acknowledgements
We thank the Alfred P. Sloan Foundation program (G-2020-13929 (DKF and MEV), G-2019-11404 (JA), G-2020-13937 (BJT), G-2020-12675 (VHG), G-2019-12365 (VHG), G-2018-11031 (RO), G-2020-13953 (RO), G-2018-11096 (JK)) for funding. We thank the entire CASA science team, including Dan Greb (NIST), Rileigh Robertson (NIST), Lisa Ng (NIST), Steve Emmerich (NIST), Art Ellison (NIST), Anna Karion (NIST), John Pettibone (NIST), Lauren Garofalo (Colorado State University), Katelyn Rediger (Colorado State University), Amy Hrdina (MIT), Amanda Gao (MIT), Matthew Goss (MIT), Erik Helstrom (MIT), Krystal Pollitt (Yale University), Elizabeth Lin (Yale University), Emily Heery (William and Mary), and Douglas Collins (Bucknell University).References
- N. E. Klepeis, W. C. Nelson, W. R. Ott, J. P. Robinson, A. M. Tsang, P. Switzer, J. V. Behar, S. C. Hern and W. H. Engelmann, J. Exposure Anal. Environ. Epidemiol., 2001, 11, 231–252 CrossRef CAS.
- A. L. Hodshire, E. Carter, J. M. Mattila, V. Ilacqua, J. Zambrana, J. P. D. Abbatt, A. Abeleira, C. Arata, P. F. DeCarlo, A. H. Goldstein, L. H. Ruiz, M. E. Vance, C. Wang and D. K. Farmer, Environ. Sci. Technol., 2022, 56, 12148–12157 CrossRef CAS.
- J. Logue, T. McKone, M. Sherman and B. Singer, Indoor air, 2011, 21, 92–109 CrossRef CAS.
- M. Shiraiwa, N. Carslaw, D. J. Tobias, M. S. Waring, D. Rim, G. Morrison, P. S. J. Lakey, M. Kruza, M. von Domaros, B. E. Cummings and Y. Won, Environ. Sci.: Processes Impacts, 2019, 21, 1240–1254 RSC.
- J. P. D. Abbatt and C. Wang, Environ. Sci.: Processes Impacts, 2020, 22, 25–48 RSC.
- A. P. Ault, V. H. Grassian, N. Carslaw, D. B. Collins, H. Destaillats, D. J. Donaldson, D. K. Farmer, J. L. Jimenez, V. F. McNeill, G. C. Morrison, R. E. O'Brien, M. Shiraiwa, M. E. Vance, J. R. Wells and W. Xiong, Chem, 2020, 6, 3203–3218 CAS.
- E. National Academies of Sciences and Medicine, Why Indoor Chemistry Matters, The National Academies Press, Washington, DC, 2022 Search PubMed.
- W. Nazaroff and A. Goldstein, Indoor air, 2015, 25, 357–361 CrossRef CAS PubMed.
- D. K. Farmer, M. E. Vance, J. P. Abbatt, A. Abeleira, M. R. Alves, C. Arata, E. Boedicker, S. Bourne, F. Cardoso-Saldaña and R. Corsi, Environ. Sci.: Processes Impacts, 2019, 21, 1280–1300 RSC.
- J. M. Mattila, C. Arata, A. Abeleira, Y. Zhou, C. Wang, E. F. Katz, A. H. Goldstein, J. P. D. Abbatt, P. F. DeCarlo, M. E. Vance and D. K. Farmer, Environ. Sci. Technol., 2022, 56, 109–118 CrossRef CAS PubMed.
- D. J. Price, D. A. Day, D. Pagonis, H. Stark, L. B. Algrim, A. V. Handschy, S. Liu, J. E. Krechmer, S. L. Miller and J. F. Hunter, Environ. Sci. Technol., 2019, 53, 13053–13063 CrossRef CAS PubMed.
- Y. Liu, P. K. Misztal, J. Xiong, Y. Tian, C. Arata, R. J. Weber, W. W. Nazaroff and A. H. Goldstein, Indoor Air, 2019, 29, 630–644 CAS.
- W. R. Simpson, J. Mao, G. J. Fochesatto, K. S. Law, P. F. DeCarlo, J. Schmale, K. A. Pratt, S. R. Arnold, J. Stutz, J. E. Dibb, J. M. Creamean, R. J. Weber, B. J. Williams, B. Alexander, L. Hu, R. J. Yokelson, M. Shiraiwa, S. Decesari, C. Anastasio, B. D'Anna, R. C. Gilliam, A. Nenes, J. M. St. Clair, B. Trost, J. H. Flynn, J. Savarino, L. D. Conner, N. Kettle, K. M. Heeringa, S. Albertin, A. Baccarini, B. Barret, M. A. Battaglia, S. Bekki, T. J. Brado, N. Brett, D. Brus, J. R. Campbell, M. Cesler-Maloney, S. Cooperdock, K. Cysneiros de Carvalho, H. Delbarre, P. J. DeMott, C. J. S. Dennehy, E. Dieudonné, K. K. Dingilian, A. Donateo, K. M. Doulgeris, K. C. Edwards, K. Fahey, T. Fang, F. Guo, L. M. D. Heinlein, A. L. Holen, D. Huff, A. Ijaz, S. Johnson, S. Kapur, D. T. Ketcherside, E. Levin, E. Lill, A. R. Moon, T. Onishi, G. Pappaccogli, R. Perkins, R. Pohorsky, J.-C. Raut, F. Ravetta, T. Roberts, E. S. Robinson, F. Scoto, V. Selimovic, M. O. Sunday, B. Temime-Roussel, X. Tian, J. Wu and Y. Yang, ACS ES&T Air, 2024, 1, 200–222 Search PubMed.
- Y. Liu, P. K. Misztal, C. Arata, C. J. Weschler, W. W. Nazaroff and A. H. Goldstein, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2018140118 CrossRef CAS.
- C. Wang, D. B. Collins, C. Arata, A. H. Goldstein, J. M. Mattila, D. K. Farmer, L. Ampollini, P. F. DeCarlo, A. Novoselac and M. E. Vance, Sci. Adv., 2020, 6, eaay8973 CrossRef CAS.
- D. B. Collins, R. F. Hems, S. Zhou, C. Wang, E. Grignon, M. Alavy, J. A. Siegel and J. P. Abbatt, Environ. Sci. Technol., 2018, 52, 12419–12427 CrossRef CAS PubMed.
- R. E. O'Brien, Y. Li, K. J. Kiland, E. F. Katz, V. W. Or, E. Legaard, E. Q. Walhout, C. Thrasher, V. H. Grassian and P. F. DeCarlo, Environ. Sci.: Processes Impacts, 2021, 23, 559–568 RSC.
- P. F. DeCarlo, A. M. Avery and M. S. Waring, Sci. Adv., 2018, 4, eaap8368 CrossRef PubMed.
- R. Sheu, C. Stönner, J. C. Ditto, T. Klüpfel, J. Williams and D. R. Gentner, Sci. Adv., 2020, 6, eaay4109 CrossRef CAS.
- D. M. Lunderberg, K. Kristensen, Y. Tian, C. Arata, P. K. Misztal, Y. Liu, N. Kreisberg, E. F. Katz, P. F. DeCarlo, S. Patel, M. E. Vance, W. W. Nazaroff and A. H. Goldstein, Environ. Sci. Technol., 2020, 54, 6751–6760 CrossRef CAS PubMed.
- P. S. Lakey, Y. Won, D. Shaw, F. F. Østerstrøm, J. Mattila, E. Reidy, B. Bottorff, C. Rosales, C. Wang and L. Ampollini, Commun. Chem., 2021, 4, 1–7 CrossRef.
- P. S. Lakey, A. Wisthaler, T. Berkemeier, T. Mikoviny, U. Pöschl and M. Shiraiwa, Indoor air, 2017, 27, 816–828 CrossRef CAS PubMed.
- D. B. Collins and D. K. Farmer, Environ. Sci. Technol., 2021, 55, 12172–12179 CrossRef CAS PubMed.
- B. E. Cummings, A. M. Avery, P. F. DeCarlo and M. S. Waring, Environ. Sci. Technol., 2021, 55, 9000–9011 CrossRef CAS PubMed.
- M. A. Pothier, E. Boedicker, J. R. Pierce, M. Vance and D. K. Farmer, Environ. Sci.: Processes Impacts, 2023, 25, 314–325 RSC.
- D. K. Farmer, Anal. Chem., 2019, 91, 3761–3767 CrossRef CAS.
- C. Arata, P. K. Misztal, Y. Tian, D. M. Lunderberg, K. Kristensen, A. Novoselac, M. E. Vance, D. K. Farmer, W. W. Nazaroff and A. H. Goldstein, Indoor air, 2021, 31, 2099–2117 CrossRef CAS PubMed.
- S. Patel, S. Sankhyan, E. K. Boedicker, P. F. DeCarlo, D. K. Farmer, A. H. Goldstein, E. F. Katz, W. W. Nazaroff, Y. Tian, J. Vanhanen and M. E. Vance, Environ. Sci. Technol., 2020, 54, 7107–7116 CrossRef CAS.
- M. Burke, A. Driscoll, S. Heft-Neal, J. Xue, J. Burney and M. Wara, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2011048118 CrossRef CAS PubMed.
- Y. Liang, D. Sengupta, M. J. Campmier, D. M. Lunderberg, J. S. Apte and A. H. Goldstein, Proc. Natl. Acad. Sci. USA, 2021, 118, e2106478118 CrossRef CAS.
- K. O’Dell, B. Ford, J. Burkhardt, S. Magzamen, S. C. Anenberg, J. Bayham, E. V. Fischer and J. R. Pierce, Environmental Research: Health, 2022, 1, 015003 Search PubMed.
- The Net Zero Energy Residential Test Facility, https://www.nist.gov/el/net-zero-energy-residential-test-facility accessed June, 2022.
- O. Farhad and B. Steven, 2013, DOI:10.6028/NIST.TN.1817, https://edu.rsc.org/download?ac=522463.
- M. Davis, W. Healy, M. Boyd, L. Ng, V. Payne, H. Skye and T. Ullah, Monitoring Techniques for the Net-Zero Energy Residential Test Facility, Report NIST TN 1854, National Institute of Standards and Technology, Gaithersburg, MD, 2014 Search PubMed.
- J. M. Mattila, P. S. Lakey, M. Shiraiwa, C. Wang, J. P. Abbatt, C. Arata, A. H. Goldstein, L. Ampollini, E. F. Katz and P. F. DeCarlo, Environ. Sci. Technol., 2020, 54, 1730–1739 CrossRef CAS.
- R. J. Weber, D. Orsini, Y. Daun, Y. N. Lee, P. J. Klotz and F. Brechtel, Aerosol Sci. Technol., 2001, 35, 718–727 CrossRef CAS.
- D. A. Orsini, Y. Ma, A. Sullivan, B. Sierau, K. Baumann and R. J. Weber, Atmos. Environ., 2003, 37, 1243–1259 CrossRef CAS.
- A. P. Sullivan, R. J. Weber, A. L. Clements, J. R. Turner, M. S. Bae and J. J. Schauer, Geophys. Res. Lett., 2004, 31, L13105 CrossRef.
- G. Isaacman, N. M. Kreisberg, L. D. Yee, D. R. Worton, A. W. H. Chan, J. A. Moss, S. V. Hering and A. H. Goldstein, Atmos. Meas. Tech., 2014, 7, 4417–4429 CrossRef.
- P. F. DeCarlo, J. R. Kimmel, A. Trimborn, M. J. Northway, J. T. Jayne, A. C. Aiken, M. Gonin, K. Fuhrer, T. Horvath, K. S. Docherty, D. R. Worsnop and J. L. Jimenez, Anal. Chem., 2006, 78, 8281–8289 CrossRef CAS.
- M. F. Link, J. Li, J. C. Ditto, H. Huynh, J. Yu, S. M. Zimmerman, K. L. Rediger, A. Shore, J. P. D. Abbatt, L. A. Garofalo, D. K. Farmer and D. Poppendieck, Environ. Sci. Technol., 2023, 57, 16446–16455 CrossRef CAS PubMed.
- D. M. Lunderberg, K. Kristensen, Y. Liu, P. K. Misztal, Y. Tian, C. Arata, R. Wernis, N. Kreisberg, W. W. Nazaroff and A. H. Goldstein, Environ. Sci. Technol., 2019, 53, 7337–7346 CrossRef CAS PubMed.
- S. M. Duncan, S. Tomaz, G. Morrison, M. Webb, J. M. Atkin, J. D. Surratt and B. J. Turpin, Environ. Sci. Technol., 2019, 53, 1812–1821 CrossRef CAS PubMed.
- H. Schwartz-Narbonne, J. P. D. Abbatt, P. F. DeCarlo, D. K. Farmer, J. M. Mattila, C. Wang, D. J. Donaldson and J. A. Siegel, Environ. Sci. Technol., 2021, 55, 10987–10993 CrossRef CAS PubMed.
- P. S. J. Lakey, A. Zuend, G. C. Morrison, T. Berkemeier, J. Wilson, C. Arata, A. H. Goldstein, K. R. Wilson, N. Wang, J. Williams, J. P. D. Abbatt and M. Shiraiwa, Environ. sci. Atmos., 2023, 3, 49–64 CAS.
- S. Zhou, C. J. Young, T. C. VandenBoer and T. F. Kahan, Environ. Sci.: Processes Impacts, 2019, 21, 1374–1383 RSC.
- W. W. Nazaroff and C. J. Weschler, Indoor Air, 2022, 32, e12942 CAS.
- H. F. Hubbard, B. K. Coleman, G. Sarwar and R. L. Corsi, Indoor Air, 2005, 15, 432–444 CrossRef CAS.
- C. Black, Y. Tesfaigzi, J. A. Bassein and L. A. Miller, Environ. Toxicol. Pharmacol., 2017, 55, 186–195 CrossRef CAS.
- J. M. Silberstein, L. E. Mael, C. R. Frischmon, E. S. Rieves, E. R. Coffey, T. Das, W. Dresser, A. C. Hatch, J. Nath, H. O. Pliszka, C. E. Reid, M. E. Vance, C. Wiedinmyer, J. A. De Gouw and M. P. Hannigan, Air Qual., Atmos. Health, 2023, 16, 1839–1850 CrossRef CAS.
- T. Salthammer, T. Schripp, S. Wientzek and M. Wensing, Chemosphere, 2014, 103, 205–211 CrossRef CAS PubMed.
- J. Li, M. F. Link, S. Pandit, M. H. Webb, K. J. Mayer, L. A. Garofalo, K. L. Rediger, D. G. Poppendieck, S. M. Zimmerman, M. E. Vance, V. H. Grassian, G. C. Morrison, B. J. Turpin and D. K. Farmer, Sci. Adv., 2023, 9, eadh8263 CrossRef CAS PubMed.
- S. Sankhyan, S. Patel, E. F. Katz, P. F. DeCarlo, D. K. Farmer, W. W. Nazaroff and M. E. Vance, Environ. Sci.: Processes Impacts, 2021, 23, 1476–1487 RSC.
- L. A. Garofalo, M. A. Pothier, E. J. Levin, T. Campos, S. M. Kreidenweis and D. K. Farmer, ACS Earth Space Chem., 2019, 3, 1237–1247 CrossRef CAS.
- W. Permar, Q. Wang, V. Selimovic, C. Wielgasz, R. J. Yokelson, R. S. Hornbrook, A. J. Hills, E. C. Apel, I.-T. Ku, Y. Zhou, B. C. Sive, A. P. Sullivan, J. L. Collett Jr, T. L. Campos, B. B. Palm, Q. Peng, J. A. Thornton, L. A. Garofalo, D. K. Farmer, S. M. Kreidenweis, E. J. T. Levin, P. J. DeMott, F. Flocke, E. V. Fischer and L. Hu, J. Geophys. Res.: Atmos., 2021, 126, e2020JD033838 CrossRef CAS.
- O. Popovicheva, A. Ivanov and M. Vojtisek, Atmosphere, 2020, 11, 319 CrossRef CAS.
- B. R. T. Simoneit, J. J. Schauer, C. G. Nolte, D. R. Oros, V. O. Elias, M. P. Fraser, W. F. Rogge and G. R. Cass, Atmos. Environ., 1999, 33, 173–182 CrossRef CAS.
- Z. Finewax, J. A. de Gouw and P. J. Ziemann, Environ. Sci. Technol., 2018, 52, 1981–1989 CrossRef CAS.
- R. Lindenmaier, S. D. Williams, R. L. Sams and T. J. Johnson, J. Phys. Chem., 2017, 121, 1195–1212 CrossRef CAS.
- T. J. Johnson, R. L. Sams, L. T. M. Profeta, S. K. Akagi, I. R. Burling, R. J. Yokelson and S. D. Williams, J. Phys. Chem., 2013, 117, 4096–4107 CrossRef CAS.
- S. J. Brey and E. V. Fischer, Environ. Sci. Technol., 2016, 50, 1288–1294 CrossRef CAS.
- P. S. Rickly, M. M. Coggon, K. C. Aikin, R. J. Alvarez, II, S. Baidar, J. B. Gilman, G. I. Gkatzelis, C. Harkins, J. He, A. Lamplugh, A. O. Langford, B. C. McDonald, J. Peischl, M. A. Robinson, A. W. Rollins, R. H. Schwantes, C. J. Senff, C. Warneke and S. S. Brown, Environ. Sci. Technol., 2023, 57, 1257–1267 CrossRef CAS.
- X. Tang, N. R. González, M. L. Russell, R. L. Maddalena, L. A. Gundel and H. Destaillats, Environ. Res., 2021, 198, 110462 CrossRef CAS PubMed.
- J. Wong, N. Carslaw, R. Zhao, S. Zhou and J. Abbatt, Indoor Air, 2017, 27, 1082–1090 CrossRef CAS.
- R. O'Brien, E. Heery, C. Shirilla, S. Sankhyan, A. Fowler and M. E. Vance, ACS ES&T Air, 2024, 1, 129–138 Search PubMed.
- J. M. Mattila, C. Arata, C. Wang, E. F. Katz, A. Abeleira, Y. Zhou, S. Zhou, A. H. Goldstein, J. P. Abbatt and P. F. DeCarlo, Environ. Sci. Technol. Lett., 2020, 7, 795–801 CrossRef CAS.
- S. Zhou, Z. Liu, Z. Wang, C. J. Young, T. C. VandenBoer, B. B. Guo, J. Zhang, N. Carslaw and T. F. Kahan, Environ. Sci. Technol., 2020, 54, 15643–15651 CrossRef CAS PubMed.
- D. Poppendieck, H. Hubbard and R. L. Corsi, Environ. Sci. Technol. Lett., 2021, 8, 320–325 CrossRef CAS.
- L. Lucattini, G. Poma, A. Covaci, J. de Boer, M. H. Lamoree and P. E. Leonards, Chemosphere, 2018, 201, 466–482 CrossRef CAS PubMed.
- R. Dal Porto, M. N. Kunz, T. Pistochini, R. L. Corsi and C. D. Cappa, Aerosol Sci. Technol., 2022, 56, 564–572 CrossRef CAS.
- M. Webb, L. Cui, G. Morrison, K. Baumann, J. D. Surratt, Z. Zhang, J. Atkin and B. J. Turpin, Environ. Sci.: Processes Impacts, 2023, 25, 1031–1048 RSC.
- M. Sleiman, L. A. Gundel, J. F. Pankow, P. Jacob, B. C. Singer and H. Destaillats, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6576–6581 CrossRef CAS.
- M. Ongwandee and P. Sawanyapanich, Indoor Air, 2012, 22, 54–63 CrossRef CAS.
- R. G. Buttery, L. C. Ling and D. J. Stern, J. Agric. Food Chem., 1997, 45, 837–843 CrossRef CAS.
- J. A. Rosati, K. A. Krebs and X. Liu, Crit. Rev. Food Sci. Nutr., 2007, 47, 701–709 CrossRef CAS PubMed.
- S. W. Kembel, J. F. Meadow, T. K. O'Connor, G. Mhuireach, D. Northcutt, J. Kline, M. Moriyama, G. Z. Brown, B. J. M. Bohannan and J. L. Green, PLoS One, 2014, 9, e87093 CrossRef PubMed.
- R. I. Adams, A. C. Bateman, H. M. Bik and J. F. Meadow, Microbiome, 2015, 3, 49 CrossRef PubMed.
- N. Balasubrahmaniam, J. King, B. Hegarty and K. Dannemiller, Moving beyond species: Fungal function in house dust provides novel targets for potential indicators of mold growth in homes, 2024, in review, DOI:10.21203/rs.3.rs-4344165/v1, https://www.researchsquare.com/article/rs-4344165/v1.
- M. J. Anderson, Austral Ecol., 2001, 26, 32–46 Search PubMed.
- H. Rintala, M. Pitkäranta and M. Täubel, in Advances in Applied Microbiology, ed. A. I. Laskin, S. Sariaslani and G. M. Gadd, Academic Press, 2012, vol. 78, pp. 75–120 Search PubMed.
- J. R. Thompson, A. Argyraki, M. Bashton, L. Bramwell, M. Crown, A. S. Hursthouse, K. Jabeen, P. Marinho Reis, A. Namdeo, A. Nelson, D. A. Pearce, S. Potgieter-Vermaak, P. E. Rasmussen, J. Wragg and J. A. Entwistle, Front. Environ. Sci., 2021, 9, 754657 CrossRef.
- K. Hashimoto and Y. Kawakami, Biocontrol Sci., 2018, 23, 215–221 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4em00209a |
| ‡ Now at: Department of Energy, Environmental, and Chemical Engineering, Washington University of St. Louis, St. Louis, MO, USA. |
| § Now at: Cooperative Institute for Research in Environmental Sciences, University of Colorado, Boulder, CO, USA and NOAA Chemical Sciences Laboratory, Boulder, CO, USA. |
| ¶ Now at: Winterthur Museum, Garden and Library, University of Delaware, Newark, DE, USA. |
| || Now at: Department of Chemistry, Southwestern Community College, Chula Vista, CA. |
| ** Now at: Colorado Department of Public Health and Environment, Denver, CO, USA. |
| This journal is © The Royal Society of Chemistry 2025 |