
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganic aerosol formation from 222 nm germicidal light: ozone-initiated vs. non-ozone pathways†
Matthew B.
Goss
 *abc and
Jesse H.
Kroll
*ad
*abc and
Jesse H.
Kroll
*ad
aDepartment of Civil and Environmental Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA. E-mail: jhkroll@mit.edu
bNow at Harvard University Center for the Environment, Harvard University, Cambridge, Massachusetts 02138, USA
cNow at John A. Paulson School of Engineering and Applied Sciences, Harvard University, Cambridge, Massachusetts 02138, USA. E-mail: mgoss@seas.harvard.edu
dDepartment of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA
First published on 17th October 2024
Abstract
Germicidal ultraviolet lamps outputting 222 nm light (GUV222) have the potential to reduce the airborne spread of disease through effective inactivation of pathogens, while remaining safe for direct human exposure. However, recent studies have identified these lamps as a source of ozone and other secondary pollutants such as secondary organic aerosol (SOA), and the health effects of these pollutants must be balanced against the benefits of pathogen inactivation. While ozone reactions are likely to account for much of this secondary indoor air pollution, 222 nm light may initiate additional non-ozone chemical processes, including the formation of other oxidants and direct photolytic reactions, which are not as well understood. This work examines the impacts of GUV222 on SOA formation and composition by comparing limonene oxidation under GUV222 and O3-only control conditions in a laboratory chamber. Differences between these experiments enable us to distinguish patterns in aerosol formation driven by ozone chemistry from those driven by other photolytic processes. These experiments also examine the influence of the addition of NO2 and nitrous acid (HONO), and investigate SOA formation in sampled outdoor air. SOA composition and yield vary only slightly with respect to GUV222vs. ozone-only conditions; NO2 and HONO photolysis do not appreciably affect the observed chemistry. In contrast, we observe consistent new particle formation under high-fluence 222 nm light (45 μW cm−2) that differs substantially from ozone-only experiments. This observed new particle formation represents an additional reason to keep GUV222 fluence rates to the lowest effective levels.
Environmental significanceGermicidal ultraviolet lamps that emit light at 222 nm (GUV222) can be a useful tool for reducing the airborne spread of disease in indoor environments, but might also negatively impact indoor air quality through the formation of ozone and particulate matter. This work demonstrates that GUV222 lamps not only drive increases in total particulate mass due to reactions of ozone with organic species, but also increase new particle formation in excess of what is caused by such reactions. The formation of ultrafine aerosol particles represents a potential health hazard in the indoor environment, and GUV222 applications should therefore keep fluence rates to the minimum effective levels to reduce negative impacts to indoor air quality. |
Introduction
The COVID-19 pandemic has increased interest in germicidal ultraviolet (GUV) light as a potential strategy for reducing the airborne spread of disease. Traditional applications of GUV light have used 254 nm mercury lamps, which effectively inactivate airborne pathogens, but pose a threat to human health if shined directly on skin and eyes. Newly available filtered KrCl excimer lamps, which emit light at 222 nm, have been reported to efficiently inactivate pathogens while still being safe for direct human exposure.1,2 Due to its shorter wavelength, the light does not penetrate the top layer of the skin or ocular tear layer, minimizing concerns about human ultraviolet light exposure.3,4However, recent work has raised concerns about the effects of 222 nm light (GUV222) on indoor air quality,5–12 in large part due to the production of ozone, a well-known human health hazard.13,14 Ozone production from GUV222, proceeding through the photolysis of O2 followed by the reaction of the resulting O atoms with O2 to form O3, is now well documented through both modeling calculations and laboratory experiments.5–8 Under laboratory conditions, GUV222 irradiation can lead to high (>100 ppb) O3 levels, but in real indoor spaces, O3 increases are generally much lower (∼6.5 ppb increase observed in an office,7 ∼5 ppb increase in a fragrant restroom,11 ∼0–10 ppb increase in a conference room12) due to the loss of ozone to indoor surfaces and relatively low average GUV222 fluence rates. These modest increases represent a potential source of concern given that the cumulative exposure to ozone is magnified by the amount of time people spend indoors;15 increases in oxidized volatile organic compounds (OVOCs) arising from O3 chemistry may enhance this potential hazard.11,16
While some prior studies have primarily focused on quantifying ozone production from GUV222,5,7 222 nm light has the potential to drive chemistry beyond O2 photolysis. As demonstrated in previous work,6,8,9 GUV222 can form the hydroxyl radical (OH), both through ozone photolysis and through the reaction of ozone with alkenes. Ozone and OH both react with volatile organic compounds, resulting in the production of OVOCs and secondary organic aerosol (SOA) particles.8,9,11 Previous laboratory work has also identified GUV222-driven new particle formation (NPF) under some conditions,8,17 which could act as a source of ultrafine particles in the indoor environment. Finally, a recent field study has characterized SOA formation and growth in the presence of GUV222 in a typical indoor space,11 demonstrating that these products of secondary chemistry, particularly SOA, can be formed under real-world conditions.
While this previous work has examined the formation of secondary pollutants other than ozone, the extent to which photochemical processes that do not directly involve ozone (referred to here as “non-ozone chemistry”) affect secondary chemistry and aerosol formation is unclear. Previous work8,9,11 demonstrates that SOA forms in the presence of GUV222, but less is known about the effects of GUV222 light on SOA yield and composition, relative to SOA formed solely from reactions with ozone. Further, new particle formation sometimes observed under 222 nm light is not understood. In addition, in our previous work,8 we suggested that the photolysis of other species, such as NO2, HONO, and photolabile organic molecules, may affect OH concentrations or radical cycling, but to our knowledge, such photolytic processes have not yet been explored.
The present work seeks to better understand non-ozone chemistry stemming from GUV222 irradiation, specifically focusing on secondary organic aerosol formation, and the potential role of trace species that can be present in indoor air. Experiments compare aerosol formation under 222 nm light and O3-only control conditions to identify differences driven by photolysis; these may be direct, for example by photolysis of an organic species, or indirect, for example from chemistry initiated by photolytically-formed OH. Using limonene as a model compound, chosen because of its common use in household cleaning products and propensity to generate SOA,18 we perform a series of experiments in two environmental chambers to identify differences in aerosol yield, composition, and tendency to form new particles. While this study does not seek to directly mimic indoor conditions (e.g., we do not include reactive surfaces), we also investigate more complex indoor mixtures by examining the influence of NOX (total NO and NO2 concentration) and HONO, often present in moderate concentrations in indoor spaces,19,20 on GUV222-derived aerosol production, and by performing experiments in which clean air is replaced by ambient outdoor air. This work focuses on the chemical processes stemming from 222 nm irradiation, and as such uses higher GUV222 fluence rates and VOC concentrations than are typically found in indoor environments, in order to enhance the distinctions between 222 nm and O3-only chemistry.
Methods
Experiments are carried out in two differently-sized Teflon chambers, described previously.8,21,22 The first (7.5 m3 in volume, referred to as the “large chamber”) is characterized by a relatively low particle wall loss rate and low GUV222 fluence rate, and is primarily used to quantify aerosol yield. The second one (0.15 m3 volume, referred to as the “mini chamber”) is used to study aerosol composition and size distribution trends due to its shorter residence time that more easily enables replicates. Both chambers are operated in “semi-batch” mode, in which clean air is continuously introduced to maintain slight positive pressure and make up for air removed through sampling.All experiments compare the effects of GUV222 with ozone-only conditions by matching ozone concentrations in the chamber for each experiment. For “GUV222” experiments, ozone is produced by the GUV222 lamp (Ushio, Care222 B1 Illuminator, peak emission at 222 nm). Average GUV222 fluence rate is estimated to be 45 μW cm−2 for the mini chamber8 and 3.9 μW cm−2 for the large chamber (see ESI†). For “ozone-only” experiments, the lamp is kept off and ozone is produced using an ozone generator (Jelight Model 610); ozone generation is tuned to match production by the GUV222 lamp by adjusting the lamp sleeve and reducing the power delivered using a Variac. Experiments are alternated between GUV222 and ozone-only conditions to avoid systematic biases in chamber conditions.
Materials and methods are broadly the same for both large (n = 7) and mini chamber experiments (n = 33). Clean air used for flushing and dilution for limonene oxidation experiments was produced by a zero-air generator (AADCO Model 737) (expts 1–13) or supplied from an ultra-zero air tank (Linde) (expts 15–32). Relative humidity for dry (<1% RH) or humid (27–45% RH) experiments is adjusted by bubbling an additional flow of clean air through Milli-Q water. Most experiments use (R)-(+)-limonene (C10H16, Sigma Aldrich) as a precursor VOC, ammonium sulfate ((NH4)2SO4, Sigma-Aldrich) for aerosol seed particles, and hexafluorobenzene (C6F6, Sigma-Aldrich) as a dilution tracer. Table 1 provides a basic overview of experiments, while specific details for each experiment are provided in Tables S1 and S2,† and more detailed Experimental methods are included in Section S.2.†
| Chamber | VOC source | Air changes per houra | Mean RH (%) | Mean starting particle conc. (#/cm3) | Dilution air sourceb | Note | Total number of experiments | Experiment numbers |
|---|---|---|---|---|---|---|---|---|
| a Measured using decay of hexafluorobenzene dilution tracer. While this was not measured for the outdoor air experiments, flow rates were held constant so the total dilution rate should match other mini chamber experiments. b AADCO = AADCO model 737 zero air generator, UZA = Linde ultra-zero air tank, ambient = outdoor air pumped through a 0.5 μm filter to remove particles. c Only one experiment was performed using 222 nm light. d This dilution rate differs from other sets of mini chamber experiments due to a small leak in the bubbler. | ||||||||
| Large | Limonene | 0.16 | 0 | 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
AADCO | 2 | 3–4 | |
| Large | Limonene | 0.16 | 35 | 21000 |
AADCO | 2 | 5–6 | |
| Large | Limonene | 0.16 | 36 | 19000 |
AADCO | HONO added | 1c | 7 |
| Mini | Limonene | 2.7d | 28 | 64000 |
AADCO | 6 | 8–13 | |
| Mini | Limonene | 3.3 | 0 | 20000 |
UZA | 6 | 15–20 | |
| Mini | Limonene | 3.2 | 35 | 24000 |
UZA | 6 | 21–26 | |
| Mini | Limonene | 3.3 | 33 | 25000 |
UZA | NOX added | 6 | 27–32 |
| Mini | Outdoor air | — | 44 | 23 | Ambient | 6 | 35–40 | |
Mini chamber experiments are carried out in two groups (expts 8–14 and expts 15–40), separated by approximately 5 months. For each mini chamber experiment, total dilution flows are maintained at 10.5 LPM, resulting in a measured dilution rate of 2.7–3.3 air changes per hour (ACH). Ozone is first allowed to reach a steady-state concentration (for NOX-free experiments: 106 ± 6 ppb (1σ)) by turning on the ozone generator or the GUV222 lamp, mounted above the chamber as in Barber et al.8 Once steady-state ozone is reached, a solution of ammonium sulfate (2 g L−1) is atomized into the chamber using an aerosol generator (TSI model 3076). This is followed by the addition of hexafluorobenzene (0.05 μL, 70 ppb), and subsequently limonene (0.1 μL, 100 ppb), initiating the experiment. This timepoint is considered t = 0. For six mini chamber experiments (expts 27–32), an additional flow of NO is added, resulting in a steady-state NOX concentration of 18.2 ± 0.7 ppb (1σ). The high levels of O3 in these experiments convert most of the added NO to NO2; since the steady-state concentration of NO is below the level of detection of our instrument (∼1 ppb), here we report only the concentrations of total NOX. For another six mini chamber experiments (expts 35–40), outdoor air is used instead of clean air, and is continuously pumped through a stainless steel 0.5 μm filter into the chamber to remove particles; no VOC, dilution tracer, or aerosol seed are added.
For large chamber experiments, total dilution flows are maintained at 20 LPM; measured dilution rate is ∼0.16 ACH. The GUV222 light is mounted outside the chamber, at the center of one of the square sides. For each experiment, ammonium sulfate (2 g L−1) is first atomized into the chamber for 9 minutes. After approximately 30 min, hexafluorobenzene (2.2 μL, 61 ppb) and limonene (6.0 μL, 119 ppb) are sequentially added to the chamber through a coated stainless-steel inlet heated to 50 and 120 °C respectively. The decays of these species are monitored for >1 h before the start of the experiment. The experiment is initiated by turning on the GUV222 lamp or the ozone generator (considered t = 0). One large chamber experiment (expt 7) uses a steady flow of HONO, resulting in a HONO concentration that ranges from 9 to 18 ppb over the course of the experiment (see ESI for more details†).
Particle-phase data are collected using a scanning mobility particle sizer (SMPS, TSI) and an aerosol mass spectrometer (AMS, Aerodyne Research, Inc.23). For large chamber experiments, AMS data are corrected for dilution and wall losses by normalizing to the ammonium sulfate concentration, and scaled such that initial aerosol seed concentration matches that measured by the SMPS (see ESI†). Time-dependent aerosol mass yield is calculated as the mass concentration of organic aerosol divided by the mass concentration of limonene reacted. For mini chamber experiments, AMS data are used primarily for elemental composition analysis;24 aerosol yields are not calculated due to the uncertainties caused by relatively high particle wall loss rates. NPF in limonene mini chamber experiments is quantified by fitting the number-weighted particle size distribution to a linear combination of lognormal distributions at each timestep. For experiments in which no NPF occurs, the data are well-represented by a single curve; for experiments in which NPF occurs, a second mode at smaller particle diameters forms. Where two (or in one case, three) modes are evident, the total particle number associated with the nucleation mode(s) is calculated based on the integral of the lognormal fit; the maximum of this value is considered to be the maximum nucleated particle concentration for purposes of comparing nucleation between experiments. Note that while no wall loss correction is applied to mini chamber data, these metrics are used only for comparing similar experiments; such comparisons should not be affected by particle deposition, which should affect the different experiments more or less equally. See the ESI† for further description of this analysis.
Gas-phase species are monitored using an additional set of online instrumentation. Ozone concentration is monitored using an ozone monitor (2B Tech). Limonene and hexafluorobenzene concentrations are monitored using a gas chromatograph with flame ionization detection (GC-FID, SRI Instruments). For mini chamber experiments, the GC-FID is started precisely at t = 4 min to ensure reproducibility between experiments, since measurements are taken only every 12 minutes. For large chamber experiments, limonene concentration is corrected for dilution based on the hexafluorobenzene time series to facilitate the calculation of aerosol yields. In some experiments (see Table S1†), trace gas measurements are supplemented with a chemiluminescence NO–NO2–NOX analyzer (NOX analyzer, Thermo Fisher Scientific) and a cavity attenuated phase shift NO2 monitor (CAPS NO2, Aerodyne Research, Inc.). The combination of these two instruments allows for quantification of NO, NO2, and HONO (via subtraction of the CAPS NO2 signal from the NOX analyzer measurement, see ESI†). For two sets of experiments, an ammonium chemical ionization mass spectrometer (NH4+ CIMS; modified PTR3 (ref. 25)) provides measurements of oxidized gas-phase organic species. While the instrument signal is unstable and uncalibrated during these experiments, it nonetheless provides qualitative insights into gas-phase products. Further instrument details and more complete description of data analysis methods are provided in the ESI.†
Results
New particle formation
Mini chamber experiments involving limonene oxidation consistently exhibit new particle formation under GUV222 irradiation; this is substantially greater than in the ozone-only experiments, often by a large margin. Fig. 1A shows results from two consecutive mini-chamber limonene experiments, one with GUV222 (expt 23) and the other with O3 addition (expt 22). Despite the fact that both have very similar ozone and limonene time series (top panels), far more NPF is observed in the GUV222 case. In the presence of GUV222, NPF occurs almost immediately after t = 0, with total particle number concentration growing to 6 × 104 cm−3. In contrast, the ozone-only experiment exhibits size distribution characteristics of SOA growth on the seed aerosol, with a much smaller nucleation mode.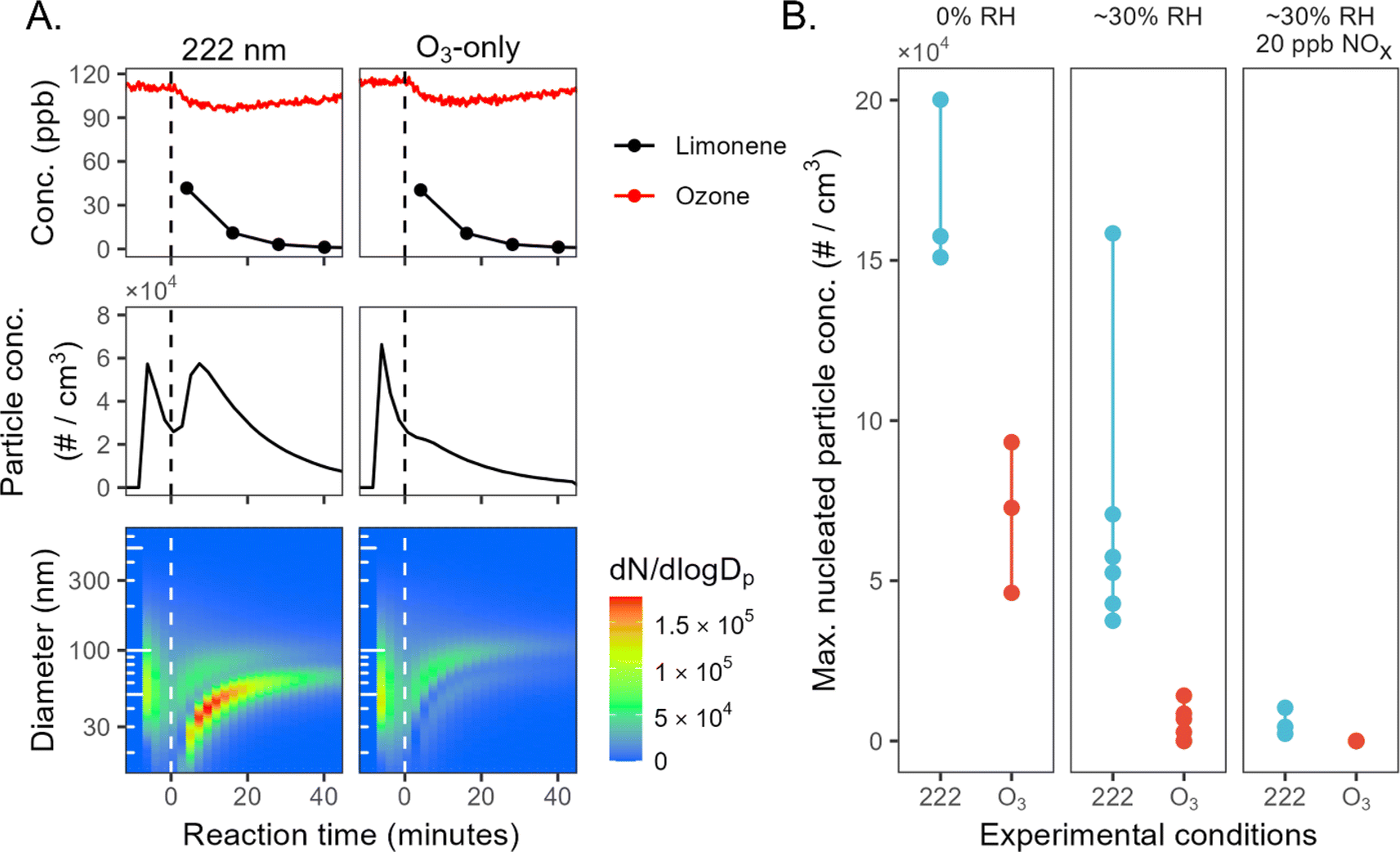 | ||
| Fig. 1 New particle formation from limonene + GUV222vs. limonene + O3. (Panel A) Particle growth and new particle formation for two example experiments (expts 22 and 23). The top panels show O3 and limonene concentrations, the middle panels show total particle number concentration over time, and the bottom panels show number-weighted size distributions over time. The spikes in particle concentration before t = 0 correspond to the addition of ammonium sulfate seed particles. Similar plots for all experiments are shown in the ESI.† (Panel B) Maximum number concentration of nucleated particles for each experiment (circles), grouped by experimental condition. See ESI† for further details on the determination of these values. | ||
Such differences in NPF occur under every chemical condition tested in the mini chamber (see Fig. S4 and S5†). Fig. 1B shows the maximum nucleated particle concentration for each experiment, allowing comparison of NPF for GUV222vs. ozone-only conditions across different humidity and NOX levels. The most nucleation occurs under dry conditions, whereas the least occurs in the presence of NOX. This is consistent with the total SOA mass concentrations in mini chamber experiments; SOA mass is generally higher under dry conditions, and lower in the presence of ∼20 ppb NO (see ESI†), suggesting that this trend may simply be controlled here by differences in SOA yield. While the maximum nucleated particle number concentration varies substantially between chamber conditions, it is always substantially greater under GUV222 conditions than in comparable ozone-only experiments.
We perform an additional set of experiments (expts 35–40) on a more complex system by filling the mini chamber with ambient air sampled continuously from outside our building. As shown in Fig. 2, new particles are formed every time the GUV222 lamp is turned on, while NPF under ozone-only conditions is almost negligible. Particles formed in the presence of GUV222 reach number concentrations as high as 3.5 × 104 particles cm−3, before the total particle number concentration drops due to coagulation. As the first nucleated mode continues to grow, a second smaller nucleation event occurs, leading to a relatively steady total particle number concentration after about one hour. In contrast, the interspersed ozone-only experiments demonstrate little obvious nucleation (max. 16–1100 particles cm−3). Under GUV222 conditions, particles grow to sufficient size to be detected as organic aerosol in the AMS (see Fig. S6†). While reaction conditions in these experiments are not identical—for example, NOX concentrations (top panels) change somewhat due to fluctuations in ambient concentrations—this cannot explain the substantial differences in NPF between the GUV222 and O3-only experiments.
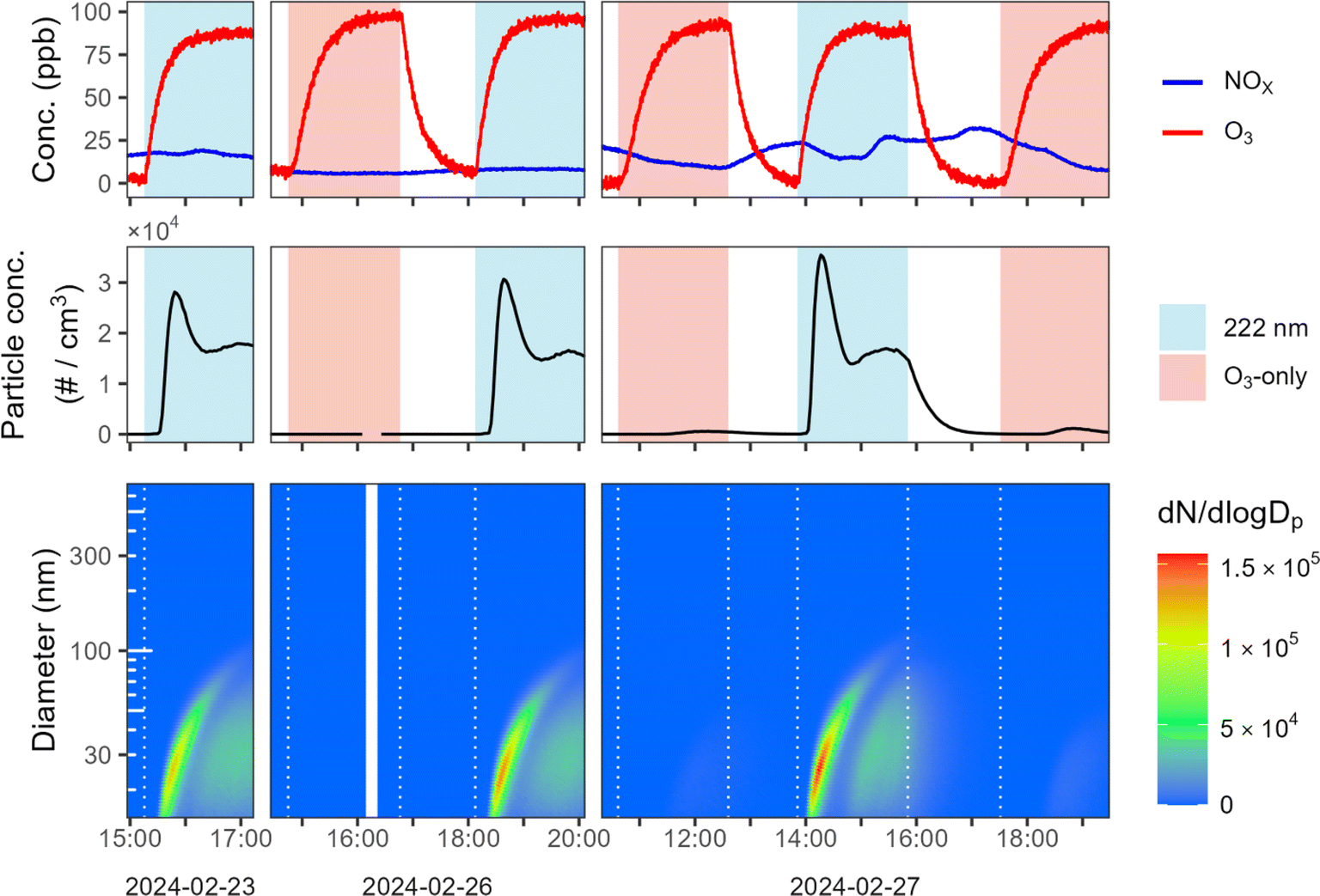 | ||
| Fig. 2 New particle formation for experiments using sampled outdoor air. Shown are O3 and NOX concentrations over time (top row), particle number concentration over time (second row), and number-weighted size distributions over time (bottom row) for six experiments in which outdoor air is introduced (expts 35–40). Experiments are carried out on three separate days. Periods when the 222 nm lamp is turned on, and when the ozone generator is turned on, are highlighted in pale blue and pale red, respectively, in the top two rows. | ||
In addition to the chamber experiments with limonene or outdoor air, several blank mini chamber experiments are run in which no VOCs (limonene or ambient species) are added to the chamber (expts 14, 33, 34) (see Fig. S7†). As in prior work,8 new particle formation occurs under 222 nm light when the mini chamber is thoroughly flushed with air from our clean air generator (expt 14). However, after replacement of the compressor for the clean air generator, such nucleation under GUV222 irradiation is not observed (expt 33). Further, nucleation does not occur during blank experiments run with ultra-zero air from a cylinder (expt 34). Thus it seems likely that oxidation of trace VOCs from the older compressor cause the NPF in earlier blank experiments. The older compressor is used only for experiments 1–14 and does not influence the interpretation of the results presented here (since GUV222 and ozone-only experiments are always run under identical conditions for each experiment set). Experiments 8–13 (mini chamber limonene oxidation) are replicated with ultra-zero air (expts 21–26); while these experiments differ slightly due to faster particle wall loss and lower seed particle concentration, they produce the same qualitative results (see Fig. S5†).
In marked contrast to mini chamber results, substantial new particle formation never occurs in large chamber experiments (see Fig. S10 and S11†). This is despite higher limonene concentrations, which might be expected to slightly increase NPF. While seed concentrations (and total condensation sink, see ESI†) are similar between experiments carried out in both chambers, the rate of formation of oxidized products is much slower in large chamber experiments due to substantially lower concentrations of ozone, particularly at the beginning of each experiment. Under these conditions, the formation of low-volatility products is likely sufficiently slow relative to the condensation sink to prevent the system from reaching the supersaturated state required for NPF. While substantial NPF is not observed in large chamber experiments, a very small (<∼200 particles cm−3) nucleation mode with particles of diameter <20 nm forms consistently in all large chamber limonene experiments. These particles do not grow in diameter and are likely driven by ozone reaction based on similarity between GUV222 and ozone-only experiments (see ESI for further discussion†). These observations of differences between chamber setups highlight the role that both the rate of VOC oxidation and the condensation sink may play in controlling NPF in indoor environments, especially given highly variable ambient indoor particle loading.26,27
Aerosol yield and composition
Given the differences in new particle formation, additional limonene chamber experiments are carried out to examine potential differences in aerosol yield and composition between GUV222 and ozone-only conditions. These are first investigated in five experiments in the large chamber (expts 3–7), since its lower surface-to-volume ratio enables better quantification of aerosol formation. Fig. 3A shows aerosol mass yield as a function of reaction time. Aerosol yields under GUV222 and ozone-only conditions are nearly identical, with the two GUV222 experiments featuring slightly lower yield by the end of the experiment. This slight difference agrees with the subtle decrease in SOA formation rate in the presence of GUV222 observed by Jenks et al.,9 and could be due to photolysis of SOA components, but may instead simply be due to experimental variability. Relative humidity appears to play a far greater role, with dry conditions increasing calculated yield by a factor as high as ∼1.5. The measured aerosol yields fall within the large range of literature values for limonene oxidation (11–109% aerosol mass yield),9,28–31 but trends in yield with respect to RH are different than those reported previously where yield was found to stay the same or even increase with increased humidity.9,30,32 When 9–18 ppb HONO is added to one experiment, aerosol yield is not appreciably different from the yield measured under standard GUV222 conditions. While the causes for discrepancies between our measurements and previous measurements are unclear, these results clearly demonstrate that the differences in the GUV222vs. ozone-only experiments are small compared to other factors controlling the mass of SOA formed.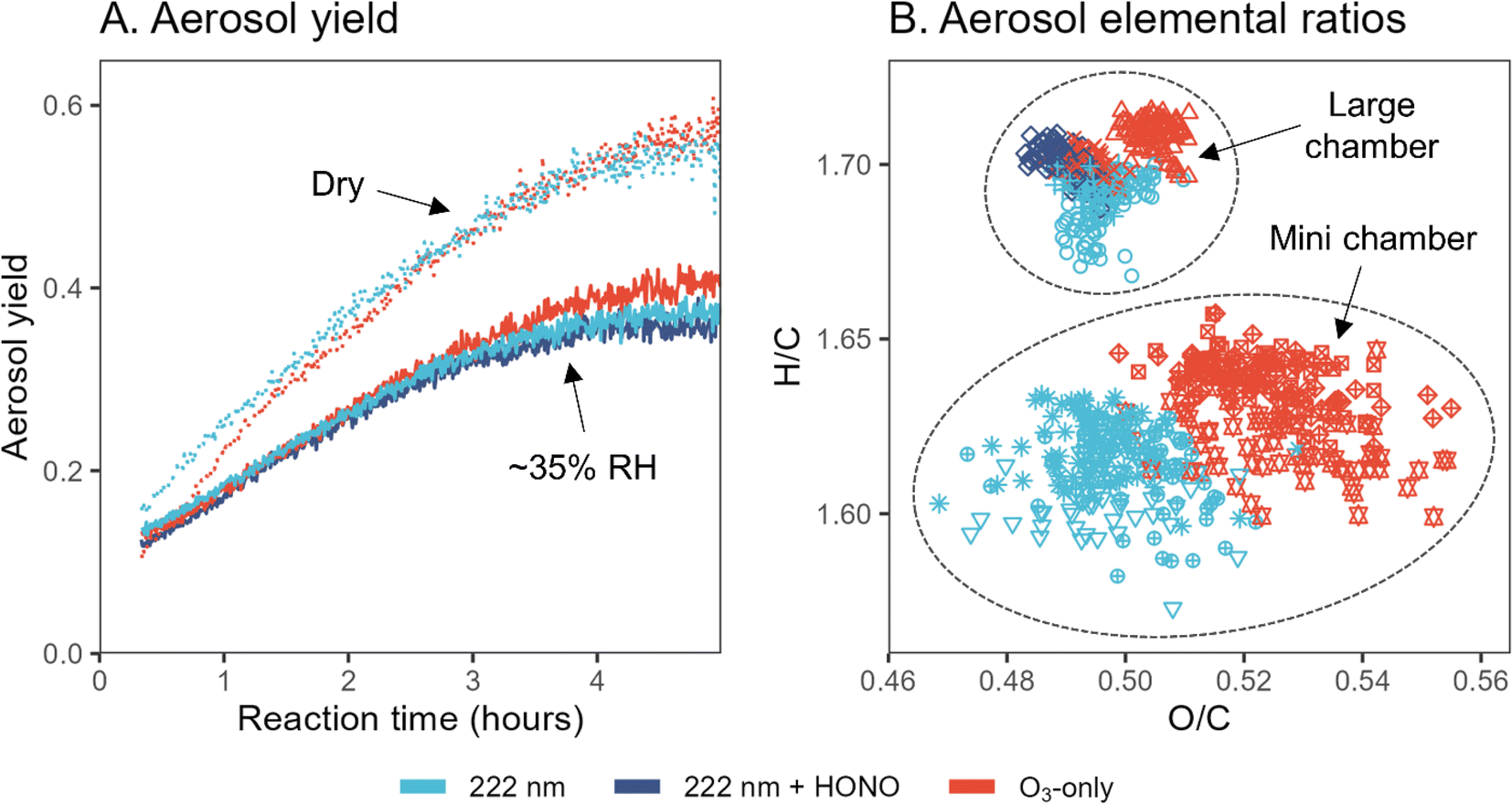 | ||
| Fig. 3 Aerosol yield and elemental composition from limonene oxidation under GUV222 and ozone-only conditions. (Panel A) Aerosol yield over time, calculated as mass concentration of SOA (μg m−3) divided by mass concentration of limonene reacted (μg m−3), for five large chamber experiments (expts 3–7). Dotted lines show ∼0% RH conditions while solid lines show results from ∼35% RH experiments. (Panel B) Van Krevelen diagram that shows elemental ratios obtained from high-resolution AMS analysis of experiments 3–7 and 8–13. Color refers to the oxidation conditions, while each point shape represents a different experiment. Points shown are for timepoints after the elemental composition has stabilized (from t = 100 to 300 min and t = 15 to 50 min for large and mini chamber experiments, respectively). Note that the axes of (B) are zoomed in substantially; the differences between experiment types, while repeatable, are quite small. See also Fig. S14† for Van Krevelen diagrams from all experiments. | ||
Aerosol yields from the mini chamber experiments are not calculated given the large uncertainties arising from rapid wall loss. As in the large chamber experiments, uncorrected organic aerosol mass is higher under dry conditions when compared to directly comparable humid experiments. When NOX is continuously added (with a steady-state level of ∼18 ppb), organic aerosol mass is substantially depressed, but this does not differ between GUV222 and ozone-only conditions (see Fig. S13†).
Fig. 3B shows elemental ratios from high-resolution AMS analysis of SOA, in both large chamber experiments (expt. 3–7) and the set of mini chamber experiments that featured the greatest AMS organic signal (expt. 8–13) (results from all mini chamber experiments are shown in Fig. S14†). Points in Van Krevelen space are shown for timepoints after the elemental composition has stabilized. All points fall within a relatively small range, but GUV222-derived aerosol features consistently lower O/C and H/C ratios than ozone-only experiments. While this effect is less clear for the large chamber experiments, the difference between GUV222 and ozone-only O/C ratios in the mini chamber is statistically significant for the set of mini chamber experiments shown (p = 0.002). This effect is also observed in other sets of mini chamber experiments (Fig. S14†), but the difference is not quite as clear, likely due to the lower AMS aerosol signal and poorer AMS peak-shape tuning. Since the differences in elemental ratios are larger in mini chamber experiments than in large chamber experiments, this effect may be related to the GUV222 fluence rate, possibly representing a chemical change driven by direct or indirect photolysis; however, further work would be required to confirm this. Ultimately, while differences between GUV222 and ozone-only elemental ratios are reproducible, they are small in magnitude, and similar in magnitude to the differences induced by using different chambers or different experimental conditions.
Discussion
Chamber experiments examining the oxidation of limonene under GUV222 and ozone-only conditions demonstrate relatively little difference in SOA yield and composition. The differences observed between GUV222 and ozone-only experiments, while reproducible, are often similar or smaller in magnitude to the magnitude of differences induced by changes in other experimental parameters (e.g., NOX, RH, chamber type). This leads us to conclude that most of the observed chemistry is driven by reactions with O3, in agreement with findings by Jenks et al.9 However, one major difference is new particle formation, which is consistently much greater in the presence of high-fluence rate 222 nm light in the mini chamber.New particle formation from the oxidation of organic species is likely to proceed through the formation of low volatility organic compounds. These may include high-mass species such as highly oxidized molecules (HOMs)33 or larger compounds with a large number of carbon atoms. However, NH4+ CIMS measurements (which were taken for only two sets of experiments; see ESI†) show no clear differences in any high mass ions, including those that might precede the formation of HOMs, between GUV222 and ozone-only conditions. Likewise, the AMS provides no evidence of dramatically differing chemical composition of the organic aerosol, with aerosol produced under GUV222 conditions actually appearing to be slightly less oxidized (Fig. 3), suggesting that HOM formation is not responsible for differences in nucleation. It is possible that even without the formation of HOMs, direct photoionization of organic molecules could influence nucleation,34 but the energy of photons at 222 nm (5.6 eV) is well below the threshold of ∼8 eV for the most easily ionizable organic molecules. This process would therefore require photons with wavelengths substantially shorter than 222 nm, which will not be the case here based on published spectra;3 ionization due to the simultaneous absorption of two photons is highly unlikely given the low photon fluxes.
Other plausible causes for the differences in new particle formation include subtle differences in reaction conditions. Nucleation may be strongly dependent on initial chamber conditions, but these experiments do not feature systematic differences in ozone, seed particle, or limonene concentration (see Fig. S2–S4†). The photolysis of ozone by GUV222 has been identified as an additional source of OH in previous studies,6,8 which could potentially impact NPF. However, mechanistic modeling using the Master Chemical Mechanism35 run in F0AM,36 suggests only modest increases in mean OH under GUV222 conditions compared to ozone-only conditions (1.7 × 106vs. 1.4 × 106 molec. cm−3) (see ESI†). Modeled OH concentrations are nearly identical in the first few minutes of the experiment when nucleation occurs, suggesting that this difference is unlikely to explain NPF. This is further supported by AMS measurements: literature chamber studies suggest that aerosol from the OH oxidation of monoterpenes features a substantially higher H/C ratio than aerosol from ozonolysis,37,38 but our measurements consistently show a slightly lower H/C ratio under GUV222, suggesting that OH oxidation is unlikely to be the primary driver of aerosol formation. Differences could also be explained by changes in radical chemistry due to the photolysis of trace NOY species. However, the addition of NOX in expts 27–32 suppresses nucleation relative to experiments with no added NOX (Fig. 1); similarly, the addition of HONO to a large chamber experiment does not impact nucleation or aerosol yield (Fig. 3). This is contrary to our earlier speculation that NOY photolysis at 222 nm may have a major impact on oxidant formation and secondary pollutant formation.8 The lack of any observed effect is likely a result of the relatively low photon flux of the GUV222 lights, which leads to photolysis rates that are substantially slower than dilution (jNO2 = 2.0 × 10−5 s−1 in the mini chamber; jHONO = 9.2 × 10−6 s−1 in the large chamber; see the ESI†). Finally, differences in other trace species such as HO2 or RO2 could also influence oxidation chemistry, but box modeling suggests that concentrations of these do not differ appreciably (see ESI†).
While our measurements do not pinpoint a mechanistic cause of increased new particle formation under GUV222 conditions, they clearly identify several patterns. First, indirect or direct photolytic processes are involved, since all variables except for light are held constant. Second, NPF likely involves gas-phase organic compounds, since it only occurs when VOCs are present (limonene, trace organics from clean air compressor, sampled ambient air). The process is unlikely to involve organic species from chamber surfaces, since no nucleation is observed in an ultra-zero air blank experiment. The concentrations of such organic precursors may be so low that they may be difficult to detect. For example, assuming the nucleated particles (diameter <20 nm) are organic, with O/C = 0.25, H/C = 2, and density = 1 g cm−3, even at their highest observed volume concentrations they would account for no more than 0.2 ppb C. Detection and characterization of such precursors might be possible via analytical techniques aimed at detection of low-volatility species, such as nitrate chemical ionization mass spectrometry39 to measure HOMs, or atmospheric pressure interface mass spectrometry,40 to detect ambient ions and charged clusters.
Conclusion
This series of laboratory experiments demonstrates that most aspects of SOA formation (e.g., yield, elemental composition, dependence on RH and NOX) in the presence of GUV222 are consistent with ozonolysis chemistry. While some measured parameters such as the aerosol O/C ratio vary reproducibly with respect to GUV222vs. ozone-only conditions, the magnitude of these differences is generally small in comparison to the changes induced by different experimental conditions.However, the major exception to this finding is the occurrence of new particle formation in the presence of high levels of GUV222, in excess of NPF observed simply from ozonolysis chemistry. While the reason for this is not clear, substantial NPF events in the presence of limonene, as well as in outdoor air pumped into the chamber, are cause for concern for indoor applications due to the relatively high concentrations of ultrafine particles formed. In real indoor environments, the likelihood of new particle formation is unclear due to competing factors. Lower average GUV222 fluence rates will result in slower oxidation chemistry and the ubiquity of surfaces may encourage particle deposition,41 both limiting NPF. However, locally high fluence rates near a GUV222 lamp might still encourage NPF or interact with surface reservoirs of semivolatile compounds in uncertain ways. Finally, ambient indoor particle loading, which can be considerably lower or higher than seed aerosol concentrations used in these experiments,26,27 may greatly impact the total condensation sink and therefore the likelihood of NPF. Indeed, recent work has detected NPF from GUV222 irradiation in a real indoor space,11 demonstrating that GUV222 driven oxidation processes can outweigh condensation sinks. Still, further work is required to fully understand this process, particularly with regard to the quantification of new particle formation as a function of 222 nm fluence rate and VOC identity.
For the purposes of deploying GUV222 lamps in indoor spaces, these results provide confirmation that most (though not all) observed chemistry follows that expected simply from reaction with ozone, consistent with earlier work.9 While ozone has serious potential as an indoor air pollutant,15 its chemistry is reasonably well understood. Our results suggest that indoor spaces with GUV222 lamps may likely be reasonably well-represented in models simply by including the lamps as an additional source of ozone, and ensuring that all downstream ozone chemistry (e.g., formation of OH, OVOCs, and SOA) is represented. Still, additional uncertainties remain, including the influence of 222 nm light on indoor surfaces and surface-bound organic species, as well as the cause of GUV222-driven new particle formation. In addition to GUV222-driven ozone production, the new particle formation observed in this work represents a further reason to keep GUV222 light intensity to the lowest effective levels when used in indoor spaces.
Data availability
Data for this article, including all processed SMPS, AMS, GC-FID, and gas-monitor data, are available on the Kroll Group publication website at http://krollgroup.mit.edu/publications.html.Author contributions
MBG collected and analyzed the data and wrote the paper. JHK aided in project design, data interpretation, and manuscript preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Frank Keutsch and Yaowei Li (Harvard University) for the use of the NH4+ CIMS, and Victoria Barber (University of California, Los Angeles) for useful discussions. This research was supported by the Training Grant in Environmental Toxicology (MIT Center for Environmental Health Sciences, NIEHS Grant # T32-ES007020 and Grant # P30-ES002109) and the MathWorks Engineering Fellowship Fund.References
- D. Welch, M. Buonanno, V. Grilj, I. Shuryak, C. Crickmore, A. W. Bigelow, G. Randers-Pehrson, G. W. Johnson and D. J. Brenner, Far-UVC light: A new tool to control the spread of airborne-mediated microbial diseases, Sci. Rep., 2018, 8, 2752 CrossRef.
- M. Buonanno, D. Welch, I. Shuryak and D. J. Brenner, Far-UVC light (222 nm) efficiently and safely inactivates airborne human coronaviruses, Sci. Rep., 2020, 10, 10285 CrossRef CAS.
- M. Buonanno, D. Welch and D. J. Brenner, Exposure of Human Skin Models to KrCl Excimer Lamps: The Impact of Optical Filtering, Photochem. Photobiol., 2021, 97, 517–523 CrossRef CAS.
- K. Sugihara, S. Kaidzu, M. Sasaki, S. Ichioka, Y. Takayanagi, H. Shimizu, I. Sano, K. Hara and M. Tanito, One-year Ocular Safety Observation of Workers and Estimations of Microorganism Inactivation Efficacy in the Room Irradiated with 222-nm Far Ultraviolet-C Lamps, Photochem. Photobiol., 2023, 99, 967–974 CrossRef CAS PubMed.
- M. F. Link, A. Shore, B. H. Hamadani and D. Poppendieck, Ozone Generation from a Germicidal Ultraviolet Lamp with Peak Emission at 222 nm, Environ. Sci. Technol. Lett., 2023, 10, 675–679 CrossRef CAS.
- Z. Peng, S. L. Miller and J. L. Jimenez, Model Evaluation of Secondary Chemistry due to Disinfection of Indoor Air with Germicidal Ultraviolet Lamps, Environ. Sci. Technol. Lett., 2023, 10, 6–13 CrossRef CAS.
- Z. Peng, D. A. Day, G. A. Symonds, O. J. Jenks, H. Stark, A. V. Handschy, J. A. De Gouw and J. L. Jimenez, Significant Production of Ozone from Germicidal UV Lights at 222 nm, Environ. Sci. Technol. Lett., 2023, 10, 668–674 CrossRef CAS.
- V. P. Barber, M. B. Goss, L. J. Franco Deloya, L. N. LeMar, Y. Li, E. Helstrom, M. Canagaratna, F. N. Keutsch and J. H. Kroll, Indoor Air Quality Implications of Germicidal 222 nm Light, Environ. Sci. Technol., 2023, 57, 15990–15998 CrossRef CAS PubMed.
- O. J. Jenks, Z. Peng, M. K. Schueneman, M. Rutherford, A. V. Handschy, D. A. Day, J. L. Jimenez and J. A. de Gouw, Effects of 222 nm Germicidal Ultraviolet Light on Aerosol and VOC Formation from Limonene, ACS ES&T Air, 2024, 1, 725–733 Search PubMed.
- Z. Liang, L. Zhou, K. Chen, Y.-H. Lin, A. C. K. Lai, P. K. H. Lee, P. H. L. Sit, R. Yin and C. K. Chan, Formation of Secondary Aerosol by 222 nm Far-UVC Irradiation on SO2, Atmos. Environ., 2024, 120559 CrossRef CAS.
- M. Link, R. Robertson, A. Shore, B. Hamadani, C. Cecelski and D. Poppendieck, Ozone Generation and Chemistry from 222 nm Germicidal Ultraviolet Light in a Fragrant Restroom, Environ. Sci.: Process. Impacts., 2024, 26, 1090–1106 CAS.
- F. Narouei, Z. Tang, S. Wang, R. Hashmi, D. Welch, S. Sethuraman, D. Brenner and V. F. McNeill, Effects of Germicidal Far-UVC on Indoor Air Quality in an Office Setting, ChemRxiv, 2024, preprint, DOI:10.26434/chemrxiv-2024-l6k59.
- M. C. Turner, M. Jerrett, C. A. Pope, D. Krewski, S. M. Gapstur, W. R. Diver, B. S. Beckerman, J. D. Marshall, J. Su, D. L. Crouse and R. T. Burnett, Long-Term Ozone Exposure and Mortality in a Large Prospective Study, Am. J. Respir. Crit. Care Med., 2016, 193, 1134–1142 CrossRef CAS.
- E. Gakidou, et al., Global, regional, and national comparative risk assessment of 84 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016, Lancet, 2017, 390, 1345–1422 CrossRef.
- W. W. Nazaroff and C. J. Weschler, Indoor ozone: Concentrations and influencing factors, Indoor Air, 2022, 32, e12942 CAS.
- C. J. Weschler and W. W. Nazaroff, Ozone Loss: A Surrogate for the Indoor Concentration of Ozone-Derived Products, Environ. Sci. Technol., 2023, 57, 13569–13578 CrossRef CAS PubMed.
- M. F. Link, R. Robertson, M. S. Claflin and D. Poppendieck, Quantification of Byproduct Formation from Portable Air Cleaners Using a Proposed Standard Test Method, Environ. Sci. Technol., 2024, 58, 7916–7923 CrossRef CAS PubMed.
- B. K. Coleman, M. M. Lunden, H. Destaillats and W. W. Nazaroff, Secondary organic aerosol from ozone-initiated reactions with terpene-rich household products, Atmos. Environ., 2008, 42, 8234–8245 CrossRef CAS.
- K. Lee, J. Xue, A. S. Geyh, H. Ozkaynak, B. P. Leaderer, C. J. Weschler and J. D. Spengler, Nitrous acid, nitrogen dioxide, and ozone concentrations in residential environments, Environ. Health Perspect., 2002, 110, 145–150 CrossRef CAS PubMed.
- C. Wang, B. Bottorff, E. Reidy, C. M. F. Rosales, D. B. Collins, A. Novoselac, D. K. Farmer, M. E. Vance, P. S. Stevens and J. P. D. Abbatt, Cooking, Bleach Cleaning, and Air Conditioning Strongly Impact Levels of HONO in a House, Environ. Sci. Technol., 2020, 54, 13488–13497 CrossRef CAS.
- J. F. Hunter, A. J. Carrasquillo, K. E. Daumit and J. H. Kroll, Secondary Organic Aerosol Formation from Acyclic, Monocyclic, and Polycyclic Alkanes, Environ. Sci. Technol., 2014, 48, 10227–10234 CrossRef CAS PubMed.
- C. Y. Lim, D. H. Hagan, M. M. Coggon, A. R. Koss, K. Sekimoto, J. de Gouw, C. Warneke, C. D. Cappa and J. H. Kroll, Secondary organic aerosol formation from the laboratory oxidation of biomass burning emissions, Atmos. Chem. Phys., 2019, 19, 12797–12809 CrossRef CAS.
- P. F. DeCarlo, J. R. Kimmel, A. Trimborn, M. J. Northway, J. T. Jayne, A. C. Aiken, M. Gonin, K. Fuhrer, T. Horvath, K. S. Docherty, D. R. Worsnop and J. L. Jimenez, Field-Deployable, High-Resolution, Time-of-Flight Aerosol Mass Spectrometer, Anal. Chem., 2006, 78, 8281–8289 CrossRef CAS PubMed.
- M. R. Canagaratna, J. L. Jimenez, J. H. Kroll, Q. Chen, S. H. Kessler, P. Massoli, L. Hildebrandt Ruiz, E. Fortner, L. R. Williams, K. R. Wilson, J. D. Surratt, N. M. Donahue, J. T. Jayne and D. R. Worsnop, Elemental ratio measurements of organic compounds using aerosol mass spectrometry: characterization, improved calibration, and implications, Atmos. Chem. Phys., 2015, 15, 253–272 CrossRef.
- A. Zaytsev, M. Breitenlechner, A. R. Koss, C. Y. Lim, J. C. Rowe, J. H. Kroll and F. N. Keutsch, Using collision-induced dissociation to constrain sensitivity of ammonia chemical ionization mass spectrometry (NH4+ CIMS) to oxygenated volatile organic compounds, Atmos. Meas. Tech., 2019, 12, 1861–1870 CrossRef CAS.
- S. Patel, S. Sankhyan, E. K. Boedicker, P. F. DeCarlo, D. K. Farmer, A. H. Goldstein, E. F. Katz, W. W. Nazaroff, Y. Tian, J. Vanhanen and M. E. Vance, Indoor Particulate Matter during HOMEChem: Concentrations, Size Distributions, and Exposures, Environ. Sci. Technol., 2020, 54, 7107–7116 CrossRef CAS PubMed.
- X. May Wu, M. G. Apte and D. H. Bennett, Indoor Particle Levels in Small- and Medium-Sized Commercial Buildings in California, Environ. Sci. Technol., 2012, 46, 12355–12363 CrossRef.
- S. Leungsakul, M. Jaoui and R. M. Kamens, Kinetic Mechanism for Predicting Secondary Organic Aerosol Formation from the Reaction of d-Limonene with Ozone, Environ. Sci. Technol., 2005, 39, 9583–9594 CrossRef CAS.
- J. Zhang, K. E. Huff Hartz, S. N. Pandis and N. M. Donahue, Secondary Organic Aerosol Formation from Limonene Ozonolysis: Homogeneous and Heterogeneous Influences as a Function of NOX, J. Phys. Chem. A, 2006, 110, 11053–11063 CrossRef CAS.
- H. Saathoff, K.-H. Naumann, O. Möhler, Å. M. Jonsson, M. Hallquist, A. Kiendler-Scharr, T. F. Mentel, R. Tillmann and U. Schurath, Temperature dependence of yields of secondary organic aerosols from the ozonolysis of α-pinene and limonene, Atmos. Chem. Phys., 2009, 9, 1551–1577 CrossRef CAS.
- X. Chen and P. K. Hopke, A chamber study of secondary organic aerosol formation by limonene ozonolysis: Chamber study of secondary organic aerosol formation, Indoor Air, 2010, 20, 320–328 CrossRef CAS.
- Y. Gong and Z. Chen, Quantification of the role of stabilized Criegee intermediates in the formation of aerosols in limonene ozonolysis, Atmos. Chem. Phys., 2021, 21, 813–829 CrossRef CAS.
- F. Bianchi, T. Kurtén, M. Riva, C. Mohr, M. P. Rissanen, P. Roldin, T. Berndt, J. D. Crounse, P. O. Wennberg, T. F. Mentel, J. Wildt, H. Junninen, T. Jokinen, M. Kulmala, D. R. Worsnop, J. A. Thornton, N. Donahue, H. G. Kjaergaard and M. Ehn, Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 2019, 119, 3472–3509 CrossRef CAS PubMed.
- A. Hirsikko, T. Nieminen, S. Gagné, K. Lehtipalo, H. E. Manninen, M. Ehn, U. Hõrrak, V.-M. Kerminen, L. Laakso, P. H. McMurry, A. Mirme, S. Mirme, T. Petäjä, H. Tammet, V. Vakkari, M. Vana and M. Kulmala, Atmospheric ions and nucleation: a review of observations, Atmos. Chem. Phys., 2011, 11, 767–798 CrossRef CAS.
- S. M. Saunders, M. E. Jenkin, R. G. Derwent and M. J. Pilling, Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 2003, 3, 161–180 CrossRef CAS.
- G. M. Wolfe, M. R. Marvin, S. J. Roberts, K. R. Travis and J. Liao, The Framework for 0-D Atmospheric Modeling (F0AM) v3.1, Geosci. Model Dev., 2016, 9, 3309–3319 CrossRef.
- D. F. Zhao, M. Kaminski, P. Schlag, H. Fuchs, I.-H. Acir, B. Bohn, R. Häseler, A. Kiendler-Scharr, F. Rohrer, R. Tillmann, M. J. Wang, R. Wegener, J. Wildt, A. Wahner and T. F. Mentel, Secondary organic aerosol formation from hydroxyl radical oxidation and ozonolysis of monoterpenes, Atmos. Chem. Phys., 2015, 15, 991–1012 CrossRef.
- J. Liu, E. L. D'Ambro, B. H. Lee, S. Schobesberger, D. M. Bell, R. A. Zaveri, A. Zelenyuk, J. A. Thornton and J. E. Shilling, Monoterpene Photooxidation in a Continuous-Flow Chamber: SOA Yields and Impacts of Oxidants, NOX, and VOC Precursors, Environ. Sci. Technol., 2022, 56, 12066–12076 CrossRef CAS PubMed.
- T. Jokinen, M. Sipilä, H. Junninen, M. Ehn, G. Lönn, J. Hakala, T. Petäjä, R. L. I. Mauldin, M. Kulmala and D. R. Worsnop, Atmospheric sulphuric acid and neutral cluster measurements using CI-APi-TOF, Atmos. Chem. Phys., 2012, 12, 4117–4125 CrossRef CAS.
- H. Junninen, M. Ehn, T. Petäjä, L. Luosujärvi, T. Kotiaho, R. Kostiainen, U. Rohner, M. Gonin, K. Fuhrer, M. Kulmala and D. R. Worsnop, A high-resolution mass spectrometer to measure atmospheric ion composition, Atmos. Meas. Tech., 2010, 3, 1039–1053 CrossRef CAS.
- W. W. Nazaroff, Indoor particle dynamics, Indoor Air, 2004, 14, 175–183 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4em00384e |
| This journal is © The Royal Society of Chemistry 2025 |