
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective catalytic hydrogenation of C2H2 from plasma-driven CH4 coupling without extra heat: mechanistic insights from micro-kinetic modelling and reactor performance†
Eduardo Morais‡
 a,
Fabio Cameli‡bc,
Georgios D. Stefanidis*bc and
Annemie Bogaerts*a
a,
Fabio Cameli‡bc,
Georgios D. Stefanidis*bc and
Annemie Bogaerts*a
aPLASMANT, Department of Chemistry, University of Antwerp, Campus Drie Eiken, Antwerp 2610, Belgium. E-mail: annemie.bogaerts@uantwerpen.be
bLaboratory for Chemical Technology, Ghent University, Tech Lane Ghent Science Park 125, Ghent, B-9052, Belgium. E-mail: georgios.stefanidis@ugent.be; gstefani@mail.ntua.gr
cSchool of Chemical Engineering, National Technical University of Athens, Iroon Polytechniou 9, 15780, Athens, Greece
First published on 16th January 2025
Abstract
We study the selective catalytic hydrogenation of C2H2, the main product from non-oxidative CH4 coupling in gas-phase plasmas, to C2H4, a cornerstone of the global chemical industry, by experiments and temperature-dependent micro-kinetic modelling. The model is validated against new experimental data from a nanosecond pulsed plasma reactor integrated with a downstream catalytic bed consisting of Pd/Al2O3. We explore the effects of varying Pd loadings (0.1, 0.5, and 1 wt%) on the catalyst activity and the C2H4/C2H6 product distribution. Consistent with the experimental data, our surface micro-kinetic model shows that while higher Pd loadings lower the catalyst activation temperature for C2H2 conversion, they also induce over-hydrogenation to C2H6 at lower temperatures and increase oligomerisation in the experiments, which are detrimental to the C2H4 yield. The model also elucidates reaction mechanisms and pathways across different temperature regimes, expanding our understanding of the hydrogenation process beyond the experimental range. Besides highlighting the importance of optimising the metal loading to balance C2H4 and C2H6 selectivity, our findings demonstrate the effective implementation of post-plasma catalysis using a simple catalyst bed heated by hot gas from the plasma region. This study opens possibilities for testing different plasma sources, catalysts, gas flow magnitude and patterns, and catalyst bed-to-plasma distances.
Broader contextThe direct conversion of methane (CH4) to ethylene (C2H4) is thermodynamically challenging yet critical, given the demand for sustainable methods to synthesise valuable base chemicals, such as ethylene – a core molecule in global industry. The integration of plasma reactors with catalysis offers a promising solution, as it provides an efficient tool for CH4 coupling into C2H2, followed by a pathway to selectively steer the reaction towards C2H4. In this study, we demonstrate this synergy by catalytically hydrogenating the C2H2 plasma-product into C2H4 using Pd catalysts activated by the hot gas stream exiting the plasma reactor, without external heating. Alongside, we developed a temperature-dependent micro-kinetic surface model, providing insights into optimising C2H4 selectivity and avoiding C2H6 and oligomerisation by-products by balancing catalyst metal loading and reaction temperature. Our results broaden the understanding of coupling plasma to downstream catalysis and open new avenues for developing electrified, scalable and energy-efficient processes for ethylene synthesis and methane valorisation. These findings highlight the potential of plasma and post-plasma catalysis to play a central role in fostering a CO2-neutral chemical industry and promoting a more sustainable future, as well as providing a framework for further research into energy and environmental (plasma) catalysis. |
1. Introduction
The emergence of plasma technologies for converting predominantly inert gases (such as CO2 and CH4) marks an important development within the efforts to shift the chemical synthesis of highly valuable light molecules, like C2H4 and CH3OH, from naphtha cracking to electrified processes with a neutral CO2 loop.1,2 The transition from traditional routes to plasma-based synthesis has positive techno-economic and environmental impacts, offering a promising solution to alleviate reliance on fossil fuels and greenhouse gas emissions.1,3–6 Plasma reactors are very flexible in terms of scale and targeted feedstock/products. They are also characterised by ideal coupling to (sometimes intermittent) renewable energy sources such as solar, wind, and hydroelectric power, which become increasingly widespread and cost-effective.7,8While plasma-based gas conversion for chemical synthesis has continuously had tangible outcomes in recent years,9–13 the low product selectivity and purity is one aspect that has challenged plasma technologies in finding large-scale industrial applications. Low selectivity, often regarded as an inherent feature of applying plasma to gas conversion, is generally ascribed to the large temperature gradients observed in plasma reactors and high reactivity of plasmas.14 The latter results in a wide variety of reactive species (at various distinct energies), which can generate many products. For instance, in pure CH4 plasmas, the reported products can range from C(s) to fully saturated C3,4 olefins, alongside H2.15,16 Distinctly, in CH4 conversion, the product distribution can be promptly correlated to the bulk gas temperature in the reactor, which is in turn determined by the energy density of the plasma source.16
On the CH4 pyrolysis front for C(s) and H2 production, Fulcheri et al.17 have been leading tireless plasma research since the 1990s and in their recent work with an arc plasma (Tgas above 2000 °C), they successfully addressed this selectivity problem, attaining >90% conversion and >95% solid carbon selectivity.9 The developed process seems robust and has already found industrial implementation with Monolith Materials using a 1 MW pilot plasma plant to co-produce 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons of carbon black and 4600 tons of hydrogen from CH4 pyrolysis per year.
000 tons of carbon black and 4600 tons of hydrogen from CH4 pyrolysis per year.
A higher degree of process control is required when CH4 valorisation is pursued by carbon coupling, such as non-oxidative CH4 coupling (NOMC), instead of cracking. Selective plasma-based synthesis of C2H4 (the most versatile light hydrocarbon, with the highest market value) at high CH4 conversions has not yet been accomplished. To date, the highest C2H4 selectivity from CH4 coupling in plasma reactors (∼50%) was reported by Delikonstantis et al.,18 with the utilisation of nanosecond pulsed discharges in a co-axial reactor with an equimolar feed of CH4 and H2. However, this level of C2H4 selectivity was only achieved at 5 bar, with C2H2 remaining the dominant product at lower pressures, as later confirmed by kinetic modelling.19 In fact, the attainment of high C2H4 selectivity at atmospheric pressure is impeded by the thermodynamic equilibrium of gas-phase CH4 shown in the diagrams in Fig. 1.
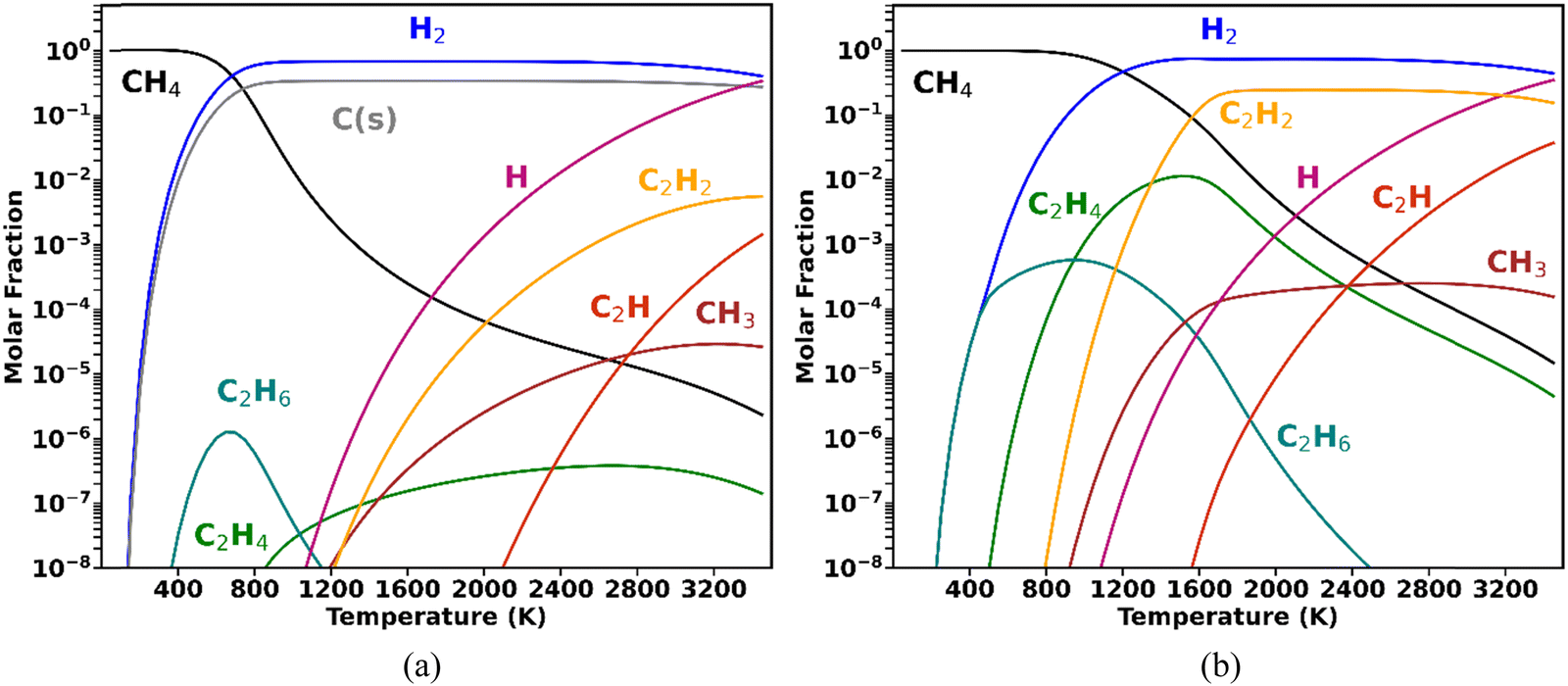 | ||
| Fig. 1 Gas-phase equilibrium composition at 1 bar, initiated with 1 mole of CH4, under two assumptions: (a) solid carbon is formed in equilibrium with the gas phase, or (b) there is no formation of a solid phase. | ||
The equilibrium compositions clearly demonstrate that when CH4 is converted under conditions favourable to solid carbon formation (Fig. 1a), the thermodynamically favoured products are C(s) and H2 (as in the work by Fulcheri et al.9), followed by C2H2, with negligible C2H4 production. On the other hand, plasma-driven CH4 pyrolysis can be performed under conditions seeking to inhibit carbon nucleation,20 which is illustrated for the ideal case in Fig. 1b (albeit some C(s) formation is inevitable in reality). In this case, the dominant products are C2H2 and H2, and although formed in appreciable concentrations, C2H4 can never become the major product and its occurrence has a very narrow temperature range.18,19
These thermodynamic trends allow for the interpretation of common experimental findings in CH4 plasmas. When a plasma operates under thermal or quasi-thermal conditions, as is the case for DC arc (employed by Monolith), gliding arcs and microwave plasmas, typically the main products observed are H2, C(s) and C2H2, with relative concentrations that depend on specific reactor configurations. Some examples can be found in ref. 9 and 21–24. Conversely, when a non-thermal plasma (such as a dielectric barrier discharge (DBD) or pulsed corona) is employed, the primary products are C2H6 (with some C2H4 generation) – mostly with low energy absorption by the gas phase, leading to poor performance in terms of CH4 conversion and energy efficiencies.25,26
Undoubtedly, this analysis reveals the essential role of catalytic C2H2 hydrogenation in plasma-based CH4 coupling for selective C2H4 synthesis with high conversion and competitive energy efficiency. The coupling of a nanosecond pulsed CH4/H2 plasma (with up to 40% C2H2 yield at an energy cost of 870 kJ mol−1)27,28 to post-plasma hydrogenation catalysis using a palladium-coated electrode structure has been performed by Cameli et al.,29 demonstrating a 60% overall C2H4 selectivity without external heat or further H2 addition. The success of this endeavour has highlighted the potential of plasma-catalyst synergy for single-pass NOMC into C2H4 in a modular fashion. Further optimisation of the structured catalyst, by employing a bimetallic Pd–Ag material, has increased the C2H4 selectivity to 76% C2H4, intensifying the process performance by lowering the downstream separation cost.30 Meanwhile, this approach shows great flexibility owing to the independent tuning of the plasma discharge and structured catalyst, which elicits research into different catalyst designs and compositions and alternative (perhaps simpler) catalytic setups, widening the scope of plasma-catalyst utilisation.
In this broader context, we have developed a temperature-dependent surface micro-kinetic model to investigate the selective hydrogenation of C2H2, synthesised from NOMC in a plate-to-plate nanosecond pulsed plasma reactor, using a downstream catalyst bed. The latter was packed with three different Pd/Al2O3 catalysts, with Pd loadings of 0.1, 0.5 and 1 wt%, which were activated by the heat created in the plasma. The combined (kinetic) modelling and experimental approach aims to explore the mechanisms of post-plasma C2H2 hydrogenation in the presence of unreacted CH4 considering the real thermal conditions in the catalyst bed downstream from the plasma zone. Building upon the current state-of-the-art,29,30 our objective is to extend the applicability of Cameli's post-plasma catalytic work by demonstrating how a classic catalyst bed can be utilised to harness plasma-generated heat and drive selective C2H2 hydrogenation. This strategy may open opportunities for the use of other metal catalysts in this process. Moreover, the new temperature-dependent surface micro-kinetic model aids in interpreting the reactivity results, providing insights into the adsorption/desorption mechanisms and reaction pathways that underlie the observed selectivity trends.
2. Experimental and computational methodology
2.1. Experimental setup
The reactor configuration has been described in detail in our previous work.29 A schematic representation of the experimental setup is reported in Fig. 2. The reactant gas consists of 100 sccm of CH4 and 100 sccm of H2 feeds (regulated by two Brooks GF40 mass flow controllers) which enter the reactor from the top of the high voltage electrode. Two parallel electrodes promote electrical breakdown of the recirculating gas. The ground electrode is composed of a 3D-printed body which hosts a stainless-steel sintered filter (AmesPore, porosity 5 μm), to prevent solid carbon deposit from entering the catalytic section downstream. Thus, the catalytic step follows the gas-phase plasma activation sequentially. An NPG-18/100k (Megaimpulse Ltd) power supply is used to ignite and sustain the nanosecond-pulsed discharge (NPD) in the plate-to-plate plasma reactor. Modulation and control of the plasma signal are attained via a waveform generator (Agilent 33220A) and an oscilloscope (Wavesurfer 10, Teledyne Lecroy). The same energy pattern is applied in all experiments, with 3000 pulses per second distributed in three bursts with a frequency of 10 kHz. The applied voltage amplitude is set at 50% of the maximum attainable by the power supply. A visual representation of the energy pattern scheme is reported in the ESI† (Fig. S1). The voltage (V) signal is used to calculate the power (P) dissipated in the discharge via a resistive coupler (RC20, Megaimpulse), which allows assessment of the forward and reflected energy (E) by measuring the voltage across the circuit with a fixed impedance (Z), as per the equation below: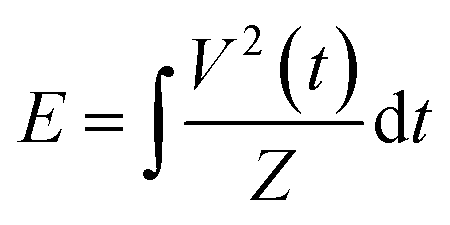
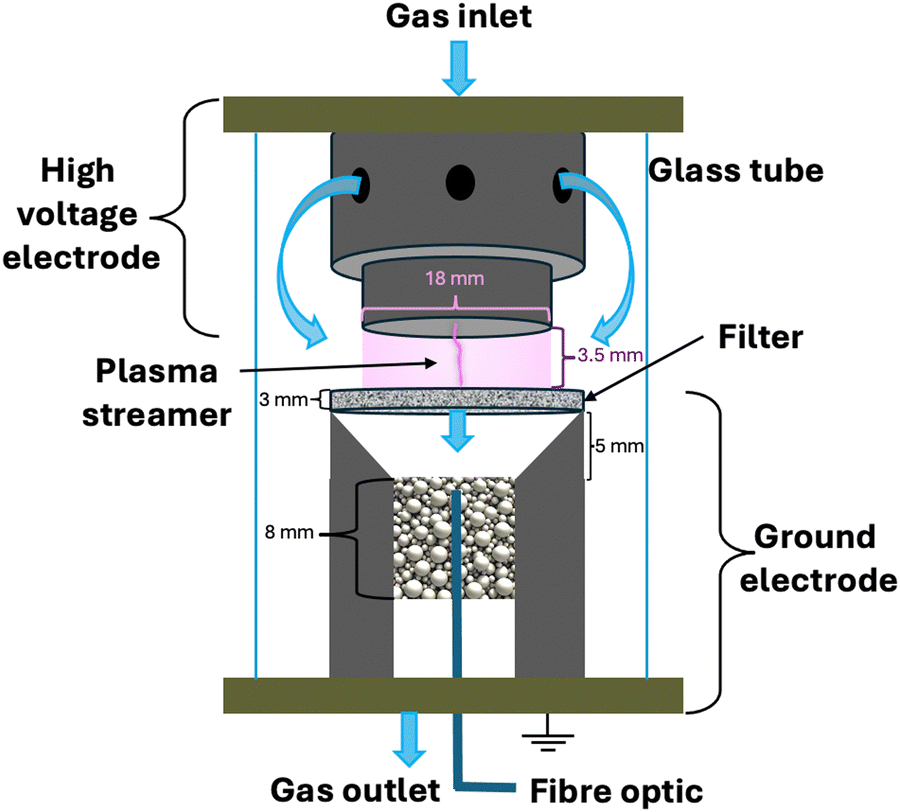 | ||
| Fig. 2 Schematic of the plate-to-plate plasma reactor with a post-plasma catalytic bed for hydrogenation. Experiments carried out with 100 sccm CH4 and 100 sccm H2, with three bursts at 10 kHz, 3000 p s−1, 50% voltage amplitude. The catalytic bed is composed of 200 mg Pd/Al2O3 and 800 mg Al2O3. | ||
2.2. Catalyst preparation and characterisation
The catalysts used in the post-plasma region were produced via incipient wetness impregnation: a solution of Pd(NO3)2 (Alpha Aesar) was used as the catalyst precursor and added to α-Al2O3 powder (Alpha Aesar, 99.95% purity, particle size: 0.25–0.45 μm, pores volume: 0.35 mL g−1). Different dilutions of the original solution containing 10 wt% Pd were prepared to attain three metal loadings of 0.1, 0.5, and 1 wt% Pd/Al2O3. Overnight drying at 120 °C and calcination at 600 °C for 6 h followed the impregnation step. Metal dispersion was assessed via pulsed H2 chemisorption (Autochem II 2920, Micromeritics) of the different catalysts. The metal load is determined via ICP-AES (iCAP 6500, Thermo Scientific) as per the ISO 11885 methodology.2.3. Downstream catalytic C2H2 hydrogenation
The post-plasma catalytic hydrogenation step is performed by packing 200 mg of catalyst, diluted in 800 mg of Al2O3, into the region downstream from the filter which serves as the ground electrode (Fig. 2). Thereby, the catalyst particles interact with the products formed via the recombination of plasma-generated radicals. Operando temperature monitoring was obtained through a FiSens fibre optic inserted in the catalyst bed. This fibre optic probe can measure multiple temperature points at 5 mm intervals along its axis. However, the length of the catalytic bed (8 mm) was only sufficient for two temperature measurements. The catalyst bed temperature was not independently regulated, as no external heating was applied during the experiments. Instead, the temperature was determined by two heat sources: the gas stream exiting the plasma region (i.e., identical heating in all experiments, given the uniform energy profile applied in the plasma), and the exothermic hydrogenation reactions occurring at the catalyst surface. Since the contribution of hydrogenation reactions to heating varies with catalyst loading, different bed temperatures were observed across the experimental conditions. The outlet stream from the system is analysed via on-line gas chromatography (3000 MicroGC, Inficon), whereby a molesieve column (10 m) with backflush (3 m, Plot U) elutes H2, N2, and CH4, whilst a Plot U column (10 m) with backflush (1 m, Plot Q) elutes C2 and C3 species. Internal standard N2 is fed directly to the GC column, to calculate the total gas volume at the outlet of the reactor, owing to the changing number of moles in the plasma coupling and post-plasma catalytic hydrogenation reactions, as outlined below. This ensures that gas expansion/contraction is properly considered for accurate appraisal of conversion and selectivity.31| 2CH4 → C2H2 + 3H2 |
| C2H2 + H2 → C2H4 + M |
| C2H4 + H2 → C2H6 + M |
2.4. Surface kinetic model
The species and reactions considered in the surface kinetic model are outlined in Table 1.
| Gas-phase species | Surface species | ||
|---|---|---|---|
| H2(g) CH4(g) C2H2(g) | H* C2H2* C2H3* C2H4* | ||
| C2H4(g) C2H6(g) | C2H5* CH3* CH2* CH* C* | ||
| Reaction | Initial state ⇌ transition state ⇌ final state | Activation energy (eV) | Reaction energy (eV) |
| r1 | H2(g) + 2 * ⇌ *–H–H–* + * ⇌ H* + H* | 0.28 | −0.83 |
| r2 | C2H2(g) + * ⇌ C2H2–* ⇌ C2H2* | 0.00 | −1.67 |
| r3 | C2H4(g) + * ⇌ C2H4–* ⇌ C2H4* | 0.00 | −0.76 |
| r4 | C2H6(g) + 2 * ⇌ *–C2H5–H–* ⇌ C2H5* + H* | 1.18 | 0.18 |
| r5 | C2H6(g) + 2 * ⇌ *–CH3–CH3–* ⇌ CH3* + CH3* | 2.89 | 0.60 |
| r6 | CH4(g) + 2 * ⇌ *–CH3–H–* ⇌ CH3* + H* | 1.29 | 0.31 |
| r7 | C2H2* + H* ⇌ *C2H2–H–* ⇌ C2H3* + * | 0.95 | 0.05 |
| r8 | C2H3* + H* ⇌ *C2H3–H–* ⇌ C2H4* + * | 0.64 | −0.48 |
| r9 | C2H4* + H* ⇌ *C2H4–H–* ⇌ C2H5* + * | 0.85 | 0.11 |
| r10 | C2H5* + * ⇌ *CH3–CH2–* ⇌ CH3* + CH2* | 2.18 | 0.74 |
| r11 | C2H4* + * ⇌ *CH2–CH2–* ⇌ CH2* + CH2* | 1.89 | 1.17 |
| r12 | C2H3* + * ⇌ *CH2–CH–* ⇌ CH2* + CH* | 1.47 | 0.34 |
| r13 | C2H2* + * ⇌ *CH–CH–* ⇌ CH* + CH* | 1.63 | 0.04 |
| r14 | CH3* + * ⇌ *CH2–H–* ⇌ CH2* + H* | 1.13 | 0.32 |
| r15 | CH2* + * ⇌ *CH–H* ⇌ CH* + H* | 0.79 | −0.35 |
| r16 | CH* + * ⇌ *C–H* ⇌ C* + H* | 1.42 | 0.38 |
| r17 | H2(g) + C2H4* + * ⇌ *–H–C2H5* ⇌ C2H5* + H* | 1.44 | −0.72 |
Our model is based on this reaction network and the Pd(111) energetics derived from density functional theory (DFT) calculations performed by Nørskov et al.32 These DFT calculations were carried out using Quantum Espresso, with the exchange–correlation contribution to the electronic energy approximated by the BEEF-vdW functional. We refer to the cited study for additional DFT details. The reaction network consists of adsorption and desorption of the gas-phase molecules – reactions r1–r6 (with H2, C2H6 and CH4 dissociating upon adsorption); hydrogenation and de-hydrogenation – reactions r7–r9, r14–r16; and surface dissociation and recombination – reactions r10–r13 and r17. In turn, the model provides quantitative information on catalyst activity and selectivity as a function of temperature. The relevant equations and formulas are presented in the following sections.
where ns is the number density of species s and t is the simulation time. The first term on the right side is related to the change in ns due to surface reactions at the catalyst, with Crs,i and Cfs,i being the stoichiometric coefficients of species s in reaction i (reverse, r, and forward, f), and ri being the reaction rate (expressed in s−1). This term must be multiplied by the total volumetric density of surface sites nsites (in cm−3, with calculation details given in Section S2 in the ESI†) to have the rate involving gas species correctly expressed in cm−3 s−1. The approach utilised to calculate the rates will be presented later in this section, following the description of the balance equation for surface species. The second and third terms represent the change in number density due to gas molecules entering and exiting the reactor, respectively. In these, ns,in is the species density at the inlet and ns,out at the outlet (note that in a CSTR model, ns,out equals the species density ns in the reactor), VCSTR is the gas volume in the reactor, and vin and vout are the volumetric flow rate entering and exiting the reactor, respectively.
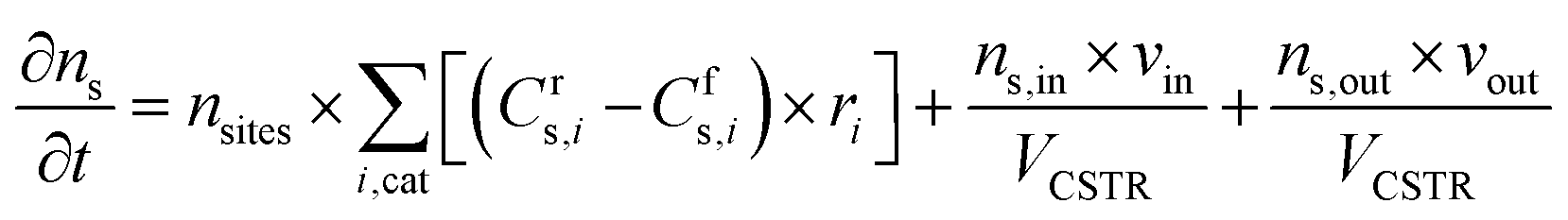
The volumetric flow rate of the exiting gas (vout) used in the equation above is calculated as follows, so that the total pressure (ptotal, expressed in Pascal here for unit consistency) in the reactor is maintained constant at ambient pressure in all simulations.
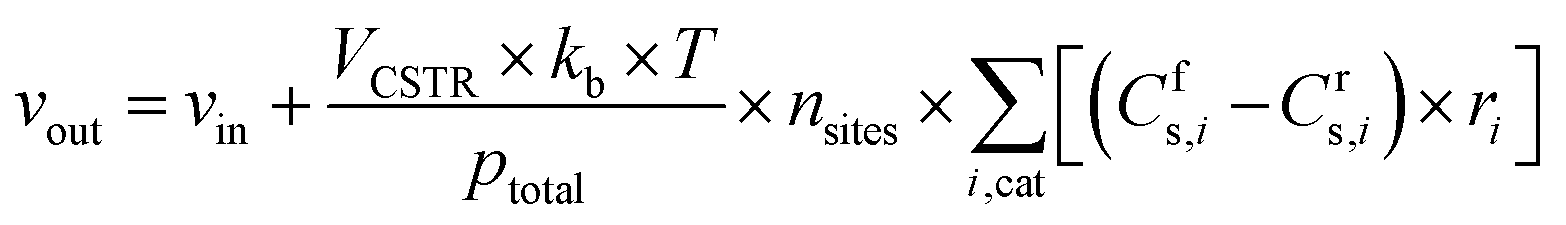
Similar to the balance equations above for the gas-phase species, the changes in the surface coverage (θs) of surface species s (Table 1) as a function of time t are described using the following balance equation, in which Cfs,i and Crs,i are the stoichiometric coefficients of species s in reaction i, with the forward (f) and reverse (r) reactions, and ri is the reaction rate of the surface reactions (again expressed in s−1).
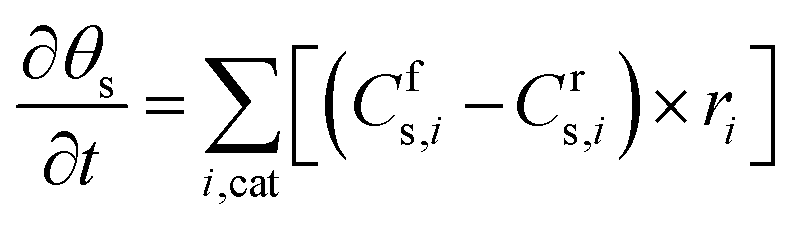
where ki,f and ki,r are the forward and reverse reaction rate coefficients, respectively, and as is the activity of species s. The latter may be the partial pressure of gas phase species (ps, divided by ptotal to maintain ki,f and ki,r in units of s−1) in surface reactions, or the fractional coverage (θs) for surface species. Note that number densities and partial pressures are interconvertible via the ideal gas law. The rate coefficients ki,f and ki,r are calculated using transition state theory with the following equation.
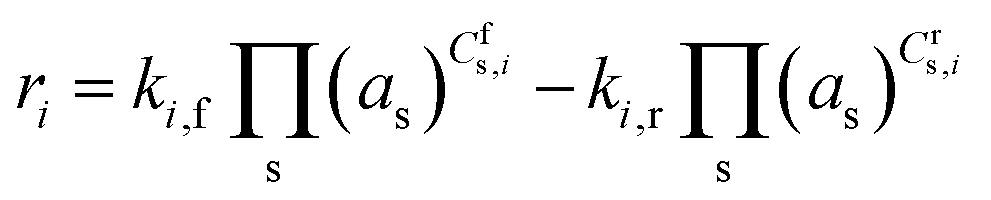
In this, h is Planck's constant, R is the universal gas constant, and ΔG‡, ΔH‡ and ΔS‡ are the differences in Gibbs free energy, enthalpy and entropy, respectively, between the initial state and the transition state (see Table 1). The inputs for enthalpy and entropy differences and their respective temperature-dependent thermodynamic corrections are discussed in the following section.
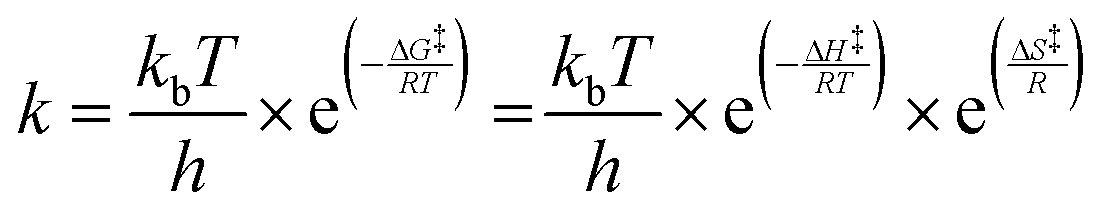
The model is solved using an in-house python code which was written for applications in post-plasma and plasma catalysis. A version of this code is quoted in Section S3 in the ESI.† The input data required to run the code include the reaction network (see Table 1), the formation enthalpies and rotational and vibrational wavenumbers of species in the model (see Table S1 in Section S4 of the ESI†), and the gas-phase species parameters for the Shomate equation (taken from the NIST database).
The temperature of the catalyst bed is also assumed to be uniform throughout the reactor and the model assumes thermal equilibrium between the gas phase and catalyst surface. While temperature gradients will exist in the reactor, their assessment would require a higher-dimensional model incorporating heat transfer mechanisms and gas flow dynamics. Such analysis, though valuable, extends beyond the capabilities of our current 0D framework, which focuses on capturing the chemical kinetic behaviour of the system.
No formation of solid products at the catalyst's surfaces (polymers and carbon black) is considered in the model. Whilst very interesting, such modelling endeavour would rely on DFT data which is presently unavailable and would require a higher dimensional model, which is not within the scope of this study. With respect to the surface reactions, the model calculates rate coefficients based on transition state theory, as explained in the above section, whilst employing DFT-derived activation barriers as input in the rate expressions. While this approach yields more accurate rates than those estimated using sticking coefficients or reaction barriers,33 it is inherently limited by the availability and quality of DFT data. In this study, all activation energies and frequencies (used to calculate entropies and temperature-dependent corrections) were extracted from the work by Nørskov et al.32 who have described the dehydrogenation of C2H6 over many close-packed metal surfaces (see Table S1 in Section S4 of the ESI†).
To account for the activity of the Pd(100) and Pd(211) facets (which may be considerable depending on nanoparticle morphology and particle size),36,37 we performed a sensitivity analysis by applying the adsorption energies for C2H2 and C2H4 on Pd(100) and Pd(211) in our micro-kinetic model (see Table S2 in the ESI†). These facets were chosen based on their respective lowest and highest reported activities in the literature.36 Due to the lack of comprehensive DFT data for all reaction species on Pd(100) and Pd(211), we retained our original reaction network developed for Pd(111) and substituted the available adsorption energies for C2H2 and C2H4. The details of this analysis can be found in Section S5 in the ESI.† The results indicate that Pd(100) is by far the least active facet, with no observable C2H2 conversion below ∼600 °C (Fig. S2a, ESI†). Conversely, Pd(211) is an overly active surface, fully hydrogenating C2H2 to C2H6 at temperatures as low as 90 °C (Fig. S2b, ESI†). These trends, albeit inherently qualitative due to the incomplete DFT datasets, do not align with our experimental results (see Fig. 3 and 4 in Section 3). Thus, we conclude that Pd(111) is the most appropriate facet to model C2H2 hydrogenation, for the conditions under study in this work.
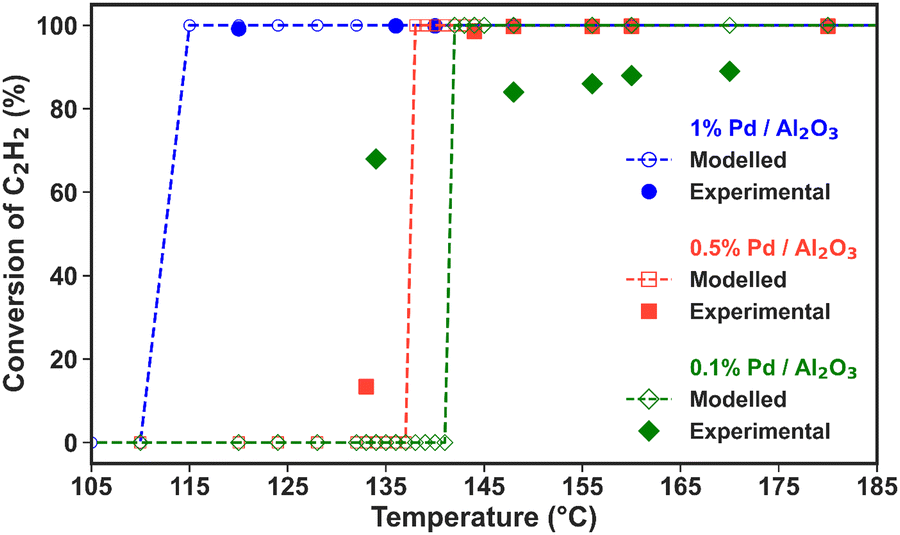 | ||
| Fig. 3 Experimental and modelled C2H2 conversion as a function of temperature (average temperature measured in the catalyst bed and assumed to be identical for solid and gas phases) for the three Pd/Al2O3 catalysts (see the legend). The number of experimental data points (solid markers) is different for each catalyst. | ||
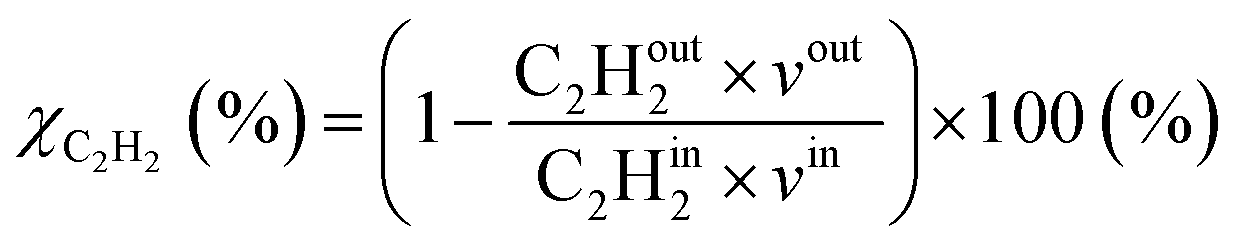
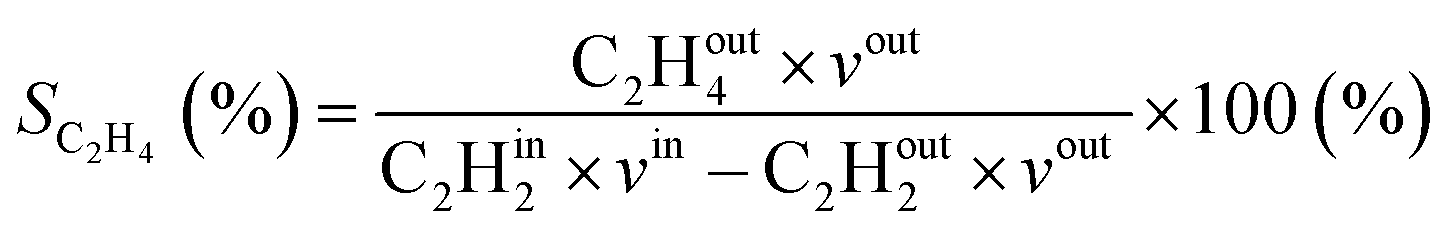
where in the experiments, C2Hout2 represents the volume fraction of C2H2 exiting the reactor (measured by GC in the coupled plasma and catalysis experiments) and vout is the outlet volumetric flow rate. Likewise, C2Hin2 represents the inlet C2H2 volume fraction (reference value, measured in the absence of catalyst, as explained above) and vin is the inlet volumetric flow rate. In the model, C2Hout2 and vout are the C2H2 density and volumetric flow rate at steady state in the outflow, C2Hin2 and vin are the initial C2H2 density and volumetric flow rate, respectively. For selectivity, C2Hout4 and C2Hout6 are GC-derived volume fractions of C2H4 and C2H6 in the outlet stream in the experiments, respectively; while in the model these are the densities of C2H4 and C2H6 in steady state, respectively (also corresponding to the outflow).
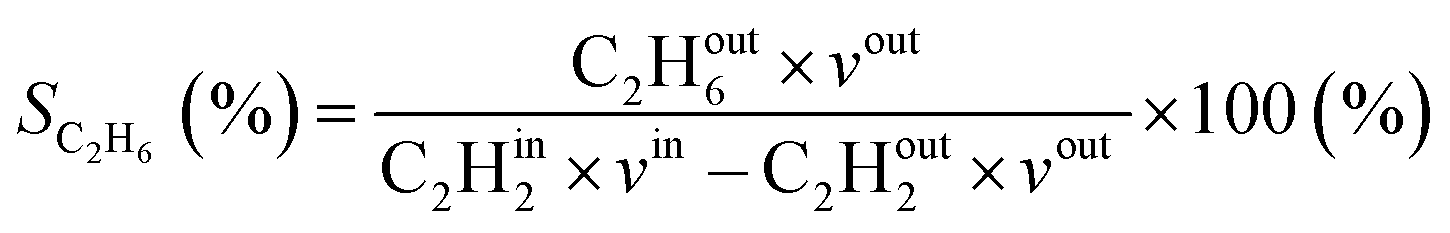
Additionally, the modelled densities are also used to calculate CH4 (SCH4) selectivity using the equation below, with CHout4 being the CH4 density in the steady state (outflow) and CHin4 being the initial CH4 density.
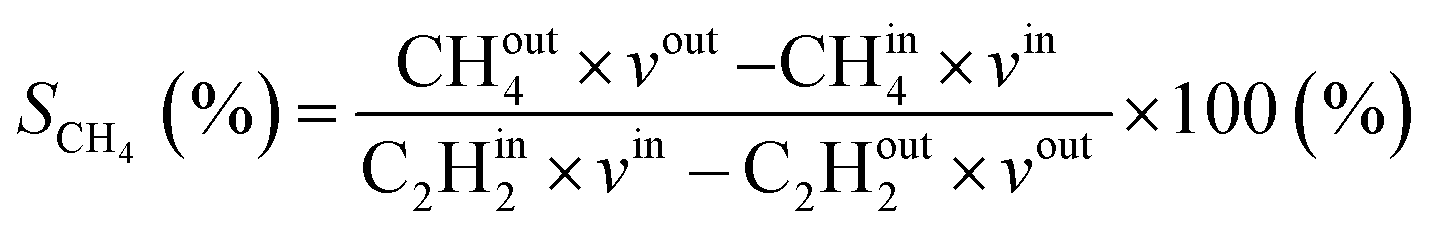
3. Results and discussion
3.1. Plasma reactor for CH4 coupling – experimental and modelled results
The gas-phase chemistry within the NPD reactor has been thoroughly characterised in our previous studies.19,27,29 A comprehensive reaction mechanism has revealed that at atmospheric pressure, CH4 coupling predominantly produces C2H2 through the stepwise dehydrogenation of C2H6, which itself is formed via the recombination of CH3 radicals.19,20,26,39 Unsurprisingly, CH3 species are the most abundant carbon-based radicals generated from CH4 dissociation via electron-impact reactions. The principal mechanism for the coupling of electron energy to gas-phase heating involves electron-impact vibrational excitation of CH4 and H2 molecules followed by rapid vibrational–translational (VT) relaxation reactions.40 The gas temperature within this NPD plasma can reach up to 1500 °C in the vicinity of the discharges, which in turn promotes dehydrogenation of C2H6 to C2H4 (but also to C2H5 and C2H3 radicals) and ultimately to C2H2.19As a result, this plasma configuration achieves a C2H2 selectivity of approximately 83% under the specified operating conditions (i.e., three bursts at 10 kHz, 3000 pulses s−1). The conversion of CH4, co-fed with an equimolar amount of H2, averages around 46%. The selectivity for C2H4 and C2H6 is about 5% and 1%, respectively. The remaining percentage is attributed to small quantities of unquantified hydrocarbon products (e.g., C3 and C4 species) and solid carbon, which amounts to about 3% of the converted CH4 in weight.
3.2. Post-plasma catalytic hydrogenation of C2H2 to C2H4
The high C2H2 selectivity attained in this NPD reactor enables effective tandem hydrogenation to C2H4, which is catalysed by the Pd/Al2O3 material placed downstream from the plasma discharge. This strategy allows for the flexible adjustment of the product distribution in a modular fashion, which can be tailored to meet oscillatory market demands. Notably, the catalytic hydrogenation reaction is solely activated by the hot gas flowing from the discharge zone, and it is self-sustained by its exothermic nature (i.e., C2H2 + H2 → C2H4, with ΔH0 = +175.9 kJ mol−1; and C2H2 + 2H2 → C2H6, with ΔH0 = +311.5 kJ mol−1). However, the three Pd loadings within the Pd/Al2O3 catalyst, along with the dispersion degree of the active metal, promote varying C2H2 conversion and C2H4/C2H6 product distribution trends within different temperature ranges. Consequently, both the reaction temperatures and kinetics may differ with the catalyst used. In the subsequent sections, we discuss the reactivity results for each catalyst within the temperature window measured in the experiments and apply our model to extrapolate the selectivity behaviour at higher and lower temperatures. Whenever possible, we compare the model predictions with our experimental data to reinforce the credibility of these temperature extrapolations.Fig. 3 also shows that the modelled results align well with the experimental data points in the region where the temperature is exclusively dictated by the plasma discharge and the exothermic heat from the hydrogenation reaction. The lack of control over the experimental temperature does not allow mapping of the catalyst activity across different temperatures in these experiments. However, these results prove that the post-plasma catalytic setup is suitable for C2H2 hydrogenation, even at low metal loading, as all catalysts attain C2H2 conversion above 90% under the experimental conditions at the steady state. The modelled trends confirm the catalysts’ high activity for C2H2 hydrogenation and can give an indication of the behaviour at higher temperatures. The modelled C2H2 conversion trends in the 80 to 750 °C temperature range can be found in Fig. S3 in the ESI† (Section S6, ESI†).
Nonetheless, the model underestimates C2H2 conversion at lower temperature for the 0.1% Pd/Al2O3 and 0.5% Pd/Al2O3 catalysts, and it suggests a sharp transition from nil to complete conversion at 137 and 142 °C, whilst experimental data show a more gradual increase. These discrepancies can be ascribed to the temperature input used in the model. The model relies on the gas temperature at the catalyst active sites, while in the experiments, the fibre optic temperature sensor is positioned along the axial axis of the catalytic bed; and it is reasonable to assume that the catalyst surface may be warmer than the surrounding gas due to the exothermic reactions occurring at the active sites. Indeed, it would be very insightful to investigate the heat transfer from the warmer catalyst surface to the gas phase and the temperature gradient in the reactor, as well as the impact of the exothermic chemical reactions, by solving an energy balance equation. However, the current model is unable to capture these effects, as the dynamics of heat transfer and gas flow cannot be accurately considered in this zero-dimensional model. While this is outside the scope of this study, in our future work, we plan the construction and application of a dedicated higher-dimensional computational fluid dynamics and surface kinetics model to explore these aspects as well.
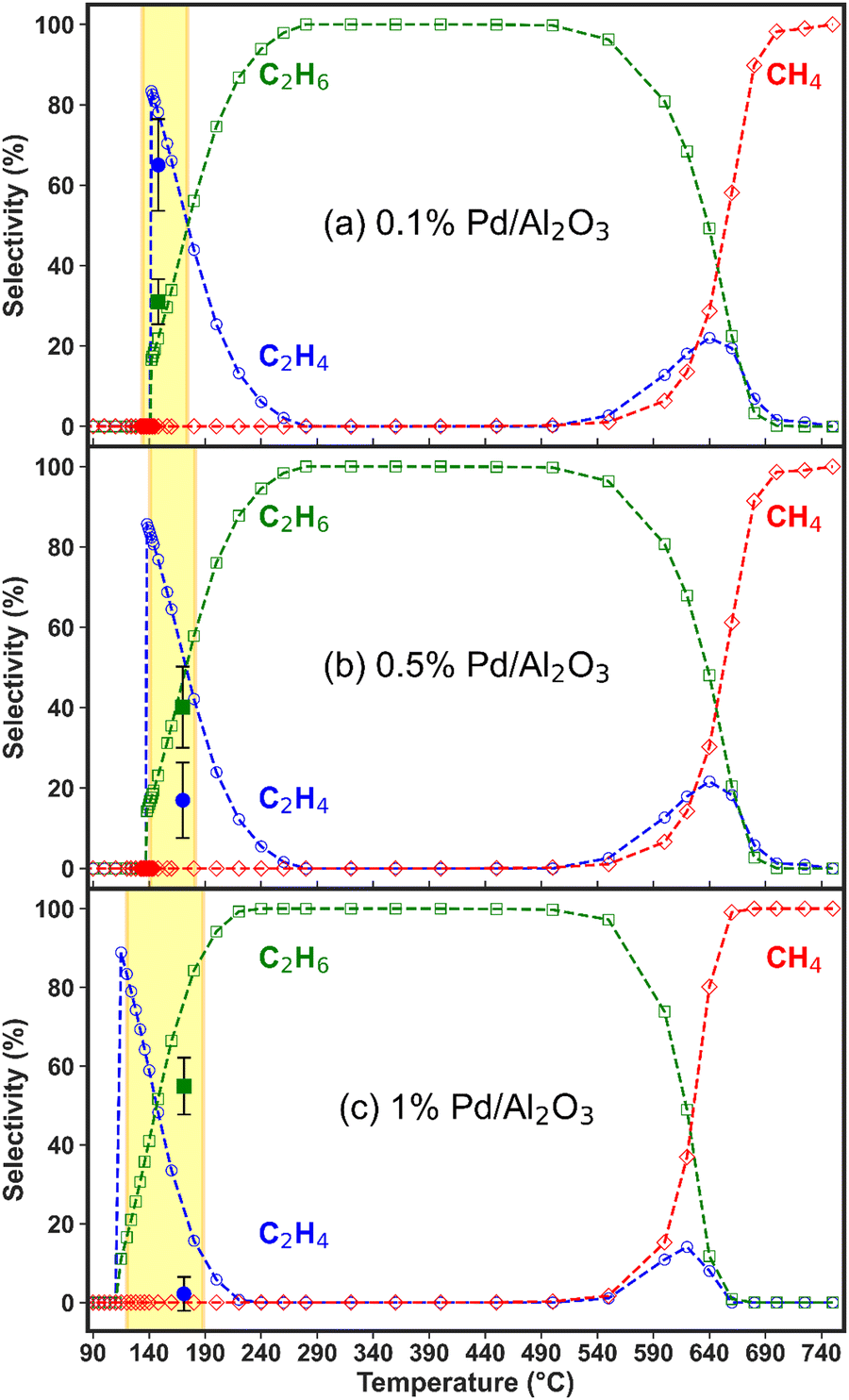 | ||
| Fig. 4 Experimental and modelled C2H4, C2H6 and CH4 selectivity as a function of temperature (average temperature measured in the catalyst bed and assumed to be identical for solid and gas phases) for the (a) 0.1%, (b) 0.5%, and (c) 1% Pd/Al2O3 catalysts. The solid markers correspond to the experimental selectivity measured within the temperature range highlighted by the shaded area (yellow rectangle) on the graph. The dashed lines and hollow markers show the trends predicted by the model. | ||
All catalysts attain near-complete C2H2 conversion in the experiments (90% for 0.1% Pd/Al2O3 and >99% for 0.5 and 1% Pd/Al2O3, see Fig. 3), which is advantageous for downstream separation steps. Thus, under these conditions, the Pd loading is the only independent variable controlling the C2H4 selectivity. Notably, the highest C2H4 yield (i.e., 12%, at 142 °C) is achieved by 0.1% Pd/Al2O3, which favours higher C2H4 selectivity despite a slightly lower C2H2 conversion.
The C2H4 yield is the major metric of successful CH4 non-oxidative coupling, as it is the most valuable product. Its maximum value is determined by the catalytic C2H2 conversion and the corresponding selectivity for C2H4, since CH4 conversion is driven solely by the plasma discharge. While the latter also affects the C2H4 yield, it does not vary with the studied Pd loadings, as the amount of C2H2 produced in the plasma region remains constant across the different Pd loadings in the post-plasma region. Similarly, the temperature of the gas exiting the plasma zone and entering the catalytic bed is identical in all experiments, as it is purely controlled by the plasma energy (uniform in all experiments). Our reactor configuration, where the catalytic bed is integrated in the post-plasma region without external heating, creates an inherent coupling between reaction temperature and catalyst loading. While the temperature of the gas exiting the plasma zone is constant across all experiments, the bed temperature varies with Pd loading due to the exothermic hydrogenation reactions. Although experiments at lower catalyst loadings could be useful to investigate incomplete conversion regimes, we focus here on conditions achieving full C2H2 conversion, which is critical for maximising C2H4 yield in industrial applications.
The modelled trend of decreasing C2H4 selectivity and increasing C2H6 selectivity upon rising temperature is qualitatively consistent for all three catalysts, with the key difference being the temperature at which C2H6 becomes the dominant product, see Fig. 4(a)–(c). This shift occurs at lower temperatures with increasing metal loading, indicating higher catalyst activity, and is detrimental for the desired overall C2H4 selectivity. Beyond this point, C2H6 selectivity becomes 100% due to C2H4 over-hydrogenation, until the temperature reaches about 500 °C, where another shift in reactivity occurs, leading to rapidly rising production of CH4 and a minor region of C2H4 formation. The thermodynamic mechanisms driving these observations are discussed in detail in the following section.
However, these higher temperatures exceed the operational range of the catalyst bed, which is limited by the temperature of the gas exiting the discharge region and the exothermic heat of the reaction. The experimental operating window of the catalyst bed (highlighted by the yellow areas in Fig. 4) corresponds to temperatures recorded over the entire hydrogenation experiment and correlate with the gas composition measured at the same acquisition time. The fast reaction kinetics results in a quick temperature rise and onset of steady-state conditions (within 10 min of plasma ignition), which does not allow the GC analysis to capture the transient composition in detail, as shown in Fig. 3. No substantial temperature increase is observed after reaching the steady-state, and all highlighted areas correspond to a relatively narrow temperature range. Given the scale difference between the modelled and experimental temperature ranges, the selectivity data from the experiments would largely overlap. For that reason, we only report one experimental point for the selectivity, as representative of the steady-state conditions.
In the experiments with the 0.1% Pd/Al2O3 catalyst, 65% C2H4 selectivity was achieved from C2H2 hydrogenation, compared to 31% C2H6 selectivity, between 140 and 175 °C (Fig. 4a), as also mentioned above. This is consistent with our previous results and other C2H2 hydrogenation reports in the literature.29,41–44 However, this state is reached within 5 min of plasma ignition and changes over longer periods, as the catalyst bed temperature rises to ∼170 °C and oligomerisation products begin to form from C2H2 conversion. This is accompanied by a drastic decline in C2 product detection in the experiments. As previously explained, oligomerisation reactions leading to solid products are not included in the model, which focuses instead on the kinetics of gaseous H2, C2H2, C2H4, C2H6, and CH4 (besides the short-lived surface species).
Nonetheless, the formation of oligomerisation by-products is the primary reason for the reduced C2 selectivity observed experimentally at higher temperatures. On this note, the carbon balance in the system drops from 91% at the start of the hydrogenation processes (at the lower end of the temperature window) to less than 70% after about 50 minutes, when the temperature of the gas-phase is expected to exceed 200 °C. Whilst these solid deposits on the catalyst surface may affect the activity, the time-on-stream data of the product gas composition shows a relatively constant trend over approximately 40 minutes (see Fig. S4 in the ESI†), suggesting that no major catalyst deactivation (via sintering, for instance) occurs.
As observed in Fig. 4(b) and (c), for both higher Pd catalyst loadings, C2H6 was detected as the primary product in the experiments, immediately after the reaction began. In fact, practically no C2H4 was detected when the 1% Pd/Al2O3 catalyst was tested, with C2H6 emerging as the sole product at ∼60% selectivity. These results are consistent with model predictions (though only at somewhat higher temperatures for the C2H4 selectivity), which suggest that C2H4 hydrogenation to C2H6 occurs at lower temperatures upon increasing Pd loading. In summary, the temperature range where C2H4 is the dominant hydrogenation product shifts to lower temperatures and narrows as the Pd loading is increased. For both catalysts, the experimental and modelled results show close alignment in terms of C2H6 selectivity, while the C2H4 selectivity is overestimated by the model in the low-temperature regime: below 240 °C for the 0.5% Pd/Al2O3 catalyst and below 210 °C for 1% Pd/Al2O3.
This discrepancy may be partially related to the difference in temperature considerations – the surface temperature input in the model versus bulk bed temperature measured in experiments, as explained above. However, the most likely factor contributing to selectivity disagreement is the extensive formation of oligomeric carbonaceous deposits (green oil)36 at full C2H2 conversion and temperatures above 170 °C, facilitated by the higher Pd loadings. Evidence supporting this mechanism is found in the calculated carbon balance of ∼76% in the experiments with both 0.5% and 1% Pd catalysts, while the hydrogen balance is greater than 91%. This indicates the formation of species with high C/H ratios, typical of oligomerisation compounds, which are not included in our model.
Additionally, an approximative evaluation of the potential impact of carbon deposition in the form of C(s) was conducted in the model by analysing the CH* + * → C* + H* reaction rate (r16). The results show that the rate of this reaction is relatively negligible across the temperature range investigated in this study (see Fig. S5 in Section S8 of the ESI†). This suggests that deposition of C(s) particles is very unlikely and cannot cause the observed differences between model predictions and the experimental data in this study. Instead, the analysis indicates that the dominant pathway is the sequential hydrogenation of CHx surface species (see Section 4), which are converted to CH4 above 500 °C (rather than dehydrogenated into solid carbon).
Also noteworthy, the formation of Pd carbide during C2H2 hydrogenation is closely associated with the deposition of carbonaceous oligomers on the catalyst surface.44 While the present model is not able to explicitly account for PdCx phases, their inclusion could, on the one hand, alter the Pd energetics of the surface reactions, leading to shifts in the model predictions. On the other hand, the model may see accumulation of PdCx species on the Pd surface, leading to lower C2H4 and (especially) C2H6 product selectivity and in turn smaller discrepancies between model and experiment. However, a detailed treatment of Pd carbide effects would require dedicated DFT data (which is not available, to the best of our knowledge) and potentially higher-dimensional modelling, beyond the scope of this study, but it represents an important direction for future research. Nevertheless, as discussed, we believe the deposition of high molecular weight oligomerisation solids on the catalyst remains the primary contributor to the disagreement in selectivity.
Finally, the morphology and size of the synthesised particles should be consistent across all tested catalysts (0.1%, 0.5%, and 1% Pd/Al2O3), as these were prepared via the same wetness impregnation method. Moreover, identical Al2O3 support particles and dilution beads were used in all experiments, further ensuring uniformity. SEM images of the spent catalysts (Fig. S6 in Section S9 of the ESI†) confirm no observable differences in particle size or morphology, indicating that these factors are not the reason for the varying catalytic performances across the three investigated Pd loadings.
Essentially, these results highlight the dual impact of higher Pd loadings: (i) they promote prompt over-hydrogenation to C2H6 by lowering the temperature at which sequential surface hydrogenation reactions occur (namely from C2H2* to C2H3* to C2H4* to C2H5* and finally C2H6; see reactions r7, r8, r9 and reverse of r4 in Table 1), and (ii) they facilitate oligomerisation reactions at lower temperatures. Therefore, optimisation of the Pd loading in these catalysts is crucial for successful coupling between the plasma and catalysis reactors, as it significantly influences the C2H4/C2H6 product distribution and the formation of unwanted solid by-products.
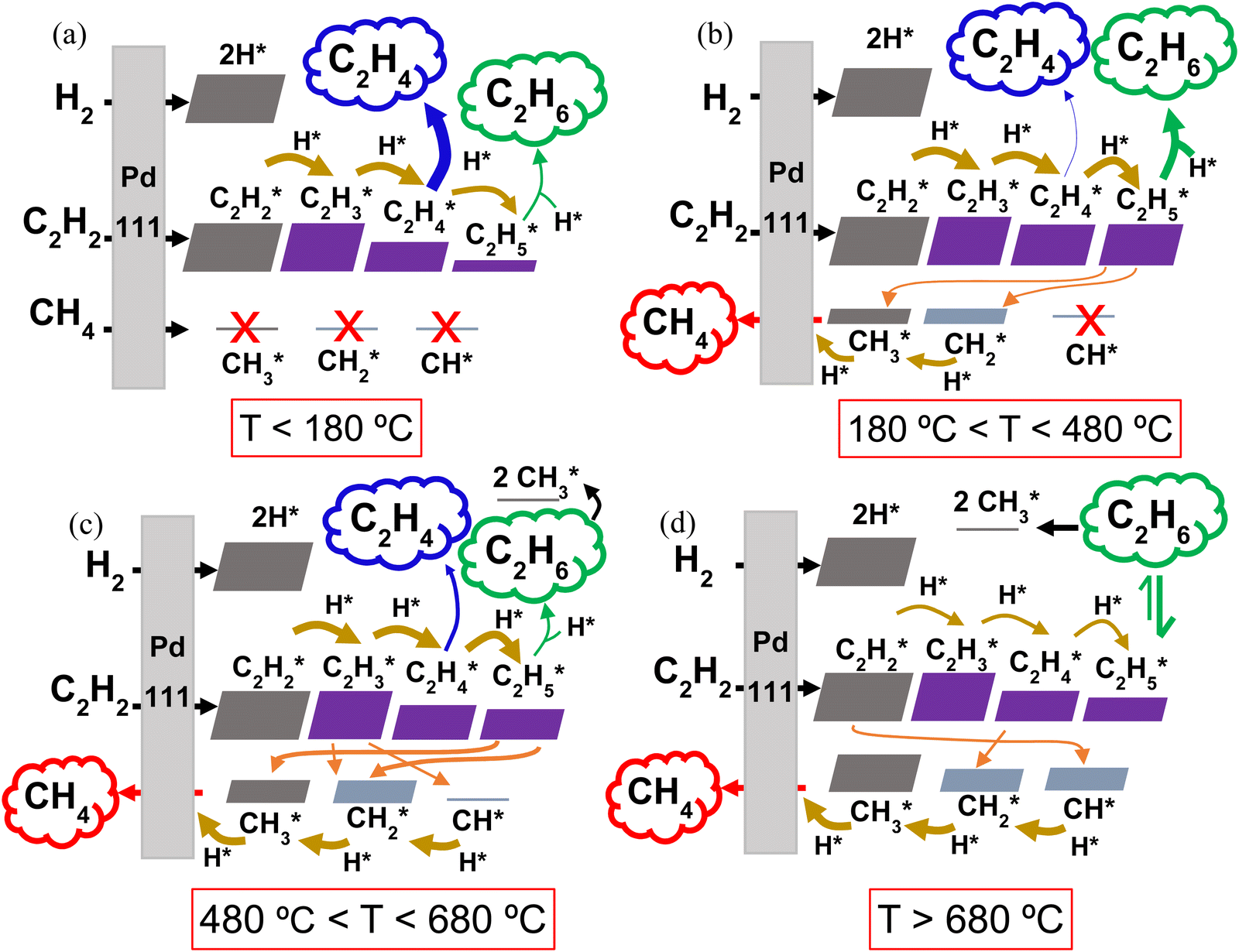 | ||
| Fig. 5 Reaction pathway diagrams illustrating the reactions between the gaseous reactants (C2H2, H2 and CH4) and the catalyst surface (a) below 180 °C, (b) between 180 and 480 °C, (c) between 480 and 680 °C and (d) above 680 °C. The resulting adsorbed surface species (marked with *) and their reactions with other surface species are also shown. The eventual desorption of the C2H4 (blue), C2H6 (green) and CH4 (red) products (with varying concentrations depending on the temperature) is indicated by the cloud-shaped text boxes. Adsorption reactions are indicated by black arrows, surface hydrogenation reactions by mustard arrows, and surface dissociation reactions by orange arrows. The thickness of the arrows and the size of the blocks are qualitatively representative of the reaction rates and species surface coverage, respectively. | ||
Within the studied temperature range, the adsorption of both H2 (into 2H*) and C2H2 (into C2H2*), onto the Pd surface occurs very rapidly, with rates approaching 1025 s−1. Following the formation of C2H2* and H*, sequential surface hydrogenation reactions (r7, r8 and r9 in Table 1) proceed, generating C2H3*, C2H4* and C2H5* species – with varying surface coverage, depending on the temperature (as shown in Fig. 5). At temperatures below 180 °C (Fig. 5a), desorption of C2H4* (reverse of reaction r3) is preferred, leading to the evolution of C2H4(g) over further hydrogenation to C2H5* (r9). This preference results in the predominance of the desired C2H4 product at low temperatures, aligning with the selectivity results observed in our experiments with the 0.1% Pd/Al2O3 catalyst. The desorption of C2H4* into C2H4(g) peaks at 144 °C, coincide with the highest C2H4 selectivity. In this temperature range, the coverage of C1 surface species remains negligible, as dissociative desorption of CH4 (r6) does not occur.
As the temperature rises, the rate of C2H4* desorption decreases, whilst the rates of C2H4* hydrogenation to C2H5* and subsequent hydrogenation to C2H6(g) (r4) increase significantly (Fig. S7 and Fig. 5b, ESI†). This behaviour was also observed by Wang et al.43 and Shi et al.44 for C2H2 hydrogenation over Cu and Au-based catalysts. As a result, the C2H6 product sees a rise in selectivity and it becomes the main product from ∼180 °C.
As the temperature is further increased, the desorption rate of C2H4* falls sharply, dropping to approximately zero around 280 °C. Simultaneously, the hydrogenation reactions to C2H5* and C2H6(g) accelerate, making C2H6 the only product seen by the model between 280 and 450 °C. These rates are plotted in Fig. S8 (ESI†). However, once the temperature reaches 450 °C, the dissociation of C2H5* into CH3* and CH2* (r10) begins to occur, as shown in Fig. 5b, competing with the hydrogenation to C2H6. In turn, this gives rise to the production of CH4(g), as CH3* undergoes surface hydrogenation (reverse of the r6 reaction, as detailed in Fig. S9, ESI†). Thus, at temperatures above 450 °C, CH4 becomes a product of C2H2 hydrogenation over this Pd catalyst, with its selectivity rising rapidly with temperature (cf. Fig. 4 above).
Interestingly, for all three Pd loadings, the model predicts the reappearance of C2H4 as a product between ∼500 and 710 °C, depending on the Pd loading (see Fig. 4 above), with a local maximum in C2H4 selectivity around 600 °C. This trend is corroborated by our rate analysis, which indicates a second region of C2H4* desorption into C2H4(g) (reverse of r3) within this temperature range (see Fig. S10 and Fig. 5c, ESI†). Concurrently, the dissociation rate of C2H5* into CH3* and CH2* (r10) steadily rises from 500 °C, and C2H3* also begins to undergo surface dissociation into CH2* and CH* (r12) (see Fig. 5c). These dissociation processes explain the sharp decline in C2H6 selectivity at elevated temperatures (cf. Fig. 4), as they compete with the hydrogenation steps required to form the C2H4 and C2H6 products. Indeed, the reaction rates of both C2H4* and C2H5* + H* (r9 and r4) dwindle with increasing temperature (Fig. S10, ESI†). Besides, the observed waning C2H6 production can also be ascribed to dissociative adsorption of C2H6(g) into 2CH3* (reverse of r5) (cf. Fig. 5c), which becomes significant from 600 °C onwards. Collectively, these reactions contribute to enhancing the catalyst surface coverage with CHx* species, ultimately resulting in the evolution of CH4(g) – which becomes the dominant product above ∼650 °C.
Beyond 680 °C, additional dissociation reactions begin to take place alongside those discussed above. These are the dissociation of C2H2* into 2CH* (r13), C2H4* into 2CH2* (r11) and C2H6(g) into C2H5* and H* (reverse of r4), as illustrated in Fig. 5d, with rates shown in Fig. S11 (ESI†). At the same time, all C2Hy* hydrogenations become slower, while the hydrogenation rates of CH* (reverse of r15) and CH2* steadily increase (cf. thickness of the arrows in Fig. 5d). As a result, the formation of C2 hydrocarbons is further weakened, becoming negligible above 700 °C. Meanwhile, the CH4 production peaks, approaching 100% selectivity at 750 °C and explaining the trend in Fig. 4.
4. Conclusions
In this work, we developed a surface micro-kinetic model that simulates the catalytic C2H2 hydrogenation to C2H4, following plasma-based non-oxidative CH4 coupling. We validated the model against experiments using an NPD plasma reactor chemically and thermally integrated with a downstream catalyst bed, employing different Pd/Al2O3 materials with Pd loadings of 0.1%, 0.5% and 1%. All three catalysts effectively convert the C2H2 present in the exiting gas stream from the plasma discharge without external heating, with conversions above 90%. By applying fixed plasma conditions, we attain a consistent CH4 conversion around 46%, and C2H2 selectivity around 83% for the plasma stage, and thus post-plasma catalytic C2H2 hydrogenation into C2H4 is the main driver of the final C2H4 yield. Our experiments show that only the 0.1% Pd/Al2O3 catalyst evolves C2H4 as the main hydrogenation product, while at higher Pd loadings, C2H6 is predominantly produced. Our model predicts a slightly different trend, with higher C2H4 selectivity for all catalysts in the low-temperature regime, though C2H6 rapidly becomes predominant above 150–180 °C. The highest C2H4 selectivity is obtained at the lowest simulated temperature (i.e., 142 °C, 135 °C and 115 °C for the 0.1%, 0.5% and 1% Pd/Al2O3 catalysts, respectively). In the experiments, other oligomerisation by-products are also observed, but they are not included in the model, which might explain the observed discrepancy.Our results also show that the exothermic nature of the hydrogenation reactions induces a temperature rise within the catalyst bed, which is detrimental to the C2H4 selectivity. This is in line with the modelled reaction mechanism, which reveals that by increasing the catalyst temperature, the C2H4* desorption rate is lowered, while further hydrogenation is favoured. As this effect is highly undesirable, the deployment of more selective catalysts (e.g., mixed metal alloys)45,46 able to fine-tune these rates may enhance the process performance.
Other key factors may influence the coupling between the plasma reactor and the catalyst bed. We believe these include the type of catalyst and support (i.e., oxides versus metal-based), the plasma source, reactor geometry, the magnitude and type of gas flow, type of filter, and the distance between the bed and discharge region. Ultimately, these factors directly impact heat transfer, and in turn the catalyst temperature, which, as shown in this work, significantly affects the selective synthesis of C2H4. Tailoring these variables allows for fine control over the catalyst bed temperature, thereby maximising C2H4 selectivity (and yield) and catalyst performance. This research opens avenues for further exploration of coupling different plasma sources and reactors with simple post-plasma catalytic setups. Potentially, the flexibility in adjusting the catalyst bed temperature through the aforementioned factors may allow for the use of cheaper and more abundant catalysts (such as Cu and Ni), which generally require higher activation temperatures.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support by the Flemish Government through the Moonshot cSBO project “Power-to-Olefins” (P2O; HBC.2020.2620), the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 810182-SCOPE ERC Synergy project), and the Methusalem grant of the University of Antwerp. Fabio Cameli acknowledges the European Union for funding through the HORIZON-WIDERA-2023-TALENTS-02 program, project 101180605.References
- I. Amghizar, L. A. Vandewalle, K. M. Van Geem and G. B. Marin, New Trends in Olefin Production, Engineering, 2017, 3, 171–178, DOI:10.1016/J.ENG.2017.02.006.
- A. Bogaerts and E. C. Neyts, Plasma Technology: An Emerging Technology for Energy Storage, ACS Energy Lett., 2018, 3, 1013–1027, DOI:10.1021/acsenergylett.8b00184.
- R. Snoeckx and A. Bogaerts, Plasma technology-a novel solution for CO2 conversion?, Chem. Soc. Rev., 2017, 46, 5805–5863, 10.1039/c6cs00066e.
- E. Delikonstantis, E. Igos, M. Augustinus, E. Benetto and G. D. Stefanidis, Life cycle assessment of plasma-assisted ethylene production from rich-in-methane gas streams, Sustainable Energy Fuels, 2020, 4, 1351–1362, 10.1039/c9se00736a.
- E. Delikonstantis, F. Cameli, M. Scapinello, V. Rosa, K. M. Van Geem and G. D. Stefanidis, Low-carbon footprint chemical manufacturing using plasma technology, Curr. Opin. Chem. Eng., 2022, 38, 100857, DOI:10.1016/j.coche.2022.100857.
- O. Mynko, I. Amghizar, D. J. Brown, L. Chen, G. B. Marin, R. F. de Alvarenga, D. C. Uslu, J. Dewulf and K. M. Van Geem, Reducing CO2 emissions of existing ethylene plants: evaluation of different revamp strategies to reduce global CO2 emission by 100 million tonnes, J. Cleaner Prod., 2022, 362, 132127, DOI:10.1016/j.jclepro.2022.132127.
- P. Lamichhane, N. Pourali, L. Scott, N. N. Tran, L. Lin, M. E. Gelonch, E. V. Rebrov and V. Hessel, Critical review: ‘Green’ ethylene production through emerging technologies, with a focus on plasma catalysis, Renewable Sustainable Energy Rev., 2024, 189, 114044, DOI:10.1016/j.rser.2023.114044.
- T. Nozaki, D. Y. Kim and X. Chen, Plasma-enabled electrification of chemical processes toward decarbonization of society, Jpn. J. Appl. Phys., 2024, 63, 030101, DOI:10.35848/1347-4065/ad280f.
- L. Fulcheri, V. J. Rohani, E. Wyse, N. Hardman and E. Dames, An energy-efficient plasma methane pyrolysis process for high yields of carbon black and hydrogen, Int. J. Hydrogen Energy, 2023, 48, 2920–2928, DOI:10.1016/j.ijhydene.2022.10.144.
- F. Girard-Sahun, O. Biondo, G. Trenchev, G. van Rooij and A. Bogaerts, Carbon bed post-plasma to enhance the CO2 conversion and remove O2 from the product stream, Chem. Eng. J., 2022, 442, 136268, DOI:10.1016/j.cej.2022.136268.
- S. Kelly and A. Bogaerts, Nitrogen fixation in an electrode-free microwave plasma, Joule, 2021, 5, 3006–3030, DOI:10.1016/j.joule.2021.09.009.
- M. Tatar, V. Vashisth, M. Iqbal, T. Butterworth, G. van Rooij and R. Andersson, Analysis of a plasma reactor performance for direct nitrogen fixation by use of three-dimensional simulations and experiments, Chem. Eng. J., 2024, 497, 154756, DOI:10.1016/j.cej.2024.154756.
- M. Scapinello, E. Delikonstantis and G. D. Stefanidis, The panorama of plasma-assisted non-oxidative methane reforming, Chem. Eng. Process. – Process Intensif., 2017, 117, 120–140, DOI:10.1016/j.cep.2017.03.024.
- A. Fridman, Plasma chemistry, Cambridge University Press, 2008, 9780521847 DOI:10.1017/CBO9780511546075.
- S. Heijkers, M. Aghaei and A. Bogaerts, Plasma-Based CH4 Conversion into Higher Hydrocarbons and H2: Modeling to Reveal the Reaction Mechanisms of Different Plasma Sources, J. Phys. Chem. C, 2020, 124, 7016–7030, DOI:10.1021/acs.jpcc.0c00082.
- M. Wnukowski, Methane Pyrolysis with the Use of Plasma: Review of Plasma Reactors and Process Products, Energies, 2023, 16, 6441, DOI:10.3390/en16186441.
- L. Fulcheri and Y. Schwob, From methane to hydrogen, carbon black and water, Int. J. Hydrogen Energy, 1995, 20, 197–202, DOI:10.1016/0360-3199(94)E0022-Q.
- M. Scapinello, E. Delikonstantis and G. D. Stefanidis, Direct methane-to-ethylene conversion in a nanosecond pulsed discharge, Fuel, 2018, 222, 705–710, DOI:10.1016/j.fuel.2018.03.017.
- E. Morais, E. Delikonstantis, M. Scapinello, G. Smith, G. D. Stefanidis and A. Bogaerts, Methane coupling in nanosecond pulsed plasmas: correlation between temperature and pressure and effects on product selectivity, Chem. Eng. J., 2023, 462, 142227, DOI:10.1016/j.cej.2023.142227.
- M. Wnukowski, A. W. van de Steeg, B. Hrycak, M. Jasiński and G. J. van Rooij, Influence of hydrogen addition on methane coupling in a moderate pressure microwave plasma, Fuel, 2021, 288, 119674, DOI:10.1016/j.fuel.2020.119674.
- K. Konno, K. Onoe, Y. Takiguchi and T. Yamaguchi, Direct Preparation of Hydrogen and Carbon Nanotubes by Microwave Plasma Decomposition of Methane over Fe/Si Activated by Biased Hydrogen Plasma, Green Sustainable Chem., 2013, 03, 19–25, DOI:10.4236/gsc.2013.31004.
- A. Khrabry, I. D. Kaganovich, Y. Barsukov, S. Raman, E. Turkoz and D. Graves, Compact and accurate chemical mechanism for methane pyrolysis with PAH growth, Int. J. Hydrogen Energy, 2024, 56, 1340–1360, DOI:10.1016/j.ijhydene.2023.12.175.
- T. Minea, D. C. M. van den Bekerom, F. J. J. Peeters, E. Zoethout, M. F. Graswinckel, M. C. M. van de Sanden, T. Cents, L. Lefferts and G. J. van Rooij, Non-oxidative methane coupling to C2 hydrocarbons in a microwave plasma reactor, Plasma Process. Polym., 2018, 15, 1–16, DOI:10.1002/ppap.201800087.
- J. R. Fincke, R. P. Anderson, T. Hyde, B. A. Detering, R. Wright, R. L. Bewley, D. C. Haggard and W. D. Swank, Plasma Thermal Conversion of Methane to Acetylene, Plasma Chem. Plasma Process., 2002, 22, 105–136, DOI:10.1023/A:1012944615974.
- N. García-Moncada, G. van Rooij, T. Cents and L. Lefferts, Catalyst-assisted DBD plasma for coupling of methane: minimizing carbon-deposits by structured reactors, Catal. Today, 2021, 369, 210–220, DOI:10.1016/J.CATTOD.2020.04.028.
- P. A. Maitre, M. S. Bieniek and P. N. Kechagiopoulos, Modelling excited species and their role on kinetic pathways in the non-oxidative coupling of methane by dielectric barrier discharge, Chem. Eng. Sci., 2021, 234, 116399, DOI:10.1016/j.ces.2020.116399.
- E. Delikonstantis, M. Scapinello, O. Van Geenhoven and G. D. Stefanidis, Nanosecond pulsed discharge-driven non-oxidative methane coupling in a plate-to-plate electrode configuration plasma reactor, Chem. Eng. J., 2020, 380, 122477, DOI:10.1016/j.cej.2019.122477.
- E. Delikonstantis, M. Scapinello and G. D. Stefanidis, Low energy cost conversion of methane to ethylene in a hybrid plasma-catalytic reactor system, Fuel Process. Technol., 2018, 176, 33–42, DOI:10.1016/J.FUPROC.2018.03.011.
- F. Cameli, M. Scapinello, E. Delikonstantis, F. Sascha Franchi, M. Ambrosetti, L. Castoldi, G. Groppi, E. Tronconi and G. D. Stefanidis, Intensification of plasma-catalytic processes via additive manufacturing. Application to non-oxidative methane coupling to ethylene, Chem. Eng. J., 2024, 482, 148720, DOI:10.1016/j.cej.2024.148720.
- F. Cameli, M. Scapinello, E. Delikonstantis and G. D. Stefanidis, Electrified methane upgrading via non-thermal plasma: intensified single-pass ethylene yield through structured bimetallic catalyst, Chem. Eng. Process. – Process Intensif., 2024, 204, 109946, DOI:10.1016/j.cep.2024.109946.
- B. Wanten, R. Vertongen, R. De Meyer and A. Bogaerts, Plasma-based CO2 conversion: How to correctly analyze the performance?, J. Energy Chem., 2023, 86, 180–196, DOI:10.1016/j.jechem.2023.07.005.
- M. H. Hansen, J. K. Nørskov and T. Bligaard, First principles micro-kinetic model of catalytic non-oxidative dehydrogenation of ethane over close-packed metallic facets, J. Catal., 2019, 374, 161–170, DOI:10.1016/j.jcat.2019.03.034.
- B. Loenders, R. Michiels and A. Bogaerts, Is a catalyst always beneficial in plasma catalysis? Insights from the many physical and chemical interactions, J. Energy Chem., 2023, 85, 501–533, DOI:10.1016/j.jechem.2023.06.016.
- H. Ma and W. F. Schneider, Plasma-catalyst modeling for materials selection: challenges and opportunities in nitrogen oxidation, J. Phys. D: Appl. Phys., 2021, 54, 12, DOI:10.1088/1361-6463/ac1bd1.
- V. Petrazzuoli, M. Rolland, V. Sassanis, V. Ngu, Y. Schuurman and L. Gamet, Numerical prediction of Péclet number in small-sized fixed bed reactors of spheres, Chem. Eng. Sci., 2021, 240, 116667, DOI:10.1016/j.ces.2021.116667.
- B. Yang, R. Burch, C. Hardacre, G. Headdock and P. Hu, Influence of surface structures, subsurface carbon and hydrogen, and surface alloying on the activity and selectivity of acetylene hydrogenation on Pd surfaces: a density functional theory study, J. Catal., 2013, 305, 264–276, DOI:10.1016/j.jcat.2013.05.027.
- A. Kordatos, K. Mohammed, R. Vakili, H. Manyar, A. Goguet, E. Gibson, M. Carravetta, P. Wells and C. K. Skylaris, Bridging the size gap between experiment and theory: large-scale DFT calculations on realistic sized Pd particles for acetylene hydrogenation, RSC Adv., 2024, 14, 27799–27808, 10.1039/d4ra03369h.
- K. Van, T. Veer, F. Reniers and A. Bogaerts, Zero-dimensional modeling of unpacked and packed bed dielectric barrier discharges: the role of vibrational kinetics in ammonia synthesis, Plasma Sources Sci. Technol., 2020, 29, 045020, DOI:10.1088/1361-6595/ab7a8a.
- S. Ravasio and C. Cavallotti, Analysis of reactivity and energy efficiency of methane conversion through non thermal plasmas, Chem. Eng. Sci., 2012, 84, 580–590, DOI:10.1016/j.ces.2012.09.012.
- E. Morais and A. Bogaerts, Modelling the dynamics of hydrogen synthesis from methane in nanosecond-pulsed plasmas, Plasma Process. Polym., 2023, 1–10, DOI:10.1002/ppap.202300149.
- R. Liu, E. Morais, D. Li, P. Liu, Q. Chen, S. Li, L. Wang, X. Gao, A. Bogaerts, H. Guo and Y. Yi, Hybrid plasma catalysis-thermal system for non-oxidative coupling of methane to ethylene and hydrogen, Chem. Eng. J., 2024, 498, 155733, DOI:10.1016/j.cej.2024.155733.
- M. Scapinello, E. Delikonstantis and G. D. Stefanidis, A study on the reaction mechanism of non-oxidative methane coupling in a nanosecond pulsed discharge reactor using isotope analysis, Chem. Eng. J., 2019, 360, 64–74, DOI:10.1016/j.cej.2018.11.161.
- S. Wang, K. Uwakwe, L. Yu, J. Ye, Y. Zhu, J. Hu, R. Chen, Z. Zhang, Z. Zhou, J. Li, Z. Xie and D. Deng, Highly efficient ethylene production via electrocatalytic hydrogenation of acetylene under mild conditions, Nat. Commun., 2021, 12, 7072, DOI:10.1038/s41467-021-27372-8.
- X. Shi, Y. Lin, L. Huang, Z. Sun, Y. Yang, X. Zhou, E. Vovk, X. Liu, X. Huang, M. Sun, S. Wei and J. Lu, Copper Catalysts in Semihydrogenation of Acetylene: From Single Atoms to Nanoparticles, ACS Catal., 2020, 10, 3495–3504, DOI:10.1021/acscatal.9b05321.
- M. Jørgensen and H. Grönbeck, Selective Acetylene Hydrogenation over Single-Atom Alloy Nanoparticles by Kinetic Monte Carlo, J. Am. Chem. Soc., 2019, 141, 8541–8549, DOI:10.1021/jacs.9b02132.
- Y. Qi, B. Wang, M. Fan, D. Li and R. Zhang, C2H2 semi-hydrogenation on the metal M (M = Cu, Ag, Au) alloyed single-atom Pd catalysts: effects of Pd coordination number and environment on the catalytic performance, Chem. Eng. Sci., 2021, 243, 116786, DOI:10.1016/j.ces.2021.116786.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ey00203b |
| ‡ Shared first authors. |
| This journal is © The Royal Society of Chemistry 2025 |