
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBromine-mediated electrochemical refinery towards tartaric acid†
Chenglin
Liang
ab,
Zhaoyu
Wen
ab,
Yuchen
Yan
ab,
Zhenghao
Mao
ab,
Na
Han
 *ab and
Yanguang
Li
*abc
*ab and
Yanguang
Li
*abc
aInstitute of Functional Nano & Soft Materials (FUNSOM), Soochow University, Suzhou, 215123, China. E-mail: yanguang@suda.edu.cn; hanna@suda.edu.cn
bJiangsu Key Laboratory for Advanced Negative Carbon Technologies, Soochow University, Suzhou 215123, China
cMacao Institute of Materials Science and Engineering (MIMSE), MUST–SUDA Joint Research Center for Advanced Functional Materials, Macau University of Science and Technology, Taipa, 999078, Macau SAR, China
First published on 28th January 2025
Abstract
Tartaric acid is an important ingredient widely used in the food, pharmaceutical, and cosmetic sectors. Over recent years, its rising industrial significance and market demand have called for more efficient and sustainable production strategies than those traditionally available. Herein, we propose bromine-mediated conversion from maleic acid—a common biomass derivative to tartaric acid. To this end, hierarchical Mo2C nanostructures are prepared as an efficient electrocatalyst for the bromine evolution reaction at close to the theoretical potential. Bromine generated at the anode then undergoes disproportionation to HOBr, and mediates the transformation of maleic acid to epoxysuccinic acid with high selectivity, which finally hydrolyzes to tartaric acid. Our demonstrated new approach circumvents the issue of product overoxidation usually encountered with direct electrochemical oxidation, and is thereby more energy efficient and atom economic.
Broader contextThe selective transformation of biomass-derived feedstocks into high-value chemicals has emerged as a prominent research focus. Tartaric acid, a valuable dicarboxylic acid with widespread applications in the food, pharmaceutical, and cosmetic industries, is traditionally produced through extraction from wine by-products or chemical oxidation. However, these methods suffer from low yields, complex processing, and significant waste generation. To address these challenges, we propose a bromine-mediated electrochemical refinery for the conversion of maleic acid, a biomass-derived platform chemical, into tartaric acid with high yield and selectivity. This approach offers a transformative approach for sustainable tartaric acid production, advancing catalytic biomass valorization and highlighting the potential of bromine-mediated refineries in green chemical manufacturing. |
1. Introduction
Tartaric acid (TTA) is an important dicarboxylic organic acid with widespread industrial applications. It is commonly used as an acidulant to adjust flavor profiles in the food industry and as a vital component in pharmaceutical formulations and polyester production. Market forecasts highlight the growing demand for TTA, predicting a compound annual growth rate of 5.1% that would increase its market value to USD 341.85 million by 2028.1 TTA is naturally found in a variety of fruits (particularly grapes) and traditionally extracted from wine fermentation by-products (Fig. 1a). However, this extraction process is usually inefficient and complex. Another method involves the oxidation of glucose with strong oxidants (such as nitric acid or NOx), but it is unfortunately limited by the inherently low yield and concomitant generation of hazardous wastes (Fig. 1b).2,3 As a result, there is a pressing need to develop more efficient and sustainable strategies for TTA production in order to meet its rising industrial demand.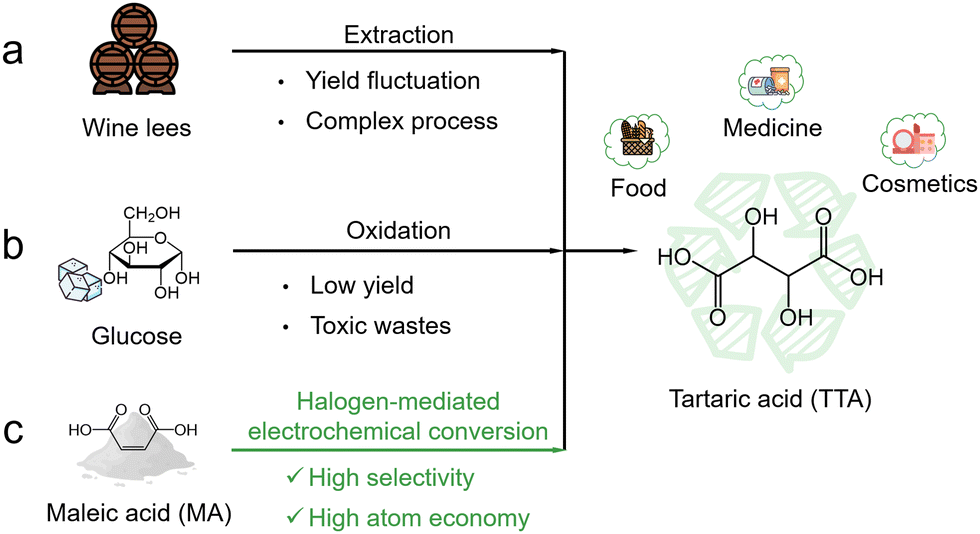 | ||
| Fig. 1 Comparison of different production routes for TTA. (a) Extraction from wine lees. (b) Glucose oxidation. (c) Halogen-mediated electrochemical conversion of MA. | ||
An electrochemical refinery powered by renewable energy presents a promising approach for converting small molecules to high-value chemicals, and plays a significant role in carbon-neutral chemical manufacturing.4–6 A plethora of examples have shown that oxidative or reductive catalytic reactions can be driven electrochemically with greater energy efficiency and atom economy.7,8 Maleic acid (MA) is a common biomass derivative, and can be used as the feedstock to the synthesis of many important chemicals. It can be epoxidized by concentrated H2O2 in the presence of catalysts such as alkali tungstate, and upon hydrolysis can be further converted to TTA.9 The direct electrochemical oxidation of MA has been studied, but it is, however, plagued by the competing water oxidation reaction and excessive C–C bond cleavage, giving rise to the formation of oxalic acid, formic acid, and even CO2.10,11 No electrochemical methodology is currently available for selectively converting MA to TTA without overoxidation to our best knowledge.
Very recently, halogen-mediated electrochemical reactions have received growing interest as a possible alternative to direct electrochemical oxidation.12 For example, chlorine and bromine have been demonstrated as the redox mediators at the anode for the oxidative epoxidation of alkenes such as ethylene and propylene, giving rise to the formation of epoxides or diols.13–15 These halogen-mediated reactions take place at significantly lower anodic potentials (hence higher energy efficiency), and exhibit improved selectivity towards target products. However, owing to the complex reaction schemes, their full potential has been inadequately explored. Following the guideline of this strategy, we here propose the bromine-mediated electrochemical conversion of MA to TTA (Fig. 1c). Upon oxidation, bromine generated at the anode is disproportionated to form hypobromous acid (HOBr) and selectively reacts with MA to form epoxysuccinic acid (ESA), which finally hydrolyzes to TTA at a high conversion rate. Such a hybrid route circumvents the undesirable overoxidation that direct oxidation reactions always encounter.
2. Results and discussion
Halogen-mediated electrochemical reactions initiate with the oxidation of halogen anions at the anode. Among them, bromine-mediated processes offer significant advantages over more commonly studied chlorine-mediated ones thanks to the lower standard redox potential of the bromine evolution reaction (BER), as well as the faster rates of subsequent disproportionation and addition reaction with alkenes.16–18 While precious metal oxides like iridium and ruthenium have traditionally served as BER electrocatalysts, their high costs and limited stability highlight the necessity for exploring other alternatives.19–21To this end, we here synthesized nanostructured Mo2C as a non-precious metal-based candidate for the BER (see ESI,† for details). During the synthesis, dopamine molecules were self-polymerized in an alkaline solution, and were chelated with heptamolybdate (Mo7O246−) ions through abundant catechol groups, giving rise to a polymeric complex (denoted as PDA-Mo).22,23 Scanning electron microscopy (SEM) images illustrate that the as-prepared PDA-Mo has a spherical nanoflower morphology, approximately 3–5 μm in diameter (Fig. S1, ESI†). Further annealing at an elevated temperature carburized the polymeric complex. The X-ray diffraction (XRD) pattern of the final product exhibits diffraction peaks assignable to those of β-Mo2C overlaid with the minor signals from graphitic carbon (Fig. 2a). Worth noting is that the final product (denoted as Mo2C NF) retains its three-dimensional hierarchical structure, featuring numerous thin nanoflakes protruding out from the central core (Fig. 2b). Transmission electron microscopy (TEM) imaging unveils that each nanoflake consists of Mo2C nanoparticles embedded within the carbonaceous support (Fig. 2c). Altering the precursor concentration affects the morphology of the product (Fig. S2, ESI†). Such a hybrid structure affords Mo2C NF with improved electrical conductivity essential to electrocatalysis as will be discussed in the following sections.
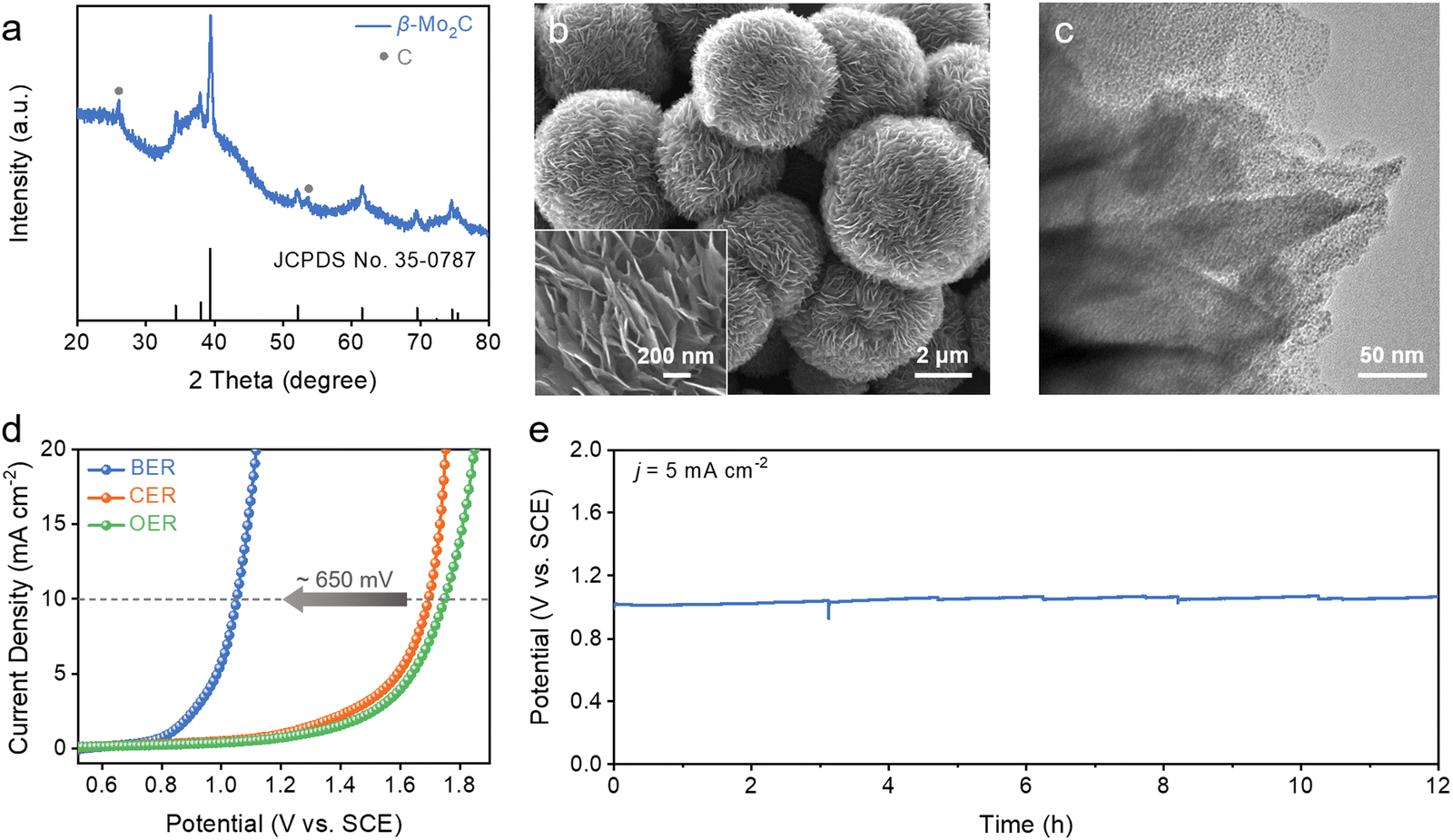 | ||
| Fig. 2 Structural characterizations and electrocatalytic BER performance of Mo2C NF. (a) XRD pattern. (b) SEM and (c) TEM image of Mo2C NF. (d) Polarization curves of the BER, CER, and OER on Mo2C NF in 0.1 M KBr, 0.1 M KCl, and 0.1 M K2SO4 solution. (e) Chronopotentiometric curve of the BER on Mo2C NF at 5 mA cm−2 in 0.1 M KBr solution. | ||
We then evaluated the electrocatalytic performance of Mo2C NF for the BER. In a bromide-free 0.1 M K2SO4 solution, only the oxygen evolution reaction (OER) takes place (Fig. 2d). It exhibits a high onset potential of 1.2 V (versus saturated calomel electrode or SCE, the same hereinafter) and small anodic current density of ∼2.5 mA cm−2 even at 1.6 V. The chlorine evolution reaction (CER) dominates in 0.1 M KCl, but it does not offer a noticeable improvement over the OER in terms of the working potential. By comparison, the BER is initiated on Mo2C NF at a significantly less anodic potential of ∼0.8 V in 0.1 M KBr, close to the theoretical value (0.83 V versus SCE). After reaction onset, the current density quickly rises with the overpotential, and reaches 10 mA cm−2 at 1.05 V, about 650 mV more negative than the OER or CER under the same current density, underscoring the thermodynamic advantage of the BER and the high electrocatalytic activity of Mo2C NF. To assess the operational stability of our catalyst, continuous electrolysis was conducted at 5 mA cm−2 in 0.1 M KBr for 12 h as illustrated in Fig. 2e. Its working potential stays around 1 V, and only slightly increases by 50 mV at the end of the evaluation. SEM characterization of Mo2C NF after the stability test reveals no discernable change of its hierarchical structure even under such a corrosive condition (Fig. S3, ESI†). Additionally, inductively coupled plasma spectroscopy (ICP) analysis of the electrolyte reveals that only 0.16 wt% of Mo is leached out from the catalyst during electrolysis. The above electrochemical measurements highlight the exceptional activity and stability of our Mo2C NF as a non-precious metal-based candidate for the BER, surpassing both carbon-based materials and many precious metal-based materials from previous studies.24–27
Building on these findings, we subsequently explored the bromine-mediated electrochemical conversion of MA. Fig. 3a summarizes the possible reaction pathways. At the anode, Br− ions are first oxidized to Br2via the BER, which then disproportionates to HOBr and HBr. Subsequently, HOBr reacts with the unsaturated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds of MA through electrophilic addition, giving rise to 2-bromo-3-hydroxysuccinic acid (BHSA). In the presence of KOH, BHSA further undergoes dehydrohalogenation to yield epoxysuccinic acid (ESA) with the concomitant regeneration of Br−. Since the reaction is not stereoselective, mixed isomers, namely cis-epoxysuccinic acid (cis-ESA) and trans-epoxysuccinic acid (trans-ESA), are resulted. Additionally, Br2 can directly react with MA through an addition reaction to form dibromosuccinic acid (DBSA) as a by-product.
C bonds of MA through electrophilic addition, giving rise to 2-bromo-3-hydroxysuccinic acid (BHSA). In the presence of KOH, BHSA further undergoes dehydrohalogenation to yield epoxysuccinic acid (ESA) with the concomitant regeneration of Br−. Since the reaction is not stereoselective, mixed isomers, namely cis-epoxysuccinic acid (cis-ESA) and trans-epoxysuccinic acid (trans-ESA), are resulted. Additionally, Br2 can directly react with MA through an addition reaction to form dibromosuccinic acid (DBSA) as a by-product.
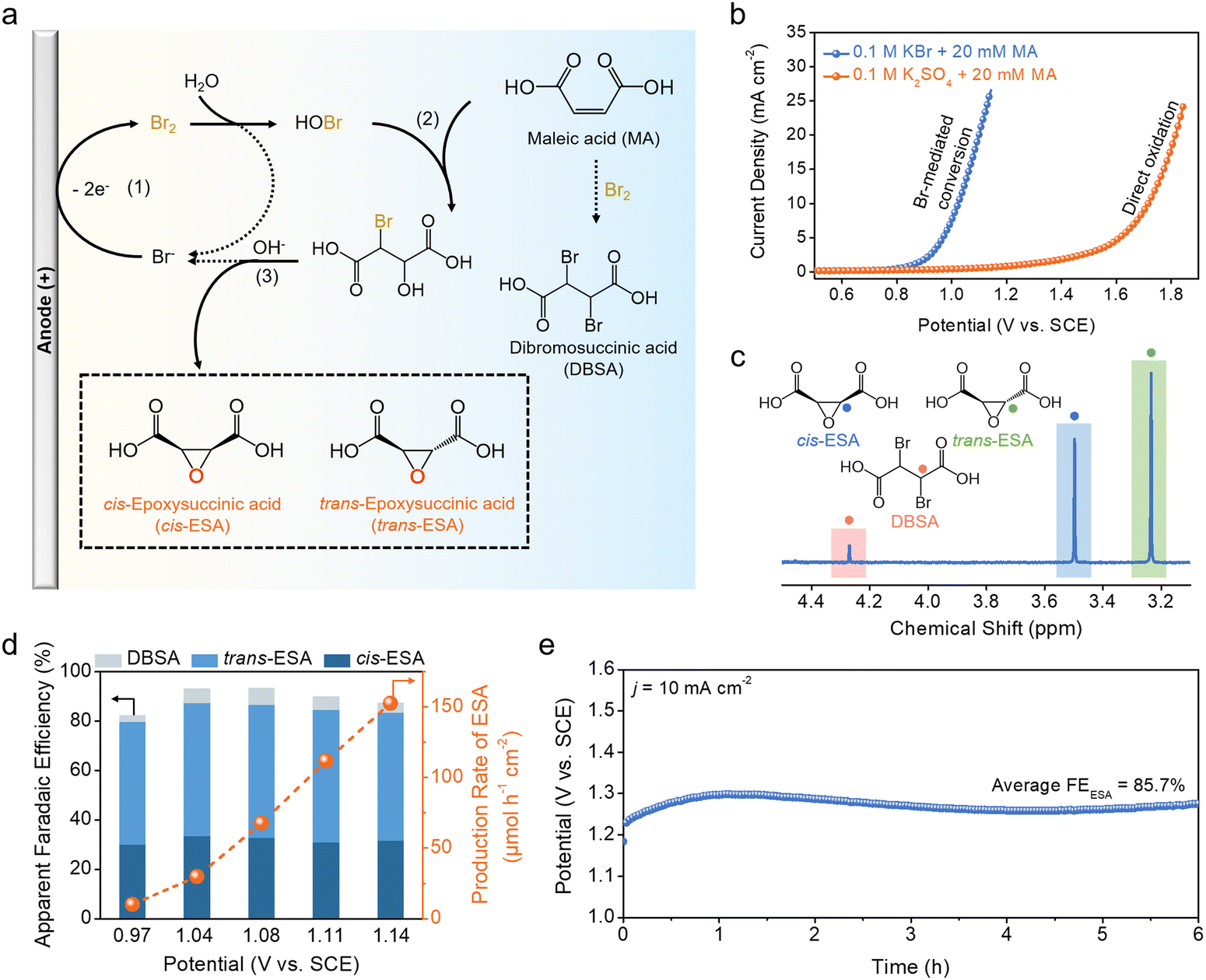 | ||
| Fig. 3 Bromine-mediated electrochemical conversion of MA to ESA. (a) Possible reaction pathways for bromine-mediated electrochemical conversion. (b) Polarization curve of bromine-mediated conversion of MA in comparison with its direct oxidation on Mo2C NF in 0.1 M KBr or K2SO4 solution, with the addition of 20 mM MA. (c) 1H NMR spectra of cis-ESA, trans-ESA and DBSA. (d) Potential-dependent apparent FEs for cis-ESA, trans-ESA and DBSA, and corresponding production rate of ESA in the solution of 0.1 M KBr and 20 mM MA. (e) Chronopotentiometric curve of bromine-mediated conversion on Mo2C NF at 10 mA cm−2 in the solution of 0.1 M KBr and 100 mM MA. | ||
Bromine-mediated electrochemical conversion was carried out in 0.1 M KBr solution with the addition of 20 mM MA. In order to neutralize the acidity of MA and facilitate the disproportionation of Br2 to HOBr, the solution was additionally adjusted with KOH until the pH of 5. The polarization curve in Fig. 3b reflects that the BER on Mo2C NF is not affected by the presence of MA. To determine the possible products from the bromine-mediated reaction, electrolysis was performed at a few selected working potentials as indicated in Fig. S4 (ESI†). At the end of each electrolysis, the anolyte was added with KOH to facilitate the dehydrohalogenation of BHSA to ESA. Liquid products were then quantitatively analyzed using 1H nuclear magnetic resonance (1H NMR) (Fig. S5, ESI†). Fig. 3c illustrates a typical NMR spectrum with chemical shifts at 3.24, 3.50, and 4.27 ppm corresponding to trans-ESA, cis-ESA, and DBSA, respectively. Their apparent faradaic efficiencies (FEs) were calculated by normalizing the charges ultimately transferred to the products over the total charges passed during the BER. As shown in Fig. 3d, the total FE for ESA exceeds 80% over the entire potential range examined, and reaches a peak value of 87.4% at 1.04 V. At the peak, trans-ESA and cis-ESA account for an FE of 53.9% and 33.5%, respectively. DBSA from the direct addition of MA with Br2 has a FE of less than 10%. The ESA production rate was also derived and shown to increase with the applied working potential, reaching up to 150 μmol h−1 cm−2 at 1.14 V. By contrast, the direct electrochemical oxidation of MA is found to yield no carbonaceous products, presumably overwhelmed by the competing OER (Fig. S6, ESI†). Moreover, the stability of the bromine-mediated conversion was assessed at 10 mA cm−2 (Fig. 3e). The anode potential remained relatively stable at 1.27 V, and the average FE for ESA over 6 h electrolysis is measured to be 85.7%.
Epoxides can hydrolyze under acidic or basic conditions to form vicinal diols. However, the hydrolysis of ESA is more challenging than common epoxides due to the stability of its oxygen ring.28 In order to accelerate this process, the above solution was acidified with HCl, and heated at 80 °C for 10 h (Fig. 4a). During the reaction, the epoxide oxygen of ESA is first protonated, resulting in the formation of a more reactive leaving group. The subsequent nucleophilic attack of the epoxide carbon by a water molecule opens the epoxide ring, and deprotonation leads to the formation of TTA. At the end of the reaction, the TTA concentration was determined using colorimetric analysis with ultraviolet-visible (UV-Vis) spectrophotometry (Fig. S7, ESI†) (see ESI,† for details).29,30 As shown in Fig. 4b, an average conversion efficiency of 75% from ESA to TTA is achieved for epoxide samples originally from the bromine-mediated conversion at different working potentials. Notably, this hydrolysis process does not involve any enzymatic or catalytic agent. Further optimization by introducing proper catalysts may improve the conversion rate, and render the whole process more efficient.
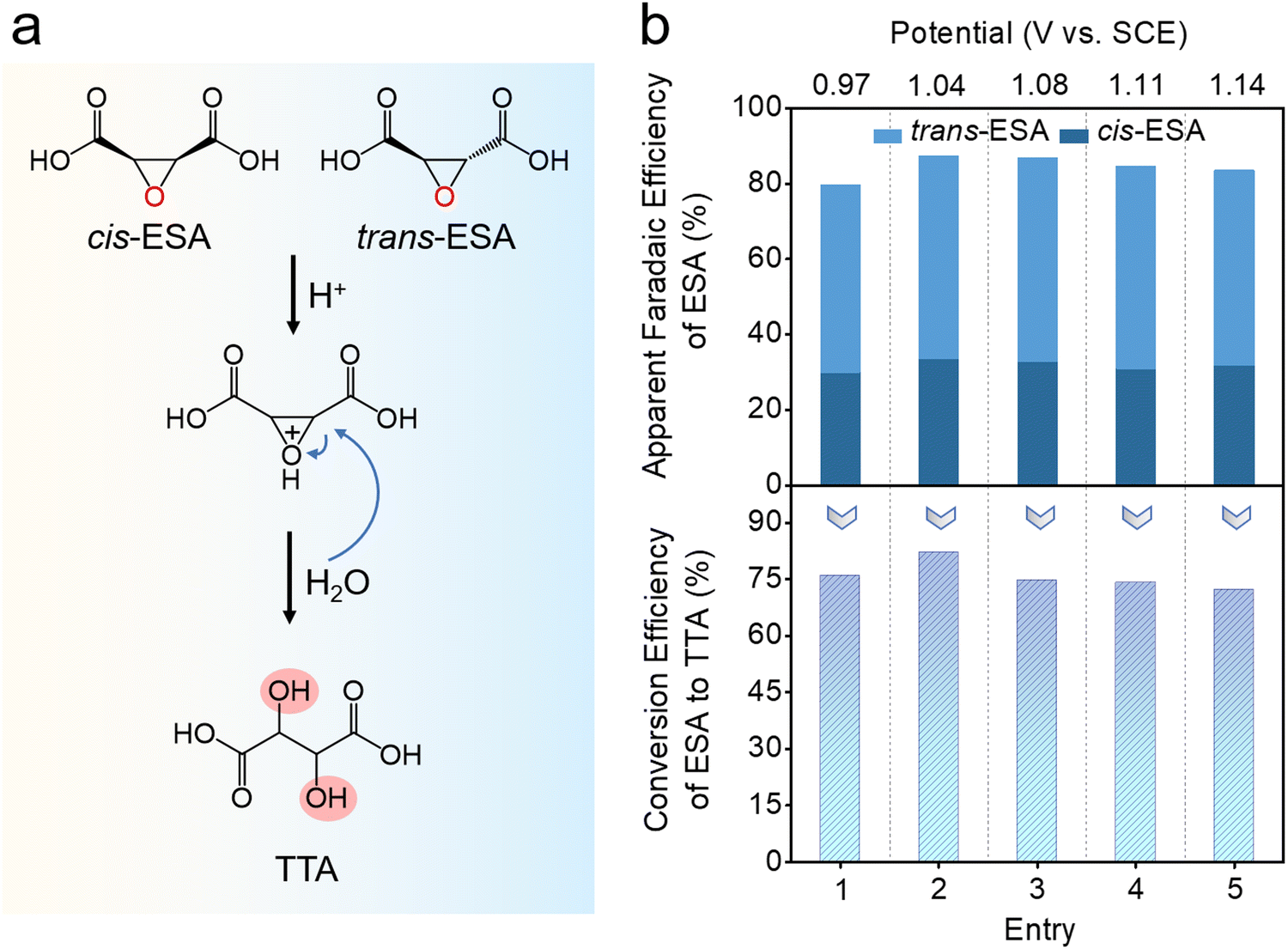 | ||
| Fig. 4 Hydrolysis conversion of ESA to TTA. (a) Reaction mechanism from ESA to TTA. (b) Conversion efficiency of five ESA samples obtained from bromine-mediated conversion at different working potentials. | ||
3. Conclusions
In summary, we proposed and demonstrated bromine-mediated conversion from MA to TTA. Mo2C NF was prepared from the self-assembly of polydopamine and Mo7O246− ions in solution, and subsequent carburization at an elevated temperature. It exhibited a flower-like hierarchical structure with β-Mo2C nanoparticles embedded in high-surface-area carbonaceous supports. In 0.1 M KBr, Mo2C NF catalyzed the BER at a potential close to the theoretical value. Bromine generated at the anode subsequently disproportionated to HOBr, and mediated the conversion of MA to ESA with high selectivity of >80% and high production rate of up to 150 μmol h−1 cm−2. Finally, the hydrolysis of ESA to TTA was achieved in acid at a high conversion efficiency of ∼75%. Compared to the direct electrochemical oxidation of MA, our new approach avoids product overoxidation (e.g. oxalic acid or formic acid). Bromine can be regenerated to continuously sustain the catalytic cycle so that the entire process is atom economic without the concomitant production of harmful waste. Our study showcases the great potential of a halogen-mediated electrochemical refinery in the future manufacturing of high value chemicals.Author contributions
Y. G. L. conceived the project and designed the experiments, N. H. supervised the project, C. L. L. synthesized the catalysts, and conducted the structure analysis and electrocatalytic studies, Z. Y. W. carried out the UV-Vis analysis, Y. C. Y. performed TEM characterizations, Z. H. M. performed product analysis. All authors discussed the results and commented on the manuscript.Data availability
The data that support the findings of this study are available within the main text and the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (52161160331, 22279084 and 52425209), the Natural Science Foundation of Jiangsu Province of China (BK20220027), the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (20KJA430002), and the Collaborative Innovation Center of Suzhou Nano Science and Technology.References
- Global tartaric acid market by source (grapes & sun-dried raisins, maleic anhydride), type (natural, synthetic), application-forecast 2024-2030, https://www.researchandmarkets.com/report/tartaric-acid#product--related-products, accessed Sep 2024.
- M. Liu, L. Lai, Z. Wang, X. Jin, J. Shen, Y. Wang, Q. Zhang, D. Zhang, Y. Sun, H. Ning, W. Wu, G. Zhang and C. Yang, Ind. Eng. Chem. Res., 2023, 62, 6052–6068 CAS.
- N. Merbouh, J. F. Thaburet, M. Ibert, F. Marsais and J. M. Bobbitt, Carbohydr. Res., 2001, 336, 75–78 CrossRef CAS PubMed.
- C. Tang, Y. Zheng, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2021, 60, 19572–19590 CrossRef CAS PubMed.
- C. Li, Y. Wang, X. Wang, T. Azam and Z.-S. Wu, Chem, 2024, 10, 2666–2699 CAS.
- A. Wiebe, T. Gieshoff, S. Mohle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS.
- G. A. Coppola, S. Pillitteri, E. V. Van der Eycken, S.-L. You and U. K. Sharma, Chem. Soc. Rev., 2022, 51, 2313–2382 RSC.
- T. H. Meyer, I. Choi, C. Tian and L. Ackermann, Chem, 2020, 6, 2484–2496 CAS.
- M. Chen and Y. Liu, Catal. Lett., 2023, 154, 1411–1419 CrossRef.
- E. Weiss, K. Groenen-Serrano, A. Savall and C. Comninellis, J. Appl. Electrochem., 2006, 37, 41–47 CrossRef.
- P. Cañizares, J. García-Gómez, J. Lobato and M. A. Rodrigo, Ind. Eng. Chem. Res., 2003, 42, 956–962 CrossRef.
- P. Zhang, T. Wang and J. Gong, CCS Chem., 2023, 5, 1028–1042 CrossRef CAS.
- W. R. Leow, Y. Lum, A. Ozden, Y. H. Wang, D. H. Nam, B. Chen, J. Wicks, T. T. Zhuang, F. W. Li, D. Sinton and E. H. Sargent, Science, 2020, 368, 1228–1233 CrossRef CAS PubMed.
- X. Liu, Z. Chen, S. Xu, G. Liu, Y. Zhu, X. Yu, L. Sun and F. Li, J. Am. Chem. Soc., 2022, 144, 19770–19777 CrossRef CAS PubMed.
- Y. Yang, X. Yuan, Q. Wang, S. Wan, C. Lin, S. Lu, Q. Zhong and K. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202314383 CrossRef CAS PubMed.
- Y. Gao and B. Zhang, Sci. Bull., 2024, 69, 1595–1597 CrossRef CAS PubMed.
- N.-N. Liang, W. Choi, D. Suk Han and H. Park, Chem. Eng. J., 2024, 494, 153042 CrossRef CAS.
- Q. Wang, C. Yang, Y. Yan, H. Yu, A. Guan, M. Kan, Q. Zhang, L. Zhang and G. Zheng, Angew. Chem., Int. Ed., 2022, 62, e202212733 CrossRef PubMed.
- J. G. Vos, A. Venugopal, W. A. Smith and M. T. M. Koper, J. Catal., 2020, 389, 99–110 CrossRef CAS.
- Y. Wang, Y. Liu, D. Wiley, S. Zhao and Z. Tang, J. Mater. Chem. A, 2021, 9, 18974–18993 RSC.
- R. K. B. Karlsson and A. Cornell, Chem. Rev., 2016, 116, 2982–3028 CrossRef CAS PubMed.
- Y. Huang, Q. Gong, X. Song, K. Feng, K. Nie, F. Zhao, Y. Wang, M. Zeng, J. Zhong and Y. Li, ACS Nano, 2016, 10, 11337–11343 CrossRef CAS PubMed.
- C. Wang, L. Sun, F. Zhang, X. Wang, Q. Sun, Y. Cheng and L. Wang, Small, 2017, 13, 1701246 CrossRef PubMed.
- M. Chi, J. Ke, Y. Liu, M. Wei, H. Li, J. Zhao, Y. Zhou, Z. Gu, Z. Geng and J. Zeng, Nat. Commun., 2024, 15, 3646 CrossRef CAS PubMed.
- J. Schalck, J. Hereijgers, W. Guffens and T. Breugelmans, Chem. Eng. J., 2022, 446, 136750 CrossRef CAS.
- Y. Gao, M. Yan, C. Cheng, H. Zhong, B.-H. Zhao, C. Liu, Y. Wu and B. Zhang, J. Am. Chem. Soc., 2023, 146, 714–722 Search PubMed.
- W. Xue, L. Quan, H. Liu, B. Yu, X. Chen, B. Y. Xia and B. You, Angew. Chem., Int. Ed., 2023, 62, e202311570 CAS.
- W. Bukowski, I. Y. Litvintsev and V. N. Sapunov, J. Chem. Technol. Biotechnol., 2004, 61, 375–379 CrossRef.
- J. M. Church and R. Blumberg, Ind. Eng. Chem. Res., 1951, 43, 1780–1786 CAS.
- K. S. Lee, D. W. Lee and J. Y. Hwang, Anal. Chem., 1968, 40, 2049–2052 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ey00259h |
| This journal is © The Royal Society of Chemistry 2025 |