
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective electroreduction of CO2 to formate by a heterogenized Ir complex using H2O as an electron/hydrogen source†
Jieun Jung *a,
Keun Woo Lee‡
a,
Naonari Sakamoto‡b,
Selvam Kaliyamoorthy‡a,
Taku Wakabayashia,
Kenji Kamadaa,
Keita Sekizawab,
Shunsuke Satob,
Tomiko M. Suzukib,
Takeshi Morikawab and
Susumu Saito*ac
*a,
Keun Woo Lee‡
a,
Naonari Sakamoto‡b,
Selvam Kaliyamoorthy‡a,
Taku Wakabayashia,
Kenji Kamadaa,
Keita Sekizawab,
Shunsuke Satob,
Tomiko M. Suzukib,
Takeshi Morikawab and
Susumu Saito*ac
aDepartment of Chemistry, Graduate School of Science, Nagoya University, Chikusa, Nagoya 464-8602, Japan. E-mail: jung.jieun.z7@f.mail.nagoya-u.ac.jp
bToyota Central R&D Laboratories, Inc, Nagakute 480-1192, Japan
cIntegrated Research Consortium on Chemical Science (IRCCS), Nagoya University, Chikusa, Nagoya 464-8602, Japan. E-mail: saito.susumu.c4@f.mail.nagoya-u.ac.jp
First published on 6th December 2024
Abstract
A newly synthesized tetradentate PNNP-coordinated iridium (Ir) complex, Mes-IrPPh2, immobilized on a carbon material, was found to be a superior catalyst for CO2 electrochemical reduction reaction (CO2ERR) to give formate, (HCOO−), allowing an operation near the theoretical potential (−0.18 V vs. RHE, pH = 7.3) in water. The combined [Mes-IrPPh2] electrode furnished HCOO− with a current density of greater than 2.2 to 7.7 mA cm−2 over −0.27 to −0.47 V vs. RHE, providing faradaic efficiencies (FE) of >90%. The outstanding robustness of the electrode attained continuous production of HCOO− up to 12.5 mmol with 2.86 μmol of Mes-IrPPh2 at −0.27 V vs. RHE over 168 h. Furthermore, solar-driven electrochemical CO2 reduction to HCOO− was also carried out in water with a Ni/Fe–Ni foam anode as a water oxidation catalyst and a silicon photovoltaic cell to achieve a solar-to-formate conversion efficiency (ηSTF) of 13.7%.
Broader contextThe electrochemical reduction of carbon dioxide (CO2) has emerged as a crucial strategy for reducing global carbon emissions and advancing sustainable energy solutions. Utilizing water as an electron donor for CO2 fixation offers a practical approach, particularly in the realm of renewable energy storage. Enhancing CO2 reduction efficiency can be achieved through the heterogenization of molecular catalysts onto electrode surfaces. However, a significant challenge remains: the long-term durability of electrochemical CO2 reduction in aqueous solutions, which often deteriorates after only a few hours. In this study, we present a novel Ir complex immobilized on a carbon material electrode to achieve both efficient and durable CO2 reduction. The heterogenization of the Ir complex notably reduces the overpotential and yields formate as the main product with a high faradaic efficiency. Remarkably, the catalyst maintains its activity for a week, demonstrating exceptional long-term stability. It is worth noting here that, when the Ir electrode was employed in solar-driven electrochemical CO2 reduction, the solar-to-formate conversion efficiency of 13.7% was achieved, highlighting its potential for scalable practical/industrial applications. |
Introduction
The electrochemical reduction of carbon dioxide (CO2) has received significant attention in recent years due to the growing demand for carbon-neutral energy carriers for the storage of renewable energy.1 Reduction of CO2 by utilizing water as an electron/hydrogen donor for the fixation of CO2 is an ideal process for practical use. Thus, the development of catalysts for CO2 reduction in aqueous solutions is the key to realizing this noble technology.2 Various materials, including semiconductors and molecular metal complexes, have been studied for photoelectrochemical3,4 and electrochemical5–7 reduction of CO2 to desired materials such as carbon monoxide (CO), formic acid, methanol, methane, and C2+ compounds. Despite substantial efforts on the development of the CO2 electrochemical reduction reaction (CO2ERR), (i) low energetic efficiency requiring a large overpotential, (ii) poor selectivity for carbonaceous products in water originating from low solubility of CO2, (iii) the competitive hydrogen evolution reaction (HER) and (iv) deactivation of the catalysts restrict this technology from practical use.8Heterogenization of molecular catalysts on electrode surfaces has emerged as a promising approach in terms of lower catalyst loading and improving electron transfer efficiency.9,10 Significant progress has been made in the functionalization of carbon surfaces of electrodes to anchor molecular catalysts as CO2ERR electrocatalysts.11–13 However, in many cases, the CO2ERR in an aqueous solution halted after no more than a few hours, and thus, its long-term durability has remained a significant challenge. Solar-driven CO2 conversion to formate (HCOO−) has also been carried out to achieve electrochemical CO2 reduction using heterogenized metal complexes in combination with photovoltaic cells.14–20 Although their solar-to-electricity conversion efficiencies are usually lower than those of the GaAs-based photovoltaic cells, silicon is known to be a more suitable material for photovoltaic cells owing to their higher cost performance. To improve solar-to-formate conversion efficiency, the cell voltage between the two poles must be reduced, and it is essential to develop an efficient electrocatalyst that can operate at a very low overpotential.
We recently reported tetradentate PNNP-coordinated iridium ((PNNP)Ir) complexes as efficient and robust photocatalysts to selectively reduce CO2 to formic acid with high reactivity in an organic solvent (with a small amount of H2O).21,22 Herein, the Ir complexes were successfully exploited to catalyze CO2 reduction in an aqueous solution through its heterogenization onto the surface of a carbon material electrode supported by carbon black and a polymer matrix.23 This heterogenized Ir complex deployed on the carbon electrode is abbreviated as [Ir-ink] in this report, wherein (1) as depicted in Scheme 1, CO2ERR at a very small overpotential of around 90 mV was achieved over the [Ir-ink] electrode (cathode) together with a platinum wire (anode) and Ag/AgCl as the counter and reference electrodes, respectively. The [Ir-ink] electrode exhibited outstanding CO2 reduction activity with a current density of 2.2 mA cm−2 at −0.27 V vs. RHE. HCOO− was produced as the main product with a high faradaic efficiency (FE) of >98% in 1 h and prolonging the reaction time to 168 h with optimal conditions generated a total amount of 12.5 mmol of HCOO− (using ca. 2.86 μmol of Ir in [Ir-ink]). (2) The developed [Ir-ink] successfully promoted electrochemical CO2 reduction to HCOO− utilizing H2O as an electron and proton source in a two-compartment cell combined with photovoltaic (PV) cells. The solar-to-formate conversion efficiency (ηSTF) reached 13.7% by combination with a Ni/Fe–Ni foam anode and a silicon PV cell.
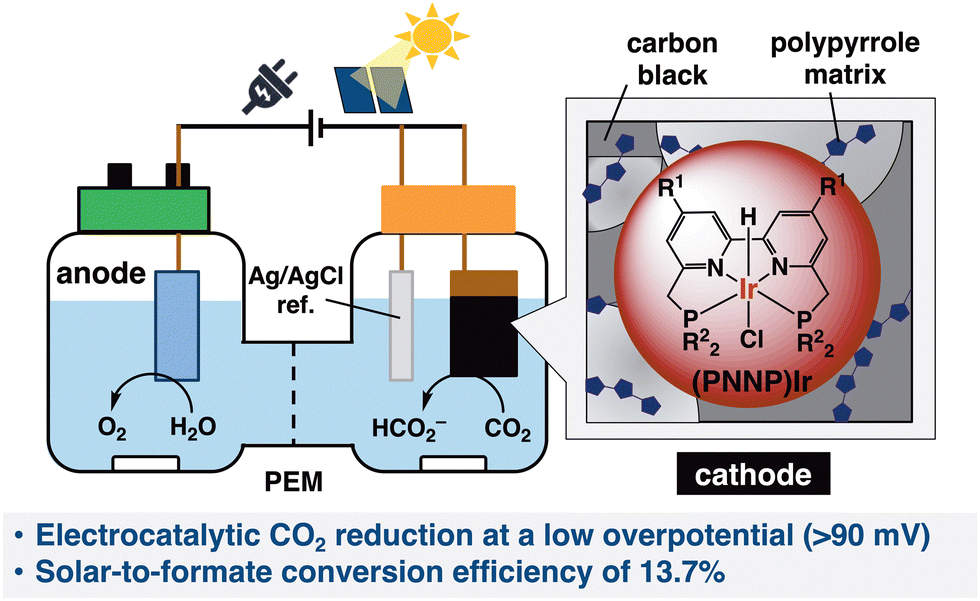 | ||
| Scheme 1 Schematic representation of a cell. | ||
Results and discussion
The (PNNP)Ir complexes were synthesized based on the previous reports22,24 and Scheme S1, ESI.† [Ir-ink] electrodes were fabricated by drop-casting a prepared Ir complex ink ((PNNP)Ir–polypyrrole–Nafion–Vulcan) on a carbon material as visualized in Scheme S2, ESI.† The ratio of (PNNP)Ir–polypyrrole–Nafion–Vulcan was recruited based on the composition of the electrodes previously prepared.20,25 The morphology of a deposited Ir complex (Fig. 1a, Mes-IrPPh2) on a carbon material ([Mes-IrPPh2], which denotes [Ir-ink] on the carbon electrode) was obtained using scanning electron microscopy (SEM) images (Fig. 1b). The element mapping images obtained via energy-dispersive X-ray spectroscopy (EDS) of the [Mes-IrPPh2] revealed a homogeneous distribution of C, P, and Ir on the plain surface, while those of the side view indicated that the Mes-IrPPh2 complex is concentrated on the outer surface of the carbon material (Fig. S1–S3, ESI†). X-ray photoelectron spectroscopy (XPS) of the [Mes-IrPPh2] electrode was further conducted to understand the electronic state of the electrode catalyst. The [Mes-IrPPh2] electrode before starting the electroreduction exhibited Ir 4f XPS peaks at 63 and 66 eV (Fig. 1c) assignable to Ir(III) (Fig. S4, ESI†)26 as the original Mes-IrPPh2 complex. Fig. 1d shows linear sweep voltammetry (LSV) results for the [Mes-IrPPh2] electrode. The small onset potential (Eon) of −0.24 V vs. RHE revealed how quickly the electrocatalyst initializes catalytic activity at such a low potential. | ||
| Fig. 1 (a) Chemical structure of Mes-IrPPh2. (b) A SEM image of the [Mes-IrPPh2] electrode. Scale bar is 500 μm. (c) Ir 4f XPS spectra of the [Mes-IrPPh2] (blue line) electrode and IrIIICl3·H2O (black line) as a reference. (d) LSV for the [Mes-IrPPh2] electrocatalyst in a CO2-saturated 0.5 M KHCO3 solution. | ||
The initial conditions for electrochemical reduction of CO2 were examined in a two-compartment cell operated at −0.37 V vs. RHE using several different PNNP-type Ir complexes (ca. 1.43 μmol of Ir in [Ir-ink]; Table S1, ESI†). Among the Ir complexes, Mes-IrPPh2 showed the most outstanding performance in achieving high product amounts and selectivity towards carbon products (HCOO− and CO; 765 μmol) with a high current density (5.49 mA cm−2). The FE for HCOO− formation reached 86%. The results indicate that introducing a bulky mesityl group and modifying the phosphine substituent of the PNNP ligand significantly enhanced HCOO− production. Replacing the cyclohexyl (Cy) group with a phenyl (Ph) group further improved selectivity by seemingly facilitating π–π/CH–π interactions with the carbon material and polypyrrole. The electrochemical properties of the Ir complexes were recorded using an Ar or CO2-saturated MeCN solution (Fig. S5, ESI†). Two redox waves were observed for Mes-IrPPh2 at −0.95 and −1.28 V vs. SCE, which were more positive than those of Mes-IrPCY2 at −1.31 V vs. SCE (Fig. S5a, ESI†). When the Ar was replaced by CO2, the catalytic current with Mes-IrPPh2 was much enhanced compared to those with Mes-IrPCY2, where the Eon for Mes-IrPPh2 (−1.17 V) was also more positive than those of Mes-IrPCY2 (−1.21 V). The results indicate that subtle modifications in the metal complex ligand structure can have a profound impact on catalytic performance (Fig. S5b, ESI†). The [Mes-IrPPh2] electrode operated at a reaction current up to 7.7 mA cm−2 over a potential of −0.07 to −0.47 V vs. RHE (Fig. 2a), achieving FEs of >90% for HCOO− production (Fig. 2b and Table S2, ESI†). When the reaction was operated at near-theoretical potential (−0.17 V vs. RHE) with a [Mes-IrPPh2] electrode, it also produced a miniscule amount of carbon products (18 μmol) with low current density (Fig. 2a, <0.5 mA cm−2). However, it is known that the activation barrier to form HCOO− can sometimes be overcome by thermal activation at room temperature.27 To acquire extra energy for inducing the catalyst, the applied potential of −0.27 V vs. RHE was selected as an optimum potential in this catalytic system. In the optimized conditions, the lack of each pivotal component (an Ir complex, carbon black, or pyrrole) resulted in the negligible formation of products, while the absence of Nafion gave small amounts of the products at −0.27 V vs. RHE for 3 h (Table S3, ESI†). Isotope-labelling experiments using 13C-labeled carbon dioxide (13CO2) in D2O were conducted to verify the carbon source of the produced HCOO− and CO. 1H NMR measurement of the reaction solution showed a doublet (J = 194.4 Hz) attributable to the hydrogen atom bound to the 13C atom of H13COO− (Fig. S6, ESI†). In the 13C NMR spectrum (Fig. S7, ESI†), the formation of H13COO− was observed together with deuterated formate, D13COO− (triplet, J = 30.3 Hz).28 Gas chromatography-mass spectrometry (GC-MS) analysis of the gaseous product also identified 13CO (m/z = 29) as the main gaseous product (Fig. S8, ESI†). The results substantiate that HCOO− and CO originated from CO2 rather than from carbon contaminants in the reaction mixture.
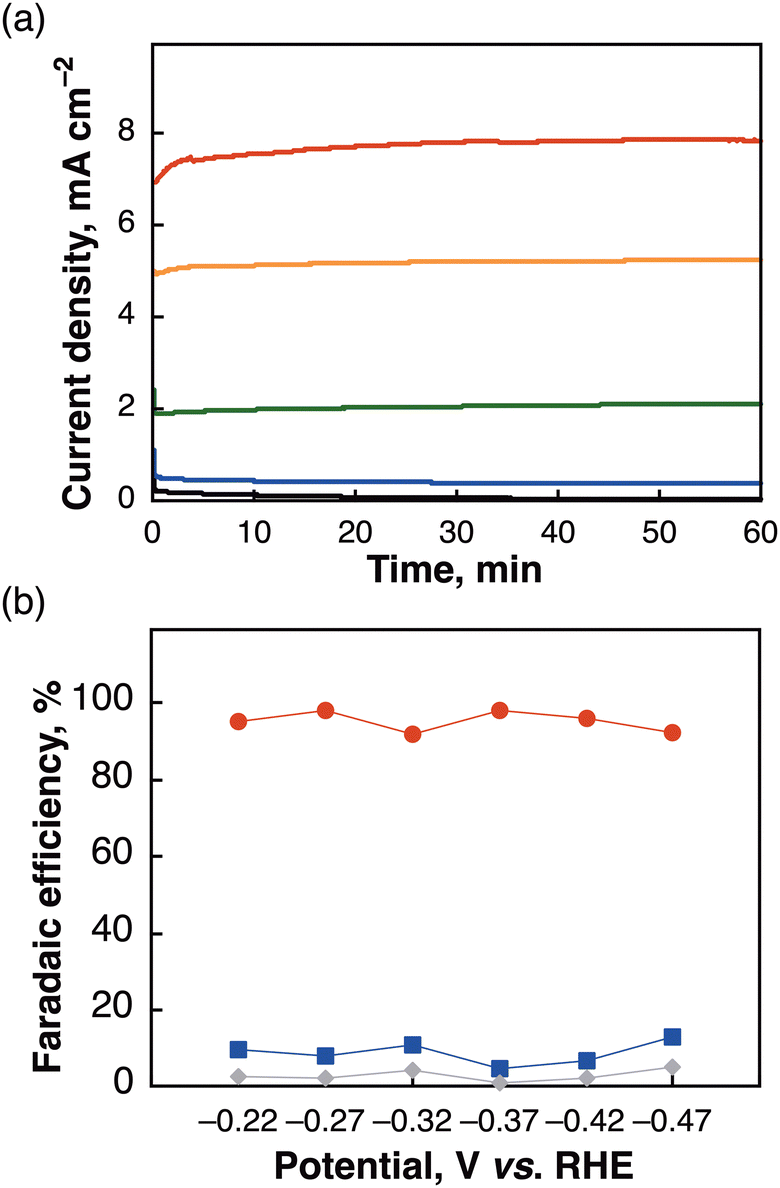 | ||
| Fig. 2 (a) Chronoamperograms over a [Mes-IrPPh2] cathode and a Pt anode for 1 h with several bias voltages [−0.07 V (black), −0.17 V (blue), −0.27 V (green), −0.37 (orange), −0.47 V (red)] and (b) FEs of the products [HCOO− (red), CO (blue), H2 (gray)] obtained during the electrocatalytic CO2 reduction using a [Mes-IrPPh2] electrode in a solution of CO2-saturated 0.5 M KHCO3. | ||
The effect of drop-casting (DC) counts of an [Ir ink] on the surface of the working electrode (cathode) was examined to optimize the amounts of the Ir catalyst (Table S4 and Fig. S9, ESI†). By increasing the count of DC from 2 to 4, 6, and 8 times, the amounts of HCOO− increased at DC = 4 and reached a plateau with DC = 6 or 8, where the current densities continued to rise, ascribable simply to the increased amount of Mes-IrPPh2 on the electrodes. In all cases, high FEs (>90%) for HCOO− formation were achieved. Thus, for subsequent experiments, the [Mes-IrPPh2] electrodes were prepared using the 4 times DC.
After fabricating the 4 times-DC [Mes-IrPPh2] electrode (containing 2.86 μmol of Mes-IrPPh2), continuous long-term electrocatalysis of CO2 at ca. −0.27 V vs. RHE was conducted over 168 h using a flow reactor for CO2 supply (Fig. 3a); however, the gaseous phase could not be analysed accurately owing to the escaping gas from the reactor. Therefore, we only evaluated the amount of non-gaseous HCOO− generated in the aqueous phase, which linearly increased over 12.5 mmol with FE of >77% by prolonging the reaction time to 168 h. To ascertain the overwhelming robustness of the [Mes-IrPPh2] catalyst, XPS measurements before and after the reaction (3 h, 15 h, and 72 h) were recorded to monitor the reaction of the electrode. The XPS analysis of the Ir 4f region revealed that the Ir metal in Mes-IrPPh2 was converted from IrIII (Fig. 3b, blue) to IrI (Fig. 3b, black) within 3 h and the IrI species remained the same over 72 h (Fig. 3b, red), consistent with the results of ESI-MS analysis (Fig. S10, ESI†). The results indicate that almost no deterioration of the catalyst occurred after an electrolysis for 72 h.
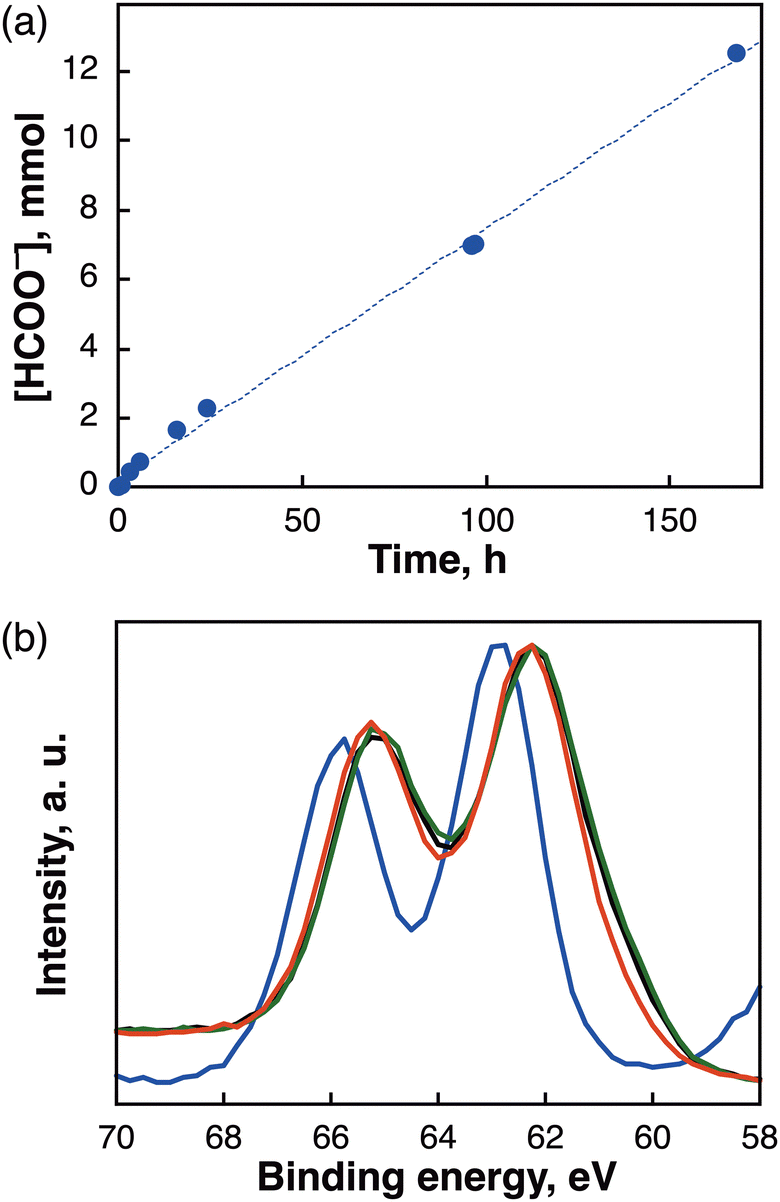 | ||
| Fig. 3 (a) Time course plots of the HCOO− obtained during the electrocatalytic CO2 reduction at −0.27 V vs. RHE using a [Mes-IrPPh2] cathode and a Pt anode in a solution of CO2-saturated 0.5 M KHCO3. (b) Ir 4f XPS spectra of a [Mes-IrPPh2] electrode [before (blue) and after electrolysis for 3 h (black), 15 h (green), 72 h (red)]. | ||
Solar-driven CO2 reduction was next examined using a two-compartment reactor composed of the [Mes-IrPPh2] electrode as a CO2 reduction catalyst (cathode), Fe/Ni–Ni foam as a water oxidation catalyst (anode),29 and a heterojunction with an intrinsic thin layer (HIT) solar cell (silicon solar cell), which served as the light absorber with a catalog-specified maximum ηPV of approximately 22.7%.30 A two-compartment reactor separated by a Nafion-117 ion-exchange membrane was used in this system, operating at −2.1 V. The CO2 reduction using the PV cell was conducted by immersing the cathode–anode conjunction system in a solution of CO2-saturated 0.5 M KHCO3 under irradiation with solar simulated light (1 sun, AM1.5) at 308 K. The time course of the FEs for different products is shown in Fig. 4, illustrating that HCOO− was generated only by the reaction of CO2 with H2O as a raw material using sunlight as an energy source. HCOO− was continuously generated during irradiation for 24 h with over 91% of FEs where CO and H2 were produced with less than 2% of FEs, respectively, and the solar-to-formate conversion efficiency (ηSTF) was calculated to be 13.7% from the rate of HCOO− generation (mmol HCOO− s−1). It is noteworthy that the ηSTF up to 13.7% in this system reached the highest level among the photoelectrochemical CO2 reduction systems14–20,31 using a molecular catalyst (Table S5, ESI†).
 | ||
| Fig. 4 Time course plots of the product [HCOO− (red), CO (blue), H2 (gray)] obtained during the CO2 electrochemical reduction using a [Mes-IrPPh2] cathode and Fe/Ni–Ni foam anode under simulated solar light irradiation. | ||
Conclusions
In this study, we demonstrated the high-performance systems for electrochemical and solar-driven electrochemical CO2 reduction in water using a PV cell through the fabrication of an effective cathode deploying a PNNP-type Ir complex as a CO2 reduction electrocatalyst. The Ir catalyst immobilized on a carbon material ([Ir-ink]: cathode) showed a superior catalytic activity for selective CO2 reduction to HCOO−, providing FEs of >98% at a constant rate, in addition to an overpotential of no more than ≥90 mV. The rate of production of HCOO− linearly increased to give 12.5 mmol (using ca. 2.86 μmol of Ir) over 168 h at −0.27 V vs. RHE, highlighting the extremely robust electrode composed of a (PNNP)Ir complex. The solar-to-formate conversion efficiency of 13.7% was achieved after 24 h irradiation yielding 7 mmol of HCOO− with FEs of >91%, in conjunction with a Ni/Fe–Ni foam anode as a water oxidation catalyst and a silicon photovoltaic cell as a photon absorber. Through a bespoke molecular design of a robust metal complex with a PNNP ligand backbone, an electrocatalyst made of hybridized molecular catalyst–bulk carbon material promoted HCOO− formation with concurrent high activity, selectivity, and stability. The present result is in good contrast to the CO2 reduction systems we have achieved using light energy and C–H bonds (electron and hydrogen donor),21,22 as well as heat energy and H–H bonds,32 which were consistently promoted by (PNNP)Ir complex catalysts. This work will surely be further advanced to hitherto unknown efficient solar-driven CO2 reduction devices using water (O–H bond) as the electron- and hydrogen donor.Author contributions
Jieun Jung supervised the project on-site and wrote the original draft of the manuscript. Keun Woo Lee, Naonari Sakamoto, and Selvam Kaliyamoorthy conducted the experiments and collected the data. Taku Wakabayashi developed the initial process for solar-driven electrochemical CO2 reduction. Kenji Kamada provided resources for the research. Keita Sekizawa handled data curation. Shunsuke Sato was responsible for the conceptualization of the study. Tomiko M. Suzuki developed the Fe/Ni–Ni foam as a water oxidation catalyst. Takeshi Morikawa was involved in funding acquisition and administrative aspects. Susumu Saito conceived and supervised the project, managed project administration and funding acquisition, and was responsible for manuscript writing, editing, and curation.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was first supported by the Asahi Glass Foundation (step-up grant to S. S.), followed by the Ministry of the Environment of the Government of Japan, and JST CREST (# JPMJCR22L2 to S. S.), Japan. This work was partially supported by a MEXT Grant-in-Aid for Transformative Research Areas (A) Green Catalysis Science (#23H04904 to S. S. and J. J.), a JSPS Grant-in-Aid for Specially Promoted Research (#23H05404 to S. S.), International Leading Research (#22K21346 to S. S.), and an Early-Career Scientist (#21K14642 to J. J.), as well as the Foundation of Public Interest of Tatematsu. We are also grateful to K. Oyama and R. Yamada for their technical support in spectroscopic measurements, K. Higuchi in high voltage electron microscope laboratory for his technical support in SEM/EDS analyses, and H. Natsume, H. Okamoto, and M. Kosaka for their assistance at the custom-made glass workshop.Notes and references
- (a) J.-P. Jones, G. K. S. Prakash and G. A. Olah, Isr J. Chem., 2014, 54, 1451–1466 CrossRef CAS; (b) S. Garg, M. Li, A. Z. Weber, L. Ge, L. Li, V. Rudolph, G. Wang and T. E. Rufford, J. Mater. Chem. A, 2020, 8, 1511–1544 RSC.
- S. Yoshino, T. Takayama, Y. Yamaguchi, A. Iwase and A. Kudo, Acc. Chem. Res., 2022, 55, 966–977 CrossRef CAS PubMed.
- A. U. Pawar, C. W. Kim, M.-T. Nguyen-Le and Y. S. Kang, ACS Sustainable Chem. Eng., 2019, 7, 7431–7455 CrossRef CAS.
- N. Nandal and S. L. Jain, Chem. Rev., 2022, 451, 214271 CAS.
- S. A. Al-Tamreh, M. H. Ibrahim, M. H. El-Naas, J. Vaes, D. Pant, A. Benamor and A. Amhamed, ChemElectroChem, 2021, 8, 3207–3220 CrossRef CAS.
- D. Ewis, M. Arsalan, M. Khaled, D. Pant, M. M. Ba-Abbad, A. Amhamed and M. H. El-Naas, Sep. Purif. Technol., 2023, 316, 123811 CrossRef CAS.
- N. Sakamoto, K. Sekizawa, S. Shirai, T. Nonaka, T. Arai, S. Sato and T. Morikawa, Nat. Catal., 2024, 7, 574–584 CrossRef CAS.
- Z. Sun, T. Ma, H. Tao, Q. Fan and B. Han, Chem, 2017, 3, 560–587 CAS.
- M. Abdinejad, M. N. Hossain and H.-B. Kraatz, RSC Adv., 2020, 10, 38013–38023 RSC.
- L. Sun, V. Reddu, A. C. Fisher and X. Wang, Energy Environ. Sci., 2020, 13, 374–403 RSC.
- P. Kang, S. Zhang, T. J. Meyer and M. Brookhart, Angew. Chem., Int. Ed., 2014, 53, 8709–8713 CrossRef CAS PubMed.
- X.-M. Hu, M. H. Rønne, S. U. Pedersen, T. Skrydstrup and K. Daasbjerg, Angew. Chem., Int. Ed., 2017, 56, 6468–6472 CrossRef CAS PubMed.
- S. Sato, K. Saita, K. Sekizawa, S. Maeda and T. Morikawa, ACS Catal., 2018, 8, 4452–4458 CrossRef CAS.
- T. Arai, S. Sato, K. Sekizawa, T. M. Suzuki and T. Morikawa, Chem. Commun., 2019, 55, 237–240 RSC.
- N. Kato, S. Mizuno, M. Shiozawa, N. Nojiri, Y. Kawai, K. Fukumoto, T. Morikawa and Y. Takeda, Joule, 2021, 5, 687–705 CrossRef CAS.
- N. Kato, Y. Takeda, Y. Kawai, N. Nojiri, M. Shiozawa, S. Mizuno, K. Yamanaka, T. Morikawa and T. Hamaguchi, ACS Sustainable Chem. Eng., 2021, 9, 16031–16037 CrossRef CAS.
- X. Chen, H. Chen, W. Zhou, Q. Zhang, Z. Yang, Z. Li, F. Yang, D. Wang, J. Ye and L. Liu, Small, 2021, 17, 2101128 CrossRef CAS PubMed.
- Z. Li, B. Sun, D. Xiao, Z. Wang, Y. Liu, Z. Zheng, P. Wang, Y. Dai, H. Cheng and B. Huang, Angew. Chem., Int. Ed., 2023, 62, e202217569 CrossRef CAS PubMed.
- X. Chen, J. Chen, H. Chen, Q. Zhang, J. Li, J. Cui, Y. Sun, D. Wang, J. Ye and L. Liu, Nat. Commun., 2023, 14, 751 CrossRef CAS PubMed.
- T. Nishi, N. Sakamoto, K. Sekizawa, T. Morikawa and S. Sato, ChemSusChem, 2024, e202401082 CrossRef PubMed.
- K. Kamada, J. Jung, T. Wakabayashi, K. Sekizawa, S. Sato, T. Morikawa, S. Fukuzumi and S. Saito, J. Am. Chem. Soc., 2020, 142, 10261–10266 CrossRef CAS PubMed.
- K. Kamada, J. Jung, Y. Kametani, T. Wakabayashi, Y. Shiota, K. Yoshizawa, S. H. Bae, M. Muraki, M. Naruto, K. Sekizawa, S. Sato, T. Morikawa and S. Saito, Chem. Commun., 2022, 58, 9218–9221 RSC.
- T. Nishi, S. Sato, K. Sekizawa, T. M. Suzuki, K. Oh-ishi, N. Takahashi, Y. Matsuoka and T. Morikawa, ChemNanoMat, 2021, 7, 596–599 CrossRef CAS.
- S. Yoshioka, S. Nimura, M. Naruto and S. Saito, Sci. Adv., 2020, 6, eabc0274 CrossRef CAS PubMed.
- T. Arai, S. Sato and T. Morikawa, Energy Environ. Sci., 2015, 8, 1998–2002 RSC.
- B. van Dijk, G. M. Rodriguez, L. Wu, J. P. Hofmann, A. Macchioni and D. G. H. Hetterscheid, ACS Catal., 2020, 10, 4398–4410 CrossRef CAS PubMed.
- S. Sato, T. Arai and T. Morikawa, Nanotechnology, 2018, 29, 034001 CrossRef PubMed.
- R. Hudson, R. de Graaf, M. Strandoo Rodin, A. Ohno, N. Lane, S. E. McGlynn, Y. M. A. Yamada, R. Nakamura, L. M. Barge, D. Braun and V. Sojo, PNAS, 2020, 117, 22873–22879 CrossRef CAS PubMed.
- S. Sato, K. Sekizawa, S. Shirai, N. Sakamoto and T. Morikawa, Sci. Adv., 2023, 9, eadh9986 CrossRef CAS PubMed.
- F. Urbain, P. Tang, N. M. Carretero, T. Andreu, L. G. Gerling, C. Voz, J. Arbiol and J. R. Morante, Energy Environ. Sci., 2017, 10, 2256–2266 RSC.
- Meanwhile, ηSTF = 14.3% was achieved using an In2S3 electrode and an applied voltage of 2.2 V very recently: Q. Zhang, J. Gao, X. Wang, J. Zeng, J. Li, Z. Wang, H. He, J. Luo, Y. Zhao, L. Zhang, M. Grätzel and X. Zhang, Joule, 2024, 8, 1–19 CrossRef.
- B. Grømer and S. Saito, Inorg. Chem., 2023, 62, 14116–14123 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ey00261j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |