
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNeighboring effects of single-atom cobalt enable high-performance CO2 photoreduction†
Wenkai Yanab,
Yajun Zhang a,
Guojun Donga and
Yingpu Bi*a
a,
Guojun Donga and
Yingpu Bi*a
aState Key Laboratory for Oxo Synthesis & Selective Oxidation, Lanzhou Institute of Chemical Physics, CAS, Lanzhou, 730000, China. E-mail: yingpubi@licp.cas.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 13th January 2025
Abstract
Herein, we demonstrate the unique neighboring effect of single-cobalt active sites anchored on BiOCl nanosheets with high CO2 photoreduction performances by combining in situ X-ray photoelectron with in situ infrared spectroscopy. More specifically, single-atom Co sites demonstrate an exceptional electron-enriched feature from adjacent Bi atoms, which facilitates the formation of *CO2–Co and *H2O–Bi species, respectively. Under light irradiation, the photoinduced electron transfer from adjacent Bi atoms to single Co active sites is favorable for the formation *COOH and *CO intermediates, accompanied by the oxidation of H2O molecules into *OH and *OOH species on Bi sites. As a result, these dynamic electronic interactions between single-atom Co and adjacent Bi sites are responsible for a record CO evolution activity of 172.6 μmol g−1 h−1 under sunlight illumination, which exceeds that of pristine BiOCl by nearly one order of magnitude. These findings provide a fundamental understanding of the intrinsic neighboring effect between single-atom sites and adjacent atoms, which should be crucial and essential for the development of high-performance single-atom catalysts.
Broader contextCarbon dioxide (CO2) concentrations have increased substantially since the beginning of the industrial revolution, rising to 420 ppm (0.04%) in 2022. Among various candidates, solar-driven photocatalytic carbon fixation has been recognized as a promising pathway for reducing CO2 emission and achieving solar-to-chemical energy storage. Recently, single-atom catalysts have attracted significant attention in photocatalytic CO2 reduction, while the intrinsic roles of single-atom active sites and adjacent atoms in the photocatalytic behaviors still remain ambiguous until now. Herein, we demonstrate a facile hydrothermal strategy for one-step anchoring of Co single atoms on BiOCl nanosheets, which exhibited a record photocatalytic CO2-to-CO production activity of 172.6 μmol g−1 h−1 under simulated sunlight, nearly one order of magnitude higher than that of pristine BiOCl samples. More importantly, we have firstly established an intrinsic correlation between photocatalytic activity and neighboring effects of Co single atoms by combining in situ X-ray photoelectron with infrared spectroscopy, which offers new insights for directing the development of high-performance single-atom catalysts. |
Large consumption of traditional fossil fuels (including coal, oil, and natural gas) leads to a significant increase of carbon dioxide (CO2) concentrations in Earth's atmosphere, resulting in seriously concerning global warming and climate change.1,2 Thereby, it is very urgent to explore and develop feasible strategies for the direct conversion of CO2 greenhouse gas into renewable fuels and chemicals.3 However, owing to the high dissociation energy of dual C
![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) O bonds in the CO2 molecule (ca. 750 kJ mol−1), its activation is difficult in traditional thermocatalysis, which generally requires high temperature and pressure conditions.4,5 Additionally, a sustainable supply of pure hydrogen would be necessary for various CO2 hydrogenation reactions, while industrial-scale hydrogen sources are mainly obtained from reforming of natural gas.6 Compared with traditional thermocatalysis, photocatalytic CO2 reduction into valuable chemicals or fuels under ambient conditions, using abundant sunlight as the only energy source and H2O as proton source, represents a promising approach towards carbon neutrality and sustainable energy supply.7,8 Among various products of CO2 reduction, carbon monoxide (CO) holds significant importance as a fundamental building block for industrial chemical manufacturing.9,10 Despite tremendous progress having been made in design and construction of various semiconductor photocatalysts, the reduction efficiency as well as selectivity for CO production are usually unsatisfactory until now. Thereby, how to achieve highly efficient CO2 photoreduction to CO is still a great challenge.11,12
O bonds in the CO2 molecule (ca. 750 kJ mol−1), its activation is difficult in traditional thermocatalysis, which generally requires high temperature and pressure conditions.4,5 Additionally, a sustainable supply of pure hydrogen would be necessary for various CO2 hydrogenation reactions, while industrial-scale hydrogen sources are mainly obtained from reforming of natural gas.6 Compared with traditional thermocatalysis, photocatalytic CO2 reduction into valuable chemicals or fuels under ambient conditions, using abundant sunlight as the only energy source and H2O as proton source, represents a promising approach towards carbon neutrality and sustainable energy supply.7,8 Among various products of CO2 reduction, carbon monoxide (CO) holds significant importance as a fundamental building block for industrial chemical manufacturing.9,10 Despite tremendous progress having been made in design and construction of various semiconductor photocatalysts, the reduction efficiency as well as selectivity for CO production are usually unsatisfactory until now. Thereby, how to achieve highly efficient CO2 photoreduction to CO is still a great challenge.11,12
Recently, single metal atoms anchored on semiconductors have been extensively reported to promote the photocatalytic performances for CO2-to-CO conversion. More specifically, a single-atom metal could serve as specific active-sites for CO2 adsorption/activation, benefiting from unsaturated coordination environments and tunable electronic structures.13,14 Moreover, a significant advantage of single-atom photocatalysts is that they are an ideal model platform for providing atomic-level insights into surface active sites and corresponding catalytic mechanisms.15,16 To date, various characterization techniques, especially ex situ electron microscopy and X-ray absorption spectroscopy, have been successfully utilized to illustrate the atomic and electronic structures of single-atom photocatalysts.17,18 However, it has been recognized that the atomic and electronic properties of single-atom photocatalysts would dynamically change under working conditions, induced by several physical or chemical effects, such as reaction conditions, photoexcitation, chemical adsorption, and intermediates.19–21 Although theoretical calculations extracted from well-defined model structures have been widely employed to illustrate the corresponding mechanisms, real single-atom photocatalysts inevitably possess specific defects, surface species, and diverse coordination under reaction conditions.22,23 These greatly hinder the atomic-level understanding and further rational design of single-atom photocatalysts with tailored activities for CO2 reduction. Therefore, a direct insight into the dynamic evolutions of their atomic and electronic structures under operation conditions is necessary, which still faces a challenge.
Herein, we demonstrate a facile and efficient hydrothermal strategy for one-step anchoring of Co single atoms on BiOCl nanosheets, which exhibited a record photocatalytic CO2-to-CO production activity of 172.6 μmol g−1 h−1 under simulated sunlight. More importantly, a fundamental understanding for the neighboring effect between single-atom Co and adjacent Bi sites during photocatalytic CO2 reduction has been firstly achieved by employing in situ XPS and in situ FTIR. More specifically, the atomically dispersed cobalt sites with the electron-attracting effects from adjacent Bi sites are favorable for CO2 adsorption and subsequent conversion to form *COOH and *CO intermediates, while the electron-deficient Bi sites facilitate H2O molecules into *OH and *OOH intermediates. Owing to these synergistic effects, highly efficient conversion of CO2 to CO has been achieved. Thereby, dynamic electron interactions of single atoms with adjacent sites should be crucial for catalyst design towards CO2 reduction.
Single-atom-cobalt-anchored BiOCl nanosheets (denoted as Co–BiOCl) were synthesized via a facile one-step hydrothermal strategy. Fig. 1A shows a typical transmission electron microscopy (TEM) image, clearly indicating that the obtained Co–BiOCl samples possess the well-defined nanosheet structure. The high-resolution TEM (HR-TEM) image shown in Fig. S1A (ESI†) reveals the well-defined single-crystalline structure, and a lattice fringe with a spacing of 0.275 nm could be well indexed to the (110) plane.24 Fig. S1B (ESI†) shows the selected area electron diffraction (SAED) pattern, and a dihedral angle of 45° could be matched well with (110) and (200) planes of tetragonal BiOCl nanosheets, indicating the [001] growth orientation of these nanosheets.25 To further confirm the distribution of single-atom cobalt in Co–BiOCl, high-angle annular dark-field scanning TEM (HAADF-STEM) was performed, as shown in Fig. 1B. Owing to the lower atomic number of Co (27) than Bi (83), a large number of individual dark spots could be evidently detected, revealing the high distribution of Co species on BiOCl nanosheet surfaces. Furthermore, the line scan profiles for HAADF intensity analysis (Fig. 1C) demonstrate that the single-atom Co substitutes Bi at tetrahedral sites, instead of being located between the lattices of BiOCl (001) facets. Moreover, energy dispersive X-ray spectroscopy (EDS) mapping further confirms the uniform distribution of Co element in Co–BiOCl (Fig. 1D). The loading amount of single-atom cobalt in the obtained Co–BiOCl samples is about 0.57 wt% (Table S1, ESI†). In addition, the coordination environment of Co atoms was further explored by X-ray absorption near-edge spectra (XANES) and extended X-ray absorption fine structure (EXAFS). As shown in Fig. 1E, compared with CoO samples, the absorption edge position shows a slight negative shift, indicating the electron transfer from Bi to Co due to the higher electronegativity of Bi (1.9) than Co (1.8). Furthermore, the Fourier transformed k3-weighted EXAFS spectrum of Co–BiOCl is quite different from those of standard Co foil and CoO (Fig. 1F), suggesting that Co should be atomically dispersed, instead of forming Co or CoO clusters/particles (Fig. S2, ESI†).26,27
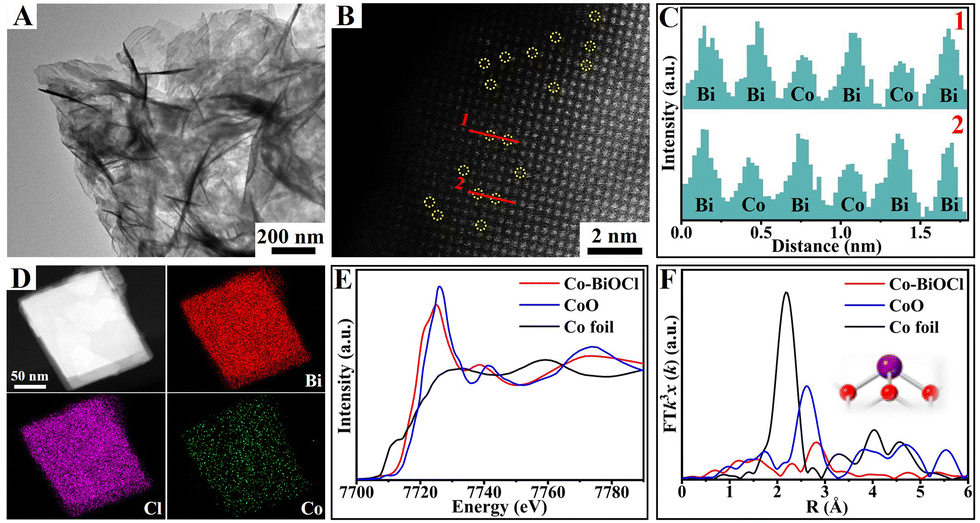 | ||
| Fig. 1 Characterizations of Co–BiOCl. (A) TEM image. (B) Aberration-corrected HAADF-STEM image and corresponding (C) atom intensity line scanning analysis. (D) Element mapping images. (E) Co K-edge XANES and (F) EXAFS spectra of Co foil, CoO and Co–BiOCl. | ||
The photocatalytic performances of Co–BiOCl samples for CO2 reduction have been explored under simulated sunlight irradiation, in which only CO2 and H2O reactants have been used. For comparison, pristine BiOCl samples have also been prepared (Fig. S3–S7, ESI†), and their photocatalytic activity has been explored under the same conditions. As shown in Fig. 2A, Co–BiOCl exhibits a much higher CO2 reduction capability for CO production than pristine BiOCl samples, and the CO production rate was calculated to be 172.6 μmol g−1 h−1 (Fig. S8, ESI†), nearly one order of magnitude more than that of pristine BiOCl samples (19.1 μmol g−1 h−1). More importantly, Co–BiOCl shows an outstanding selectivity (>99%) toward CO2-to-CO conversion (Fig. S9, ESI†). The apparent quantum yield (AQY) of CO2 to CO has also been determined, as shown in Fig. S10 (ESI†), and an AQY value of 0.81% has been obtained at a wavelength of 365 nm. Fig. 2B shows the photocatalytic CO2 reduction stability of Co–BiOCl. During five cycling experiments, the CO production rates generally remained constant, and no evident change could be detected (Fig. S11–S13, ESI†). Meanwhile, the 13C-labeled isotope experiment has been performed by using 13CO2 gas as feedstock to validate the carbon source of CO product. As shown in Fig. 2C, the dominant peaks of 13CO2 (m/z = 45.1) and 13CO (m/z = 29.1) could be evidently detected by gas chromatography-mass spectrometry (GC-MS). On the basis of the above results, it can be concluded that the formation of CO product should result from photocatalytic CO2 reduction. In addition to cobalt, other metals anchored on BiOCl nanosheets, including Ni, Cu, and Zn, could also be achieved via this facile one-step method (Fig. S14–S18, ESI†). As shown in Fig. 2D, their photocatalytic performances for CO2 reduction have also been effectively improved compared with pristine BiOCl, confirming the universal applicability for metal anchoring on BiOCl nanosheets and significantly promoting the CO2 reduction activity. Furthermore, photoelectrochemical (PEC) measurements have been performed on both pristine BiOCl and Co–BiOCl to further evaluate their photogenerated charge separation and migration ability. As shown in Fig. 2E, compared with pristine BiOCl, the photocurrent density of Co–BiOCl has been drastically enhanced, indicating the anchoring of single-atom Co could effectively promote charge separation and accelerate charge transfer. Moreover, Fig. 2F shows the electrochemical impedance spectroscopy (EIS) results, and Co–BiOCl demonstrates lower charge transport resistances and higher electro-conductibility than pristine BiOCl samples.28 On the basis of the above photocatalytic and PEC results, it could be concluded that the rational anchoring of single-atom metal active sites on semiconductors should be a feasible strategy for significantly promoting the photocatalytic activity for CO2 reduction.
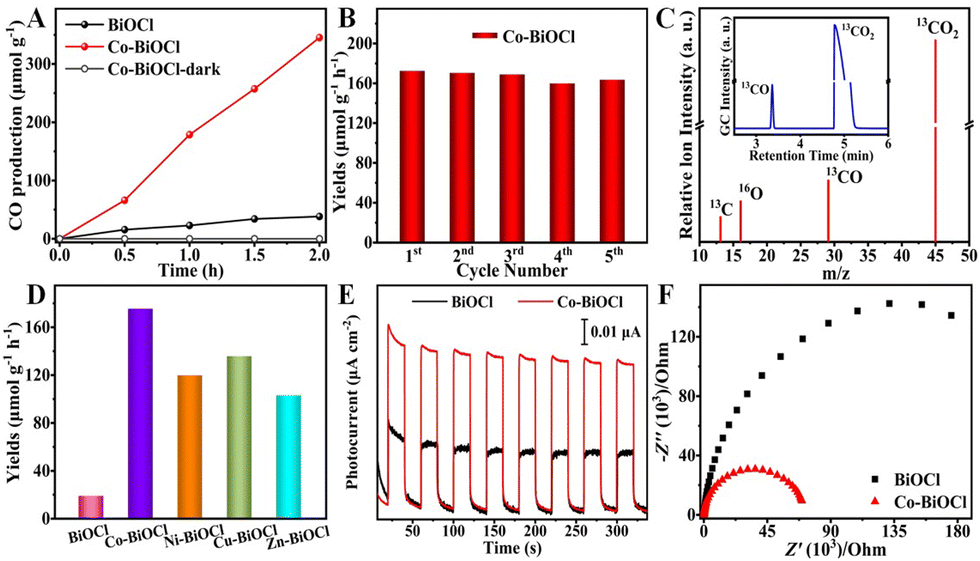 | ||
| Fig. 2 (A) Photocatalytic CO2 activities of as-prepared photocatalysts. (B) Cycling tests of Co–BiOCl sample. (C) Mass spectrum of the 13CO2 photoreduction products over Co–BiOCl (inset image: GC signal). (D) Comparative presentation of CO evolution rates. (E) i–t curves and (F) EIS curves of the as-obtained photocatalysts. | ||
Furthermore, the charge separation capability has been firstly explored by steady-state photoluminescence (PL) spectroscopy, as shown in Fig. S19 (ESI†). It could be clearly seen that compared with pristine BiOCl samples, the PL peak intensity of Co–BiOCl has been significantly reduced, indicating that the anchoring of Co single atoms could effectively restrain the recombination of photogenerated charge carriers in BiOCl nanosheets. In addition to the steady-state PL spectra, time-resolved PL (TR-PL) spectra have also been measured to probe interfacial charge carrier dynamics under excited state (Fig. S20, ESI†). The average carrier lifetime over Co–BiOCl is calculated to be 9.46 ns, which is much longer than that of pristine BiOCl (7.78 ns).29,30 Moreover, Kelvin probe force microscopy (KPFM) has been performed to explore the photoinduced surface potential changes. Fig. 3A and B show the typical surface potential mapping of Co–BiOCl under darkness and light irradiation, respectively. To quantitatively explore the surface potential changes, the line-scanning surface potentials of Co–BiOCl and BiOCl have been measured. As shown in Fig. 3C, Co–BiOCl displays a dramatic surface potential change of ∼54 mV under darkness and light irradiation, clearly revealing the efficient charge separation on the surfaces of Co–BiOCl.31,32 In contrast, the pristine BiOCl samples only exhibit a surface potential change of ∼16 mV (Fig. S21, ESI†). Subsequently, scanning photoelectrochemical microscopy (SPECM) has also been conducted to further investigate the surface photoactive properties of both Co–BiOCl and BiOCl. As shown in Fig. 3D, a relatively high photocurrent is detected in Co–BiOCl regions under light illumination, while the photocurrent is drastically reduced in the BiOCl regions, indicating that the Co–BiOCl catalyst exhibits much higher charge separation and transfer capability than BiOCl samples.33,34 Furthermore, the crystal structure evolutions of Co single atoms have been measured by using in situ X-ray diffraction. As shown in Fig. S22 (ESI†), the characteristic diffraction peaks of Co–BiOCl slightly shifted toward the low-value direction under light illumination, indicating the increases of the interlayer lattice spacing (Fig. 3F), which should be attributed to the photoinduced electronic redistribution and bond changes in the interior of Co–BiOCl.35
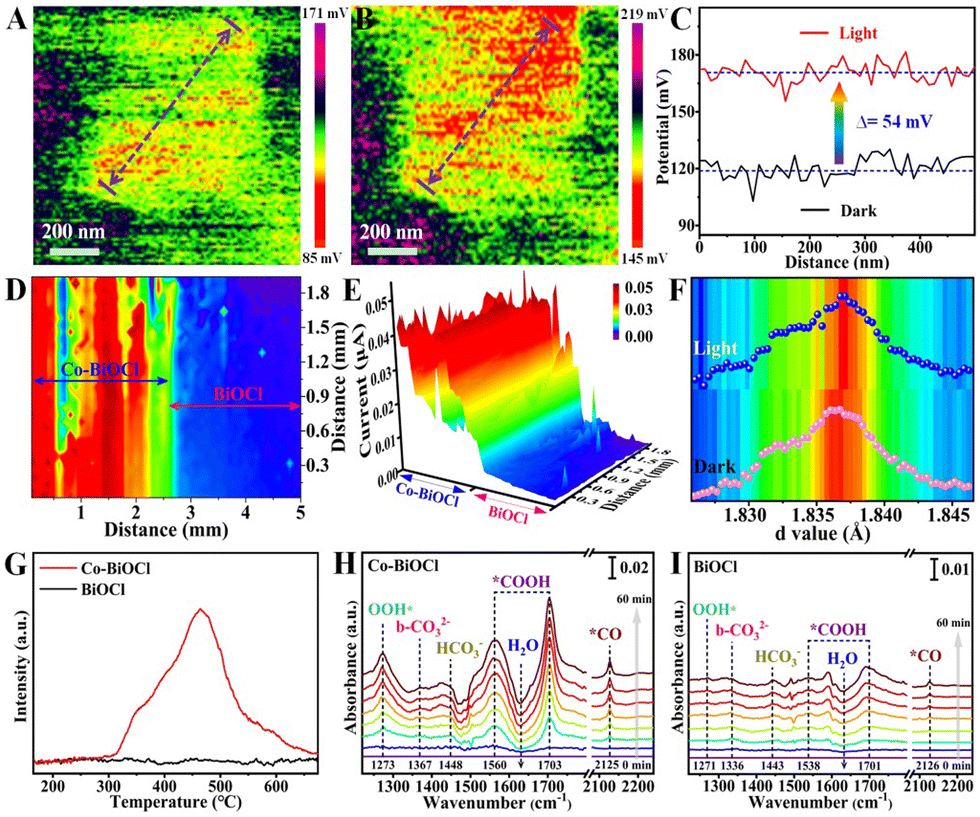 | ||
| Fig. 3 KPFM mappings of the surface potential distribution of Co–BiOCl under darkness (A) and light irradiation (B). (C) The line-scanning surface potential changes under darkness and light irradiation. (D) Top and (E) side view of SPECM surface photocurrents under light irradiation. (F) The layer spacing changes of Co–BiOCl before and after light irradiation. (G) CO2-TPD curves of pristine BiOCl and Co–BiOCl. In situ FTIR spectra of CO2 reduction on Co–BiOCl (H) and pristine BiOCl (I). | ||
In addition to the surface charge distribution and structure evolutions, the CO2 adsorption properties have been explored by CO2 temperature-programmed desorption (CO2-TPD).36 As shown in Fig. 3G, the CO2-TPD curves clearly reveal that pristine BiOCl exhibits a relatively poor capability for CO2 adsorption, primarily suffering from insufficient surface active sites. Notably, the introduction of Co single atoms could significantly enhance the chemisorption capability of CO2, and a dominant adsorption peak at ∼465 °C has been detected for Co–BiOCl samples. Additionally, in situ Fourier transform infrared reflection (IS-FTIR) has been conducted to further explore the reactant adsorption and intermediate formations during photocatalytic CO2 reduction. Under darkness, Co–BiOCl shows much higher CO2 absorption peaks than pristine BiOCl samples (Fig. S25 and S26, ESI†). Upon light irradiation on Co–BiOCl, the new peaks at ∼1560 and ∼1703 cm−1 assigned to *COOH intermediates, critical intermediates for CO2 conversion to CO, are remarkably increased with increasing irradiation time, and the infrared peaks of *CO intermediates at ∼2125 cm−1 could also be detected.37,38 Meanwhile, two broad peaks at ∼3535 cm−1 and ∼1273 cm−1 could be indexed to OH* and OOH* intermediates, respectively, resulting from the activation and dissociation of H2O molecules at ∼1640 cm−1 (Fig. 3H and Fig. S27, ESI†).39,40 Compared with Co–BiOCl, the FTIR peak changes of reaction intermediates for pristine BiOCl are unobvious (Fig. 3I and Fig. S28, ESI†), demonstrating its relatively poor activation and conversion capability for both CO2 and H2O molecules. On the basis of the above results, it could be concluded that the Co single atoms anchored on BiOCl nanosheets could significantly promote the adsorption/activation of both CO2 and H2O and facilitate the formation *COOH and *OOH intermediates.
To further elucidate the underlying mechanisms responsible for the high photocatalytic activity of Co–BiOCl towards CO2-to-CO conversion, in situ X-ray photoelectron spectroscopy (IS-XPS) has been employed to explore the dynamic evolutions of electronic structures and chemical bonds during the CO2 photoreduction process.41,42 Note that before CO2 and H2O adsorption, the evident Bi shoulder peaks located at high binding energy (BE) have been detected for Co–BiOCl samples (Fig. 4), while pristine BiOCl only demonstrates the standard Bi3+ peaks (Fig. S30, ESI†). This phenomenon clearly reveals that the Co single atoms anchored on BiOCl nanosheets could attract electrons from the surrounding Bi atoms, leading to the formation of surface Bi sites with high valences.43 After CO2 and H2O adsorption, two characteristic peaks at 288.3 eV and 286.4 eV could be obviously detected in the C 1s spectrum, which could be well indexed to *CO2 and *CO intermediates due to CO2 adsorption and dissociation. Interestingly, the ratios of high-valence Bi species in Bi 4f spectra have been evidently reduced, accompanied by increased Co3+/Co2+ ratios. It was considered that the adsorption of H2O molecules should be located on the electron-deficient Bi(3+x)+ sites, and their electron injection effects lead to the decrease of high-valence Bi sites. In contrast, the CO2 molecules should be located on the electron-enriched Co sites, and the electron-attracting effects result in the increase of high-valence Co species. Under light irradiation, the intensity of *CO2 peaks is significantly decreased, while that of *CO peaks is obviously increased, indicating the further activation of CO2 molecules on Co–BiOCl surfaces (Fig. 4A). Notably, the high-valence Bi(3+x)+ peaks have remarkably increased (Fig. 4B), attributed to the activation of H2O molecules into *OH intermediates on surface Bi active sites (Fig. S31, ESI†). Note that owing to the formation of *CO intermediates on single-atom Co active sites, the ratio of Co3+/Co2+ has been correspondingly decreased (Fig. 4C). Related IS-XPS studies of pristine BiOCl have also been carried out under the same conditions, as shown in Fig. S30 (ESI†). The CO2 adsorption and dissociation are much less than for Co–BiOCl samples in the C 1s spectrum, which is consistent with the poor adsorption and activation of CO2 shown in CO2-TPD and FTIR spectra. Additionally, no evident changes of Bi 4f peaks under light irradiation could be observed, further confirming the single-atom Co active sites for promoting the photocatalytic CO2 reduction activity by adsorbing and activating CO2 molecules.
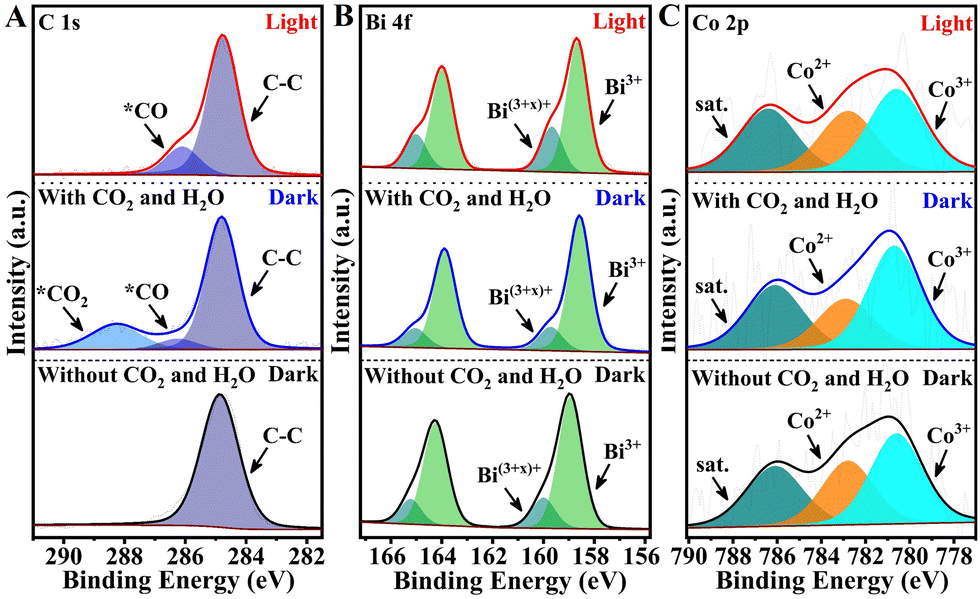 | ||
| Fig. 4 High-resolution IS-XPS spectra of C 1s (A), Bi 4f (B) and Co 2p (C) on single-atom Co–BiOCl tested in darkness and under illumination. | ||
Considering the above in situ characterization results, a possible mechanism has been proposed to elucidate CO2 reduction to CO over Co–BiOCl during the photocatalytic process (Scheme 1). Note that single-atom Co sites on BiOCl nanosheets demonstrate an exceptional electron-enriched feature from adjacent Bi atoms, which could serve as efficient active sites for facilitating the adsorption of CO2 molecules. Simultaneously, owing to the electron transfer to single-atom Co sites (Fig. S32, ESI†), the adjacent Bi atoms with high-valence states possess strong adsorption capability for H2O molecules. Under light irradiation, the electron injection from water molecules into the surface Bi active sites and single-atom Co sites facilitates the absorbed *CO2 interacting with the proton to form COOH* intermediates. Furthermore, the *COOH located on single-atom Co active sites could be further reduced via combining with another proton and electron, and the *CO desorption towards CO formation is finally achieved, accompanied by the activation of H2O molecules into *OH and *OOH intermediates on surface Bi active sites. Accordingly, owing to the electron-enriched single-atom Co sites and electron-deficient Bi sites for efficient adsorption/activation for H2O and CO2 molecules, a significant improvement of photocatalytic reactivity for CO2 reduction to CO product has been achieved on the Co–BiOCl surfaces. Thereby, the combination of in situ spectroscopy techniques could be a powerful strategy to correlate electronic structure of single-atom active sites with photocatalytic activity, which should be a feasible pathway for directing the rational design of CO2 photoreduction catalysts.
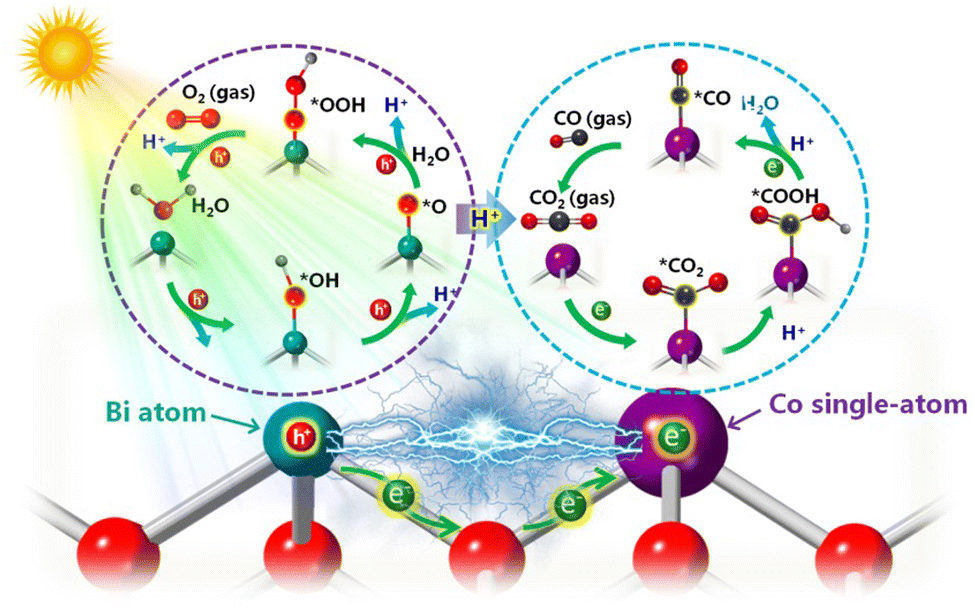 | ||
| Scheme 1 Schematic diagram of the neighboring effect for CO2 photoreduction over the Co–BiOCl sample. | ||
In summary, we have successfully constructed Co single atoms on BiOCl nanosheets by a one-step hydrothermal method, which exhibited an excellent activity and selectivity for CO2 photoreduction to CO by utilizing H2O as the proton source. The CO production rate could reach up to 172.6 μmol g−1 h−1, a more than 9-times enhancement compared with pristine BiOCl. More importantly, the atomic-level identification of the neighboring effect of single-atom cobalt active sites anchored on BiOCl nanosheets during photocatalytic CO2 reduction has been achieved by employing IS-XPS and IS-FTIR. The single-atom Co demonstrates an exceptional electron-enriched feature from adjacent Bi atoms, which could serve as efficient active sites for facilitating the adsorption/activation of CO2 molecules. Additionally, electron-deficient Bi sites could effectively promote H2O adsorption and conversion into *OH and *OOH intermediates. Thereby, Co single atoms anchored on BiOCl nanosheets not only significantly facilitate the local charge separation and adsorption/activation of CO2, but also tailor the electronic structure of adjacent Bi atoms for promoting H2O molecule dissociation.
Author contributions
W. K. Y. and Y. P. B. conceived and designed experiments. W. K. Y. performed the sample synthesis and activity measurements. Y. J. Z. performed the XPS measurement and G. J. D. performed the TEM characterizations. W. K. Y. and Y. P. B. wrote the manuscript. All authors reviewed the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The work was supported by the National Natural Science Foundation of China (21832005, 22372181, 22072168, 22002175), Major Program of the Lanzhou Institute of Chemical Physics, CAS (no. ZYFZFX-3), Major Science and Technology Projects in Gansu Province (22ZD6GA003), the CAS “Light of West China” Program and West Light Foundation of the Chinese Academy of Sciences (xbzg-zdsys-202209).References
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87–97 CrossRef CAS PubMed.
- H. He, R. J. Kramer, B. J. Soden and N. Jeevanjee, Science, 2023, 382, 1051–1056 CrossRef CAS PubMed.
- X. Wu, Y. Li, G. Zhang, H. Chen, J. Li, K. Wang, Y. Pan, Y. Zhao, Y. Sun and Y. Xie, J. Am. Chem. Soc., 2019, 141, 5267–5274 CrossRef CAS PubMed.
- G. Chen, G. I. N. Waterhouse, R. Shi, J. Zhao, Z. Li, L. Wu, C. Tung and T. Zhang, Angew. Chem., Int. Ed., 2019, 58, 17528–17551 CrossRef CAS PubMed.
- R. W. Dorner, D. R. Hardy, F. W. Williams and H. D. Willauer, Energy Environ. Sci., 2010, 3, 884–890 RSC.
- L. C. Buelens, V. V. Galvita, H. Poelman, C. Detavernier and G. B. Marin, Science, 2016, 354, 449–452 CrossRef CAS PubMed.
- Y. Yin, W. Jing, H. Qiu, F. Wang, Y. Liu and L. Guo, EES. Catal., 2023, 1, 755–764 RSC.
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS PubMed.
- R. T. Rashid, Y. Chen, X. Liu, F. A. Chowdhury, M. Liu, J. Song, Z. Mi and B. Zhou, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2121174119 CrossRef CAS PubMed.
- Y. Wang, E. Chen and J. Tang, ACS Catal., 2022, 12, 7300–7316 CrossRef CAS PubMed.
- J. Fu, K. Jiang, X. Qiu, J. Yu and M. Liu, Mater. Today, 2020, 32, 222–243 CrossRef CAS.
- X. Zhu, H. Xu, J. Liu, C. Bi, J. Tian, K. Zhong, B. Wang, P. Ding, X. Wang, P. K. Chu, H. Xu and J. Ding, Adv. Sci., 2023, 10, 2307192 CrossRef CAS PubMed.
- S. Hu, P. Qiao, X. Yi, Y. Lei, H. Hu, J. Ye and D. Wang, Angew. Chem., Int. Ed., 2023, 62, e202304585 Search PubMed.
- C. Ding, X. Lu, B. Tao, L. Yang, X. Xu, L. Tang, H. Chi, Y. Yang, D. M. Meira, L. Wang, X. Zhu, S. Li, Y. Zhou and Z. Zou, Adv. Funct. Mater., 2023, 33, 2302824 CrossRef CAS.
- R. Lang, X. Du, Y. Huang, X. Jiang, Q. Zhang, Y. Guo, K. Liu, B. Qiao, A. Wang and T. Zhang, Chem. Rev., 2020, 120, 11986–12043 CrossRef CAS PubMed.
- C. Gao, J. Low, R. Long, T. Kong, J. Zhu and Y. Xiong, Chem. Rev., 2020, 120, 12175–12216 CrossRef CAS PubMed.
- L. Yang, Z. Chen, Q. Cao, H. Liao, J. Gao, L. Zhang, W. Wei, H. Li and J. Lu, Adv. Mater., 2023, 36, 2306758 CrossRef PubMed.
- P. Chen, B. Lei, X. Dong, H. Wang, J. Sheng, W. Cui, J. Li, Y. Sun, Z. Wang and F. Dong, ACS Nano, 2020, 14, 15841–15852 CrossRef CAS PubMed.
- Z. Teng, Q. Zhang, H. Yang, K. Kato, W. Yang, Y. Lu, S. Liu, C. Wang, A. Yamakata, C. Su, B. Liu and T. Ohno, Nat. Catal., 2021, 4, 374–384 CrossRef CAS.
- M. Li, S. Wu, D. Liu, Z. Ye, L. Wang, M. Kan, Z. Ye, M. Khan and J. Zhang, J. Am. Chem. Soc., 2024, 146, 15538–15548 CrossRef CAS PubMed.
- C. Han, R. Qi, R. Sun, K. Fan, B. Johannessen, D. Qi, S. Cao and J. Xu, Appl. Catal., B, 2023, 320, 121954 CrossRef CAS.
- Z. Xue, J. Yang, L. Ma, H. Li, L. Luo, K. Ji, Z. Li, X. Kong, M. Shao, L. Zheng, M. Xu and H. Duan, ACS Catal., 2024, 14, 249–261 CrossRef CAS.
- S. Mo, X. Zhao, S. Li, L. Huang, X. Zhao, Q. Ran, M. Zhang, R. Peng, Y. Zhang, X. Zhou, Y. Fan, Q. Xie, Y. Guo, D. Ye and Y. Chen, Angew. Chem., Int. Ed., 2023, 62, e202313868 CrossRef CAS PubMed.
- D. Cui, L. Wang, K. Xu, L. Ren, L. Wang, Y. Yu, Y. Du and W. Hao, J. Mater. Chem. A, 2018, 6, 2193 RSC.
- F. Guo, C. Mao, C. Liang, P. Xing, L. Yu, Y. Shi, S. Cao, F. Wang, X. Liu, Z. Ai and L. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202314243 Search PubMed.
- J. Di, C. Chen, S. Yang, S. Chen, M. Duan, J. Xiong, C. Zhu, R. Long, W. Hao, Z. Chi, H. Chen, Y. Weng, J. Xia, L. Song, S. Li, H. Li and Z. Liu, Nat. Commun., 2019, 10, 2840 CrossRef PubMed.
- C. Gao, S. Chen, Y. Wang, J. Wang, X. Zheng, J. Zhu, L. Song, W. Zhang and Y. Xiong, Adv. Mater., 2018, 30, 1704624 CrossRef PubMed.
- Y. Tian, R. Wang, S. Deng, Y. Tao, W. Dai, Q. Zheng, C. Huang, C. Xie, Q. Zeng, J. Lin and H. Chen, Nano Lett., 2023, 23, 10914–10921 CrossRef CAS PubMed.
- H. Li, B. Zhu, B. Cheng, G. Luo, J. Xu and S. Cao, J. Mater. Sci. Technol., 2023, 161, 192–200 CrossRef CAS.
- S. Bera, A. Patra, S. Shyamal, D. Nasipuri and N. Pradhan, ACS Energy Lett., 2022, 7, 3015–3023 CrossRef CAS.
- Y. Lu, M. Chen, T. Huang, Y. Huang, J. Cao, H. Li, W. Ho and S. C. Lee, Environ. Sci.: Nano, 2021, 8, 1927 RSC.
- G. Sun, J. Zhang, B. Cheng, H. Yu, J. Yu and J. Xu, Chem. Eng. J., 2023, 476, 146818 Search PubMed.
- C. H. Ryu and H. Ren, Nano Lett., 2024, 24, 6112–6116 Search PubMed.
- L. Yan, G. Dong, X. Huang, Y. Zhang and Y. Bi, Appl. Catal., B, 2024, 345, 123682 Search PubMed.
- H. Tsai, R. Asadpour, J. Blancon, C. C. Stoumpos, O. Durand, J. W. Strzalka, B. Chen, R. Verduzco, P. M. Ajayan, S. Tretiak, J. Even, M. A. Alam, M. G. Kanatzidis, W. Nie and A. D. Mohite, Science, 2018, 360, 67–70 CrossRef CAS PubMed.
- Y. Li, Y. Wang, J. Li, M. Qi, M. Conte, Z. Tang and Y. Xu, ACS Catal., 2024, 14, 657–669 CrossRef CAS.
- X. Xiong, C. Mao, Z. Yang, Q. Zhang, G. I. N. Waterhouse, L. Gu and T. Zhang, Adv. Energy Mater., 2020, 10, 2002928 Search PubMed.
- Y. Li, Y. Xue, X. Gao, L. Wang, X. Liu, Z. Wang and S. Shen, Adv. Funct. Mater., 2024, 34, 2312634 CrossRef CAS.
- Y. Xu, D. Wu, Q. Zhang, P. Rao, P. Deng, M. Tang, J. Li, Y. Hua, C. Wang, S. Zhong, C. Jia, Z. Liu, Y. Shen, L. Gu, X. Tian and Q. Liu, Nat. Commun., 2024, 15, 564 CrossRef CAS PubMed.
- B. Su, Y. Kong, S. Wang, S. Zou, W. Lin, Y. Fang, Y. Hou, G. Zhang, H. Zhang and X. Wang, J. Am. Chem. Soc., 2023, 145, 27415 CrossRef CAS PubMed.
- Y. Lan, Y. Zhang, X. Huang and Y. Bi, Angew. Chem., Int. Ed., 2024, 63, e202407736 CrossRef CAS PubMed.
- J. An, S. Ge, G. Wang and H. Fu, Energy Environ. Sci., 2024, 17, 5039–5047 RSC.
- L. Zhao, J. Bian, X. Zhang, L. Bai, L. Xu, Y. Qu, Z. Li, Y. Li and L. Jiang, Adv. Mater., 2022, 34, 2205303 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, and additional data containing, TEM, XRD, and XPS measurements. See DOI: https://doi.org/10.1039/d4ey00274a |
| This journal is © The Royal Society of Chemistry 2025 |