
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBreaking the selectivity-activity seesaw in ethane oxidative dehydrogenation via the synergetic effects of doping and electrochemical activation†
Xiang Sun‡
a,
Benchi Chen‡a,
Heejae Yang‡b,
Mengzhen Zhoua,
Nian Zhangc,
Yunkyung Kimd,
Wonyoung Lee ef,
Jeong Woo Han*d,
Zhang Linag and
Yan Chen*a
ef,
Jeong Woo Han*d,
Zhang Linag and
Yan Chen*a
aGuangdong Provincial Key Laboratory of Solid Wastes Pollution Control and Recycling, School of Environment and Energy, South China University of Technology, Guangzhou, 510006, China. E-mail: escheny@scut.edu.cn
bDepartment of Chemical Engineering, Pohang University of Science and Technology (POSTECH), Pohang, Gyeongbuk 37673, Republic of Korea
cShanghai Synchrotron Radiation Facility, Shanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai, 201204, China
dDepartment of Materials Science and Engineering, Research Institute of Advanced Materials, Seoul National University, Seoul 08826, Republic of Korea. E-mail: jwhan98@snu.ac.kr
eSKKU Institute of Energy Science and Technology (SIEST), Sungkyunkwan University, Suwon, Gyeonggi-do 16419, Republic of Korea
fSchool of Mechanical Engineering, Sungkyunkwan University, Suwon, Gyeonggi-do 16419, Republic of Korea
gChinese National Engineering Research Center for Control & Treatment of Heavy Metal Pollution, School of Metallurgy and Environment, Central South University State Key Laboratory of Functional Materials for Informatics, Shanghai Institute of Microsystem and Information Technology, Chinese Academy of Sciences, Shanghai, 200050, China
First published on 28th March 2025
Abstract
The oxidative dehydrogenation (ODH) of alkanes using a solid oxide electrolysis cell (SOEC) has attracted worldwide attention as an efficient method for producing ethylene. Nevertheless, it remains challenging to achieve both a high alkane conversion rate and high ethylene selectivity. In this work, we demonstrate that the combination of doping and electrochemical activation can break this activity–selectivity seesaw and achieve a high ethylene yield. Using Sr2Ti0.8(Co1.2−xFex)O6−δ with different dopants as model electrodes, we show that increasing the Fe content efficiently lowers the oxygen activity by weakening metal–oxygen covalency, downshifting O 2p-band relative to the Fermi level, and increasing the oxygen vacancy formation energy. Such changes result in a lower ethane conversion rate but higher ethylene selectivity for Sr2Ti0.8Fe1.2O6−δ (STF) compared to electrodes with higher Co content. By increasing the applied potential, we can effectively increase the conversion rate of ethane without sacrificing too much ethylene selectivity. Ultimately, the SOEC with STF anode achieves an ethylene yield of up to 71% at 800 °C at 1.2 V with CO2 as the oxidant on the cathode side, which is among the highest documented. The insights gained from this study knowledge can guide the rational design of high-temperature electrochemical devices for other small molecule conversion reactions.
Broader contextAlkene is one of the most important raw materials for modern industry. Currently, the main sources of alkenes are the conversion of alkanes via direct cracking, dehydrogenation, or oxidative dehydrogenation reactions (ODH). By introducing an oxidant into the dehydrogenation process, the alkane ODH reactions can occur at lower temperatures, and coke formation can be avoided due to the presence of the oxidant. A solid oxide electrolysis cell (SOEC) has emerged as a potential platform for the ODH reactions of alkanes. Driven by electrochemical potential, oxygen from CO2 or O2 at the cathode of the SOEC passes through the solid electrolyte to the anode surface, which actively reacts with alkanes to produce olefins. Nevertheless, highly active catalysts tend to over-oxidize the dehydrogenated species to CO or CO2, leading to low olefin selectivity. Here, we show that the synergistic effect of doping and electrochemical activation is an effective way to break the selectivity-activity seesaw, achieving high conversion while maintaining high selectivity. This principle can be used to guide the rational design of high-temperature electrochemical devices for other small molecule conversion reactions. |
Introduction
Ethylene is one of the most important industrial organic chemicals and serves as the building block for the petrochemical industry. The production of ethylene from steam-cracking crude oil is an energy-intensive process due to its high operating temperatures.1 With the large-scale exploitation of shale gas, the dehydrogenation of light alkanes (C1–C4) has emerged as an alternative approach to produce olefins.2 However, the direct dehydrogenation of light alkanes is thermodynamically unfavorable and must be carried out at extremely high temperatures with limited product yields. By introducing an oxidant into the dehydrogenation process, alkane oxidative dehydrogenation (ODH) reactions can occur at lower temperatures and coke formation can be avoided due to the presence of the oxidant.3 Significant efforts have been devoted to developing high-performance catalysts for alkane ODH reactions4–6 with high activity to break the C–H bond, enabling dehydrogenation.7 Nevertheless, highly active catalysts tend to over-oxidize the dehydrogenated species to CO or CO2, leading to low olefin selectivity.7–10 To achieve a high yield of ethylene through alkane ODH reactions, it is essential to find a way to break this selectivity-activity seesaw.The alkane ODH process is reported to be strongly related to the characteristics of surface oxygens.4,11,12 For instance, electrophilic oxygen (O−, O2−, and O22−) can lead to over-oxidation of ethane,13 while nucleophilic oxygen (O2−) tends to promote selective conversion of ethane to ethylene.14 Tian et al. attributed the high activity of Ce-doped SrFeO3−δ catalysts in the ethane ODH reaction to the high proportion of active oxygen species in the near-surface region.7 Zhu et al. doped Sn, Ti, and W into nickel oxide to reduce highly reactive oxygen species on the surface, thereby improving ethylene selectivity.10 These works demonstrate that modulating surface oxygen presents an efficient approach to enhancing the performance of alkane ODH reactions.
Solid oxide electrolysis cells (SOEC) have emerged as a promising means for the ODH reaction of alkanes. Driven by electrochemical potential, oxygen from CO2, H2O or O2 on the cathode of the SOEC passes through the solid electrolyte to the anode surface, where it actively reacts with alkane to produce olefins. Perovskite-oxide-based anode materials, such as La0.6Sr0.4Co0.2Fe0.8O3−δ–Sm0.2Ce0.8O2−δ, Sr2Fe2−xMoxO6−δ, Sr1.95Fe1.4Co0.1Mo0.5O6−δ, and La0.2Sr0.8TiO3±σClσ, have shown promising performance.4,6,15,16 Interestingly, Sun et al.17 recently reported that the applied potential drives the formation of highly active oxygen species, effectively promoting the dehydrogenation process. All this pioneering work highlights the electrochemical potential in SOEC as a promising tuning knob for modulating ODH kinetics.
In this study, we achieved an outstanding ethane oxidative dehydrogenation performance on a doped SrTiO3 electrode by combining cation doping and electrochemical activation. Electrode materials with different dopants, including Sr2Ti0.8Co1.2O6−δ (STC), Sr2Ti0.8Co0.6Fe0.6O6−δ (STCF), and Sr2Ti0.8Fe1.2O6−δ (STF), were prepared as SOEC anodes for the ethane ODH reaction. The combination of advanced spectroscopic techniques and density functional theory (DFT) calculations revealed that increasing the Fe/Co ratio lowers the oxygen activity by presenting weaker metal–oxygen covalency, a lower O 2p band center position relative to the Fermi level, and higher energies for oxygen vacancy formation. DFT results predicted that such a change in oxygen activity with the Fe/Co ratio could result in lower dehydrogenation energy (ΔG1) and a larger difference between ethylene desorption free energy and ethylene dehydrogenation free energy (ΔGdesorption − ΔG3 (eV)), which would eventually lead to lower activity and higher selectivity for electrodes with high Fe content. Consistent with theoretical calculations, we found that as the Fe content in the electrode increased, the ethane conversion rate decreased while the ethylene selectivity increased. Further applying an electrochemical potential effectively increased the conversion rate of ethane without sacrificing too much ethylene selectivity. Consequently, the SOEC with the STF electrode achieved an ethylene yield of up to 71% at 800 °C with CO2 as the oxidant on the cathode side, which is among the highest levels reported in the literature. This study reveals the critical role of material surface oxygen activity in the ethane ODH reaction. The methodology of combining doping and electrochemical activation can be applied to other high-temperature electrochemical small-molecule conversions.
Results and discussion
Modulated oxygen characteristics probed by spectroscopic techniques
We first demonstrated experimentally that the oxygen characteristics of the electrode can be effectively modulated by cation doping. Three electrode materials, Sr2Ti0.8CoO6−δ (STC), Sr2Ti0.8Co0.6Fe0.6O6−δ (STCF), and Sr2Ti0.8FeO6−δ (STF), with different dopants at the B site, were synthesized as model electrodes (Fig. S1 and S2, ESI†).To avoid the influence of the complex structure on the characterization of oxygen activity, STC, STCF, and STF thin-film model systems were prepared on yttria-stabilized zirconia (YSZ) substrates using pulsed laser technology (PLD), with gadolinia-doped ceria (GDC) as the intermediate layer and embedded Au as an internal reference (Fig. 1a). The prepared films were highly textured in the (001) crystal orientation. The obtained thin film has a flat surface with a root mean square (RMS) roughness value of 0.182 nm (Fig. 1c).
 | ||
| Fig. 1 (a) Schematic of pulsed laser deposition and thin film model system; (b) HRXRD diagram of the thin film systems STC, STCF, and STF deposited by PLD. The sapphire peak is from the sample holder used for XRD measurement. (c) AFM image of an STF thin film. | ||
Having obtained a model thin film with a well-defined structure, the oxygen characteristics of different samples were studied using several advanced spectroscopic techniques.
The electronic structure of oxygen and the valence state of transition metals were first probed by soft X-ray absorption spectroscopy (XAS) technique in total electron yield (TEY) mode. Fig. 2a shows the O K-edge XAS spectra of these samples. The peak located near 530 eV corresponds to the hybridization of the O 2p energy band with the transition metal 3d energy band (O 2p–TM 3d), whereas the peaks located around 535 eV and 540–545 eV are attributed to the hybridization of the O 2p energy band with the Sr 4d energy band (O 2p–Sr 4d) and the TM 4sp energy band (O 2p–TM 4 sp), respectively.18,19
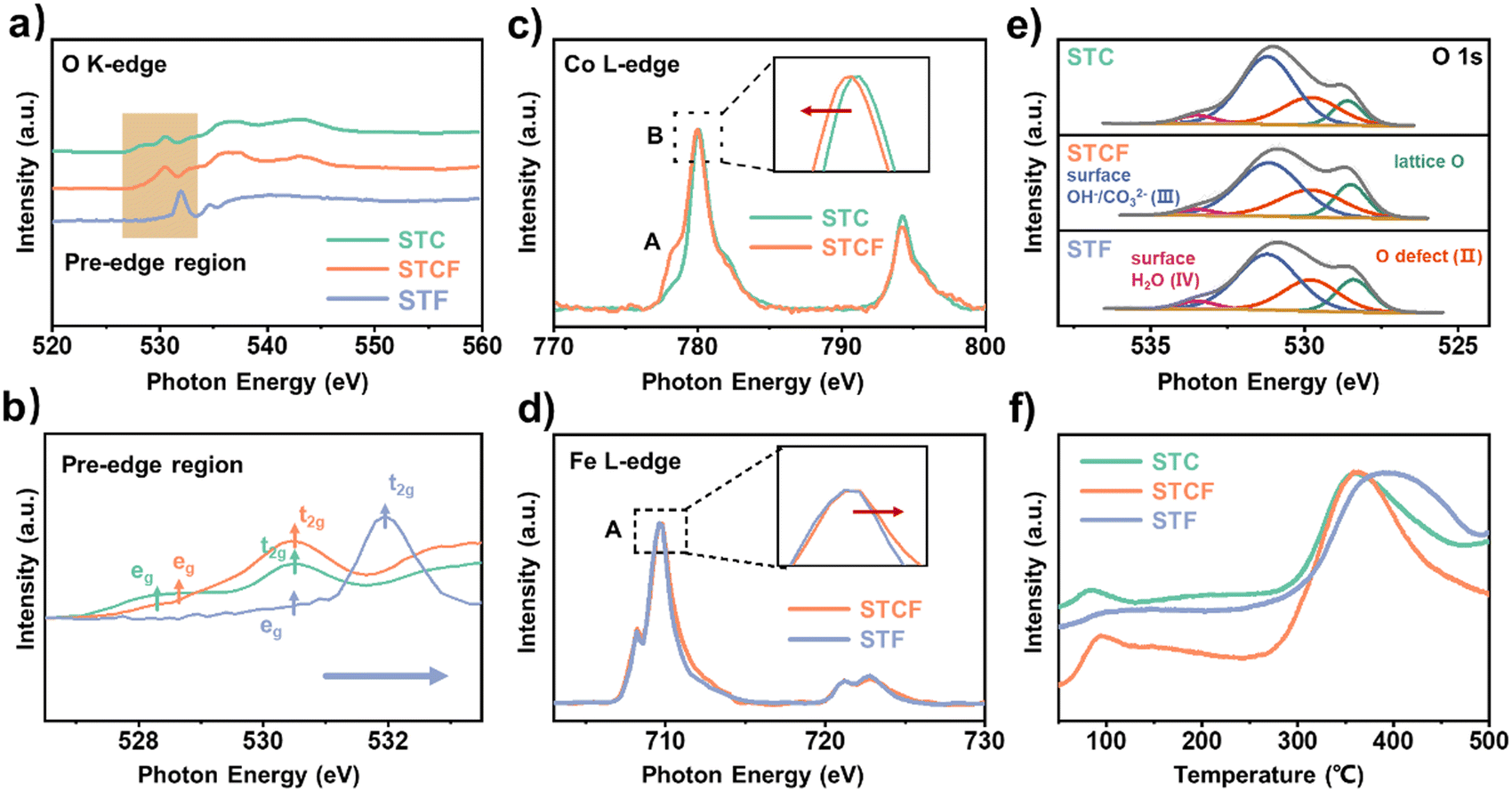 | ||
| Fig. 2 (a) O K-edge; (b) pre-edge region in the O-K edge (shadowed region); (c) Co L-edge; and (d) Fe L-edge soft X-ray absorption spectroscopies (sXAS); (e) XPS spectra of O 1s; and (f) O2-TPD of STC, STCF, and STF samples. | ||
As the Fe doping level increases, the pre-edge peak (marked by yellow) shifts to higher photon energy, and its intensity decreases, indicating a weakening of metal–oxygen covalency.20 Particularly, we observed strong absorption peaks starting at ∼527 eV and 529.5 eV in the pre-edge region of the STC sample (green arrow), which are associated with the eg and t2g state.21 Interestingly, the features corresponding to the eg state decreased while those associated with the t2g state increased with Fe content (Fig. 2b). Such changes in the pre-edge region imply a decrease in the number of O 2p ligand holes (O(2−δ)−) in these materials as Fe content increases.22,23 Previous studies21,24,25 have shown that a high metal–oxygen covalency promotes the transfer of electrons between the cation sites and the oxygen ligands and facilitates the formation of oxygen ligand holes. Therefore, the weakened metal–oxygen covalency in STF is the reason for the inhibited formation of oxygenated ligand holes. These O K-edge XAS spectroscopy results indicate that as the Fe/Co ratio at the B site increases, the metal–oxygen covalency is weakened, resulting in fewer O 2p ligand holes observed in the materials. These changes may significantly affect the oxygen activities of the electrode surface, as will be discussed in detail in the following section.
The changes in the electronic structures of Fe and Co can also provide critical information about the oxygen properties of the different materials. For the XAS spectra of the Co L-edge (Fig. 2c), the characteristic peak of Co2+ (A) was significantly enhanced with the increase of the doped Fe/Co stoichiometric ratio, while the position of the B peak was shifted to a lower photon energy. This result indicates that replacing Co with Fe at the B site leads to a decrease in Co's valence state.26 In contrast, the valence state of Fe in STF was slightly higher than that of Fe in STCF, which can be seen from the shift of peak A in Fe L-edge XAS spectra for STCF compared to STF (Fig. 2d).27 These results suggest that the increased Fe/Co ratio at the B site leads to electron redistribution in the lattice, with electron transfer from Fe to Co through the Co–O–Fe motif.28 Such electron redistribution is likely the reason for the weakened metal–oxygen covalence with the Fe dopant.29,30
The oxygen chemical environments in the three samples were further analyzed by X-ray photoelectron spectroscopy (XPS) (Fig. 2e). The O 1s spectra were deconvoluted into four components: lattice oxygen at 528.4 eV (lattice O, O2−, peak I), highly oxidizable oxygen (O22−/O2−) associated with oxygen vacancies at 529.8 eV (Odefect, peak II), surface-adsorbed oxygen containing hydroxyl or carbonate at 531.3 eV (surface O, peak III), and adsorbed molecular water at 533.5 eV (peak IV).31,32 The number of surface oxygen species (surface O, peak III) on STF was found to be lower than that of STCF and STC. The contents of surface-adsorbed oxygen peaks were quantified as 58.4%, 51.2%, and 50.2% for STC, STCF, and STF, respectively (Table S1, ESI†). Consistently, we observed lower contents of non-lattice-bound Sr (Sr–OH or Sr–CO3) on the STF sample compared to STC (Fig. S3a and b, ESI†).33 This result suggested that the increase in Fe content decreased the surface-adsorbed oxygen species, which are believed to be highly active oxygen species for oxidative dehydrogenation reactions.7,17,34
The dynamics of oxygen loss from various samples were further compared using oxygen-temperature-programmed desorption (O2-TPD) profiles (Fig. 2f). A clear peak near 100 °C was observed in all three samples, which corresponds to the desorption of oxygen-containing species on the surface. This desorption peak of STC and STCF was more intense than that of STF, implying more adsorbed surface oxygen species.35 This result was consistent with the higher surface oxygen species peak (surface O, peak III) observed in the XPS results (Fig. 2e). The peak with high intensity located between 300–500 °C was related to the formation of oxygen vacancies in the lattice. As shown in Fig. 2f, the onset temperature of this peak shifted to a higher value as the Fe/Co ratios increased, implying that oxygen vacancies are harder to form in samples with higher Fe doping levels.36 Electron paramagnetic resonance (EPR) measurements were used to confirm the difference in oxygen vacancy content between the materials with the highest levels of cobalt and iron doping (Fig. S4, ESI†). The STC samples exhibited higher intensity, indicating a higher oxygen vacancy content. As shown in Fig. S5 and Table S1 (ESI†), we also observed a higher intensity of the oxygen defects peak relative to the lattice oxygen peak for the STC samples. All these results consistently implied a decrease in oxygen vacancy concentrations with increasing Fe content. N2-TG results are shown in Fig. S6 (ESI†); the reduction in the mass of the material above 300 °C is attributed to the loss of lattice oxygen.37 From 300 °C to 900 °C, the weight losses were 4.12%, 3.01%, and 2.02% for STC, STCF, and STF, respectively (Fig. S6, ESI†). As the Fe/Co ratio increased, the material lost less lattice oxygen. This indicates a lower lattice oxygen activity and possibly a lower susceptibility to oxygen vacancies for samples with higher Fe contents. These results also suggest that the Fe/Co ratio can modulate the adsorption of oxygen on the surface as well as the formation of oxygen vacancies in the lattice.
To summarize the spectroscopic results, as the Fe/Co ratio at the B site of SrTiO3 increased, the electrode materials exhibited lower oxygen activity by presenting weaker metal–oxygen covalency, fewer O 2p holes, smaller amounts of surface-active species, and higher oxygen vacancy formation energies.
Modulated oxygen characteristics probed by DFT calculation
To reveal the influence of Fe/Co stoichiometric ratios on oxygen activity and the ethane ODH reaction process, we further carried out DFT calculations on the electronic structure. Three model systems were constructed for the DFT calculations, with detailed structures shown in Fig. S7 (ESI†).According to the projected density of states (PDOS) analysis (Fig. 3a and b), the O 2p-band center (εp) position relative to the Fermi level follows the trend of STC (−1.46 eV) > STCF (−1.55 eV) > STF (−1.65 eV). The smallest value for STF indicates that the center of the O 2p-band is farthest from the Fermi level. In previous studies, a closer position of the O 2p band center relative to the Fermi level tends to predict an increase in the oxidation capacity of the lattice oxygen.38 These results imply that electron transfer from the O 2p-band to the Fermi level is harder for the sample with higher Fe content, which is consistent with the fewer O 2p ligand holes observed in the spectroscopic results (Fig. 2a and b). The shift of the O 2p-band further away from the Fermi level, due to the increasing Fe/Co ratio, also results in a less electrophilic lattice oxgyen,39,40 which may inhibit the binding of H* and C2H5* to the lattice oxygen and hinder the transfer of electrons from the bound reaction intermediates.38
 | ||
| Fig. 3 (a) Projected density of states (PDOS) diagram; (b) the O 2p-band centre diagram; (c) calculation of surface electronic structure of oxygen; and (d) oxygen vacancy formation energy for the surface in three model systems (STC, STCF, and STF). | ||
We further carried out a Bader charge analysis of the oxygen site in the phase with different Fe/Co ratios (Fig. 3c and Fig. S8, S9a, ESI†). The obtained charge densities follow the trend of STC > STCF > STF for both surface oxygen and bulk oxygen. The smaller value for the phase with high Fe content represents a relatively less electron-deficient state. This is consistent with the fewer O 2p-ligand holes (O(2−δ)−) observed in these materials with higher Fe content, as indicated by the adsorption spectroscopy results (Fig. 2a and b). The most negative charge on the lattice oxygen of STF materials, which makes it the least electrophilic, may lead to less radical adsorption (H* and C2H5*) during the surface ODH reaction.41
Oxygen vacancy formation energy was calculated for the surface (Fig. 3d) and bulk phases (Fig. S8 and S9b, ESI†). The surface oxygen vacancy formation energies were −1.21 eV for STC, −0.91 eV for STCF, and −0.21 eV for STF. The bulk phase oxygen vacancy formation energies were also found to be lowest for STC and highest for STF. This is consistent with our experimental results shown in Fig. 2f and Fig. S4 (ESI†).
The downshift of the O 2p band away from the Fermi level, lower charge density on the oxygen sites, and increased oxygen vacancy formation energy all consistently suggest that the lattice oxygen activity decreases with an increasing Fe dopant at the B site, which aligns with the experimental results above.
Prediction of ODH performance based on DFT calculations
Having confirmed that the oxygen activity can be effectively changed by varying the dopant at the B site, we further investigated the potential impact of such changes in oxygen activity on the ODH reaction pathway.First, the free energy diagrams of ODH reaction on different samples were determined by DFT calculations. The ODH reactions of ethane consist of several dehydrogenation steps (Fig. S10, ESI†): the first and second dehydrogenation steps (C2H6 (g) + * → C2H5* + H*, C2H5* + H* → C2H4* + 2H*) and the third dehydrogenation step (C2H4* + 2H* → C2H3* + 3H*) or the C2H4 desorption step (C2H4* + 2H* → C2H4 (g) + 2H*). In the reaction pathway diagrams (Fig. 4a), solid lines represent the energies for the dehydrogenation steps, and dashed lines show the results for the ethylene desorption step. The first dehydrogenation free energy (ΔG1) followed the order of STC (−0.91 eV) < STCF (−0.62 eV) < STF (−0.49 eV) (Fig. 4b). This result implies that hydrogen is more easily stripped from the ethane molecule on STC samples than on those with higher Fe content at the B site,42 indicating degradation in activity toward ethane conversion with Fe doping at the B site.
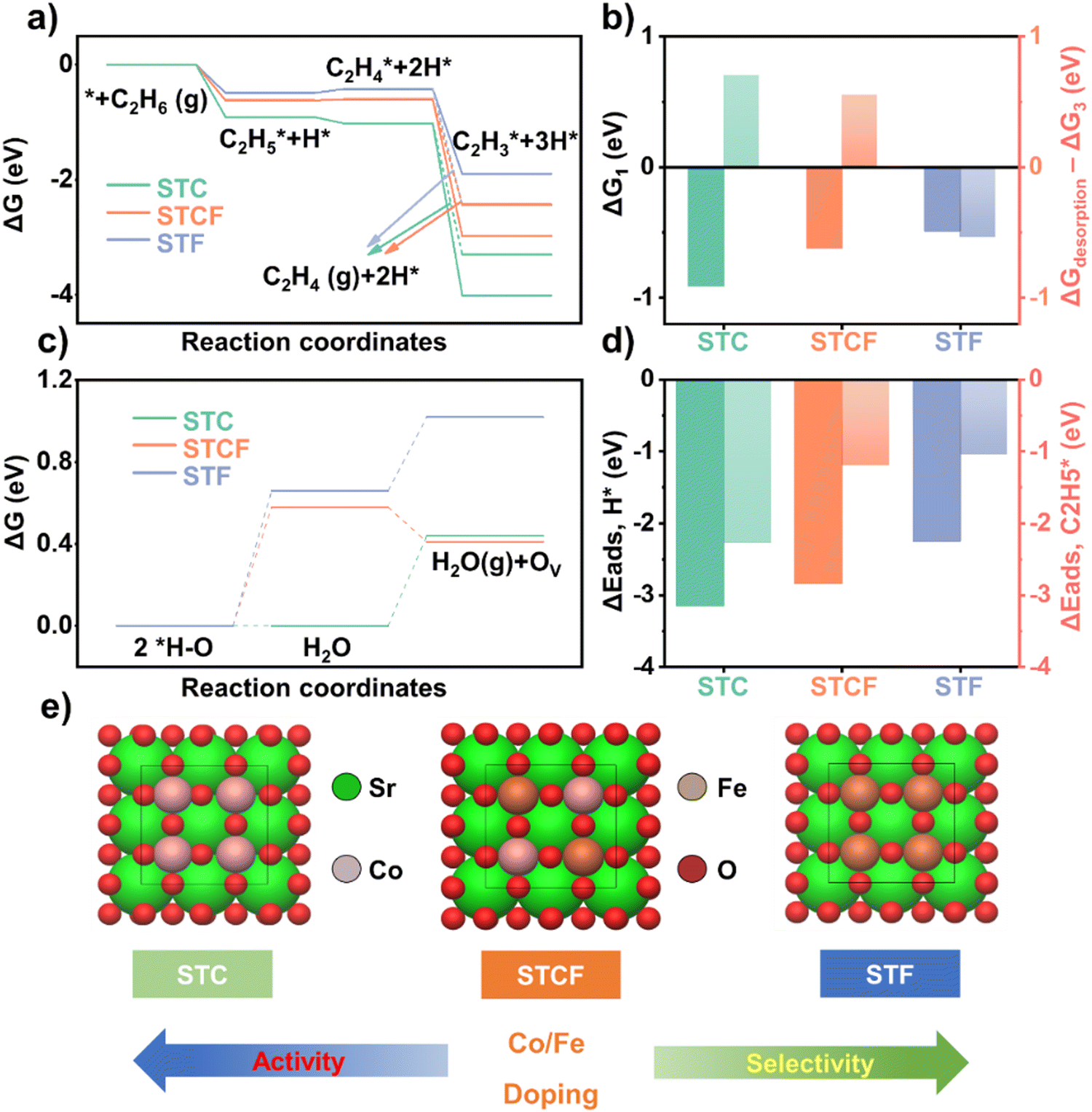 | ||
| Fig. 4 (a) Free energy diagrams of the ethane ODH reaction (solid line: dehydrogenation step, dashed line: desorption step); (b) the first dehydrogenation energy (ΔG1) and the difference between ethylene desorption free energy and ethylene dehydrogenation free energy (ΔGdesorption − ΔG3 (eV)); (c) the adsorption energies of H (ΔEads, H*) and C2H5 (ΔEads, C2H5*) (d) the free energy diagrams of H2O formation and desorption in the three model systems (STC, STCF, and STF); and (e) the three model systems. | ||
The differences between the ethylene desorption free energy and the third dehydrogenation free energy (ΔGdesorption − ΔG3) were evaluated. This value is representative of ethylene selectivity because timely desorption of ethylene is essential to avoid the further generation of over-oxidized products like COx. The ΔGdesorption − ΔG3 value decreased with Fe content, i.e., STC (0.7 eV) > STCF (0.55 eV) > STF (−0.54 eV) (Fig. 4b). This trend indicates that as the Fe dopant level increases, the intermediate C2H4* tends to desorb to form ethylene rather than further dehydrogenate to form other by-products, such as CO or CO2, which could result in enhanced selectivity toward ethylene.
We further evaluated the adsorption capacity of different material surfaces for ethane or reaction intermediates by comparing the binding strength of the reaction intermediates on the material surfaces. In the first dehydrogenation step, C2H6(g) is dissociatively adsorbed (C2H6(g) + * → C2H5* + H*) (Fig. 4a). The adsorption energies of H* and C2H5* strongly influence the dehydrogenation activity of the materials for ethane. The adsorption energy of H* (ΔEads,![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) H*) of STC (3.15 eV) was much more negative than that of STCF (−2.84 eV) and STF (−2.25 eV) (Fig. 4d). The adsorption energy of C2H5* followed by the same trend: STC (−2.28 eV) < STCF (−1.2 eV) < STF (−1.04 eV) (Fig. 4d). As the Fe/Co ratios increased, the adsorption of the two intermediates in the dehydrogenation step weakened, further implying lower activity toward the ethane dehydrogenation process on the STF surface. During the ethane dehydrogenation steps, the desorbed hydrogen can be adsorbed onto the lattice oxygen to form hydroxyl groups, which then react to form H2O. The bond strength of the formed H2O to the surface can influence the further dehydrogenation process. In the STC, STCF, and STF models, the free energy of H2O formation (2H* − Olattice → H2Olattice + Olattice) and desorption steps (H2Olattice → H2O(g) + Ov) (Fig. S11, ESI†) were calculated. For STF and STCF, the reaction free energy of H2O formation was higher than that of H2O desorption, while STC showed the opposite trend (Fig. 4c). STC has the lowest reaction free energy of H2O formation (0.00 eV), followed by STCF (0.58 eV) and STF (0.66 eV) (Fig. 4c). The free energy of H2O desorption for STC is lower than that of H2O formation for both STF and STCF. STC is superior in thermodynamic H2O formation and desorption processes, which is consistent with the oxygen vacancy formation energy results. Based on the free energy diagram results (Fig. 4a and c), it was predicted that the activity toward ethane dehydrogenation decreased with Fe content, while the selectivity toward ethene increased with Fe dopant levels.
H*) of STC (3.15 eV) was much more negative than that of STCF (−2.84 eV) and STF (−2.25 eV) (Fig. 4d). The adsorption energy of C2H5* followed by the same trend: STC (−2.28 eV) < STCF (−1.2 eV) < STF (−1.04 eV) (Fig. 4d). As the Fe/Co ratios increased, the adsorption of the two intermediates in the dehydrogenation step weakened, further implying lower activity toward the ethane dehydrogenation process on the STF surface. During the ethane dehydrogenation steps, the desorbed hydrogen can be adsorbed onto the lattice oxygen to form hydroxyl groups, which then react to form H2O. The bond strength of the formed H2O to the surface can influence the further dehydrogenation process. In the STC, STCF, and STF models, the free energy of H2O formation (2H* − Olattice → H2Olattice + Olattice) and desorption steps (H2Olattice → H2O(g) + Ov) (Fig. S11, ESI†) were calculated. For STF and STCF, the reaction free energy of H2O formation was higher than that of H2O desorption, while STC showed the opposite trend (Fig. 4c). STC has the lowest reaction free energy of H2O formation (0.00 eV), followed by STCF (0.58 eV) and STF (0.66 eV) (Fig. 4c). The free energy of H2O desorption for STC is lower than that of H2O formation for both STF and STCF. STC is superior in thermodynamic H2O formation and desorption processes, which is consistent with the oxygen vacancy formation energy results. Based on the free energy diagram results (Fig. 4a and c), it was predicted that the activity toward ethane dehydrogenation decreased with Fe content, while the selectivity toward ethene increased with Fe dopant levels.
The weakened metal–oxygen covalency and fewer O-ligand holes inhibited the adsorption of ethane and reaction intermediates to the lattice oxygen by hindering electron transfer from reaction intermediates.24,25 Additionally, the lower amount of surface-adsorbed oxygen species and oxygen vacancy content for the samples with high Fe content (Fig. 2e, f, and Fig. S4, ESI†) may decrease ethane conversion but prevent the over-oxidation of ethane. All these factors combined resulted in a decreased conversation rate but enhanced selectivity for the ODH reaction, as shown in Fig. 4e.
Modulating ODH performance via doping and electrochemical activation
To verify the prediction about the impact of Fe content on the conversion rate and selectivity of ODH activity, we conducted electrochemical tests to evaluate the conversion performance. An electrolyte-supported cell was constructed using La0.8Sr0.2Ga0.83Mg0.17O3−σ (LSGM) as the electrolyte, with three different materials (STC, STCF, and STF) as anodes and STCF as the cathode. While ethane gas was injected into the anode for the ODH reaction, CO2 gas was introduced to the cathode side as the oxygen ion source (Fig. 5a and Fig. S12, ESI†). When an electrochemical potential was applied, CO2 was reduced to CO, and ethane underwent an ODH reaction to produce ethylene. The fabricated cell showed a typical three-layer structure with a well-bonded porous anode layer to the electrolyte-supported structure (inset in Fig. 5b and Fig. S13, ESI†). The electrochemical impedance spectra (EIS) results showed that the ohmic impedance and the polarization impedance of the three anode cells did not have noticeable differences (Fig. 5b and c), indicating similar electrochemical properties.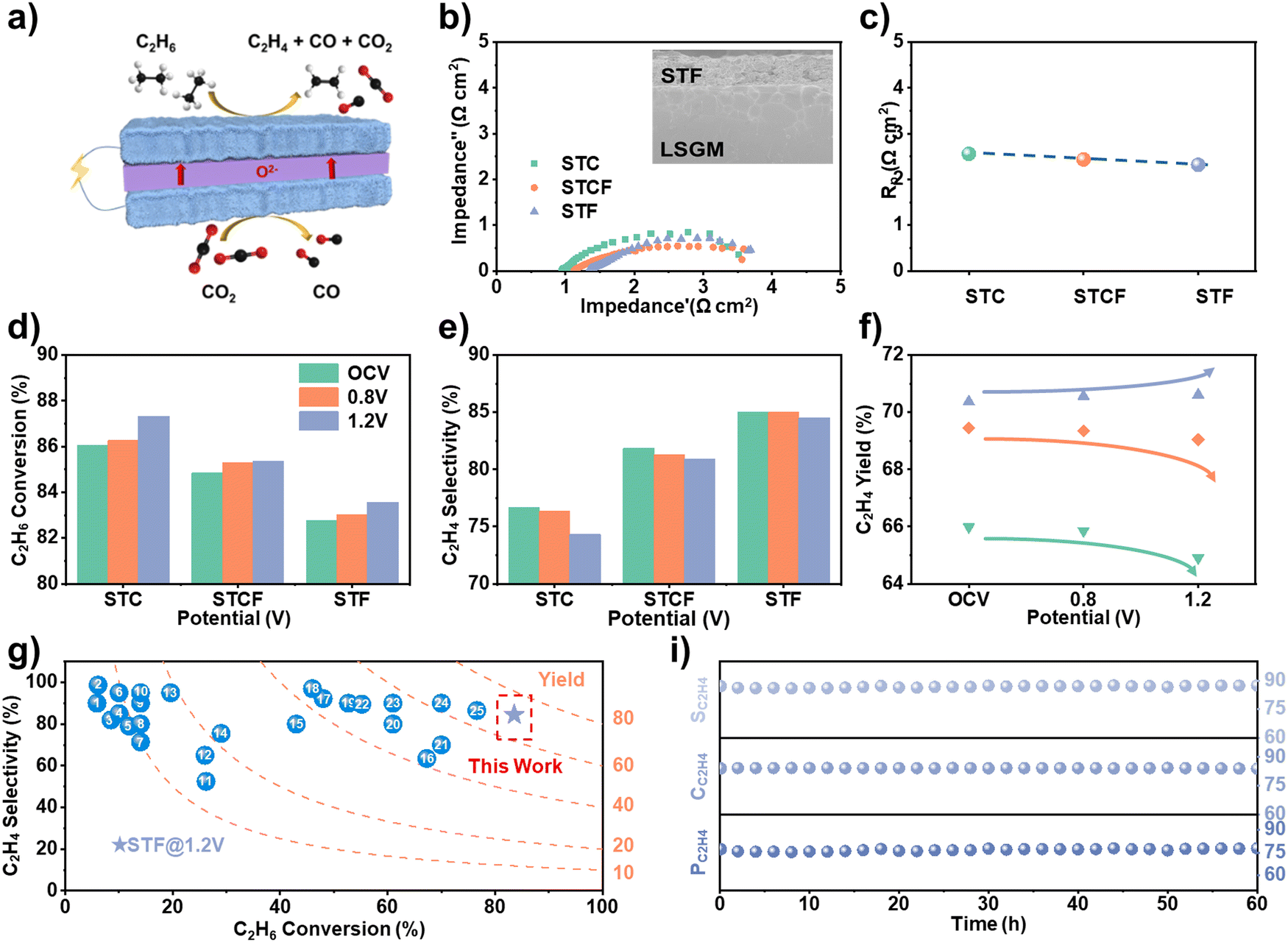 | ||
| Fig. 5 (a) The coupling of electrochemical oxidative dehydrogenation and CO2 electrolysis using SOEC; EIS curve (the inset figure shows the SEM images of the STF anode and electrolyte (LSGM) interface) and (b) polarization resistance (Rp) (c) at 1.2 V, (d) C2H6 conversion rate, (e) C2H4 selectivity, (f) C2H4 yield, (g) comparison of the performance as an oxidant in this work with other reported results (Numbers in the figure refer to entries in Table S2, ESI†) (i) C2H6 conversion (CC2H6), C2H4 percentage, and selectivity (PC2H4 and SC2H4) versus electrolysis time of cell with STF as anode at 0.8 V with a 20%C2H6/Ar flow rate of 20 mL min−1. | ||
The ethane ODH performance of different anodes (STC, STCF, and STF) was systematically evaluated by analyzing the reaction products using online gas chromatography. The conversion rate and ethylene selectivity for STC, STCF, and STF under different potential conditions are shown in Fig. 5d, e. Consistent with the theoretical predictions (Fig. 4e), while the conversion rate decreased with Fe content, the selectivity increased under all applied potentials (Fig. 5d and e). The lowest oxygen activity of STF (Fig. 2 and 3) resulted in the lowest activity toward the dehydrogenation process, but it prevented the deep oxidation of ethane to generate carbon oxides (Fig. S14, ESI†). Therefore, STF exhibited the lowest conversion rate and the highest ethylene selectivity (Fig. 5d).
We further found that applying electrochemical potential could effectively modulate the conversion performance. In our previous work,17 we employed synchrotron-based spectroscopy to investigate the evolution of oxygen species on the anode surface under electrochemical conditions. Our findings revealed the formation of highly reactive oxygen species upon the application of electrochemical potential. DFT calculations indicated that these oxygen species promote ethane conversion while suppressing ethylene selectivity. As the potential increased, the ethane conversion rate increased, while ethylene selectivity decreased (Fig. 5d, e and Fig. S14, ESI†) for all three different cells. These results are attributed to the formation of active oxygen species by pumping oxygen from the cathode through electrochemical potential, leading to high C–H bond activation, which also leads to deep oxidative dehydrogenation and the formation of COx.9 Interestingly, we observed an increase in the ethylene yield for STF sample under applied potential, and an opposite trend was noted for STCF and STC (Fig. 5f). The cell with STF as the anode had the highest ethylene yield of 71%, which is among the highest values reported in the literature under similar conditions (Fig. 5g and Table S2, ESI†). Such high conversion performance was sustained for long-term operation, with negligible changes in C2H6 conversion rate (CC2H6), C2H4 percentage, and selectivity (PC2H4 and SC2H4) as shown in Fig. 5i.
Correspondingly, the XRD patterns of the anode side of the STF after electrolysis (Fig. S15a, ESI†) showed that the anode material retains the original phase structure without change. Raman spectroscopy results (Fig. S15b, ESI†) imply no carbon deposition on the surface. Therefore, we consider that all converted ethane is in the form of gaseous products (COx, C2H4, etc.) and that all carbon is fully used. To further investigate the reaction properties of the materials in the electrolysis tests, we performed XPS tests on the anode surface after electrolysis. The percentage of lattice oxygen in the three anode materials, STC, STCF, and STF, increased in order after a short-term electrolysis reaction (Fig. S15c, ESI†). The STC surface contains only non-lattice-bound Sr (Srnon-lattice), while the lattice-bound Sr (Srlattice) on the STCF and STF surfaces increases sequentially (Fig. S15d, ESI†), which is consistent with the increase in the lattice oxygen. This is also an indication that the lattice oxygen of STC is more active. All these results consistently indicate that the cell with STF anode under applied potential exhibited superior activity and stability.
We have consistently demonstrated that by simply applying doping or potential, the conversion rate and selectivity cannot be improved simultaneously. This means a seesaw effect is generally observed, i.e., an increase in ethane conversion is often accompanied by a decrease in ethylene selectivity, and vice versa.6,16,17 Interestingly, the ethylene yield, which is the most important factor for the ODH reactions, is essentially dependent on the balance between conversion and selectivity.
The oxidative dehydrogenation of ethane, which is strongly influenced by the oxygen activity of the electrode, faces a fundamental challenge: achieving high selectivity and high conversion simultaneously is difficult. The variations in selectivity and conversion observed for ethane oxidative dehydrogenation on STC, STCF, and STF anodes can be attributed to differences in their oxygen activities, as demonstrated by spectroscopic techniques and DFT. STF samples with a high Fe dopant level exhibit low oxygen activity, resulting in low ethane conversion but high ethylene selectivity. By applying an electric potential, we can enhance the conversion rate without significantly compromising selectivity, thereby improving ethylene yield on STF samples (Fig. 6). This ability to balance activity and selectivity through strategic dopant selection and applied potential represents the synergistic effect of breaking the activity and selectivity seesaw we mentioned previously.
 | ||
| Fig. 6 Breaking the selectivity-activity seesaw in ethane ODH. | ||
Consequently, by utilizing the synergistic effect of doping and electrochemical activation, we optimized the balance between activity and selectivity for STF. The ethylene yields increased with the applied electrochemical potential (Fig. 5f), leading to excellent ODH performance.
Conclusion
The oxygen activity of three electrode materials with different Co/Fe stoichiometric ratios (STC, STCF, STF) have been systematically investigated by using advanced spectroscopic techniques combined with DFT calculations. The increase of Fe content was found to decrease the oxygen activity of the electrode by presenting weakened covalency of the metal with oxygen, lower O 2p-band center relative to the Fermi energy, and increased oxygen vacancy formation energy. DFT calculations further that these changes in oxygen activity associated with the Fe/Co ratio resulted in a reduction of the ethane dehydrogenation energy (ΔG1) and an enhancement of the disparity between the free energy of ethylene desorption and that of ethylene dehydrogenation (ΔGdesorption − ΔG3 (eV)). This could ultimately lead to a decrease in ethane conversion and an increase in ethylene selectivity with Fe dopant level. Results of the DFT calculations were confirmed experimentally by constructing electrolyte-supported SOEC with the ethane dehydrogenation reaction on three different electrodes and using CO2 as the oxygen source on the counter electrode. As the Fe doping in the electrode increases, the ethane conversion decreases while the ethylene selectivity increases. Interestingly, by increasing the applied potential, we can effectively increase the ethane conversion without overly sacrificing the ethylene selectivity. Finally, with the synergistic effect of doping and electrochemical activation, SOEC with the STF anode achieved 71% ethylene yield at 800 °C at 1.2 V, which was among the highest in the literature for ethane ODH reaction with CO2 as the oxygen source. The results of this work provide critical insight into the role of oxygen activity on the ODH reactions. The methodology of combining doping and electrochemical activation can be applied to other high-temperature electrochemical devices for energy and environmental applications.Author contributions
Yan Chen and Jeong Woo Han conceived and supervised the research project. Xiang Sun, Benchi Chen carried out materials synthesis, electrochemical measurement, and material characterization. Heejae Yang and Yunkyung Kim carried out the DFT calculations. Mengzhen Zhou and Nian Zhang performed XAS characterizations. Wonyoung Lee and Zhang Lin conducted some characterization analysis. All the authors participate in the manuscript writing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R and D Program of China (2024YFE0101500) and the Guangdong Provincial Science and Technology Program Project (2023A0505050096); Beamline 02B02 of the Shanghai Synchrotron Radiation Facility, which is supported by ME2 project under contract from the National Natural Science Foundation of China (11227902). This work was financially supported by the Carbon Neutral Industrial Strategic Technology Development Program (RS-2023-00261088) funded by the Ministry of Trade, Industry & Energy (MOTIE, Korea). This work also was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (2022R1A2C3012372). Prof. Y. Chen acknowledges the support of “TCL Young Scholars Program”.Notes and references
- Y. Gao, L. Neal, D. Ding, W. Wu, C. Baroi, A. M. Gaffney and F. Li, ACS Catal., 2019, 9, 8592–8621 CAS.
- Y. Dai, X. Gao, Q. Wang, X. Wan, C. Zhou and Y. Yang, Chem. Soc. Rev., 2021, 50, 5590–5630 CAS.
- Y. Gao, L. M. Neal and F. Li, ACS Catal., 2016, 6, 7293–7302 CAS.
- Y. Song, L. Lin, W. Feng, X. Zhang, Q. Dong, X. Li, H. Lv, Q. Liu, F. Yang, Z. Liu, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2019, 58, 16043–16046 CAS.
- X. He, X. Huang, H. Sun and L. Gan, Int. J. Hydrogen Energy, 2024, 79, 1030–1036 CAS.
- J. Kim, M. Ferree, S. Gunduz, A. C. Co and U. S. Ozkan, Electrochim. Acta, 2023, 461 Search PubMed.
- X. Tian, C. Zheng and H. Zhao, Appl. Catal., B, 2022, 303, 120894 CAS.
- L. Ye, Z. Shang and K. Xie, Angew. Chem., Int. Ed., 2022, 61, e202207211 CrossRef CAS PubMed.
- L. Ye, X. Duan and K. Xie, Angew. Chem., Int. Ed., 2021, 60, 21746–21750 CAS.
- H. Zhu, D. C. Rosenfeld, M. Harb, D. H. Anjum, M. N. Hedhili, S. Ould-Chikh and J.-M. Basset, ACS Catal., 2016, 6, 2852–2866 CrossRef CAS.
- E. Heracleous and A. Lemonidou, J. Catal., 2006, 237, 175–189 Search PubMed.
- S. Najari, S. Saeidi, P. Concepcion, D. D. Dionysiou, S. K. Bhargava, A. F. Lee and K. Wilson, Chem. Soc. Rev., 2021, 50, 4564–4605 Search PubMed.
- J. Li, P. Yu, J. Xie, J. Liu, Z. Wang, C. Wu, J. Rong, H. Liu and D. Su, ACS Catal., 2017, 7, 7305–7311 Search PubMed.
- J. F. Durán-Pérez, J. G. Rivera de la Cruz and C. O. Castillo-Araiza, Chem. Eng. J., 2023, 470 Search PubMed.
- M. Qin, S. Zhang, W. Sun, C. Xu, J. Qiao, Z. Wang, S. Zhen and K. Sun, Ceram. Int., 2023, 49, 30178–30186 CrossRef CAS.
- D. Dogu, K. E. Meyer, A. Fuller, S. Gunduz, D. J. Deka, N. Kramer, A. C. Co and U. S. Ozkan, Appl. Catal., B, 2018, 227, 90–101 CrossRef CAS.
- X. Sun, H. Yang, B. Chen, M. Zhou, Y. Ye, X. Li, H. Zhang, B. Yu, Y. Li, J. W. Han and Y. Chen, ACS Catal., 2024, 14, 5827–5837 CrossRef CAS.
- Z. Gong, W. Zhong, Z. He, C. Jia, D. Zhou, N. Zhang, X. Kang and Y. Chen, Catal. Today, 2022, 402, 259–265 CrossRef CAS.
- M. Zhou, J. Liu, Y. Ye, X. Sun, H. Chen, D. Zhou, Y. Yin, N. Zhang, Y. Ling, F. Ciucci and Y. Chen, Small, 2021, 17 Search PubMed.
- Y. Zhu, Q. Lin, Z. Hu, Y. Chen, Y. Yin, H. A. Tahini, H. J. Lin, C. T. Chen, X. Zhang, Z. Shao and H. Wang, Small, 2020, 16 Search PubMed.
- C. Jia, X. Xiang, J. Zhang, Z. He, Z. Gong, H. Chen, N. Zhang, X. Wang, S. Zhao and Y. Chen, Adv. Funct. Mater., 2023, 33 Search PubMed.
- N. Zhang, X. Feng, D. Rao, X. Deng, L. Cai, B. Qiu, R. Long, Y. Xiong, Y. Lu and Y. Chai, Nat. Commun., 2020, 11, 4066 CrossRef CAS PubMed.
- J. Shan, T. Ling, K. Davey, Y. Zheng and S.-Z. Qiao, Adv. Mater., 2019, 31, 1900510 Search PubMed.
- Y. Zhu, Q. Lin, Z. Hu, Y. Chen, Y. Yin, H. A. Tahini, H.-J. Lin, C.-T. Chen, X. Zhang, Z. Shao and H. Wang, Small, 2020, 16, 2001204 CrossRef CAS PubMed.
- J. T. Mefford, X. Rong, A. M. Abakumov, W. G. Hardin, S. Dai, A. M. Kolpak, K. P. Johnston and K. J. Stevenson, Nat. Commun., 2016, 7, 11053 CrossRef CAS PubMed.
- S. Yang, X. Xiang, Z. He, W. Zhong, C. Jia, Z. Gong, N. Zhang, S. Zhao and Y. Chen, Chem. Eng. J., 2023, 457, 141344 CrossRef CAS.
- Z. Wang, Y. Xiao, Y. Zhang, Y. Wang, X. Wang, F. Wang and T. He, Chem. Eng. J., 2024, 490, 151911 CrossRef CAS.
- W. T. Hong, K. A. Stoerzinger, Y.-L. Lee, L. Giordano, A. Grimaud, A. M. Johnson, J. Hwang, E. J. Crumlin, W. Yang and Y. Shao-Horn, Energy Environ. Sci., 2017, 10, 2190–2200 RSC.
- Z. He, J. Zhang, Z. Gong, H. Lei, D. Zhou, N. Zhang, W. Mai, S. Zhao and Y. Chen, Nat. Commun., 2022, 13, 2191 CrossRef CAS PubMed.
- Y. Y. Chin, Z. Hu, H. J. Lin, S. Agrestini, J. Weinen, C. Martin, S. Hébert, A. Maignan, A. Tanaka, J. C. Cezar, N. B. Brookes, Y. F. Liao, K. D. Tsuei, C. T. Chen, D. I. Khomskii and L. H. Tjeng, Phys. Rev. B, 2019, 100 Search PubMed.
- Y. Zhu, L. Zhang, B. Zhao, H. Chen, X. Liu, R. Zhao, X. Wang, J. Liu, Y. Chen and M. Liu, Adv. Funct. Mater., 2019, 29, 1901783 CrossRef.
- Q. Liu, Y. Zhu, Z. He, S. Jin and Y. Chen, Int. J. Hydrogen Energy, 2020, 45, 22808–22816 CrossRef CAS.
- Y. Chen, D. D. Fong, F. W. Herbert, J. Rault, J. P. Rueff, N. Tsvetkov and B. Yildiz, Chem. Mater., 2018, 30, 3359–3371 CrossRef CAS.
- M. Hye Jeong, D. Hyung Lee, J. Won Moon, J. Sun, J. Soon Choi, D. Sik Hong, C.-H. Chung and J. Wook Bae, Chem. Eng. J., 2022, 433, 134621 CrossRef CAS.
- E. Siebert, C. Roux, A. Boréave, F. Gaillard and P. Vernoux, Solid State Ionics, 2011, 183, 40–47 CrossRef CAS.
- X. Lin, S. Li, H. He, Z. Wu, J. Wu, L. Chen, D. Ye and M. Fu, Appl. Catal., B, 2018, 223, 91–102 CrossRef CAS.
- H. Lu, L. Zhu, J. P. Kim, S. H. Son and J. H. Park, Solid State Ionics, 2012, 209–210, 24–29 CrossRef CAS.
- J. Hwang, R. R. Rao, L. Giordano, K. Akkiraju, X. R. Wang, E. J. Crumlin, H. Bluhm and Y. Shao-Horn, Nat. Catal., 2021, 4, 663–673 CrossRef CAS.
- N. Zhang and Y. Chai, Energy Environ. Sci., 2021, 14, 4647–4671 RSC.
- T. Z. Wu, S. N. Sun, J. J. Song, S. B. Xi, Y. H. Du, B. Chen, W. A. Sasangka, H. B. Liao, C. L. Gan, G. G. Scherer, L. Zeng, H. J. Wang, H. Li, A. Grimaud and Z. J. Xu, Nat. Catal., 2019, 2, 763–772 CrossRef CAS.
- V. Fung, Z. Wu and D. E. Jiang, J. Phys. Chem. Lett., 2018, 9, 6321–6325 CrossRef CAS PubMed.
- D. Ding, Y. Zhang, W. Wu, D. Chen, M. Liu and T. He, Energy Environ. Sci., 2018, 11, 1710–1716 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ey00012b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |