
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePlasma catalysis: what is needed to create synergy?
Joran Van Turnhout a,
Kevin Rouwenhorstbc,
Leon Lefferts*b and
Annemie Bogaerts*a
a,
Kevin Rouwenhorstbc,
Leon Lefferts*b and
Annemie Bogaerts*a
aResearch group PLASMANT, Department of Chemistry, University of Antwerp, Universiteitsplein 1, 2610 Antwerpen, Belgium. E-mail: annemie.bogaerts@uantwerpen.be
bCatalytic Processes & Materials, MESA+ Institute for Nanotechnology, Department of Science & Technology, University of Twente, Enschede, The Netherlands. E-mail: l.lefferts@utwente.nl
cAmmonia Energy Association, Brooklyn, NY, USA
First published on 2nd April 2025
Abstract
Plasma catalysis is gaining increasing interest for the synthesis of chemicals and fuels, but the underlying mechanisms are still far from understood. This hampers plasma–catalyst synergy. Indeed, there is not enough insight into the optimal catalyst material tailored to the plasma environment, and vice versa, in the optimal plasma conditions for the catalyst needs. Furthermore, plasma catalysis suffers from energy losses via backward reactions, and probably most importantly, there is a clear need for improved plasma reactor design with better contact between plasma and catalyst. In this paper, we describe these critical limitations and suggest possible solutions. In addition, we stress the importance of correct measurements and consistent reporting, and finally we also propose other promising plasma–material combinations beyond the strict definition of catalysts. We hope this opinion paper can help to make progress in this booming research field.
Broader contextPlasma catalysis, initially mainly used for VOC removal, has received an ever-increasing amount of interest for gas conversions such as CO2 and CH4 conversion, and NH3 and NOx synthesis. Plasma couples particularly well with renewable energy sources, because of how rapidly it is switched on and off again and thus could prove useful for energy storage into liquid fuels such as CH3OH (methanol). However, in contrast to its applications for VOC removal, the plasma applications for gas conversions are at a much lower TRL, mainly because of low energy efficiencies and low product selectivities. Thus, there is a clear need for better insights into the current limitations. In this paper, we aim to identify the critical limitations in the field, and where possible, we suggest what is needed to overcome them, to make plasma catalysis an alternative to the present thermo-catalytic systems, while being competitive with other sustainable alternatives. |
1. Introduction
Plasma catalysis is gaining increasing interest for various applications, for air pollution control (i.e., removing low concentrations of harmful components, like volatile organic compounds (VOCs), particulate matter and NOx, from the air1–7), and for sustainable chemistry (such as CO2 and CH4 conversion, including CO2 hydrogenation, partial oxidation or dry reforming of CH4 (DRM) to produce syngas, higher hydrocarbons or oxygenates, as well as NH3 and NOx synthesis from N2 and H2 or air, respectively8–14).The first application field is already at high technology readiness level (TRL), with commercial devices available for many years, especially for VOC removal (see e.g., the 2020 plasma catalysis roadmap for more details1), and the main metric is conversion of VOCs, rather than energy efficiency. In contrast, the second application field is at much lower TRL,1 and still faces several challenges, such as limited energy efficiency, limited product yield, and limited product selectivity. The main reason is that the underlying mechanisms are far from understood.1,4,7,15–17 Indeed, while plasma–catalyst synergy is often reported (for instance18–22), in other cases it is not observed (for instance23–26). Hence, there is a need for better insights into the current limitations, and especially how to overcome them, in order to make significant progress in this emerging research field.
This paper aims to identify the critical limitations in the field of plasma catalysis for sustainable chemistry applications, and where possible, we also suggest what is needed to solve the limitations. We believe the main limitations are: (i) lack of insight in the optimal catalyst material tailored to the plasma environment, leading to trial-and-error experiments often based on insights from thermal catalysis, (ii) the plasma conditions not being tuned to the catalyst needs and thereby suboptimal plasma activation of molecules, (iii) the need for improved plasma reactor design with better contact between plasma and catalyst, (iv) the needs for correct measurements and consistent reporting of the obtained results, and (v) energy losses via the backward reactions, both thermo-catalytic as well as plasma-enhanced, and related to this, the need to think out of the box.
These aspects will be discussed in the following sections. We will only focus on in-plasma catalysis, where the catalyst is placed inside the plasma reactor, typically performed with dielectric barrier discharge (DBD) plasmas, because of their relatively low operating temperatures, required for the catalyst stability. Indeed, this combination is the most straightforward for plasma–catalyst synergy, due to the direct contact between reactive (short-lived) plasma species (or other plasma components, like the electric field) and the catalyst surface. However, it is not necessarily the best combination. Indeed, placing a catalyst post-plasma, as is investigated for warm plasmas, might be more beneficial, because the hot plasma gas can be used to thermally activate the catalyst. Therefore, at the end of the paper, we will also discuss some other plasma–material combinations (in a broader sense than only catalysts) that are promising to create synergy, also in post-plasma configuration.
2. Lack of insight into the optimal catalyst material
Many researchers, certainly in the past, used catalysts active for thermal catalysis in their plasma setup, such as nickel-based catalysts for DRM.19,27 However, plasma creates many reactive species, as well as an electric field, which may interact with the catalyst, so the best catalysts in thermal catalysis are not necessarily the best in plasma catalysis. While there is general awareness of the difference, and of the need to design catalysts tailored to the plasma environment, there is still a clear lack of insight into which catalysts would be most suitable in plasma catalysis.2.1. Metal catalysts act as radical scavengers – insights from computer modeling
Loenders et al.28 developed a coupled chemical kinetics model for plasma-catalytic DRM, describing both the plasma and catalyst surface chemistry, and studied the effect of different metal catalysts. Note that this model only focuses on chemistry and hence does not consider the physical effects of introducing a catalyst material into a DBD reactor. The model suggests that metal catalysts do not improve the performance, because they act as radical scavengers. Indeed, this is not unexpected: radicals are readily adsorbed at a catalyst surface, which is not a problem if they would react to the desired products (e.g., CO, H2, oxygenates). However, the model reveals that the radicals rather react back into the reactants (CO2 and CH4, in case of DRM). Furthermore, as the radicals easily adsorb at the catalyst surface, their density inside the plasma significantly drops, compared to plasma without catalyst. Hence, also the reaction rates inside the plasma, aiming to produce value-added compounds, drop upon implementing a catalyst, resulting in a net lower production as compared to plasma without catalyst.Fig. 1 presents the net CH3OH production rates from CO2/CH4 mixtures, calculated by the model of Loenders et al.,28 for plasma-only (without catalyst), or combined with a Rh, Cu or Ag catalyst, for a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CO2/CH4 mixing ratio. The blue bars represent the total production rates, which in the case of plasma-only is given by the plasma production rate. It is clearly higher than for the combination with any of the three catalysts. Indeed, the radicals are adsorbed at the catalyst surface, but only a small fraction reacts to CH3OH (cf. orange bars, and note the logarithmic scale of the y-axis). At the same time, the radical density inside the plasma drops due to the radicals being scavenged at the catalyst surface, so the CH3OH formation rates inside the plasma are also lower than in plasma-only (cf. the purple bars), and the sum of both (i.e., the blue bars in Fig. 1) are significantly lower than in plasma-only.
:1 CO2/CH4 mixing ratio. The blue bars represent the total production rates, which in the case of plasma-only is given by the plasma production rate. It is clearly higher than for the combination with any of the three catalysts. Indeed, the radicals are adsorbed at the catalyst surface, but only a small fraction reacts to CH3OH (cf. orange bars, and note the logarithmic scale of the y-axis). At the same time, the radical density inside the plasma drops due to the radicals being scavenged at the catalyst surface, so the CH3OH formation rates inside the plasma are also lower than in plasma-only (cf. the purple bars), and the sum of both (i.e., the blue bars in Fig. 1) are significantly lower than in plasma-only.
 | ||
| Fig. 1 Calculated net CH3OH production rate at steady state (t = 106 s) for plasma without catalyst, or combined with a Rh, Cu or Ag catalyst, for a 1:1 CO2/CH4 mixing ratio, at a total pressure of 1 bar and a temperature of 500 K. The color bars indicate the total reaction rate, the rate on the catalyst surface and inside the plasma (see legend). Note that the rates are logarithmically scaled. The model predicts that the CH3OH production rate in plasma-only is higher than when combined with any of the catalysts investigated. See details in text. Reproduced from ref. 28 with permission from Elsevier, copyright 2023. | ||
This poses a fundamental problem for plasma catalysis, because radicals are the most important plasma species in DBD reactors (especially at high plasma powers), which are the main plasma sources used in plasma catalysis. Hence, according to this model, a metal catalyst rather acts as “anti-catalyst” in plasma catalysis, resulting in lower performance than in plasma without catalysts.
It should be noted that the model of Loenders et al. simplifies reality, as it only models the surface reactions occurring on a single, ideal metal facet (Ag(111), Cu(111) and Rh(111), respectively), thus ignoring, to some extent, the complexity of a real heterogenous catalyst. Indeed, the model does not account for e.g. surface defects, different facets, or metal–support interactions, and assumes that the catalyst does not undergo chemical modifications such as oxidation. Moreover, it only focuses on the chemical kinetics, and it assumes perfect contact between the catalyst and the plasma, and therefore does not account yet for mass transport to and from the catalyst surface. If the radicals have a lifetime that is too short, they might even not be able to diffuse to the catalyst surface within their lifetime, unless they would be formed at/near the catalyst surface (see further). Thus, the model likely overestimates the effect of surface reactions. Regardless, the effect of radical adsorption on the transition metals seems to be either negligible or negative in case of CH3OH production from CH4 and CO2. It should be noted, however, that radical scavenging can have a net positive effect for other plasma-catalytic reactions, such as for NH3 synthesis, where the surface coverage with Hads is high because H2 easily adsorbs dissociatively on the metal surface even in the absence of plasma. In this case, N or NHx radicals in the plasma can readily react with adsorbed H, forming NH3.29–32
2.2. Which catalysts can work in optimal synergy with the reactive plasma environment?
The question thus arises: which catalysts are needed, that are better tailored to the reactive plasma environment, to realize plasma–catalyst synergy? Based on the above, it may be interesting to consider catalysts other than transition metals.Would metal oxide catalysts, for example, show a different behavior, i.e., not scavenging radicals? Indeed, similar to their use in thermal catalysis, metal oxides are reported to enhance the oxidation of hydrocarbons to CO2 in plasma-catalytic systems.33 Moreover, Patil et al.34 illustrated that the presence of supported metal oxides enhances nitrogen fixation, but attribute this effect to the facilitation of microdischarges, rather than a chemical interaction with the plasma-activated species. The authors further suggest that increasing the temperature may be necessary to unlock new surface pathways.
An interesting feature of multivalent metal oxides is their ability to accommodate oxygen vacancies (OV), formed by oxidizing a reactant, inducing partial (superficial) reduction or lattice distortions of the catalyst, according to the Mars van Krevelen (MvK) mechanism. The reverse MvK mechanism is particularly interesting for CO2 activation, where CO2 is activated by filling the OV, forming CO, as illustrated in Fig. 2. Parastaev et al.35 proposed, based on in situ DRIFTS experiments, that the role of OV for thermo-catalytic CO2 hydrogenation on a Co/CeZrO4 catalyst indeed lies in the enhancement of the reduction of CO2 through a formate pathway, in which formates are formed on OV at the metal–support interface. Meanwhile, the authors suggest that for the plasma-catalytic system, these formates do not play a role in CO2 hydrogenation, unless the temperature is increased substantially. On the other hand, Ning et al.36 report an enhanced CO2 conversion upon loading with Cu/CeZrO4 compared to the empty DBD reactor and propose a reaction mechanism involving (i) a hydrogen spillover from the copper nanoparticles to CO2 adsorbed on OV near the support–metal interface, and (ii) a reaction following the Eley–Rideal mechanism, in which excited H species directly react with CO2 adsorbed on OV. The latter is of particular interest, as this plasma-enabled mechanism would not require the presence of a transition metal on the catalyst surface. However, the contribution of this mechanism seems to be limited, as the performance of the reactor filled with the CeZrO4 support is similar to the performance of the empty reactor.
 | ||
| Fig. 2 Possible mechanisms involving oxygen vacancies (OV) in plasma-catalytic CO2 splitting on a multivalent metal oxide (in grey). (i) Shows the scavenging of O atoms from the plasma by OV on the metal oxide, preventing them from the backwards reaction with CO. (ii) Shows the splitting of CO2 adsorbed on an OV at the metal oxide surface, thus the reverse Mars van Krevelen mechanism. In both cases, the catalytic cycle is closed by the formation of O2 at the oxide surface. | ||
Golubev et al.37 found that CO2 splitting in a DBD plasma is enhanced by introducing MgO–CeO2 catalysts and that the CO2 dissociation improves with an increasing CeO2 fraction. The authors assigned this activity to surface reactions on the OV of CeO2, although no direct proof of surface species is provided. Similarly, Ashford et al.38 reported enhanced CO2 splitting on ceria-promoted iron oxide catalysts in plasma, attributing this to surface reactions involving OV, although the mechanism remains speculative. We believe metal oxides with OV can have a beneficial role for CO2 splitting along two mechanisms (see Fig. 2): (i) O radicals can be scavenged from the plasma by OV (preventing reactions to form CO2 from CO and O radicals), or (ii) via dissociation of CO2 adsorbed on the vacancy forming CO in gas phase while the O atom combines with the OV, incorporating the O into the lattice directly. In both cases, O2 is formed in a next step by generating two new OV, closing the catalytic cycle.
An alternative explanation for the plasma-catalytic activity for CO2 hydrogenation can be the formation of H radicals, which can react with the metal oxides to form water and a reduced metallic surface. Further thermo-catalytic and plasma-catalytic reactions can then occur on the metallic surface. Similarly, in oxidative environments, it is important to realize that transition metals can be oxidized to their respective oxides, while nitrides and carbides can be formed in the presence of activated nitrogen or carbon-containing molecules.
Based on the above, we believe that the use of metal oxides in plasma catalysis, like in thermal catalysis, may be beneficial for oxidation reactions. As always in plasma catalysis, special attention should be given to the influence of metal oxides on the physical properties of the plasma. Importantly, these physical properties should be reported as a function of time, as in situ reduction of these metal oxides may alter the dielectric properties of the material over the course of the experiment. Ideally, the oxidation state of the metal is monitored in situ, as for example done by Gibson et al.,39 who performed in situ XAFS experiments in a DBD reactor, providing information on the bulk oxidation state. The possible role of OV for CO2 activation in plasma remains largely unclear, as the current literature either suggests a limited role of OV on the surface reactions, or suggests mechanisms mainly based on structure–activity correlations. In situ techniques, like in situ FTIR, but also in situ XPS, can greatly help elucidate the role of OV in plasma catalysis. Indeed, on one hand, the possible role of OV in the formation of surface species in plasma-catalytic systems remains unclear, while on the other hand it is largely unknown to what extent OV are formed, filled (especially by reactive O radicals formed in the plasma), and regenerated in these systems. For example, Parastaev et al.35 found that OV are not formed on a CeZrO4 support exposed to pulsed H2 plasma without the presence of cobalt. Instead, hydroxyl (OH) groups are formed. Also, plasmas are highly heterogeneous in time,40 making it difficult to draw conclusions regarding OV density linked to plasma species. Moreover, the effect of OV on the physical properties of the plasma remains thus far unexplored.
In addition to redox properties, oxides can also contain acid and/or base surface sites. Recently, the use of zeolites for plasma-catalytic applications has gained traction. Xu et al.41 reported the good performance of Ru-loaded ZSM-5 nanostructures for CO2 methanation, which they attribute to enhanced Ru dispersion. Additionally, they find that the positioning of Ru nanoparticles (on the inside or outside of the zeolite framework) determines the accessibility of the nanoparticles to the reactive plasma species. Fan et al.42 show the use of a Ni/HZSM-5 catalyst for DRM and claim that the selectivity can be tuned towards alcohols by enhancing the Lewis acid sites with strong acidity. Conversely, the presence of Brønsted acid sites and Ni2+ species leads to relatively high acetic acid selectivities. Similar findings are reported by Wang et al.,43 who studied the plasma-catalytic DRM on HZSM-5 and 13X zeolites.
The use of zeolites for plasma catalysis, however, seems somewhat counterintuitive. Indeed, while zeolites are known for their complex microstructures and relatively controlled acidity (both in terms of number of active sites as well as acid strength), computer modeling has predicted that plasma streamers cannot penetrate inside pores smaller than the so-called Debye length44–46 (which is typically above 500 nm for typical plasma catalysis (DBD) conditions), and thus, reactive plasma species cannot be generated inside such small pores. Moreover, diffusion of these reactive species into small pores is also very limited (see more detailed discussion in Section 4.1 below). Hence, one could wonder if the complex, ordered microstructure of zeolites is necessary for obtaining the above results. In this regard, we propose the consideration of amorphous silica-alumina (ASA) as catalysts in future studies. Indeed, Coumans et al.47 showed that the Brønsted (and Lewis) acidity of ASA can be tuned, negating the need for structured microporosity found in zeolites. Evaluating the activity of ASA in comparison to zeolite benchmarks may clarify the potential role of Brønsted or Lewis acid sites in the plasma-catalytic activity of zeolites and may simultaneously enhance the surface area available to plasma-generated reactive species. Note that many oxides and mixed oxides, in addition to alumina-silicates, have acid–base properties48 and the performance in plasma catalysis may be explored. Especially, macroporous acidic materials would be worthwhile exploring.
At this stage, however, the behavior of catalysts other than metal surfaces cannot be investigated with chemical kinetic plasma–catalyst models, such as the one developed by Loenders et al.,28 because the necessary input data for such models (typically obtained from density functional theory) is lacking. Experimentally, however, these materials can easily be tested, though the interpretation of the obtained experimental results is often not straightforward. In Section 5, we discuss some important considerations for plasma-catalytic experiments in more detail.
3. The need to tune plasma conditions to the catalyst needs
On the other hand, we can also reformulate the question posed in Section 2.2: which plasma conditions are needed to work in optimal synergy with typical (metal) catalysts? The obvious answer would be: conditions that exploit other plasma species, rather than plasma radicals, such as (electronically and vibrationally) excited gas molecules.3.1. Are excited molecules more suitable to create plasma–catalyst synergy?
The reason why (electronically and vibrationally) excited gas molecules could be a more obvious choice to explore plasma–catalyst synergy is because these species can reduce the energy barrier for dissociative adsorption at the catalyst surface, as compared to molecules in the ground state, and therefore enhance the reaction rates, compared to thermal catalysis. For example, Juurlink et al.49 found that CH4 molecules excited to ν = 1 of the ν3 C–H stretching vibration are up to 1600 times more reactive on Ni(100) than CH4 in the ground vibrational state. This is indeed why thermal catalysis researchers show interest in plasma catalysis, as efficiently distributing energy into reactive vibrationally excited states, or electronically excited states, could greatly decrease the activation energy of a catalytic reaction. For electronically excited states, to the best of our knowledge, there is no existing literature on their potential role in surface chemistry. Therefore, we will focus on vibrational excitation in this section, while not ruling out a potential role of electronic excitation.Some authors have explicitly studied the role of vibrationally excited molecules in plasma catalysis.29,50–52 Mehta et al.51,52 and Engelmann et al.29 demonstrated computationally that vibrationally excited molecules can increase NH3 synthesis rates on materials that are kinetically limited by N2 dissociation, and for this reason, that the optimal catalytic material in plasma catalysis can be different from thermal catalysis, as well as that NH3 yields can exceed equilibrium limits at low temperatures. On the other hand, Engelmann et al.29 also demonstrated that at practical DBD conditions, radicals appear more important than vibrationally excited molecules. Engelmann et al.50 further explored the role of vibrationally excited CH4 on the non-oxidative coupling of CH4 using microkinetic modelling, and found that vibrationally excited species are essential for enhancing the selectivity towards ethylene, especially on Pt, Rh and Pd surfaces.
Besides their ability to reduce the energy barrier for dissociative adsorption, creating electronically and especially vibrationally excited gas molecules requires less energy than creating radicals, because the latter often occurs through electron impact excitation to higher electronically excited levels, followed by dissociation.14 Simply stated, making radicals is too energetically costly: the required energy for this is largely wasted, certainly if these radicals will recombine at the catalyst surface, generating heat.
This was demonstrated by Rouwenhorst et al.,53 who classified plasma-catalytic NH3 synthesis in DBD plasma into four possible mechanisms: (a) plasma-phase NH3 synthesis, (b) surface-enhanced plasma-driven NH3 synthesis, (c) plasma-enhanced semi-catalytic NH3 synthesis (also including Eley–Rideal reactions), and (d) plasma-enhanced catalytic NH3 synthesis (only including Langmuir–Hinshelwood pathways), see Fig. 3. This classification was based on whether dissociation (and thus: radical creation) of both N2 and H2, only N2, or neither N2 nor H2 occurs in the plasma phase. When N2 is dissociated in the plasma, hence creating radicals, the theoretical minimum energy required for NH3 production was calculated to be 0.47 MJ per mol-NH3.53 On the other hand, a better energy efficiency can be reached if plasma only promotes dissociation of N2 on the catalytic surface upon excitation, instead of dissociating N2 in the plasma. Upon vibrational activation, the N2 dissociation barrier on Ruthenium can be decreased by about 70 kJ mol−1,53 which is equivalent to an energy cost of 0.035 MJ per mol-NH3.
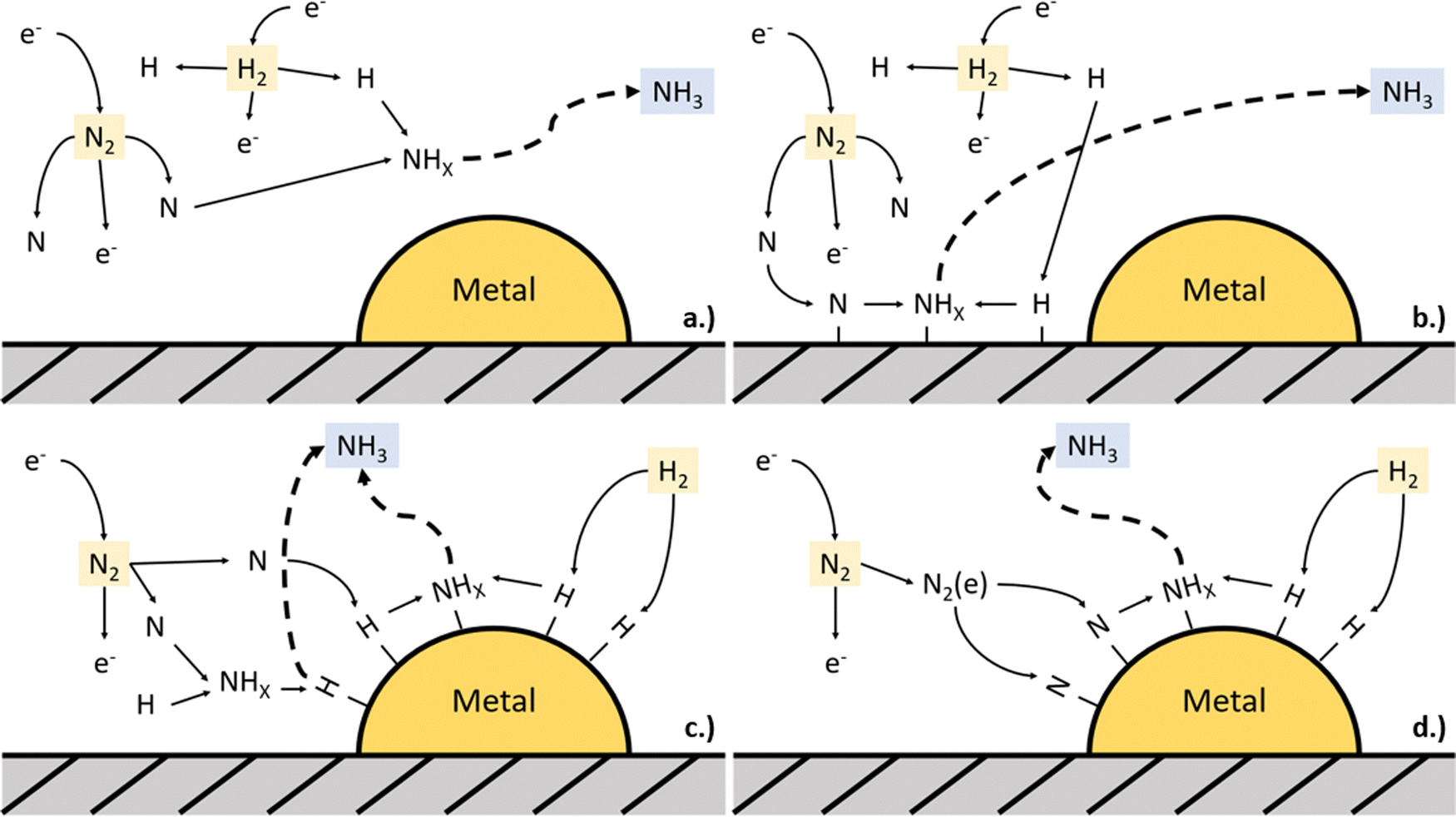 | ||
| Fig. 3 Possible mechanisms for plasma-catalytic NH3 synthesis: (a) plasma-phase NH3 synthesis, (b) surface-enhanced plasma-driven NH3 synthesis, (c) plasma-enhanced semi-catalytic NH3 synthesis (also including Eley–Rideal reactions), and (d) plasma-enhanced catalytic NH3 synthesis (only including Langmuir–Hinshelwood pathways). | ||
This is similar to values reached for a large-scale Haber–Bosch process (0.025–0.043 MJ per mol-NH3, ammonia synthesis loop only). If no heat losses occur and only N2 is vibrationally activated for NH3 synthesis, plasma catalysis may be competitive with a small-scale, container-sized Haber–Bosch process (compatible with renewable energy sources).11,54 It should be noted that additional energy will be required for separation in case of the plasma-catalytic process, which adds to the energy requirement for NH3 synthesis.55
In general, the energy cost of DBD reactors is nowadays still too high for plasma catalysis to be competitive with other technologies, e.g., a factor 4–5 for CO2 splitting and DRM,14 a factor 5–10 for NH3 synthesis,11 and a factor 5–8 for NOx synthesis.56 This can indeed be attributed to the dominant formation of radicals, which requires too much energy. In case of N2, the bond dissociation energy is 945 kJ mol−1. Furthermore, the dominant formation of radicals leads to high gas-phase activity, inhibiting potential selectivity gains by plasma catalysis. This high energy cost will hamper industrial implementation. However, even in case of mild plasma-activation of N2, it will be difficult for plasma-catalytic NH3 synthesis to compete with thermal catalysis, as the reaction is exothermic,55 implying that any energy input from the plasma will be lost as heat.
A critical note is, however, needed. Recent experimental work by Bayer et al.57 shows that, for the investigated conditions, NH3 production is (i) enhanced by introducing a catalyst material (Ag, Fe and Fe), and (ii) this enhancement can be attributed to surface-mediated reactions involving N radicals, and not vibrationally excited N2. Later work by the authors58 shows that the limited contribution of vibrationally excited N2 is not due to their low prevalence, but rather because they are rapidly quenched due to vibrational relaxation at the catalyst surface. Only vibrationally excited species with sufficiently high energy (depending on the catalyst material) can undergo dissociative adsorption faster than vibrational relaxation. In the applied plasma jet setup, in which the catalyst is not placed in the discharge region, but rather downstream (i.e. in the afterglow), the density of these high-energy vibrationally excited species was found to be (much) lower than the density of N radicals. Thus, the contribution of the latter towards catalytic reactions is (much) more significant than that of the former. In DBDs, the distance between the catalyst and the plasma (<1 mm, ideally ≪1 mm, see also below) is significantly smaller than in the setup studied by Bayer et al. (5 mm), limiting vibrational relaxation in the gas-phase and thus possibly enhancing the densities of high-energy vibrationally excited species. Nevertheless, this work shows that not the total density of vibrationally excited species is relevant for plasma catalysis, but rather the density of vibrationally excited species with sufficiently high energy to overcome vibrational relaxation at the catalyst surface. Vibrational relaxation at the catalyst surface should be considered when modelling the interaction between vibrationally excited species and a catalyst surface.
In short, enhancing the contribution of excited molecules to plasma-catalytic surface reactions could be beneficial for plasma–catalyst synergy, reducing the energy barrier for dissociative adsorption with relatively mild energy demand as compared to dissociation in the plasma. Consequently, also the energy efficiency of the system would be enhanced.
3.2. How can we maximize (vibrational) excitation in the plasma?
As mentioned in Section 2.1 above, DBD plasmas mainly produce radicals, while electronically and especially vibrationally excited gas molecules are typically less prominent. The reason is that DBD plasmas are characterized by a relatively high reduced electric field (i.e., electric field divided by the gas number density (E/N), expressed in Td, where 1 Td = 10−21 V m2). Indeed, typical reduced electric field values in DBD plasmas are above 100–200 Td, giving rise to relatively high electron energies, typically at least several eV, which is most suitable for electron impact dissociation to radicals, while vibrational excitation requires lower electron energies (order of 1 eV). Electronic excitation is also more prevalent than vibrational excitation at these higher electron energies, but electronically excited molecules quickly relax to the ground state (or lower excited levels) by emission of radiation, unless they are in metastable levels. In the following discussion, we will therefore mainly focus on the potential of vibrationally excited molecules, although we do not want to rule out the potential benefit of electronically excited molecules.We could maximize the population of vibrationally excited molecules by trying to tune E/N in DBDs to lower values, in the order of 50 Td.14 In theory, this can be realized by applying a lower voltage (creating a weaker electric field: E = V/d). This was again demonstrated by Rouwenhorst et al.,53 for plasma-catalytic NH3 synthesis, where a relatively low power resulted in a catalytic effect, and a lower energy cost. Indeed, the authors observed that the catalyst was more active in plasma catalysis in the case of Ru supported on more basic oxide supports, which is a characteristic phenomenon for Ru-based catalysts in thermal catalysis, due to an enhancement in N2 dissociation activity on the catalyst.
A critical note is, however, needed. At low power, the NH3 yield will be low, so the overall performance of plasma catalysis might be too limited, or a compromise may be needed to optimize both NH3 yield and energy cost. Furthermore, a practical issue arises when trying to reduce E. Indeed, a minimum voltage is required to sustain the discharge (Vsust), effectively forming a lower limit for E. This Vsust can be expected to decrease with rising temperature, lower gas flow rate, lower pressure, and smaller discharge gaps. It also depends on the composition of the gas filling the discharge gap. Interestingly, Sheng et al.59 found that, at 5 kPa, Vsust can be drastically reduced by increasing the frequency. Indeed, increasing the working frequency from 12 kHz to 100 kHz reduced the sustaining voltage almost threefold. Thus, power supply operation at high frequencies may be beneficial for reducing E/N, enhancing the population of vibrationally excited species.
It should be noted that the breakdown voltage is higher than the voltage required to sustain the plasma, allowing for a reduction in applied voltage after ignition, but both voltages are typically within the same order of magnitude.
Another option to reduce E/N is to increase the gas number density (N), by either increasing the pressure, or by decreasing the temperature (following ideal gas law). However, as mentioned above, increasing the pressure typically increases Vsust, so this is not a straightforward option to lower E/N. Likewise, a lower temperature also increases Vsust. Moreover, catalysts generally become less active upon decreasing the temperature, further questioning this strategy. Finally, pressure and temperature can also impact vibrational–translational (VT) relaxation, which is much more prominent at higher pressure and temperature, thus reducing the population of vibrational levels. Therefore, increasing the pressure would have a detrimental effect on the vibrational population, while a lower temperature could be beneficial (see also below). Clearly, the options to reduce E/N in a DBD are limited.
An alternative would be to develop completely new reactor designs, being closer to the concept of warm plasmas, such as gliding arc, microwave, and atmospheric pressure glow discharge. These are indeed characterized by lower E/N (order of 50 Td). In such warm plasmas, vibrational excitation is thus more important, as schematically illustrated in Fig. 4 (top panel), which shows that most of the electron energy goes into electron impact vibrational excitation for E/N values below ca. 50 Td (hence, typical for warm plasmas), while electronic excitation, ionization and dissociation take over at higher E/N (typical for DBDs). Warm plasmas also exhibit much lower energy costs for gas conversion applications,14,60–62 at least for endothermic reactions (hence not for NH3 synthesis11). However, the vibrationally excited molecules easily relax back to the ground state upon collision with gas molecules (VT relaxation), thereby increasing the gas temperature, which is typically too high (order of 3000 K or more) for catalyst implementation inside the plasma. Hence, warm plasmas are not suitable for in-plasma catalysis. Nevertheless, they can be used for post-plasma catalysis, where the hot gas can thermally activate the catalysts.63–66 However, in this case, reactive plasma species do not reach the catalyst, but there would only be thermal activation of the catalyst. Thus, the catalysis mechanism itself is in line here with that of thermal catalysis.
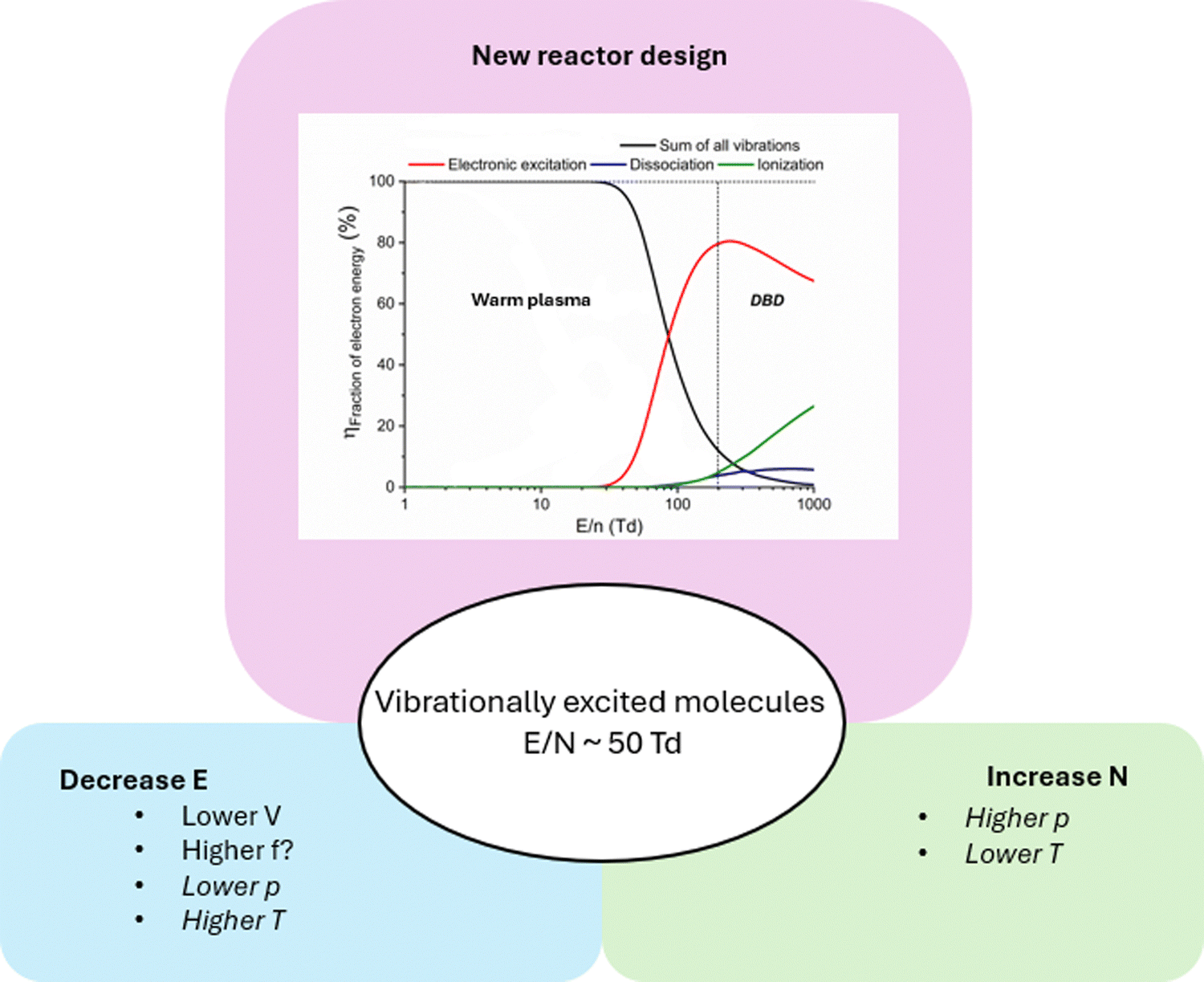 | ||
| Fig. 4 Schematic representation of how to increase the population of vibrationally excited molecules in plasma catalysis. The graph under “new reactor design” illustrates that warm plasmas (with E/N around 50 Td) would be more suitable to promote vibrational excitation, and was adapted from ref. 14 with permission from Royal Society of Chemistry, copyright 2017. | ||
In conclusion, we believe there is a need to design an intermediate type of plasma, in between DBD and warm plasmas, with E/N around 50 Td, so that (i) vibrational excitation is important, but (ii) gas heating is minimized, by avoiding that the vibrationally excited levels would quickly relax to the ground state. The reason why the latter should be avoided is twofold. First, it allows the vibrationally excited molecules to still be exploited for plasma catalysis. Second, if vibrationally excited molecules retain their energy longer, less energy is converted into heat, helping maintain a lower gas temperature. If the gas temperature can remain below 1000 K, catalysts can be directly inserted in the plasma, for direct plasma–catalyst synergy without the risk of thermal damage. Fig. 4 summarizes how a lower E/N (order of 50 Td) could in principle be realized, by either lowering E or by increasing N, although some parameters (pressure, p, and temperature, T) affect E and N in the opposite way, showing why it is not so straightforward to realize such conditions.
3.3. Should we target other plasma components to create more plasma–catalyst synergy?
Plasma produces other components as well, besides radicals and (electronically and vibrationally) excited levels, such as ions and electrons, which can give rise to catalyst surface charging, and thus modify the electronic structure of the catalyst material during operation via band structure shifts or work function changes, potentially altering the reactivity of the surface. For example, for CO2 activation, negative charging of the catalyst surface can alter the adsorption process by (i) shifting the antibonding states of CO2 toward the valence band, (ii) increasing the polarization effects and (iii) changing the adsorption site of the molecule.67Furthermore, plasma is also characterized by an electric field that can interact with a catalyst surface. Although research on the effects of electric fields and surface charging remains limited, Bal and Jafarzadeh et al.67–69 have investigated these effects through DFT simulations, providing valuable insights. For instance, charging a dielectric Al2O3 surface loaded with a single metal atom (Ti, Ni or Cu) was found to activate a CO2 molecule upon adsorption.68 Also, the adsorption energy of CO2 on TiO2-supported Ni5 and Cu5 catalyst clusters was found to rise upon charging,67 and the effect of both charging and electric fields on the adsorption and activation of CO2 on various Cu-surfaces was investigated.69 Additionally, Mangolini and coworkers showed by temperature-programmed desorption measurements coupled to in situ DRIFTS that plasma reduces the effective binding energy of CO on Pt surface, and their DFT simulations also revealed the role of plasma-induced charging and electric fields in this process.70 However, Rouwenhorst and Lefferts showed that the electric fields in plasmas are negligible versus alkali promoters on Ru-based catalysts for NH3 synthesis.71 For reference, alkali promoters typically have an electric field in the range 0.5–1.0 V Å−1,72 while modelling and imaging studies indicate electric fields of only 10−4–10−3 V Å−1 for plasmas.44,73–75
Finally, the possible role of photons (emitted due to relaxation of electronically excited gas species) or high-energy electron-induced electronic excitation of the catalyst surface should be considered, especially in the case of semi-conductors. The intensity of the UV light emitted by plasma is generally considered to be insufficient to activate semi-conductors to a significant extent.76–78 However, the band gap of semi-conductors is typically within the same range as the average electron energy in DBD plasma, so that the creation of electron–hole pairs may be induced by these high-energy electrons. This would enable mechanisms not unlike the ones found in photocatalysis. While previous studies suggest the occurrence of this effect,78,79 its existence has, to the best of our knowledge, not yet been unequivocally shown. Nevertheless, a better understanding of these effects, both on the structure and composition of catalysts, and validated with experiments, would be beneficial to potentially exploit these plasma components for creating plasma–catalyst synergy.
3.4. Does plasma catalysis benefit from elevated temperatures?
Several studies have previously reported a temperature dependence of apparent plasma–catalyst synergy, raising the question whether external heating is required to obtain real plasma–catalyst synergy. Nozaki et al.18 demonstrated improved plasma catalysis performance for steam reforming of CH4 (SRM), as the CH4 conversion in DBD plasma with Ni/SiO2 catalyst was higher than the sum of plasma-only and thermal catalysis, in the temperature range between 673–873 K.18 Furthermore, Kim et al.80 reported a temperature dependence of plasma–catalyst synergy for DRM, showing synergy at temperatures above 600 K. The authors attributed the observed synergy to vibrationally excited CH4 molecules, lowering the activation energy for CH4 dissociative adsorption, and thus increasing the surface reaction rates.80 Other reports showing apparent plasma–catalyst synergy also predominantly involve experiments at elevated temperatures.20–22The question thus arises: do these reports show that external heating is required to obtain plasma–catalyst synergy, or can the results be explained by plasma-induced overheating of the catalytic surface? Indeed, while DBD plasma is often considered to be around room temperature, in reality the system is locally heated, especially in the vicinity of (intense) microdischarges. Thus, the surface temperature can locally surpass the externally applied temperature in so-called “hot spots”. Although recently several authors developed methods to monitor the temperature of a catalyst surface in a DBD in situ,35,39,81 DBD plasma is notoriously inhomogeneous in both time and space. This means that to provide accurate data on the surface temperature in a DBD, in situ measurements should be both highly time- and space-resolved, adding additional complexity. At present, in situ temperature measurements are limited to providing a temperature averaged over a relatively long timescale (much longer than the lifetime of microdischarges) and over a relatively large volume (much larger than the volume exposed to a microdischarge). Therefore, we believe that at present, it is impossible to rule out the contribution of plasma-induced overheating to these observed synergies. Attributing these reported synergies to plasma–surface interactions, like enhanced dissociative adsorption of vibrationally excited CH4 on transition metals, is therefore, in our opinion, not yet properly substantiated, especially considering the limited prevalence of these species in atmospheric pressure DBDs, as discussed in Section 3.2.
Regardless, we believe that to obtain plasma–catalyst synergy, it is sensible to operate at elevated temperatures. Indeed, low operating temperatures may be problematic for achieving plasma–catalyst synergy, as many elementary surface reactions, which lie at the basis of catalysis, require elevated temperatures to take place. Indeed, while non-thermal plasma can enhance the often rate-limiting activation of reactants, potentially altering the rate-limiting step of a reaction, the kinetic barrier of subsequent surface reactions remains, as illustrated in Fig. 5. For example, Rouwenhorst et al.82 showed that for NH3 synthesis on Ru-based catalysts at temperatures below 200 °C, the hydrogenation steps at the surface and/or NH3 desorption become rate-limiting instead of N2 activation, even if N radicals are supplied to the surface. In other words, while non-thermal plasma can supply the catalyst surface with an abundance of reactive species, the subsequent reactions at the surface (including desorption) will still require elevated temperatures, unless they are barrierless. It should be noted that non-thermal plasma can potentially also alter other elementary reaction steps to some extent. For example, it has been suggested that electron-induced desorption of surface adsorbates can play a role in plasma catalysis,15 although to the best of our knowledge, this has not yet been experimentally shown. Eley–Rideal reactions could also contribute to low-temperature surface activity, but recent work by Michiels et al.83 shows that their prevalence in plasma-catalytic systems is often greatly overstated.
 | ||
| Fig. 5 Potential energy scheme of an arbitrary exothermic, heterogeneously catalyzed reaction with activated adsorption. The full line represents the barrier of the corresponding gas phase reaction, while the dotted lines represent the barriers of the subsequent elementary reactions at the catalyst surface. Plasma activation of the reactants is shown by the purple dashed line, showing a reduction in the barrier towards adsorption and an increase in potential energy of the reactants. However, subsequent barriers remain the same, showing the need for elevated temperature for these elementary reactions to take place. | ||
It thus becomes apparent that while non-thermal plasma can potentially lower the activation energy of a catalytic reaction, it does not fully remove the need for external heating if plasma–catalyst synergy is desired. In plasma-catalytic experiments at room temperature, plasma chemistry can be expected to dominate, and plasma-catalytic effects are likely to be largely physical in nature (see Section 4.2).
It should be noted that increasing the temperature of the plasma-catalytic system can have additional effects, which may not be negligible. Firstly, a higher temperature could lead to a reduced E/N, when the system is operated at constant power and frequency. Indeed, in this case, the operating voltage drops, lowering E/N. However, with increasing temperature, at constant pressure, the gas number density decreases, enhancing E/N. The influence of higher temperature on the prevalence of vibrationally excited species is therefore not straightforward. Secondly, an important consideration is that the electron recombination rate coefficient depends on the gas temperature. This effect can largely be resolved by consistent experimental design, in which a catalyst should always be compared to a reference tested at the same temperature. Finally, the diffusion coefficient of reactive species is proportional to temperature to the power 1.5, and their recombination rate coefficient also depends on temperature, so that the interaction between reactive species and the surface may be altered at higher temperatures (see also Section 4.1).
4. Plasma chemistry is too dominant
4.1. Limited contact between plasma and catalyst – need for more efficient packing geometry
We can describe the space that is covered by reactive species formed in a streamer by a cylindrical volume with a certain length L (i.e. the discharge length), and a radius R, with the streamer being at the center of the cylindrical volume, as illustrated in Fig. 6. For species i, given a certain discharge length L, a diffusion time τi, and a diffusion coefficient Di, the radius of this volume can be estimated by eqn (1),85 in which J0 is the first zero of the zero order Bessel function (≈2.405). This radius thus represents the distance travelled by species i perpendicular to the streamer.
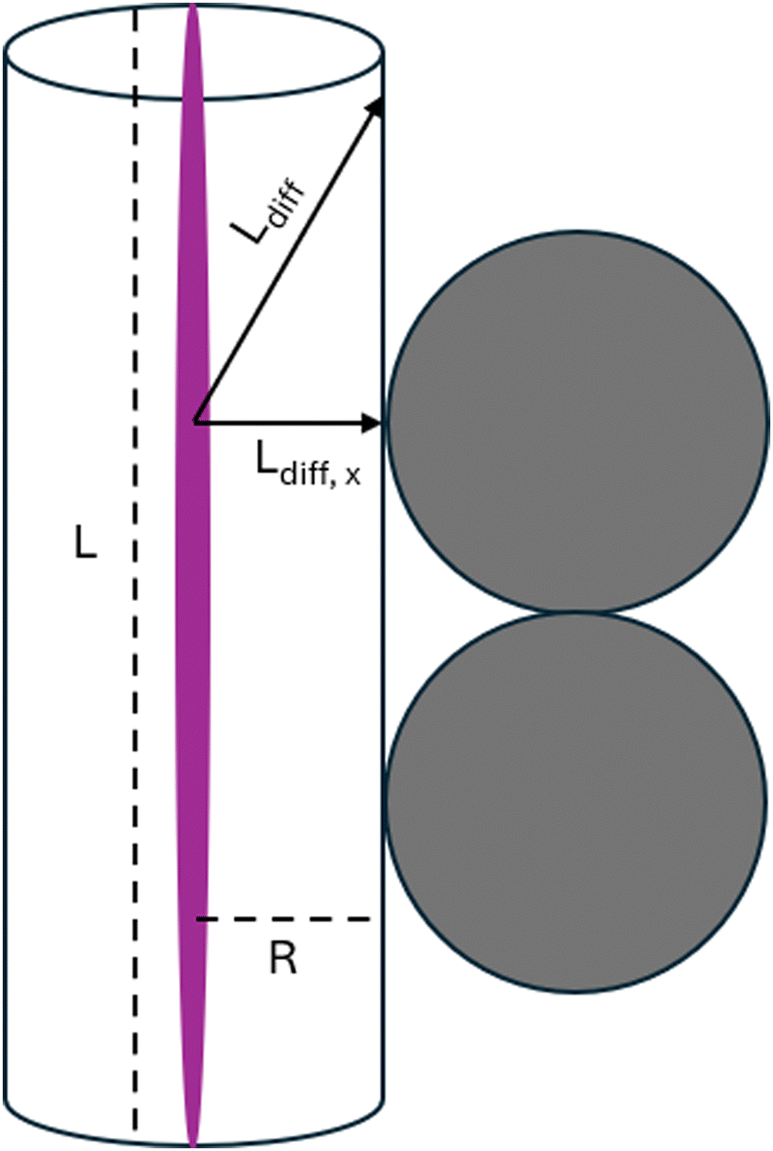 | ||
| Fig. 6 Representation of the space covered by reactive species generated in a streamer as a cylindrical volume with length L and radius R. An example of the total diffusion length (Ldiff) of a reactive species is shown in the figure, along with its direction. The contribution of the direction perpendicular to the streamer to this diffusion length, and thus the maximum allowed distance between the streamer and the catalytic surface, is given by Ldiff,x, which is equal to R. | ||
Di is typically in the order of 10−5 m2 s−1 at 300 K. Moreover, typical DBD reactors used for gas conversion have discharge gaps in the mm range, so we take L = 10−3 m. Finally, we take τi equal to the lifetime of the species; while it is impossible to generalize the latter for all reactive species formed in DBD plasma, we estimate lifetimes to be in the order of 10−4 s at 300 K, based on modelling work for the lifetime of O radicals at 2000 K.86 Inserting these values in equation 1 yields an R of 8 × 10−5 m.
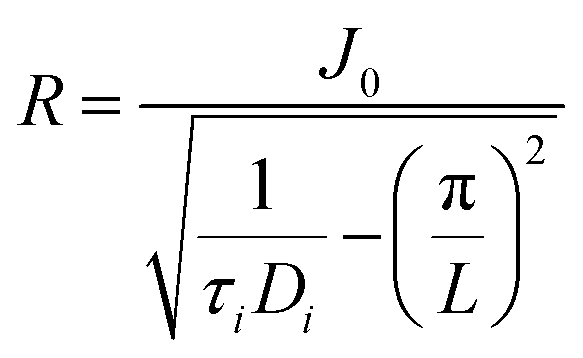 | (1) |
In other words, reactive species are estimated to be present up to a distance of 8 × 10−5 m perpendicular to the streamer. Hence, reactive species generated at a distance >8 × 10−5 m from the surface can be expected to mainly react in the plasma phase, thus contributing to the plasma chemistry. Note that this “diffusion length” (in fact it is the contribution to the total diffusion length perpendicular to the streamer) is of a similar order of magnitude as estimated by Kim et al.,87 who applied the Einstein–Smoluchowski relation in one dimension. It is thus clear that, near room temperature, the void space diameter should be smaller than 100 μm to promote the surface reactions with respect to the plasma chemistry.
Note that the above is only a rough estimate, used to provide an order of magnitude of the diffusion length rather than the exact value. The lifetime of reactive species can greatly differ from the lifetime we estimated above. For example, Jiang and Bruggeman88 showed that for the plasma-catalytic oxidation of CH4, the CH3O2 radical may play a significant role in the formation of CH3OH. The lifetime was estimated to be in the order of 10−3 s, i.e. an order of magnitude larger than our estimate. However, it should be noted that the authors applied a plasma jet, in which the catalyst is exposed to the afterglow of the plasma, rather than positioned in the discharge region, so that the mechanism will inherently favor long-lived species. We believe, however, that short-lived reactive species, particularly vibrationally excited species, are of vital importance to create plasma-catalytic synergy, as discussed above, justifying our lower estimate. In fact, for vibrationally excited species, our calculated R is likely an overestimation, as they are typically characterized by very short lifetimes. For example, the lifetime of vibrationally excited CO2 molecules in a low pressure (6.7 mbar) CO2 glow discharge was estimated to be in the order of 10−3 s.89 At atmospheric pressure, the VT relaxation will be much faster, and thus the lifetime much shorter. We estimate the lifetime of vibrationally excited CO2 at atmospheric pressure to be in the order of 10−5 s, based on calculations using the same chemistry and modelling approach as used in Tsonev et al.,89 but for atmospheric pressure. Using this estimation, we obtain an R value of 2 × 10−5 m, suggesting the need for a void space of 20 μm or lower.
Moreover, the available surface area in plasma catalysis is inherently (much) smaller than in thermal catalysis using porous support particles. Indeed, as discussed in Section 2.2, reactive plasma species cannot be generated in small catalyst pores, and diffusion from the plasma to inside the pores is limited due to the short lifetimes of these species, with a diffusion length similar to the diffusion length estimated at the outer surface of packed bed particles. That means that only a thin outer layer of a porous particle is accessible to activated species and any active sites deeper in the porous particles cannot contribute.
An option could be to use smaller beads, which will also reduce the void space. Wang et al.93 demonstrated that (sub)micrometer SiO2 spheres (with and without supported Ni catalyst) in the range between 120 and 2390 nm yielded significant performance improvement in a packed bed DBD used for DRM, with the best performance reached for the 740 nm spheres (with 5 wt% Ni), resulting in CO2 and CH4 conversions of 44 and 55%, and an energy yield of 0.271 mmol kJ−1, compared to 20%, 27%, and 0.116 mmol kJ−1 for plasma-only, at the same flow rate. Such improvement is typically not seen for the more common mm-sized spheres and might indicate that smaller bead sizes are indeed more effective, due to the reduced void space. On the other hand, care must be taken not to have significant pressure build-up due to a too dense packing (which could also explain the reported enhanced activity in Wang et al.93) and that the void sizes are still large enough for plasma streamers to propagate between the electrodes (i.e. in the range of the Debye length). The latter is not easily verifiable, as often opaque reactors and/or electrodes make it impossible to visually observe the catalytic bed. Fig. 7 gives a schematic representation of the importance of the multi-scale morphology of the catalyst bed for the interaction with activated plasma species.
 | ||
| Fig. 7 Left: Various geometrical considerations for plasma–catalyst interaction, such as the inter-particle void distance, the catalyst pore diameter, and the diffusion length. Right: Criteria for direct interaction inside catalyst pores. Reproduced from ref. 87 with permission from Elsevier, copyright 2015. | ||
Another possible solution could be to use catalyst/support materials with wide enough pores (preferably above 1 μm) so that plasma can be created inside the pores. Catalyst/support structures usually aim at maximal surface area to maximize the number of active sites per unit volume in catalytic reactors, and therefore, macro-porous structures are less common in catalysis. Nevertheless, many macroporous materials are available for other applications, e.g. for inorganic membranes, and several techniques have been described in literature, e.g. based on templating techniques.94 Wang et al. recently managed to prepare 3D porous Cu and CuO catalysts with different pore sizes up to 2 μm, using templates based on uniform SiO2 particles (10–2000 nm), and applied them to plasma-catalytic DRM.95
It is also worth exploring the potential of surface DBDs, as they create plasma directly on the surface. Furthermore, we hypothesize that they give rise to a more uniform plasma, thus with enhanced contact between plasma and catalyst surface. Grid-like electrodes could be used, as for example shown by Xie et al.103 or Jakob et al.104 Ideally, the catalyst could be deposited within the electrode grid, and the grid spacing should be optimized in order to ensure optimal contact between plasma and catalyst. The dead volume of such a reactor should be minimal to ensure the gas flow is effectively treated by the plasma (see e.g. Di et al.105).
Note that a packed bed DBD reactor has the disadvantage that the packing can lead to lower conversions (at the same gas flow rate) due to a reduction in reaction volume. In this regard, several authors proposed a different way of introducing the catalyst into the reactor, namely as a coating on the reactor wall or on the electrode. García-Moncada et al.106 coated μm-thin layers of Pd/Al2O3 on the wall of a DBD reactor for the coupling of CH4. They found a 200% increase in selectivity towards higher hydrocarbons as compared to the blank reactor, while CH4 conversions barely decrease. Moreover, they reported an enhanced hydrocarbon selectivity compared to the reactor partly packed with the same catalyst in powder form. More recently, Gregory et al.107 applied a similar method for CO2 hydrogenation. They coated the inner wall of a quartz tube with Ir/TiO2 and used a helical inner electrode to enhance the contact between reactive plasma species and reactor wall. The authors reported an enhancement in CH4 selectivity for the coated reactor as compared to the blank reactor with a factor of 1.5, along with enhanced CO2 conversion, but did not compare the results to a packed-bed DBD. Similarly, Peters et al.108 coated their electrode with a catalyst layer, showing enhanced decomposition towards CO2 in the oxidation of n-butane, with minor changes in conversion, as compared to the uncoated electrode.
While these studies show that coating the electrode or the wall of a DBD reactor can enhance the selectivity of the system, while retaining (or even increasing) conversions, there is at present no reason to assume that these systems work inherently better than packed bed DBDs. Indeed, the former have the advantage of maximizing the reaction volume and thus conversion, but we believe these systems are limited because they inherently allow for a large contribution of non-selective plasma chemistry to occur at current, relatively large gap sizes. In our opinion, the main goal of plasma catalysis would be to enhance the selectivity of a system with respect to plasma-only, rather than increasing the conversion(s), and in that respect optimizing the relative contribution of surface reactions would be essential. Hence, we believe such reactors can only work if the void space is sufficiently small (in the μm range as discussed above, but larger than the Debye length).
Finally, Gao et al.115 compared various metal–supported catalysts (Ni–CuO, Co–CuO and NiCo–CuO) in a DBD for CO2 splitting, as well as with a plasma-only system, and reported a synergistic interaction between plasma and NiCo–CuO catalyst. Based on OES and ICCD imaging, the authors revealed that the NiCo–CuO catalyst improves the plasma uniformity and modifies the plasma energy distribution, promoting the formation of excited molecules and their subsequent catalytic reactions on the NiCo–Cu surface. Hence, this example illustrates how the choice of catalyst can affect the plasma uniformity (thereby possibly improving the contact between plasma and catalyst surface, cf. above) and can also promote the formation of excited species, which can work in better synergy with the catalyst (see Section 3.1 above).
4.2. Chemical catalytic effects are masked by the physical effects of the catalysts
An important consequence of the plasma chemistry being dominant is that the effects of the catalyst (or rather: material) on the discharge behavior, and thus on the plasma chemistry, overshadow any possible catalytic effect. This was demonstrated by Ndayirinde et al.,91 for plasma-catalytic NH3 synthesis, where the metal catalyst coating on the dielectric beads acts as plasma modifier rather than as real chemical catalyst.A similar conclusion was also reported by Navascués et al.,119 and by De Meyer et al.92 The latter compared two catalyst synthesis methods, i.e., wet impregnation and spray coating, and demonstrated how the catalyst synthesis method changes the catalyst coverage on the beads, which in turn affects the plasma behavior. Specifically, the authors showed that the so-called microdischarge quantity (which is defined based on both the number of microdischarges and their intensity) drops dramatically upon catalyst coating (especially for the spray-coated catalysts), while the discharge areal fraction (i.e., a measure for the fraction of the reactor volume filled with plasma) increases, both demonstrating the evolution from filamentary to uniform plasma (see Fig. 8).
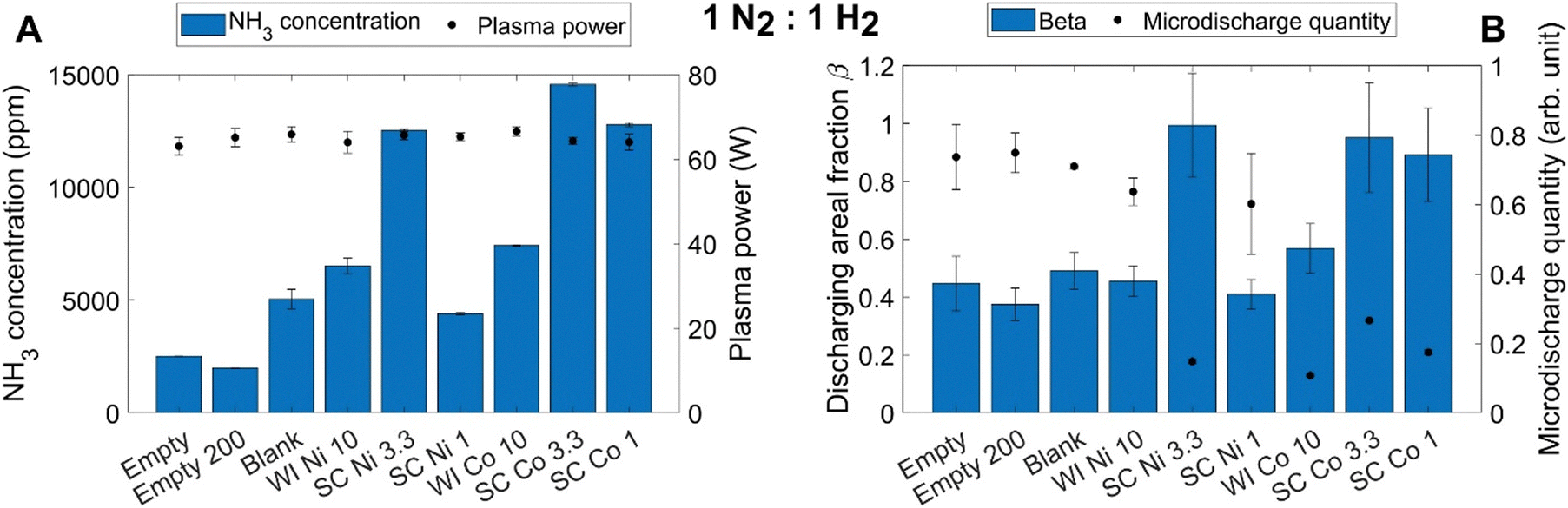 | ||
| Fig. 8 Effect of catalyst synthesis method (wet impregnation (WI) vs. spray coating (SC)) and catalyst loading (10, 3.3 and 1 wt%) on the produced NH3 concentration and measured plasma power (A), as well as on the discharging areal fraction β and microdischarge quantity (B), for a N2:H2 packed bed DBD plasma, with gas mixing ratio of 1:1 and total gas flow rate of 100 mL min−1. Comparison is also made with the data for an empty DBD reactor (at same flow rate of 100 mL min−1 and at a flow rate of 200 mL min−1, corresponding to the same residence time as in the packed bed DBD) and with a packed bed with blank Al2O3 beads (first three data points in A and B). Reproduced from ref. 92 with permission from Elsevier, copyright 2023. | ||
It is clear from this figure that the DBD plasma discharge characteristics have a significant impact on the overall performance (i.e., produced NH3 concentration), despite employing similar catalytic materials (Ni or Co, with two different catalyst synthesis methods). The spray-coated (SC) Ni catalyst with 3.3 wt% loading, and the SC Co with 3.3 and 1 wt% loading are characterized by the highest discharging areal fraction and lowest microdischarge quantity, representing the most uniform plasma, and they give rise to the highest NH3 concentration formed. The latter is in line with model predictions, which indeed revealed that NH3 gets destroyed inside microdischarge filaments,120 and thus, a more uniform plasma yields a higher net NH3 production. Hence, the catalyst materials (and synthesis method) have a physical effect on the plasma behavior, which in turn affects the plasma chemistry, and maybe this physical effect masks the possible chemical catalytic effects. Similar results were shown for DRM, but in this case, increasing the microdischarge quantity enhanced the performance, as intense microdischarges are required to activate the reactants,92 again in line with model predictions.121 Thus, the desired physical effect is reaction-dependent, suggesting that for efficient use of plasma catalysis, it may be essential to align both the physical and possible chemical effects, so that both work in synergy.
We believe many groups are not yet fully aware of this effect. Hence, it is important that researchers always analyze the plasma electrical characteristics (i.e., current–voltage profiles, obtained with oscilloscopes and probes with sufficient time-resolution and bandwidth, as well as Lissajous figures) to account for this effect. Only if the plasma electrical behavior is the same, the real chemical-catalytic effects of catalysts can be compared.
5. The need for correct measurements, standardization and consistent reporting
It is clear from above that plasma catalysis still faces fundamental challenges. Nevertheless, many groups have reported excellent results, in particular for the synthesis of oxygenates.26,43,122–130 However, similar experiments in other labs were not able to obtain such high oxygenate yields. Thus, the question arises: is there a fundamental reason that can explain these excellent results, compared to other reports that do not report plasma–catalyst synergy? In general, we believe that in our research field practical problems and discrepancy in observations and interpretation should be discussed more critically, also when results seem less favorable for practical applications, so that other researchers can learn from it.An important example is the quality of the data on conversion and formation of products. This depends on the quality of the analysis methods used for measuring the conversion of reactants, as well as the formation of products. In catalysis research, it is a good habit to report the mass balance, calculating the number of atoms entering and leaving the reactor. This is usually done for C and N. Balances on O and H are more difficult because quantitative analysis of H2O is usually not possible. In case water is not converted or formed, also balances on H and O are possible. In catalysis research, usually a mass balance closure within 95% is accepted as a sign that the experiment is not affected by e.g. analysis errors or material loss via leakages. This causes more challenges when many different products are formed, compared to the selective conversion to only one or two products.
Because analysis methods result in concentrations in feed and product mixture, also accurate data on the flow rates of both streams is required. The flow rate of the feed stream is usually controlled by mass flow controllers. The flow rate of the product stream, however, can be different and needs to be measured in case of gas expansion or contraction. Indeed, when the number of moles before and after a reaction changes (e.g., in DRM: CO2 + CH4 → 2CO + 2H2), the volumetric flow rate will also change, which affects the measurement of conversion and product yields, as explained in detail in other work.131,132 As demonstrated by Wanten, Vertongen et al.,132 many formulas circulate in literature, and when not correctly accounting for gas expansion, they can overestimate (but also underestimate) the conversion and product yields, and therefore also energy cost and energy efficiency.
Mass balances become even more difficult when products form that condensate, resulting in two product streams, i.e. products in the gas phase and products in a separate liquid phase. In that case, reliable information of concentrations in both phases, as well as on the amount of gas phase and liquid phase is required. The amount of gas phase can be obtained via the flow rate. The amount of liquid formed is usually determined based on weight. This is all rather troublesome and therefore in thermal catalysis research, formation of liquid products is prevented, if possible, by using online gas analysis (e.g. online gas chromatography) while heating the lines between the outlet of the reactor and the online GC, again preventing condensation of e.g. oxygenates.133–138
Unfortunately, offline measurements are typically reported in the plasma catalysis community when oxygenates are formed, condensing the oxygenates in a cold trap, which are analyzed post factum.26,43,122–130 In our (PLASMANT) group, this approach was also used before,139 because online measurements of oxygenates were not possible at that time. Indeed, such measurements are not straightforward with every GC, especially when they are equipped for the analysis of permanent gases. It is important to explicitly state the measurement method, because offline measurements on condensed products affect the quality of the data. Unfortunately, the weight of liquid products, and sometimes even the flow rate of the product gas stream, are not reported in plasma-catalytic studies with product condensation. Thus, mass balances are missing, endangering the reliability of the data. Moreover, using this methodology, authors are inherently comparing a steady-state measurement (online gas analysis) with a cumulative measurement (offline liquid analysis from cold trap). Even more serious is the fact that sometimes, the product distribution in the liquid phase is used to calculate a “selectivity” to a specific product, based on exclusively products in the liquid phase, without mentioning that this has no meaning in terms of the absolute selectivity to that product, as gas phase products are denied in the calculation. This highlights the need for greater transparency in reporting methodologies to ensure reproducibility and comparability across studies. Additionally, the protocols used for the offline measurements are often not adequately described. We believe that to correctly quantify oxygenates, online measurements with a rigorous check on the mass balance (instead of just assuming a closed mass balance) should become the standard, adopting the methods generally used in research on thermal catalysis. For improving our collective understanding of the underlying processes, it is vital that such yields should be adequately reported, especially when seemingly spectacular results are reported, as is increasingly the case in recent literature.26,43,122–130
In general, there is a need for more standardization in plasma catalysis research, so that insights from one study can also be applied to other studies, to make general progress in our field, and to improve reproducibility of experiments. Indeed, at present, we are often limited to activity testing in packed bed DBDs in which multiple effects are confounded, providing information about the system, rather than about catalytic activity. In this regard, we explicitly wish to refer the reader to a recent publication by Lefferts,140 in which the author provides important considerations for obtaining correct intrinsic kinetic data in (plasma-)catalytic systems. This is essential when comparing experimental results with microkinetic models.
We believe the field may benefit from simpler plasma catalysis setups (e.g., planar DBD with catalyst coated onto the dielectric material and/or electrode, instead of packed-bed DBD), useful to gain more insights into structure–activity relationships by in situ/operando diagnostics, and facilitating a one-on-one comparison with microkinetic models. For a better understanding of the potential role of surface reactions, it is vital that the use of in situ/operando techniques is not limited to steady-state conditions, but that transient conditions are also considered, for example by using isotope switches.141,142 Currently, claims about plasma-catalytic reaction mechanisms are often made based on steady-state in situ FTIR experiments, while these experiments do not allow for distinguishing between surface intermediates and spectator species. In contrast, in situ/operando experiments at transient conditions can separate spectator species from surface intermediates, so that they, in combination with other techniques like SSITKA (steady-state isotopic transient kinetic analysis), can help elucidate the reaction mechanisms. Although the reactors applicable for in situ/operando spectroscopy are usually poor chemical reactors, the detailed insights obtained can be useful to identify limitations and further improve the performance of plasma-catalytic systems.
We again want to stress the importance of a holistic approach to plasma catalysis, in which both the chemical and physical effects of a catalyst material are considered, along with their potential interplay. We believe that plasma is still too often seen as a black box by researchers in our field, while it is clear that physical effects cannot be decoupled from chemical effects. For plasma-specific considerations on standardization of diagnostics, computations, reporting and plasma sources, all of which are vital to improve the reproducibility of experiments from one lab to another, we want to refer the reader to the work of Alves et al.143
Naturally, the need for standardization should go hand in hand with comprehensive and consistent reporting of all experimental conditions. Indeed, a wide variability in reactor setups exists. Among others, the reactor design should be fully described, including electrode materials, discharge gap widths and dielectric barrier materials, and where possible, pictures should be included along with schematics. The working frequency, as well as the plasma power (not the applied PSU power) should be provided, along with recorded current and voltage profiles. Regarding the catalyst material, the synthesis method should be described in detail, with special attention to pre-treatments, such as drying and ex situ reduction. Indeed, while in thermal catalysis the catalyst is often reduced and/or activated in situ, most plasma-catalytic setups are not suitable to reach the high temperatures required for this, forcing researchers to perform these pre-treatments before loading the catalyst into the reactor, or neglecting them altogether. Alternatively, in situ reduction by H2/Ar plasma is performed, although little information is available on its efficacy. Catalyst characterization should be done after the same pretreatment, as well as after the plasma catalysis experiment, without exposure to ambient. Moreover, special attention should go to catalyst shaping and morphology, as this can greatly impact the experimental results. In short, we believe that describing the experimental details is particularly important in our field, enabling unequivocal and exact reproduction by other researchers.
Finally, correct and transparent reporting is also crucial for modeling of plasma catalysis. This includes careful analysis of the modeling input data (e.g., rate coefficients), as the modeling output critically depends on the input. Rate coefficients of gas-phase reactions are characterized by some uncertainties, and good practice should take these into account when evaluating the output. This was illustrated by some authors, applying a Monte Carlo procedure for selecting the rate coefficients within their range of uncertainty, demonstrating that the modeling results are subject to accumulated uncertainties, and cannot be used for quantitative predictions, but only to explain qualitative trends.144–146 When modeling plasma–catalyst surface interactions, the risk of incorrect predictions is even larger, as the input data are either based on sticking coefficients, which are too approximate, or on DFT data, but this DFT data is subject to larger uncertainties than often realized (e.g., too approximate density functionals, inconsistent data sets, or simply unavailability of data, leading to rough approximations). Moreover, DFT results are sometimes used to explain plasma catalysis experimental data, but there is a large gap between the atomic scale and plasma catalysis reactor scale, where many other effects come into play, such as changes in discharge characteristics due to a catalyst packing, as explained earlier.
6. Thinking “out of the box”: the combination of plasma with other materials
Besides the combination of plasma with catalysts, we believe that other materials can also be interesting in combination with plasma, more specifically sorption materials, scavenging materials or membranes, for separation purposes. Indeed, they would allow for the removal of products, suppressing thermo-catalytic, plasma-catalytic, and gas phase product decomposition, improving the energy efficiency. This is conceptually illustrated in Fig. 9, using a metaphor for plasma catalysis introduced by Lefferts.140 Indeed, an endergonic reaction can be illustrated as pumping water to a higher level. The pump is equivalent to plasma enabling the reaction, while the leak in the upper reservoir represents losses caused by the backward reaction. The left panel represents plasma catalysis without any separation, while the right-hand panel represents plasma catalysis with integrated separation of the product in the third, leak-tight reservoir. The consequence is that the size of the leak to the lower container is decreased, and the energy efficiency is increased. | ||
| Fig. 9 Pumping water as a schematic model for plasma catalysis of an endergonic reaction with a significant leak representing the backward reaction (left panel) and plasma catalysis with integrated separation of product, decreasing losses via the backward reaction (right panel). Expanded version of a figure published by Lefferts.140 | ||
Indeed, in thermal catalysis, the reverse reaction will take place when approaching thermodynamic equilibrium. Once the conversion is at equilibrium, e.g. by increasing the contact time in a fixed bed reactor, the forward reaction rate is equal to the reverse reaction rate, resulting in a net-zero rate. This has no consequences for the energy efficiency because the zero rate implies no heat generation or consumption. As explained in detail by Lefferts,140 reverse reactions will occur even more in case of plasma catalysis, because not only the thermal backward reaction sets in, but also this reaction is enhanced by plasma activation of the product molecules. In this case, energy efficiency deteriorates because of the plasma energy input, while decreasing the conversion, showing the potential benefit of in situ product separation.
With respect to sorption materials, Rouwenhorst et al.147 reported NH3 protection, by shielding it from decomposition in the plasma, upon sorption inside the pores (of zeolite 4A) that cannot be reached by plasma, followed by desorption after the plasma is turned off. This results in much higher NH3 yields, i.e., a factor two compared to without using an adsorbent. Such “shielding protection” was also claimed by Wang et al.148 using mesoporous MCM-41, but without providing clear evidence because thermal desorption of NH3 was not considered. Indeed, protective adsorption of ammonia results in a decrease of the ammonia concentration in the product mixture, compared to an experiment with a catalyst with the same rate of formation of ammonia, without protective adsorption of ammonia. This decrease will be observed if the catalyst is not yet saturated with ammonia, and once it is saturated the same steady state ammonia production will be achieved with both catalysts. Protective adsorption can suppress ammonia decomposition exclusively if ammonia desorption and flushing out of the reactor is done in absence of plasma. Instead, Wang et al.148 reported the opposite effect, and operate in continuous mode. Nevertheless, these examples show the potential of rational catalyst/material design, based on insights into the mechanisms, i.e., that NH3 gets destroyed by plasma microdischarges and that plasma streamers cannot penetrate into catalyst pores when they are smaller than ca. 500 nm, both obtained by modeling.45,120 We believe such in situ product removal can also be interesting for other plasma catalysis applications, such as for CH3OH production, to avoid product decomposition in the plasma phase, which is indeed identified as a limitation in plasma catalysis.
A similar approach can be utilized for plasma-based NOx synthesis from N2 and O2. Rouwenhorst et al.56 demonstrated that the energy yield could be improved by a factor 15 upon adsorbing NOx on MgO, followed by desorption after the plasma is turned off. This “shielding” thus limits the reverse Zeldovich mechanism in the plasma, in which NO would react back into N2 and O2.
Interesting to note is also the work by Li et al., who combined a DBD plasma with solid sorbents packed inside the reactor, for one-step plasma-based CO2 capture and utilization.149–151 Hence, in this innovative concept, the sorbents are not used to remove the products from plasma (or to protect them from being decomposed in the plasma), but for carbon capture, followed by desorption and conversion inside the plasma. Similarly, Giammaria et al.152 demonstrated a synergistic effect for CaCO3 decomposition to CaO and the reverse water gas shift reaction in a DBD plasma. CaO can then be used again for CO2 capture.
For separation purposes, the combination of DBD plasma with solid oxide electrolyser cell (SOEC) in a hybrid reactor was reported already about 10 years ago for CO2 splitting.153 The SOEC was able to remove the oxygen from the plasma region, thereby avoiding the backreactions from CO into CO2, and thus increasing the overall CO2 conversion. Even more, this setup, using Co–Mo catalyst supported on a quartz substrate inside the hybrid reactor, allowed to synthesize carbon nanotubes, based on CO2 as carbon source.153
Specifically for separation, several interesting papers have recently been published, combining plasma with membranes. Studies are reported for cold plasmas (DBD), such as for CO2/CH4 separation based on zeolites,154 or H2 separation with Pd-based membranes,155–159 but also for warm plasmas, where the hot effluent gas can activate the membrane, such as for O2 removal with perovskite-based membranes.160,161
Besides membranes, also other materials can be placed in/after a plasma reactor, to remove one of the reaction products, and thus shifting the equilibrium to the right. For instance, Delikostantis et al.162 reported a successful example of plasma-assisted chemical looping, by placing a nanostructured CeO2/Fe2O3 oxygen scavenger post-plasma, which suppresses the recombination of CO with O atoms, by capturing the latter. They were able to reach an overall CO2 conversion at the reactor outlet of ca. 29%. According to chemical equilibrium calculations, such conversion values can only be achieved above 2775 K, hence above the operating temperature in these experiments. Therefore, the authors concluded that plasma with post-plasma scavenging materials can significantly overcome chemical equilibrium limits.
A similar example of plasma-assisted chemical looping applied to CO2 splitting was recently presented by Long et al.,163 based on Ce0.7Zr0.3O2 oxygen carrier, yielding 84% CO2 conversion and no O2 in the outlet stream. The authors reported a significant drop in the temperature needed for the chemical looping process, from 650–1000 °C in conventional chemical looping, to only 320 °C, suggesting a clear synergy with the plasma process.
More in general, the concept of chemical looping could also be interesting to overcome the limitations of plasma catalysis, i.e., by decoupling reactant consumption and product generation stages, so that products can be collected without downstream separation, and destruction of products in the gas phase is limited.164,165
Inspired by this concept, Sharma et al.166 combined plasma with proton-conducting SOEC for NH3 production from N2 and H2O. In this innovative concept, plasma is used to activate N2 (at the cathode), which reacts towards NH3 with hydrogen species that are produced by water oxidation over the anode and transported through the proton-conducting membrane towards the cathode. Other concepts, combining plasma-based N2 fixation to NOx with some kind of catalytic and/or electrochemical reduction to NH3 have also been developed in recent years.167–172
Veng et al. recently presented a membrane-DBD reactor, using a porous Al2O3 membrane as dielectric barrier and as distributor of H2, leading to much higher NH3 production than using pre-mixed N2 and H2.173 The membrane was surrounded by catalyst powder on porous glass wool support filling the plasma region. The authors used electrical, optical and spectroscopic diagnostics, and also Fourier-Transform Infrared spectroscopy, and concluded that the glass wool support suppresses microdischarges, leading to higher NH3 production, because NH3 is typically destroyed in the microdischarges, as explained above.120
Finally, the combination of warm plasma with a post-plasma carbon bed results in successful oxygen scavenging (e.g., ref. 174–178). Indeed, the produced O and O2 from CO2 conversion react with the carbon atoms, avoiding their recombination with CO. Furthermore, the fraction of CO2 that is not converted by the plasma can also react with the carbon atoms if the carbon bed temperature is above 1000 K, through the reverse Boudouard reaction, thereby producing more CO. For instance, Girard-Sahun et al. could enhance both the overall CO2 conversion and energy efficiency by a factor two, while the CO production was even three times higher, and nearly all O2 was removed from the product mix, which significantly reduces separation costs.176 Finally, both Biondo et al.,178 and O’Modhrain et al.177 developed an improved setup, reaching higher temperature at the catalyst bed, resulting in even higher conversion and energy efficiency. The carbon sourcing is a key factor determining the sustainability for a carbon bed as oxygen scavenger.
All these examples show that combining plasma with (sorption, membrane, scavenging) materials is promising, indicating indeed that we should think “out of the box”, as there is a lot of potential in plasma–material combinations, beyond the classical plasma catalysis concept in the strict sense.
7. Outlook
It has been argued numerous times that plasma catalysis is especially suitable for small scale operation, related to fast changes in capacity, which is especially important in view of intermittent availability of green electricity. We want to share our thoughts about what type of conversions for the synthesis of chemicals and fuels would be most promising with this in mind.• In general, we believe that plasma catalysis, in the broader sense, is most promising aiming at endergonic reactions (ΔG > 0), for which thermal catalysis is not suitable; instead, plasma catalysis is directly competing with electro-catalysis and photo-catalysis. However, plasma catalysis is easier to scale than electro-catalysis, because the latter has the disadvantage that reactions can take place only at the surface of electrodes with limited surface area. Typical examples are CO2 and H2O dissociation and NOx synthesis from N2 and O2, as well as CH4 coupling, CH4 pyrolysis and H2S dissociation. Note that these reactions can be turned into exergonic reactions, the next category to discuss, by using extremely high temperatures.
• The second most promising application would be exergonic endothermic reactions (ΔG < 0, ΔH > 0). Energy losses in the plasma can be used to deliver the required heat, in addition to enhancing rates via plasma catalysis. The main competition for this approach is electrical heating of thermal-catalytic endothermic reactions, which is already at higher TRL. Reforming reactions, including RWGS and DRM, as well as NH3 cracking, belong to this category. Note that the subdivision between the first and second group is a bit arbitrary, and depends on the operating temperature: at higher temperature, the reactions of group 1 would shift to group 2 as well. However, catalysts cannot operate at very high temperatures because adsorption of reactants is thermodynamically impossible.
• Exergonic exothermic reactions (ΔG < 0, ΔH < 0) are less likely to be successful. Possible advantages would be to operate at milder conditions compared to thermal catalysis, which might lead to an advantage in capital cost (CAPEX) and operating cost (OPEX), but only if the energy efficiency is at least similar to the thermal catalytic competition. A typical example is NH3 synthesis, but so far, the energy efficiency is an order of magnitude too low. Energy loss in the plasma further increases the cost for removing not only the reaction heat but also the heat caused by plasma energy loss. In case heat integration is possible at the production location, the capital cost will increase for the required heat exchangers.
In general, we want to stress that energy efficiency is the Achilles heel for plasma catalysis to produce chemicals and fuels, especially when keeping in mind that high product concentrations are required. The consequence is that product decomposition will occur significantly, devastating energy efficiency. Therefore, as indicated in previous section, integration of conversion and separation is needed and the field of plasma catalysis should seek interaction with the research community on process intensification.179,180 Furthermore, chemistry intensification is an opportunity for plasma-based conversion technology for production, i.e. replacing multiple conversion steps with one single conversion. A typical example is the production of nitric acid (HNO3): replacement of the sequence “H2 production + NH3 synthesis + NH3 oxidation” by a single step, i.e., “NOx production from air”.
The latter process is typically carried out in warm plasmas, such as gliding arc, microwave, and atmospheric pressure glow discharges, where the chemistry is mainly thermal, but it is much more efficient than in cold plasmas, like DBD, typically used in plasma catalysis, as briefly explained in Section 3.2. This was not only demonstrated for NOx production from air, but also e.g., for CO2 splitting, DRM, and NH3 cracking.14,181,182 The reason why these warm plasmas are promising for energy-efficient production of chemical and fuels is because the heating does not occur through the walls, but from the gas itself, making it an easy way of heating to very high temperatures. For instance, a recent techno-economic analysis for CO2 conversion in gliding arc plasma with carbon bed reported that the energy cost was 43% less than for electrolysis and conventional CO2 conversion methods.183 However, as mentioned in Section 3.2, the gas temperature in these warm plasmas is in the order of 3000 K and more, which is too high for catalyst implementation inside the plasma. Therefore, these warm plasmas typically operate without catalysts, although they can also be operated with post-plasma catalysts that are thermally activated, which we believe is an interesting application of plasma catalysis, worth to be further explored.
Overall, plasma reactor design, considered as a chemical reactor, as well as design of the entire chemical process, including the required separations and possibly heat integration, is essential to realize plasma-catalytic production of chemicals and fuels. From a chemical engineering point of view, the concentration of targeted molecules in the product mixture should be as high as possible to limit costs of separation and recycling of unconverted reactants. This makes the need to integrate plasma(-catalytic) conversion with separation even more urgent.
Industrial thermal catalysis has been studied and optimized for more than a century, while plasma catalysis research, especially for the synthesis of chemicals and fuels, is quite new. It is obvious that it takes time to optimize and increase the level of understanding of plasma catalysis, requiring more fundamental insights, systematic studies and critical analysis. Then, plasma catalysis can be an interesting supplement to the existing conversion technologies that will be needed to electrify the chemical industry. In that respect, plasma catalysis should not be compared to today's technologies based on fossil feedstock and energy. Instead, plasma catalysis should be compared to alternative technologies based on green electricity, including electro-catalysis, photo-catalysis, processes based on green H2 and electrical heating of endothermic conversions.
8. Conclusion
Clearly, there are several hurdles to take in plasma catalysis for the production of chemicals and fuels, before it becomes a mature technology ready for industrial implementation. There is a need for more insight into the most suitable catalysts tailored to the plasma environment, and/or for changes in plasma reactor design to reach optimal plasma–catalyst interaction. Moreover, the effect of the packing material in a DBD on the plasma physics should be considered and may be even exploited for some reactions. The critical role of mass transport in plasma catalysis, and the need to improve plasma-catalytic reactor design (instead of just empirical reactor optimization) was recently also highlighted by Bayer et al.84Designing the optimal catalyst should focus both on shaping (to allow sufficient contact between plasma species and catalyst surface and ensure the plasma chemistry does not dominate the surface chemistry) and on composition/structure (tuning the catalyst to the plasma environment, to avoid radical scavenging by metal catalysts in case of DRM, or exploiting the role of OVs in metal oxides). On the other hand, also the plasma reactor design needs improvement to maximize plasma–catalyst contact, as well as to tune the plasma conditions towards lower E/N. In this way, we could maximize vibrational excitation, which requires less energy than radical production, thus reducing the energy cost of plasma catalysis, which is nowadays still too high for the production of chemicals and fuels. Importantly, that would also strengthen the conceptual approach of plasma catalysis, as dissociative adsorption on the catalyst surface would remain an elementary reaction, like in thermal catalysis, but now enhanced by vibrational excitation. In fact, we believe the optimal plasma catalysis reactor should be in between DBD and warm plasmas, i.e., with reduced electric fields around 50 Td (for maximum vibrational excitation) but gas temperatures below 1000 K, to allow the direct implementation of catalysts (i.e., in-plasma catalysis), for optimal plasma–catalyst synergy.
Finally, we should not just focus on plasma catalysis in a strict sense, but more in general on the combination of plasma with materials, like adsorbents or membranes, to protect the products from being decomposed in the plasma, or to remove them from the plasma. Integration of plasma(-catalytic) conversion with separation technology can suppress product decomposition. We presented some success stories from literature, e.g., based on sorption materials, SOEC, membranes, chemical looping materials and post-plasma carbon beds.
In conclusion, we believe plasma catalysis, or better, plasma–material interaction, has a lot of potential, especially for endergonic and endothermic reactions, but more fundamental research is needed to understand the synergy. This will require multi-disciplinary research, because besides the chemical (catalytic) effects, also physical effects of the catalyst (or supports) on the plasma behavior must be considered.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 810182 – SCOPE ERC Synergy project), as well as by the Horizon Europe (HE) Research and Innovation Action (RIA) project ALCHEMHY (grant agreement 101177996). We would like to explicitly thank Anne Tiehuis for her work on some of the figures in this manuscript, Ivan Tsonev for his advice during the writing of this manuscript, and Robin De Meyer and Omar Biondo for their advice during the reviewing process.References
- A. Bogaerts, X. Tu, J. C. Whitehead, G. Centi, L. Lefferts, O. Guaitella, F. Azzolina-Jury, H.-H. Kim, A. B. Murphy, W. F. Schneider, T. Nozaki, J. C. Hicks, A. Rousseau, F. Thevenet, A. Khacef and M. Carreon, The 2020 plasma catalysis roadmap, J. Phys. D: Appl. Phys., 2020, 53, 443001 CAS.
- H. L. Chen, H. M. Lee, S. H. Chen, M. B. Chang, S. J. Yu and S. N. Li, Removal of volatile organic compounds by single-stage and two-stage plasma catalysis systems: a review of the performance enhancement mechanisms, current status, and suitable applications, Environ. Sci. Technol., 2009, 43(7), 2216–2227 CAS.
- H. H. Kim, Nonthermal Plasma Processing for Air-Pollution Control: A Historical Review, Current Issues, and Future Prospects, Plasma Processes Polym., 2004, 1(2), 91–110 CAS.
- E. C. Neyts, K. K. Ostrikov, M. K. Sunkara and A. Bogaerts, Plasma Catalysis: Synergistic Effects at the Nanoscale, Chem. Rev., 2015, 115(24), 13408–13446 CAS.
- J. Van Durme, J. Dewulf, C. Leys and H. Van Langenhove, Combining non-thermal plasma with heterogeneous catalysis in waste gas treatment: a review, Appl. Catal., B, 2008, 78(3–4), 324–333 CAS.
- A. M. Vandenbroucke, R. Morent, N. De Geyter and C. Leys, Non-thermal plasmas for non-catalytic and catalytic VOC abatement, J. Hazard. Mater., 2011, 195, 30–54 CAS.
- X. Tu, J. C. Whitehead and T. Nozaki, Plasma Catalysis Fundamentals and Applications, Springer Ser. At., Opt., Plasma Phys., 2014, 106, 1–19 Search PubMed.
- A. Bogaerts and E. C. Neyts, Plasma Technology: An Emerging Technology for Energy Storage, ACS Energy Lett., 2018, 3(4), 1013–1027 CAS.
- J. Hong, S. Prawer and A. B. Murphy, Plasma Catalysis as an Alternative Route for Ammonia Production: Status, Mechanisms, and Prospects for Progress, ACS Sustainable Chem. Eng., 2017, 6(1), 15–31 Search PubMed.
- S. Liu, L. R. Winter and J. G. Chen, Review of Plasma-Assisted Catalysis for Selective Generation of Oxygenates from CO2 and CH4, ACS Catal., 2020, 10(4), 2855–2871 CAS.
- K. H. R. Rouwenhorst, Y. Engelmann, K. van ‘t Veer, R. S. Postma, A. Bogaerts and L. Lefferts, Plasma-driven catalysis: green ammonia synthesis with intermittent electricity, Green Chem., 2020, 22(19), 6258–6287 CAS.
- K. H. R. Rouwenhorst, F. Jardali, A. Bogaerts and L. Lefferts, From the Birkeland-Eyde process towards energy-efficient plasma-based NO(X) synthesis: a techno-economic analysis, Energy Environ. Sci., 2021, 14(5), 2520–2534 CAS.
- M. Scapinello, E. Delikonstantis and G. D. Stefanidis, The panorama of plasma-assisted non-oxidative methane reforming, Chem. Eng. Process., 2017, 117, 120–140 CAS.
- R. Snoeckx and A. Bogaerts, Plasma technology – a novel solution for CO(2) conversion?, Chem. Soc. Rev., 2017, 46(19), 5805–5863 CAS.
- A. Bogaerts, E. C. Neyts, O. Guaitella and A. B. Murphy, Foundations of plasma catalysis for environmental applications, Plasma Sources Sci. Technol., 2022, 31, 053002 Search PubMed.
- E. C. Neyts and A. Bogaerts, Understanding plasma catalysis through modelling and simulation—a review, J. Phys. D: Appl. Phys., 2014, 47, 224010 CrossRef.
- J. C. Whitehead, Plasma–catalysis: the known knowns, the known unknowns and the unknown unknowns, J. Phys. D: Appl. Phys., 2016, 49, 243001 CrossRef.
- T. Nozaki, N. Muto, S. Kado and K. Okazaki, Dissociation of vibrationally excited methane on Ni catalyst, Catal. Today, 2004, 89(1–2), 57–65 CrossRef CAS.
- X. Tu and J. C. Whitehead, Plasma-catalytic dry reforming of methane in an atmospheric dielectric barrier discharge: understanding the synergistic effect at low temperature, Appl. Catal., B, 2012, 125, 439–448 CrossRef CAS.
- Q. Wang, B.-H. Yan, Y. Jin and Y. Cheng, Dry Reforming of Methane in a Dielectric Barrier Discharge Reactor with Ni/Al2O3 Catalyst: Interaction of Catalyst and Plasma, Energy Fuels, 2009, 23(8), 4196–4201 CrossRef CAS.
- A.-J. Zhang, A.-M. Zhu, J. Guo, Y. Xu and C. Shi, Conversion of greenhouse gases into syngas via combined effects of discharge activation and catalysis, Chem. Eng. J., 2010, 156(3), 601–606 CrossRef CAS.
- F. Ahmad, E. C. Lovell, H. Masood, P. J. Cullen, K. K. Ostrikov, J. A. Scott and R. Amal, Low-Temperature CO2 Methanation: Synergistic Effects in Plasma-Ni Hybrid Catalytic System, ACS Sustainable Chem. Eng., 2020, 8(4), 1888–1898 CrossRef CAS.
- J. A. Andersen, J. M. Christensen, M. Østberg, A. Bogaerts and A. D. Jensen, Plasma—catalytic dry reforming of methane: screening of catalytic materials in a coaxial packed-bed DBD reactor, Chem. Eng. J., 2020, 397, 125519 CrossRef CAS.
- J. Sentek, K. Krawczyk, M. Młotek, M. Kalczewska, T. Kroker, T. Kolb, A. Schenk, K.-H. Gericke and K. Schmidt-Szałowski, Plasma-catalytic methane conversion with carbon dioxide in dielectric barrier discharges, Appl. Catal., B, 2010, 94(1–2), 19–26 CrossRef CAS.
- A. Wang, J. H. Harrhy, S. Meng, P. He, L. Liu and H. Song, Nonthermal plasma-catalytic conversion of biogas to liquid chemicals with low coke formation, Energy Convers. Manage., 2019, 191, 93–101 CrossRef CAS.
- L. Wang, Y. Yi, C. Wu, H. Guo and X. Tu, One-Step Reforming of CO(2) and CH(4) into High-Value Liquid Chemicals and Fuels at Room Temperature by Plasma-Driven Catalysis, Angew. Chem., Int. Ed., 2017, 56(44), 13679–13683 CrossRef CAS PubMed.
- K. Zhang, T. Mukhriza, X. Liu, P. P. Greco and E. Chiremba, A study on CO2 and CH4 conversion to synthesis gas and higher hydrocarbons by the combination of catalysts and dielectric-barrier discharges, Appl. Catal., A, 2015, 502, 138–149 CrossRef CAS.
- B. Loenders, R. Michiels and A. Bogaerts, Is a catalyst always beneficial in plasma catalysis? Insights from the many physical and chemical interactions, J. Energy Chem., 2023, 85, 501–533 CrossRef CAS.
- Y. Engelmann, K. van ’t Veer, Y. Gorbanev, E. C. Neyts, W. F. Schneider and A. Bogaerts, Plasma Catalysis for Ammonia Synthesis: A Microkinetic Modeling Study on the Contributions of Eley–Rideal Reactions, ACS Sustainable Chem. Eng., 2021, 9(39), 13151–13163 CrossRef CAS.
- K. H. R. Rouwenhorstm and L. Lefferts, Plasma-catalytic Ammonia Synthesis via Eley–Rideal Reactions: A Kinetic Analysis, ChemCatChem, 2023, 15(12), e202300078 CrossRef CAS.
- Y. Gorbanev, Y. Engelmann, K. van ’t Veer, E. Vlasov, C. Ndayirinde, Y. Yi, S. Bals and A. Bogaerts, Al2O3-Supported Transition Metals for Plasma-Catalytic NH3 Synthesis in a DBD Plasma: Metal Activity and Insights into Mechanisms, Catalysts, 2021, 11(10), 1230 CrossRef CAS.
- S. Yamijala, G. Nava, Z. A. Ali, D. Beretta, B. M. Wong and L. Mangolini, Harnessing Plasma Environments for Ammonia Catalysis: Mechanistic Insights from Experiments and Large-Scale Ab Initio Molecular Dynamics, J. Phys. Chem. Lett., 2020, 11(24), 10469–10475 CrossRef CAS.
- M. S. Gandhi and Y. S. Mok, Non-thermal plasma-catalytic decomposition of volatile organic compounds using alumina supported metal oxide nanoparticles, Surf. Coat. Technol., 2014, 259, 12–19 CAS.
- B. S. Patil, N. Cherkasov, J. Lang, A. O. Ibhadon, V. Hessel and Q. Wang, Low temperature plasma-catalytic NOx synthesis in a packed DBD reactor: effect of support materials and supported active metal oxides, Appl. Catal., B, 2016, 194, 123–133 CAS.
- A. Parastaev, N. Kosinov and E. J. M. Hensen, Mechanistic study of catalytic CO2 hydrogenation in a plasma by operando DRIFT spectroscopy, J. Phys. D: Appl. Phys., 2021, 54, 264004 CAS.
- Z. Ning, L. Wen, R. Li, K. Xin, P. Liu, L. Liu, Y. Sun, Y. Zhu and P. Ning, Oxygen vacancy-enriched Cu/CeO2–ZrO2 catalyst with highly dispersed Cu0 towards plasma catalytic advanced CO2 utilization, J. Cleaner Prod., 2024, 442, 141010 CAS.
- O. V. Golubev, P. S. Il’chuk, A. A. Sadovnikov and A. L. Maximov, Carbon Dioxide Utilization Using Plasma Reactor Packed with Magnesia-Ceria Catalysts with Various Morphology, Petroleum Chem., 2023, 63(9), 1097–1109 CrossRef CAS.
- B. Ashford, Y. Wang, C.-K. Poh, L. Chen and X. Tu, Plasma-catalytic conversion of CO2 to CO over binary metal oxide catalysts at low temperatures, Appl. Catal., B, 2020, 276, 119110 CrossRef CAS.
- E. K. Gibson, C. E. Stere, B. Curran-McAteer, W. Jones, G. Cibin, D. Gianolio, A. Goguet, P. P. Wells, C. R. A. Catlow, P. Collier, P. Hinde and C. Hardacre, Probing the Role of a Non-Thermal Plasma (NTP) in the Hybrid NTP Catalytic Oxidation of Methane, Angew. Chem., Int. Ed., 2017, 56(32), 9351–9355 CrossRef CAS PubMed.
- K. van ‘t Veer, S. van Alphen, A. Remy, Y. Gorbanev, N. De Geyter, R. Snyders, F. Reniers and A. Bogaerts, Spatially and temporally non-uniform plasmas: microdischarges from the perspective of molecules in a packed bed plasma reactor, J. Phys. D: Appl. Phys., 2021, 54, 174002 CrossRef.
- S. Xu, P. Dugkhuntod, S. Ding, Y. Zhang, P. Gosalvitr, S. Chen, J. Huang, S. Klinyod, S. Chansai, C. Hardacre, C. Wattanakit and X. Fan, Product selectivity controlled by the nano-environment of Ru/ZSM-5 catalysts in nonthermal plasma catalytic CO2 hydrogenation, Appl. Catal., B, 2024, 348, 123826 CrossRef CAS.
- L. Fan, Y. Wang, X. Zhai, Q. Yin, J. Zhang, Y. Zhu and L. Wang, Production of Oxygenates from CH4/CO2 Plasma Reaction Assisted by Ni/HZSM-5 Catalyst, Plasma Chem. Plasma Process., 2023, 43(6), 1979–1998 Search PubMed.
- Y. Wang, L. Fan, H. Xu, X. Du, H. Xiao, J. Qian, Y. Zhu, X. Tu and L. Wang, Insight into the synthesis of alcohols and acids in plasma-driven conversion of CO2 and CH4 over copper-based catalysts, Appl. Catal., B, 2022, 315, 121583 CrossRef CAS.
- Y.-R. Zhang, K. Van Laer, E. C. Neyts and A. Bogaerts, Can plasma be formed in catalyst pores? A modeling investigation, Appl. Catal., B, 2016, 185, 56–67 Search PubMed.
- Q.-Z. Zhang and A. Bogaerts, Propagation of a plasma streamer in catalyst pores, Plasma Sources Sci. Technol., 2018, 27, 035009 Search PubMed.
- Q.-Z. Zhang, W.-Z. Wang and A. Bogaerts, Importance of surface charging during plasma streamer propagation in catalyst pores, Plasma Sources Sci. Technol., 2018, 27, 065009 CrossRef.
- F. Coumans, A. Bolshakov, R. C. J. van de Poll, D. Anastasiadou, B. Mezari and E. J. M. Hensen, Improving the performance of ASA in the DAC of 2,5-DMF and ethylene, Catal. Sci. Technol., 2023, 13(24), 6959–6967 Search PubMed.
- R. A. van Santen, Mechanisms in Heterogeneous Catalysis, World Scientific, Europe, 2022, vol. 22 Search PubMed.
- L. B. F. Juurlink, P. R. McCabe, R. R. Smith, C. L. DiCologero and A. L. Utz, Eigenstate-Resolved Studies of Gas-Surface Reactivity:CH4(ν3) Dissociation on Ni(100), Phys. Rev. Lett., 1999, 83(4), 868–871 CAS.
- Y. Engelmann, P. Mehta, E. C. Neyts, W. F. Schneider and A. Bogaerts, Predicted Influence of Plasma Activation on Nonoxidative Coupling of Methane on Transition Metal Catalysts, ACS Sustainable Chem. Eng., 2020, 8(15), 6043–6054 Search PubMed.
- P. Mehta, P. Barboun, F. A. Herrera, J. Kim, P. Rumbach, D. B. Go, J. C. Hicks and W. F. Schneider, Overcoming ammonia synthesis scaling relations with plasma-enabled catalysis, Nat. Catal., 2018, 1(4), 269–275 Search PubMed.
- P. Mehta, P. M. Barboun, Y. Engelmann, D. B. Go, A. Bogaerts, W. F. Schneider and J. C. Hicks, Plasma-Catalytic Ammonia Synthesis beyond the Equilibrium Limit, ACS Catal., 2020, 10(12), 6726–6734 CrossRef CAS.
- K. H. R. Rouwenhorst, H.-H. Kim and L. Lefferts, Vibrationally Excited Activation of N2 in Plasma-Enhanced Catalytic Ammonia Synthesis: A Kinetic Analysis, ACS Sustainable Chem. Eng., 2019, 7(20), 17515–17522 CrossRef CAS.
- H.-H. Kim, Y. Teramoto, A. Ogata, H. Takagi and T. Nanba, Plasma Catalysis for Environmental Treatment and Energy Applications, Plasma Chem. Plasma Process., 2015, 36(1), 45–72 CrossRef.
- K. H. R. Rouwenhorst and L. Lefferts, Feasibility Study of Plasma-Catalytic Ammonia Synthesis for Energy Storage Applications, Catalysts, 2020, 10(9), 999 CrossRef CAS.
- K. H. R. Rouwenhorst, S. Tabak and L. Lefferts, Improving the energy yield of plasma-based NOx synthesis with in situ adsorption, React. Chem. Eng., 2024, 9(3), 528–531 RSC.
- B. N. Bayer, P. J. Bruggeman and A. Bhan, Species, Pathways, and Timescales for NH3 Formation by Low-Temperature Atmospheric Pressure Plasma Catalysis, ACS Catal., 2023, 13(4), 2619–2630 CrossRef CAS.
- B. N. Bayer, S. Raskar, I. V. Adamovich, P. J. Bruggeman and A. Bhan, Availability and reactivity of N2(v) for NH3 synthesis by plasma catalysis, Plasma Sources Sci. Technol., 2023, 32, 125005 CrossRef CAS.
- Z. Sheng, Y. Watanabe, H.-H. Kim, S. Yao and T. Nozaki, Plasma-enabled mode-selective activation of CH4 for dry reforming: first touch on the kinetic analysis, Chem. Eng. J., 2020, 399, 125751 CrossRef CAS.
- A. Bogaerts and G. Centi, Plasma Technology for CO2 Conversion: A Personal Perspective on Prospects and Gaps, Front. Energy Res., 2020, 8, 111 CrossRef.
- S. Kelly and A. Bogaerts, Nitrogen fixation in an electrode-free microwave plasma, Joule, 2021, 5(11), 3006–3030 CrossRef CAS.
- M. Ramakers, G. Trenchev, S. Heijkers, W. Wang and A. Bogaerts, Gliding Arc Plasmatron: Providing an Alternative Method for Carbon Dioxide Conversion, ChemSusChem, 2017, 10(12), 2642–2652 CrossRef CAS PubMed.
- K. Li, J.-L. Liu, X.-S. Li, X. Zhu and A.-M. Zhu, Warm plasma catalytic reforming of biogas in a heat-insulated reactor: dramatic energy efficiency and catalyst auto-reduction, Chem. Eng. J., 2016, 288, 671–679 CrossRef CAS.
- J.-L. Liu, Z. Li, J.-H. Liu, K. Li, H.-Y. Lian, X.-S. Li, X. Zhu and A.-M. Zhu, Warm-plasma catalytic reduction of CO2 with CH4, Catal. Today, 2019, 330, 54–60 CrossRef CAS.
- W. Xu, L. C. Buelens, V. V. Galvita, A. Bogaerts and V. Meynen, Improving the performance of gliding arc plasma-catalytic dry reforming via a new post-plasma tubular catalyst bed, J. CO2 Util., 2024, 83, 102820 CrossRef CAS.
- E. Delikonstantis, M. Scapinello and G. D. Stefanidis, Low energy cost conversion of methane to ethylene in a hybrid plasma-catalytic reactor system, Fuel Process. Technol., 2018, 176, 33–42 CrossRef CAS.
- A. Jafarzadeh, K. M. Bal, A. Bogaerts and E. C. Neyts, CO2 Activation on TiO2-Supported Cu5 and Ni5 Nanoclusters: Effect of Plasma-Induced Surface Charging, J. Phys. Chem. C, 2019, 123(11), 6516–6525 CrossRef CAS.
- K. M. Bal, S. Huygh, A. Bogaerts and E. C. Neyts, Effect of plasma-induced surface charging on catalytic processes: application to CO2 activation, Plasma Sources Sci. Technol., 2018, 27, 024001 CrossRef.
- A. Jafarzadeh, K. M. Bal, A. Bogaerts and E. C. Neyts, Activation of CO2 on Copper Surfaces: The Synergy between Electric Field, Surface Morphology, and Excess Electrons, J. Phys. Chem. C, 2020, 124(12), 6747–6755 CrossRef CAS.
- M. Kim, S. Biswas, I. Barraza Alvarez, P. Christopher, B. M. Wong and L. Mangolini, Nonthermal Plasma Activation of Adsorbates: The Case of CO on Pt, JACS Au, 2024, 4(8), 2979–2988 CrossRef CAS PubMed.
- K. H. R. Rouwenhorst and L. Lefferts, On the mechanism for the plasma-activated N2 dissociation on Ru surfaces, J. Phys. D: Appl. Phys., 2021, 54, 393002 CrossRef CAS.
- J. J. Mortensen, B. Hammer and J. K. Nørskovi, A theoretical study of adsorbate–adsorbate interactions on Ru(0001), Surf. Sci., 1998, 414(3), 315–329 CrossRef CAS.
- K. Van Laer and A. Bogaerts, Fluid modelling of a packed bed dielectric barrier discharge plasma reactor, Plasma Sources Sci. Technol., 2016, 25, 015002 CrossRef.
- K. Van Laer and A. Bogaerts, Influence of Gap Size and Dielectric Constant of the Packing Material on the Plasma Behaviour in a Packed Bed DBD Reactor: A Fluid Modelling Study, Plasma Processes Polym., 2016, 14(4–5), 1600129 Search PubMed.
- E. Slikboer, K. Acharya, A. Sobota, E. Garcia-Caurel and O. Guaitella, Revealing Plasma-Surface Interaction at Atmospheric Pressure: Imaging of Electric Field and Temperature inside the Targeted Material, Sci. Rep., 2020, 10(1), 2712 CrossRef CAS PubMed.
- H. H. Kim and A. Ogata, Nonthermal plasma activates catalyst: from current understanding and future prospects, Eur. Phys. J.: Appl. Phys., 2011, 55, 13806 CrossRef.
- O. Guaitella, F. Thevenet, E. Puzenat, C. Guillard and A. Rousseau, C2H2 oxidation by plasma/TiO2 combination: influence of the porosity, and photocatalytic mechanisms under plasma exposure, Appl. Catal., B, 2008, 80(3–4), 296–305 CrossRef CAS.
- A. E. Wallis, J. C. Whitehead and K. Zhang, Plasma-assisted catalysis for the destruction of CFC-12 in atmospheric pressure gas streams using TiO2, Catal. Lett., 2007, 113(1–2), 29–33 CrossRef CAS.
- D. Mei, X. Zhu, Y.-L. He, J. D. Yan and X. Tu, Plasma-assisted conversion of CO2 in a dielectric barrier discharge reactor: understanding the effect of packing materials, Plasma Sources Sci. Technol., 2014, 24, 015011 Search PubMed.
- J. Kim, M. S. Abbott, D. B. Go and J. C. Hicks, Enhancing C–H Bond Activation of Methane via Temperature-Controlled, Catalyst–Plasma Interactions, ACS Energy Lett., 2016, 1(1), 94–99 CrossRef CAS.
- J. Van Turnhout, D. Aceto, A. Travert, P. Bazin, F. Thibault-Starzyk, A. Bogaerts and F. Azzolina-Jury, Observation of surface species in plasma-catalytic dry reforming of methane in a novel atmospheric pressure dielectric barrier discharge in situ IR cell, Catal. Sci. Technol., 2022, 12(22), 6676–6686 Search PubMed.
- K. H. R. Rouwenhorst, H. G. B. Burbach, D. W. Vogel, J. Núñez Paulí, B. Geerdink and L. Lefferts, Plasma-catalytic ammonia synthesis beyond thermal equilibrium on Ru-based catalysts in non-thermal plasma, Catal. Sci. Technol., 2021, 11(8), 2834–2843 Search PubMed.
- R. Michiels, N. Gerrits, E. Neyts and A. Bogaerts, Plasma Catalysis Modeling: How Ideal Is Atomic Hydrogen for Eley-Rideal?, J. Phys. Chem. C, 2024, 128(27), 11196–11209 CrossRef CAS.
- B. N. Bayer, P. J. Bruggeman and A. Bhan, NO formation by N2/O2 plasma catalysis: the impact of surface reactions, gas-phase reactions, and mass transport, Chem. Eng. J., 2024, 482, 149041 CrossRef CAS.
- V. Guerra, A. Tejero-del-Caz, C. D. Pintassilgo and L. L. Alves, Modelling N2–O2 plasmas: volume and surface kinetics, Plasma Sources Sci. Technol., 2019, 28, 073001 CrossRef CAS.
- M. Albrechts, I. Tsonev and A. Bogaerts, Investigation of O atom kinetics in O2 plasma and its afterglow, Plasma Sources Sci. Technol., 2024, 33, 045017 CrossRef CAS.
- H.-H. Kim, Y. Teramoto, N. Negishi and A. Ogata, A multidisciplinary approach to understand the interactions of nonthermal plasma and catalyst: a review, Catal. Today, 2015, 256, 13–22 CAS.
- J. Jiang and P. J. Bruggeman, Tuning plasma parameters to control reactive species fluxes to substrates in the context of plasma catalysis, J. Phys. D: Appl. Phys., 2021, 54, 214005 CrossRef CAS.
- I. Tsonev, O. Biondo and A. Bogaerts, Simulation of a pulsed CO2 plasma based on a six-temperature energy approach, Plasma Sources Sci. Technol., 2025, 34, 015014 CAS.
- I. Michielsen, Y. Uytdenhouwen, J. Pype, B. Michielsen, J. Mertens, F. Reniers, V. Meynen and A. Bogaerts, CO2 dissociation in a packed bed DBD reactor: first steps towards a better understanding of plasma catalysis, Chem. Eng. J., 2017, 326, 477–488 CAS.
- C. Ndayirinde, Y. Gorbanev, R.-G. Ciocarlan, R. De Meyer, A. Smets, E. Vlasov, S. Bals, P. Cool and A. Bogaerts, Plasma-catalytic ammonia synthesis: packed catalysts act as plasma modifiers, Catal. Today, 2023, 419, 114156 CAS.
- R. De Meyer, Y. Gorbanev, R.-G. Ciocarlan, P. Cool, S. Bals and A. Bogaerts, Importance of plasma discharge characteristics in plasma catalysis: dry reforming of methane vs. ammonia synthesis, Chem. Eng. J., 2024, 488, 150838 CAS.
- J. Wang, K. Zhang, M. Mertens, A. Bogaerts and V. Meynen, Plasma-based dry reforming of methane in a dielectric barrier discharge reactor: importance of uniform (sub)micron packings/catalysts to enhance the performance, Appl. Catal., B, 2023, 337, 122977 CAS.
- M. T. Colomer, Special Issue “Design, Synthesis and Applications of Macroporous, Mesoporous, and Microporous Materials”, Int. J. Mol. Sci., 2024, 25(13), 7127 CAS.
- J. Wang, K. Zhang, A. Bogaerts and V. Meynen, 3D porous catalysts for Plasma-Catalytic dry reforming of Methane: How does the pore size affect the Plasma-Catalytic Performance?, Chem. Eng. J., 2023, 464, 142574 CrossRef CAS.
- Q.-Z. Zhang and A. Bogaerts, Plasma streamer propagation in structured catalysts, Plasma Sources Sci. Technol., 2018, 27(10), 105013 CrossRef.
- S. Sato, K. Hensel, H. Hayashi, K. Takashima and A. Mizuno, Honeycomb discharge for diesel exhaust cleaning, J. Electrost., 2009, 67(2–3), 77–83 CrossRef CAS.
- K. Hensel, S. Sato and A. Mizuno, Sliding Discharge Inside Glass Capillaries, IEEE Trans. Plasma Sci., 2008, 36(4), 1282–1283 CAS.
- K. Hensel, Microdischarges in ceramic foams and honeycombs, Eur. Phys. J. D, 2009, 54(2), 141–148 CrossRef CAS.
- A. Mizuno, Generation of non-thermal plasma combined with catalysts and their application in environmental technology, Catal. Today, 2013, 211, 2–8 CrossRef CAS.
- D. B. Nguyen, S. Shirjana, M. M. Hossain, I. Heo and Y. S. Mok, Effective generation of atmospheric pressure plasma in a sandwich-type honeycomb monolith reactor by humidity control, Chem. Eng. J., 2020, 401, 125970 CrossRef CAS.
- D. Khunda, S. Li, N. Cherkasov, A. Chaffee and E. V. Rebrov, Scaling Down the Great Egypt Pyramids to Enhance CO2 Splitting in a Micro DBD Reactor, Plasma Chem. Plasma Process., 2023, 43(6), 2017–2034 CAS.
- S. Xie, Y. He, D. Yuan, Z. Wang, S. Kumar, Y. Zhu and K. Cen, The effects of gas flow pattern on the generation of ozone in surface dielectric barrier discharge, Plasma Sci. Technol., 2019, 21, 055505 CrossRef CAS.
- H. Jakob, M. Paliwoda, J. L. Rovey and M. Kim, Surface DBD plasma microbubble reactor for degrading methylene blue, Phys. Scr., 2023, 98, 025603 CrossRef CAS.
- L.-B. Di, X.-S. Li, C. Shi, Y. Xu, D.-Z. Zhao and A.-M. Zhu, Atmospheric-pressure plasma CVD of TiO2 photocatalytic films using surface dielectric barrier discharge, J. Phys. D: Appl. Phys., 2009, 42, 032001 CrossRef.
- N. García-Moncada, G. van Rooij, T. Cents and L. Lefferts, Catalyst-assisted DBD plasma for coupling of methane: minimizing carbon-deposits by structured reactors, Catal. Today, 2021, 369, 210–220 CrossRef.
- J. W. Gregory, N. Pourali, Y. Gong, R. I. Walton, V. Hessel and E. V. Rebrov, Development of an Ir/TiO2 catalytic coating for plasma assisted hydrogenation of CO2 to CH4, Appl. Catal., A, 2024, 675, 119639 CrossRef CAS.
- N. Peters, L. Schücke, K. Ollegott, C. Oberste-Beulmann, P. Awakowicz and M. Muhler, Catalyst-enhanced plasma oxidation of n-butane over α-MnO2 in a temperature-controlled twin surface dielectric barrier discharge reactor, Plasma Processes Polym., 2021, 18(4), 2000127 CrossRef CAS.
- Z. Falkenstein, Influence of ultraviolet illumination on microdischarge behavior in dry and humid N2, O2, air, and Ar/O2: the Joshi effect, J. Appl. Phys., 1997, 81(9), 5975–5979 CrossRef CAS.
- Y. Ji, D. Peng, X. Yuan, Z. Wang, Y. Zhang, R. Li, H. Huang, J. Li, D. Ye and J. Wu, Enhancing One-Step Methanol Production from Methane in a Double Dielectric Barrier Discharge Reactor with Glow-like Discharge, Ind. Eng. Chem. Res., 2024, 63(40), 17025–17037 CrossRef CAS.
- W. Zhou, D. Zhang, X. Duan, X. Zhu, F. Liu and Z. Fang, Effect of dielectric material on the uniformity of nanosecond pulsed dielectric barrier discharge, Plasma Sci. Technol., 2024, 26, 094008 CrossRef CAS.
- T. Shao, R. Wang, C. Zhang and P. Yan, Atmospheric–pressure pulsed discharges and plasmas: mechanism, characteristics and applications, High Voltage, 2018, 3(1), 14–20 CrossRef.
- M. Kogoma and S. Okazaki, Raising of ozone formation efficiency in a homogeneous glow discharge plasma at atmospheric pressure, J. Phys. D: Appl. Phys., 1994, 27(9), 1985–1987 CrossRef CAS.
- J. R. Roth, S. Nourgostar and T. A. Bonds, The One Atmosphere Uniform Glow Discharge Plasma (OAUGDP)—A Platform Technology for the 21st Century, IEEE Trans. Plasma Sci., 2007, 35(2), 233–250 CrossRef.
- Y. Gao, R. Zhou, B. Chen, L. Xiao, X. Zhao, J. Sun, R. Zhou, J. Zhang and Z. Liu, Plasma Catalysis-Driven Decomposition of CO2: Optimizing Energy Distribution with NiCo–CuO Catalyst, ACS Sustainable Chem. Eng., 2024, 12(29), 10993–11005 CrossRef CAS.
- X. Feng, H. Liu, C. He, Z. Shen and T. Wang, Synergistic effects and mechanism of a non-thermal plasma catalysis system in volatile organic compound removal: a review, Catal. Sci. Technol., 2018, 8(4), 936–954 RSC.
- B. Jiang, K. Xu, J. Li, H. Lu, X. Fei, X. Yao, S. Yao and Z. Wu, Effect of supports on plasma catalytic decomposition of toluene using in situ plasma DRIFTS, J. Hazard. Mater., 2021, 405, 124203 CrossRef CAS PubMed.
- R. Liu, H. Song, B. Li, X. Li and T. Zhu, Simultaneous removal of toluene and styrene by non-thermal plasma-catalysis: effect of VOCs interaction and system configuration, Chemosphere, 2021, 263, 127893 CrossRef CAS PubMed.
- P. Navascues, J. Garrido-Garcia, J. Cotrino, A. R. Gonzalez-Elipe and A. Gomez-Ramirez, Incorporation of a Metal Catalyst for the Ammonia Synthesis in a Ferroelectric Packed-Bed Plasma Reactor: Does It Really Matter?, ACS Sustainable Chem. Eng., 2023, 11(9), 3621–3632 CrossRef CAS PubMed.
- K. van ‘t Veer, Y. Engelmann, F. Reniers and A. Bogaerts, Plasma-Catalytic Ammonia Synthesis in a DBD Plasma: Role of Microdischarges and Their Afterglows, J. Phys. Chem. C, 2020, 124(42), 22871–22883 CrossRef.
- R. Snoeckx, R. Aerts, X. Tu and A. Bogaerts, Plasma-Based Dry Reforming: A Computational Study Ranging from the Nanoseconds to Seconds Time Scale, J. Phys. Chem. C, 2013, 117(10), 4957–4970 CAS.
- L. Wang, Y. Yi, H. Guo and X. Tu, Atmospheric Pressure and Room Temperature Synthesis of Methanol through Plasma-Catalytic Hydrogenation of CO2, ACS Catal., 2017, 8(1), 90–100 CrossRef.
- L. Wang, Y. Wang, L. Fan, H. Xu, B. Liu, J. Zhang, Y. Zhu and X. Tu, Direct conversion of CH4 and CO2 to alcohols using plasma catalysis over Cu/Al(OH)3 catalysts, Chem. Eng. J., 2023, 466, 143347 CAS.
- D. Li, V. Rohani, F. Fabry, A. Parakkulam Ramaswamy, M. Sennour and L. Fulcheri, Direct conversion of CO2 and CH4 into liquid chemicals by plasma-catalysis, Appl. Catal., B, 2020, 261, 118228 CAS.
- J. Li, L. Dou, Y. Gao, X. Hei, F. Yu and T. Shao, Revealing the active sites of the structured Ni-based catalysts for one-step CO2/CH4 conversion into oxygenates by plasma-catalysis, J. CO2 Util., 2021, 52, 101675 CAS.
- M. Ronda-Lloret, Y. Wang, P. Oulego, G. Rothenberg, X. Tu and N. R. Shiju, CO(2) Hydrogenation at Atmospheric Pressure and Low Temperature Using Plasma-Enhanced Catalysis over Supported Cobalt Oxide Catalysts, ACS Sustainable Chem. Eng., 2020, 8(47), 17397–17407 CrossRef CAS PubMed.
- Z. Cui, S. Meng, Y. Yi, A. Jafarzadeh, S. Li, E. C. Neyts, Y. Hao, L. Li, X. Zhang, X. Wang and A. Bogaerts, Plasma-Catalytic Methanol Synthesis from CO2 Hydrogenation over a Supported Cu Cluster Catalyst: Insights into the Reaction Mechanism, ACS Catal., 2022, 12(2), 1326–1337 CrossRef CAS.
- S. Meng, L. Wu, M. Liu, Z. Cui, Q. Chen, S. Li, J. Yan, L. Wang, X. Wang, J. Qian, H. Guo, J. Niu, A. Bogaerts and Y. Yi, Plasma-driven CO2 hydrogenation to CH3OH over Fe2O3/γ-Al2O3 catalyst, AIChE J., 2023, 69(10), e18154 CrossRef CAS.
- R. K. Masumbuko, N. Kobayashi, Y. Itaya and A. Suami, Enhanced methanol selectivity and synthesis in a non-catalytic dielectric barrier discharge (DBD) plasma reactor, Chem. Eng. Sci., 2024, 287, 119698 CrossRef CAS.
- Q. Chen, S. Meng, R. Liu, X. Zhai, X. Wang, L. Wang, H. Guo and Y. Yi, Plasma-catalytic CO2 hydrogenation to methanol over CuO–MgO/Beta catalyst with high selectivity, Appl. Catal., B, 2024, 342, 123422 CrossRef CAS.
- N. Pinhão, A. Moura, J. B. Branco and J. Neves, Influence of gas expansion on process parameters in non-thermal plasma plug-flow reactors: a study applied to dry reforming of methane, Int. J. Hydrogen Energy, 2016, 41(22), 9245–9255 CrossRef.
- B. Wanten, R. Vertongen, R. De Meyer and A. Bogaerts, Plasma-based CO2 conversion: How to correctly analyze the performance?, J. Energy Chem., 2023, 86, 180–196 CrossRef CAS.
- J. Zhu, D. Ciolca, L. Liu, A. Parastaev, N. Kosinov and E. J. M. Hensen, Flame Synthesis of Cu/ZnO–CeO(2) Catalysts: Synergistic Metal–Support Interactions Promote CH(3)OH Selectivity in CO(2) Hydrogenation, ACS Catal., 2021, 11(8), 4880–4892 CrossRef CAS PubMed.
- B. Liang, J. Ma, X. Su, C. Yang, H. Duan, H. Zhou, S. Deng, L. Li and Y. Huang, Investigation on Deactivation of Cu/ZnO/Al2O3 Catalyst for CO2 Hydrogenation to Methanol, Ind. Eng. Chem. Res., 2019, 58(21), 9030–9037 CrossRef CAS.
- J. Wu, M. Saito, M. Takeuchi and T. Watanabe, The stability of Cu/ZnO-based catalysts in methanol synthesis from a CO2-rich feed and from a CO-rich feed, Appl. Catal., A, 2001, 218(1–2), 235–240 CrossRef CAS.
- Y. Wang, S. Kattel, W. Gao, K. Li, P. Liu, J. G. Chen and H. Wang, Exploring the ternary interactions in Cu–ZnO–ZrO(2) catalysts for efficient CO(2) hydrogenation to methanol, Nat. Commun., 2019, 10(1), 1166 CrossRef PubMed.
- F. Arena, G. Italiano, K. Barbera, S. Bordiga, G. Bonura, L. Spadaro and F. Frusteri, Solid-state interactions, adsorption sites and functionality of Cu–ZnO/ZrO2 catalysts in the CO2 hydrogenation to CH3OH, Appl. Catal., A, 2008, 350(1), 16–23 CrossRef CAS.
- P. Gao, F. Li, N. Zhao, F. Xiao, W. Wei, L. Zhong and Y. Sun, Influence of modifier (Mn, La, Ce, Zr and Y) on the performance of Cu/Zn/Al catalysts via hydrotalcite-like precursors for CO2 hydrogenation to methanol, Appl. Catal., A, 2013, 468, 442–452 CrossRef CAS.
- S. Li, J. Sun, Y. Gorbanev, K. van’t Veer, B. Loenders, Y. Yi, T. Kenis, Q. Chen and A. Bogaerts, Plasma-Assisted Dry Reforming of CH4: How Small Amounts of O2 Addition Can Drastically Enhance the Oxygenate Production—Experiments and Insights from Plasma Chemical Kinetics Modeling, ACS Sustainable Chem. Eng., 2023, 11(42), 15373–15384 CrossRef CAS.
- L. Lefferts, Leveraging Expertise in Thermal Catalysis to Understand Plasma Catalysis, Angew. Chem., Int. Ed., 2024, 136(10), e202305322 CrossRef.
- P. Navascués, J. M. Obrero-Pérez, J. Cotrino, A. R. González-Elipe and A. Gómez-Ramírez, Isotope Labelling for Reaction Mechanism Analysis in DBD Plasma Processes, Catalysts, 2019, 9(1), 45 Search PubMed.
- A. Parastaev, W. F. L. M. Hoeben, B. E. J. M. van Heesch, N. Kosinov and E. J. M. Hensen, Temperature-programmed plasma surface reaction: an approach to determine plasma-catalytic performance, Appl. Catal., B, 2018, 239, 168–177 CAS.
- L. L. Alves, M. M. Becker, J. van Dijk, T. Gans, D. B. Go, K. Stapelmann, J. Tennyson, M. M. Turner and M. J. Kushner, Foundations of plasma standards, Plasma Sources Sci. Technol., 2023, 32, 023001 Search PubMed.
- M. M. Turner, Uncertainty and error in complex plasma chemistry models, Plasma Sources Sci. Technol., 2015, 24, 035027 Search PubMed.
- A. Berthelot and A. Bogaerts, Modeling of CO2 plasma: effect of uncertainties in the plasma chemistry, Plasma Sources Sci. Technol., 2017, 26, 115002 Search PubMed.
- W. Wang, A. Berthelot, Q. Zhang and A. Bogaerts, Modelling of plasma-based dry reforming: how do uncertainties in the input data affect the calculation results?, J. Phys. D: Appl. Phys., 2018, 51, 204003 Search PubMed.
- K. H. R. Rouwenhorst, S. Mani and L. Lefferts, Improving the Energy Yield of Plasma-Based Ammonia Synthesis with In Situ Adsorption, ACS Sustainable Chem. Eng., 2022, 10(6), 1994–2000 CAS.
- Y. Wang, W. Yang, S. Xu, S. Zhao, G. Chen, A. Weidenkaff, C. Hardacre, X. Fan, J. Huang and X. Tu, Shielding Protection by Mesoporous Catalysts for Improving Plasma-Catalytic Ambient Ammonia Synthesis, J. Am. Chem. Soc., 2022, 144(27), 12020–12031 CAS.
- S. Li, M. Ongis, G. Manzolini and F. Gallucci, Non-thermal plasma-assisted capture and conversion of CO2, Chem. Eng. J., 2021, 410, 128335 CrossRef CAS.
- S. Li and F. Gallucci, CO2 capture and activation with a plasma-sorbent system, Chem. Eng. J., 2022, 430, 132979 CrossRef CAS.
- R. Vertongen, G. De Felice, H. van den Bogaard, F. Gallucci, A. Bogaerts and S. Li, Sorption-Enhanced Dry Reforming of Methane in a DBD Plasma Reactor for Single-Stage Carbon Capture and Utilization, ACS Sustainable Chem. Eng., 2024, 12(29), 10841–10853 CrossRef CAS PubMed.
- G. Giammaria and L. Lefferts, Synergy between dielectric barrier discharge plasma and calcium oxide for reverse water gas shift, Chem. Eng. J., 2020, 392, 123806 CrossRef CAS.
- S. Mori, N. Matsuura, L. L. Tun and M. Suzuki, Direct Synthesis of Carbon Nanotubes from Only CO2 by a Hybrid Reactor of Dielectric Barrier Discharge and Solid Oxide Electrolyser Cell, Plasma Chem. Plasma Process., 2015, 36(1), 231–239 CrossRef.
- F. Gorky, V. Storr, G. Jones, A. Nambo, J. B. Jasinski and M. L. Carreon, Performance and Enhanced Efficiency Induced by Cold Plasma on SAPO-34 Membranes for CO(2) and CH(4) Mixtures, Membranes, 2024, 14(8), 178 CrossRef CAS PubMed.
- S. Kambara, Y. Hayakawa, Y. Inoue and T. Miura, Hydrogen Production from Ammonia Using a Plasma Membrane Reactor, J. Sustainable Dev. Energy, Water Environ. Syst., 2016, 4(2), 193–202 CrossRef.
- M. El-Shafie, S. Kambara and Y. Hayakawa, Energy and exergy analysis of hydrogen production from ammonia decomposition systems using non-thermal plasma, Int. J. Hydrogen Energy, 2021, 46(57), 29361–29375 CrossRef CAS.
- M. El-Shafie, Hydrogen separation using palladium-based membranes: assessment of H2 separation in a catalytic plasma membrane reactor, Int. J. Energy Res., 2021, 46(3), 3572–3587 CrossRef.
- Y. Hayakawa, T. Miura, K. Shizuya, S. Wakazono, K. Tokunaga and S. Kambara, Hydrogen production system combined with a catalytic reactor and a plasma membrane reactor from ammonia, Int. J. Hydrogen Energy, 2019, 44(20), 9987–9993 CrossRef CAS.
- Y. Hayakawa, S. Kambara and T. Miura, Hydrogen production from ammonia by the plasma membrane reactor, Int. J. Hydrogen Energy, 2020, 45(56), 32082–32088 CrossRef CAS.
- R. Antunes, K. Wiegers, A. Hecimovic, C. K. Kiefer, S. Buchberger, A. Meindl, T. Schiestel, A. Schulz, M. Walker and U. Fantz, Proof of Concept for O2 Removal with Multiple LCCF Membranes Accommodated in the Effluent of a CO2 Plasma Torch, ACS Sustainable Chem. Eng., 2023, 11(44), 15984–15993 Search PubMed.
- A. Rashid, H. Lim, D. Plaz, G. Escobar Cano, M. Bresser, K. S. Wiegers, G. Confalonieri, S. Baek, G. Chen, A. Feldhoff, A. Schulz, A. Weidenkaff and M. Widenmeyer, Hydrogen-Tolerant La(0.6)Ca(0.4)Co(0.2)Fe(0.8)O(3−d) Oxygen Transport Membranes from Ultrasonic Spray Synthesis for Plasma-Assisted CO(2) Conversion, Membranes, 2023, 13(11), 875 Search PubMed.
- E. Delikonstantis, M. Scapinello, V. Singh, H. Poelman, C. Montesano, L. M. Martini, P. Tosi, G. B. Marin, K. M. Van Geem, V. V. Galvita and G. D. Stefanidis, Exceeding Equilibrium CO2 Conversion by Plasma-Assisted Chemical Looping, ACS Energy Lett., 2022, 7(6), 1896–1902 CrossRef CAS.
- Y. Long, X. Wang, H. Zhang, K. Wang, W. L. Ong, A. Bogaerts, K. Li, C. Lu, X. Li, J. Yan, X. Tu and H. Zhang, Plasma Chemical Looping: Unlocking High-Efficiency CO(2) Conversion to Clean CO at Mild Temperatures, JACS Au, 2024, 4(7), 2462–2473 CrossRef CAS PubMed.
- P. M. Barboun, H. O. Otor, H. Ma, A. Goswami, W. F. Schneider and J. C. Hicks, Plasma-Catalyst Reactivity Control of Surface Nitrogen Species through Plasma-Temperature-Programmed Hydrogenation to Ammonia, ACS Sustainable Chem. Eng., 2022, 10(48), 15741–15748 CrossRef CAS.
- D. J. Haycock, R. J. Clarke, D. B. Go, W. F. Schneider and J. C. Hicks, Fundamental insights and emerging opportunities in plasma catalysis for light alkane conversion, Curr. Opin. Green Sustainable Chem., 2025, 51, 100987 CrossRef.
- R. K. Sharma, H. Patel, U. Mushtaq, V. Kyriakou, G. Zafeiropoulos, F. Peeters, S. Welzel, M. C. M. van de Sanden and M. N. Tsampas, Plasma Activated Electrochemical Ammonia Synthesis from Nitrogen and Water, ACS Energy Lett., 2020, 6(2), 313–319 CrossRef.
- L. Hollevoet, F. Jardali, Y. Gorbanev, J. Creel, A. Bogaerts and J. A. Martens, Towards Green Ammonia Synthesis through Plasma-Driven Nitrogen Oxidation and Catalytic Reduction, Angew. Chem., 2020, 132(52), 24033–24037 CrossRef.
- L. Hollevoet, E. Vervloessem, Y. Gorbanev, A. Nikiforov, N. De Geyter, A. Bogaerts and J. A. Martens, Energy-Efficient Small-Scale Ammonia Synthesis Process with Plasma-Enabled Nitrogen Oxidation and Catalytic Reduction of Adsorbed NO(x), ChemSusChem, 2022, 15(10), e202102526 CrossRef CAS PubMed.
- J. Sun, D. Alam, R. Daiyan, H. Masood, T. Zhang, R. Zhou, P. J. Cullen, E. C. Lovell, A. Jalili and R. Amal, A hybrid plasma electrocatalytic process for sustainable ammonia production, Energy Environ. Sci., 2021, 14(2), 865–872 RSC.
- L. Li, C. Tang, X. Cui, Y. Zheng, X. Wang, H. Xu, S. Zhang, T. Shao, K. Davey and S. Z. Qiao, Efficient Nitrogen Fixation to Ammonia through Integration of Plasma Oxidation with Electrocatalytic Reduction, Angew. Chem., Int. Ed., 2021, 133(25), 14250–14256 CrossRef.
- A. Wu, J. Yang, B. Xu, X.-Y. Wu, Y. Wang, X. Lv, Y. Ma, A. Xu, J. Zheng, Q. Tan, Y. Peng, Z. Qi, H. Qi, J. Li, Y. Wang, J. Harding, X. Tu, A. Wang, J. Yan and X. Li, Direct ammonia synthesis from the air via gliding arc plasma integrated with single atom electrocatalysis, Appl. Catal., B, 2021, 299, 120667 CrossRef CAS.
- I. Muzammil, Y.-N. Kim, H. Kang, D. K. Dinh, S. Choi, C. Jung, Y.-H. Song, E. Kim, J. M. Kim and D. H. Lee, Plasma Catalyst-Integrated System for Ammonia Production from H2O and N2 at Atmospheric Pressure, ACS Energy Lett., 2021, 6(8), 3004–3010 CrossRef CAS.
- V. Veng, S. A. Ibrahim, B. Tabu, E. Simasiku, J. Landis, J. H. Mack, F. Che and J. P. Trelles, Ammonia Synthesis via Membrane Dielectric-Barrier Discharge Reactor Integrated with Metal Catalyst, Plasma Chem. Plasma Process., 2024, 44(6), 2031–2055 CrossRef CAS.
- Z. Li, T. Yang, S. Yuan, Y. Yin, E. J. Devid, Q. Huang, D. Auerbach and A. W. Kleyn, Boudouard reaction driven by thermal plasma for efficient CO2 conversion and energy storage, J. Energy Chem., 2020, 45, 128–134 Search PubMed.
- J. Huang, H. Zhang, Q. Tan, L. Li, R. Xu, Z. Xu and X. Li, Enhanced conversion of CO2 into O2-free fuel gas via the Boudouard reaction with biochar in an atmospheric plasmatron, J. CO2 Util., 2021, 45, 101429 CAS.
- F. Girard-Sahun, O. Biondo, G. Trenchev, G. van Rooij and A. Bogaerts, Carbon bed post-plasma to enhance the CO2 conversion and remove O2 from the product stream, Chem. Eng. J., 2022, 442, 136268 CAS.
- C. O’Modhrain, Y. Gorbanev and A. Bogaerts, Post-plasma carbon bed design for CO2 conversion: does size and insulation matter?, J. Energy Chem., 2025, 104, 312–323 CrossRef.
- O. Biondo, K. Wang, H. Zhang and A. Bogaerts, Coupling a CO2 plasma with a carbon bed: the closer the better, Chem. Eng. J., 2025, 507, 160190 CAS.
- A. I. Stankiewicz and J. A. Moulijn, Process intensification: transforming chemical engineering, Chem. Eng. Prog., 2000, 96(1), 22–34 CAS.
- A. I. Stankiewicz and P. Yan, 110th Anniversary: The Missing Link Unearthed: Materials and Process Intensification, Ind. Eng. Chem. Res., 2019, 58(22), 9212–9222 CAS.
- I. Fedirchyk, I. Tsonev, R. Quiroz Marnef and A. Bogaerts, Plasma-assisted NH3 cracking in warm plasma reactors for green H2 production, Chem. Eng. J., 2024, 499, 155946 CAS.
- I. Tsonev, H. Ahmadi Eshtehardi, M.-P. Delplancke and A. Bogaerts, Importance of geometric effects in scaling up energy-efficient plasma-based nitrogen fixation, Sustainable Energy Fuels, 2024, 8(10), 2191–2209 CAS.
- J. Osorio-Tejada, M. Escriba-Gelonch, R. Vertongen, A. Bogaerts and V. Hessel, CO(2) conversion to CO via plasma and electrolysis: a techno-economic and energy cost analysis, Energy Environ. Sci., 2024, 17(16), 5833–5853 CAS.
| This journal is © The Royal Society of Chemistry 2025 |