
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvancements in dinitrogen activation for catalytic breakthroughs
Vamsi Vikram Gande†
a,
Nishithan C. Kani†a,
Ishita Goyal†a,
Rohit Chauhan† a,
Yancun Qi†a,
Samuel A. Olusegun†b,
Joseph A. Gauthier*b and
Meenesh R. Singh*a
a,
Yancun Qi†a,
Samuel A. Olusegun†b,
Joseph A. Gauthier*b and
Meenesh R. Singh*a
aDepartment of Chemical Engineering, University of Illinois Chicago, 929 W. Taylor St., Chicago, Illinois 60607, USA. E-mail: mrsingh@uic.edu; Tel: +1 (312) 413-7673
bDepartment of Chemical Engineering, Texas Tech University, Lubbock, Texas 79409, USA. E-mail: joe.gauthier@ttu.edu; Tel: +1 806 742 3552
First published on 24th April 2025
Abstract
Activation and catalytic transformation of dinitrogen (N2) remains a grand challenge at the intersection of global food security, sustainable energy, and chemical manufacturing. The remarkable strength of the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond poses formidable thermodynamic and kinetic barriers, driving reliance on the century-old Haber–Bosch process-an energy-intensive route that consumes substantial fossil fuels. Recent advances underscore a growing shift toward alternative strategies, including biological and enzymatic pathways inspired by nitrogenase, homogeneous catalysis through transition-metal complexes, plasma-assisted reactions leveraging high-energy species, and diverse electrochemical or thermo-electrochemical methods integrating renewable power. Key breakthroughs in catalyst design, from metal nitrides and single-atom catalysts to next-generation perovskite oxides, highlight the importance of targeted bond weakening, electron back-donation, and multi-electron/proton transfer steps. Concurrently, mechanistic insights gleaned from in situ spectroscopy, density functional theory, and machine learning-guided screening are refining our understanding of molecular orbital interactions and reaction intermediates. Looking ahead, the N2 activation field seeks to unite high efficiency with lower energy footprints by tailoring catalysts for mild conditions, exploring hydrogen sources beyond conventional H2, and adopting process intensification strategies to curb carbon emissions. By bridging fundamental discoveries with scalable engineering, future research should aim to deliver cost-effective, low-carbon nitrogen fixation, reshaping the global nitrogen economy and paving the way toward sustainable ammonia production and novel nitrogen-based chemicals.
N bond poses formidable thermodynamic and kinetic barriers, driving reliance on the century-old Haber–Bosch process-an energy-intensive route that consumes substantial fossil fuels. Recent advances underscore a growing shift toward alternative strategies, including biological and enzymatic pathways inspired by nitrogenase, homogeneous catalysis through transition-metal complexes, plasma-assisted reactions leveraging high-energy species, and diverse electrochemical or thermo-electrochemical methods integrating renewable power. Key breakthroughs in catalyst design, from metal nitrides and single-atom catalysts to next-generation perovskite oxides, highlight the importance of targeted bond weakening, electron back-donation, and multi-electron/proton transfer steps. Concurrently, mechanistic insights gleaned from in situ spectroscopy, density functional theory, and machine learning-guided screening are refining our understanding of molecular orbital interactions and reaction intermediates. Looking ahead, the N2 activation field seeks to unite high efficiency with lower energy footprints by tailoring catalysts for mild conditions, exploring hydrogen sources beyond conventional H2, and adopting process intensification strategies to curb carbon emissions. By bridging fundamental discoveries with scalable engineering, future research should aim to deliver cost-effective, low-carbon nitrogen fixation, reshaping the global nitrogen economy and paving the way toward sustainable ammonia production and novel nitrogen-based chemicals.
Broader contextDinitrogen (N2) activation is at the heart of a sustainable future, influencing global agriculture, energy, and industrial chemistry. The current state-of-the-art, dominated by the Haber–Bosch process, is energy-intensive and heavily reliant on fossil fuels, contributing to high carbon emissions. The imperative to transition toward low-carbon, energy-efficient nitrogen fixation technologies has sparked interest in alternative pathways, including biological, electrochemical, plasma-assisted, and thermochemical approaches. These emerging methods promise greener nitrogen fixation by leveraging advances in catalysis, mechanistic understanding, and computational modeling. Recent breakthroughs in catalyst design—such as single-atom catalysts, transition-metal nitrides, and perovskite oxides—have enabled new strategies for breaking the strong NN bond under mild conditions. Additionally, in situ spectroscopy, density functional theory (DFT), and machine learning are refining our understanding of catalytic pathways, helping to optimize activity and selectivity. Exploring alternative hydrogen sources, integrating renewable energy, and designing multi-functional catalytic systems will be crucial for next-generation nitrogen activation. This review aligns with global efforts to decarbonize chemical manufacturing and reduce dependence on fossil fuels. By bridging molecular-level insights with scalable engineering solutions, advancements in N2 activation can pave the way for low-energy ammonia synthesis and novel nitrogen-based chemicals, reshaping the global nitrogen economy.
|
1. Introduction
Nitrogen, which constitutes most of the Earth's atmosphere, predominantly exists as dinitrogen (N2), a molecule characterized by its exceptional stability due to the strength of the triple bond between nitrogen atoms (∼9.79![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) eV). This strong bond presents significant challenges for nitrogen activation, converting N2 into reactive or biologically valuable forms such as ammonia (NH3).1 The inert nature of N2 arises from both thermodynamic and kinetic factors. Thermodynamically, the formation of nitrogen-containing compounds, such as nitrogen oxides, is challenging due to their positive standard free energies of formation, making them less stable than N2 itself. Kinetically, N2 activation is sluggish in reactions with most reagents, necessitating high energy input or catalytic intervention. A prominent example is the industrial synthesis of NH3 from N2 and H2, which requires a highly reactive catalyst and elevated temperatures to overcome these barriers. Addressing the challenge of N2 activation is crucial for advancing sustainable agriculture, as NH3 and NO3− are cornerstones of fertilizers, and their production is currently reliant on energy-intensive processes like the Haber–Bosch method. This review highlights the importance of N2 activation, explores the challenges involved, and discusses emerging methods to facilitate its chemical transformation.
eV). This strong bond presents significant challenges for nitrogen activation, converting N2 into reactive or biologically valuable forms such as ammonia (NH3).1 The inert nature of N2 arises from both thermodynamic and kinetic factors. Thermodynamically, the formation of nitrogen-containing compounds, such as nitrogen oxides, is challenging due to their positive standard free energies of formation, making them less stable than N2 itself. Kinetically, N2 activation is sluggish in reactions with most reagents, necessitating high energy input or catalytic intervention. A prominent example is the industrial synthesis of NH3 from N2 and H2, which requires a highly reactive catalyst and elevated temperatures to overcome these barriers. Addressing the challenge of N2 activation is crucial for advancing sustainable agriculture, as NH3 and NO3− are cornerstones of fertilizers, and their production is currently reliant on energy-intensive processes like the Haber–Bosch method. This review highlights the importance of N2 activation, explores the challenges involved, and discusses emerging methods to facilitate its chemical transformation.
1.1. Need for N2 activation (i.e., to form NH3 and nitrogen oxyanions)
Nitrogen activation is essential for converting atmospheric N2 into biologically accessible forms such as nitrogen oxyanions (e.g., nitrates (NO3−)) and NH3. This process is critical for several reasons, as outlined below, along with some historical significance:The standard free energy formation of ammonia is −16.48 kJ mol−1, and thus, the equilibrium constant of the reaction.
 | (1) |

This equilibrium constant suggests a favorable synthesis for NH3 at room temperature if equilibrium were achievable at reasonable timescales. However, the extremely stable N–N bond leads to essentially zero forward rate of reaction in an uncatalyzed process at standard conditions despite the favorable equilibrium constant.3
While the well-established and productive Haber–Bosch process is not expected to be replaced in the immediate future, the reliance on d-block metals as catalysts for the N2 reduction reaction (N2RR) in both industry and nature has prompted the exploration of alternative strategies involving transition metals since the mid-1960s.5 The critical role of ammonia in sustaining human life and societal progress has driven the quest to discover the requirements for efficient NN splitting under mild conditions with minimal energy input. Beyond the conventional use of NH3, it has recently garnered attention as a promising green fuel for fuel cells, elevating academic and industrial interest in achieving its fully sustainable production.
1.2. Challenges inhibiting N2 activation
Despite the clear need for fixed nitrogen as a source of fertilizer and myriad nitrogen-containing chemical products, large-scale nitrogen activation remains a challenge that requires substantial fossil-derived energy and capital costs. Although the Haber process, given by eqn (1), is exergonic at ambient conditions,6 state-of-the-art facilities require operation at extreme temperatures and pressure to achieve high production rates. At the heart of this challenge is the NN triple bond, among the most stable bonds in nature, with a dissociation energy of nearly 10 eV.7 The high stability of the bond in N2 can be understood by its orbital arrangement, illustrated below in Fig. 1. Each atom has five valence electrons in the 2s–2p orbitals. When these orbitals combine, they form eight molecular orbitals: four bonding (two σ and two π) and four antibonding (two σ* and two π*), as shown in Fig. 1.
 | ||
| Fig. 1 Molecular orbital diagram for the N2 molecule. | ||
The N2 molecule has a total bond order of three, with electrons shared in both σ and π orbitals, a characteristic unique to N among diatomic elements. In addition to its high bond dissociation energy, N2 has over a 10 eV gap between the highest occupied molecular orbital and the lowest unoccupied molecular orbital under standard conditions.8 The triple bond encompasses one sigma (σ) bond and two pi (π) bonds, with the sigma bond formed by a head-on overlap of atomic orbitals and the pi bonds resulting from lateral overlap.
In N2, the molecular orbitals include the highest occupied molecular orbital (HOMO), housing electrons in the sigma bond (σ), and the lowest unoccupied molecular orbital (LUMO), involving the π* antibonding orbitals. The activation of N2 involves overcoming the high bond dissociation energy associated with the triple bond – a challenging process usually addressed industrially with the assistance of transition metals acting as catalysts. Initiating the nitrogen reduction reaction (NRR) is thermodynamically challenging due to several factors like dominant side reaction (HER), the large HOMO–LUMO gap that inhibits e− transfer,8 the high enthalpy change needed for proton transfer,9 and the significant energy requirement to break the NN triple bond.10,11 Transition metals, commonly used as catalysts, play a critical role in facilitating the activation of N2. The metal catalyst provides a site for interaction with the electrons of the N2 molecule, and the metal's d orbitals interact specifically with the π* antibonding orbitals. Through back donation, electrons from the metal catalyst are donated into the antibonding orbitals of N2, thereby weakening the triple bond by reducing the electron density between the nitrogen atoms.
This interaction leads to the redistribution of electron density within the N2 molecule, making the triple bond more susceptible to cleavage. During the activation process, intermediate species are formed, representing transitional stages in breaking the N2 bond. These intermediates contribute to the eventual formation of nitrogen-containing compounds, showcasing the catalytic role of transition metals in driving the activation process. Understanding the principles of molecular orbital theory and the role of transition metal catalysts is pivotal for unraveling the complex activation of N2. Such insights are not only fundamental to the understanding of chemical processes but also have practical applications. In this review, we discuss the aspects of the N2 activation mechanism and the different methods of N2 activation.
2. Methods for N2 activation
Although molecular N2 is abundant in nature, making up nearly 80% of the atmosphere, fixed nitrogen is far less common due to the N2 molecule's very high stability. Naturally occurring fixed nitrogen is typically the result of complex biological pathways or high-energy plasma (i.e., lightning strikes). Anthropogenic nitrogen activation was a topic of intense scientific interest in the late 19th century due to its importance in food production, chemical synthesis pathways to critical commodity chemicals, and relative rarity in nature. Over the past century, numerous strategies have been developed for efficient artificial nitrogen activation, varying widely in energy efficiency and output. In Fig. 2, we illustrate one efficiency metric comparing anthropogenic and biogenic nitrogen activation methods by plotting energy consumption as a function of the production rate. While the Haber–Bosch reaction is energy-intensive, the optimal heat recovery systems allow the overall energy consumption to be <20 GJ per ton NH3. The other methods are currently more energy-intensive and have lower production rates. Table 1 shows the optimal and reported energy requirements of different ammonia production technologies. | ||
| Fig. 2 Illustration of energy consumption vs. production rate for several methods of nitrogen activation.12–15 | ||
| Energy requirement (GJ per tNH3) | ||
|---|---|---|
| Reported | Potential | |
| a About 199 GJ per tNH3 is required as direct solar energy. | ||
| Brown ammonia | ||
| Steam methane reforming (SMR) | 26 | 26 |
| Naptha | 35 | — |
| Heavy fuel oil | 38 | — |
| Coal | 42 | — |
| Blue ammonia | ||
| SMR with CCS (carbon capture and storage) | 33 | 27 |
| Coal with CCS (carbon capture and storage) | 57 | — |
| eSMR | — | 26 |
| Green ammonia | ||
| Low temperature electrolysis | 33 | 31 |
| High-temperature electrolysis | — | 26 |
| Others | ||
| Benchmark electrolysis-based Haber–Bosch process | 33 | 26 |
| Non-thermal plasma technology | 155 | 60–70 |
| Electrochemical and photochemical synthesis | 135 | 27–29 |
| Electrochemical synthesis | 135 | 27–29 |
| Photochemical synthesis | — | 200a |
| Electro-thermochemical looping | 64 | 55 |
| Homogeneous catalysis | 900 | 159 |
In the following sections, we categorize the anthropogenic and biogenic strategies for nitrogen activation and provide mechanistic details, challenges, and potential for improvement.
2.1. Thermochemical N2 activation
Thermochemical processes were among the first anthropogenic sources of fixed nitrogen. Today, thermochemical processes, primarily the Haber–Bosch process, account for the vast majority of fixed anthropogenic nitrogen at a rate of over 200 million tons of ammonia per year.16 Most of this ammonia is converted into fertilizer and is necessary for sustaining our population, nearing 8 billion people. As such, the discovery of the Haber process has been hailed as the most important innovation of the 20th century.17 In this section, we begin by discussing the background and history leading to the creation of the Haber process because understanding the context behind its development facilitates understanding the challenges we face as we search for a more sustainable alternative. We then discuss the mechanism underlying the Haber process and end with a future outlook for thermochemical processes.| CaC2 + N2 → CaCN2 + C | (2) |
The process was operated at an extremely high temperature, around 1000 °C, achieved using electrical resistive heating. The tremendous quantities of electricity required to maintain this high temperature limited its operation geographically to areas such as Norway, where dedicated hydroelectric power plants were constructed to supply the needed electricity.21 Although highly energy inefficient, the Frank–Caro process was exceptionally important as a proof-of-concept: it demonstrated that anthropogenic dinitrogen activation via thermo-chemistry was possible on industrial scales. Interest in a more energy-efficient process grew, and in 1901, Le Chatelier discovered what would be later known as the Haber process,22 in which gaseous nitrogen and hydrogen react to form ammonia at high temperature and pressure over an iron catalyst. Le Chatelier abandoned the research after an explosion in the high-pressure reactor nearly killed an assistant. The process was rediscovered by Haber in 1905.19 After lab-scale production was demonstrated with an osmium catalyst in Haber's laboratory, BASF purchased rights to the process, with scale-up of the process assigned to Carl Bosch. Owing to its scarcity and tendency to deactivate over time, Alwin Mittasch was tasked with finding a cheaper alternative to the osmium catalyst. After thousands of attempts, Mittasch discovered the promoted iron catalyst that is still used in some ammonia production facilities today.23
N bond.24 At high temperatures, the reaction Gibbs free energy can shift from negative (spontaneous) to positive (non-spontaneous), even though the reaction enthalpy remains exothermic. This occurs because the negative reaction entropy contributes more significantly to the Gibbs free energy at elevated temperatures. High pressure is then required to favor the forward reaction by countering the entropy-related effects and ensuring a reasonable reaction rate. Fig. 3(a) shows the thermodynamic equilibrium between N2/H2 and NH3 for the Haber–Bosch process. In this manner, the Haber–Bosch equilibrium refers to the chemical equilibrium established in the synthesis of NH3 from N2 and H2 gases shown in eqn (1). This reaction is exothermic (ΔH < 0). It involves a reduction in entropy due to the decrease in gas molecules. As a result, the equilibrium is favored by low temperatures and high pressures, according to Le Chatelier's principle. Industrial processes use moderately high temperatures (400–500 °C) to maintain a practical reaction rate, as lower temperatures significantly slow the kinetics. High pressures (150–300 atm) are applied to push the equilibrium towards NH3 production. Catalysts, such as iron-based compounds, are utilized to overcome kinetic barriers and enhance the reaction rate under these conditions. The catalytic process and its mechanism are illustrated in Fig. 3(b).
 | ||
| Fig. 3 (a) Thermodynamic equilibrium between N2/H2 and NH3 (Haber–Bosch equilibrium); (b) animated illustration of the Haber–Bosch process, which occurs over a promoted iron catalyst at high temperature and pressure. | ||
The mechanism of the Haber process on Fe catalysts was largely unraveled by Gerhard Ertl in a series of papers that leveraged low energy electron diffraction (LEED) to estimate sticking coefficients for various intermediate steps along the process,25–29 partly leading to his receipt of the Nobel Prize in 2007. The mechanism with individual reaction steps is given below in eqn (3)–(10). The rate-determining step is associated with the activation of bound dinitrogen, i.e., eqn (4).
| N2 + * → *N2 | (3) |
| *N2 + * → 2*N | (4) |
| H2 + * → *H2 | (5) |
| *H2 + * → 2*H | (6) |
| *N + *H → *NH + * | (7) |
| *NH + *H → *NH2 + * | (8) |
| *NH2 + *H → *NH3 + * | (9) |
| *NH3 → NH3(g) | (10) |
Later investigations into the Haber process by Nørskov and Chorkendorff demonstrated that activity likely is dominated by step sites30 instead of the hypothesized terrace sites.29 Further analysis of the role of the alkali cation promoter has, in time, revealed that the transition state of N2 activation is stabilized through a dipole–field effect, and the promoter simultaneously reduces the effect of oxygen poisoning and modulates the binding energies of intermediates further down the pathway.24,31–33
Dinitrogen can undergo direct reactions with certain metallic elements. For example, the reaction of dinitrogen with lithium yields lithium nitride (Li3N) which proceeds slowly at room temperature but rapidly at 250 °C.3 Similarly, dinitrogen reacts with alkaline earth metals to form nitrides like Mg3N2, which occurs rapidly at temperatures above 500 °C. At even higher temperatures, dinitrogen reacts with elements such as boron, aluminum, silicon, and various transition metals. Dinitrogen is generally inert around 25 °C, it exhibits direct reactivity with several elements at elevated temperatures.3
Nitrogen forms a wide range of nitrides categorized as ionic, covalent, or interstitial. Elements like lithium, alkaline earth metals, zinc, and cadmium from nitrides containing the N3-ion yield ammonia upon hydrolysis:
| Li3N + 3H2O → 3Li+ + 3OH− + NH3 | (11) |
| Ca3N2 + 6H2O → 3Ca2+ + 6OH− + 2NH3 | (12) |
Nitrogen compounds with elements from groups III, IV, and V are typically covalently bonded nitrides, including BN, Si3N4, and P3N5.
When nitrogen reacts with finely divided transition metals, it produces interstitial nitrides like W2N, TiN, and Mo2N. These nitrides feature nitrogen atoms within the metallic lattice, sharing similarities with interstitial carbides in hardness, high melting points, electrical conductivity, non-ideal stoichiometry, and chemical inertness.
2.1.3.1. Improvement in thermocatalysts. The industrial Haber–Bosch process poses significant challenges, i.e., high pressures and temperatures. A corresponding equation of thermocatalytic N2 activation suggests that reducing the reaction temperature would be beneficial. If the reaction is conducted at a comparatively low temperature, the rate of reaction of N2 activation reduces significantly. Therefore, extensive efforts have been made to facilitate this reaction in moderate conditions while maintaining the reaction rate.
Compared to industrial processes, milder conditions can be used to interchange nitrogen atoms between nitrogen molecules and lattice nitrogen. This exchange facilitates the splitting of nitrogen bonds. Additional strategies are needed to reduce the reaction barrier and further enhance the performance of thermocatalytic N2 activation in ambient conditions. While Ru-based materials have an advantage over Fe-based materials due to less NH3 poisoning, ruthenium catalysts are susceptible to hydrogen poisoning because of the strong binding of hydrogen atoms. Hosono and co-workers34 proposed a mechanism of NH3 production using ruthenium supported on the C12A7:e− substrate, which allows for reversible release and storage of hydrogen atoms, thereby preventing hydrogen poisoning. Metal hydrides, such as TiH2, LiH, BaH2, and CaH2, have also been crucial in low-temperature thermocatalytic nitrogen activation processes. These hydrides create energy-favored pathways for thermocatalytic NH3 production in milder environments. Despite the progress made, some thermocatalytic processes still rely on high pressure, which poses a disadvantage due to the need for a source of H2 gas and N2.35–37 Table 2 consists of a few thermocatalysts, which showed great performances for N2 activation under mild conditions. In addition, the effect of spin promotion on producing NH3 is studied, and the probable structure of active sites with various promoters is determined. Furthermore, two factors that influence catalytic activity were observed. Firstly, an electrostatic relation occurs between the N–N dissociation transition state and the adsorbed promoter. Additionally, it reported a novel promoter effect in magnetic catalysts, significantly reducing activation energy.38 These findings open opportunities for discovering new types of thermocatalysts for NH3 synthesis.
| Catalyst | Temperature | Remark | Ref. |
|---|---|---|---|
| Ba–Co/C | 320–440 °C | The NH3 concentration rises almost three times. | 39 |
| Ru-loaded C12A7:e− | 360–400 °C | The electrode C12A7:e− is a stable and effective electron donor, acting as a reversible hydrogen-storage material to improve catalytic activity in NH3 synthesis. | 34 |
| Ru/Ca2N:e− | 300–340 °C | Showed superior catalytic functioning for low-temperature NH3 synthesis compared to heterogeneous catalysts. | 40 |
| Ru/BaO–CaH2 | 200–350 °C | NH3 synthesis at low temperatures with minimal activation energy (41 kJ mol−1). | 41 |
| BaH2–Co/CNTs | 250–350 °C | Exceptional catalytic activity at low temperatures, ≈20 and 2.5 times greater than the substantially active Cs–Ru/MgO catalyst. | 37 |
| Cr–LiH, Mn–LiH, Fe–LiH and Co–LiH | 150–300 °C | The collaboration between transition metals and LiH establishes an energy-efficient route for NH3 synthesis at moderate conditions. | 42 |
| Ni/LaN | 350 °C | LaN catalyst with Ni loading demonstrates activity surpassing traditional Ni- and Co-based catalysts, equivalent to Ru-based catalysts. | 43 |
| CeN, LaN, and YN, | 150–400 °C | Rare earth metal nitrides assist effectively as supports or catalysts in NH3 synthesis, with nitrogen-vacancy generation significantly influencing catalytic performance. | 44 |
| Ru/Ca(NH2)2 | 250–350 °C | The excellent catalytic performance is credited to the development of a dense array of flat-shaped ruthenium nanoparticles and the electron-promoting influence of Ca(NH2)2. | 35 |
| Ru/CexZr1−xO2 | 390 °C | Enhanced electronic metal–support interactions increased NH3 synthesis catalytic activity and reduced the apparent reaction activation energy. | 45 |
| Ru/C in Cs | 250–400 °C | Dosing pure metallic Cs in situ proves significantly more effective for NH3 synthesis than conventionally synthesized Ru catalysts, which are promoted ex situ with Cs. | 46 |
High activity and long-term stability can be achieved by surface modification or alloying. This can potentially reduce the susceptibility to hydrogen poisoning by altering the electronic and geometric properties of the Ru surface, thus enhancing both catalytic activity and resistance to poisoning. Alternatively, the surface can modify hydrogen adsorption properties, making it less prone to poisoning. For example, combining Ru with oxide supports like Al2O3 or TiO2 can improve stability. These modifications can help reduce the adsorption of hydrogen molecules on Ru while maintaining high catalytic activity for NH3 synthesis. Electrostatically polar MgO(111) to replace nonpolar MgO as the support can significantly alleviate the hydrogen poisoning and facilitate an unprecedented ammonia production rate by its high intrinsic proton capture ability.47 Similarly, other refractory metals, or oxide supports, can be explored as future work.
2.1.3.2. More sustainable sources of hydrogen. Today, ammonia producers exhibit greater operational expertise in hydrogen production than all other segments of the hydrogen-producing industry combined.48 This knowledge is crucial for their success in competitive markets, as it can determine profitability. NH3 producers are now seeking a new type of hydrogen, one that is decarbonized, sustainable, and renewable. While there will be numerous significant markets for renewable hydrogen in the future, the immediate focus lies on renewable fertilizers, which present a practical and substantial market opportunity that far outweighs existing markets in scale. The production of green hydrogen through low-temperature water electrolysis powered by renewable energy is a critical step toward rapidly reducing carbon emissions in the industrial sector, particularly in ammonia synthesis via the Haber–Bosch process. Several challenges currently hinder green hydrogen production's economic viability, including high costs, scalability, energy efficiency, etc. Despite these obstacles, this technology is expected to decarbonize chemical transformation processes.
To further enhance its potential, researchers are exploring direct seawater electrolysis, which utilizes abundant seawater resources and eliminates the need for high-purity water, addressing one of the key limitations of conventional electrolysis methods. The world's oceans hold an almost limitless supply of seawater, whereas freshwater is becoming an increasingly scarce resource. Direct seawater electrolysis is an emerging technology for converting and storing electricity into hydrogen in this global context. It proves most valuable in areas with abundant renewable electricity, limited freshwater access, and ample seawater resources. It is also advantageous for offshore hydrogen and mobile maritime-based applications, such as powering unmanned underwater vehicles for offshore maintenance.49,50 There are few environmental and economic incentives to invest in research and development for current direct seawater electrolysis technology. The costs associated with commercial water electrolysis are significantly higher compared to seawater reverse osmosis (SWRO). The operating and capital costs of SWRO are trivial, resulting in a minimal enhancement in the levelized cost of hydrogen (less than $0.1 per kg of H2) and CO2 releases (less than 0.1%) in a seawater reverse osmosis-proton exchange membrane coupled process.49 Promising catalysts have been identified for anodes and cathodes whereas novel separators and advanced membrane concepts are crucial for enhancing the solidity of direct seawater electrolyzer devices.51
The steam-methane reforming (SMR) process is another significant process for producing hydrogen. The production of significant chemicals like NH3 and hydrogen contributes significantly to CO2 emissions due to reliance on hydrocarbon combustion for heating during synthesis. SMR is responsible for approximately 50% of the global hydrogen supply and is one of the most significant endothermic processes. The production of hydrogen alone is estimated to contribute 3% of the emissions of CO2.52 The Chorkendorff and Mortensen52 research group presented a highly efficient reformer driven by electricity, utilizing direct resistive (ohmic) heating. This scalable technology is suitable for industrial conditions and capacities. The direct interaction between the electric heat source and the catalyst allows precise reaction control and eliminates thermal constraints. This innovative approach removes the need for a fired section, significantly reducing reactor volume, waste-heat streams, and CO2 emissions. This breakthrough technology gives existing industrial reformers a competitive edge by enabling the production of environment friendly hydrogen to synthesize crucial chemicals like NH3. Electrifying SMR opens up new possibilities for reactor design, scalability, and implementation.52
Despite the limitations of SMR in terms of economics and the environment, no existing renewable power sources, including electrolysis-based hydrogen, can rival the large-scale H2 production achieved by SMR. Another option for H2 production is the pyrolysis of CH4, which does not generate CO2 (i.e., CH4(g) → C(s) + 2H2(g) ΔH° = 74 kJ mol−1).53 Although this method produces only half the amount of H2/mole of CH4 associated with SMR, it requires significantly less energy contribution and produces solid carbon instead of CO2. This solid carbon can be safely deposited indefinitely and even utilized in various applications such as electrodes or additives in materials like asphalt, concrete, and rubber. Furthermore, straight CH4 pyrolysis can be carried out in a comparatively simple and potentially cost-effective commercial process, requiring only a single reaction step. Most downstream processes can tolerate small amounts of unconverted CH4, making it suitable for applications like NH3 production or use in fuel cells. On the other hand, carbon oxides produced by the SMR process can be detrimental to catalysts and must be eliminated.53 The McFarland research group53 has developed a catalytic molten metal method for direct methane conversion into hydrogen and carbon separation. The insoluble carbon of this system rises to the surface for easy removal. The optimal alloy composition, 73% Bi and 27% Ni, enables pure H2 production without emitting CO2 or by-products. In addition, Palmer et al.54 and Kang et al.55 synthesized molten Bi–Cu alloy and molten MnCl2–KCl catalyst, respectively, for CH4 pyrolysis to produce CO2-free H2.
2.2. Biological and enzymatic N2 activation
Biological N2 activation is a crucial natural phenomenon, possibly second to photosynthesis. The vitality of many natural ecological systems relies significantly on the biological conversion of atmospheric N2 into forms that plants and microbes can utilize for growth, all without modern fertilizers or animal waste. Various prokaryotic microbes, including bacteria, archaea, cyanobacteria, and actinomycete Frankia, orchestrate this conversion.56 Fig. 4 shows the classification of nitrogen-fixing microorganisms. The organisms involved in nitrogen activation can either function independently as free-living entities or form associations of varying complexes with other plants, microbes, and animals. These associations range from free connections, such as associative symbioses, to intricate symbiotic relationships (Fig. 5), where precise molecular transfer occurs between the host plant and the bacterium, accompanied by allocated physiological actions. The core of the system is the prokaryotic microorganism that hosts the nitrogenase enzyme complex, which facilitates nitrogen activation by converting N2 into NH3. Dinitrogen activation is an exclusive capability of certain prokaryotes. Therefore, when encountering claims about “nitrogen-fixing” plants, it's crucial to recognize that they achieve this through their partnership with prokaryotic organisms, not independently. Organisms that rely on atmospheric N2 as their primary nitrogen source crucial for growth are diazotrophs. The term “diazo” pertains to N2, and “trophs” signifies nourishing, a terminology introduced by Burns and Hardy in 1975.57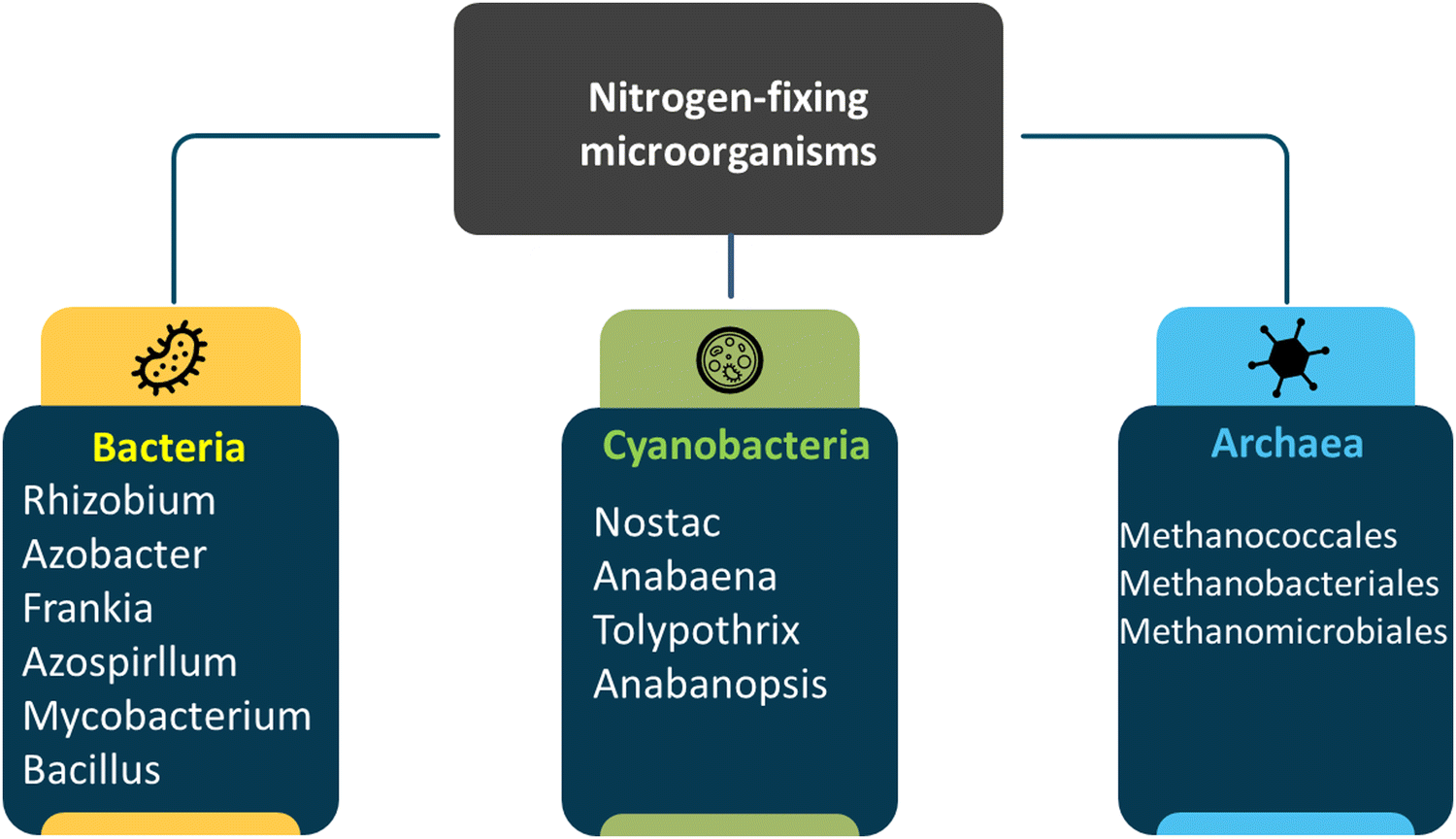 | ||
| Fig. 4 Classification of nitrogen-fixing microorganism.58 | ||
 | ||
| Fig. 5 The exclusive mediation of biological nitrogen activation occurs through free-living diazotrophs prokaryotic and a diverse array of plant-microbe connections with variable degrees of complexity.56 | ||
The Haber–Bosch process contributes approximately 95 million metric tons per year to global nitrogen supplies, requiring substantial energy inputs, typically as natural gas (Fig. 5). This quantity is overshadowed by the universal impact of fixed nitrogen obtained within biological nitrogen activation. Pinpointing precise assessments for biological N2 activation proves challenging.59 Fowler et al.60 propose that approximately one-third of human-caused nitrogen contribution into terrestrial ecosystems originates from biological nitrogen activation (58 million metric tons ± 50%), while the remaining two-thirds come through fertilizer involvements (120 million metric tons ± 10%). The difficulty in obtaining accurate measurements is emphasized by the wide range of values observed for biological N2 activation. Symbiotic N2 activation constitutes the primary source of biological input, but associative and nonsymbiotic N2 activation also play a notable role in crops like sugarcane and sorghum, particularly in ecosystems where nitrogen is a growth-limiting factor. Rice cultivation, characterized by submerged soils, obtains substantial benefits from free-living cyanobacteria and diazotrophic bacteria. These microorganisms thrive in favorable habitats within flooded soils, such as modest oxygen concentrations and root exudates.59,60
Biological N2 activation offers a cost-effective substitution for expensive nitrogen from NH3-based fertilizers. Sustaining high-profit agricultural practices in the USA and other regions solely through animal manure and biological nitrogen activation poses significant challenges. The increasing global population necessitates production systems that judiciously incorporate both artificial and, wherever feasible, natural fertilizers. Leguminous plants, alongside further nitrogen fixing methods such as the Azolla–Anabaena interaction and potentially naturally transformed vegetation (still in development), will remain integral to ecological agricultural processes. N2-fixing plants also significantly benefit the restoration of concerned or depleted soils. These have multiple purposes, acting as natural manures, outstanding cover crops, and fodder for cattle. Utilizing nitrogen-fixing crops has the added perspective to mitigate groundwater pollution with NO3− and minimize the emission of N-based “greenhouse gases”. This multifaceted approach underscores the importance of integrating various strategies to ensure agricultural productivity and environmental sustainability.59–61
All these microbes share a common trait—they generate the complex enzyme known as nitrogenase, facilitating biological nitrogen activation. The term “nitrogenase complex” is aptly applied as it comprises two components of protein, consisting of various subunits, respectively, and accommodates various metallic clusters crucial for catalyzing the reaction of dinitrogen to NH3.62 Table 3 represents the nomenclature for nitrogenase enzyme complexes. Fig. 6 illustrates the understanding of the nitrogenase enzyme complex. Unique for its dual proteins, nitrogenase complexes comprise the MoFe and protein known as dinitrogenase and dinitrogenase reductase, respectively. Susceptible to oxygen, it contains Fe and Mo, or V. Activation requires Mg2+ and involves the conversion of ATP to ADP. Inhibition by ADP occurs, yet it converts N2 and various molecules having triple bonds. Remarkably, it reduces protons to dihydrogen even in the presence of dinitrogen.
| Nomenclature | Nitrogenase complexes |
|---|---|
| Nitrogenase | The overall complex |
| Dinitrogenase | The MoFe protein, which conventionally receives its name based on its substrate (in this case, dinitrogen), plays a crucial role in nitrogen activation. |
| Dinitrogenase reductase | The Fe protein (the consensus is that its role is to reduce dinitrogenase) |
 | ||
| Fig. 6 The nitrogenase enzyme complex is composed of two protein constituents, i.e., dinitrogenase reductase and dinitrogenase.56,63 | ||
The biological nitrogen activation reaction utilizing nitrogenase is depicted in Fig. 6. For all e− transfer from dinitrogenase reductase into dinitrogenase, two MgATPs are necessary. The written reaction demands 16 ATP molecules (112 kcal). An estimated 20–30 MgATP may be required under usual operational conditions, as the procedure is less effective than in optimal lab conditions.64 Over several years, a consensus has gradually emerged on the universal nitrogenase model.
In summary, the nitrogenase mechanism involves dinitrogenase reductase (i.e., Fe-based protein) receiving e− from a donor with low-redox, binding two MgATP. It then transfers e− separately to dinitrogenase (i.e., MoFe-based protein). This complex facilitates electron transfer and hydrolyzing of two MgATP to MgADP + Pi. After dissociation, the process repeats. Once dinitrogenase accumulates adequate e−, it combines N2; then, it is reduced into NH4+. The dinitrogenase continues the cycle by accepting extra e− from dinitrogenase reductase.
In each nitrogen activation cycle, dinitrogenase reductase and dinitrogenase combine, hydrolysis of MgATP occurs, and an e− is shifted. The rate-limiting step is dissociating the MoFe protein–Fe protein complex. Notably slow, the nitrogenase complex takes 1.25 s to convert one enzyme molecule into two NH3 molecules. To accomplish this, the two proteins must undergo a process of association and dissociation eight times to catalyze the reduction of a single dinitrogen molecule.65 Due to its sluggish nature, nitrogen-fixing bacteria must produce a substantial amount of the protein. Nitrogenase can make up 10% cell's proteins, with recorded concentrations reaching ≈40%.65 In the overall reaction illustrated in Fig. 6, it could be noted that for each 8e− transmitted by the nitrogenase complex, the 2e− is utilized in H2 production. Hydrogen production is essential for nitrogen activation, with each molecule of H2 (requiring 4 MgATP) supporting the activation of one N2 molecule, which is then converted into 2NH3 molecules (requiring 12 MgATP). Consequently, 25% of the MgATP energy is wasted in the production of H2, whereas certain diazotrophs understand hydrogenase, enabling oxidation of specific H2 and producing a reduced e− carrier or MgATP. This regained energy can be harnessed during the nitrogen activation process, compensating for the small amount of energy initially lost.
All “nitrogenases” consist of a dinitrogenase and dinitrogenase reductase component. Nitrogenases 2 and 3 generate more gaseous hydrogen compared to nitrogenase 1. Instead of converting acetylene to ethylene, they transform it into ethane. In the 1990s, a new putative nitrogenase was reported in Streptomyces thermoautotrophicus, a thermophilic and chemoautotrophic actinomycete.67,68 This unique nitrogenase, not inhibited by CO like others, comprised of three proteins: dinitrogenase, carbon monoxide dehydrogenase and, superoxide oxidoreductase, accountable for the N2-activation. The enzyme-containing Mn served as a superoxide dismutase, supplying e− to the dinitrogenase. Remarkably, this nitrogenase, specified as a superoxide-dependent nitrogenase, not only resisted O2 poisoning but also required O2 in its reaction mechanism.67
In 2016, MacKellar and colleagues, in a collaborative effort across seven laboratories globally, reported conclusive evidence that S. thermoautotrophicus cannot fix nitrogen.69 Despite working with the original isolate and two extra strains, they couldn't validate the combination of 15N-labeled N2 (15N2) in cell constituents. The authors suggested reclassifying species H1, UBT1, and P1-2 as a non-diazotrophic, non-streptomycete, and facultative chemolithoautotroph. Based on their findings, it was strongly concluded that the presence of the previously suggested oxygen-tolerant nitrogenase is highly improbable.69 With >99% nucleotide identity to Hydrogenibacillus schegelii, a carbon-monoxide oxidizing bacterium, the statement of nitrogen activation by S. thermoautotrophicus awaits further verification.
The discovery that N2 can be fixed without the involvement of molybdenum is remarkable. Before discovering alternative nitrogenases, scientists considered molybdenum an obligatory requirement for N2 activation. The prevalence of microorganisms with nitrogenase 1 (the enzyme-containing Mo) might be attributed to researchers consistently using Mo-containing enzymes for isolating nitrogen-fixing bacteria. It remains uncertain whether other nitrogenases are widespread. The exploration of alternative nitrogenase systems is in its early stages, and numerous fundamental questions are yet to be answered.56
 | ||
| Fig. 7 (a) Two theoretical pathways for N2 photocleavage, as proposed by Fischler and von Gustorf, involving an Fe-η1-N2 complex;70 (b) reduction of N2 to NH3 in nitrogenase complex (reproduced with permission from Elsevier. Copyright © 2012 Elsevier).72 | ||
In the conventional biological process of N2 activation through nitrogenase enzymes, compelling evidence suggests that electron back-donation to π* orbitals facilitates the adsorption and activation of N2 on the Fe–Mo–S cofactor within the FeMo–protein. The peripheral Fe protein then hydrolyzes MgATP to provide the necessary energy and electrons for the subsequent multiple proton–electron transfer (PET) processes leading to NH3 generation.73,74 Throughout these reduction processes, the Fe–protein scaffold around the cofactor functions as a coordination buffer sphere, utilizing H-bonding and redox-active groups to ensure a smooth PET process with a low energy barrier (Fig. 7(b)).75,76 Significantly, the active centers capable of adsorbing N2, such as transition metals Mo and Fe, along with efficient e− transfer pathways to the antibonding π* orbitals of N2, are essential for NN bond cleavage in NRR. Although the initial stage of NRR is challenging, the subsequent PET processes are thermodynamically favorable.10
The reduction of N2 to NH3 by the nitrogenase enzyme involves several crucial steps, with the FeMo-cofactor playing a vital role in weakening the NN bond and facilitating electron transfer. N2 binds to the FeMo-cofactor, where interactions with the metal atoms (such as Fe and Mo) in the cofactor transfer electron density into the antibonding orbitals (π*) of N2. This reduces the bond order and makes N2 more reactive. Furthermore, the back-donation mechanism involves metal atoms, particularly Mo and Fe, donating electrons from their d-orbitals into the π* orbitals of N2, further weakening the triple bond and facilitating its cleavage. The reduction process is mediated by the stepwise transfer of electrons from the Fe protein to the MoFe protein, and ultimately to the N2 molecule, with each electron transfer often coupled with protonation (addition of H+ ions). As electrons and protons are added, the electronic structure of the intermediates like diazene (N2H2) and hydrazine (N2H4) changes, leading to further weakening of the NN bond and gradual formation of N–H bonds. The final steps in the reduction process involve the complete reduction of N2 to NH3. Each protonation step consists of the addition of H+ to form N–H bonds, accompanied by corresponding e− transfers to neutralize the added protons. The stabilization of these intermediates and the stepwise reduction pathway culminated in the production of NH3.77–80
| Feature | Abiotic or biotic | Effect on N2 activation |
|---|---|---|
| Energy/carbon source | Abiotic/biotic | • The primary controlling factor for nitrogen activation in free-living soil diazotrophs is the absence of an abundant supply of available organic carbon. |
| • This is due to the requirement for substantial amounts of ATP to support nitrogenase activity. | ||
| O2 | Abiotic | • In most cases, nitrogenase is conclusively reduced when exposed to oxygen. |
| • For aerobes, the most vigorous nitrogen activation occurs when oxygen levels are significantly reduced, and for anaerobes and facultative anaerobes, oxygen must generally be absent. | ||
| Combined N (NO3−, NH4+, etc.) | Abiotic | • Combined nitrogen in the soil, such as NH4+, NO3−, and organic nitrogen compounds (like amino acids, etc.), strongly regulates nitrogenase activity. |
| Competition | Biotic | • Nitrogen-fixing microbes face competition with other soil bacteria for carbon sources, among other resources. |
| • It is widely accepted that diazotrophs comprise 1% to 10% of the cultivable microbial population. | ||
| Others | Biotic/abiotic | • Like other soil microbes, diazotrophs are vulnerable to predation by protozoa and bacterial virus lysis (bacteriophages). |
| • Factors such as temperature, pH, and trace component availability also influence their survival. |
Environmental factors significantly impact rhizobial diversity, shaped by microbial species' adaptability to prevailing conditions.81,82 Environmental conditions influence both the rhizobial response and the host, resulting in a decline in the efficiency of symbiotic N2 activation.83 The extent of this reduction is contingent on how microorganisms thrive, live, and survive in diverse conditions, considering the dynamic nature of stress influenced by collective conditions of salinity, temperature, pesticides, soil pH, drought, or nutrient deficiency.84 This symbiotic process is influenced by prominent factors shown in Fig. 8.
 | ||
| Fig. 8 Factors significantly influencing the efficiency of biological nitrogen activation in symbiosis. | ||
Nitrogenase is the only known biological enzyme responsible for the biogenic reduction of atmospheric N2 to NH3 at ambient temperature and pressure.85 Nitrogenase is produced by certain diazotrophic microorganisms, such as cyanobacteria, also known as blue-green bacteria, and rhizobacteria, a symbiotic diazotroph associated with root nodules of legume plants.65,86 The nitrogenase family has three known isozymes, Mo-nitrogenase, V-nitrogenase, and Fe-nitrogenase, with Mo-nitrogenase being the most widespread and largest contributor to the nitrogen cycle and the best-studied among three isozymes.87 Nitrogenase is usually an assembly of two components; the electron-delivery Fe–protein, which is responsible for the supply of electrons, and a catalytic FeM (M = Mo, V, Fe) protein which uses the electrons provided to reduce N2 to NH3.77,80,88,89 A schematic diagram of N2 activation in nitrogenase is shown in Fig. 9, respectively. The overall reaction for nitrogenase catalyzed N2 activation can be written as:
| N2 + 8H+ + 8e− + 16ATP → 2NH3 + H2 + 16ADP + 16Pi | (13) |
 | ||
| Fig. 9 N2 activation in N cycle. | ||
In the reaction above, ATP represents adenosine triphosphate, ADP and Pi represent adenosine diphosphate and inorganic phosphate, respectively. ATP provides the necessary energy to drive the various enzymatic reactions in nitrogen activation from the free energy released when ATP is converted to ADP and Pi.
Biological N2 activation operates under ambient conditions but requires a substantial energy input in the form of ATP, with approximately 16 ATP molecules needed per N2 molecule reduced to NH3 shown in eqn (13). In biological N2 activation, six out of eight electrons are used to reduce N2, while the remaining two contribute to the unavoidable production of H2, resulting in an electron efficiency of 75%. ATP, often referred to as the cell's energy currency, can be used to express the energy demand of N2 activation in conventional units. The hydrolysis of ATP to ADP under biological conditions has been measured at 55 kJ mol−1 and 48–63 kJ mol−1.90,91 Assuming similar energy is needed for ATP regeneration, the total energy required to fix ½N2 molecule is estimated at 0.38 MJ mol−1, which is slightly lower than the 0.48 MJ mol−1 used in the Haber–Bosch process.92 More precise calculations by Alberty research group,93 considering factors like pH, ionic strength, and Mg2+ concentration, have shown that the Gibbs free energy for ammonia formation varies between 63.2 and 180 kJ mol−1, further emphasizing the complexity of biological nitrogen fixation efficiency. This translates to an energy requirement of 0.38–0.77 MJ mol−1 NH3, depending on ATP synthesis efficiency and cellular metabolism. In comparison, the Haber–Bosch process, despite its high temperature (400–500 °C) and pressure (150–300 bar), consumes approximately 0.48 MJ mol−1 NH3, making it slightly more energy-efficient in direct thermodynamic terms. Biological N2 activation avoids fossil fuel dependency and integrates with natural ecosystems, making it more sustainable. Despite its ATP cost, biological N2 activation remains crucial in agriculture, particularly for legume-based cropping systems, which reduce reliance on synthetic fertilizers and associated environmental impacts.62,92
From an industrial standpoint, biological N2 activation can be evaluated based on energy efficiency, considering inherent energy losses in metabolic processes. The oxidation of one glucose molecule is commonly estimated to generate up to 36 ATP molecules, though recent studies suggest a more accurate value of 29.85 ATP per glucose. Given that the complete oxidation of glucose releases approximately 2.87 MJ mol−1, each ATP molecule effectively stores around 96 kJ mol−1. This translates to an estimated 0.77 MJ mol−1 of NH3 produced via biological fixation. Real-world efficiency may be lower, with some studies indicating that 20–30 ATP molecules may be required instead of the theoretical minimum of 16 ATP. Additionally, nitrogenase enzymes exhibit varying electron efficiencies: Mo-nitrogenase (70%), V-nitrogenase (40%), and Fe-nitrogenase (20%), further influencing overall energy use. Comparatively, biological N2 activation approaches the energy efficiency of the Haber–Bosch process, though nitrogen-fixing bacteria tend to activate this pathway only when no alternative nitrogen sources are available. The key environmental advantage of biological N2 activation lies in its self-regulating nature, relying on renewable carbon sources like carbohydrates rather than fossil fuels. Enhancing natural plant-microbe symbiosis could reduce reliance on synthetic fertilizers and mitigate associated inefficiencies. Additionally, biological N2 activation presents opportunities for sustainable waste management, where agricultural or forestry by-products serve as feedstocks for nitrogen fixation, further integrating biological processes into circular economy models.62,92
Over the years, two major competing pathways have been proposed: the “distal” and the “alternating” pathway.96–98 These reaction pathways for N2 reduction begin with the Janus state (E4). In the distal pathway utilized by N2 fixation in inorganic Mo complexes and suggested to apply in reaction at Mo of FeMo, the terminal nitrogen of N2 is hydrogenated in three steps until the first NH3 is liberated, then the remaining nitrido-N is hydrogenated three more times to yield the second NH3. In the alternating pathway, one hydrogen is added to the terminal nitrogen; then, one hydrogen is added to the nitrogen directly bonded to the metal. This alternating pattern continues until NH3 is released.85,99,100
| Distal pathway | Alternating pathway |
|---|---|
| FeM–N2 + H + e− → FeM–N–NH | FeM–N2 + H + e− → FeM–N–NH |
| FeM–N–NH + H + e− → FeM–N–NH2 | FeM–N–NH + H + e− → FeM–NH–NH |
| FeM–N–NH2 + H + e− → FeM–N–NH3 | FeM–NH–NH + H + e− → FeM–NH–NH2 |
| FeM–N + H + e− → FeM–NH | FeM–NH–NH2 + H + e− → FeM–NH2–NH2 |
| FeM–NH + H + e− → FeM–NH2 | FeM–NH2–NH2 + H + e− → FeM–NH2 + NH3 |
| FeM–NH2 + H + e− → FeM + NH3 | FeM–NH2 + H + e− → FeM + NH3 |
2.3. Homogeneous and metallocomplex N2 activation
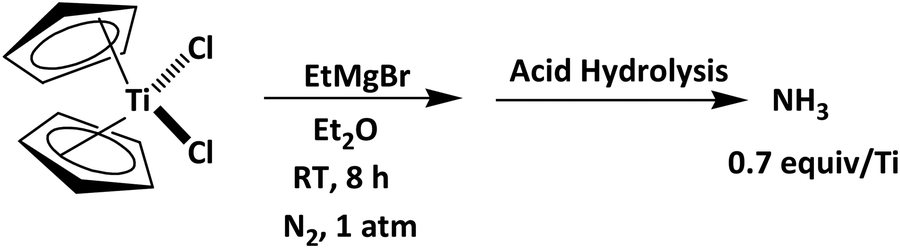 | ||
| Scheme 1 N2 activation by transition-metal complexes in moderate reaction operating conditions applying EtMgBr and Cp2TiCl2.102 | ||
 | ||
| Scheme 2 Catalytic silylation of N2 with the help of transition metals catalysts.103 | ||
A significant breakthrough occurred when the molecular [Ru(NH3)5(N2)]2+ was synthesized, challenging the belief that N2 was too stable to react in modest conditions. This breakthrough sparked hope for developing a transition metal mimicking nitrogenase in a homogeneous system.106 Transition metal complexes, like the aforementioned [Ru(NH3)5(N2)]2+, are considered excellent catalysts due to their unique configuration that allows for easy interchange of oxidation states. Inspired by the enzyme nitrogenase structure, chemists have explored Fe and Mo-based metal complexes, such as trans-[Mo(N2)2(depf)2], for catalytic N2 activation.107 For instance, trans-[Mo(N2)2(depf)2] demonstrated N2 catalysis in the presence of Me3SiCl and metallic Na as a stoichiometric reductant.108 The N(SiMe3)3 intermediate, synthesized in the process, was treated with hydrogen chloride. This treatment released ammonium chloride (NH4Cl) and completed the catalytic cycle.109 Other studies have shown that transient molybdenum species can dissociate N2 and form undesired terminal nitride complexes after N2 cleavage.108 To address this issue, late transition metal complexes with weak coordination to N2 were designed, such as [HIPTN3N]Mo and [Mo(PNP)(N2)2]2(μ-N2). These complexes were found to activate N2 insufficiently but still continuously produce NH3.110
In a different approach, Schrock and Yandulov developed a Mo-based homogeneous catalytic system for converting N2 to NH3. This system involved [LutH][BAr4] as the proton source and decamethylchromocene as the electron source.111 Exploring the captivating realm of nitrogen activation, this intricate mermaid code delves into the complexities of catalytic cycles and complexes. It sheds light on the groundbreaking discovery and development of iron-based systems that have successfully converted N2 into NH3, marking a significant breakthrough in recent years. The code discusses the synthesis of diverse Fe-based complexes with various ligands, each contributing to the advancement of the nitrogen transfer reaction cycle. Noteworthy achievements include the complete stoichiometric N2 activation in 2011 and the design of a tris(phosphine)borane-supported iron complex that catalyzes the reduction of N2 to NH3 under mild conditions. It also touches upon the investigation of Fe coordination complexes to gain insights into the potential role of the interstitial C atom in the FeMo cofactor. Furthermore, the code highlights the growing interest in multinuclear uranium and iron complexes for N2 cleavage and binding due to their remarkable capability to weaken and break the N2 triple bond. Ultimately, this mermaid code offers a comprehensive overview of the advancements, challenges, and potential future targets in the field of N2 activation.108
Apart from the Mo- and Fe-based complexes mentioned earlier, other well-precise transition metal complexes have been developed as effective homogeneous types of catalysts for N2 activation. Nishibayashi's research group developed co-nitrogen complexes with an anionic polyvinylpyrrolidone-type (PVPtype) pincer ligand. These complexes exhibited remarkable activity in directly converting N2 gas into NH3 under mild conditions due to stabilizing the PVP ligand to the Co center in various oxidation states.112 Mock and his colleagues also demonstrated that Cr–N2 complexes act as mediators for N2 reduction through three routes at room temperature.113 Nishibayashi et al.114 also successfully fabricated V–N2 complexes, representing the first examples of V-catalyzed N2 conversion into NH3 in modest conditions. Moreover, Mizobe and his team reported the isolation of the first cubane-type ruthenium metal complex to bind N2. They estimated the catalytic cycle of the pathways for the N2 activation with the help of DFT.115 These outcomes provide valuable understanding for the design of new homogeneous catalysts for N2 activation. It is worth noting that separating the generated products poses a challenge and requires more streamlined operations.
Homogeneous electrocatalysts used for nitrogen reduction typically consist of molecular complexes with metal centers of transition metals. These catalysts are dissipated in acidic medium electrolytes and undergo reduction on the electrode surface during reactions where the electrodes remain inactive. The cyclic reaction pathway of homogeneous electrocatalytic reactions involves the coordination of reactants by metal centers, storing charges attained from electrodes, and the conversion of reactants into products. The products are discharged, and the catalysts revert to their initial pattern. In addition, Chatt et al.116 reported the protolysis of a N2 complex cis-[W(N2)2(PMe2Ph)4], inspired the investigations of molecular complexes with an [MP4] core (i.e., M = W or Mo) for nitrogen reduction. The [MP4] complexes have been used as mediators for the cyclic conversion of dinitrogen to NH3.117 W/Mo complexes containing N2 ([M(N2)2(dppe)2], M = W or Mo) or hydrazido(2-) ligands ([M(X)(NNH2)(dppe)2]X, M = W or Mo, X = BF4, Br, or HSO4) have reported NH3 production rates ranging from 1.3% to 35.8% per mol of the complexes.117 These complexes rarely produce NH3 exclusively, and by-products like N2H4 and N2H2 have been occasionally detected. The ligands organized to the metal centers and the arrangement of the molecular complexes are believed to influence the selectivity towards different types of products. On Mo(III)–Mg2+–R3P–phospholipid, for example, both hydrazine and NH3 were synthesized at a ratio of 1/10.105
In 1989, Furuya et al.118 discovered that the metal phthalocyanine catalyzes electrochemical nitrogen reduction in an ambient environment. When Fe-phthalocyanine was used as the catalyst, a faradaic efficiency of approximately 1.6% for the NH3 production rate was accomplished at −0.6 V vs. RHE during the initial electrolysis stage. The anode was a platinized gas-diffusion electrode, and the electrolyte was 1 M Na2SO4. Fe-phthalocyanine proved unstable in the reaction conditions, with the faradaic efficiency dropping sharply to <0.1% after approximately 10 minutes of electrolysis. The stability and efficiency of phthalocyanine-based catalysts were found to be dependent on the center metals of the phthalocyanines and electrolytes. A mixture of KOH and KHCO3 as electrolytes enhanced Fe-phthalocyanine's faradaic efficiency and stability, compared to Na2SO4 as an electrolyte. Among the series of metal phthalocyanine catalysts tested, Sn-phthalocyanine exhibited the highest stability and activity.118 The estimated faradaic efficiencies (FE) were ranked as follows: H (a free base phthalocyanine) > Fe, Ti > Pd > Co > Pt > In, Ni > Pb > Cu > Zn > Sn, Ga > Pb.119
Active catalysts for electrochemical nitrogen reduction reaction have been reported in the form of a group of Sacconi-type tetradentate ligands, abbreviated as P3E, where three phosphine donors (P) are attached to a central atom over an ortho-phenylene linker (E = C, B, Si).120 Notably, iron-based Sacconi-type tetradentate ligands, known as P3EFe (E = C, B, Si), have demonstrated the ability to synthesize NH3 in the presence of dinitrogen. Specifically, it has been reported that P3BFe can yield 64 equivalents of NH3/Fe site, while P3CFe can produce 47 equivalents of NH3 per Fe site. These ligands have also shown good permanence during various reaction series.120 Conventional and in situ characterization techniques have recommended an electrochemical dinitrogen introduction progression reaction mechanism in which P3BFe–N2− acts as the catalyst and the Fe-borohydrido-hydride complex serves as the supporting state. Similarly, P3ECo (E = C, B, Si) complexes have demonstrated the capability to promote the activation and binding of dinitrogen in electrochemical nitrogen reduction reactions.120
Catalytic N2 reduction into silylamines has been accomplished in modest reaction conditions with the help of various metal complexes such as Mn, Re, Ti, Cr, W, Ir, Ni, Co, U, and Rh. Co derivatives have exhibited superior catalytic activity among these metal complexes, producing up to 320 equivalents of silylamines/catalyst or 270 equivalents/metal. Meanwhile, the yield of silylamine has been constrained to 50% due to the formation of silicon-containing by-products. Although silyl radicals are commonly believed to be the reactive species in the catalytic reaction, experimental evidence is lacking.101 Further mechanistic studies are needed to improve more effective reaction methods. Developing selective and efficient metal complex catalytic techniques for synthesizing valuable N-containing composites is the subsequent focus of research in this field.
 | ||
| Fig. 10 (a) Binding schemes for dinitrogen to a metal (M), and (b) primary coordination approaches of N2. | ||
Among these modes, the terminal end-on mode is the most prevalent in catalytic systems. In contrast, the bridging side-on and terminal modes generally demonstrate higher reactivity than bridging end-on N2. After the coordination of N2, substrate reduction can proceed through two different pathways: an associative mechanism or a dissociative mechanism.
2.3.2.1. Dissociative mechanism for N2 activation. The N–N triple bond undergoes direct reductive dissociation, which necessitates a significant energy input to permeate the high kinetic barrier of the reaction. This process can occur under extreme Haber–Bosch conditions, leading to the creation of surface-bound Fe–nitrides. These nitrides are subsequently rapidly hydrogenated, resulting in the release of NH3.121 Several molecular complexes can also facilitate direct N2 cleavage, forming distinct molecular nitrides. It is worth noting that only one known instance of a homogeneous catalyst operates through this particular mechanism.
Cummins et al.122 reported metal-assisted N2 reductive scission. They used a MoIII-trisamide complex [(NR,Ar)3Mo] to coordinate N2, forming a μ-η1:η1-N2 bridged dimer at low-temperature Fig. 11(a). After warming, the N2 split to yield two terminal MoVI–nitride complexes. This process was facilitated by the π10 (end-on) electronic configuration of the Mo2N2 core, as recommended by DFT studies. Unlike other metal–nitride complexes, the Cummins MVI–nitride adduct was too stable to liberate NH3 through protonation. Later, Nishibayashi's group discovered that the PNPH,tBuMoI3 complex, supported by a pincer ligand, can directly produce a MoIV–nitride adduct by adding CoCp*2 under N2 atmosphere Fig. 11(b). This complex also acts as an effective catalyst for reducing N2 to NH3.123 Kinetic and DFT studies recommend that the rate-determining step in this catalytic process is the direct “proton-unassisted” dinitrogen splitting reaction, which is unique compared to other reported catalytic systems. The nitride adduct can release NH3 and regenerate a Mo–N2 adduct by accepting electrons and protons. Iodide co-ligands play a role in the distinct reactivity of PNPH,tBuMoI3 compared to other complexes. These iodides stabilize a transient N2-bridged MoI dimer, which further splits into MoIV–nitrides, rather than forming a m-N2 bridged dinuclear Mo0 complex. In the case of PPPMo, no Mo0–N2 dimers are detected, suggesting a dissociative N2 reduction mechanism.
 | ||
| Fig. 11 Examples of dissociative mechanism for N2 activation. Reproduced with permission from John Wiley & Sons. Copyright © 1999–2023 John Wiley & Sons.124 | ||
In addition, N2 cleavage was achieved through collaboration between multiple metal centers studied by Holland and Murray research groups in 2011 and 2015, respectively (Fig. 11(c) and (d)).125,126 Holland demonstrated that the b-diketiminate dinuclear [(b-diketim)FeIICl]2 complex reacts with KC8 under a N2 environment to develop a dinitride-bridged tetranuclear complex.126 The rational steric bulk of the supplementary ligand and the interaction between the Fe-bound nitrides and the K+ cation are crucial in facilitating N2 rupture. Likewise, Murray illustrated a triiron(II) complex assisted by a b-diketiminate cyclophane ligand, [(chpb-diketim)FeII3Br3], which also splits N2 after reduction with the help of KC8. The subsequent trinuclear mixed valence FeII/III complex comprises three protonated N-atom bridges, with the source of protons not yet fully understood. Adding acids to the N-atom bridged complexes developed by Holland and Murray, (sub)stoichiometric quantities of NH3 can be released, although the process is non-catalytic.
As mentioned earlier, the studies emphasize the necessity of two or more metal centers to facilitate N2 splitting through a dissociative pathway. This requirement arises because a single metal center cannot provide the six electrons needed, and the involvement of more than two metal atoms complicates the catalytic process's advancement. Protonation, which is likely to remove the N-atom bridges, could potentially lead to the collapse of the polynuclear structure, making the initial complex's regeneration difficult. Thus, achieving a catalytic process becomes quite challenging in such cases.
2.3.2.2. Associative mechanism for N2 activation. The N2 reduction mechanism in nitrogenase and many metal complexes (shown in Fig. 12) includes the stepwise addition of electrons and protons to split the N
N bond and establish N–H bonds.77 This hydrogenative cleavage likely supports an associative mechanism, which allows for N–N bond rupture without the involvement of a neighboring metal center. This mechanism helps to reduce the kinetic barrier for the reduction of N2. Notably, when the proton and electron transfer steps co-occur, either through hydrogen-atom transfer (HAT) or proton-coupled electron transfer (PCET), the formation of N–H bonds is facilitated without high-energy intermediates.124
 | ||
| Fig. 12 Metal complexes for N2 activation through its reduction (X = C, B, Si; Br, R = H; R1 = H, tBu, Me, OMe, Ph, Fc, Me3Si; Ad, R2 = tBu; R3 = H, Me; R4 = tBu, Cy; Ar = 2,6-diisopropylphenyl). Reproduced with permission from John Wiley & Sons. Copyright © 1999–2023 John Wiley & Sons.124 | ||
Given negligible kinetic barriers for HAT/PCET reactions, the thermodynamic feasibility of every individual N–H bond configuration via HAT/PCET can be assessed. This involves comparing the intensity of the N–H bond, which is established during the NH3 production process through the D–H bond from the HC donor (D), as shown in eqn (14):
| D–H + [N] = [N–H] + D | (14) |
Eqn (14) defines [N] as the nitrogen substrate (e.g., M–NH2 or M–N2), with [N–H] representing the subsequent HC adduct (e.g., M–NH3 or M–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–H). The intensity of N–H and D–H bonds can be assessed through bond dissociation enthalpies (BDE) or free energies of bond dissociation, incorporating the effects of entropy.124 For a successful PCET/HAT, an N–H bond forms only if the D–H bond is ineffective, i.e., when BDED–H is lower than BDEN–H. The stability of the N–H bond depends on the comparative strength of these bonds. The H atom donor's nature is crucial, and the three primary options include (i) H2 (BDEH–H = 104 kcal mol−1), (ii) metal-hydrides, or (iii) a sequence of reducing agents and acids.
N–H). The intensity of N–H and D–H bonds can be assessed through bond dissociation enthalpies (BDE) or free energies of bond dissociation, incorporating the effects of entropy.124 For a successful PCET/HAT, an N–H bond forms only if the D–H bond is ineffective, i.e., when BDED–H is lower than BDEN–H. The stability of the N–H bond depends on the comparative strength of these bonds. The H atom donor's nature is crucial, and the three primary options include (i) H2 (BDEH–H = 104 kcal mol−1), (ii) metal-hydrides, or (iii) a sequence of reducing agents and acids.
Utilizing distinct electron and proton donors can trigger a multi-site PCET pathway, forming a transient species, i.e., D–H, that may serve as a potential source of HC. In such instances, the adequate bond dissociation energy (i.e.,  ) can be determined through experimental or theoretical means. This involves incorporating the acid's dissociation constant (pKa) and the reductant's standard reduction potential (E0 vs. Fc+/Fc) into the Bordwell equation (eqn (15), where C is a constant dependent on solvent). Consider the case where a potent acid (with a low value of pKa) is paired with a robust reducing agent (having a low value of E0). This combination results in a diminished
) can be determined through experimental or theoretical means. This involves incorporating the acid's dissociation constant (pKa) and the reductant's standard reduction potential (E0 vs. Fc+/Fc) into the Bordwell equation (eqn (15), where C is a constant dependent on solvent). Consider the case where a potent acid (with a low value of pKa) is paired with a robust reducing agent (having a low value of E0). This combination results in a diminished  value, anticipating both (i) the sequential creation of N–H bonds from N2 and (ii) a concurrent enhancement in the energy expenditure of the reaction.124
value, anticipating both (i) the sequential creation of N–H bonds from N2 and (ii) a concurrent enhancement in the energy expenditure of the reaction.124
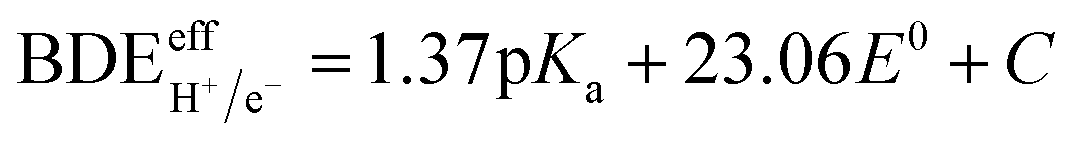 | (15) |
In terms of mechanism, both pathways, i.e., dissociative and associative, have proven effective for complex metal systems. While the dissociative reduction of N2 necessitates the collaboration of multiple metal centers, the associative pathway minimizes the kinetic requirements by facilitating multiple hydrogen atom additions to the metal–N2 adduct. The associative route appears more manageable, allowing for the promotion of PCET events, and is advantageous in avoiding the formation of unreactive multimetallic N-bridged products.
2.4. Thermal and non-thermal plasma mediated N2 activation
Plasma N2 fixation was inspired by lightning and thunder, which have enough energy and heat to break the strong bond in nitrogen gas that supports nitrogen molecules to contact with oxygen, forming NOx. | ||
| Fig. 13 (a) Plasma-activated nitrogen activation. Figure has been reproduced from ref. 128 with permission from Elsevier, © 2020 Elsevier. (b) Process scheme for the Birkeland–Eyde industrial nitrogen-activation process.127 Figure has been reproduced from ref. 127 with permission from the Royal Society of Chemistry, © 2021 openly licensed via CC BY 3.0. | ||
In the B–E process, air quickly flows through the electric arc furnace and is heated to 3000 °C by using a high electric voltage alternating current between two water-cooled copper electrodes. At 3000 °C and 1 atm, nitrogen in the air combines with oxygen to reach the maximum 6.5% NO concentration. Next, the cooling chamber decreases the temperature to 1100 °C to avoid the reverse reaction.127 The remaining heat is gathered in the waste heat boilers for future use. Then, the gases in the cooling chamber are moved to a large oxidation chamber for oxidizing NO to NO2. Since the absorption rate increases with decreasing temperature, the temperature of NO and NO2 mixture is dropped from 1100 °C to around 200 °C. The temperature further decreases to 50 °C before NOx enters the absorption tower. Finally, inside the absorption tower, NO2 is absorbed in water to produce a solution that contains approximately 30% concentrated nitric acid. The remaining NOX enters the alkaline absorption columns for further absorption, and 3% of NOX escapes the atmosphere even after the second absorption.
The first commercial B–E process was built in Niagara Falls, as the waterfall could produce enough hydronic power for the process. According to the study of Rouwenhorst's and Patil's team, the plant consumes 175-ton air to obtain 1-ton N. The energy consumption reaches 15 MW h per ton of nitric acid, which indicates that the B–E process is highly energy intensive. Numerous improvements have been found in the process, including using a 50–50% mixture of N2 and O2, preheating the reaction gas, and increasing the pressure in the furnace. Further studies need to be done to reduce energy consumption, use catalysts, and save heat energy to keep plasma-based nitric oxide production commercially reliable.
| e− + N2 → e− + N2* | (16) |
| O + N2* → NO + N* | (17) |
| O2 + N* → NO + O | (18) |
During NTP (Fig. 14a) for NH3 synthesis, the reaction depends on the plasma discharge to dissociate the reactants and produce NH3 with catalysts. Different sources, such as microwave and dielectric barrier discharge, can be utilized to form plasma essential for the synthesis. Finally, the NH3 product will be collected, and the unreacted reactants will be recycled (Fig. 14b).129 Generally, NTP refers to any plasma that is not in thermodynamic equilibrium. This can occur either when the ion temperature differs from the electron temperature or when the velocity distribution of one of the species deviates from a Maxwell–Boltzmann distribution. The most notable feature is that the temperature of the electrons can be several orders of magnitude higher than that of the surrounding gas. Therefore, electrons in NTP are promising high-energy species for activating reactants. The energy from their electronic vibration modes can be utilized in nitrogen activation reactions to achieve the significant activation energy required. Compared to thermal plasma N2 activation, the NTP has lower temperature and pressure and relatively clean, carbon-free production, making NTP environmentally friendly recently. Additional research is required due to the small scale and lower efficiency.
 | ||
| Fig. 14 (a) Summative scheme of the main reactions between molecules and atoms. Figure has been reproduced from ref. 133 with permission from Wiley © 2019 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (b) A flow diagram of the non-thermal plasma NH3 synthesis process.129 Figure has been reproduced from ref. 129 with permission from Elsevier © 2018 Elsevier. | ||
To achieve high energy efficiency in N2 fixation, Rusanov et al.130 identified several crucial experimental conditions. First, a substantial amount of vibrationally excited N2 molecules must be generated using non-thermal plasma, with electron temperatures reaching at least 10000 K. Second, the process should be conducted under moderate pressure, typically between 0.1 and 130 kPa. Third, the temperature of the resulting gas must remain low to prevent equilibrium-driven limitations that would otherwise reduce product yield and efficiency. Azizov et al.134 demonstrated nitrogen oxidation via microwave-induced plasma, reporting an energy consumption of 0.29 MJ mol−1, which remains higher than that of the Haber–Bosch process. Vibrational excitation lowers activation barriers in gas-phase reactions and heterogeneous catalysis, where catalysts enhance plasma-driven N2 activation by activating oxygen or hydrogen. Eremin et al.135 achieved the highest efficiency using a MoO3 catalyst, reducing energy consumption by 35% to 0.86 MJ mol−1. However, plasma-catalyst interactions remain unclear, as excited nitrogen species often lose energy before reaching the catalyst. Van Durme et al.136 proposed that microscopic plasma channels near catalyst surfaces improve efficiency. A key challenge is the volatility of MoO3, which can vaporize in plasma, acting as a gas-phase oxidation catalyst. Understanding these interactions is essential for optimizing catalyst performance in nitrogen fixation.
Nonthermal plasma N2 activation presents environmental and efficiency advantages by utilizing widely available raw materials like air and electricity while generating minimal by-products. Theoretical calculations suggest that the energy consumption for nitrogen oxidation could be as low as 0.2 MJ mol−1, making it potentially over 2.5 times more efficient than the Haber–Bosch process.92,130 Several challenges remain before this method can be industrially viable, including improving energy efficiency, understanding plasma-catalyst interactions, and optimizing catalyst stability for long-term operation. For future advancements in N2 activation, non-thermal plasma stands out as a promising approach, as it can circumvent thermodynamic constraints by selectively channeling energy into a specific reaction pathway. Efficiency is maximized when sufficient vibrationally excited N2 forms, gas temperatures remain low to prevent decomposition, and reactions proceed along optimal routes. This targeted energy distribution enables more effective N2 activation than conventional methods.92
 | ||
| Fig. 15 (a) Scheme of molecular orbits of N2 and O2, reproduced form ref. 137 with permission from Springer Nature, Copyright © 2018, Higher Education Press and Springer-Verlag GmbH Germany, part of Springer Nature (b) schematic showing the fundamental processes in N2 plasma. Reproduced form ref. 138 with permission from Elsevier, © 2023 licensed via CC BY-NC-ND 4.0. | ||
Non-thermal plasma N2 activation presents several practical barriers to reactor design and cost analysis. While this method operates at lower temperatures and pressures than the Haber–Bosch process, challenges include nitrogen fixation efficiency and preventing back reactions that decompose ammonia post-synthesis. Reactor design must optimize plasma discharge to enhance nitrogen dissociation and ammonia formation while integrating rapid separation mechanisms to avoid decomposition. The cost structure also plays a crucial role, as non-thermal plasma technology can lower capital expenditures by eliminating the need for high-pressure systems. In contrast, operational expenses remain high due to energy requirements. Efficient catalysts with vigorous plasma synergistic activity, such as ruthenium-based materials with promoters, are essential to improving ammonia yield and reducing energy consumption. Additionally, integrating this technology with renewable energy sources, such as wind or solar, presents an opportunity for decentralized and sustainable ammonia production. Further research is needed to enhance energy efficiency, scale-up feasibility, and overall economic viability.129
2.5. Electrochemical N2 activation
Electrochemical N2 activation (reduction and oxidation) is attractive as excess renewable energy can produce valuable chemicals decentralized by using air and water as feedstocks.139–142 In theory, N2 can be electrochemically reduced to NH3 and N2H4. Also, N2 can be electrochemically oxidized to various NOx products such as NO, NO2, N2O4, N2O5, N2O, and HNO3. Table 5 denotes the electrochemical reactions with standard equilibrium potentials for various N2 oxidation and reduction products. Though thermodynamically possible to reduce or oxidize N2 electrochemically, it is challenging due to the competing hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) shown in Table 6. The standard equilibrium potential of the NH3 formation reaction from N2 is very close to HER. Hydrazine formation reaction has a slightly higher overpotential. The N2 oxidation reactions (NORs) have a standard thermodynamic equilibrium potential higher than the OER's. N2 is in the gas phase with very low solubility in water, making it even more challenging to activate N2 in aqueous media. Furthermore, N2 is inert with very high dissociation energy of the NN (941 kJ mol−1), N2 does not have a permanent dipole, has low proton affinity (493.8 kJ mol−1), has negative electron affinity (−1.90 eV), has high ionization potential (15.84 eV), and has a large energy gap between the (highest occupied molecular orbital) HOMO and (lowest unoccupied molecular orbital) LUMO levels. Hence, it is highly challenging to activate N2 in aqueous medium not just electrochemically but also thermochemically.
| S. no. | Electrochemical reaction | Standard equilibrium potential (E0, V vs. RHE) | Product |
|---|---|---|---|
| 1 | ½N2(g) + 3H+ + 3e− → NH3(g) | −0.042 | Ammonia |
| 2 | N2(g) + 4H+ + 4e− → N2H4(l) | −0.39 | Hydrazine |
| 3 | ½N2(g) + H2O → 2H+ + 2e− + NO(g) | 1.68 | Nitric oxide |
| 4 | ½N2(g) + 2H2O → 4H+ + 4e− + NO2(g) | 1.36 | Nitrogen dioxide |
| 5 | N2(g) + 4H2O → 8H+ + 8e− + N2O4(g) | 1.36 | Dinitrogen tetroxide |
| 6 | N2(g) + 5H2O → 10H+ + 10e− + N2O5(g) | 1.35 | Dinitrogen pentoxide |
| 7 | N2(g) + H2O → 2H+ + 2e− + N2O(g) | 1.77 | Nitrous oxide |
| 8 | ½N2(g) + 3H2O → 5H+ + 5e− + HNO3(l) | 1.31 | Nitric acid |
| S. no. | Electrochemical reaction | Standard equilibrium potential (E0, V vs. RHE) | Product |
|---|---|---|---|
| 1 | 2H+ + 2e− → H2(g) | 0 | Hydrogen |
| 2 | 2H2O(l) → 4H+ + 4e− + O2(g) | 1.23 | Oxygen |
Electrocatalysts that are active, selective, and stable for NORs and NRRs and are inactive towards HER and OER are needed. Experimental screening of candidate catalysts would be a trial-and-error approach, and it is also time-consuming. Computational methods could provide an opportunity to discover and design candidate catalysts that are selective towards NORs and NRRs. The most active catalysts predicted theoretically for N2 reduction to NH3 are Mo,143,144 Fe, Rh, and Ru, but these catalysts are more active for HER, and hence, the NH3 faradaic efficiency will be very low. Interestingly, Ru143 instead of Fe on MoS2 was experimentally discovered as an active catalyst for ENRR, as shown in Fig. 16A. On the other hand, early transition metal catalyst surfaces such as Sc, Y, Ti, and Zr bind the H-adatoms weakly in comparison to N-adatoms. Hence, they were predicted to produce more NH3 than H2 at a high negative bias of −1 V to −1.5 V vs. SHE. The conclusion was made based on the binding energies of N-adatoms, and several other factors were neglected, such as the solvation effects, potential dependent activation barriers, and lateral interactions among the adsorbates. There are no reliable experimental studies that reported high NH3 FE in comparison to H2 on Sc, Y, Ti, and Zr.
 | ||
| Fig. 16 (A) Ru/MoS2 catalyzes the eNRR process, with the Ru cluster serving as the active site for nitrogen reduction, while the sulfur vacancy (S-vacancy) in MoS2 functions as the hydrogenation site.143 Figure has been reproduced from ref. 143 with permission from American Chemical Society Copyright © 2019 American Chemical Society. (B) NRR pathway on the surface of VN0.7O0.45 through a Mars–van Krevelen (MvK) mechanism, along with the catalyst deactivation process.145 Figure has been reproduced from ref. 145 with permission from American Chemical Society Copyright © 2018, American Chemical Society. | ||
Similar trends were observed on transition metal nanoclusters with Fe, Mo, and Ru on top of the volcano when an associative mechanism was considered for the N2 reduction,146 but these catalysts were more active for HER than NRR. Montaya et al. performed DFT calculations on various transition metal surfaces and found that the linear scaling between the adsorption energies of two main intermediates of NRR, such as N2H and NH2, would need at least −0.5 V. All transition metal catalysts suffer from this scaling relation, and an ideal catalyst should break the scaling relation by selectively stabilizing N2H or by destabilizing NH2 to have higher activity and selectivity towards NH3. Bimetallic catalysts or high entropy alloys that simultaneously bind N2H and NH2 onto multiple sites could overcome this problem. Anand et al.147 investigated the NOR mechanisms on a suitable OER catalyst, IrO2, and a poor OER catalyst, TiO2, and found that NOR activity is poor on the studied catalysts. In a recent study, Olusegun et al.148 reported that dynamic potential control could be a promising scheme to improve NOR activity by potentially suppressing the oxygen evolution reaction (OER) and facilitating N2 adsorption. As of now, there are no catalysts predicted by the theory that can activate N2 electrochemically in an aqueous medium. Experimentally, numerous catalysts, including their oxides, nitrides, sulfides, carbides, and phosphides of various morphologies, have been tested for N2 reduction to NH3 in aqueous medium. One good example was demonstrated by vanadium nitride (VN) NPs, where surface VN0.7O0.45 acted as the active phase,145 as shown in Fig. 16B. A detailed review of them is provided by Qing et al.149 None of the published works report an NH3 current density > −1 mA cm−2. Most of the works are not reproducible as the NH3 quantification protocols are not rigorous, and the N2 feed used for the reactions is not pure and has some forms of reactive nitrogen, such as NOx and NH3, which leads to false positives for electrochemical N2 reduction activity. Similar challenges exist for the N2 oxidation.
The commonly used NH3 quantification methods in the literature are Nessler's reagent and indophenol blue methods. Both methods are highly unreliable as they are sensitive to pH, counter-cation or anion interferences, and time. Using these methods could lead to the over-estimation of the formed NH3. These methods are economical and easily accessible across labs. Hence, the methods must be benchmarked with 1H-NMR for reliable quantification. The calibration graphs must be prepared using the electrolyte used, and quantification must be performed at constant pH to ensure the reliability of the method. The electrocatalysts can leach into the solution, interfere with the quantification method, and lead to an overestimation of NH3. Therefore, N-15 isotope labeling experiments have to be performed for at least two different conditions, and the measured N15H3 should match within the error bars (±5%) with the N-14 experiments. It is a known fact that the N-containing compounds in the catalyst, support, and electrolyte could produce NH3 and lead to a false positive. Hence, steps should be undertaken to avoid false positives. Andersen et al.150 provide a rigorous quantification procedure for electrochemical N2 reduction experiments. NOx contamination, either in the form of gaseous forms (NO, NO2, and N2O) or ionic forms (NO2− and NO3−), could lead to false positives of N2 reduction or oxidation. Choi et al.151 provide a detailed protocol for avoiding NOx contamination in the reactant feed, electrolyte, and electrochemical setups.
 | ||
| Fig. 17 (A) Competitive hydrogen evolution reaction (HER) on the electrocatalyst surface suppresses the electrochemical nitrogen reduction reaction (eNRR), reducing selectivity toward nitrogen molecules and resulting in low faradaic efficiency and decreased NH3 production.152 (B) The highly inert nature of the nitrogen molecule is attributed to its triple bond structure, which contributes to its significant stability.152 (A) and (B) Have been reproduced from ref. 152 with permission from Elsevier Copyright © 2024 Elsevier. (C) Reaction scheme representing various eNRR mechanisms: a dissociative, b alternating associative, c distal associative, d and MvK mechanism.153 Figure has been reproduced from ref. 153 with permission from Springer Nature, Copyrigh © 2023, openly licensed via CC BY 4.0. (D) Sabatier plot showing the optimal conditions for N2 adsorption on the catalyst surface (M), where the interaction is balanced to avoid hindering product dissociation or limiting N2 activation. Figure has been reproduced from ref. 152 with permission from Elsevier Copyright © 2024 Elsevier. | ||
2.5.1.1. Dissociative mechanism. The dissociative mechanism involves the direct cleavage of the N
N triple bond before hydrogenation occurs. The nitrogen molecule adsorbs onto the catalyst surface in this pathway, where the strong triple bond is broken into two nitrogen atoms. These nitrogen atoms then undergo stepwise protonation to form NH3. This mechanism is thermodynamically demanding due to the high bond dissociation energy of N2 (941 kJ mol−1).154 Transition metals like ruthenium (Ru) and iron (Fe), which exhibit strong nitrogen binding energies, are effective for this pathway as they can stabilize the intermediate nitrogen atoms during reduction.36,139,152,153,155,156 The dissociative mechanism is less favorable under ambient conditions because of the significant energy required to break the NN bond. The kinetic barrier for this process is high, often necessitating elevated temperatures or pressures, as in the traditional Haber–Bosch process. Thus, while effective at catalyzing NH3 synthesis, catalysts that follow the dissociative pathway may struggle to achieve high efficiencies in mild conditions without significant advancements in catalyst design.152
2.5.1.2. Associative mechanism. The associative mechanism retains the N
N bond during the initial steps of the reaction, allowing the molecule to undergo sequential proton–electron transfer events. This pathway is particularly suitable for ambient conditions as it avoids the high energy barrier associated with triple-bond cleavage. It distributes activation energy across intermediate steps, enabling nitrogen reduction at ambient conditions. Effective nitrogen adsorption on the catalyst surface is essential, guided by the Sabatier principle, which emphasizes an optimal interaction strength of neither too strong nor too weak between the catalyst and nitrogen molecule, as shown in Fig. 17D. The associative mechanism is further categorized into two sub-pathways:157,158• Associative alternating pathway: hydrogenation alternates between the two nitrogen atoms. Each atom sequentially gains protons and electrons until NH3 is formed on both ends. This balanced hydrogenation process ensures that both nitrogen atoms remain partially bonded until the final stages of the reaction.111,159
• Associative distal pathway: hydrogenation occurs primarily on one nitrogen atom until the first NH3 molecule is formed and released. The second nitrogen atom is then protonated to form another NH3 molecule. This stepwise approach reduces interactions between intermediates and may be advantageous for certain catalyst configurations.160–162
The associative mechanism generally exhibits lower kinetic barriers than the dissociative pathway, making it more feasible under mild conditions. Electrocatalysts that promote the end-on adsorption of N2, such as transition metals with optimized d-orbital interactions, are well-suited for this mechanism.152,153
2.5.1.3. Mars–van Krevelen (MvK) mechanism. The Mars–van Krevelen (MvK)163 mechanism involves nitrogen atoms that are already part of the catalyst's structure, such as transition metal nitrides (TMNs). In this pathway, the nitrogen atoms on the catalyst surface are first protonated to form NH3, leaving behind vacancies on the surface, as shown in Fig. 18A. These vacancies are subsequently replenished by nitrogen gas from the surrounding environment. Alternatively, nitrogen from the bulk of the material migrates to the surface to fill the vacancies, enabling further reduction cycles.164 While the MvK mechanism is efficient in utilizing surface nitrogen, the migration of bulk nitrogen to the surface can degrade the catalyst structure over time, impacting stability and reusability. Catalysts following this mechanism are typically robust and suitable for continuous operation, but maintaining structural integrity remains challenging. The MvK mechanism is particularly relevant for nitrogen-rich materials, where surface activity can be replenished.165,166
 | ||
| Fig. 18 (A) Schematic representation of the Mars–van Krevelen mechanism for eNRR on the molybdenum nitride catalyst surface.152 (B) Typical density functional theory (DFT) graph for electrochemical nitrogen reduction reaction.152 (A) and (B) Have been reproduced from ref. 152 with permission from Elsevier Copyright © 2024 Elsevier. (C) LiMAS experimental set up representation and proposed mechanism.167 Figure has been reproduced from ref. 167 with permission from American Chemical Society Copyright © 2024 American Chemical Society. (D) The electrochemical nitrogen reduction reaction pathway facilitated by lithium.152 Figure has been reproduced from ref. 152 with permission from Elsevier Copyright © 2024 Elsevier. | ||
2.5.1.4. Lithium-mediated NH3 synthesis (LiMAS). The lithium-mediated NH3 synthesis (LiMAS) mechanism is a unique approach that utilizes non-aqueous electrolytes and lithium as an intermediate as shown in Fig. 18C and D. In this process, lithium reacts with N2 to form lithium nitride (Li3N).152,167–170 The nitride is then protonated to produce NH3, regenerating lithium for subsequent reactions. This pathway effectively suppresses the hydrogen evolution reaction (HER) because it operates in non-aqueous media, where protons are supplied by sacrificial donors like alcohol. LiMAS offers high faradaic efficiency, and NH3 yields due to the absence of competing HER, but it faces scalability and energy efficiency challenges. The process is energetically intensive and relies on sacrificial reagents, which limits its practicality for large-scale applications. Moreover, the regeneration of lithium adds complexity to the overall system, requiring further innovation to make it commercially viable. Each eNRR mechanism has distinct advantages and challenges, influenced by the nature of the catalyst, electrolyte, and operating conditions. While the dissociative and associative mechanisms are widely explored in aqueous systems, the MvK and LiMAS pathways offer unique opportunities in specific configurations. Advancing the understanding of these mechanisms is essential for developing catalysts and systems capable of overcoming the intrinsic challenges of N2 activation and achieving efficient, scalable NH3 synthesis.152,153,171
2.5.2.1. Reaction mechanism of NRR on heterogeneous catalysts. Similar to homogeneous catalysts, heterogeneous catalysts predominantly follow the associative mechanism for nitrogen reduction reactions. Consequently, most theoretical studies have focused on this pathway. Unlike homogeneous catalysts that primarily involve single-metal sites or small clusters, metal surfaces in heterogeneous catalysis can dissociate the N–N bond without requiring simultaneous proton and electron addition.172
A computational study on NRR over ruthenium (Ru) surfaces revealed that multiple pathways involving different N–N bond dissociation steps can achieve activity comparable to the associative mechanism, with similar limiting potentials.172,173 This finding highlights the necessity of considering alternative reaction pathways beyond the conventional associative model. For instance, intermediates such as *NNH2, *NHNH2, and *NH2NH2 can undergo dissociation to yield *N + *NH2, *NH + *NH2, and *NH2 + *NH2, respectively, with low activation energy (0.20–0.52 eV). These steps, absent in the traditional associative mechanism, were also confirmed through in situ surface-enhanced infrared absorption spectroscopy (SEIRAS), which detected N2H2 (diazene) as an intermediate. This compound can either decompose or be further protonated to ammonia, supporting the argument that additional reaction pathways must be considered to fully understand NRR mechanisms on heterogeneous catalysts.173
To design more efficient NRR catalysts, it is crucial to consider the overall reaction equilibrium and limiting steps. The conversion of N2 to NH3 requires six proton–electron pairs:172
| N2 + 6 (H+ + e−) → 2NH3 (E° = 0.05 V vs. NHE) | (19) |
Although forming NH3 from N2 and H2 is slightly exothermic (with a Gibbs free energy change of −16.4 kJ mol−1), NRR is hindered by the negative equilibrium potentials of its intermediates, particularly the first protonation step forming *N2H.172,174,175 Theoretical studies and volcano plots indicate that this step is the thermodynamic bottleneck for most catalysts. Catalysts that stabilize *N2H enhance NRR performance, whereas those that destabilize *NH2 help lower the overpotential.172,174,175 For weak nitrogen-binding metal surfaces, the limiting step is the initial protonation (*N2 → *N2H), while for strong nitrogen-binding metals, the final protonation (*NH2 → NH3) is the most challenging step.172,174,175
Recent research suggests an alternative surface-hydrogenation mechanism for noble-metal catalysts like palladium (Pd) and gold (Au).176 In this mechanism, nitrogen activation occurs through hydrogen atoms adsorbed on the catalyst surface rather than directly on the metal. Theoretical calculations indicate that forming N2H2 via surface hydrogenation (N2 + 2H → *N2H2) requires lower activation energy compared to the conventional *NNH formation (N2 + [H+ + e−] → *NNH).176 This pathway is energetically favorable, leading to NRR at reduced overpotentials. Once *N2H2 is formed, subsequent protonation steps proceed exothermically, further supporting this alternative pathway for efficient NRR on noble metals.172,176
2.5.2.2. NRR mechanism on transition metal nitrides. The nitrogen reduction reaction on transition metal nitrides follows a different pathway than pure or alloyed transition metals.172 Unlike metal surfaces, transition metal nitrides have pre-existing nitrogen atoms incorporated into their structure. This enables them to participate in nitrogen reduction through both the direct reduction of N2 gas and the reduction of surface nitride species.172,177
This process follows the Mars–van Krevelen mechanism, a well-established concept in catalytic oxidation on transition metal oxides. This mechanism initially reduces surface-bound nitrogen atoms to ammonia, creating nitrogen vacancies.172 Subsequently, atmospheric N2 replenishes these vacancies, restoring the surface nitride composition and enabling further ammonia synthesis.172
This mechanism operates based on the redox cycling of the catalyst surface, where oxygen vacancies play an important role in facilitating the reaction. In the first step, an oxygen atom from the catalyst's surface is removed (often due to interaction with N2 or other species in the environment), creating an oxygen vacancy on the surface. This oxygen vacancy allows for further reaction steps. The nitrogen molecule then adsorbs onto the catalyst's surface at the location of the oxygen vacancy. The interaction between N2 and the active site (the oxygen vacancy) facilitates breaking the NN triple bond, which is a key step in N2 reduction. Once adsorbed, the nitrogen undergoes reduction, where electrons are transferred to the adsorbed N2 molecule, which is reduced to form ammonia.47,152,178
Computational studies by Skúlason and colleagues identified several transition metal nitrides, including chromium nitride (CrN) and vanadium nitride (VN), as promising catalysts for electrochemical nitrogen reduction via this mechanism.175 Density functional theory (DFT) calculations suggest that these materials facilitate nitrogen reduction at low potentials. More recently, a combination of 15N isotope labeling experiments and DFT simulations provided strong evidence that VN follows the Mars–van Krevelen mechanism for ammonia synthesis.145
2.5.2.3. High-entropy alloys (HEAs) for nitrogen reduction reaction (NRR). High-entropy alloys (HEAs) catalysts have been proposed as promising strategies for nitrogen reduction reaction (NRR) to overcome the scaling relations between N2H and NH2 adsorption energies; their effectiveness remains contingent mainly on specific elemental compositions and local electronic structures. Christensen et al.179 investigation of AuCoFeMoRu HEAs demonstrates that these materials can, to some extent, circumvent traditional scaling constraints by introducing a heterogeneous distribution of adsorption sites. Their findings suggest that a complete decoupling of transition-state and final-state energetics remains fundamentally restricted. Microkinetic modeling reveals that although specific HEA compositions exhibit up to a twofold improvement over monometallic surfaces, these enhancements are incremental rather than groundbreaking.179
The assumption that HEAs can universally overcome linear scaling relationships is an oversimplification. Research on high-entropy catalysts for NRR suggests that although their atomic diversity results in a broader range of adsorption energies, they do not fundamentally break the thermodynamic constraints imposed by scaling relations. Instead of entirely decoupling key reaction intermediates, these materials expand the energetic distribution, indicating that while HEAs and bimetallic catalysts provide avenues for incremental performance gains, they do not constitute a definitive solution to the scaling limitation problem. This raises a crucial critique of the prevailing discourse in the field. HEAs and bimetallic catalysts are often credited with capabilities that are overstated in theoretical studies and lack robust experimental confirmation. Consequently, rather than considering these materials as a conclusive breakthrough, future research should prioritize a more systematic approach integrating experimental validation, refined electronic structure tuning, and operando characterization to rigorously evaluate their catalytic viability.179,180
In addition, optimizing N2 adsorption and activation while suppressing the competing HER remains a central challenge. Theoretical studies suggest that Fe, Mo, Ru, and Rh are promising catalytic centers for NRR; their strong hydrogen adsorption frequently results in HER side reactions, reducing selectivity. Incorporating transition metals such as Zr, Ti, Y, and Sc has been proposed as a strategy to modulate adsorption energetics and mitigate HER competition. RuFeCoNiCu HEAs have demonstrated significant NH3 production at low overpotentials, achieving notable activity at 0.05 V versus the reversible hydrogen electrode (RHE). DFT calculations indicate that Fe when integrated within a multi-component system, serves as an optimal N2 activation site, while Ni–Ru and Co–Cu interactions enhance surface hydrogenation at minimal overpotentials. Furthermore, boron doping in FeCoNiCuPd HEAs has been shown to fine-tune electronic structures, leading to a dual-phase material with enhanced electrochemical NRR performance, boasting a FE of ≈40% and NH3 production rates of ≈25 μmol h−1 cm−2. These findings highlight the potential of compositional tuning in HEAs to push the boundaries of conventional catalytic limitations and improve efficiency and selectivity in ammonia synthesis. Continued experimental validation and advanced theoretical modeling remain imperative to assess their practical viability.180–182
2.5.2.4. In situ and operando studies. In situ surface-enhanced infrared absorption spectroscopy (SEIRAS) has been employed to identify reaction intermediates and elucidate the NRR mechanism on various catalysts. For example, SEIRAS studies on Au thin films detected IR bands corresponding to H–N–H bending, –NH2 wagging, and N–N stretching of *N2Hy (3 ≤ y ≤ 4) species, confirming the presence of an alternative associative pathway involving N2H4 as an intermediate.158 In contrast, SEIRAS analysis of Ru surfaces in acidic media revealed IR signals of *N2Hx (0 ≤ x ≤ 2) species, with an N
N stretching mode appearing at potentials below 0.2 V in 0.1 M HClO4, indicating the involvement of N2H2 as the key intermediate.183 These signals were absent in alkaline electrolytes, suggesting poor N2 adsorption on Ru under such conditions. Unlike Ru, Rh exhibited strong IR signals corresponding to end-on *N2Hx species in alkaline solutions, indicating an associative two-electron transfer pathway where N2H2 decomposes to form NH3.184 Similar SEIRAS studies on NiFe–MoS2 nanocubes,185 Fe-doped ReS2,186 and Al-doped Co3O4187 have further supported associative NRR mechanisms, while phosphorus-doped carbon nanotubes were found to follow a distal pathway.172In situ Raman spectroscopy has been widely utilized to identify reaction intermediates and elucidate the N2 reduction pathway. For instance, Raman analysis of Re2MnS6 revealed a prominent peak at 658 cm−1, corresponding to the N–H stretching mode of *NH2–NH2 species, confirming that the NRR follows an associative alternating pathway facilitated by dual-metal sites.172,188 In contrast, ReS2 exhibited a blue-shifted peak at 709 cm−1, attributed to *NNH2, suggesting a distal pathway over single-metal active sites. Similarly, Bi-based metal–organic frameworks underwent electrochemical transformation into Bi nanoparticles at potentials more negative than −0.5 V, as evidenced by in situ Raman spectroscopy,189 indicating the formation of active Bi species during NRR. Given that Raman spectroscopy relies on laser excitation, which may influence intermediate formation, near-infrared lasers are often preferred to minimize fluorescence and optical interference from catalysts.172
Online differential electrochemical mass spectrometry (DEMS) enables real-time monitoring of volatile intermediates and products during the NRR process, providing direct insights into the reaction mechanism.172,189 The presence of mass-to-charge ratio (m/z) signals at 27, 30, 31, and 33 confirms the formation of N2H4. In contrast, the signal at 15 is attributed to either NH3 or N2H4. Although NH3 and water exhibit overlapping signals (m/z = 17 and 16, respectively), the NH3 signal varies with applied potential, distinguishing it from water. Recently, Shao et al.184 identified N2H+ (m/z = 29) alongside H2 (m/z = 2) at potentials below −0.3 V on Rh, while N2H2+ (m/z = 30) and N2H3+ (m/z = 31) were undetectable, suggesting a two-step reaction pathway involving an initial two-electron transfer to form N2H2, which subsequently decomposes in KOH electrolyte to generate NH3.172,184
Operando X-ray absorption spectroscopy (XAS) provides information on the electronic structure, coordination environment, and oxidation state of catalysts during NRR.172 XAS studies on VN revealed that the position and intensity of the vanadium K-edge white line remained constant at −0.1 and −0.2 V (versus RHE).145 A pre-edge peak at 5468.4 eV indicated the formation of oxynitride species (VN0.7O0.45), which gradually weakened over time, suggesting its transformation to VN. This conversion accelerated with increasing overpotential, reaching 57.8% after 1 hour at −0.2 V. These findings support the hypothesis that VN0.7O0.45 acts as the active site for NRR, with its gradual reduction to VN leading to catalyst deactivation.145,172
In situ X-ray photoelectron spectroscopy (XPS) enables real-time tracking of catalyst valence states and adsorbed intermediates during NRR. Valov et al.190 demonstrated electrochemical activation of N2 at the interface between an iridium microelectrode and (111)-oriented 9.5 mol% yttria-stabilized zirconia (YSZ) at 450 °C and 10−5 Pa. Under cathodic voltages more negative than −1.5 V, a broad N 1s XPS peak at 397–398 eV emerged, which deconvoluted into three components: an N3− ion (397.2 eV) and N2 species in more positive oxidation states (397.7 and 398.6 eV).172,190 The reversibility of the N 1s peak suggested that the reduced nitrogen species accumulated only in the top monolayers and desorbed upon voltage removal. At potentials below −2.0 V, reduction of the solid electrolyte was observed, as indicated by the emergence of lower formal charge Zr 3d and Y 3d components.172,190
The decomposition of NH3 into N2 and H2 also presents a significant challenge in NH3 synthesis, as it competes with the forward reaction and reduces overall yield. This endothermic reaction becomes increasingly favorable at high temperatures, necessitating effective inhibition strategies to maintain ammonia concentrations. The mechanism of NH3 decomposition involves sequential dehydrogenation steps, where NH3 dissociates into NH2, NH, and ultimately atomic nitrogen, recombining to form N2, while H atoms form H2. The efficiency of these steps is primarily dictated by catalyst properties, particularly the adsorption characteristics of nitrogen species. Several strategies have been proposed to mitigate ammonia decomposition. Modifying the catalyst surface by incorporating elements with strong nitrogen affinity, such as certain high-entropy alloys (HEAs), has been shown to stabilize nitrogen intermediates and suppress N2 formation. Optimizing reaction conditions, such as lowering reaction temperatures, increasing system pressure, and maintaining a high H2 partial pressure, can shift the equilibrium toward ammonia synthesis, thereby reducing decomposition rates. The introduction of promoters like potassium (K) has also been found to enhance catalytic performance for NH3 synthesis while selectively inhibiting decomposition pathways. Recent studies have further demonstrated that tuning catalyst supports, such as Ni-based materials with enhanced metal–support interactions and oxygen vacancies, can significantly impact NH3 decomposition kinetics. The design of HEA catalysts with optimized compositions has exhibited high catalytic activity for ammonia synthesis while concurrently suppressing its reverse reaction. These findings underscore the importance of an integrated approach combining catalyst engineering, reaction condition optimization, and selective inhibition strategies to effectively counteract NH3 decomposition and improve ammonia synthesis efficiency.194,195
Innovative electrolyte systems that optimize proton availability and suppress HER are crucial. Non-aqueous systems and hybrid approaches involving ionic liquids or sacrificial proton donors can provide an alternative to traditional aqueous setups, offering improved FEs and NH3 yields. Future work should explore such systems' scalability and environmental impact to ensure commercial viability. Understanding the fundamental mechanisms of eNRR, including dissociative, associative, and Mars–van Krevelen pathways, is essential for tailoring reaction conditions. Real-time spectroscopic and electrochemical techniques can provide deeper insights into intermediate species and reaction kinetics, guiding the rational design of more effective systems.152,153
Moreover, Li-mediated nitrogen reduction reaction faces challenges such as poor energy efficiency, notably lower than the traditional Haber–Bosch process, due to lithium's highly reducing electroplating potential (∼−3.04 V vs. SHE). Additionally, the long-term stability of Li-NRR is compromised by electrolyte degradation, which affects lithium recovery, and the high cost of lithium salts makes the process economically unfeasible. To enhance energy efficiency, exploring metals with lower electroplating potentials could offer significant improvements. Li-NRR proceeds through electrochemical and chemical steps, with lithium's unique properties, such as stable nitride decomposition and an optimal SEI, contributing to its excellent selectivity. Key criteria for effective NH3 electrosynthesis mediators include spontaneous nitride formation, stable surface nitrogen vacancies, exergonic binding of N2, solubility of mediator salts in non-aqueous electrolytes, and facile nitrogen diffusion. Calcium and magnesium were identified as promising alternatives, meeting these criteria alongside lithium. Calcium (∼−2.87 V vs. SHE) has shown favorable performance, with a faradaic efficiency (FE) of 50% for NH3 production using calcium perchlorate and dimethoxy ethane as the electrolyte.170 Magnesium has also demonstrated potential, with an FE of 27%170 in preliminary studies, although recent work by Krebsz et al. reported a lower FE of 7%.196
Integrating eNRR with renewable energy sources such as solar, wind, or hydropower could enhance its sustainability. Coupling eNRR with CO2 reduction or other value-added processes can create multifunctional systems that optimize resource utilization. For instance, hybrid systems that simultaneously address environmental challenges like CO2 sequestration and NH3 synthesis could yield synergistic benefits. Scaling up eNRR technologies to meet industrial demands requires addressing technical bottlenecks like mass transfer limitations and operational stability. Designing continuous flow reactors, improving catalyst durability, and optimizing reaction conditions will be critical. Lifecycle assessments and cost analyses will help ensure the developed processes are economically competitive and environmentally sustainable. The potential applications of eNRR extend beyond NH3 synthesis. Emerging technologies like metal–N2 batteries highlight the versatility of nitrogen reduction in energy storage and conversion systems.152,153 Future research could explore innovative applications of N2 activation in producing other nitrogen-based compounds and energy carriers.
2.6. Thermo-electrochemical N2 activation
The predominant approach in industrial NH3 synthesis involves the Haber–Bosch process, where NH3 is generated through the chemical reaction of N2 and H2 under the influence of iron or ruthenium-based catalysts.197 Generally, the Haber–Bosch process with Fe-based catalysts operates at 300–500 °C temperatures and 130–170 bar pressures.198 To achieve NH3 synthesis under milder conditions, Ru-based catalysts have been used to operate the Haber–Bosch process at relatively low pressures (≤100 bar).159 Despite these efforts, the thermodynamic conversion remains low (around 10–15%), consuming 1–2% of the worldwide annual energy.199As the global demand for NH3 continues to rise, driven by a decrease in fossil fuels and a simultaneous increase in greenhouse gas emissions, there is a growing imperative to develop more sustainable and cost-effective methodologies for NH3 synthesis.200 Recognizing the limitations of the Haber–Bosch process, recent research endeavors are increasingly directed toward optimizing NH3 production across various temperature conditions. The experimental studies are categorized based on their operating temperature into three groups: (a) high temperature (above 500 °C), (b) intermediate temperature (between 500 °C and 100 °C), and (c) low temperature (below 100 °C).200 Currently, most of the research in NH3 production is concentrated on achieving low-temperature synthesis (below 100 °C) in water-based solutions.201 Studies also explore intermediate-temperature methods (from 200 °C to 500 °C) using molten salt electrolytes and high-temperature approaches (above 400 °C) with solid-state electrolytes.200 The high-temperature route could be significant by reducing the energy required due to lower kinetic barriers and improving the efficiency of NH3 formation by enhancing catalytic activity. Both solid-state electrolytes, characterized by their robustness and stability, and molten electrolytes, known for their high ionic conductivity, play pivotal roles in facilitating effective electrochemical pathways at elevated temperatures for NH3 synthesis.200 Fig. 19 provides a temperature-classified map offering detailed insights.202 The red region (above 500 °C) in the center of Fig. 19(A) shows a narrow range of current densities, largely independent of efficiency, which in some cases approaches the practical threshold. The blue region (below 100 °C) features points clustered at lower efficiencies (below 20%) and fails to reach the reaction rates achieved at higher temperatures. Meanwhile, the orange region (100–500 °C) is more scattered, indicating the potential for achieving high energy efficiency or high current densities, though not both simultaneously.
 | ||
| Fig. 19 Performance maps of electrocatalytic nitrogen reduction reactions across temperature regimes. (A) Current density versus energy efficiency, with the DOE target zone highlighted in green. (B) Energy efficiency as a function of temperature, with the mixed water-splitting Haber–Bosch process (HBH2O) indicated in gray. Background color intensity reflects data density from kernel density analysis. Figures have been reproduced from ref. 202 with permission from Elsevier Copyright © 2018 Elsevier Inc. | ||
 | ||
| Fig. 20 (A) Relationship between NH3 yield and varying temperature and pressure.41,206 (B) Arrhenius plots illustrating NH3 synthesis kinetics over a commercial Fe catalyst. (A) and (B) Have been reproduced from ref. 206 with permission from Springer Nature, Copyright © 2020, openly licensed via CC BY 4.0. (C) Possible mechanism of N2 activation over Ru/Ce0.5La0.5O1.75_650red.210 Figure has been reproduced from ref. 211 with permission from the Royal Society of Chemistry, Copyrigh © 2018, openly licensed via CC BY 3.0. | ||
Most of the reactions at the anode and cathode involved gaseous H2 and N2, respectively. Pd-containing catalysts, especially Ag–Pd electrodes, exhibited high reaction rates and FE.200 The introduction of water vapor (steam) was found to enhance protonic conductivity in perovskite electrolytes. When barium cerate-based electrolytes operated with wet hydrogen over the anode, higher reaction rates and FEs were observed.212,213
In contrast to the industrial process, the electrochemical synthesis process bypasses the need for purified gaseous H2, relying on protons (H+) conducted through the solid electrolyte. This approach opens the possibility of using any hydrogen-containing compound, eliminating the extensive purification required in traditional methods.200 Notably, studies demonstrated the feasibility of solid state ammonia synthesis (SSAS) from steam and nitrogen using a solid electrolyte cell with an Ag–Ru/MgO catalyst (cathode).211 Fig. 20(C) illustrates the two modes of N2 adsorption:210 direct interaction, where N2 adsorbs on Ru atoms strongly coupled with the reduced support through strong metal–support interaction (SMSI), and indirect interaction, where N2 adsorbs on Ru atoms with weak coupling to the reduced support. Moreover, an oxygen-ion (O2−) conductor can be employed for SSAS, synthesizing NH3 from gaseous nitrogen and steam. Reaction rates in O2− cells were notably lower than those in proton-conducting cells, potentially due to oxygen-containing compounds at the cathode. The use of gaseous fuel at the anode in O2− cells could potentially reduce energy requirements for in situ hydrogen production.211
In industrial processes, a substantial portion of the overall cost is associated with preparing and purifying hydrogen feed gas, making the electrochemical synthesis approach an attractive alternative. Specific studies, such as Wang et al.,209 reported high reaction rates in SSAS using different electrolytes and hydrogen sources (natural gas, CH4).
Several issues in NH3 synthesis have been identified, such as limitations in proton transport rates and catalyst modification due to ion spillover from the solid electrolyte.205,214 The utilization of water as a proton source resolves problems related to impurity-induced catalyst poisoning.215 Electrochemical NH3 synthesis from steam and N2 via solid-state proton-conducting electrolytes, exemplified by SrCe0.95Yb0.05O3−δ, displayed promising outcomes, producing NH3 at notable rates.211 Reducing the operating temperature to 400 °C significantly boosted NH3 production from wet N2, although concerns regarding proton conductivity and resistance loss emerged.216 Thinner electrolyte membranes are crucial for practical applications, given that bulk proton conductors are relatively thick and sintered at high temperatures. Thin proton-conducting membranes showed reduced resistance and improved NH3 formation rates.212 The electrochemical NH3 synthesis process using solid electrolytes exhibits significant potential, especially when employing high-temperature protonic conductors. There are several challenges also which persist regarding temperature optimization and membrane thickness.205,207–209,212,217
Although this adjustment yielded reduced NH3 yields, hinting at potential NH3 reactions with oxygen ions.221 Subsequent studies introduced nanoparticle catalysts within molten NaOH/KOH electrolytes, enhancing NH3 formation rates albeit with voltage considerations and catalyst particle size being key factors determining activity.200,222 Parallel advancements explored composite electrolytes, combining solid oxides with eutectic mixtures of alkali metal salts, primarily aiming to enhance ionic conductivity and lower operating temperatures for hydrogen fuel cells within the 400 °C to 800 °C range.200 Amar et al. assessed these composite electrolytes for solid-state NH3 synthesis (SSAS) in cell configurations akin to earlier designs.223 Their exploration involving a mixture of (Li/Na/K)2CO3 and LiAlO2 revealed varied catalysts and hydrogen sources, identifying Co3Mo3N as the most promising catalyst, achieving significant rates at 450 °C albeit with relatively lower efficiencies, primarily attributed to rapid hydrogen evolution.223 Noteworthy enhancements in reaction rates were observed with alterations in electrolyte composition, emphasizing the potential of (Li,Na,K)2CO3-SDC as an electrolyte, showcasing an augmented reaction rate compared to prior electrolytes.203,218–221,224
Low-temperature experiments for NH3 synthesis relied on either gaseous H2228,229 or H2O as the hydrogen source, following similar cell configurations as depicted in Fig. 16 and 17, respectively. Varied cathode materials were explored, and noteworthy high reaction rates were observed on mixed oxide and perovskite cathodes.229,230 For instance, Xu et al. reported a notably high rate of 1.13 × 10−8 mol s−1 cm−2 and an exceptional faradaic efficiency of 90.4% using an SFCN electrode.231 Although Lan et al. employed Pt electrodes and operated at 25 °C, achieving a reaction rate and faradaic efficiency not among the highest (1.14 × 10−9 mol s−1 cm−2 and 0.55, respectively), their work significantly contributed to advancing the electrochemical synthesis of NH3 by utilizing abundant reactants (water and air) at ambient conditions (25 °C and atmospheric pressure).232
N triple bond to yield isolated N atoms, which then react with H atoms to form NH3 molecules. In the associative pathway, hydrogenation occurs while the N2 molecule remains bonded, either in end-on or side-on configurations, to active sites.233,234 The end-on configuration involves bonding of one N atom of N2 to active sites, whereas in the side-on configuration, both N atoms bond to active sites.235 The end-on pathway offers distal and alternating pathways for NH3 production, while the side-on pathway primarily follows an enzymatic pathway.233,234 In the distal pathway of the end-on configuration, proton–electron pairs initially attack the N atom furthest from the active site, leading to N2 reduction to NH3.235 The key step involves facilitating electron exchange between N2 and the catalyst, where N2 initially donates electrons from its bonding orbitals to generate NH3 and subsequently attacks the other N atom of N2.235 In the alternating pathway, proton–electron pairs alternately attack both N atoms of N2, resulting in sequential NH3 release.235 The enzymatic pathway parallels the alternating pathway in its hydrogenation process.233,234In electrochemical NRR, ATP-hydrolysis electrons are replaced by those from an external electrode. Regardless of the method, essential steps include N2 adsorption, activation, hydrogenation, and NH3 desorption.235 Effective N2 adsorption in electrochemical NRR involves selecting catalysts with favorable active sites, similar to the biological FeMo cofactor, and other transition metals known for their N2 adsorption and activation capabilities using d-orbital electrons.236,237 Additionally, strategies such as vacancy engineering, edge adsorption, heterojunction, and heteroatoms doping can enhance N2 adsorption by redistributing catalyst charge density.235,238 Furthermore, catalysts with high specific surface areas enhance N2 adsorption by providing more exposed active sites.235,239 The concentration of N2 in the reaction solvent also affects adsorption rates through gas–liquid–solid interface exchange.235,240
N2 activation typically involves a donation-acceptance process, often requiring a negative electrode potential to inject electrons into N2's antibonding π* orbital.108 Other methods include photo-induced and plasmon-driven hot electrons for electron injection.234,241 At this activation step, a free energy increase often occurs, involving *N2 or *N2H formation.234 During hydrogenation, the catalytic environment influences pathways and orders, yielding products like NH3, N2H4, and H2.108 NH3 desorption, crucial for catalytic cycle efficiency, is facilitated by destabilizing *NH2 on strong-binding sites, reducing free energy during *NH3 formation and desorption.234
Produced NH3 readily dissolves in water, facilitating active site release for subsequent cycles. Ensuring electron exchange throughout the NRR process necessitates a conductive catalyst substrate to support continuous electron injection to active sites.108,242 Fig. 21 presents various electrochemical setups for NH3 synthesis, each utilizing different approaches to nitrogen reduction. Panel A shows a solid-state proton-conducting cell that produces NH3 from steam (H2O) and nitrogen (N2). Panel B5,242 depicts an electrochemical membrane reactor where NH3 is synthesized using methane (CH4), steam (H2O), and nitrogen (N2). Panel C243 illustrates a molten salt (NaOH-KOH) cell that facilitates NH3 production through the reaction of hydrogen (H2) with N3− ions. In panel D, a hydrochloric acid (HCl)244 cell is shown, where NH3 is synthesized on a VN/TM (VN on titanium mesh) catalyst via the Mars–van Krevelen mechanism. Finally, panel E presents a KOH cell catalyzed by metal–organic frameworks (MOFs) made of Fe, Cu, and Co,245 highlighting a diverse range of electrolyte and catalyst designs aimed at enhancing NH3 synthesis efficiency. These schematics underscore the evolving strategies across different reaction environments for sustainable NH3 production.
 | ||
| Fig. 21 (A) Schematic of a solid-state proton-conducting cell showing the process of NH3 synthesis using steam (H2O) and N2.242 (B) Schematic of an electrochemical membrane reactor illustrating NH3 synthesis using CH4, H2O, and N2.242 (C) Schematic of a molten salt (NaOH-KOH) cell where NH3 is produced from the reaction of H2 with N3− ions.242,243 (D) Schematic of a HCl cell where NH3 is produced on a VN/(titanium mesh) (VN/TM) catalyst via a Mars–van Krevelen mechanism.242,244 (E) Diagram of the KOH cell where NH3 production was catalyzed by metal–organic frameworks (MOFs) of Fe, Cu, and Co.242 Figures have been reproduced with permission from ref. 242 © 2019 by the authors. Licensee MDPI, Basel, Switzerland, openly licensed via CC BY 3.0. | ||
Addressing practical challenges includes overcoming low NH3 yield and Faraday efficiency (FE). Challenges include the competitive hydrogen evolution reaction (HER) at similar standard potentials, hindering electron availability for NRR.246 High energy barriers associated with N-related intermediates during hydrogenation or NH3 desorption can also impede active sites and promote HER.235 Catalyst properties such as intrinsic activity, porosity, specific surface area, and substrate conductivity also influence NH3 production efficiency.
Advancing this field requires a collaborative effort encompassing both theoretical and practical dimensions. A thorough comprehension of the high-temperature electrocatalytic NH3 synthesis mechanism across diverse electrocatalysts is imperative. This involves conducting experimental and theoretical studies to explore reaction pathways on various active surfaces, from precious metals to metal nitrides and perovskite oxides. Leveraging advanced characterization techniques like in situ/in operando spectroscopy can offer deeper insights into these processes, and innovative electrode design is also paramount. Relying on precious metal-based electrodes might be rethought in favor of novel electrocatalysts that can sustain high activity while suppressing dominant hydrogen evolution reactions. Computational studies hint at transition metal nitrides significantly impacting nitrogen reduction for NH3 synthesis via specific mechanisms.254 Resolving debates around nitrogen source origins requires meticulous isotope labeling experiments, clarifying whether reported production values stem from potential contamination or the materials themselves.
3. Summary and overall outlook
This review offers an in-depth exploration of the challenges and recent advancements in nitrogen activation, focusing on the activation and catalytic transformation of atmospheric N2 into biologically and industrially valuable compounds, such as NH3 and nitrates. Nitrogen activation is central to global agriculture, chemical production, and ecological balance, as it provides reactive nitrogen essential for fertilizers, fine chemicals, and biological processes. The Haber–Bosch process, a century-old innovation, continues to dominate industrial nitrogen activation due to its efficiency and scalability. Its high energy consumption, reliance on fossil fuels, and significant carbon emissions have driven research into alternative methods. Emerging approaches are being developed to address these limitations, including biological nitrogen activation, plasma-assisted processes, electrochemical and thermochemical methods, and homogeneous catalysis.Biological nitrogen activation, mediated by nitrogenase enzymes, operates under ambient conditions and provides a sustainable pathway for nitrogen conversion. The process is inherently energy-intensive, as it requires substantial ATP to overcome the stability of the NN triple bond. Similarly, mechanistic insights into catalytic nitrogen activation reveal the complexity of breaking the triple bond, with transition metals playing a pivotal role by facilitating back-donation of electrons into nitrogen's antibonding orbitals. Novel catalyst designs, such as Ru-based systems and metal hydrides, can help reduce the reaction barriers. Furthermore, homogeneous catalytic systems inspired by enzymatic structures have demonstrated significant progress, with molybdenum- and iron-based complexes achieving selective N2 reduction under mild conditions.
A significant focus in recent research has been on advancing sustainable hydrogen sources to support nitrogen activation processes. Innovations in methane pyrolysis and direct seawater electrolysis aim to reduce carbon emissions associated with hydrogen production. These technologies provide greener alternatives and integrate well with emerging electrochemical nitrogen activation methods. Non-thermal plasma techniques have also garnered attention for their ability to activate nitrogen under atmospheric conditions, offering a potential route for decentralized, non-thermal nitrogen activation. Achieving selectivity and minimizing by-products in alternative catalytic methods remain active research areas. Optimizing catalytic systems to reduce reaction conditions, improving the energy efficiency of hydrogen production, and leveraging the insights from biological nitrogen activation are critical steps for progress.
4. Conclusions
This review highlights the different dinitrogen (N2) activation methods that offer a transformative pathway for developing energy-efficient ammonia synthesis and other nitrogen-based chemical products. We have seen that N2's inertness stems from its high bond dissociation energy, large HOMO–LUMO gap, and minimal polarity factors demanding extraordinary reaction conditions or specialized catalysts to achieve practical conversion into valuable products such as ammonia, nitrates, urea, or other nitrogen-containing compounds. The substantial energy requirements of Haber–Bosch process, its reliance on fossil-derived hydrogen, and inevitable CO2 emissions have stimulated extensive research into alternative pathways and catalysts.We surveyed an array of emerging strategies for nitrogen fixation using nitrogenase enzymes, homogeneous and metallocomplex routes leveraging transition-metal complexes, plasma-based activation inspired by lightning, electrochemical approaches driven by renewable electricity, and integrated thermo-electrochemical processes that combine the best of both high-temperature and electrochemical methods. Each route presents specific benefits and challenges: biological systems can circumvent harsh industrial conditions but face scalability limits; homogeneous catalysts enable selective transformations yet often struggle with product separation; plasma processes provide high reactivity but can be energy-intensive; electrochemical methods offer greener, on-demand synthesis but require breakthroughs in catalyst selectivity to overcome competing side reactions such as the hydrogen evolution reaction; and thermo-electrochemical platforms aim to capitalize on robust materials and high protonic conductivity but must still balance reaction kinetics and energy costs.
Collectively, these insights highlight that no single method offers a universal solution. Instead, future progress will depend on converging multiple innovations: designing catalysts that selectively favor N2 activation over undesired reactions, enhancing active-site density via advanced nanostructures or single-atom approaches, coupling reaction systems with renewable energy sources, improving our mechanistic understanding of elementary reaction steps through in situ/operando techniques, and employing rigorous protocols (including isotope labeling) to validate performance. Successful breakthroughs in these areas could ultimately transform nitrogen fixation into a more sustainable, decentralized, and energy-efficient process. As research accelerates in catalysis, materials science, and process engineering, the collective goal remains clear: to unlock the full potential of N2 as a readily available feedstock for vital chemicals and clean energy carriers, thereby contributing to a more resilient and sustainable global economy.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This material is based on the work performed in the Materials and Systems Engineering Laboratory at the University of Illinois Chicago (UIC) and the Gauthier Laboratory at Texas Tech University (TTU). The authors used ‘Generative AI on text selection’ by ‘Grammarly’ to improve readability and language of the article. This material is based upon work supported by the National Science Foundation (NSF) Engineering Research Center for Advancing Sustainable and Distributed Fertilizer Production (CASFER) under Grant No. 2133576. S. A. O. and J. A. G. gratefully acknowledge support from The Welch Foundation under Grant NumberD-2188-20240404.References
- H. Iriawan, S. Z. Andersen, X. Zhang, B. M. Comer, J. Barrio, P. Chen, A. J. Medford, I. E. Stephens, I. Chorkendorff and Y. Shao-Horn, Nat. Rev. Methods Primers, 2021, 1, 56 CrossRef CAS.
- B. Chang, H. Zhang, S. Sun and G. Zhang, Carbon Energy, 2024, 6, e491 CrossRef CAS.
- University Chemistry, 4/E, Pearson Education, 2009.
- A. O'Connor, The Undergraduate Historical Journal at UC Merced, 2014, 2 Search PubMed.
- A. Rovaletti, L. De Gioia, C. Greco and F. Arrigoni, Dalton Trans., 2023, 52, 7966–7974 RSC.
- J. D. Cox, D. D. Wagman and V. A. Medvedev, CODATA—Key Values for Thermodynamics, aus der Reihe: CODATA, Series on Thermodynamic Properties, in Berichte der Bunsengesellschaft für physikalische Chemie, Hemisphere Publishing Corporation, New York, Washington, Philadelphia, London, 1989 DOI:10.1002/bbpc.19900940121.
- B. L. Oberholtzer, Publications of the National Bureau of Standards, U.S. Department of Commerce, 1970, p. 378.
- H.-P. Jia and E. A. Quadrelli, Chem. Soc. Rev., 2014, 43, 547–564 RSC.
- D.-Y. Hwang and A. M. Mebel, J. Phys. Chem. A, 2003, 107, 2865–2874 CrossRef CAS.
- Y. Huang, D. D. Babu, Z. Peng and Y. Wang, Adv. Sci., 2020, 7, 1902390 CrossRef CAS PubMed.
- A. Shilov, Russ. Chem. Bull., 2003, 52, 2555–2562 CrossRef CAS.
- K. H. R. Rouwenhorst, P. M. Krzywda, N. E. Benes, G. Mul and L. Lefferts, Techno-Economic Challenges of Green Ammonia as an Energy Vector, 2021, pp. 41–83 Search PubMed.
- C. Smith, A. K. Hill and L. Torrente-Murciano, Energy Environ. Sci., 2020, 13, 331–344 RSC.
- T. D. Rapson and C. C. Wood, Catalysts, 2022, 12, 844 CrossRef CAS.
- R. K. Sharma, H. Patel, U. Mushtaq, V. Kyriakou, G. Zafeiropoulos, F. Peeters, S. Welzel, M. C. Van De Sanden and M. N. Tsampas, ACS Energy Lett., 2020, 6, 313–319 CrossRef.
- M. K. Sosna, Y. A. Sokolinskii, D. Khudyakov and A. Lapidus, Solid Fuel Chem., 2022, 56, 456–461 CrossRef CAS.
- V. Smil, Nature, 1999, 400, 415 CrossRef CAS.
- J. G. Speight, Kirk-Othmer Encycl. Chem. Technol., 2000 DOI:10.1002/0471238961.0701190519160509.a01.
- F. Haber, Nobel Lect., 1920, 2, 1 Search PubMed.
- A. Frank and N. Caro, DE88363, 1895.
- The Rjukan–Notodden Industrial World Heritage Site, https://ra.brage.unit.no/ra-xmlui/bitstream/handle/11250/177016/Verdensarv_norsk_industriarv_submission_nov_2009.pdf?sequence=1&isAllowed=y.
- A. Silverman, J. Chem. Educ., 1938, 15, 289 CrossRef.
- R. Oesper, J. Chem. Educ., 1948, 25, 531 CrossRef CAS.
- A. Vojvodic, A. J. Medford, F. Studt, F. Abild-Pedersen, T. S. Khan, T. Bligaard and J. Nørskov, Chem. Phys. Lett., 2014, 598, 108–112 CrossRef CAS.
- G. Ertl, Angew. Chem., Int. Ed., 2008, 47, 3524–3535 CrossRef CAS PubMed.
- T. Zambelli, J. Trost, J. Wintterlin and G. Ertl, Phys. Rev. Lett., 1996, 76, 795 CrossRef CAS PubMed.
- R. Imbihl, R. J. Behm, G. Ertl and W. Moritz, Surf. Sci., 1982, 123, 129–140 CrossRef CAS.
- F. Bozso, G. Ertl, M. Grunze and M. Weiss, J. Catal., 1977, 49, 18–41 CrossRef CAS.
- G. Ertl, Catal. Rev.:Sci. Eng., 1980, 21, 201–223 CrossRef CAS.
- S. Dahl, A. Logadottir, R. Egeberg, J. Larsen, I. Chorkendorff, E. Törnqvist and J. K. Nørskov, Phys. Rev. Lett., 1999, 83, 1814 CrossRef.
- G. Ertl, M. Weiss and S. Lee, Chem. Phys. Lett., 1979, 60, 391–394 CrossRef CAS.
- B. A. Rohr, A. R. Singh and J. K. Nørskov, J. Catal., 2019, 372, 33–38 CrossRef CAS.
- G. P. Connor and P. L. Holland, Catal. Today, 2017, 286, 21–40 CrossRef CAS PubMed.
- M. Kitano, Y. Inoue, Y. Yamazaki, F. Hayashi, S. Kanbara, S. Matsuishi, T. Yokoyama, S.-W. Kim, M. Hara and H. Hosono, Nat. Chem., 2012, 4, 934–940 CrossRef CAS PubMed.
- Y. Inoue, M. Kitano, K. Kishida, H. Abe, Y. Niwa, M. Sasase, Y. Fujita, H. Ishikawa, T. Yokoyama and M. Hara, ACS Catal., 2016, 6, 7577–7584 CrossRef CAS.
- Y. Kobayashi, Y. Tang, T. Kageyama, H. Yamashita, N. Masuda, S. Hosokawa and H. Kageyama, J. Am. Chem. Soc., 2017, 139, 18240–18246 CrossRef CAS PubMed.
- W. Gao, P. Wang, J. Guo, F. Chang, T. He, Q. Wang, G. Wu and P. Chen, ACS Catal., 2017, 7, 3654–3661 CrossRef CAS.
- A. Cao, V. J. Bukas, V. Shadravan, Z. Wang, H. Li, J. Kibsgaard, I. Chorkendorff and J. K. Nørskov, Nat. Commun., 2022, 13, 2382 CrossRef CAS PubMed.
- S. Hagen, R. Barfod, R. Fehrmann, C. J. Jacobsen, H. T. Teunissen, K. Ståhl and I. Chorkendorff, Chem. Commun., 2002, 1206–1207 RSC.
- M. Kitano, Y. Inoue, H. Ishikawa, K. Yamagata, T. Nakao, T. Tada, S. Matsuishi, T. Yokoyama, M. Hara and H. Hosono, Chem. Sci., 2016, 7, 4036–4043 RSC.
- M. Hattori, T. Mori, T. Arai, Y. Inoue, M. Sasase, T. Tada, M. Kitano, T. Yokoyama, M. Hara and H. Hosono, ACS Catal., 2018, 8, 10977–10984 CrossRef CAS.
- P. Wang, F. Chang, W. Gao, J. Guo, G. Wu, T. He and P. Chen, Nat. Chem., 2017, 9, 64–70 CrossRef CAS PubMed.
- T.-N. Ye, S.-W. Park, Y. Lu, J. Li, M. Sasase, M. Kitano, T. Tada and H. Hosono, Nature, 2020, 583, 391–395 CrossRef CAS PubMed.
- T.-N. Ye, S.-W. Park, Y. Lu, J. Li, M. Sasase, M. Kitano and H. Hosono, J. Am. Chem. Soc., 2020, 142, 14374–14383 CrossRef CAS PubMed.
- Z. Ma, X. Xiong, C. Song, B. Hu and W. Zhang, RSC Adv., 2016, 6, 51106–51110 RSC.
- V. Shadravan, A. Cao, V. J. Bukas, M. K. Grønborg, C. D. Damsgaard, Z. Wang, J. Kibsgaard, J. K. Nørskov and I. Chorkendorff, Energy Environ. Sci., 2022, 15, 3310–3320 RSC.
- S. Wu, Y.-K. Peng, T.-Y. Chen, J. Mo, A. Large, I. McPherson, H.-L. Chou, I. Wilkinson, F. Venturini and D. Grinter, ACS Catal., 2020, 10, 5614–5622 CrossRef CAS.
- T. Brown, Chem. Eng. Prog., 2019, 115, 47–53 CAS.
- M. Khan, T. Al-Attas, S. Roy, M. M. Rahman, N. Ghaffour, V. Thangadurai, S. Larter, J. Hu, P. M. Ajayan and M. G. Kibria, Energy Environ. Sci., 2021, 14, 4831–4839 RSC.
- M. F. Lagadec and A. Grimaud, Nat. Mater., 2020, 19, 1140–1150 CrossRef CAS PubMed.
- S. r Dresp, F. Dionigi, M. Klingenhof and P. Strasser, ACS Energy Lett., 2019, 4, 933–942 CrossRef CAS.
- S. T. Wismann, J. S. Engbæk, S. B. Vendelbo, F. B. Bendixen, W. L. Eriksen, K. Aasberg-Petersen, C. Frandsen, I. Chorkendorff and P. M. Mortensen, Science, 2019, 364, 756–759 CrossRef CAS PubMed.
- D. C. Upham, V. Agarwal, A. Khechfe, Z. R. Snodgrass, M. J. Gordon, H. Metiu and E. W. McFarland, Science, 2017, 358, 917–921 CrossRef CAS PubMed.
- C. Palmer, M. Tarazkar, H. H. Kristoffersen, J. Gelinas, M. J. Gordon, E. W. McFarland and H. Metiu, ACS Catal., 2019, 9, 8337–8345 CrossRef CAS.
- D. Kang, N. Rahimi, M. J. Gordon, H. Metiu and E. W. McFarland, Appl. Catal., B, 2019, 254, 659–666 CrossRef CAS.
- D. A. Zuberer, Principles and applications of soil microbiology, Elsevier, 2021, pp. 423–453 Search PubMed.
- R. C. Burns and R. W. F. Hardy, Nitrogen Fixation in Bacteria and Higher Plants, Molecular Biology, Biochemistry and Biophysics, Springer Berlin Heidelberg, Germany, 2012 Search PubMed.
- A. Adeniyi, I. Bello, T. Mukaila, N. C. Sarker and A. Hammed, BioTech, 2023, 12, 41 CAS.
- C. C. Cleveland, A. R. Townsend, D. S. Schimel, H. Fisher, R. W. Howarth, L. O. Hedin, S. S. Perakis, E. F. Latty, J. C. Von Fischer and A. Elseroad, Global Biogeochem. Cycles, 1999, 13, 623–645 CrossRef CAS.
- D. Fowler, M. Coyle, U. Skiba, M. A. Sutton, J. N. Cape, S. Reis, L. J. Sheppard, A. Jenkins, B. Grizzetti and J. N. Galloway, Philos. Trans. R. Soc., B, 2013, 368, 20130164 CrossRef PubMed.
- J. K. Ladha, M. B. Peoples, P. M. Reddy, J. C. Biswas, A. Bennett, M. L. Jat and T. J. Krupnik, Field Crops Res., 2022, 283, 108541 CrossRef PubMed.
- F. J. de Bruijn and M. Hungria, Good Microbes Med., Food Prod., Biotechnol., Biorem., Agric., 2022, 466–475 CAS.
- N. Rascio and N. La Rocca, Encyclopedia of ecology, Elsevier, 2008, vol. 1, pp. 412–419 Search PubMed.
- R. H. Burris and G. P. Roberts, Annu. Rev. Nutr., 1993, 13, 317–335 CrossRef CAS PubMed.
- J. R. Postgate, Nitrogen fixation, Cambridge University Press, 1998 Search PubMed.
- J. Young and G. Stacey, in Phylogenetic Classification of Nitrogen Nitrogen-¢ xing Organism, ed. G. Stacey, R. H. Burris and H. J. Evans, 1992, pp. 43–86 Search PubMed.
- D. Gadkari, Catalysts for Nitrogen Fixation: Nitrogenases, Relevant Chemical Models and Commercial Processes, Springer, 2004, pp. 309–332 Search PubMed.
- M. Ribbe, D. Gadkari and O. Meyer, J. Biol. Chem., 1997, 272, 26627–26633 CrossRef CAS PubMed.
- D. MacKellar, L. Lieber, J. S. Norman, A. Bolger, C. Tobin, J. W. Murray, M. Oksaksin, R. L. Chang, T. J. Ford and P. Q. Nguyen, Sci. Rep., 2016, 6, 20086 CrossRef CAS PubMed.
- V. Krewald, Dalton Trans., 2018, 47, 10320–10329 RSC.
- I. Fischler and E. K. von Gustorf, Naturwissenschaften, 1975, 62, 63–70 CrossRef CAS.
- L. C. Seefeldt, B. M. Hoffman and D. R. Dean, Curr. Opin. Chem. Biol., 2012, 16, 19–25 CrossRef CAS PubMed.
- R. Cai and S. D. Minteer, ACS Energy Lett., 2018, 3, 2736–2742 CrossRef CAS.
- R. Bjornsson, F. A. Lima, T. Spatzal, T. Weyhermueller, P. Glatzel, E. Bill, O. Einsle, F. Neese and S. DeBeer, Chem. Sci., 2014, 5, 3096–3103 RSC.
- S. M. Keable, O. A. Zadvornyy, L. E. Johnson, B. Ginovska, A. J. Rasmussen, K. Danyal, B. J. Eilers, G. A. Prussia, A. X. LeVan and S. Raugei, J. Biol. Chem., 2018, 293, 9629–9635 CrossRef CAS PubMed.
- D. Sippel and O. Einsle, Nat. Chem. Biol., 2017, 13, 956–960 CrossRef CAS PubMed.
- B. M. Hoffman, D. Lukoyanov, Z.-Y. Yang, D. R. Dean and L. C. Seefeldt, Chem. Rev., 2014, 114, 4041–4062 CrossRef CAS PubMed.
- T. Spatzal, M. Aksoyoglu, L. Zhang, S. L. Andrade, E. Schleicher, S. Weber, D. C. Rees and O. Einsle, Science, 2011, 334, 940 CrossRef CAS PubMed.
- O. Einsle and D. C. Rees, Chem. Rev., 2020, 120, 4969–5004 CrossRef CAS PubMed.
- B. K. Burgess and D. J. Lowe, Chem. Rev., 1996, 96, 2983–3012 CrossRef CAS PubMed.
- G. Koskey, S. W. Mburu, J. M. Kimiti, O. Ombori, J. M. Maingi and E. M. Njeru, Front. Microbiol., 2018, 9, 968 CrossRef PubMed.
- H. B. Vuong, P. H. Thrall and L. G. Barrett, J. Ecol., 2017, 105, 540–548 CrossRef CAS.
- M. Laranjo and S. Oliveira, Antonie van Leeuwenhoek, 2011, 99, 651–662 CrossRef CAS PubMed.
- R. K. Goyal, M. A. Schmidt and M. F. Hynes, Microorganisms, 2021, 9, 125 CrossRef CAS PubMed.
- J. Yang, Catalysts, 2019, 9, 939 CrossRef CAS.
- F. B. Simpson and R. H. Burris, Science, 1984, 224, 1095–1097 CrossRef CAS PubMed.
- Q. Wang, Y. Guan, J. Guo and P. Chen, Cell Rep. Phys. Sci., 2022, 3, 100779 CrossRef CAS.
- F. A. Tezcan, J. T. Kaiser, J. B. Howard and D. C. Rees, J. Am. Chem. Soc., 2015, 137, 146–149 CrossRef CAS PubMed.
- L. C. Seefeldt, Z.-Y. Yang, D. A. Lukoyanov, D. F. Harris, D. R. Dean, S. Raugei and B. M. Hoffman, Chem. Rev., 2020, 120, 5082–5106 CrossRef CAS PubMed.
- J. Fiolet, A. Baartscheer, C. Schumacher, R. Coronel and H. Ter Welle, J. Mol. Cell. Cardiol., 1984, 16, 1023–1036 CrossRef CAS PubMed.
- H. Kammermeier, P. Schmidt and E. Jüngling, J. Mol. Cell. Cardiol., 1982, 14, 267–277 CrossRef CAS PubMed.
- N. Cherkasov, A. Ibhadon and P. Fitzpatrick, Chem. Eng. Process., 2015, 90, 24–33 CrossRef CAS.
- R. A. Alberty, Arch. Biochem. Biophys., 2001, 389, 94–109 CrossRef CAS PubMed.
- R. N. Thorneley and D. Lowe, Biochem. J., 1984, 224, 887–894 CrossRef CAS PubMed.
- B. M. Hoffman, D. Lukoyanov, D. R. Dean and L. C. Seefeldt, Acc. Chem. Res., 2013, 46, 587–595 CrossRef CAS PubMed.
- B. M. Hoffman, D. R. Dean and L. C. Seefeldt, Acc. Chem. Res., 2009, 42, 609–619 CrossRef CAS PubMed.
- F. Neese, Angew. Chem., Int. Ed., 2006, 45, 196–199 CrossRef CAS PubMed.
- B. Hinnemann and J. K. Nørskov, Top. Catal., 2006, 37, 55–70 CrossRef CAS.
- J. Kästner and P. E. Blöchl, J. Am. Chem. Soc., 2007, 129, 2998–3006 CrossRef PubMed.
- R. R. Schrock, Acc. Chem. Res., 2005, 38, 955–962 CrossRef CAS PubMed.
- S. Kuriyama and Y. Nishibayashi, Tetrahedron, 2021, 83, 131986 CrossRef CAS.
- M. Vol'Pin and V. Shur, Nature, 1966, 209, 1236 CrossRef.
- K. Shiina, J. Am. Chem. Soc., 1972, 94, 9266–9267 CrossRef CAS.
- E.-Y. Jeong, C.-Y. Yoo, C. H. Jung, J. H. Park, Y. C. Park, J.-N. Kim, S.-G. Oh, Y. Woo and H. C. Yoon, ACS Sustainable Chem. Eng., 2017, 5, 9662–9666 CrossRef CAS.
- H. Liu, L. Wei, F. Liu, Z. Pei, J. Shi, Z.-J. Wang, D. He and Y. Chen, ACS Catal., 2019, 9, 5245–5267 CrossRef CAS.
- H. Basch, D. G. Musaev, K. Morokuma, M. D. Fryzuk, J. B. Love, W. W. Seidel, A. Albinati, T. F. Koetzle, W. T. Klooster and S. A. Mason, J. Am. Chem. Soc., 1999, 121, 523–528 CrossRef CAS.
- I. Čorić and P. L. Holland, J. Am. Chem. Soc., 2016, 138, 7200–7211 CrossRef PubMed.
- S. Wang, F. Ichihara, H. Pang, H. Chen and J. Ye, Adv. Funct. Mater., 2018, 28, 1803309 CrossRef.
- Y. Nishibayashi, Dalton Trans., 2012, 41, 7447–7453 RSC.
- K. Arashiba, Y. Miyake and Y. Nishibayashi, Nat. Chem., 2011, 3, 120–125 CrossRef CAS PubMed.
- D. V. Yandulov and R. R. Schrock, Science, 2003, 301, 76–78 CrossRef CAS PubMed.
- S. Kuriyama, K. Arashiba, H. Tanaka, Y. Matsuo, K. Nakajima, K. Yoshizawa and Y. Nishibayashi, Angew. Chem., Int. Ed., 2016, 55, 14291–14295 CrossRef CAS PubMed.
- A. J. Kendall, S. I. Johnson, R. M. Bullock and M. T. Mock, J. Am. Chem. Soc., 2018, 140, 2528–2536 CrossRef CAS PubMed.
- Y. Sekiguchi, K. Arashiba, H. Tanaka, A. Eizawa, K. Nakajima, K. Yoshizawa and Y. Nishibayashi, Angew. Chem., Int. Ed., 2018, 57, 9064–9068 CrossRef CAS PubMed.
- K. Hashizume, Y. Mizobe and M. Hidai, Organometallics, 1996, 15, 3303–3309 CrossRef CAS.
- J. Chatt, A. Pearman and R. Richards, Nature, 1975, 253, 39–40 CrossRef CAS.
- J. Y. Becker and S. Avraham, J. Electroanal. Chem. Interfacial Electrochem., 1990, 280, 119–127 CrossRef CAS.
- N. Furuya and H. Yoshiba, J. Electroanal. Chem. Interfacial Electrochem., 1989, 263, 171–174 CrossRef CAS.
- N. Furuya and H. Yoshiba, J. Electroanal. Chem. Interfacial Electrochem., 1989, 272, 263–266 CrossRef CAS.
- T. J. Del Castillo, N. B. Thompson and J. C. Peters, J. Am. Chem. Soc., 2016, 138, 5341–5350 CrossRef CAS PubMed.
- T. Li, S. Han, Y. Wang, J. Zhou, B. Zhang and Y. Yu, Angew. Chem., Int. Ed., 2023, 62, e202217411 CrossRef CAS PubMed.
- C. E. Laplaza and C. C. Cummins, Science, 1995, 268, 861–863 CrossRef CAS PubMed.
- K. Arashiba, A. Eizawa, H. Tanaka, K. Nakajima, K. Yoshizawa and Y. Nishibayashi, Bull. Chem. Soc. Jpn., 2017, 90, 1111–1118 CrossRef CAS.
- Y. Roux, C. Duboc and M. Gennari, ChemPhysChem, 2017, 18, 2606–2617 CrossRef CAS PubMed.
- Y. Lee, F. T. Sloane, G. Blondin, K. A. Abboud, R. García-Serres and L. J. Murray, Angew. Chem., Int. Ed., 2015, 54, 1499–1503 CrossRef CAS PubMed.
- M. M. Rodriguez, E. Bill, W. W. Brennessel and P. L. Holland, Science, 2011, 334, 780–783 CrossRef CAS PubMed.
- K. H. Rouwenhorst, F. Jardali, A. Bogaerts and L. Lefferts, Energy Environ. Sci., 2021, 14, 2520–2534 RSC.
- L. R. Winter and J. G. Chen, Joule, 2021, 5, 300–315 CrossRef CAS.
- P. Peng, P. Chen, C. Schiappacasse, N. Zhou, E. Anderson, D. Chen, J. Liu, Y. Cheng, R. Hatzenbeller and M. Addy, J. Cleaner Prod., 2018, 177, 597–609 CrossRef CAS.
- V. D. Rusanov, A. Fridman and G. V. E. Sholin, Sov. Phys.-Usp., 1981, 24, 447 CrossRef.
- T. Grigoreva, A. Levitskii, S. Macheret, L. Polak, V. Rusanov and A. Fridman, High Energy Chem., 1984, 18, 268–272 Search PubMed.
- A. Gicquel, S. Cavadias and J. Amouroux, J. Phys. D:Appl. Phys., 1986, 19, 2013 CrossRef CAS.
- P. Peng, C. Schiappacasse, N. Zhou, M. Addy, Y. Cheng, Y. Zhang, K. Ding, Y. Wang, P. Chen and R. Ruan, ChemSusChem, 2019, 12, 3702–3712 CrossRef CAS PubMed.
- R. Azizov, V. Zhivotov, M. Krotov, V. Rusanov, Y. Tarasov and A. Fridma, Khim. Vys. Energ., 1980, 14, 366–368 CAS.
- E. Eremin, V. Belova and A. Mal'tsev, Zh. Fiz. Khim., 1978, 52, 1679–1682 CAS.
- J. Van Durme, J. Dewulf, C. Leys and H. Van Langenhove, Appl. Catal., B, 2008, 78, 324–333 CrossRef CAS.
- Y. He, Z. Chen, Z. Li, G. Niu and J. Tang, Front. Optoelectron., 2018, 11, 92–96 CrossRef.
- X. Zhao and Y. Tian, Cell Rep. Phys. Sci., 2023, 4, 101618 CrossRef CAS.
- D. Liu, H. Yan, J. Lin, S. Lu, Y. Xie, X. Peng, S. Liang and L. Jiang, Chem. Eng. Sci., 2024, 283, 119448 CrossRef CAS.
- J. Lin, X. Lin, S. Lu, W. Liao, T. Qi, S. Liang, Z.-J. Zhao and L. Jiang, Chem. Eng. Sci., 2024, 300, 120664 CrossRef CAS.
- Z. Sun, J. Lin, S. Lu, Y. Li, T. Qi, X. Peng, S. Liang and L. Jiang, Langmuir, 2024, 40, 5469–5478 CrossRef CAS PubMed.
- W. Liao, L. Qi, Y. Wang, J. Qin, G. Liu, S. Liang, H. He and L. Jiang, Adv. Funct. Mater., 2021, 31, 2009151 CrossRef CAS.
- B. H. R. Suryanto, D. Wang, L. M. Azofra, M. Harb, L. Cavallo, R. Jalili, D. R. G. Mitchell, M. Chatti and D. R. MacFarlane, ACS Energy Lett., 2019, 4, 430–435 CrossRef CAS.
- W. Liao, K. Xie, L. Liu, X. Wang, Y. Luo, S. Liang, F. Liu and L. Jiang, J. Energy Chem., 2021, 62, 359–366 CrossRef CAS.
- X. Yang, J. Nash, J. Anibal, M. Dunwell, S. Kattel, E. Stavitski, K. Attenkofer, J. G. Chen, Y. Yan and B. Xu, J. Am. Chem. Soc., 2018, 140, 13387–13391 CrossRef CAS PubMed.
- H. Fang, D. Liu, Y. Luo, Y. Zhou, S. Liang, X. Wang, B. Lin and L. Jiang, ACS Catal., 2022, 12, 3938–3954 CrossRef CAS.
- M. Anand, C. S. Abraham and J. K. Nørskov, Chem. Sci., 2021, 12, 6442–6448 RSC.
- S. A. Olusegun, Y. Qi, N. C. Kani, M. R. Singh and J. A. Gauthier, ACS Catal., 2024, 14, 16885–16896 CrossRef CAS.
- G. Qing, R. Ghazfar, S. T. Jackowski, F. Habibzadeh, M. M. Ashtiani, C.-P. Chen, M. R. Smith III and T. W. Hamann, Chem. Rev., 2020, 120, 5437–5516 CrossRef CAS PubMed.
- S. Z. Andersen, V. Čolić, S. Yang, J. A. Schwalbe, A. C. Nielander, J. M. McEnaney, K. Enemark-Rasmussen, J. G. Baker, A. R. Singh and B. A. Rohr, Nature, 2019, 570, 504–508 CrossRef CAS PubMed.
- J. Choi, B. H. Suryanto, D. Wang, H.-L. Du, R. Y. Hodgetts, F. M. Ferrero Vallana, D. R. MacFarlane and A. N. Simonov, Nat. Commun., 2020, 11, 5546 CrossRef CAS PubMed.
- A. U. Shetty and R. Sankannavar, J. Energy Chem., 2024, 92, 681–697 CrossRef CAS.
- S. C. Jesudass, S. Surendran, J. Y. Kim, T.-Y. An, G. Janani, T.-H. Kim, J. K. Kim and U. Sim, Electrochem. Energy Rev., 2023, 6, 27 CrossRef CAS.
- C. Hu, X. Chen, J. Jin, Y. Han, S. Chen, H. Ju, J. Cai, Y. Qiu, C. Gao, C. Wang, Z. Qi, R. Long, L. Song, Z. Liu and Y. Xiong, J. Am. Chem. Soc., 2019, 141, 7807–7814 CrossRef CAS PubMed.
- K.-i Aika, Catal. Today, 2017, 286, 14–20 CrossRef CAS.
- Y. Kobayashi, M. Kitano, S. Kawamura, T. Yokoyama and H. Hosono, Catal.:Sci. Technol., 2017, 7, 47–50 RSC.
- C. D. Zeinalipour-Yazdi, J. S. J. Hargreaves and C. R. A. Catlow, J. Phys. Chem. C, 2018, 122, 6078–6082 CrossRef CAS.
- Y. Yao, S. Zhu, H. Wang, H. Li and M. Shao, J. Am. Chem. Soc., 2018, 140, 1496–1501 CrossRef CAS PubMed.
- M. A. Shipman and M. D. Symes, Catal. Today, 2017, 286, 57–68 CrossRef CAS.
- C. Yang, Y. Zhu, J. Liu, Y. Qin, H. Wang, H. Liu, Y. Chen, Z. Zhang and W. Hu, Nano Energy, 2020, 77, 105126 CrossRef CAS.
- X. Cui, T. Cheng and Q. Zhang, Adv. Energy Mater., 2018, 8, 1800369 CrossRef.
- J. Kibsgaard, J. K. Nørskov and I. Chorkendorff, ACS Energy Lett., 2019, 4, 2986–2988 CrossRef CAS.
- P. Mars and D. W. van Krevelen, Chem. Eng. Sci., 1954, 3, 41–59 CrossRef CAS.
- H. Y. Kim and G. Henkelman, J. Phys. Chem. Lett., 2013, 4, 216–221 CrossRef CAS PubMed.
- S. U. Haq, M. Aghajamali and H. Hassanzadeh, RSC Adv., 2021, 11, 24387–24397 RSC.
- C. D. Zeinalipour-Yazdi, J. S. J. Hargreaves and C. R. A. Catlow, J. Phys. Chem. C, 2015, 119, 28368–28376 CrossRef CAS.
- N. C. Kani, I. Goyal, J. A. Gauthier, W. Shields, M. Shields and M. R. Singh, ACS Appl. Mater. Interfaces, 2024, 16, 16203–16212 CrossRef CAS PubMed.
- K. Kim, S. J. Lee, D. Y. Kim, C. Y. Yoo, J. W. Choi, J. N. Kim, Y. Woo, H. C. Yoon and J. I. Han, ChemSusChem, 2018, 11, 120–124 CrossRef CAS PubMed.
- A. Tsuneto, A. Kudo and T. Sakata, J. Electroanal. Chem., 1994, 367, 183–188 CrossRef CAS.
- I. Goyal, N. C. Kani, S. A. Olusegun, S. Chinnabattigalla, R. R. Bhawnani, K. D. Glusac, A. R. Singh, J. A. Gauthier and M. R. Singh, ACS Energy Lett., 2024, 9, 4188–4195 CrossRef CAS.
- W. Liao, H.-X. Liu, L. Qi, S. Liang, Y. Luo, F. Liu, X. Wang, C.-R. Chang, J. Zhang and L. Jiang, Cell Rep. Phys. Sci., 2021, 2, 100557 CrossRef CAS.
- H. Shen, C. Choi, J. Masa, X. Li, J. Qiu, Y. Jung and Z. Sun, Chem, 2021, 7, 1708–1754 CAS.
- S. Back and Y. Jung, Phys. Chem. Chem. Phys., 2016, 18, 9161–9166 RSC.
- C. J. M. van der Ham, M. T. M. Koper and D. G. H. Hetterscheid, Chem. Soc. Rev., 2014, 43, 5183–5191 RSC.
- E. Skúlason, T. Bligaard, S. Gudmundsdóttir, F. Studt, J. Rossmeisl, F. Abild-Pedersen, T. Vegge, H. Jónsson and J. K. Nørskov, Phys. Chem. Chem. Phys., 2012, 14, 1235–1245 RSC.
- C. Ling, Y. Zhang, Q. Li, X. Bai, L. Shi and J. Wang, J. Am. Chem. Soc., 2019, 141, 18264–18270 CrossRef CAS PubMed.
- Y. Abghoui, A. L. Garden, V. F. Hlynsson, S. Björgvinsdóttir, H. Ólafsdóttir and E. Skúlason, Phys. Chem. Chem. Phys., 2015, 17, 4909–4918 RSC.
- S. Y. Park, Y. J. Jang and D. H. Youn, Catalysts, 2023, 13, 639 CrossRef CAS.
- O. Christensen, A.-I. Hutu, H. H. Kristoffersen and J. Rossmeisl, J. Catal., 2024, 436, 115572 CrossRef CAS.
- L. Sun, K. Wen, G. Li, X. Zhang, X. Zeng, B. Johannessen and S. Zhang, ACS Mater. Au, 2024, 4, 547–556 CrossRef CAS PubMed.
- D. Zhang, H. Zhao, X. Wu, Y. Deng, Z. Wang, Y. Han, H. Li, Y. Shi, X. Chen and S. Li, Adv. Funct. Mater., 2021, 31, 2006939 CrossRef CAS.
- Y. Wen, W. Zhang, X. Wang, S. Lu, F. Duan, H. Zhu and M. Du, Chem. Commun., 2023, 59, 13371–13374 RSC.
- Y. Yao, H. Wang, X.-Z. Yuan, H. Li and M. Shao, ACS Energy Lett., 2019, 4, 1336–1341 CrossRef CAS.
- Y. Yao, S. Zhu, H. Wang, H. Li and M. Shao, Angew. Chem., Int. Ed., 2020, 59, 10479–10483 CrossRef CAS PubMed.
- L. Zeng, X. Li, S. Chen, J. Wen, W. Huang and A. Chen, J. Mater. Chem. A, 2020, 8, 7339–7349 RSC.
- F. Lai, N. Chen, X. Ye, G. He, W. Zong, K. B. Holt, B. Pan, I. P. Parkin, T. Liu and R. Chen, Adv. Funct. Mater., 2020, 30, 1907376 CrossRef CAS.
- X.-W. Lv, Y. Liu, R. Hao, W. Tian and Z.-Y. Yuan, ACS Appl. Mater. Interfaces, 2020, 12, 17502–17508 CrossRef CAS PubMed.
- Y. Fu, T. Li, G. Zhou, J. Guo, Y. Ao, Y. Hu, J. Shen, L. Liu and X. Wu, Nano Lett., 2020, 20, 4960–4967 CrossRef CAS PubMed.
- D. Yao, C. Tang, L. Li, B. Xia, A. Vasileff, H. Jin, Y. Zhang and S.-Z. Qiao, Adv. Energy Mater., 2020, 10, 2001289 CrossRef CAS.
- I. Valov, B. Luerssen, E. Mutoro, L. Gregoratti, R. A. De Souza, T. Bredow, S. Günther, A. Barinov, P. Dudin, M. Martin and J. Janek, Phys. Chem. Chem. Phys., 2011, 13, 3394–3410 RSC.
- R. Tort, A. Bagger, O. Westhead, Y. Kondo, A. Khobnya, A. Winiwarter, B. J. Davies, A. Walsh, Y. Katayama and Y. Yamada, ACS Catal., 2023, 13, 14513–14522 CrossRef CAS PubMed.
- H. Jin, S. S. Kim, S. Venkateshalu, J. Lee, K. Lee and K. Jin, Adv. Sci., 2023, 10, 2300951 CrossRef CAS PubMed.
- Z. Chen, C. Liu, L. Sun and T. Wang, ACS Catal., 2022, 12, 8936–8975 CrossRef CAS.
- I. Lucentini, X. Garcia, X. Vendrell and J. Llorca, Ind. Eng. Chem. Res., 2021, 60, 18560–18611 CrossRef CAS.
- D. Shlyapin, V. Borisov, V. Temerev, K. Iost, Z. Fedorova and P. Snytnikov, Kinet. Catal., 2023, 64, 815–825 CrossRef CAS.
- M. Krebsz, R. Y. Hodgetts, S. Johnston, C. K. Nguyen, Y. Hora, D. R. MacFarlane and A. N. Simonov, Energy Environ. Sci., 2024, 17, 4481–4487 RSC.
- Y. Bicer and I. Dincer, J. Electrochem. Soc., 2017, 164, H5036 CrossRef CAS.
- H. Liu, Chin. J. Catal., 2014, 35, 1619–1640 CrossRef CAS.
- I. Rossetti, N. Pernicone and L. Forni, Catal. Today, 2005, 102–103, 219–224 CrossRef CAS.
- V. Kyriakou, I. Garagounis, E. Vasileiou, A. Vourros and M. Stoukides, Catal. Today, 2017, 286, 2–13 CrossRef CAS.
- X. Guo, Y. Zhu and T. Ma, J. Energy Chem., 2017, 26, 1107–1116 CrossRef.
- A. J. Martín, T. Shinagawa and J. Pérez-Ramírez, Chem, 2019, 5, 263–283 Search PubMed.
- R. Zhao, H. Xie, L. Chang, X. Zhang, X. Zhu, X. Tong, T. Wang, Y. Luo, P. Wei, Z. Wang and X. Sun, EnergyChem, 2019, 1, 100011 CrossRef.
- Á. B. Höskuldsson, Y. Abghoui, A. B. Gunnarsdóttir and E. Skúlason, ACS Sustainable Chem. Eng., 2017, 5, 10327–10333 CrossRef.
- C. G. Yiokari, G. E. Pitselis, D. G. Polydoros, A. D. Katsaounis and C. G. Vayenas, J. Phys. Chem. A, 2000, 104, 10600–10602 CrossRef CAS.
- M. Hattori, S. Iijima, T. Nakao, H. Hosono and M. Hara, Nat. Commun., 2020, 11, 2001 CrossRef CAS PubMed.
- G. Marnellos and M. Stoukides, Science, 1998, 282, 98–100 CrossRef CAS PubMed.
- B. H. Wang, R. Q. Liu, J. D. Wang, Z. J. Li and Y. H. Xie, Chin. J. Inorg. Chem., 2005, 21, 1551–1555 CAS.
- B. H. Wang, J. D. Wang, R. Q. Liu, Y. H. Xie and Z. J. Li, J. Solid State Electrochem., 2006, 11, 27–31 CrossRef CAS.
- Y. Ogura, K. Sato, S. I. Miyahara, Y. Kawano, T. Toriyama, T. Yamamoto, S. Matsumura, S. Hosokawa and K. Nagaoka, Chem. Sci., 2018, 9, 2230–2237 RSC.
- A. Skodra and M. Stoukides, Solid State Ionics, 2009, 180, 1332–1336 CrossRef CAS.
- W. B. Wang, X. B. Cao, W. J. Gao, F. Zhang, H. T. Wang and G. L. Ma, J. Membr. Sci., 2010, 360, 397–403 CrossRef CAS.
- C.-H. Chen, S.-J. Chang, S.-P. Chang, M.-J. Li, I. C. Chen, T.-J. Hsueh and C.-L. Hsu, Chem. Phys. Lett., 2009, 476, 69–72 CrossRef CAS.
- C. G. Vayenas, M. M. Jaksic, S. I. Bebelis and S. G. Neophytides, in Modern Aspects of Electrochemistry, ed. J. O. M. Bockris, B. E. Conway and R. E. White, Springer US, Boston, MA, 1996, vol. 29, pp. 57–202 DOI:10.1007/978-1-4613-0327-5_2.
- S. Giddey, S. P. S. Badwal and A. Kulkarni, Int. J. Hydrogen Energy, 2013, 38, 14576–14594 CrossRef CAS.
- C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 7231–7234 CrossRef CAS PubMed.
- Á. B. Höskuldsson, Y. Abghoui, A. B. Gunnarsdóttir and E. Skúlason, ACS Sustainable Chem. Eng., 2017, 5, 10327–10333 CrossRef.
- T. Murakami, T. Nishikiori, T. Nohira and Y. Ito, J. Am. Chem. Soc., 2003, 125, 334–335 CrossRef CAS PubMed.
- T. Murakami, T. Nohira, Y. H. Ogata and Y. Ito, Electrochem. Solid-State Lett., 2005, 8, D12–D14 CrossRef CAS.
- T. Murakami, T. Nohira, T. Goto, Y. H. Ogata and Y. Ito, Electrochim. Acta, 2005, 50, 5423–5426 CrossRef CAS.
- T. Murakami, T. Nohira, Y. Araki, T. Goto, R. Hagiwara and Y. H. Ogata, Electrochem. Solid-State Lett., 2007, 10, E4–E6 CrossRef CAS.
- S. Licht, B. Cui, B. Wang, F. F. Li, J. Lau and S. Liu, Science, 2014, 345, 637–640 CrossRef CAS PubMed.
- I. A. Amar, R. Lan, C. T. G. Petit and S. Tao, Int. J. Electrochem. Sci., 2015, 10, 3757–3766 CrossRef CAS.
- I. A. Amar, C. T. G. Petit, L. Zhang, R. Lan, P. J. Skabara and S. Tao, Solid State Ionics, 2011, 201, 94–100 CrossRef CAS.
- I. Garagounis, V. Kyriakou, A. Skodra, E. Vasileiou and M. Stoukides, Front. Energy Res., 2014, 2 DOI:10.3389/fenrg.2014.00001.
- I. A. Amar, R. Lan, C. T. G. Petit and S. Tao, J. Solid State Electrochem., 2011, 15, 1845–1860 CrossRef CAS.
- V. Kordali, G. Kyriacou and C. Lambrou, Chem. Commun., 2000, 1673–1674, 10.1039/b004885m.
- J. N. Renner, L. F. Greenlee, A. M. Herring and K. E. Ayers, Electrochem. Soc. Interface, 2015, 24, 51–57 CrossRef CAS.
- Z. Zhang, Z. Zhong and R. Liu, J. Rare Earths, 2010, 28, 556–559 CrossRef CAS.
- R. Liu and G. Xu, Chin. J. Chem., 2010, 28, 139–142 CrossRef CAS.
- G. Xu, R. Liu and J. Wang, Sci. China, Ser. B:Chem., 2009, 52, 1171–1175 CrossRef CAS.
- R. Lan, J. T. S. Irvine and S. Tao, Sci. Rep., 2013, 3, 1145 CrossRef PubMed.
- C. Ling, X. Bai, Y. Ouyang, A. Du and J. Wang, J. Phys. Chem. C, 2018, 122, 16842–16847 CrossRef CAS.
- Y. Wan, J. Xu and R. Lv, Mater. Today, 2019, 27, 69–90 CrossRef CAS.
- B. Ma, H. Zhao, T. S. Li, Q. Liu, Y. Luo, C. Li, S. Lu, A. M. Asiri, D. Ma and X. Sun, Nano Res., 2020, 14, 1–15 Search PubMed.
- J. S. Anderson, G. E. Cutsail, III, J. Rittle, B. A. Connor, W. A. Gunderson, L. Zhang, B. M. Hoffman and J. C. Peters, J. Am. Chem. Soc., 2015, 137, 7803–7809 CrossRef CAS PubMed.
- Z. Wang, F. Gong, L. Zhang, R. Wang, L. Ji, Q. Liu, Y. Luo, H. Guo, Y. Li, P. Gao, X. Shi, B. Li, B. Tang and X. Sun, Adv. Sci., 2019, 6, 1801182 CrossRef PubMed.
- F. Lai, J. Feng, X. Ye, W. Zong, G. He, C. Yang, W. Wang, Y.-E. Miao, B. Pan, W. Yan, T. Liu and I. P. Parkin, J. Mater. Chem. A, 2020, 8, 1652–1659 RSC.
- W. Tong, B. Huang, P. Wang, Q. Shao and X. Huang, Natl. Sci. Rev., 2020, 8, nwaa088 CrossRef PubMed.
- M. Ali, F. Zhou, K. Chen, C. Kotzur, C. Xiao, L. Bourgeois, X. Zhang and D. R. MacFarlane, Nat. Commun., 2016, 7, 11335 CrossRef CAS PubMed.
- C. Ling, X. Niu, Q. Li, A. Du and J. Wang, J. Am. Chem. Soc., 2018, 140, 14161–14168 CrossRef CAS PubMed.
- I. Garagounis, A. Vourros, D. Stoukides, D. Dasopoulos and M. Stoukides, Membranes, 2019, 9, 112 CrossRef CAS PubMed.
- Y. Bicer and I. Dincer, J. Electrochem. Soc., 2017, 164, H5036–H5042 CrossRef CAS.
- R. Zhang, Y. Zhang, X. Ren, Y. Luo, G. Cui, A. M. Asiri, B. Zheng and X.-P. Sun, ACS Sustainable Chem. Eng., 2018, 6, 9545–9549 CrossRef CAS.
- X. Zhao, F. Yin, N. Liu, G. Li, T. Fan and B. Chen, J. Mater. Sci., 2017, 52, 1–11 CrossRef.
- L. Zhang, G.-F. Chen, L.-X. Ding and H. Wang, Chem. – Eur. J., 2019, 25, 12464–12485 CrossRef CAS PubMed.
- H. Ishaq and C. Crawford, Energy Convers. Manage., 2024, 300, 117869 CrossRef CAS.
- A. E. Yüzbaşıoğlu, A. H. Tatarhan and A. O. Gezerman, Heliyon, 2021, 7, e08257 CrossRef PubMed.
- C. A. Fernandez, O. Chapman, M. A. Brown, C. E. Alvarez-Pugliese and M. C. Hatzell, Environ. Sci. Technol., 2024, 58, 6964–6977 CrossRef CAS PubMed.
- A. M. Mohamed, I. G. Economou and Y. Bicer, Curr. Opin. Green Sustainable Chem., 2024, 100947 CrossRef CAS.
- F. Dominguez-Flores and M. M. Melander, Curr. Opin. Electrochem., 2022, 36, 101110 CrossRef CAS.
- J. Resasco, F. Abild-Pedersen, C. Hahn, Z. Bao, M. T. Koper and T. F. Jaramillo, Nat. Catal., 2022, 5, 374–381 CrossRef.
- Y. Yang, J. Wang, Y. Shu, Y. Ji, H. Dong and Y. Li, Phys. Chem. Chem. Phys., 2022, 24, 8591–8603 RSC.
- S. Gunduz, D. J. Deka and U. S. Ozkan, J. Catal., 2020, 387, 207–216 CrossRef CAS.
Footnote |
| † Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |