
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDisassembling and reassembling perovskites for oxygen electrocatalysis
Gao Chen
 *a,
Yubo Chenbc,
Zezhou Lind,
Ting Chene,
Dongsheng Genga,
Yanping Zhu
*e,
Wei Wangf and
Wei Zhou
*fg
*a,
Yubo Chenbc,
Zezhou Lind,
Ting Chene,
Dongsheng Genga,
Yanping Zhu
*e,
Wei Wangf and
Wei Zhou
*fg
aJiangsu Key Laboratory of New Energy Devices and Interface Science, School of Chemistry and Materials Science, Nanjing University of Information Science and Technology, Nanjing 210044, P. R. China. E-mail: gaochen@nuist.edu.cn
bHydrogen Energy Institute, Zhejiang University, Hangzhou 310027, P. R. China
cInstitute of Advanced Equipment, College of Energy Engineering, Zhejiang University, Hangzhou 310027, P. R. China
dDepartment of Applied Physics and Research Institute for Smart Energy, The Hong Kong Polytechnic University, Hong Kong, People's Republic of China
eCollege of Materials Science and Technology, Nanjing University of Aeronautics and Astronautics, Nanjing, People's Republic of China. E-mail: yanping.zhu@nuaa.edu.cn
fState Key Laboratory of Materials-Oriented Chemical Engineering, College of Chemical Engineering, Nanjing Tech University, Nanjing 211800, P. R. China. E-mail: zhouwei1982@njtech.edu.cn
gSuzhou Laboratory, Suzhou 215000, P. R. China
First published on 13th May 2025
Abstract
Perovskite oxides (ABO3) are widely studied in oxygen electrocatalysis due to their simple synthesis routes, rich compositions, adjustable crystal/electronic structures, and high intrinsic activities. Despite these advantages, high calcination temperatures usually lead to agglomeration of perovskite materials, greatly reducing atomic utilization. Moreover, the different element features of A/B cations generally make easy enrichment of surface A-sites, and such surface deviation from the ideal structure would impede the precise illustration of structure–activity relationships for electrocatalysis. Up to now, various strategies have been developed to tackle the above issues, through which significant progress in both catalytic performance and underlying catalytic mechanisms has been achieved. Here we summarize those optimization methods as “disassembling and reassembling perovskites” and concisely review related studies and findings in terms of the fundamental understanding of approaches and the applications in oxygen electrocatalysis. Three typical methods, including physical, chemical, and electrochemical, are introduced with their effects on perovskite structures/catalytic mechanisms thoroughly discussed. Finally, four scientific issues regarding disassembling and reassembling perovskites are proposed for future studies. We aim to raise the community's awareness of this emerging approach and hope it could contribute to material design for applications beyond oxygen electrocatalysis.
 Gao Chen | Gao Chen is a Professor in the School of Chemistry and Materials Science, Nanjing University of Information Science and Technology (NUIST). After obtaining his PhD degree from Nanjing Tech University, he received postdoc training at Nanyang Technology University and The Hong Kong Polytechnic University. His research interests cover designing high-efficiency electrocatalysts by disassembling and reassembling perovskites, as well as elucidating the fundamental connections between materials and reaction mechanisms in electrocatalysis. |
 Yanping Zhu | Yanping Zhu received her PhD in Chemical Engineering from Nanjing Tech University. From 2018 to 2023, she worked as a postdoctoral fellow at National Taiwan University and The Hong Kong Polytechnic University. In 2024, she joined the College of Materials Science and Technology at the Nanjing University of Aeronautics and Astronautics. Her research interests include developing advanced materials related to electrocatalysis and in situ methodologies toward various chemical reactions. |
 Wei Zhou | Wei Zhou received his PhD from Nanjing Tech University in 2009, focusing on the design and synthesis of high-performance cathode materials for solid oxide fuel cells (SOFCs). In 2010, he was awarded an Australian Postdoctoral Fellowship and joined the University of Queensland, where he was later promoted to a Research Fellow in 2013. In 2015, he returned to Nanjing Tech University as a Professor. His research interests encompass the development of advanced materials for SOFCs and electrocatalysts for oxygen reduction, water splitting, and CO2 reduction reactions. |
Broader contextPerovskite oxides (ABO3) have emerged as pivotal materials in oxygen electrocatalysis, underpinning critical energy conversion and storage technologies such as water electrolysis, fuel cells, and green electrochemical synthesis. Their compositional flexibility, tunable electronic structures, and high intrinsic activity position them as promising alternatives to noble metal catalysts. However, as-synthesized perovskites face inherent limitations, including low atomic utilization due to bulk particle formation at high synthesis temperatures and surface enrichment of inactive A-site cations, which obscure active B–O motifs and hinder precise structure–activity relationships. Addressing these challenges is vital to advancing sustainable energy systems and achieving global decarbonization goals. This work introduces ‘disassembling and reassembling perovskites’ as a transformative strategy to overcome these limitations. By leveraging physical, chemical, and electrochemical methods, the original perovskite structure is strategically broken down and reconfigured to enhance geometric and intrinsic activity, expose active sites, and clarify catalytic mechanisms. These approaches not only optimize oxygen evolution and reduction reactions but also unveil dynamic surface reconstruction phenomena and paracrystalline motifs, offering fresh insights into catalyst design. By bridging fundamental research with scalable engineering solutions, this work paves the way for next-generation materials that balance efficiency, stability, and cost-effectiveness in sustainable energy technologies. |
1. Introduction
Oxygen electrocatalysis, encompassing the oxygen evolution reaction (OER) and oxygen reduction reaction (ORR), is fundamental to energy conversion and storage devices as well as green electrochemical synthesis with zero carbon emissions.1–3 For example, the OER serves as the anode half-reaction in hydrogen production via water electrolysis, while the ORR functions as the cathode half-reaction in fuel cells and the electrochemical synthesis of hydrogen peroxide. Developing efficient and cost-effective oxygen electrocatalysts is crucial for enhancing catalytic conversion efficiency and reducing cost.4,5Perovskite oxides (with the structural formula ABO3) have served as pivotal materials in oxygen electrocatalysis for over four decades,6 owing to their multifaceted merits such as straightforward synthesis methods, diverse compositions, tunable crystal and electronic structures, and high intrinsic activities.7–9 Most perovskite oxides are synthesized by solid-phase and sol–gel processes, followed by calcination processes at high temperatures to form the perovskite phase. Some perovskites can be synthesized directly using hydrothermal methods, omitting further calcination. Approximately 90% of the elements in the periodic table can be incorporated into the perovskite structure. The substitution of A or B site elements allows precise tuning of crystalline and electronic structures. For example, by fine-tuning the constituent elements and their proportions, the Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF) perovskite shows a unity filling in the eg orbital, thus exhibiting the highest OER activity among a series of perovskites,10 from which the use of perovskites as oxygen electrocatalysts has exploded, leading to significant breakthroughs in oxygen electrocatalysis. These include establishing eg filling10 and lattice oxygen mechanisms,11,12 unveiling dynamic surface reconstruction,13 defining stability numbers,14 and finding paracrystalline motifs on reconstructed iridium perovskites.15
Despite these achievements, two main issues of perovskite oxides limit their further advancement. First, due to the high phase formation temperature (700–1200 °C), perovskite oxides usually exist as bulk particles, confining most active sites within the particles thus preventing their participation in surface oxygen electrocatalysis, resulting in low atom utilization.16 Additionally, the intrinsic property difference between A and B cations makes easy enrichment of surface A-sites, leading to surface deviations from the ideal perovskite structure, posing challenges to the availability of the B site as well as the precise establishment of structure–activity relationship.17,18
In order to meet the higher requirements brought about by quick development of oxygen electrocatalysis, perovskite oxides have been tailored by various strategies to address the above mentioned issues, for which significant progress has been achieved. Here we unified those tailoring methods as “disassembling and reassembling perovskites”19–21 and gave a comprehensive review of disassembling and reassembling perovskites for oxygen electrocatalysis. After defining disassembling and reassembling perovskites, advantages brought about by such tailoring are generalized. The materials discussed include traditional perovskites, layered perovskites, anti-perovskites, and non-oxide perovskites, spanning applications in acidic and alkaline OERs (from room temperature to 80 °C) as well as the alkaline ORR at room temperature. We classify those approaches into three categories: physical, chemical and electrochemical methods. The corresponding effects of each method on the perovskite structure, catalyst performance and the underlying catalytic mechanisms are discussed in detail, after which potential challenges and future directions regarding disassembling and reassembling perovskites for oxygen electrocatalysis are proposed.
2. Fundamental understanding of disassembling and reassembling perovskites
The perovskite family adopts an ABO3 structure, where A-site cations (Sr2+, La3+, etc.) occupy the voids within the framework of corner-sharing BO6 octahedra, while the B-site transition metals (Co4+, Fe3+, etc.) occupy the octahedral center. Disassembling and reassembling perovskites involve breaking down the original structure of perovskites through specific methods and then recombining certain or all parts to form the target products. Whether the final product is perovskite or not depends on the method used. Experiencing such modification processes, the as-obtained materials usually have obvious advantages when participating in oxygen electrocatalysis including,(i) Increasing geometrical activity by decreasing the size of the bulk perovskite to the nanoscale or the even atomic level. This is crucial for mitigating the adverse effects associated with the high phase-formation temperature of perovskites.
(ii) Enhancing intrinsic activity by manipulating component elements: removing inactive A-site elements while exposing active B–O octahedra with unusual local structure or abundant unsaturated coordination.
(iii) Precise illustration of structure–activity relationship by exposing specific lattice planes of perovskites enables more precise prediction of the relation between catalyst structure and catalytic performance.
3. Approaches used for disassembling and reassembling perovskites
Typically, the approaches utilized for disassembling and reassembling perovskites can be classified into three categories: physical, chemical, and electrochemical methods (Fig. 1). | ||
| Fig. 1 Schematic illustration of the strategies for disassembling and reassembling perovskites. D&R perovskites: disassembling and reassembling perovskites. | ||
(i) Physical methods: it generally refers to utilizing specific equipment to physically break the original perovskite structure and transform it into the desired states. The commonly used physical methods include pulsed laser deposition (PLD), magnetron sputtering, plasma etching, and ball milling.22–24
(ii) Chemical methods: through chemical reactions with perovskites using atmosphere or solution as medium, selective separation and rebuilding of perovskite components can be realized. The commonly used chemical approaches include exsolution, exfoliation, acid leaching, and chemical dissolution–deposition methods.25–27
(iii) Electrochemical methods: these methods are based on surface reconstruction of perovskites during electrocatalysis. Knowing that perovskites are rich in compositional elements, different components of perovskites exhibit different leach-deposition behaviors under the same pH and potential conditions, which could be predicted by the Pourbaix diagram. Desired disassembling and/or reassembling on perovskites can be realized by tuning composition, pH value of electrolyte and the applied potentials.28
4. Applications in oxygen electrocatalysis
According to previous studies, after disassembling and reassembling perovskites, the obtained catalyst materials are endowed with new properties, which in turn lead to satisfactory catalytic behaviors and help gain deep insights into the underlying mechanism of oxygen electrocatalysis. This section is organized by three typical categories of approaches for disassembling and reassembling perovskites. Features, advantages, corresponding effects and the applicability of each method are provided.4.1. Physical methods and corresponding effects
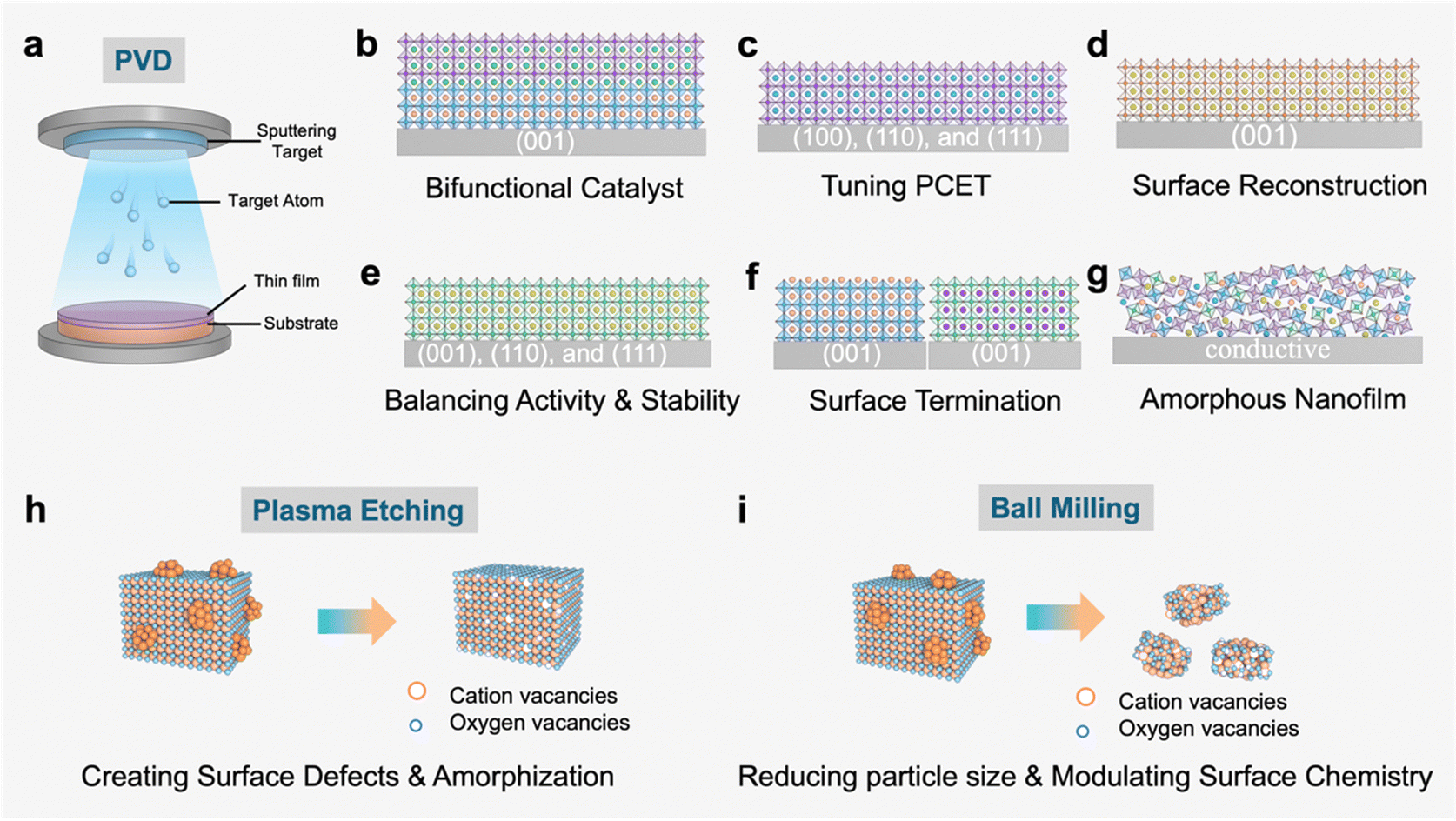 | ||
| Fig. 2 Schematic illustration of physical strategies for disassembling and reassembling perovskites. (a) The physical vapor deposition (PVD) approach. (b)–(g) Examples of PVD approaches. (h) The plasma etching approach. (i) The ball milling approach. PCET: proton-coupled electron transfer. | ||
4.1.1.1. Enhancing bifunctional oxygen electrocatalysis. BSCF and manganite perovskites are among the most active catalysts for the OER and ORR, respectively.10,29 The Shao-Horn group utilized PLD to fabricate a two-layer perovskite composed of BSCF and La0.8Sr0.2MnO3−δ (LSMO) on a (001)-oriented Nb-doped SrTiO3 (NSTO) substrate (Fig. 2b).30 The fabrication of (001) oriented BSCF|LSMO|NSTO films involved two-steps. First, a 16 nm layer of LSMO was deposited on the NSTO substrate. BSCF layers with varying thicknesses (1, 2, 5, 10, and 20 nm) were subsequently deposited on the LSMO substrate, achieving BSCF surface coverages of 20%, 28%, 48%, 91%, and 94%, respectively. Electrochemical analysis revealed that these two-layer perovskite films with tunable BSCF coverage (20–91%) exhibit bifunctional oxygen electrocatalysis, where BSCF domains drive the OER kinetics while LSMO regions govern the ORR activity. As a result, the combined BSCF|LSMO|NSTO catalyst shows a comparable OER activity to BSCF powder and superior ORR activity to LSMO powder. Moreover, the BSCF decoration was confirmed to enhance the surface stability of the ORR. Furthermore, it is necessary to mention that LSMO can bring down the Schottky barrier between BSCF and NSTO, facilitating the charge transfer at the solid–solid interface. The introduction of a buffer layer is able to tailor band bending and covalency of surface near oxygen bonds.31,32 For more details, please refer to the high-impact review paper published by the Risch research group on epitaxial perovskites for the OER.23
4.1.1.2. Tuning proton-coupled electron transfer (PCET) in the OER. The OER generally involves a PCET step. Modulating the PCET process can speed up the rate of the OER.33 In a study conducted by Liu and co-workers, the PCET process was modified by tuning crystal orientation of PrBa0.5Sr0.5Co1.5Fe0.5O5+δ (PBSCF) through the PLD method (Fig. 2c).34 PBSCF thin films with well-defined orientation of (100), (110), and (111) planes are prepared on an LaAlO3 single crystal substrate. Electrochemical measurements and DFT calculations consistently verified that the OER activity of PBSCF thin films follows the order of (100) > (110) > (111) planes. The outstanding activity of the (100) plane is attributed to the facilitated deprotonation process, leading to an effective PCET during the OER. The most direct evidence stems from near-ambient pressure X-ray photoelectron spectroscopy (APXPS), which definitively reveals that water deprotonation proceeds with the highest efficiency on the (100) surface. This study highlights the modification of the OER process on perovskite-based catalysts prepared by the PLD method.
4.1.1.3. Uncovering surface reconstruction. Iridium (Ir) oxide is the only known material that can operate stably in the acidic electrolyte for the OER.35 Due to the high-cost and scarcity of noble-metal Ir, a large amount of studies have focused on improving activity while lowering the content of Ir. In 2016, Jaramillo and coworkers prepared a SrIrO3 perovskite thin film by PLD and successfully uncovered its reconstruction behavior in the acidic electrolyte (Fig. 2d).36 The electrochemical test shows a continuously increased OER activity, which is much higher than its theoretical performance predicted from DFT calculations. Then atomic force microscopy (AFM) records the evolution of the SrIrO3 surface from flat to rough. Moreover, the relative quantity of surface Sr determined by X-ray photoelectron spectroscopy (XPS) is much less than that in the subsurface layer. Based on the above observations, it is concluded that during the OER, the SrIrO3 perovskite would in situ transform into the IrOx/SrIrO3 catalyst owing to the leaching of Sr. Specifically, it is difficult to capture such direct evidence of surface reconstruction in bulk perovskites.
Weber and coworkers systematically mapped the degradation pathways in a representative Co-based perovskite, i.e., La0.6Sr0.4CoO3−δ, during alkaline OER operation, uncovering the complex surface-mediated activation–deactivation process.37 A multiscale microscopy and spectroscopy approach reveals that the OER activity arises from the in situ formed CoO(OH) active layers, whereas La(OH)3 passivation drives performance decay. Specifically, Sr leaching facilitates the creation of metastable mixed-phase interfaces, while Co depletion from the self-assembled CoO(OH) active layer coupled with La(OH)3 accumulation ultimately leads to a gradual decrease in OER activity. This landmark study establishes the critical role of lanthanide-alkaline earth cation synergy in controlling reconstruction equilibria that dictate both activity and stability in alkaline OER catalysis.
4.1.1.4. Investigating links between activity and stability. Due to the relatively lower price and higher activity of Ru, researchers have attempted to replace Ir by Ru when designing OER catalysts.38 Nevertheless, Ru based oxides suffer from severe degradation under OER conditions, so balancing activity and stability of Ru-based catalysts becomes the target pursued for the practical applications.39 In 2013, with the help of magnetron sputtering, the Markovic group first prepared single-crystal SrRuO3 nanofilms with three typical orientations40 and found the OER activity following the order of (001) < (110) < (111) planes, which is inverse to their stability trend (Fig. 2e). Following this study, modification methods are employed to further improve the activity or stability of the most stable SrRuO3(001) nanofilms. For instance, the phase-transformed SrRuO3 (001) nanofilm induced by Ru/O vacancies results in a 30% decrease in the overpotential for the OER.41 Furthermore, an ultrathin coating of SrTiO3 on the SrRuO3 nanofilms prepared through PLD demonstrated much better OER stability but a slight decrease in activity.42
Epitaxial stabilization of SrRuO3 beneath a two-unit-cell SrTiO3 overlayer achieves atomic-scale passivation, effectively suppressing corrosion of the metastable SrRuO3 phase during the OER while maintaining interfacial charge transfer capabilities. PLD preparation provides convenience for investigating links between the activity and stability of perovskite catalysts. For further insights into balancing activity and stability, we recommend referring to the papers published by research groups of Bauemer and Stroerzinger.43,44
4.1.1.5. Controlling surface terminated cations. The surface of bulk perovskites is often enriched with A-site cations owing to their lower surface energy compared to B-site cations.45,46 The surface enrichment of A-site cations would hinder the involvement of active B–O sites during the OER, thus suppressing the activity of perovskite catalysts. For example, although LaNiO3 (LNO) is a highly efficient OER catalyst from theoretical prediction, its actual performance is far from satisfactory due to the easy enrichment of inert La at the surface.47–52 Notably, the PVD technique can realize the regulation of surface terminated cations, adjusting catalytic behaviors. As shown in Fig. 2f, Baeumer et al. used the PVD technique to obtain La-terminated LaNiO3 (La-LNO) and Ni-terminated LaNiO3 (Ni-LNO) nanofilms (001).53 The Ni-LNO sample shows a much better OER performance due to the easy formation of the disordered NiO2 layer on the catalyst surface during the OER, which is inaccessible in the La-LNO counterpart. This highlights the distinct merit of the PVD method in finely controlling surface terminated cations to reduce activity loss resulting from inert-cation enrichment on the perovskite surface, which is difficult to realize in bulk perovskite materials. Subsequent work conducted by Baeumer and colleagues systematically investigated the crystallographic anisotropy of OER activity in LNO, revealing a distinct crystal-facet-dependent trend: LNO (111) > LNO (110) > LNO (001).54 This hierarchy was attributed to the preferential formation of reconstructed oxyhydroxide-like NiOO(H) active species on the LNO (111) surface, resulting in optimized binding energy of reaction intermediates for enhanced water oxidation kinetics.
4.1.1.6. Amorphous nanofilms. Amorphous materials with an unsaturated coordination environment usually lead to enhanced OER activity.55,56 In 2017, we utilized magnetron sputtering to prepare a series of amorphous BSCF nanofilms with varied thickness (1, 5, 10, 15, and 20 nm) at room temperature (Fig. 2g).57 The mass activity of BSCF increased by 2 orders of magnitude owing to the largely increased atom utilization. Moreover, the intrinsic OER activity of amorphous BSCF nanofilms shows a volcano trend as a function of the film thickness, which is well-correlated with the electronic configuration of Co/Fe active sites. Following this work, we further prepared a series of perovskite-derived amorphous nanofilms via room temperature magnetron sputtering,58 from which a universal rule is obtained, namely, the amorphization of perovskites accelerates the in situ surface reconstruction during water splitting regardless of the initial crystal structures and compositions of perovskites. Generally, amorphous nanofilms prepared by magnetron sputtering have significant advantages such as improving mass activity, fine-tuning electronic configuration and accelerating reconstruction during the OER.
4.1.2.1. Creating surface cation vacancies. Perovskite hydroxide refers to a class of materials that have a perovskite-like crystal structure and contain hydroxide (OH−) ions instead of oxygen ions (O2−) in traditional perovskites. Sometimes perovskite hydroxides can be formed without A-site cations. In 2018, Wang and co-workers adopted Ar plasma to disassemble and reassemble SnCo0.9Fe0.1(OH)6 perovskite hydroxides.63 X-ray absorption spectral (XAS) analysis confirms that Sn vacancies are preferentially formed compared to Co and Fe, which is attributed to the lower lattice energy and weaker chemical bond in Sn(OH)4. Significantly, the introduced Sn vacancies help expose more active CoFe sites and improve electron conductivity, consequently enhancing the OER performance. In addition, the specific surface area and the hydrophilicity of SnCo0.9Fe0.1(OH)6 perovskite hydroxides are greatly improved, providing extra benefits to the OER. Later, a solution plasma was utilized to disassemble and reassemble the SnCo(OH)6 perovskite hydroxides.64 It is revealed that the OER activity of SnCo(OH)6 perovskite hydroxide after solution plasma is related to the surface composition, which is determined by the pH of the solution used.
4.1.2.2. Creating oxygen vacancies. Oxygen vacancy is a key parameter to regulate the electrocatalytic performance of perovskite materials.61 In 2018, Gui et al. utilized Ar plasma and created abundant oxygen vacancies on the surface of La0.7Sr0.3CoO3−δ.65 The introduction of oxygen vacancies in La0.7Sr0.3CoO3−δ was shown to significantly enhance its OER performance, with marked improvements in catalytic activity, reaction kinetics, and long-term stability, likely mediated by vacancy-induced electronic structure modulation. In 2019, He et al. adopted a different H2/Ar plasma to treat Bi0.1(Ba0.5Sr0.5)0.9Co0.8Fe0.2O3−δ and obtained a unique structure with a crystalline core and an amorphous shell.66 The formation of an amorphous shell is likely attributable to the reductive H2 gas, resulting in the generation of much more oxygen vacancies and thus the degradation of the surface structure. This oxygen-vacancy-rich core–shell structure demonstrated a 1.6-fold improvement in intrinsic OER activity, which is ascribed to the enhanced exposure of electrochemically active oxygen sites. Furthermore, by adjusting operation parameters, Liu's group67 successfully created oxygen vacancies with various concentrations on PrBa0.5Sr0.5Co1.5Fe0.5O5+δ (PBSCF) perovskite thin films using H2/Ar plasma. This study reveals that oxygen vacancies can enhance OH− adsorption and reduce the theoretical energy barrier for O* formation on the surface, thereby accelerating OER kinetics. However, excessive oxygen vacancies also create a larger bandgap and depress the O 2p band center in PBSCF, which could potentially impede OER kinetics. By carefully balancing these competing effects, oxygen-deficient PBSCF demonstrates improved OER activity. Another highlight of this work is the combination of PLD and plasma etching for disassembling and reassembling perovskites, which offers the reference for future studies.
4.2. Chemical methods and corresponding effects
 | ||
| Fig. 3 Schematic illustration of chemical strategies for disassembling and reassembling perovskites. (a) The exsolution approach. (b) The exfoliation approach. (c) The acid dissolution approach. (d) The selective dissolution and deposition approaches. SMSI: strong metal–support interaction. | ||
4.2.1.1. A-site: strong metal support interaction (SMSI). A-site cations are mostly alkali or rare earth metals with poor oxygen electrocatalytic performance. In a few cases, electrochemically active metals can be incorporated into the A site of perovskites. Silver ions (Ag+) with a large radius often exist in the A-site of perovskites. After being treated with H2/N2, the single-phase Pr0.475Ba0.475Ag0.05MnO3−δ (PBAMO3) perovskite was transformed into an A-site deficient layered perovskite structure decorated with Ag NPs (denoted as Ag-PBMO5).73 As expected, the interaction between surface Ag NPs and the perovskite matrix is intensified (SMSI), leading to facile electron transfer and ion migration during the ORR. As a result, the Ag-PBMO5 catalyst outperforms its counterparts, i.e., PBMO5 catalyst, Pr0.5Ba0.5MnO3−δ catalyst, and physically mixed Ag + PBMO5 composite. Coincidentally, Irvine, a pioneer in the field of perovskite exsolution, studied the exsolution of Ni NPs from the A site of the (Ca0.92Ni0.04)TiO2.96 perovskite.74 Compared to its counterpart exsolving Ni NPs from the B site of the perovskite, the Ni NPs in the A-site have a smaller size and larger population and specific surface area, as well as intensified SMSI with the perovskite matrix, leading to a better OER activity.
4.2.1.2. B-site: reorganizes surface active sites. As mentioned above, the surface of perovskites is often enriched with A-site cations, blocking the electrochemically active B-site cations. Exsolution of B-site cations could facilitate the exposure of active sites that are originally located in the subsurface layer, forming new nanostructures. For instance, an A-deficient double perovskite, (PrBa0.8Ca0.2)0.95(Co1.5Fe0.5)0.95Co0.05O5+δ, was designed for the facile exsolution of Co/CoOx NPs.75 Benefiting from the tailored nanostructures at the surface, the obtained electrocatalyst shows great promise when catalyzing the ORR/OER under alkaline conditions. Furthermore, a lanthana-anchored CoFe alloy catalyst was obtained from LaCo0.8Fe0.2O3−δ (LCF) by applying a harsher treatment (700 °C for 3 h in 5% H2/Ar).76 Operando XAS analysis demonstrates the transformation of CoFe species into CoFe hydroxides during the OER process. The lanthana support helps promote such transformation, contributing to an enhanced OER activity. Nevertheless, the exsolution process under a reduction atmosphere poses a safety risk. In light of this, Shao et al. reported an organic ligand-facilitated in situ exsolution method under an inert N2 atmosphere and successfully grew CoFe nanoalloys over the BSCF perovskite, and the newly formed catalyst showed much improved OER and ORR performances.77
4.2.2.1. Aurivillius compounds. Aurivillius compounds are a class of layered perovskite-like materials with the general formula (Bi2O2)2+(Am−1BmO3m+1)2−. These compounds feature a unique structure consisting of alternating layers of (Bi2O2)2+ and perovskite-like (Am−1BmO3m+1)2− slabs. The layered structure makes them suitable for exfoliation, allowing the production of two-dimensional materials with promising oxygen electrocatalysis performance. Specifically, CoBi2O2F4 is among the Aurivillius family.78 The two main building units are [CoF6] octahedra and [BiO4F4] distorted cubes. The corner-shared [CoF6] octahedra form a [CoF4]∞ layer and the [BiO4F4] cubes form a [BiO2F2]∞ layer. Using isopropanol as the solvent,79 the exfoliation took place at the relatively weak interlayer Bi–F bonds. The exfoliation is confirmed by the corresponding SEM images in which the thickness of exfoliated CoBi2O2F4 is only 20 to 30 nm. In addition, the exfoliation process also leads to enlarged d-spacing along z-axis dangling bonds on the edge. These features greatly improve the accessibility of Co active sites involved in the OER, leading to a decrease of 26 mV in overpotential to achieve 10 mA cm−2.
4.2.2.2. Ruddlesden–Popper perovskites. Ruddlesden–Popper (RP) perovskites are a family of layered perovskite materials with the general formula An+1BnO3n+1 (AO·n(ABO3)), where A is typically a larger cation such as rare earth or alkaline earth metal and B is a smaller transition metal cation.80 The integer n represents the number of perovskite layers between the rock-salt layers. The structure of RP perovskite consists of n layers of corner-sharing octahedra (ABO3 layers) separated by rock-salt layers (AO). Such layered structure makes them suitable candidates for exfoliation, allowing the production of two-dimensional materials with promising oxygen electrocatalysis performance. Among them, Sr2IrO4 is a typical RP perovskite. As described in the study of the Grimaud group, the immersion of Sr2IrO4 in acidic solution could produce protonated iridate HxIrOy via cation exchange.81 Then the protonated iridate was stirred in a tetrabutylammonium hydroxide (TBAOH) solution to insert TBA+ inside the structure, followed by sonication to finally complete the exfoliation. The largely increased specific surface area of the obtained HxIrOy contributes obviously boosted geometrical and mass activity, while the reversible exchange of protons ensures improved catalytic stability. Another study from the Zou group realized the exfoliation of Sr2IrO4 without damaging the perovskite structure.82 The 2D morphology of protonated perovskite nanosheets enables the straightforward assembly of a low-iridium-loading catalyst film, delivering a mass activity 10-fold higher than commercial IrO2, while maintaining comparable iridium dissolution rates under OER conditions. A thermochemical dehydration method may provide a more facile route for manipulating protons in perovskites without degrading the crystal quality of perovskites.83
4.2.2.3. Dion–Jacobson perovskites. Dion–Jacobson (DJ) perovskites are a class of layered perovskite materials with the general formula A′An−1BnO3n+1, where A′ refers to a large organic or inorganic cation, A includes smaller cations such as alkali or alkaline earth metal, and B is a transition metal cation.84 The integer n represents the number of perovskite layers between the A′ cation layers. In DJ perovskites, the structure consists of n layers of corner-sharing BO6 octahedra (perovskite layers) separated by a single layer of A′ cations. The typical ion-exchange and subsequent exfoliation treatments used on the above RP perovskites are also applicable to bulk DJ perovskites for making An−1BnO3n+1 nano-slabs. For instance, bulk DJ perovskite KCa2Nb3−xTaxO10 (x = 0, 0.5, 1, 1.5) can be exfoliated by sequentially subjecting to HNO3 and TBAOH solutions.85 The exfoliated catalyst was demonstrated to enhance H2O2 generation by increasing exposure of active sites, diminishing interfacial resistance and promoting charge transport.
4.2.3.1. Creating A-site defects. The first case is concerning slight dissolution of A-site cations, which is achieved on the La0.6Sr0.4Co0.8Fe0.2O3 (LSCF) perovskite by finely controlling the acid treating time.87 A 6-hour treatment in 0.2 M HNO3 solution leads to the predominant dissolution of Sr2+ and slight dissolution of La/Co cations. Originated from the optimized surface with rich A-site defects, enlarged surface area and a large amount of active oxygen species, the mass activity and specific activity of the treated LSCF catalysts when catalyzing the OER were 6-fold and 2.6-fold higher than those of the pristine LSCF, respectively.
4.2.3.2. Reconstruction of B–O motifs. In the report from the Francisco group, when exposed to HNO3 solution, the surface A-site cations of the LSMO perovskite were dissolved while the remaining Mn–O motifs transformed into MnOx simultaneously, thus generating the MnOx/LSMO composite catalyst enriched with Mn at the surface.88 The resulting catalyst shows improved ORR performance compared to the pristine LSMO owing to the relatively higher specific surface area and much more active sites (Mn and O). Another example is Ir-based perovskites.89 Through treatment in strong acid solution, the surface Ba cations of the 9R-BaIrO3 perovskite are dissolved with uniform IrOx particles (1 nm) generated on the surface. The resulting IrOx/9R-BaIrO3 catalyst exhibits the highest iridium mass activity (168 A g−1) among the reported iridium-based Ir perovskites for the acidic OER, which is ascribed to the abundant higher-valence Ir at the catalyst surface and the enhanced metallic conductivity.
4.2.4.1. Regulating the catalyst interface. Antiperovskites refer to a class of materials with the perovskite structure but reversed arrangement of cations and anions.90 Owing to the flexibility in element composition and high electronic conductivity, antiperovskites have shown great potential in oxygen electrocatalysis. However, similar to the traditional perovskite, they also suffer from bulk characteristics resulting from sintering. In 2018, our group treated the Cu-excess CuNNi3 antiperovskite (CuNNi3 + Cu) with acidic FeCl3 solution (pH = 2.3),91 during which the excessive Cu particles can be oxidized to soluble Cu2+ ions, thus creating rich pores. Besides, metallic Cu and Ni in CuNNi3 can be etched to form soluble Cu2+ and Ni2+ ions. Owing to the continuous consumption of H+, the hydrolysis of these metal ions was promoted, leading to the deposition of amorphous FeNiCu hydroxide on the antiperovskite. The obtained core–shell composite material with a well-tailored interface shows a dramatically decreased overpotential of 300 mV to achieve 10 mA cm−2. Following this research, Cui et al. adopted a similar approach to optimize ZnNNi3 antiperovskites,92 after which an obvious reduction in overpotential was realized on the fabricated electrode in operation of the OER.
4.2.4.2. Turning bulk perovskites into amorphous NPs. Using LN as a model material, we showed how FeCl3 solution turns bulk LN perovskites into amorphous Ni–Fe based hydroxides with unique local structures.93 The key processes are listed as follows: (i) the acidic FeCl3 solution selectively dissolves alkaline La3+ with the depletion of H+ (pH value gets larger); (ii) simultaneously, the corner-shared Ni3+–O octahedra change into edge-shared Ni3+–O octahedra; and (iii) the larger pH will promote the hydrolysis of Fe3+ to form FeOOH surrounded at the interstitial sites of Ni–O octahedra. As a result, the pristine bulk LN perovskite is turned into amorphous Ni–Fe-based NPs with distinguished local structures and boosted OER activity/stability.
4.2.4.3. Tailoring surface cation configuration. During the dissolution and deposition processes, surface cation configuration of catalysts can be well tailored, which plays critical roles in determining the final catalytic performance. La2NiO4 (L2N) is a typical RP perovskite with an La-enriched surface.94 We utilized FeCl3 solution with various concentrations to study its influence on the RP perovskite structure.95 When the concentration is low, a considerable amount of inert La remains, blocking active Ni sites and preventing the formation of Ni–Fe pairs. Meanwhile, excessive concentration of FeCl3 solution would not only dissolve all La cations at the surface but also induce significant accumulation of FeOOH, which buries the more active Ni–Fe pairs. As expected, FeCl3 treatment on La2NiO4 with the optimal solution concentration leads to the desirable surface cation configuration. Both the OER activity and stability of treated samples show a volcano-shaped dependence on the atomic ratio of (Ni + Fe)/La at the surface. The optimized catalyst with the (Ni + Fe)/La value close to unity demonstrates the best OER activity/stability among all RP based perovskites thus far.
4.3. Electrochemical approaches and corresponding effects
Electrochemical leaching is a scientific term describing the process during which a soluble component is separated or extracted from its carrier/substance and transferred into electrolyte driven by electro-potentials. The Pourbaix diagrams (potential–pH diagrams) provide precise information for element leaching in aqueous systems based on equilibrium thermodynamics.28 When exposed to applied potentials, metastable species characterized by fragile bonds in the initial structure and solubility in electrolyte would easily detach from the perovskite lattice and dissolve into the electrolyte.28 This leads to the disruption of the original perovskite structure, the creation of defects and the greater exposure of the solid–liquid interface, namely, surface reconstruction (Fig. 4). Generally, electrochemical leaching is more prevalent in complex oxides such as perovskites owing to their rich elemental composition and diverse chemical properties. In the following sections, disassembling and reassembling perovskites through electrochemical leaching and reconstruction are summarized based on distinct electrochemical reactions. | ||
| Fig. 4 Schematic illustration of the electrochemical strategy for disassembling and reassembling perovskites. | ||
Besides, many studies focus on using iridium-based perovskites as OER catalysts to reduce the Ir content and utilize component leaching to form the more active Ir motifs. The Xu group reported the surface reconstruction of B-site doped SrIrO3 perovskites by non-noble metals.107,108 The B-site substitution further reduces the content of Ir, thus reducing the price of the catalyst. What's more, the non-noble B-site cations leach out together with the A-site Sr cations, further promoting the degree of surface reconstruction. Specifically, the SrCo0.9Ir0.1O3 perovskite shows obvious surface reconstruction on its surface region.107 The reconstructed corner-shared and under-coordinated IrOx octahedra are responsible for the exceptionally high OER activity. A further study shows that the A-site cation leaching generates more electrochemical surface area for the reaction, and additional leaching of B-site induces the formation of a honeycomb-like IrOxHy phase, significantly enhancing the intrinsic activity.108 The reconstructed IrOxHy motifs show an activity improvement by approximately two orders of magnitude compared to the pristine counterpart. For more related studies please refer to ref. 109–111.
5. Conclusion and perspectives
In this minireview, we give a summary regarding disassembling and reassembling perovskites including their definition, benefits, approaches, and applications in oxygen electrocatalysis. Representative examples illustrate how the disassembling and reassembling perovskites effectively tackle the major challenges faced by original perovskite catalysts. The disassembling and reassembling strategy aims to engineer electrochemical active sites through two synergistic approaches: precisely tailoring intrinsic active sites within perovskite architectures, while strategically generating extrinsic active sites in perovskite-derived matrices. Notably, even the pre-engineered active sites in parent perovskites may experience dynamic transformations during electrocatalytic operation, ultimately evolving into emergent active sites through structural restructuring. Significant breakthroughs including the revealing of dynamic surface reconstruction, proposal of stability number, and the findings about paracrystalline motifs on reconstructed iridium oxides are highlighted. Among the three methods of disassembling and reassembling perovskites, the physical method allows more precise control of structure properties, making it suitable for fundamental studies. The chemical method is straightforward and effective and holds promise for large-scale catalyst production. Significantly, the electrochemical pathway refers to critical processes applied on catalysts under operational conditions, which hold great importance and promise for future related investigations. Despite significant advances, several scientific issues still remain in this emerging field, particularly concerning the electrochemical method:(1) Amorphous or paracrystalline phase?
For a long time, new species generated from electrochemical reconstruction are considered amorphous by default. However, very recently, the newly generated IrOx species in situ formed on Ir-based perovskites are identified paracrystalline with short-to-medium-range-order structure rather than the amorphous phase.15 This revelation enables theoretical modeling for corresponding DFT calculations, which is difficult to realize for amorphous cases. It is thus imperative to re-evaluate the reconstructed process and products for perovskite catalysts, which would help establish more accurate structure–activity relationships advancing the unveiling of reaction mechanisms and promoting practical applications for hydrogen production.
(2) Deteriorated interfaces between perovskites and ionomers
Disassembling and reassembling perovskites by the electrochemical method generally involves in situ leaching and redeposition processes, which are often accompanied by evolution of morphology or reduction of particle size. However, accordingly the catalytic activity often follows the trend that first increases and then decreases. The activity decay may result from the deteriorated interface between catalyst and ionomer, which would reduce ion conductivity, decrease electron transfer efficiency, and increase electrochemical resistance. The underlying mechanism needs to be investigated and corresponding strategies must be developed to prevent the interfacial degradation, so as to ensure long-term operation when enjoying the benefits of disassembling and reassembling perovskites.
(3) Directed by artificial intelligence (AI)
Predicting highly efficient catalyst materials using AI should result in a higher efficiency than that obtained using the traditional trial and error methods. However, considering the existence of electrochemical reconstruction, candidate materials predicted by AI at present should act as pre-catalysts in oxygen electrocatalysis, but not real-catalysts. As we have summarized in the electrochemical methods for disassembling and reassembling perovskites, there are remarkable property differences between pristine perovskite and electrochemically reconstructed products. From this perspective, if AI prediction is based on real-catalysts rather than pre-catalysts, it would give us a more accurate direction when developing target catalysts.
(4) Extending applications beyond oxygen electrocatalysis
Perovskite materials have been widely applied in the field of energy conversion and storage. Besides oxygen electrocatalysis, large surface area and favorable structure are also essential properties for other electrochemical processes. Considering the various strategies and obvious advantages when disassembling and reassembling perovskites, it should bring more opportunities in extending applications beyond oxygen electrocatalysis. For example, in carbon electrocatalysis (CO2 reduction and organics conversion), dynamically restructured interfaces enable multi-carbon pathway control via metastable intermediate stabilization. For nitrogen cycling applications (N2 fixation and nitrate reduction), the multi-component active sites can be responsible for nitrogen dissociation and protonation, respectively, breaking scaling relations through spatially decoupled reaction steps.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the Program for Jiangsu Specially-Appointed Professors (R2023T05), the Startup Foundation for Introducing Talent of NUIST (2024R078), the Natural Science Foundation of Jiangsu Province (BK20240707, BK20241425), and the National Natural Science Foundation of China (No. 22278203).References
- Y. Chen, D. J. Zheng, Z. J. Xu and Y. Shao-Horn, Nat. Sustainability, 2024, 7, 371 CrossRef.
- H. Yang, X. Han, A. I. Douka, L. Huang, L. Gong, C. Xia, H. S. Park and B. Y. Xia, Adv. Funct. Mater., 2021, 31, 2007602 CrossRef CAS.
- Z. Wang, Y.-R. Zheng, I. Chorkendorff and J. K. Nørskov, ACS Energy Lett., 2020, 5, 2905 CrossRef CAS.
- W. T. Hong, M. Risch, K. A. Stoerzinger, A. Grimaud, J. Suntivich and Y. Shao-Horn, Energy Environ. Sci., 2015, 8, 1404 RSC.
- P. Gayen, S. Saha and V. Ramani, Acc. Chem. Res., 2022, 55, 2191 CrossRef CAS PubMed.
- J. Hwang, R. R. Rao, L. Giordano, Y. Katayama, Y. Yu and Y. Shao-Horn, Science, 2017, 358, 751 CrossRef CAS PubMed.
- J. Moon, W. Beker, M. Siek, J. Kim, H. S. Lee, T. Hyeon and B. A. Grzybowski, Nat. Mater., 2024, 23, 108 CrossRef CAS PubMed.
- C. Shang, X. Xiao and Q. Xu, Coord. Chem. Rev., 2023, 485, 215109 CrossRef CAS.
- X. Liang, W. Yan, Y. Yu, K. Zhang, W. An, H. Chen, Y. Zou, X. Zhao and X. Zou, Angew. Chem., Int. Ed., 2023, 62, 202311606 CrossRef PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383 CrossRef CAS PubMed.
- J. T. Mefford, X. Rong, A. M. Abakumov, W. G. Hardin, S. Dai, A. M. Kolpak, K. P. Johnston and K. J. Stevenson, Nat. Commun., 2016, 7, 11053 CrossRef CAS PubMed.
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y. L. Lee, L. Giordano, K. A. Stoerzinger, M. T. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 457 CrossRef CAS PubMed.
- E. Fabbri, M. Nachtegaal, T. Binninger, X. Cheng, B. J. Kim, J. Durst, F. Bozza, T. Graule, R. Schäublin, L. Wiles and M. Pertoso, Nat. Mater., 2017, 16, 925 CrossRef CAS PubMed.
- S. Geiger, O. Kasian, M. Ledendecker, E. Pizzutilo, A. M. Mingers, W. T. Fu, O. Diaz-Morales, Z. Li, T. Oellers, L. Fruchter and A. Ludwig, Nat. Catal., 2018, 1, 508 CrossRef CAS.
- B. Lu, C. Wahl, R. dos Reis, J. Edgington, X. K. Lu, R. Li, M. E. Sweers, B. Ruggiero, G. K. K. Gunasooriya, V. Dravid and L. C. Seitz, Nat. Catal., 2024, 7, 868 CrossRef CAS.
- X. Xu, W. Wang, W. Zhou and Z. Shao, Small Methods, 2018, 2, 1800071 CrossRef.
- B. Koo, K. Kim, J. K. Kim, H. Kwon, J. W. Han and W. Jung, Joule, 2018, 2, 1476 CrossRef CAS.
- Y. Li, W. Zhang, Y. Zheng, J. Chen, B. Yu, Y. Chen and M. Liu, Chem. Soc. Rev., 2017, 46, 6345 RSC.
- H. J. Song, H. Yoon, B. Ju and D.-W. Kim, Adv. Energy Mater., 2021, 11, 2002428 CrossRef CAS.
- C. Wu, X. Wang, Y. Tang, H. Zhong, X. Zhang, A. Zou, J. Zhu, C. Diao, S. Xi, J. Xue and J. Wu, Angew. Chem., Int. Ed., 2023, 52, 202218599 Search PubMed.
- C. H. Park, H. Lee, J.-S. Choi, T. G. Yun, Y. Lim, H. B. Bae and S. Y. Chung, Adv. Mater., 2024, 36, 2403392 CrossRef CAS PubMed.
- H. Sun, H. Kim, X. Xu, L. Fei, W. Jung and Z. Shao, Renewables, 2023, 1, 21 CrossRef.
- D. Antipin and M. Risch, J. Phys. Energy, 2020, 2, 032003 CrossRef CAS.
- Z. Sun, W. Fan and T. Lin, J. Mater. Chem. A, 2023, 11, 24982 Search PubMed.
- H. Guo, Y. Yang, G. Yang, X. Cao, N. Yan, Z. Li, E. Chen, L. Tang, M. Peng, L. Shi, S. Xie, H. Tao, C. Xu, Y. Zhu., X. Fu, Y. Pan, N. Chen, J. Lin, X. Tu, Z. Shao and Y. Sun, ACS Catal., 2023, 13, 5007 Search PubMed.
- Y. Ebina, T. Sasaki and M. Watanabe, Solid State Ionics, 2002, 151, 177 CrossRef CAS.
- H. Li, Y. Chen, J. Z. Y. Seow, C. Liu, A. C. Fisher, J. W. Ager and Z. J. Xu, Small Sci., 2022, 2, 2100048 CrossRef CAS PubMed.
- G. Chen, Y. Zhu, S. She, Z. Lin, H. Sun and H. Huang, InfoMat, 2024, 6, e12609 Search PubMed.
- A. Grimaud, C. E. Carlton, M. Risch, W. T. Hong, K. J. May and Y. Shao-Horn, J. Phys. Chem. C, 2013, 117, 259262 CrossRef.
- M. Risch, K. A. Stoerzinger, S. Maruyama, W. T. Hong, I. Takeuchi and Y. Shao-Horn, J. Am. Chem. Soc., 2014, 136, 5229 CrossRef CAS PubMed.
- J. D. Baniecki, H. Yamaguchi, C. Harnagea, D. Ricinschi, Z. Gu, J. E. Spanier, T. Yamazaki and H. Aso, Adv. Energy Mater., 2019, 9, 1803846 CrossRef.
- L. Heymann, M. L. Weber, M. Wohlgemuth, M. Risch, R. Dittmann, C. Baeumer and F. Gunkel, ACS Appl. Mater. Interfaces, 2022, 14, 14129–14136 CrossRef CAS PubMed.
- D. G. Nocera, J. Am. Chem. Soc., 2022, 144, 1069 Search PubMed.
- Y. Zhu, Z. He, Y. Choi, H. Chen, X. Li, B. Zhao, Y. Yu, H. Zhang, K. A. Stoerzinger, Z. Feng, Y. Chen and M. Liu, Nat. Commun., 2020, 11, 4299 CrossRef CAS PubMed.
- C. Wei, Z. Wang, K. Otani, D. Hochfilzer, K. Zhang, R. Nielsen, I. Chorkendorff and J. Kibsgaard, ACS Catal., 2023, 13, 14058 Search PubMed.
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, H. Y. Hwang, J. K. Norskov and T. F. Jaramillo, Science, 2016, 353, 1011 CrossRef CAS PubMed.
- M. L. Weber, G. Lole, A. Kormanyos, A. Schwiers, L. Heymann, F. D. Speck, T. Meyer, R. Dittmann, S. Cherevko, C. Jooss, C. Baeumer and F. Gunkel, J. Am. Chem. Soc., 2022, 144, 17966–17979 CrossRef CAS PubMed.
- S. Li, S. Zhao, F. Hu, L. Li, J. Ren, L. Jiao, S. Ramakrishna and S. Peng, Prog. Mater. Sci., 2024, 145, 101294 CrossRef CAS.
- N. Danilovic, R. Subbaraman, K. C. Chang, S. H. Chang, Y. Kang, J. Snyder, A. P. Paulikas, D. Strmcnik, Y. T. Kim, D. Myers and V. R. Stamenkovic, Angew. Chem., Int. Ed., 2014, 53, 14016 CrossRef CAS PubMed.
- S. H. Chang, N. Danilovic, K.-C. Chang, R. Subbaraman, A. P. Paulikas, D. D. Fong, M. J. Highland, P. M. Baldo, V. R. Stamenkovic, J. W. Freeland, J. A. Eastman and N. M. Markovic, Nat. Commun., 2014, 5, 4191 Search PubMed.
- S. A. Lee, S. Oh, J.-Y. Hwang, M. Choi, C. Youn, J. W. Kim, S. H. Chang, S. Woo, J.-S. Bae, S. Park, Y.-M. Kim, S. Lee, T. Choi, S. W. Kim and W. S. Choi, Energy Environ. Sci., 2017, 10, 924 RSC.
- A. R. Akbashev, L. Zhang, J. T. Mefford, J. Park, B. Butz, H. Luftman, W. C. Chueh and A. Vojvodic, Energy Environ. Sci., 2018, 11, 1762 RSC.
- M. Wohlgemuth, M. L. Weber, L. Heymann, C. Baeumer and F. Gunkel, Front. Chem., 2022, 10, 913419 CrossRef CAS PubMed.
- M. Vitale-Sullivan and K. A. Stoerzinger, Curr. Opin. Electrochem., 2023, 39, 101252 CrossRef CAS.
- B. Koo, K. Kim, J. K. Kim, H. Kwon, J. W. Han and W. Jung, Joule, 2018, 2, 1476 Search PubMed.
- Y. Li, W. Zhang, Y. Zheng, J. Chen, B. Yu, Y. Chen and M. Liu, Chem. Soc. Rev., 2017, 46, 6345 RSC.
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159 CrossRef CAS.
- J. S. Yoo, X. Rong, Y. Liu and A. M. Kolpak, ACS Catal., 2018, 8, 4628 CrossRef CAS.
- W. Zhou and J. Sunarso, J. Phys. Chem. Lett., 2013, 4, 2982 CrossRef CAS.
- J. Chen, J. Wu, Y. Liu, X. Hu and D. Geng, Phys. Status Solidi A, 2018, 215, 1800380 CrossRef.
- N. I. Kim, J. Lee, S. Jin, J. Park, J.-Y. Jeong, J. Lee, Y. Kim, C. Kim and S. M. Choi, Small Methods, 2024, 8, 2400284 Search PubMed.
- J. R. Petrie, V. R. Cooper, J. W. Freeland, T. L. Meyer, Z. Zhang, D. A. Lutterman and H. N. Lee, J. Am. Chem. Soc., 2016, 138, 2488 Search PubMed.
- C. Baeumer, J. Li, Q. Lu, A. Y.-L. Liang, L. Jin, H. P. Martins, T. Duchoň, M. Glöβ, S. M. Gericke, M. A. Wohlgemuth, M. Giesen, E. E. Penn, R. Dittmann, F. Gunkel, R. Waser, M. Bajdich, S. Nemšák, J. T. Mefford and W. C. Chueh, Nat. Mater., 2021, 20, 674 Search PubMed.
- A. Füngerlings, M. Wohlgemuth, D. Antipin, E. van der Minne, E. M. Kiens, J. Villalobos, M. Risch, F. Gunkel, R. Pentcheva and C. Baeumer, Nat. Commun., 2023, 14, 8284 Search PubMed.
- J. Kang, X. Yang, Q. Hu, Z. Cai, L. M. Liu and L. Guo, Chem. Rev., 2023, 123, 8859 Search PubMed.
- H. Sun, X. Xu, G. Chen and Z. Shao, Carbon Energy, 2024, 6, e595 Search PubMed.
- G. Chen, W. Zhou, D. Guan, J. Sunarso, Y. Zhu, X. Hu, W. Zhang and Z. Shao, Sci. Adv., 2017, 3, 1603206 Search PubMed.
- G. Chen, Z. Hu, Y. Zhu, B. Gu, Y. Zhong, H.-J. Lin, C.-T. Chen, W. Zhou and Z. Shao, Adv. Mater., 2018, 30, 1804333 Search PubMed.
- K. Wang, J. Zhou, L. Fu, Y. Kang, Z. Zhou, Y. Cheng, K. Wu and Y. Yamauchi, Small, 2024, 20, 2404239 Search PubMed.
- H. He, D. Huang, W. Pang, D. Sun, Q. Wang, Y. Tang, X. Ji, Z. Guo and H. Wang, Adv. Mater., 2018, 30, 1801013 Search PubMed.
- D. Yan, Y. Li, J. Huo, R. Chen, L. Dai and S. Wang, Adv. Mater., 2017, 29, 1606459 Search PubMed.
- S. Anantharaj and S. Noda, Small, 2020, 16, 1905779 CrossRef CAS PubMed.
- D. Chen, M. Qiao, Y.-R. Lu, L. Hao, D. Liu, C.-L. Dong, Y. Li and S. Wang, Angew. Chem., Int. Ed., 2018, 57, 8691 Search PubMed.
- M. Narahara, S. Y. Lee, K. Sasaki, K. Fukushima, K. Tanaka, S. Chae, X. Hu, G. Panomsuwan and T. Ishizaki, Sustainable Energy Fuels, 2023, 7, 2582 Search PubMed.
- Y. Lu, A. Ma, Y. Yu, R. Tan, C. Liu, P. Zhang, D. Liu and J. Gui, ACS Sustainable Chem. Eng., 2018, 7, 2906 Search PubMed.
- J. Sun, Z. Zhang, Y. Gong, H. Wang, R. Wang, L. Zhao and B. He, Sci. Rep., 2019, 9, 4210 Search PubMed.
- Y. Zhu, L. Zhang, B. Zhao, H. Chen, X. Liu, R. Zhao, X. Wang, J. Liu, Y. Chen and M. Liu, Adv. Funct. Mater., 2019, 29, 1901783 Search PubMed.
- G. Chen, Z. Hu, Y. Zhu, Z.-G. Chen, Y. Zhong, H.-J. Lin, C.-T. Chen, L. H. Tjeng, W. Zhou and Z. Shao, J. Mater. Chem. A, 2018, 6, 9854 Search PubMed.
- Y. Zhu, W. Zhou, Z.-G. Chen, Y. Chen, C. Su, M. O. Tadé and Z. Shao, Angew. Chem., Int. Ed., 2015, 54, 3897 CrossRef CAS PubMed.
- M. Risch, A. Grimaud, K. J. May, K. A. Stoerzinger, T. J. Chen, A. N. Mansour and Y. Shao-Horn, J. Phys. Chem. C, 2013, 117, 8628 Search PubMed.
- M. Lu, Y. Zheng, Y. Hu, B. Huang, D. Ji, M. Sun, J. Li, Y. Peng, R. Si, P. Xi and C.-H. Yan, Sci. Adv., 2022, 8, 3563 Search PubMed.
- X. Sun, H. Chen, Y. Yin, M. T. Curnan, J. W. Han, Y. Chen and Z. Ma, Small, 2021, 17, 2005383 CrossRef CAS PubMed.
- Y.-Q. Zhang, H.-B. Tao, J. Liu, Y.-F. Sun, J. Chen, B. Hua, T. Thundat and J.-L. Luo, Nano Energy, 2017, 38, 392 CrossRef CAS.
- S. Zuo, Y. Liao, C. Wang, A. B. Naden and J. T. Irvine, Small, 2024, 20, 2308867 Search PubMed.
- B. Hua, M. Li, Y.-F. Sun, Y.-Q. Zhang, N. Yan, J. Chen, T. Thundat, J. Li and J.-L. Luo, Nano Energy, 2017, 32, 247 Search PubMed.
- S. Song, J. Zhou, X. Su, Y. Wang, J. Li, L. Zhang, G. Xiao, C. Guan, R. Liu, S. Chen and H. J. Lin, Energy Environ. Sci., 2018, 11, 2945 RSC.
- Y. Arafat, M. R. Azhar, Y. Zhong, R. O'Hayre, M. O. Tadé and Z. Shao, J. Mater. Chem. A, 2023, 11, 12856 RSC.
- E. Mitoudi-Vagourdi, S. Müllner, P. Lemmens, R. K. Kremer and M. Johnsson, Inorg. Chem., 2018, 57, 9115 CrossRef CAS PubMed.
- X. Yu, E. Mitoudi-Vagourdi and M. Johnsson, ChemCatChem, 2019, 11, 6105 Search PubMed.
- C. Cao, C. Shang, X. Li, Y. Wang, C. Liu, X. Wang, S. Zhou and J. Zeng, Nano Lett., 2020, 20, 2837 CrossRef CAS PubMed.
- R. Zhang, P. E. Pearce, V. Pimenta, J. Cabana, H. Li, D. Alves Dalla Corte, A. M. Abakumov, G. Rousse, D. Giaume, M. Deschamps and A. Grimaud, Chem. Mater., 2020, 32, 3499 CrossRef CAS.
- H. Chen, L. Shi, K. Sun, K. Zhang, Q. Liu, J. Ge, X. Liang, B. Tian, Y. Huang, Z. Shi, Z. Wang, W. Zhang, M. Liu and X. Zou, ACS Catal., 2022, 12, 8658 Search PubMed.
- H. Chen, Z. Xu, L. Wei, M. Dong, Y. Hu, Y. Lu, N. Zhang, J. Wu and Q. Lu, J. Mater. Chem. A, 2024, 12, 23658 Search PubMed.
- H. Fukuoka, T. Isami and S. Yamanaka, J. Solid State Chem., 2000, 151, 40 CrossRef CAS.
- X. Yang, Y. Gao, X. Xu, W. Xu, D. Wang, B. Luo, D. Liu, T. Liang and B. Wang, Nano Res., 2024, 17, 4934 Search PubMed.
- W. Bian, W. Wu, B. Wang, W. Tang, M. Zhou, C. Jin, H. Ding, W. Fan, Y. Dong, J. Li and D. Ding, Nature, 2022, 604, 479 Search PubMed.
- W. Guo, L. Cui, H. Xu and C. Gong, Appl. Surf. Sci., 2020, 529, 147165 Search PubMed.
- J. Yu, D. Chen, M. Saccoccio, K. Lam and F. Ciucci, ChemElectroChem, 2018, 5, 1105 Search PubMed.
- N. Li, L. Cai, C. Wang, Y. Lin, J. Huang, H. Sheng, H. Pan, W. Zhang, Q. Ji, H. Duan, W. Hu, W. Zhang, F. Hu, H. Tan, Z. Sun, B. Song, S. Jin and W. Yan, J. Am. Chem. Soc., 2021, 143, 18001 CrossRef CAS PubMed.
- K. T. Lai, I. Antonyshyn, Y. Prots and M. Valldor, J. Am. Chem. Soc., 2017, 139, 9645 CrossRef CAS PubMed.
- Y. Zhu, G. Chen, Y. Zhong, Y. Chen, N. Ma, W. Zhou and Z. Shao, Nat. Commun., 2018, 9, 1 CrossRef PubMed.
- J. Zhang, J. Liu, L. Zhang, J. Ke, C. Zhong, Y. Tu, L. Wang, H. Song, L. Du, Z. Zhang and Z. Cui, ACS Catal., 2023, 13, 5043 Search PubMed.
- G. Chen, Y. Zhu, H. M. Chen, Z. Hu, S.-F. Hung, N. Ma, J. Dai, H.-J. Lin, C.-T. Chen, W. Zhou and Z. Shao, Adv. Mater., 2019, 31, 1900883 CrossRef PubMed.
- J. Wu, S. S. Pramana, S. J. Skinner, J. A. Kilner and A. P. Horsfield, J. Mater. Chem. A, 2015, 3, 23760 RSC.
- Y. Li, G. Chen, H. C. Chen, Y. Zhu, L. Fei, L. Xu, T. Liu, J. Dai, H. Huang, W. Zhou and Z. Shao, Energy Environ. Sci., 2023, 16, 3331 RSC.
- G. Ou, C. Yang, Y. Liang, N. Hussain, B. Ge, K. Huang, Y. Xu, H. Wei, R. Zhang and H. Wu, Small Methods, 2019, 3, 1800279 CrossRef.
- L. Gui, G. Pan, X. Ma, M. You, B. He, Z. Yang, J. Sun, W. Zhou, J. Xu and L. Zhao, Appl. Surf. Sci., 2021, 543, 148817 CrossRef CAS.
- F. Song and X. Hu, Nat. Commun., 2014, 5, 4477 CrossRef CAS PubMed.
- M. Yu, K. S. Belthle, C. Tüysüz and H. Tüysüz, J. Mater. Chem. A, 2019, 7, 23130 Search PubMed.
- Y. Li, G. Chen, Y. Zhu, Z. Hu, T.-S. Chan, S. She, J. Dai, W. Zhou and Z. Shao, Adv. Funct. Mater., 2021, 31, 2103569 Search PubMed.
- G. Ou, Y. Xu, B. Wen, R. Lin, B. Ge, Y. Tang, Y. Liang, C. Yang, K. Huang, D. Zu, R. Yu, W. Chen, J. Li, H. Wu, L.-M. Liu and Y. Li, Nat. Commun., 2018, 9, 1302 CrossRef PubMed.
- W. Zhou, R. Ran and Z. Shao, J. Power Sources, 2009, 192, 231 CrossRef CAS.
- Z. Shao and S. M. Haile, Nature, 2004, 431, 170 CrossRef CAS PubMed.
- P. P. Lopes, D. Y. Chung, X. Rui, H. Zheng, H. He, P. Farinazzo Bergamo Dias Martins, D. Strmcnik, V. R. Stamenkovic, P. Zapol, J. F. Mitchell, R. F. Klie and N. M. Markovic, J. Am. Chem. Soc., 2021, 143, 2741 CrossRef CAS PubMed.
- O. Diaz-Morales, S. Raaijman, R. Kortlever, P. J. Kooyman, T. Wezendonk, J. Gascon, W. T. Fu and M. T. M. Koper, Nat. Commun., 2016, 7, 12363 Search PubMed.
- R. Zhang, N. Dubouis, M. Ben Osman, W. Yin, M. T. Sougrati, D. A. D. Corte, D. Giaume and A. Grimaud, Angew. Chem., Int. Ed., 2019, 58, 4571 CrossRef CAS PubMed.
- Y. Chen, H. Li, J. Wang, Y. Du, S. Xi, Y. Sun, M. Sherburne, J. W. Ager III, A. C. Fisher and Z. J. Xu, Nat. Commun., 2019, 10, 572 Search PubMed.
- Y. Chen, Y. Sun, M. Wang, J. Wang, H. Li, S. Xi, C. Wei, P. Xi, G. E. Sterbinsky, J. W. Freeland, A. C. Fisher, J. W. Ager III, Z. Feng and Z. J. Xu, Sci. Adv., 2021, 7, 1788 Search PubMed.
- L. An, C. Wei, M. Lu, H. Liu, Y. Chen, G. G. Scherer, A. C. Fisher, P. Xi, Z. J. Xu and C.-H. Yan, Adv. Mater., 2021, 33, 2006328 Search PubMed.
- H. J. Song, H. Yoon, B. Ju and D.-W. Kim, Adv. Energy Mater., 2021, 11, 2002428 Search PubMed.
- Z. Lei, T. Wang, B. Zhao, W. Cai, Y. Liu, S. Jiao, Q. Li, R. Cao and M. Liu, Adv. Energy Mater., 2020, 10, 2000478 Search PubMed.
- Y. Lu, Q. Shen, Q. Yu, F. Zhang, G. Li and W. Zhang, J. Phys. Chem. C, 2016, 120, 28712 CrossRef CAS.
- G. Chen, Y. Zhu, Y. Ying, Y. Yao, Z. Hu, D. Zu, Z. Lin, C. W. Pao, Y. C. Zhang, L. Li, Y. Zhu and H. Huang, Matter, 2024, 7, 2265 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |