
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN, Fe co-incorporated CoO nanoarray enhanced by magnetic field for efficient water oxidation†
Keke Huanga,
Yaotian Yan*a,
Yaqiang Yua,
Taili Yanga,
Liang Qiaob,
Jinchun Tuc,
Jiehe Suia,
Wei Caia,
Shude Liu *d and
Xiaohang Zheng*a
*d and
Xiaohang Zheng*a
aSchool of Materials Science and Engineering, Harbin Institute of Technology, Harbin 150001, China. E-mail: zhengxiaohang@hit.edu.cn; ytyanhit@hit.edu.cn
bKey Laboratory of Materials Design and Quantum Simulation, College of Science, Changchun University, Changchun 130022, China
cState Key Laboratory of Marine Resource Utilization in South China Sea, College of Materials and Chemical Engineering, Hainan University, Haikou 570228, China
dEngineering Research Center of Technical Textile, Ministry of Education, College of Textiles, Donghua University, Shanghai 201620, China. E-mail: sdliu@dhu.edu.cn
First published on 20th May 2025
Abstract
CoO, as a typical water oxidation electrocatalyst, has gradually entered the bottleneck stage of performance modulation through composition optimization. Herein, the N, Fe co-bonded CoO was achieved by N plasma, which suggests further potential to be enhanced by a magnetic field during oxygen evolution reaction (OER) electrocatalysis. N atoms are a bridge for bonding Fe and Co centers, which serve as a fast channel for electron transfer. N, Fe co-doping decreases the electron density around Co2+ centers, which increases the unpaired electrons for electron acceptors. As a result, the intrinsic OER activities are boosted, which is further beneficial for amplifying the magnetic enhancement effect. The best performance emerges under a parallel magnetic field with 420 mT intensity, which results in a lowered overpotential of 217 mV and a Tafel slope of 25.1 mV dec−1 in alkaline media. The magnetic enhancement comes from the magnetohydrodynamic effect and the escape energy barrier reduction of the paramagnetic triplet state of O2. The magnetic enhancement effect would be amplified when the catalytic current becomes larger (magnetic current is 8 mA and 22 mA under 500 mA and 1000 mA total current, respectively). This work provides an in-depth insight into the magnetic enhancing mechanism and a highly feasible strategy for coupling heteroatoms with the magnetic field to operate and break through the bottleneck of non-noble electrocatalysis performance.
Broader contextExploring an efficient strategy for water oxidation electrocatalysts is vital for implementing distributed mobile energy (fuel cell, hydrogen refueling station, hydrogen energy vehicles, etc.). CoO is considered a potential metal compound as an OER electrocatalyst, due to rich Co crustal abundance, low materials cost, and multiple valence states. However, the conductivity of CoO is usually insufficient, and the intrinsic activities for OER are far from ideal. Much progress has been made in modulating nanosized cobalt oxides, where compositional modification strategies, including the incorporation of heteroatoms, are a typical mainstream advanced technology. However, the OER performance modulation effect induced by composition optimization gradually enters the bottleneck stage. Magnetic field assisting catalytic process, as a typical external field control method, has the potential for breaking through electrocatalysis performance limitations beyond composition optimization. The coupling of magnetic field assistance and heteroatoms incorporation will pave the way for breaking the bottleneck of catalytic performance and developing advanced, cost-effective catalytic electrodes and devices. However, few research studies have explored this topic yet. |
1. Introduction
Water oxidation electrocatalysis (OER) is vital in implementing distributed mobile energy in fuel cells, hydrogen refueling stations, and hydrogen energy vehicles.1 However, the OER comprises four electron transfer steps, whose slow kinetics limit the total efficiency of related devices.2,3 Therefore, high-performance electrocatalysts were demanded to facilitate the OER process. The commonly used electrocatalysts are IrO2 and RuO2, whose scarcity and high-cost block commercial applications.4 Therefore, high-efficiency non-noble metal electrocatalysts were urged to be explored, which has become a common concern for related fields.5Among these, 3d-transition-metal oxides, typically CoO-based compounds, have attracted interest as potential OER electrocatalysts due to rich Co crustal abundance, low materials cost, and multiple valence states.6–8 The unique di-μ-oxo bridged Co–Co coordination benefits the deprotonation process, where the formation of active oxygen ligands is promoted for boosting OER.9 However, the conductivity of CoO is usually insufficient, and the intrinsic activities for OER are far from ideal states.10,11 Many efforts have been devoted to compositional design to address the above issue, such as defect engineering,12 phase transition,13 and doping,14 which gradually enter the bottleneck stage for modulating OER performances. External magnetic fields have recently been reported to be highly effective for controlling fluid behavior and regulating ionic chemical reactions.15–17 Electrocatalytic water oxidation involves adsorbing and transforming a series of intermediates (OH−, O2−, OOH3−, and O2, etc.). Importantly, the nature of the paramagnetic triplet state of molecular oxygen means that the parallel electron spin directions in adjacent adsorbed oxygen (*O) would lower the formation energy barrier of the O2 product. Considering that the adsorption of *O on catalysts follows the spin conservation rule between *O and active sites, unifying the spin direction of the catalyst surface would enable the induced spin parallel to *O to boost O2 formation.18
Previous researchers, such as Xu et al.9,19–22 and Galán-Mascarós et al.,16 have made significant breakthroughs in interactions between external magnetic fields and magnetic materials, revealing that the external magnetic field could influence the spin orientation of solid surfaces. The stronger the magnetic properties of a solid, the more favorable it is for spin regulation. However, CoO is a weak magnetic substance,23 which means the external magnetic field has very limited effects on surface spin regulation. Utilizing a strong magnetic Fe element to incorporate CoO may be a promising way to enhance the effect of the external magnetic field on surface spin. However, the fine manipulation of high valence Fe3+ cations on the oxide surface remains a challenge because the electrostatic repulsion of Co and Fe cations results in unstable surface incorporation. Until recently, a few excellent studies about Fe–N coordination modes on carbon materials suggest that the half-filling p orbital in N induces rich valence states and bonding properties compared with O.24–27 This inspires us that the N ligand can bridge spin-polarized Fe cations on the CoO surface. Therefore, coupling magnetic fields with compositionally modified electrocatalysts is expected to regulate the intermediates’ adsorption and transformation behaviors, thus making breakthroughs in overcoming the bottleneck of OER performances. However, few attempts have been made in such aspects, and the integration of N and Fe heteroatoms with a huge electronegativity difference on the CoO surface remains a big challenge.
To address the above concerns, a high-energy N atmosphere generated by plasma was utilized to anchor the Fe cations on CoO to form N–Fe–CoO nanoarrays, which suggests further potential to be enhanced by a magnetic field during OER. N atoms are a bridge for bonding Fe and Co centers, which serve as a fast channel for electron transfer. N, Fe co-doping decreases the electron density around Co2+ centers, which increases the unpaired electrons and holes for electron acceptors in the valence band. As a result, the intrinsic OER activities are boosted, which is further beneficial for amplifying the magnetic enhancement effect. The best performance emerges under a parallel magnetic field with 420 mT intensity, which results in a lowered overpotential of 217 mV and a Tafel slope of 25.1 mV dec−1 in alkaline media.
2. Results and discussion
As shown in Fig. 1a, CoO prepared by hydrothermal is grown on carbon cloth in a nanosheet structure, with an edge size of ∼2 microns, but the thickness is extremely thin at the nanometer level. The morphology of Fe–CoO in Fig. 1b is the same as the size of CoO, indicating that the Fe3+ ions treatment does not change the morphology of the sample. In addition, the N–Fe–CoO suggests a similar morphology to CoO in Fig. 1c, indicating that the N-plasma would not destroy the surface lamellar structure. Further, to determine the composition of the sample, the XRD test was used to analyze the phase of the sample, as shown in Fig. 1d. As can be seen from the figure, the diffraction peaks of the samples correspond to the standard CoO phase (JCPDS card no. 43-1004). No other peak positions were found, indicating that the CoO electrode derived by the hydrothermal method is a pure phase. The peak positions of the N–Fe–CoO spectrum are consistent with those of CoO, and there is no other diffraction peak of Fe or N-based compounds, which indicates that Fe and N atoms exist in the form of doping rather than forming compounds. Moreover, the results of element mapping (Fig. S2, ESI†) and X-ray photoelectron spectroscopy (XPS) XPS survey spectra (Fig. S3, ESI†) reveal that the Co, O, Fe, and N elements in N–Fe–CoO are evenly distributed in the nanoarray, and the atomic percentage of N and Fe is about 10.9% and 1.9%. Notably, the diffraction peak is not sharp, which means a low crystallinity of the CoO phase. The low crystallinity often means the rich unsaturated coordination atoms, which are favorable for serving as electrocatalytic OER. | ||
| Fig. 1 The microstructures of (a) CoO, (b) Fe–CoO, (c) N–Fe–CoO. (d) The XRD patterns of CoO and N–Fe–CoO samples, (e) the pair distribution function (PDF) analysis for CoO and N–Fe–CoO nanosheets (derived under CuKa1 radiation, λ = 1.54056 Å). | ||
To further confirm that the N and Fe heteroatoms are successfully introduced into CoO, the pair distribution function (PDF) analysis is conducted (Fig. 1e). The introduction of N and Fe atoms in CoO is expected to influence the bond length of surrounding coordination atoms. The first peak represents the shortest interaction distance of the atoms in the material, where the N–Fe–CoO nanosheets suggest a larger interaction distance than the CoO nanosheets. This is also consistent with the structural relaxation result by density functional theory (DFT) calculation (Fig. S4, ESI†), where the bond length of the Co–N pair (∼2.5 Å) and the Fe–O pair (∼2.0 Å) is larger than the Co–O pair (1.8 Å). The atoms’ interaction distance would cause a change in surface chemical states and electronic structure, which can be confirmed by XPS in the next section.
The TEM images show more detailed information about the microstructure of CoO and N–Fe–CoO nanosheets (Fig. 2). Compared with CoO (Fig. 2a), the surface of N–Fe–CoO is rougher (Fig. 2c), which results from the N plasma etching. Furthermore, the high-resolution TEM (HRTEM) images (Fig. 2b and d) show well-defined lattice fringes with d-spacings of ∼0.20 nm and ∼0.28 nm, which closely correspond to the (200) and (111) planes of CoO (JCPDS card no. 43-1004). Importantly, the sub-nanometer interface emerges after introducing N and Fe heteroatoms into CoO (marked by a blue dashed line), which indicates that the bonding process of N and Fe atoms on the CoO surface is accompanied by slight lattice reconstruction.
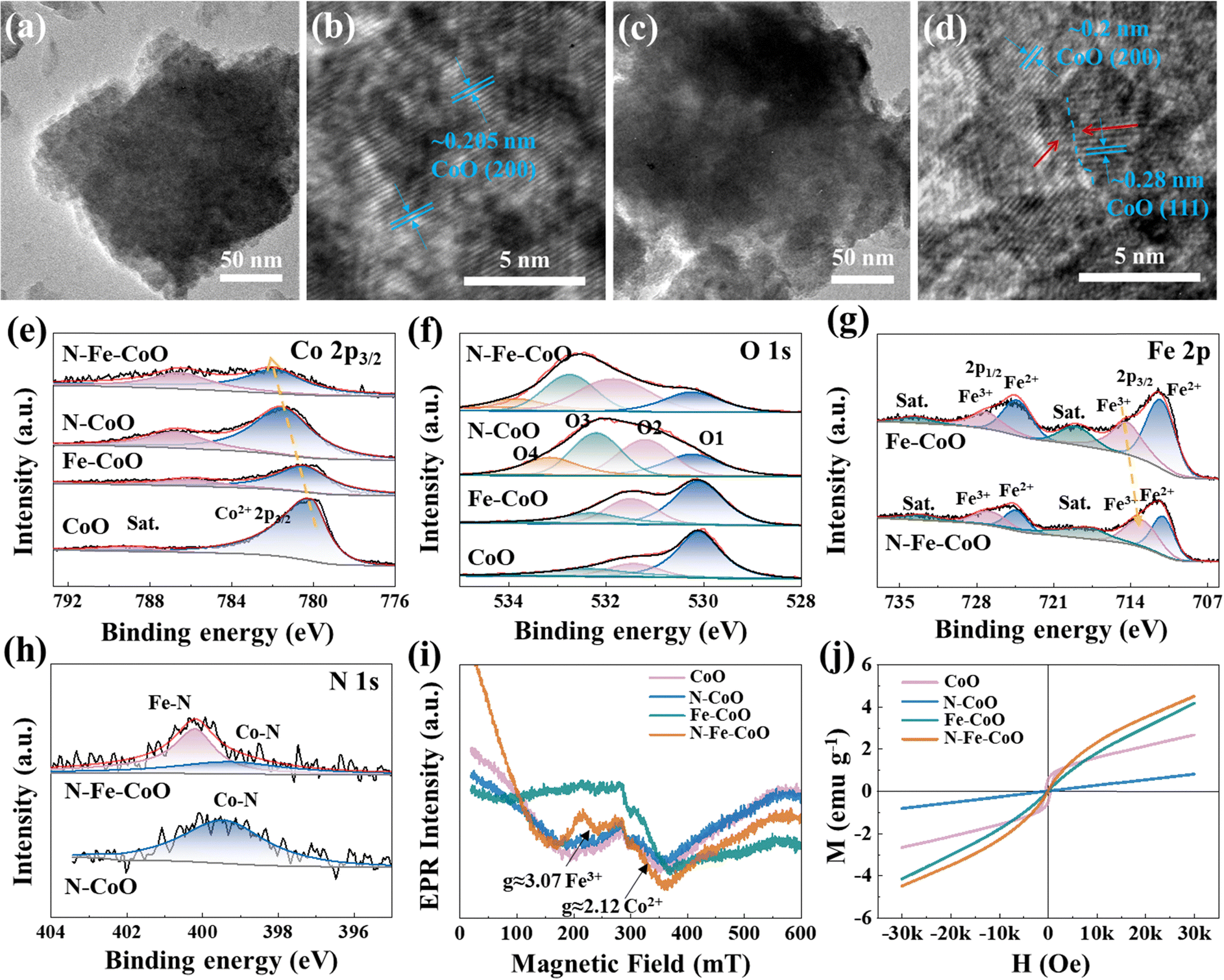 | ||
| Fig. 2 The blue dashed line marks the sub-nanometer interface for the TEM characterizations of (a) and (b) CoO and (c) and (d) N–Fe–CoO samples. The (e) Co 2p XPS spectra and (f) O 1s spectra of as-prepared samples, (g) the Fe 2p XPS spectra of Fe–CoO and N–Fe–CoO samples, (h) the N 1s XPS spectra of N–CoO and N–Fe–CoO samples, and the (i) EPR spectra (ν = 9.84 GHz) and (j) magnetic hysteresis loops of CoO, N–CoO, Fe–CoO, and N–Fe–CoO samples. | ||
XPS was employed to investigate the synthesized catalysts’ chemical states and surface electronic structures. As shown in Fig. 2e, the Co 2p3/2 peak at 780.1 eV in CoO nanosheets is attributed to Co2+.28,29 In Fe–CoO nanosheets, the Co2+ 2p3/2 peak shifts 0.4 eV towards a higher binding energy compared to CoO, suggesting that the strong electrostatic attraction of Fe3+ makes the outer electrons of Co atoms more likely to be released, reducing the electron density around Co centers. Meanwhile, compared to pure CoO, the binding energy of Co2+ 2p3/2 in N–CoO shows an increase of 1.4 eV. This shift implies that N-doping significantly alters the electronic structure of Co2+ centers, indicating a potential chemical bonding interaction between Co and N elements. N–Fe–CoO nanosheets exhibit the highest Co2+ 2p3/2 binding energy (781.9 eV), and the binding energy shift relative to CoO is approximately equal to the sum of the shifts for N–CoO and Fe–CoO, confirming the synergistic effect of N and Fe dopants. Besides, the Fe 2p spectra of Fe–CoO and N–Fe–CoO nanosheets are depicted in Fig. 2g, where six peaks are identified. Excluding the low-intensity satellite peak, the peaks at 711.3 eV and 724.4 eV in Fe–CoO are assigned to Fe2+, while those at 714.3 eV and 727.2 eV correspond to Fe3+.30,31 For N–Fe–CoO, the peaks at 711.0 eV and 724.4 eV are attributed to Fe2+, and those at 713.6 eV and 726.8 eV to Fe3+. The binding energy of Fe 2p3/2 is shifted towards lower energy by ∼0.2 eV compared to the Fe–CoO electrode, which indicates that the doping of N leads to an increase in electron density near the Fe atoms. This confirms the electrons transferring from the Co center to the Fe center.
Furthermore, as shown in Fig. 2i, the EPR spectra of CoO, N–CoO, Fe–CoO, and N–Fe–CoO show a very broad line with a g ≈ 2.12 that can be attributed to the high-spin Co2+ (three unpaired electrons).32–34 In general, the intensity of the EPR signal is positively correlated with the number of unpaired electrons. Notably, the N–Fe–CoO sample exhibits the highest EPR signal intensity, suggesting that the number of unpaired electrons at the Co center increases following N and Fe co-doping. In addition, it can be observed that the EPR signal strength of N–CoO is slightly smaller than that of CoO, indicating that introducing N may reduce Co's unpaired electron density by forming Co–N covalent bonds. Additionally, the Fe–CoO and N–Fe–CoO exhibit a distinct new peak at g ≈ 3.07 in their EPR spectra (Fig. 2i), which can be assigned to the spin of Fe3+ (five unpaired electrons).35 This observation indicates that Fe doping introduces unpaired electrons into the CoO system. Considering that the OER follows the following pathways:36
| * + OH− → *OH + e−, |
| *OH → *O + H+ + e−, |
| *O + OH− → *OOH + e−, |
| *OOH → * + O2 + H+ + e−, |
The process comprises potential-consuming 4-electron-consuming transfer steps, where the catalyst surface serves as an electron acceptor. The increase of unpaired electrons benefits from increasing electron acceptors on the electrode surface. Besides, the unpaired electrons are favorable for responding to an external magnetic field, which is expected to boost the OER process.
The O 1s spectra (Fig. 2f) of the samples were performed to investigate the surface defects of the catalysts. The O 1s spectra of CoO and Fe–CoO were fitted with three peaks corresponding to lattice oxygen (O1, 530.1 eV), defect sites with low oxygen coordination (O2, 531.4 eV), and hydroxyl groups or surface-adsorbed oxygen (O3, 532.3 eV).37,38 Additionally, the O 1s spectra of N–CoO and N–Fe–CoO reveal additional peaks at 533.2 eV and 533.4 eV, respectively, assigned to adsorbed water molecules (O4) on the surface. It is worth noting that the peak ratio of O2 and O3 increases after the electrode is doped with Fe and N. Importantly, the actual proportion of oxygen vacancies can be approximated by the peak area ratio of O2/(O1 + O2 + O3). According to the theoretical calculation, the O2 ratios of CoO, Fe–CoO, N–CoO, and N–Fe–CoO are 16.8%, 27.3%, 35.5%, and 42.7%, respectively. The results show that N plasma will produce more oxygen vacancy defects on the electrode surface. This is also one of the reasons for the improvement of OER activity, i.e., oxygen vacancy can change the active species’ adsorption energy and enhance the catalysts’ intrinsic activity. Fig. 2h shows the N 1s spectra of the N–CoO and N–Fe–CoO electrodes. Only one obvious peak at 399.5 eV is identified from the N–CoO electrode, which is attributed to the Co–N bond.39 However, two peaks can be observed in the N–Fe–CoO electrode; the peak near 399.3 eV can be attributed to the Co–N bond, and the peak around 400.2 eV can be attributed to the Fe–N bond.40 This indicates that N doping is a bridge for bonding Co and Fe centers, which benefits from accelerating electron flow and charge transfer during OER.
The magnetic hysteresis loops of CoO, N–CoO, and N–Fe–CoO at room temperature are also included, as shown in Fig. 2j. It is evident that N–CoO and N–Fe–CoO exhibit negligible hysteresis, indicating paramagnetic behavior (Fig. S5, ESI†).41 In contrast, CoO shows a distinct hysteresis loop, characteristic of ferromagnetism.42 The hysteresis loop trend shows that the saturation magnetization of N–CoO is lower than that of CoO. Combined with the EPR results, it can be speculated that this reduction may be attributed to the formation of covalent bonds between Co and N (Co–N), which decreases the density of unpaired electrons in Co and consequently reduces its magnetic moment contribution. Conversely, Fe–CoO demonstrates significantly higher saturation magnetization than CoO. This enhancement is likely due to the incorporation of Fe3+ ions, which introduce many unpaired electrons into the CoO matrix following Fe doping. Notably, N–Fe–CoO may demonstrate a higher degree of magnetization compared to Fe–CoO. This observation diverges from the trend of saturated magnetization when N is doped in CoO, suggesting that the increase in saturated magnetization following N doping in Fe–CoO is closely associated with the interaction between Fe and N.
To explore the effects of magnetic field direction on OER performances of as-prepared electrodes, herein, three magnetic field directions relative to the electrode surface are tested: 90° (vertical), 45°, and 0° (parallel). The polarization curves are shown in Fig. 3a. Compared with the N–Fe–CoO electrode without a magnetic field, the overpotential of the N–Fe–CoO electrode with a magnetic field is improved. As the angle between the magnetic field and the electrode surface decreases, the overpotential decreases gradually. At 10 mA cm−2 current density, the electrode overpotential was 221 mV without a magnetic field, 220 mV when the magnetic field was perpendicular to the electrode surface, and 219 mV when the azimuth angle was 45°. The lowest overpotential of 217 mV emerged when the magnetic field was parallel to the electrode surface. According to the electrochemical impedance spectrum (EIS) in Fig. 3b, the solution impedance and charge transfer resistance (Rct) (reflected by semicircle diameter) are reduced after the magnetic field is applied. Then, the effect of magnetic field intensity on the behavior of the N–Fe–CoO electrode on oxygen evolution was investigated. During the test, the magnetic field is kept parallel to the electrode surface, and the magnetic field strength is changed by adjusting the distance between the magnet and the electrode. The polarization and EIS curves are shown in Fig. 3c and d. The starting position value of the impedance spectrum first increases and then gradually decreases with the increase of the magnetic field strength, and the minimum value emerges when the magnetic field strength is 420 mT. This indicates that the magnetic field affects the movement of the ions in solution. Based on this consideration, the polarization curves without IR correction can more truly reflect the effects of magnetic field strength on the OER performance. As shown in Fig. 3c, when the magnetic field intensity is 303 mT and 330 mT, the overpotential does not change; when the magnetic field increases to 370 mT, the overpotential decreases, and the overpotential continues to decrease as the magnetic field continues to increase.
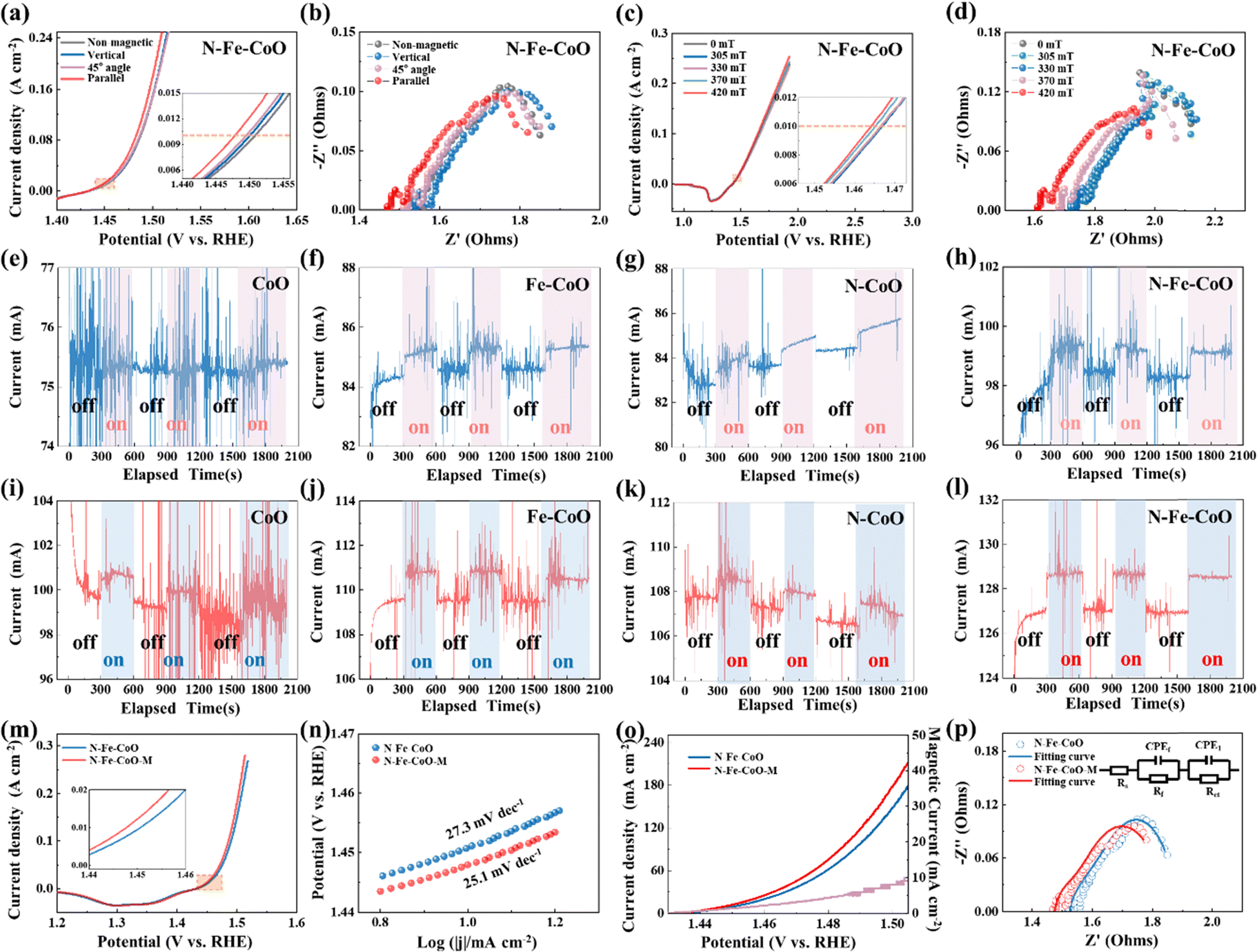 | ||
| Fig. 3 (a) OER polarization curves and (b) electrochemical impedance spectroscopy of N–Fe–CoO under magnetic fields of different directions. The (c) OER polarization curves and (d) electrochemical impedance spectroscopy (EIS) of N–Fe–CoO under magnetic fields with different induction intensities. The chronoamperometry curves were stimulated by a parallel magnetic field under 0.75 V vs. Hg/HgO reference electrode of (e) CoO, (f) Fe–CoO, (g) N–CoO, and (h) N–Fe–CoO. The chronoamperometry curves were stimulated by a parallel magnetic field under 0.8 V vs. Hg/HgO reference electrode of (i) CoO, (j) Fe–CoO, (k) N–CoO, and (l) N–Fe–CoO. The comparison of (m) polarization curves, (n) Tafel slopes, (o) magnetically enhanced current curve, and (p) EIS with the corresponding fitting of the N–Fe–CoO sample with and without a magnetic field. | ||
To investigate the enhancing mechanism of the magnetic field on OER more accurately, the N–Fe–CoO electrode is tested by the chronoamperometry method. During the test, the magnetic field is applied (on) and removed (off) to determine the enhancement effect of the magnetic field. The constant voltages of 0.75 V and 0.8 V (relative to the reference electrode) are utilized. The time interval is unified as 300 s when there is no magnetic field to ensure the stability of the current, and then the magnetic field is applied to maintain the current for 300 s. This cycle is repeated three times to ensure that the magnetic field enhancement effect is not accidental. As the patterns under potentials of 0.75 V (Fig. 3e–h) and 0.8 V (Fig. 3i–l) are similar, the following statements are based typically on 0.8 V potential. The initial current of the CoO electrode is ∼99 mA, and applying a magnetic field induces a significant change in current, with a variation of ∼1 mA, referred to as the “magnetic current” in subsequent analysis. For the Fe–CoO electrode, the initial current is slightly higher at ∼109.5 mA. Under magnetic field enhancement, the current rises instantaneously to ∼111 mA, resulting in a magnetic current of ∼1.5 mA, marginally higher than that observed for the CoO electrode. The N–CoO electrode exhibits an initial stable current comparable to the Fe–CoO electrode. Applying a magnetic field also induces a noticeable change, with a magnetic current of ∼1.5 mA. Notably, the N–Fe–CoO electrode demonstrates the highest initial current among the four electrodes, reaching ∼127 mA. This observation confirms that co-doping with nitrogen (N) and iron (Fe) optimizes the electrocatalytic activity of CoO. Under magnetic field conditions, the current of the N–Fe–CoO electrode further increases to ∼129 mA, with a magnetic current of ∼2 mA, indicating the most pronounced magnetic enhancement effect among the tested electrodes.
The following bar chart reveals the relationship between the magnetic current and the initial electrode current (Fig. S6, ESI†). Both the magnetic current and initial current of Fe–CoO and N–CoO increase under the same potential, which indicates that the N doping or Fe doping benefits the magnetic enhancement effect on OER. The magnetic current of N–Fe–CoO nanosheets is highest under the same potential, which confirms that the co-doping of N and Fe introduces the highest magnetic field enhancement for OER. To further analyze the enhancement effect of the magnetic field on the N–Fe–CoO electrode, the OER performance without a magnetic field and with the optimum parameter of the magnetic field (parallel to the electrode surface and the intensity is maximum) was analyzed in detail. As shown in Fig. 3m, the polarization curves showed that the overpotential at 10 mA cm−2 was improved by 4 mV, from 221 mV to 217 mV. The Tafel slope obtained by processing the polarization curve is shown in Fig. 3n. The Tafel slope under the magnetic field is 25.1 mV dec−1, smaller than the Tafel slope value of 27.3 mV dec−1 without the magnetic field. The magnetic field will accelerate the kinetics of the OER reaction, which means that the improvement of current density can be amplified to a great degree when the magnetic field is applied at higher potential or current conditions. This is confirmed by Fig. 3o, where the magnetic current density gradually increases with potential increase. At 1.505 V (vs. RHE), the enhanced magnetic current density is 10 mA cm−2. Such improvement will be of great significance in practical applications. The EIS was further fitted,43,44 as shown in Fig. 3p, the solution resistance Rs under the magnetic field was 1.466 Ω, 0.05 Ω lower than those without the magnetic field of 1.516 Ω. The Rct decreased from 0.173 Ω to 0.113 Ω. This indicates that the magnetic field promotes the material transport in solution and accelerates the charge transfer at the electrolysis interface.45
The catalytic reaction process can be divided into an electrode reaction and a liquid phase mass transfer. Electrode reactions involve complex REDOX reactions, while liquid phase mass transfer continuously transports reactants from the solution to the electrode surface. Thus, introducing a magnetic field will be a combined effect, affecting the electron reaction on the one hand and the transport of matter on the other. When the electric and magnetic fields exist simultaneously in the electrocatalytic system, the charged particles in the solution will be affected by both fields. The charged ions migrate directionally under the action of a voltage. When the direction of movement is orthogonal to the magnetic field component, the magnetic field will exert the Lorentz force. At this time, charged ions will change their original direction of movement, thus generating convection around the electrode, which is called the magnetohydrodynamic effect (MHD). During the catalytic reaction, the solution near the electrode surface leads to concentration polarization due to a concentration difference (diffusion layer). According to the Butler–Volmer equation, the concentration overpotential can be expressed as:

The relationship between limiting current Id and diffusion layer thickness δ is given by Fick's first law:
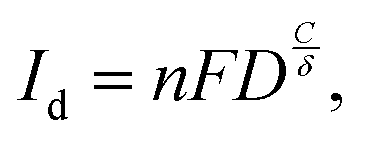
Fig. 4a describes the CoO electrode's OER performance enhancement mechanism by a magnetic field. The magnetic field, electric field, and Lorentz force are distributed in three dimensions and are orthogonal. When the electrode is undergoing OER, a magnetic field parallel to the surface of the electrode (orthogonal to the direction of the electric field) is applied externally. Under the magnetic field, the charged particles (OH−) in the electrolyte will be subjected to the Lorentz force, as shown in the figure, which will cause the original motion direction to change and drive the fluid convection. Magnetic fluid acts on the electrode surface to accelerate the escape of gas molecules. Thus, concentration polarization, ohmic polarization, and activation polarization can be effectively reduced. Based on this mechanism, it is easy to understand the influence of magnetic field direction and strength on electrochemical performance. When the magnetic field is perpendicular to the electrode surface, the macroscopic solution has no MHD effect. However, there may be a small microscopic MHD effect on the electrode surface, reducing the overpotential. With the change of angle, the magnetic field component orthogonal to the electric field becomes larger and larger, the magnetic field strength becomes larger and larger, and the MHD effect becomes stronger and stronger, so the overpotential improvement effect becomes more and more obvious.
 | ||
| Fig. 4 (a) Schematic diagram of magnetic field enhancement mechanism for mass transfer during OER, (b) the established (200) atomic model for CoO and N–Fe–CoO, (c) the electronic energy band structure, and (d) total density of states (TDOS) of N–Fe–CoO, (e) the isosurface of atomic deformation electron density distribution of N–Fe–CoO, the red arrow marks the local charge distribution after N, Fe introduction, (f) the sequence diagram of O2 molecule escape process, (g) the adsorption of O2 with or without a magnetic field, (h) the escape free energy of O2 molecule without and with magnetic field. | ||
The magnetic enhancement effect of pure-phase CoO and contrast samples can also be explained. At the same voltage, different catalysts have different intrinsic activities, resulting in different degrees of reaction. Less active electrocatalysts, such as CoO, have fewer bubbles on the surface and do not restrict the electrocatalytic reaction. However, the number of bubbles is excessive for electrodes with high catalytic activity (such as N–Fe–CoO). Without a magnetic field, it cannot be removed in time, becoming an obstacle to oxygen evolution. Therefore, after applying a magnetic field, the MHD effect can effectively alleviate this phenomenon so that the overpotential greatly improves. Further exploration of the effect of the magnetic field on the intrinsic activity of the N–Fe–CoO electrode is conducted through DFT calculations.
The atomic model of the (200) plane is established (Fig. 4b) according to the HRTEM characterization result shown in Fig. 2. The N–Fe–CoO suggests typical metallic energy band properties (Fig. 4c), where the obvious total density of states (TDOS) emerges near the Fermi level (Fig. 4d). This indicates the N, Fe incorporation ensures good electronic transmission properties. The isosurface of atomic deformation electron density distribution is calculated in Fig. 4e, and the red area and yellow area represent the surrounding electrons increasing and decreasing, respectively, induced by N, Fe coupling. The electrons around the Co and Fe centers decrease, while those around the O and N centers increase. The N, Fe coupling in CoO caused some electrons to delocalize from Co/Fe centers to O/N centers. Thus, the average valence of Co and Fe atoms becomes higher, consistent with XPS analysis in Fig. 3. The positive shift of average valence benefits from forming a hole channel to accept electrons from the KOH electrolyte, as reported in previous research.46 The simulation of the magnetic field effect on the catalytic process is based on the oxygen evolution mechanism shown in Fig. 4f, where adjacent *O binds to form the final O2 product. When there is no magnetic field, the electron spin directions in N–Fe–CoO are chaotic, and the electron spin directions of adjacent *O are opposite (Fig. 4g). While applying a magnetic field, the electron spin directions are forced to be consistent. Therefore, the electron spin directions of adjacent *O are the same. The escape free energy of the O2 molecule is calculated in Fig. 4h, which demonstrates that the magnetic field reduces the energy barrier for O2 molecules to escape (from ∼4.2 eV on CoO to ∼1.5 eV on N–Fe–CoO), facilitating the OER process.
Based on the understanding of the above mechanism, the magnetic response currents of the N–Fe–CoO electrode (2 cm × 4 cm) were tested at currents of 100 mA, 500 mA (high current), and 1000 mA (industrial current), respectively. The test results are shown in Fig. 5. When the electrode's current is 100 mA, the magnetic enhancement current is 2 mA. When the current is 500 mA, the magnetic enhancement current is 8 mA. When the current is 1 A, the magnetic enhanced current can reach 22 mA, showing a relatively ideal expected effect, which proves its potential in industrial production. In order to prove the MHD effect, we designed two different methods. If the MHD effect exists, the gas bubbles will be driven and accelerated to detach. Therefore, one can determine the MHD effect by monitoring the rate of bubble detachment before and after applying a magnetic field during the catalytic process. Fig. 5d shows the assembly method, and Fig. 5e shows that when non-magnetic platinum plates are used as electrodes, the situation before and after applying a magnetic field under constant potential. The peak spacing reflects the speed of bubble detachment. The peak spacing decreases after applying a magnetic field, indicating that bubble detachment accelerates. Therefore, the MHD effect exists. Moreover, the non-magnetic carbon cloth substrate also suggests an obvious enhancement of catalytic performance (Fig. 5f), which also suggests the existence of the MHD effect. In addition, the magnetic field enhancement effect and the stability of the catalyst electrode in the magnetic field were investigated. The electrode was tested at a 10 mA cm−2 current density for 12 h. As shown in Fig. 5g, the catalyst overpotential suggests a stable potential during the work for 12 h under the magnetic field. Comparing the potential values of the electrodes tested without a magnetic field, the magnetic field enhancement remained stable within 12 h. The N–Fe–CoO electrocatalyst has bright commercial value, and its performance can be further enhanced by the magnetic field, which provides a new reference for industrial production.
 | ||
| Fig. 5 The chronoamperometry curves of (a) 100 mA, (b) 500 mA and (c) 1000 mA stimulated by 420 mT parallel magnetic field; (d) MHD effect mechanism diagram; (e) the chronoamperometry curves of pure Pt foil electrode under the zero field or magnetic field (420 mT); (f) the chronoamperometry curves of pure carbon cloth electrode under the zero field or magnetic field (420 mT); (g) the chronopotentiometry analysis under 10 mA cm−2 with and without magnetic field. | ||
3. Experiment section
3.1 Setup of magnetic field enhanced electrocatalysis system
The electrocatalytic performance of the N–Fe–CoO electrode was studied under a magnetic field. An electromagnet (WD-80, TINDUN) was used as a magnetic field generator. The intensity and orientation of the magnetic field can be easily tuned by adjusting the direction and the distance between the magnet and the samples (Fig. S1, ESI†). During the test, the magnetic field is applied near the working electrode, as shown in the schematic diagram of Fig. 6a. The relationship between the magnetic induction intensity and distance from the magnet surface is shown in Fig. 6b, where a linear relationship can be observed, which is the reference for adjusting the magnetic induction intensity at the electrode. As for the direction, Fig. 6c suggests the reference of the parallel direction. | ||
| Fig. 6 (a) The illustration of electrolysis of water for oxygen evolution assisted by an external magnetic field; (b) the magnetic induction intensity versus magnet distance function curve; (c) the reference of parallel direction. | ||
3.2 Synthesis of CoO nanosheets
The carbon cloth was first treated with concentrated nitric acid at 100 °C for 3 h to generate hydrophilicity, then washed with deionized water and ethanol several times and dried in air. The treated carbon cloth was placed into a mixed solution containing 2.3 g Co(NO3)2·6H2O, 3.2 g hexamethylenetetramine, and 60 mL deionized water. The mixture was then transferred to the reaction kettle and kept at 120 °C for 6 h. The obtained samples were washed several times with deionized water and ethanol and then dried at 60 °C. The above samples were placed in a tubular furnace and heated to 250 °C at a rate of 2 °C min−1 under an Ar atmosphere for 2 h. Finally, pure phase CoO is obtained.3.3 Synthesis of N–Fe–CoO nanosheets
Firstly, the prepared CoO nanosheet electrode (2 cm × 5 cm) was prepared, and then it was immersed in the FeCl3·6H2O solution (4 mmol in 30 mL deionized water). After 30 s immersion treatment, the electrode sheet was completely infiltrated into the solution. Then, the electrode was removed and placed into the plasma enhanced chemical vapor deposition (PECVD) device for plasma interaction under the nitrogen plasma atmosphere. Experimental parameters are as follows: the experimental temperature is 300 °C, the N2 atmosphere flow is 50 sccm, the plasma reflector power is 300 W, and the pressure is 0.5 torr under low vacuum conditions. For ease of presentation, the prepared sample is denoted as N–Fe–CoO.3.4 Synthesis of N–CoO sample
To compare and show the doping effect of the N element, a sample without Fe immersion treatment was prepared without changing other experimental parameters, which was abbreviated as N–CoO here.3.5 Synthesis of Fe–CoO sample
To compare and show the doping effect of the Fe element, a sample without N plasma treatment was prepared without changing other experimental parameters, which was abbreviated as Fe–CoO here.4. Conclusion
The N, Fe co-bonded CoO was simultaneously achieved through a one-step high-energy N plasma atmosphere, which suggests a bright potential to be enhanced by a magnetic field during OER electrocatalysis. N atoms are a bridge for bonding Fe and Co centers, providing a fast channel for electron transfer. The coupling of N and Fe co-doping decreases the electron density around Co2+ centers, which increases the unpaired electrons for electron acceptors. As a result, the intrinsic OER activities are boosted, which is further favorable for amplifying the magnetic enhancement effect. The best performance emerges under a parallel magnetic field with 420 mT intensity, which results in a lowered overpotential of 217 mV and a Tafel slope of 25.1 mV dec−1 in alkaline media. The magnetic enhancement comes from the magnetohydrodynamic effect and the escape energy barrier reduction of the paramagnetic triplet state O2, which benefits from lowering both charge and mass transfer resistance. The magnetic enhancement effect would be amplified when the catalytic current becomes larger (magnetic current is 8 mA and 22 mA under 500 mA and 1000 mA total current, respectively). The current method affords unique advantages as an OER activity amplifier and is feasible for industrial operation.Data availability
The data supporting this study's findings are available in the main text and ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially sponsored by National Natural Science Foundation of China (Grant No. 52471199 and No. 52302223), the Postdoctoral Fellowship Program of CPSF under Grant Number GZB20240949, Natural Science Foundation of Heilongjiang Province (LH2024E031), Heilongjiang Postdoctoral Fund (LBH-Z24174), State Key Laboratory of Precision Welding & Joining of Materials and Structures (24-R-01), Heilongjiang Touyan Team Program, Heilongjiang Province Science and Technology Special Fund (2024ZXDXA19), Hainan Province Science and Technology Special Fund (No. ZDYF2024SHFZ082). Thanks to the HIT Center of Analysis, Measurement, and Computing and Dr Zheng Zhen for the electron microscope test analysis.References
- H. Tüysüz, Acc. Chem. Res., 2024, 57, 558–567 Search PubMed.
- X. H. Xie, L. Du, L. T. Yon, S. Y. Park, Y. Qiu, J. Sokolowski, W. Wang and Y. Y. Shao, Adv. Funct. Mater., 2022, 32, 2110036 CrossRef CAS.
- X. R. Ren, Y. Y. Zhai, N. Yang, B. L. Wang and S. Z. Liu, Adv. Funct. Mater., 2024, 34, 2401610 CrossRef CAS.
- F. L. Lyu, Q. F. Wang, S. M. Choi and Y. D. Yin, Small, 2019, 15, 1804201 CrossRef PubMed.
- M. N. Lakhan, A. Hanan, A. Hussain, I. A. Soomro, Y. Wang, M. Ahmed, U. Aftab, H. Y. Sun and H. Arandiyan, Chem. Commun., 2024, 60, 5104–5135 RSC.
- Q. M. Sun, Y. W. Zhao, X. D. Yu, C. Zhang and S. X. Xing, J. Electrochem. Soc., 2022, 169, 060537 CrossRef CAS.
- M. S. Kim, M. A. Abbas, R. Thota and J. H. Bang, J. Mater. Chem. A, 2019, 7, 26557–26565 RSC.
- J. X. Guo, D. Y. Yan, K. W. Qiu, C. Mu, D. Jiao, J. Mao, H. Wang and T. Ling, J. Energy Chem., 2019, 37, 143–147 CrossRef.
- T. Z. Wu, X. Ren, Y. M. Sun, S. N. Sun, G. Y. Xian, G. G. Scherer, A. C. Fisher, D. Mandler, J. W. Ager, A. Grimaud, J. L. Wang, C. M. Shen, H. T. Yang, J. Gracia, H. J. Gao and Z. C. J. Xu, Nat. Commun., 2021, 12, 3634 CrossRef CAS PubMed.
- R. Q. Li, P. F. Hu, M. Miao, Y. L. Li, X. F. Jiang, Q. Wu, Z. Meng, Z. Hu, Y. Bando and X. B. Wang, J. Mater. Chem. A, 2018, 6, 24767–24772 RSC.
- X. M. Fan, Y. Y. Fan, X. Zhang, L. Tang and J. X. Guo, J. Alloys Compd., 2021, 877, 160279 CrossRef CAS.
- J. F. Xie, H. Zhang, S. Li, R. X. Wang, X. Sun, M. Zhou, J. F. Zhou, X. W. Lou and Y. Xie, Adv. Mater., 2013, 25, 5807 CrossRef CAS PubMed.
- J. Hu, S. W. Li, J. Y. Chu, S. Q. Niu, J. Wang, Y. C. Du, Z. H. Li, X. J. Han and P. Xu, ACS Catal., 2019, 9, 10705–10711 CrossRef CAS.
- H. Y. Jin, X. Liu, S. M. Chen, A. Vasileff, L. Q. Li, Y. Jiao, L. Song, Y. Zheng and S. Z. Qiao, ACS Energy Lett., 2019, 4, 805–810 CrossRef CAS.
- J. D. Yao, W. J. Huang, W. Fang, M. Kuang, N. Jia, H. Ren, D. B. Liu, C. D. Lv, C. T. Liu, J. W. Xu and Q. Y. Yan, Small Methods, 2020, 4, 2000494 CrossRef CAS.
- F. A. Garcés-Pineda, M. Blasco-Ahicart, D. Nieto-Castro, N. López and J. R. Galán-Mascarós, Nat. Energy, 2019, 4, 519–525 CrossRef.
- Y. Y. Zhang, C. Liang, J. Wu, H. Liu, B. Zhang, Z. X. Jiang, S. W. Li and P. Xu, ACS Appl. Energy Mater., 2020, 3, 10303–10316 CrossRef CAS.
- Z. Fang, W. T. Zhao, T. Shen, D. P. Qiu, Y. C. Lv, X. M. Hou and Y. L. Hou, Precis. Chem., 2023, 1, 395–417 CrossRef CAS.
- Y. M. Sun, X. Ren, S. N. Sun, Z. Liu, S. B. Xi and Z. C. J. Xu, Angew. Chem., Int. Ed., 2021, 60, 14536–14544 CrossRef CAS PubMed.
- Y. M. Sun, S. N. Sun, H. T. Yang, S. B. Xi, J. Gracia and Z. C. J. Xu, Adv. Mater., 2020, 32, 202003297 Search PubMed.
- T. Wang, J. Y. Wang, Y. M. Sun, Y. Duan, S. N. Sun, X. Hu, S. B. Xi, Y. H. Du, C. Wang and Z. C. J. Xu, Appl. Catal., B, 2019, 256, 117844 CrossRef CAS.
- X. Ren, T. Z. Wu, Y. M. Sun, Y. Li, G. Y. Xian, X. H. Liu, C. M. Shen, J. Gracia, H. J. Gao, H. T. Yang and Z. J. Xu, Nat. Commun., 2021, 12, 2608 CrossRef CAS PubMed.
- G. Li, P. Wang, C. Y. Li, Z. Fang, M. He, W. W. Wang, X. L. Yuan, H. Li, P. G. Li and Z. X. Li, Mater. Res. Lett., 2022, 10, 744–753 CrossRef CAS.
- T. Marshall-Roth, N. J. Libretto, A. T. Wrobel, K. J. Anderton, M. L. Pegis, N. D. Ricke, T. Van Voorhis, J. T. Miller and Y. Surendranath, Nat. Commun., 2020, 11, 5283 CrossRef CAS PubMed.
- C. Q. Li, C. S. He, F. Z. Sun, M. C. Wang, J. H. Wang and Y. Q. Lin, ACS Appl. Nano Mater., 2018, 1, 1801–1810 CrossRef CAS.
- R. Jiang, L. Li, T. Sheng, G. F. Hu, Y. G. Chen and L. Y. Wang, J. Am. Chem. Soc., 2018, 140, 11594–11598 CrossRef CAS PubMed.
- L. Jiao, J. K. Li, L. L. Richard, Q. Sun, T. Stracensky, E. R. Liu, M. T. Sougrati, Z. P. Zhao, F. Yang, S. C. Zhong, H. Xu, S. Mukerjee, Y. Huang, D. A. Cullen, J. H. Park, M. Ferrandon, D. J. Myers, F. Jaouen and Q. Y. Jia, Nat. Mater., 2021, 20, 1385 CrossRef CAS PubMed.
- Y. Y. Li, L. Zhang, J. H. Peng, W. Zhang and K. Peng, J. Power Sources, 2019, 433, 226704 CrossRef CAS.
- R. Zhang, Y. Lu, L. Wei, Z. G. Fang, C. H. Lu, Y. R. Ni, Z. Z. Xu, S. Y. Tao and P. W. Li, J. Mater. Sci.:Mater. Electron., 2015, 26, 9941–9948 CrossRef CAS.
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M. J. Cheng, D. Sokaras, T. C. Weng, R. Alonso-Mori, R. C. Davis, J. R. Bargar, J. K. Norskov, A. Nilsson and A. T. Bell, J. Am. Chem. Soc., 2015, 137, 1305–1313 CrossRef CAS PubMed.
- B. M. Hunter, N. B. Thompson, A. M. Muller, G. R. Rossman, M. G. Hill, J. R. Winkler and H. B. Gray, Joule, 2018, 2, 747–763 CrossRef CAS.
- N. L. A. Rodin, M. R. Sahar and F. Mohd-Noor, J. Magn. Magn. Mater., 2020, 496, 165931 CrossRef.
- M. Al-Ghoul, H. El-Rassy, T. Coradin and T. Mokalled, J. Cryst. Growth, 2010, 312, 856–862 CrossRef CAS.
- S. K. Misra, S. I. Andronenko, S. S. Rao, J. Chess and A. Punnoose, J. Magn. Magn. Mater., 2015, 394, 138–142 CrossRef CAS.
- E. Balan, T. Allard, B. Boizot, G. Morin and J. P. Muller, Clay Clay Miner., 1999, 47, 605–616 CrossRef CAS.
- Y. L. Pan, X. M. Xu, Y. J. Zhong, L. Ge, Y. B. Chen, J. P. M. Veder, D. Q. Guan, R. O'Hayre, M. R. Li, G. X. Wang, H. Wang, W. Zhou and Z. P. Shao, Nat. Commun., 2020, 11, 2002 CrossRef CAS PubMed.
- L. Y. Wu, Q. L. Wu, Y. Han, D. D. Zhang, R. R. Zhang, N. Song, X. F. Wu, J. R. Zeng, P. Yuan, J. Chen, A. J. Du, K. K. Huang and X. D. Yao, Adv. Mater., 2024, 36, 2401857 CrossRef CAS PubMed.
- J. Yang, S. Y. Hu, Y. R. Fang, S. Hoang, L. Li, W. W. Yang, Z. F. Liang, J. Wu, J. P. Hu, W. Xiao, C. Q. Pan, Z. Luo, J. Ding, L. Z. Zhang and Y. B. Guo, ACS Catal., 2019, 9, 9751–9763 CrossRef CAS.
- X. L. Hu, X. Chen, X. Li and C. H. Xu, Adv. Funct. Mater., 2024, 34, 2316699 CrossRef CAS.
- Q. A. Song, M. Li, X. S. Hou, J. C. Li, Z. J. Dong, S. Zhang, L. Yang and X. Liu, Appl. Catal., B, 2022, 317, 121721 CrossRef CAS.
- J. G. Zhao, M. Yang and Z. H. Hua, J. Magn. Magn. Mater., 2014, 371, 10–13 CrossRef CAS.
- A. Tomou, D. Gournis, I. Panagiotopoulos, Y. Huang, G. C. Hadjipanayis and B. J. Kooi, J. Appl. Phys., 2006, 99, 123915 CrossRef.
- Y. T. Yan, P. C. Wang, J. H. Lin, J. Cao and J. L. Qi, J. Energy Chem., 2021, 58, 446–462 CrossRef CAS.
- Y. T. Yan, J. H. Lin, T. Liu, B. S. Liu, B. Wang, L. Qiao, J. C. Tu, J. Cao and J. L. Qi, Corros. Sci., 2022, 200, 110231 CrossRef CAS.
- Y. T. Yan, J. H. Lin, T. X. Xu, B. S. Liu, K. K. Huang, L. Qiao, S. D. Liu, J. Cao, S. C. Jun, Y. Yamauchi and J. L. Qi, Adv. Energy Mater., 2022, 12, 2200434 CrossRef CAS.
- Y. Yan, K. Huang, J. Lin, T. Yang, P. Wang, L. Qiao, W. Cai and X. Zheng, Appl. Catal., B, 2023, 330, 122595 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ey00040h |
| This journal is © The Royal Society of Chemistry 2025 |