
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect conversion of CO2 to aromatics based on the coupling strategy and multi-functional catalysis
Chang Liu
a,
Yangdong Wang
*a,
Lin Zhanga,
Junjie Sua,
Su Liua,
Haibo Zhoua,
Wenqian Jiaoa and
Zaiku Xie
 *ab
*ab
aState Key Laboratory of Green Chemical Engineering and Industrial Catalysis, SINOPEC Shanghai Research Institute of Petrochemical Technology Co., Ltd, 201208 Shanghai, P. R. China. E-mail: wangyd.sshy@sinopec.com; xzk@sinopec.com
bChina Petrochemical Corporation (SINOPEC Group), 100728 Beijing, P. R. China
First published on 9th April 2025
Abstract
As fundamental chemicals and building blocks for the modern chemical industry, aromatics possess a huge market demand. The direct and atom-economic conversion of CO2 to aromatics holds the potential to diminish the reliance on petroleum resources and provides a viable approach towards a net-zero chemical industry. The key lies in the implementation of the highly efficient coupling catalysis strategy and utilization of multi-functional catalysts. In this review, recent advances in the direct conversion of CO2 to aromatics via the methanol-mediated pathway and the modified Fischer–Tropsch synthesis route are comprehensively discussed, including an in-depth analysis of the tandem reaction mechanism and bifunctional catalysts, which consist of metal-based materials (including metals, metal oxides, or metal carbides) and zeolites. Furthermore, several novel catalytic pathways, involving coupling CO2 conversion with reactions such as CO hydrogenation, aromatic alkylation, or alkane aromatization, are also elaborated. Subsequently, the coupling effect of multi-functional catalysis, as well as the influence of the proximity between catalytic components, is explored. Moreover, the revealing and construction of the spatial pathway for tandem reactions, which enable the spatio-temporal coupling of multi-functional catalytic systems, are addressed. The challenges and potential directions for the further development of the direct CO2-to-aromatics conversion technology are finally proposed.
 Chang Liu | Dr Chang Liu received her BS and MS in chemical engineering from Tsinghua University. She joined the SINOPEC Shanghai Research Institute of Petrochemical Technology in 2015, during which she received her PhD in physical chemistry from Fudan University. She is now working as an associate research fellow. Her research interests focus on the design and synthesis of multi-functional catalytic materials, and the application of the coupling catalysis strategy in C1 utilization and production of olefins and aromatics. |
 Yangdong Wang | Prof. Yangdong Wang got his PhD degree in physical chemistry from Nanjing University in 2000. He then joined the SINOPEC Shanghai Research Institute of Petrochemical Technology. He is currently the chief expert of SRIPT. His research interests focus on the coupling catalysis strategy and industrial catalysis in petrochemical, coal chemical, and green chemistry technologies, including metathesis of C4 olefins, hydrogenation of CO and CO2 to chemicals, and green catalytic oxidation. |
 Zaiku Xie | Prof. Zaiku Xie received his PhD from the East China University of Science and Technology. He joined SINOPEC in 1985 and is currently the chief scientist of the Sinopec Group. He was the chief scientist on porous catalytic materials in the National Basic Research Development Program of China from 2003 to 2013. He is an academician of the Chinese Academy of Sciences and a Fellow of the Royal Society of Chemistry. His research interests are mainly focused on the preparation, characterization, and industrial applications of novel zeolite-based catalytic materials. |
Broader contextThe conversion and utilization of CO2 is an important pathway for carbon cycling and a pivotal aspect of sustainable energy solutions, representing a cutting-edge area of academic and technological research. Among diverse reaction paths, CO2 hydrogenation to aromatics stands as an alternative approach for production of fundamental chemicals. This process involves a complex tandem reaction network, including C–O activation, C–H bond formation, and C–C coupling, and can be realized utilizing the coupling strategy and multi-functional catalysis. However, uncertainty remains concerning which pathway holds the greatest potential for industrialization. Therefore, a systematic and rigorous analysis of existing advancements is necessary to clarify directions for future progress. Regarding the coupling catalytic strategy, the underlying coupling mechanism, particularly the spatio-temporal effects involving a temporal sequence of reactions across a spatial arrangement of catalytic sites, constitutes the cornerstone of regulating the catalytic performance in CO2 conversion. This review summarizes and analyzes research progress in the direct conversion of CO2 into aromatics based on the coupling strategy and multi-functional catalysis, focusing on the construction of spatio-temporal coupling and directional spatial pathways for tandem reactions. With the continuous development and expansion of the coupling strategy, the realm of CO2 conversion will undoubtedly demonstrate renewed vigor and vitality. |
1. Introduction
As indicated by the International Energy Agency, global energy-related carbon dioxide (CO2) emissions have been steadily growing over the past five decades and reached a new record of 37.4 Gt in 2023 (Fig. 1).1,2 Consequently, the emerging environmental issues, particularly global warming and climate change, are threatening the long-term human survival. Hence, it has become a universal consensus to reduce CO2 emissions, and countries such as China have made their net-zero commitments. The key to achieving the net-zero goal lies in the innovation and development of novel pathways for carbon conversion and carbon cycling with high atom economy. Aromatics are pivotal platform chemicals and monomers for synthetic materials, serving as the building blocks for the chemical and materials industries, with a big market. Thus, the conversion of CO2 into aromatics using renewable “green hydrogen” has emerged as a promising approach to optimize carbon resource utilization and close the carbon cycle. Compared with the traditional indirect route consisting of the CO2-to-methanol and methanol-to-aromatics steps, the novel direct route for CO2-to-aromatics conversion requires a shorter process and improves the theoretical hydrogen atom economy by approximately 11–20%, making it more technically competitive.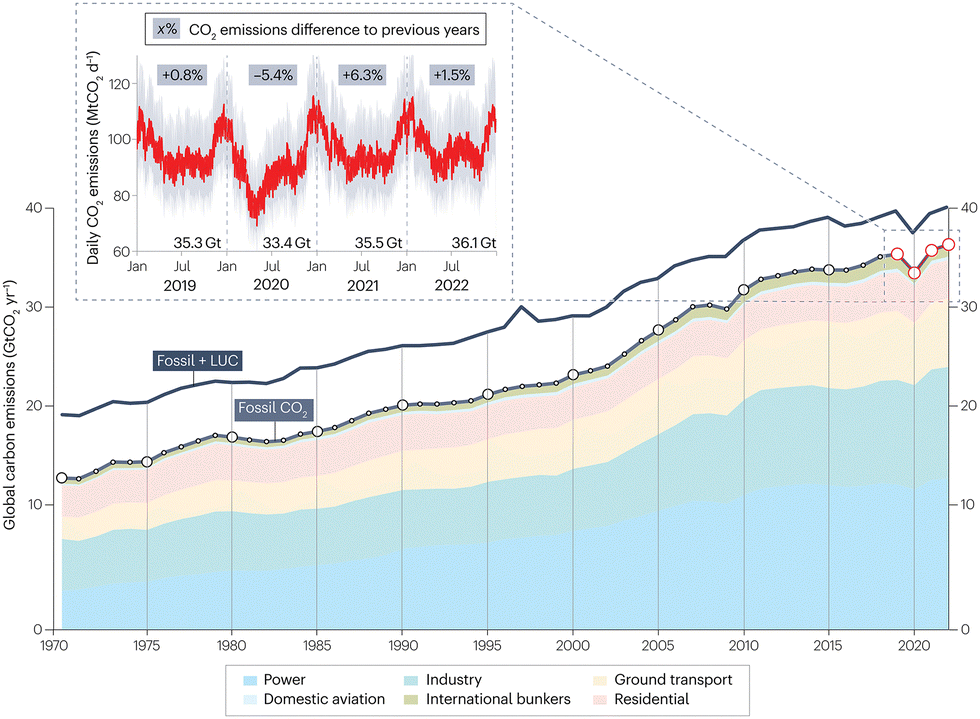 | ||
| Fig. 1 Global CO2 emissions during 1970–2022. Reprinted with permission from Liu et al. Copyright (2023), Springer Nature Limited.2 | ||
The direct conversion of CO2 to aromatics involves the coupling of diverse reaction steps utilizing multi-functional catalysts. In 2016, Bao et al. developed a novel bifunctional catalyst that integrates metal oxides, which are active for CO hydrogenation, with zeolites possessing carbon chain growth activity. This metal oxide/zeolite (OXZEO) catalyst realizes the direct conversion of syngas (CO/H2) into light olefins with an 80% selectivity for C=2–C=4, surpassing the Anderson–Schulz–Flory (ASF) limitation of approximately 58% for C2–C4 in traditional Fischer–Tropsch synthesis reactions. This coupling reaction strategy using bifunctional catalysts further establishes a crucial foundation for the direct conversion of CO2 to aromatics, a field of research that has drawn great attention in recent years. Extensive research has been conducted utilizing metal-based materials (including metals, metal oxides, or metal carbides) and zeolites via the methanol-mediated or the modified Fischer–Tropsch synthesis pathways. For the methanol-mediated pathway, metal oxide catalysts exhibiting exceptional CO2 activation and restricted carbon chain growth capabilities, such as Zn–Zr and Zn–Cr binary oxides and zeolite catalysts with tailored acidity, especially ZSM-5, have been developed. Most of these catalysts achieve a CO2 conversion below 30% and an aromatic selectivity of approximately 80%. Based on the modified Fischer–Tropsch pathway, Fe-based catalysts, including NaZn–Fe and Cu–Fe, have been integrated with the ZSM-5 zeolite. However, a broad product distribution is obtained, due to the relatively uncontrollable carbon chain growth on the surface of the Fe-based catalyst. While CO2 conversion can exceed 40%, the aromatic selectivity mostly remains below 60%. As a highly active and extensively studied research field, comprehensive reviews have been published on CO2-to-hydrocarbon conversion via the methanol-mediated or the modified Fischer–Tropsch synthesis routes.3–9 With the literature corpus expanding and new coupling catalytic routes emerging, this review summarizes recent advancements in the direct conversion of CO2 into aromatics based on the coupling strategy and multi-functional catalysis. Beyond the methanol-mediated or the modified Fischer–Tropsch synthesis routes, new routes integrating CO2 conversion with processes, such as CO hydrogenation, aromatic alkylation, or alkane aromatization, are also elaborated. From the new perspective of spatio-temporal coupling catalysis, the underlying coupling effects are clarified, encompassing the impact of spatial proximity between catalytic components. Thus, the rational design principles for multi-functional catalysts are proposed, emphasizing the construction of spatio-temporal coupling and directional spatial pathways for tandem reactions. Then, the trends in CO2-to-aromatics technology, focusing on the coupling catalytic strategy and spatio-temporal pathway for tandem reactions, are analyzed.
2. Direct conversion of CO2 to aromatics through the methanol-mediated pathway
2.1. Reaction mechanism
In the process of direct conversion of CO2 to aromatics through the methanol-mediated pathway, CO2 undergoes activation followed by hydrogenation to form the methanol intermediate on the surface of the metal oxide catalyst. Then, the methanol intermediate is transferred to and catalyzed by the zeolite catalyst, resulting in its transformation into aromatics. The corresponding reaction equations are eqn (1) and (2):4CO2 hydrogenation to methanol:
| CO2 + 3H2 → CH3OH + H2O | (1) |
Conversion of methanol to aromatics (MTA):
| nCH3OH → CnH2n/CnH2n+2 → C6+nH6+2n | (2) |
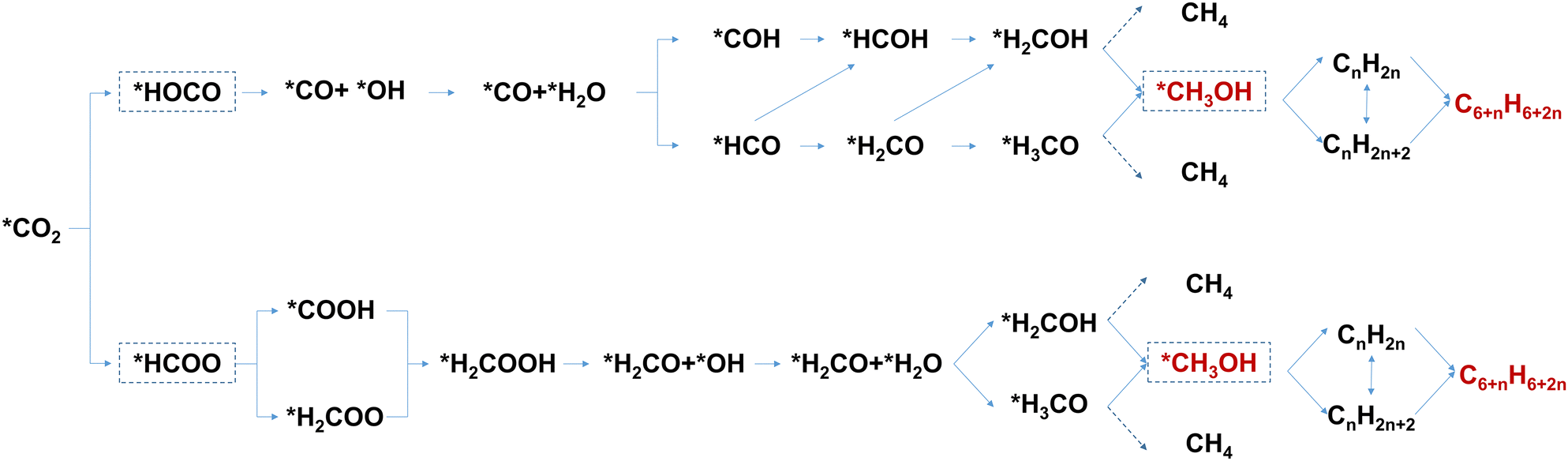 | ||
| Fig. 2 Reaction pathways for conversion of CO2-to-aromatics via the methanol-mediated route. Reproduced with permission from Wang et al. Copyright (2021), Elsevier Inc.4 | ||
 | ||
| Fig. 3 (A) The induction, steady-state and deactivation stages. Reprinted with permission from Dai et al. Copyright (2022), Wiley-VCH GmbH.11 (B) The modified hydrocarbon pool mechanism, i.e., the dual-cycle mechanism of the methanol conversion reaction in zeolites. Reprinted with permission from Erichsen et al. Copyright (2013), Elsevier B.V.16 | ||
The aromatic products are mainly generated by the aromatization of olefins. Specifically, the olefins, which are the primary products of the hydrocarbon pool, further undergo a series of reactions, including oligomerization, cyclization, and hydrogen transfer/dehydrogenation over the acid sites of zeolites, and are ultimately converted into aromatics.27–31 Depending on the catalytic system employed, the olefin aromatization reaction proceeds along two distinct pathways. In the Brønsted acid (zeolite) catalytic system, the reaction predominantly follows the hydrogen transfer pathway (Fig. 4A).32–34 Initially, C=2–C=3 olefins are protonated and oligomerized into C=4–C=10 olefins, which are subsequently transformed into dienes and alkanes through hydrogen transfer. Then, the cyclization (intramolecular oligomerization) of dienes results in the formation of cyclic olefins, which undergo further hydrogen transfer to produce cyclic dienes. Aromatics are finally obtained through hydrogen transfer of the cyclic dienes.35,36 Characterized by multiple hydrogen transfer reaction steps, this pathway generates a large amount of alkanes as by-products. However, due to their low reactivity, these alkanes are difficult to further convert into aromatics, thereby limiting the aromatic selectivity of this reaction pathway.37 Upon the introduction of a metallic or Lewis acid center into the Brønsted acid zeolite, the synergy between Lewis and Brønsted acid sites shifts the reaction from the hydrogen transfer pathway to the dehydrogenation pathway (Fig. 4B), greatly enhancing the reaction activity and aromatic selectivity.32–34
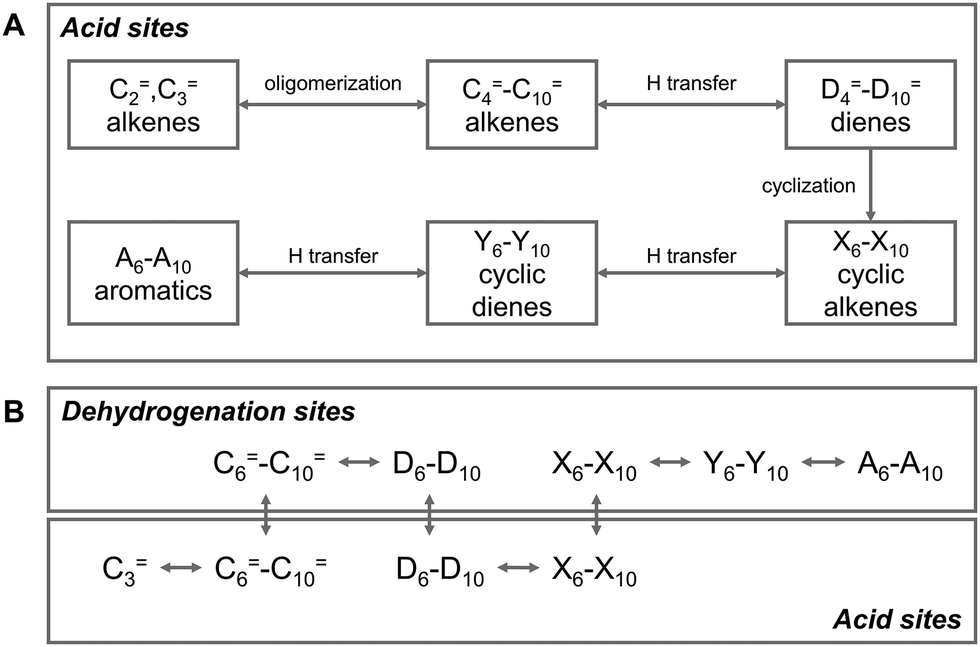 | ||
| Fig. 4 The aromatization of olefins over (A) ZSM-5 and (B) metal-ZSM-5. Reproduced with permission from Lukyanov et al. Copyright (1995), American Chemical Society33 and Caeiro et al. Copyright (2006), Elsevier B.V.34 | ||
Until recently, the hydrocarbon pool mechanism and olefin aromatization mechanism have been successfully employed to elucidate the CO/CO2-to-olefins/aromatics/gasoline reactions via the methanol-mediated pathway.30,38–43 However, significant differences exist between the coupling reaction of CO2-to-aromatics and the methanol conversion reaction, including the co-existence of two distinct catalytic components namely metal oxides and zeolites, the high-pressure environment of CO2 and H2, varying concentrations of methanol as either an intermediate or a reactant, and potential differences in the structure and composition of hydrocarbon pool species. These factors collectively contribute to variations in the reaction mechanisms. Specifically, Yang et al. recently proposed an initial aldol-cycle, which is closely linked to the dual-cycle with the participation of both Brønsted and Lewis acid sites (Fig. 5A).44 Arslan et al. identified oxygenated hydrocarbon pool species in the CO/CO2-to-aromatics reaction and put forward an “aldol-aromatic” mechanism (Fig. 5B).45 According to this mechanism, in the metal oxide/zeolite bifunctional catalytic system, species such as CH3O* and CHxO* generated on the metal oxide surface are transferred to and converted into aldol condensates via the aldol reaction pathway within the zeolite. The aldol species are then transformed into cyclic oxygenates, namely phenols, ketones, and cyclic aldehydes, through intermolecular or intramolecular cyclization. Ultimately, these cyclic oxygenates are converted into single-ring polymethylbenzene through the “aldol–phenol–aromatic” cycle, which is referred to as the “aldol–aromatic” mechanism.
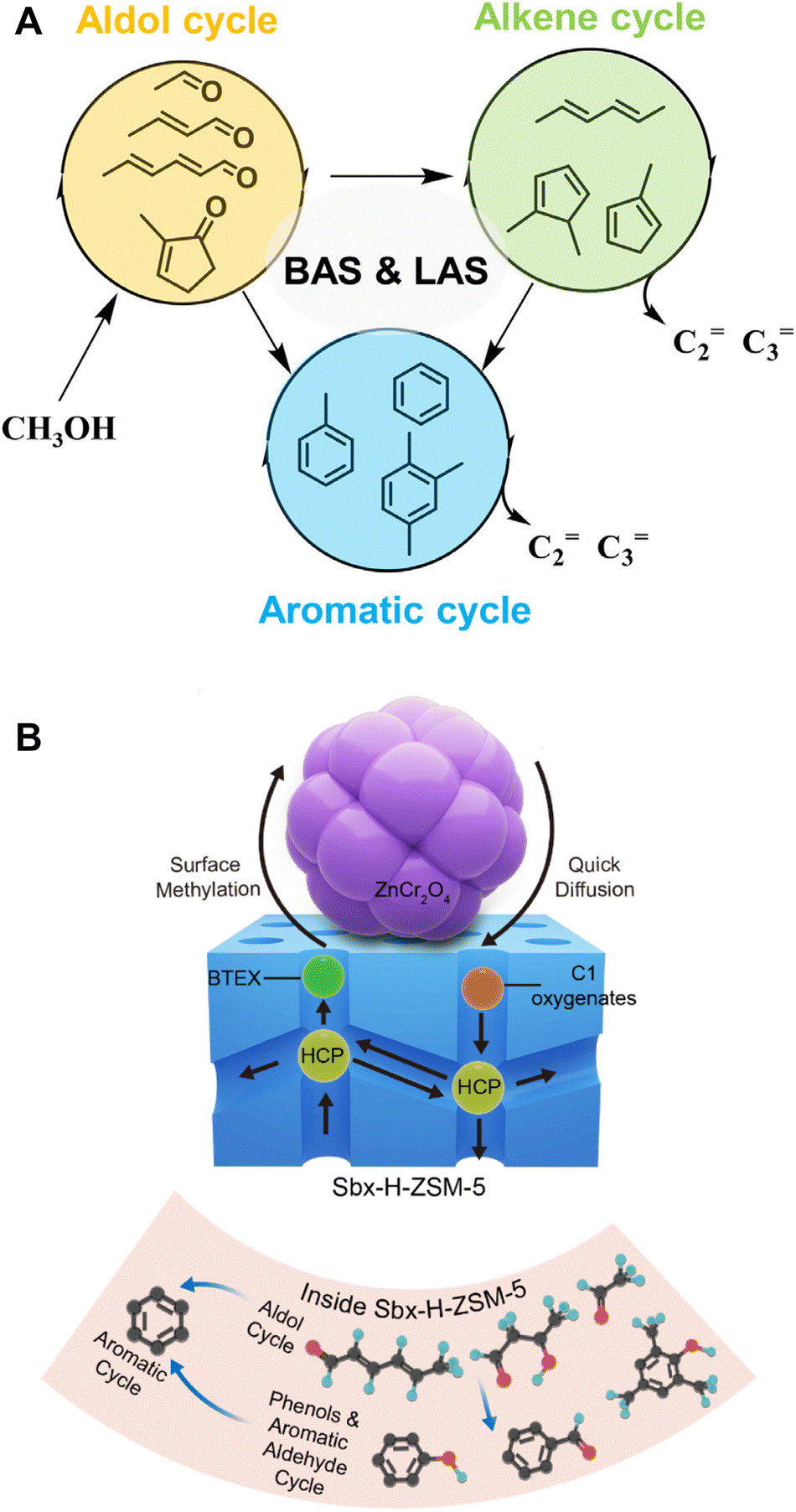 | ||
| Fig. 5 (A) The “aldol–alkene–aromatic” triple-cycle mechanism. Reprinted with permission from Yang et al. Copyright (2022), KeAi.44 (B) The “aldol–phenol–aromatic” cycle, i.e., the “aldol–aromatic” mechanism. Reproduced with permission from Arslan et al. Copyright (2022), American Chemical Society.45 | ||
Alongside the main reaction sequence of CO2 conversion to methanol and subsequently to aromatics, this tandem reaction is accompanied by a series of side reactions. On the one hand, through the RWGS reaction, CO2 can react with H2 and generate H2O and CO on the metal oxide surface, which reduces the carbon conversion efficiency. The existence of H2O may even induce structural changes in both the metal oxides and zeolite catalysts. On the other hand, long-chain products may either crack into smaller hydrocarbons, which is unfavorable for aromatization,42,46–48 or undergo intensive condensation, leading to coke deposition and catalyst deactivation.42,48,49 Typical deactivating species and their formation routes in the methanol conversion reaction system have been proposed, as shown in Fig. 6.22,50–52 Other side reactions, such as isomerization and alkylation, are correlated with the spatial positioning of zeolite acid sites and significantly influence the aromatic distribution.41,43,53–56 Furthermore, deep hydrogenation of intermediates or products into alkanes also occurs, either through the activated hydrogen species on the metal oxides, or through hydrogen transfer over the Brønsted acid sites of zeolites.30,49,57
 | ||
| Fig. 6 Typical deactivating species and their formation routes. (A) Dihydro-trimethylnaphthalene and coke species resulting from the rearrangement of heptamethylbenzene ions. (B) Naphthalene generated by the reaction between aromatics and cyclopentenyl cations. (C) Formaldehyde generated from hydrogen transfer or dehydrogenation of methanol, and coke formation resulting from alkylation of olefins and aromatics by formaldehyde. Reprinted with permission from Bjørgen et al. Copyright (2003), Elsevier Science (USA),22 Wang et al. Copyright (2020), Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim,51 Hwang et al. Copyright (2019), American Chemical Society,52 and Li et al. Copyright (2021), The Authors.50 | ||
2.2. Metal oxide/zeolite bifunctional catalysts
| Catalyst | Temperature (°C) | Pressure (MPa) | Feedstock | Space velocity (mL g−1 h−1) | CO2 conv. (%) | CO Sel. (%) | Aro. Sel. (%) | Aro. STY (mmol C g−1 h−1) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Cr2O3/H-ZSM-5 | 350 | 3 | H2/CO2 = 3 | 1200 | 33.6 | 41.2 | 70.5 | 1.87 | 55 |
| Cr2O3 + Zn–ZSM-5@SiO2 | 350 | 3 | H2/CO2/Ar = 70.25/26.5/3.25 | 1200 | 22.1 | 35.1 | 70.1 | 1.43 | 54 |
| ZnCrOx-ZnZSM-5 | 320 | 5 | H2/CO2/N2 = 72/24/4 | 2000 | 19.9 | 70.2 | 56.5 | 0.75 | 58 |
| ZnCrOx/ZSM-5 | 330 | 3 | H2/CO2 = 3 | 3000 | 17.5 | 38.1 | 64.6 | 2.34 | 43 |
| ZnCr2O4/ZSM-5 | 275 | 2 | H2/CO2 = 3 | 300 | 17.4 | 90.7 | 79.9 | 0.04 | 45 |
| ZnCr2O4/ZSM-5 | 350 | 2 | H2/CO2 = 3 | 300 | 37.48 | 85 | 48.1 | 0.09 | 45 |
| ZnAlOx&ZSM-5 | 320 | 3 | H2/CO2/Ar = 3/1/0.2 | 2000 | 9.1 | 57.4 | 73.9 | 0.61 | 41 |
| ZrO2–Cr/ZSM-5@SiO2 | 360 | 4 | H2/CO2/Ar = 71.95/24.02/4.03 | 1200 | 13.9 | 29 | 76.8 | 0.97 | 56 |
| ZnO/ZrO2-ZSM-5 | 340 | 3 | H2/CO2 = 3 | 4800 | 9.1 | 42.5 | 70 | 1.96 | 59 |
| ZnZrO/HZSM-5 | 320 | 4 | H2/CO2/Ar = 72/24/4 | 1200 | 14.1 | 44 | 73 | 0.77 | 60 |
| ZnZrOx/1Mg-3T-ZSM-5 | 320 | 2 | H2/CO2 = 3 | 4800 | 6.6 | 32.1 | ∼65 | 1.56 | 61 |
| ZnO–ZrO2 Aerogels/H-ZSM-5 | 340 | 4 | H2/CO2 = 3 | 7200 | 16 | 34.3 | 76 | 6.42 | 62 |
| ZnZrOx/ZSM-5 | 315 | 3 | H2/CO2/N2 = 72/24/4 | 1020 | 17.5 | 23.8 | 60.3 | 0.88 | 53 |
| 6% Zn-UIO-662/Z5 | 320 | 3 | H2/CO2 = 3 | 4800 | 21.2 | 32.8 | 88.8 | 6.78 | 63 |
| In2O3–ZnZrOx/ZSM-5 | 320 | 3 | H2/CO2 = 3 | 4000 | 22.4 | 11 | 92.1 | 8.20 | 64 |
| Ga–ZnZrOx/ZSM-5 | 340 | 3 | H2/CO2 = 3 | 6000 | 14.7 | 23.6 | 88.1 | 6.63 | 65 |
Chromium oxide (Cr2O3) and spinel-structured oxides including ZnCrOx and ZnAlOx. As demonstrated in the study conducted by Wang et al., the abundant oxygen vacancies in Cr2O3 nanoparticles promote the activation of C–O bonds, thereby facilitating the formation of CO2* intermediates and the production of methanol through the formate pathway.54,55 Enhancing oxygen vacancy formation is a common strategy for modifying metal oxide catalysts, which can be achieved through the synthesis of structures with high porosity and defect content or through the doping/loading of another metal as a promoter. For example, the incorporation of Zn into Cr2O3 not only enhances the adsorption and dissociation of H2 at the Zn–O site, but also accelerates the formation of oxygen defects. The formation of oxygen vacancies is particularly favored in non-stoichiometric spinel structures, which exhibit a notably random distribution of metallic cations.66–70 Furthermore, pretreatment with H2 can remove specific oxygen atoms and further facilitate the formation of surface oxygen defects, thereby enhancing both CO2 adsorption and conversion (Fig. 7A–J).43 ZnCrOx, as the first commercial methanol synthesis catalyst, demonstrates excellent resistance to sulfide impurities and high activity for CO/CO2 hydrogenation at elevated temperatures. Recently, ZnCrOx has also exhibited outstanding performance in syngas66,71–73 and CO2 conversion reaction systems.43,58 During the transformation of CO2 over ZnCrOx, intermediate species such as HCOO* and CO32−* are formed, ultimately leading to the formation of methanol, as evidenced by in situ characterization.43,58 ZnAlOx, another typical spinel-structured metal oxide, has also been employed in the CO2-to-aromatics reaction (Fig. 7K–N).41 By combining a nano-ZnAlOx spinel with nano-ZSM-5 having a high Si/Al ratio, a selectivity of 73.9% for aromatics can be achieved. In this bifunctional catalytic system, CO2 is hydrogenated to formate species at the Zn2+ sites of ZnAlOx and subsequently hydrogenated into methanol. Afterwards, methanol is dehydrated at the AlOx sites to form dimethyl ether, which is identified as another potential intermediate besides methanol.
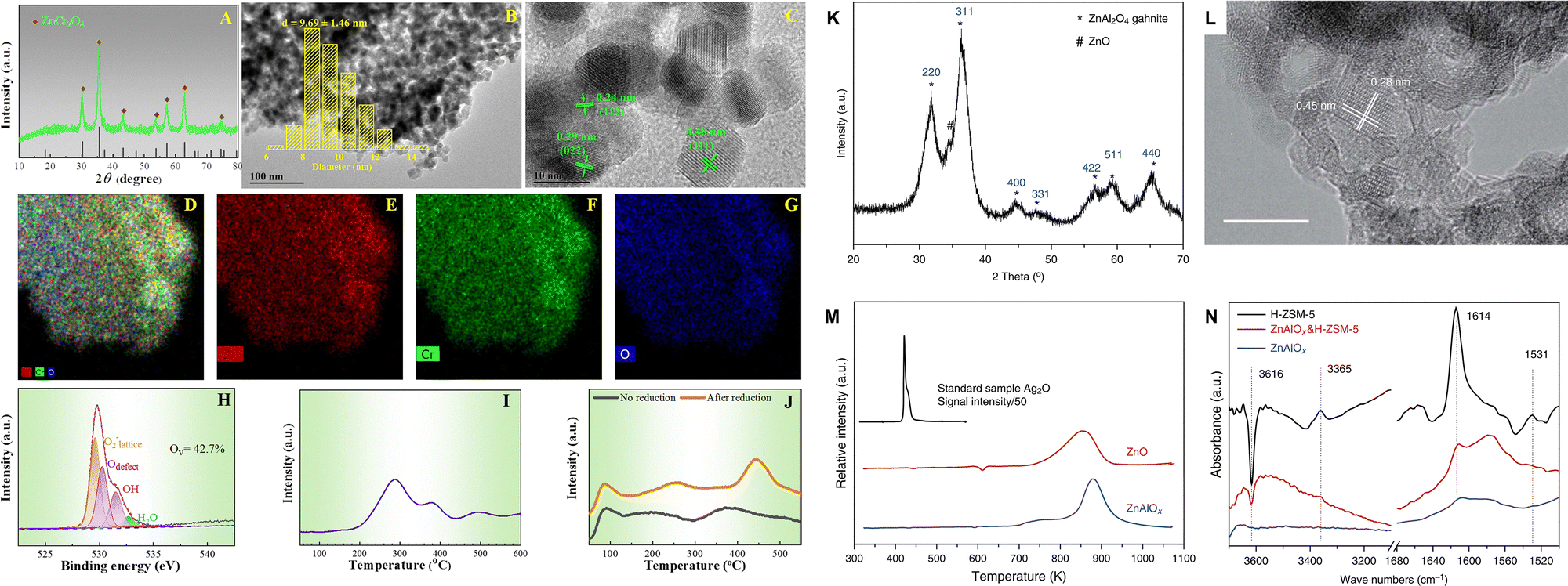 | ||
| Fig. 7 Morphology, structure and property of the ZnCrOx and ZnAlOx spinels. (A) XRD pattern, (B) TEM image, (C) HR-TEM image, (D)–(G) STEM-EDX elemental mapping, (H) O 1s XPS spectra, (I) H2-TPR profile, and (J) CO2-TPD profile of the ZnCrOx oxide. Reprinted with permission from Guo et al. Copyright (2024), American Chemical Society,43 (K) XRD pattern, (L) HR-TEM image with a scale bar of 10 nm, (M) H2-TPR profiles normalized by weight and the relative intensity of the standard sample Ag2O was divided by 50, (N) FTIR subtraction spectra relative to adsorption of DTBPy of the ZnAlOx oxide. Reprinted with permission from Ni et al. Copyright (2018), The Authors.41 | ||
Metal oxides with solid solution structure. The ZnO–ZrO2 solid solution has shown excellent performance in the CO2 hydrogenation to methanol, facilitated by adjacent Zn–Zr dual-sites, which simultaneously activate H2 and CO2, respectively (Fig. 8A–D).74 It has also been employed in the CO2-to-aromatics reaction after combining with ZSM-5.53,59,60,62 For example, Li et al. achieved a 73% selectivity to aromatic at a 14% CO2 conversion over ZnZrO/ZSM-5.60 The CHxO species derived from the activation and reaction of CO2 and H2 over ZnZrO have been determined as the intermediates in the tandem reaction, according to a series of characterization methods, including in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS), chemical trapping, and the in situ capture of CHxO by SBA-15 functionalized with an –NH2 group. Zhou et al. reported the synthesis of a ZnO–ZrO2 aerogel using a combined sol–gel and subsequent supercritical drying method (Fig. 8E).62 Compared with the ZnZrOx oxides prepared by methods of impregnation, co-precipitation, or hard templating, the ZnO–ZrO2 aerogel has a loose structure characterized by the co-existence of mesopores and macropores, leading to a higher surface area and a larger amount of oxygen vacancies (Fig. 8F–K). A strong correlation between the oxygen vacancy density and the formation rate of methanol is further demonstrated (Fig. 8L), thereby confirming the surface oxygen vacancies as the active sites for the adsorption and activation of CO2. H2–D2 exchange experiments indicate Zn as the active sites for H2 dissociation, and the synergy between ZrO2 and ZnO within the ZnO–ZrO2 solid solution is then proposed. By modification of the ZnZrOx solid solution with indium or gallium, the formation of oxygen vacancies is promoted and the adsorption and activation of CO2 are therefore enhanced.64,65 Moreover, the synergistic sites of Zn–Zr can also be constructed from metal–organic frameworks (MOFs). Tian et al. utilized the Zr-containing UIO-66 as a carrier and introduced Zn, Ga, and In by the physical mixing method.63 Among all, Zn-UIO-66 exhibits superior performance in the conversion of CO2 to aromatics when coupled with ZSM-5, achieving a 21.2% CO2 conversion and an 84.9% selectivity to benzene, toluene, and xylene (BTX). As shown in Table 1, the Cr- and Al- based metal oxides are characterized by a high CO selectivity of 35–91%, while the Zr-based metal oxides show a significantly lower CO selectivity of 10–45%, thus showing superiority in the spacetime yield (STY) of aromatics.
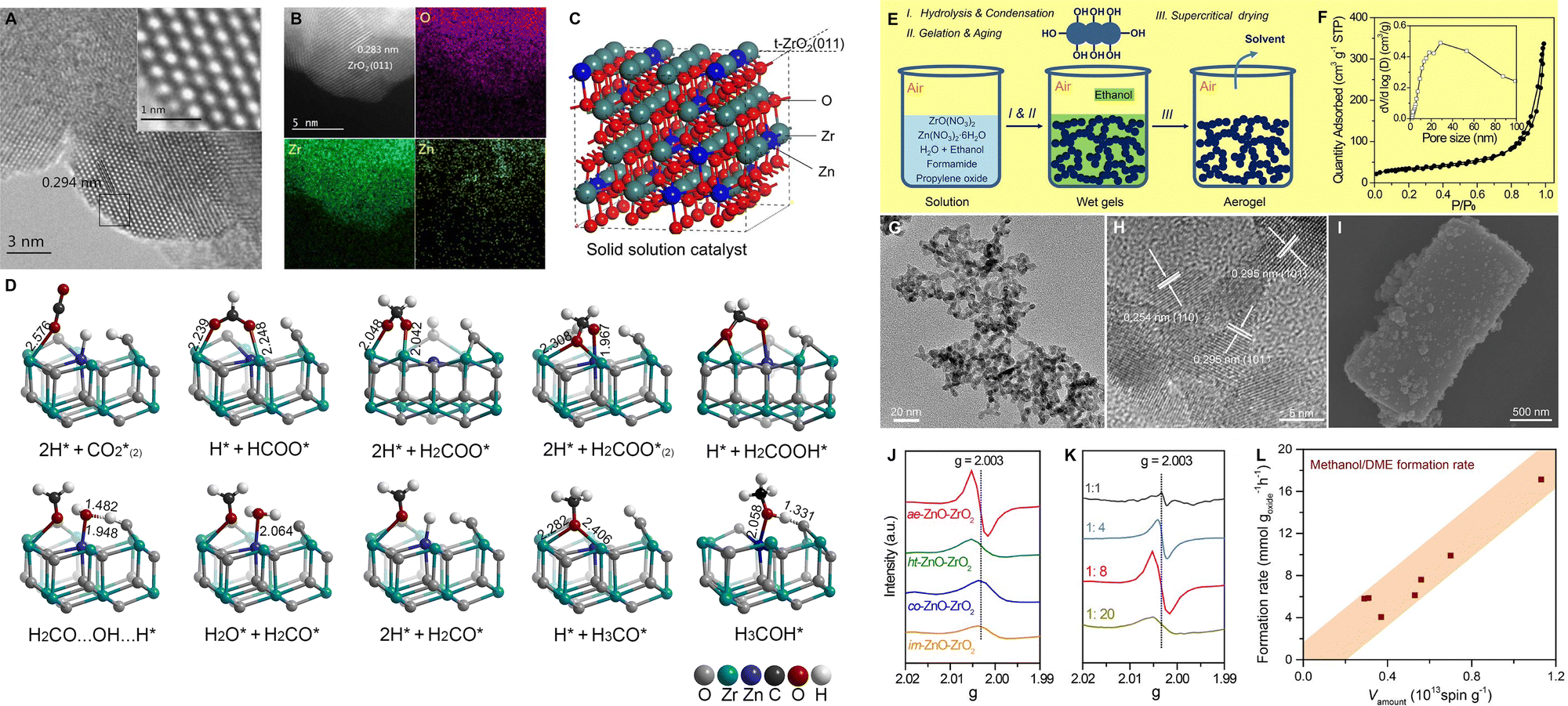 | ||
| Fig. 8 (A) HRTEM, (B) aberration corrected scanning TEM-high-angle annular dark-field images and elemental distribution of 13% ZnO–ZrO2, (C) schematic description of the ZnO–ZrO2 solid solution catalyst mode and (D) CO2 hydrogenation reaction path on the (101) surface of the tetragonal ZnO–ZrO2 model by DFT calculations. Reprinted with permission from Wang et al. Copyright (2017), the Authors,74 (E) preparation of the ZnO–ZrO2 aerogel by the sol–gel and subsequent supercritical drying methods, (F) N2 physisorption analysis of the ZnO–ZrO2 aerogel after calcination, (G) TEM, (H) HRTEM, and (I) SEM images. EPR spectra of ZnO–ZrO2 catalysts with (J) different preparation methods and (K) Zn/Zr ratios. (L) The correlation between oxygen vacancy density and the formation rate of methanol. Reprinted with permission from Zhou et al. Copyright (2020), American Chemical Society.62 | ||
The strength, density and type of acid sites. As the rate-determining step in the formation of aromatics, the dehydrogenation and cyclization reactions exhibit high activation energies, e.g. 207 and 230 kJ mol−1 for dehydrogenation of ethane and butane, respectively.75 Thus, a high acid strength of the active site is required. The acid strength of silicon–aluminum zeolites, including ZSM-5, is strongly correlated with their Si/Al ratio.76,77 On the one hand, the Si–OH–Al bridging hydroxyl groups, which compensate for the negative charge of the AlO4 tetrahedron, are the source of Brønsted acidity. On the other hand, the electron donating effect of the AlO4 tetrahedron may weaken the acid strength of the bridging hydroxyls, particularly at a high content of adjacent aluminum, or equivalently, at a relatively low Si/Al ratio.76,77 Consequently, with the increase of the Si/Al ratio, the acid density decreases, whereas the acid strength especially the Brønsted acid strength intensifies. A series of studies have investigated the effect of zeolite acidity on the catalytic performance of ZnZrOx/ZSM-5 and ZnCrOx/ZSM-5 in the CO2-to-aromatics reaction.43,58–60 The non-acidic silicalite-1 zeolite shows negligible carbon chain growth activity.59 The combination of relatively low acid density and high acid strength at a high Si/Al ratio promotes the formation of C5+ products, particularly aromatics.58 However, excessive acidity will exacerbate the deep hydrogenation of olefins and inhibit the carbon chain growth and aromatization reaction, thus leading to the formation of alkane by-products (Fig. 9A and B).58 In addition to adjusting the acid strength and density through the Si/Al ratio, introducing metals, such as Zn, as Lewis acid sites into ZSM-5 by methods like ion exchange, can facilitate the dehydrogenation and cyclization of olefins. This process is assisted by the dehydrogenation activity of Lewis acids and their synergy with adjacent Brønsted acid sites.54,58,78 Wang et al. modified the Cr2O3/ZSM-5 bifunctional catalyst by doping ZSM-5 with Zn ions, leading to the formation of (ZnOH)+ Lewis acid sites and precise manipulation of the B/L acid distribution to enhance the synergy between Brønsted and Lewis acid sites, which achieves a para-xylene selectivity of 38.7% at a CO2 conversion of 22.1% (Fig. 9C–F).54
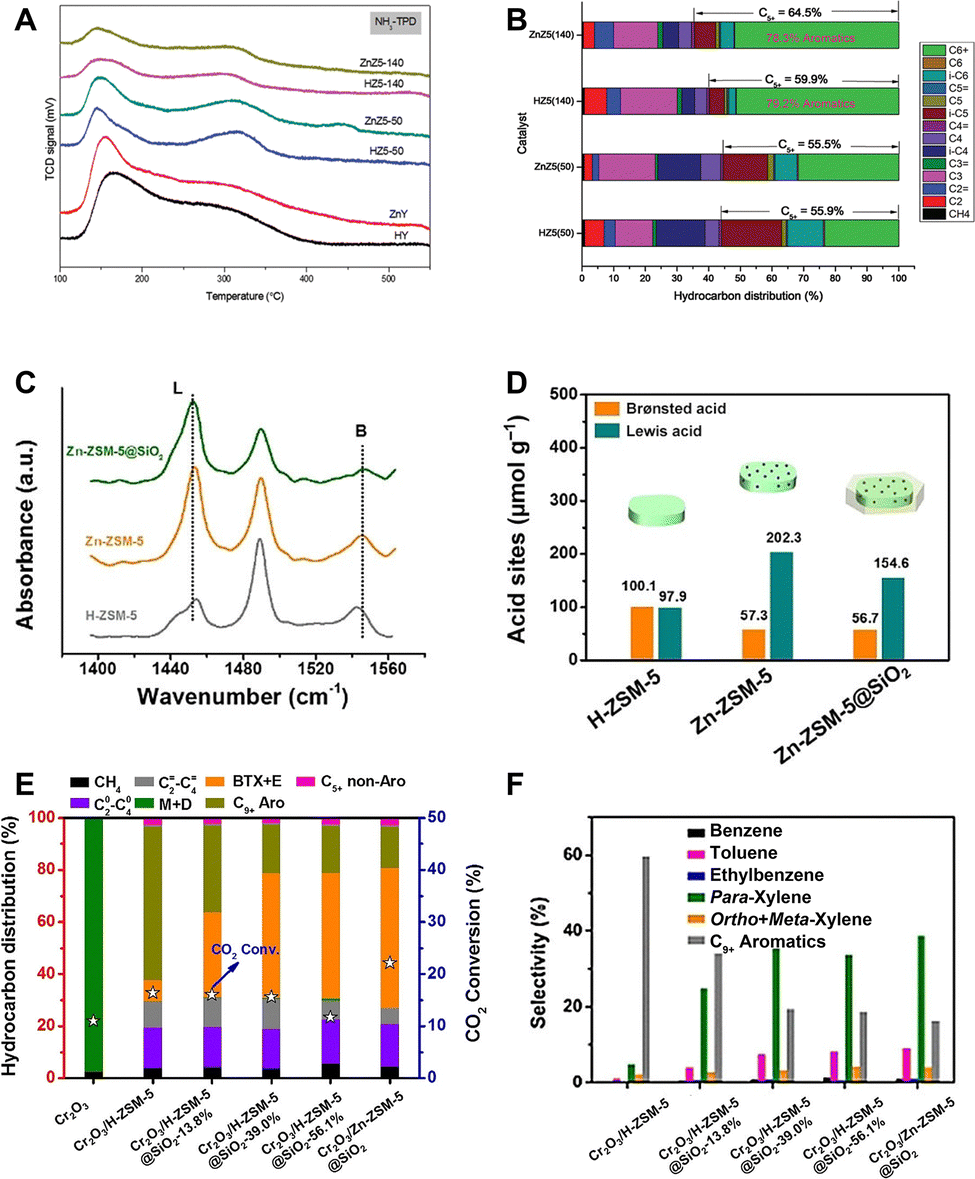 | ||
| Fig. 9 (A) NH3-TPD profiles of ZSM-5 samples with various Si/Al ratios, and with and without Zn-exchange, and (B) their catalytic performances when combined with ZnCrOx. Reprinted with permission from Zhang et al. Copyright(2019), The Royal Society of Chemistry.58 (C) FTIR spectra of adsorbed pyridine and (D) densities of B and L acid sites on H-ZSM-5, Zn-ZSM-5, and Zn-ZSM-5@SiO2. (E) and (F) Catalytic performances of Zn modified ZSM-5. Reprinted with permission from Wang et al. Copyright (2019), Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.54 | ||
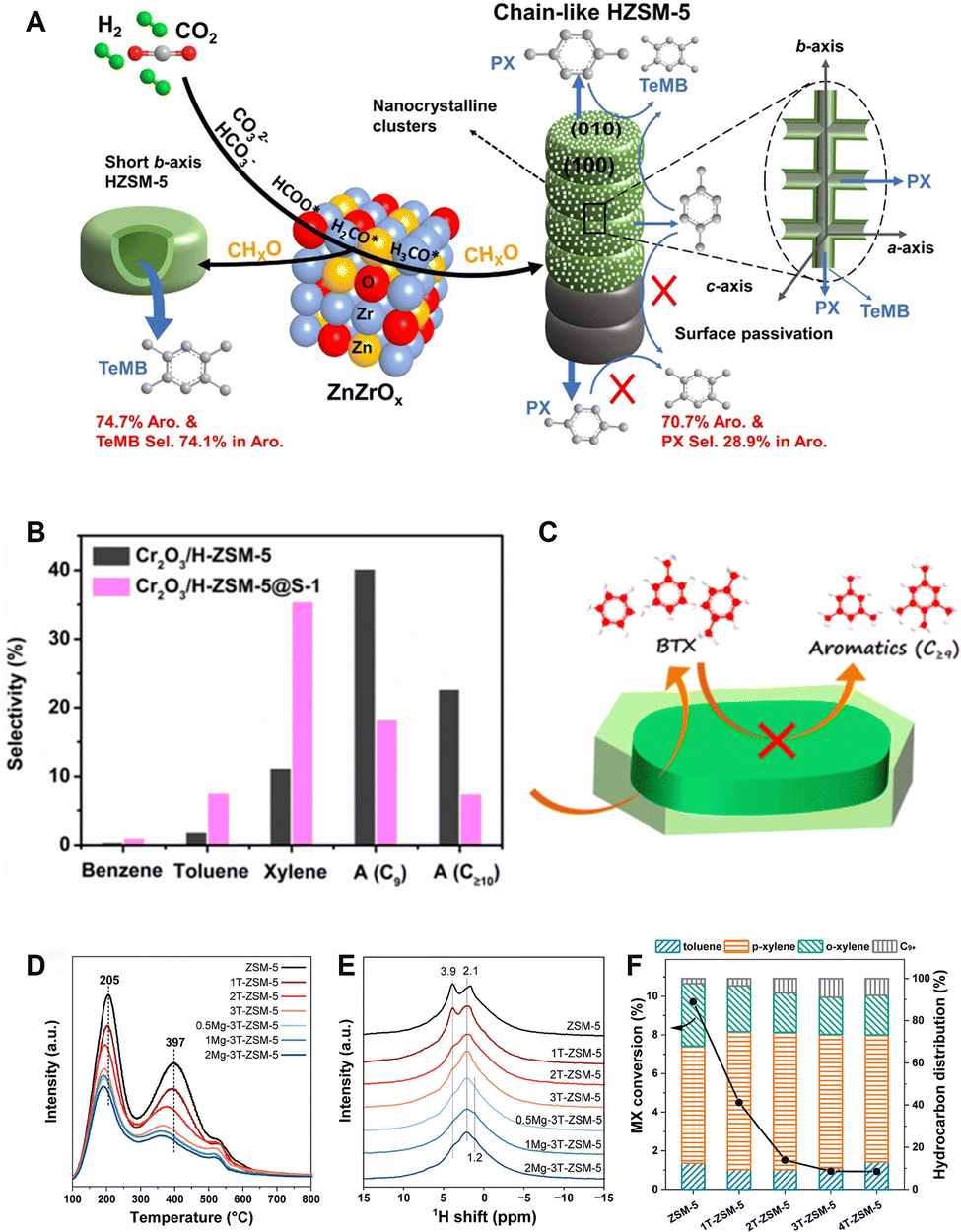
The spatial positioning of acid sites. The spatial distribution of acid sites is another crucial factor influencing catalytic performance, in addition to the strength, density and type of acid sites. Liu et al. identified the b-axis length of ZSM-5 as the key morphology parameter governing aromatic distribution in the syngas-to-aromatics reaction. The strong linear correlation is revealed between the reciprocal of the b-axis length (1/b) and SC9+/A, an indicator of the relative activity of alkylation to aromatization.79 Wang et al. applied the ZSM-5 zeolite with various morphologies to the CO2-to-aromatics reaction in combination with ZnZrOx.53 The chain-like ZSM-5 with a large b-axis size and low external acidity obtains a high selectivity to para-xylene, while the hollow ZSM-5 with a much smaller b-axis size and higher external acidity favors the formation of tetramethylbenzene. The selectivity to xylene and tetramethylbenzene reaches 28.9% and 74.1%, respectively (Fig. 10A). Furthermore, the external acidity of ZSM-5 can be partially eliminated by silanization via chemical liquid deposition or epitaxial growth of an inert silicalite-1 shell (Fig. 10B and C).55 Additionally, the opening of the 10-MR pores can be narrowed by MgO modification (Fig. 10D–F).61 Both methods enhance the shape-selectivity and inhibit side reactions, such as alkylation and isomerization, thereby contributing to an optimized aromatic distribution, especially a high fraction of light aromatics.41,54–56,61 For example, Wang et al. synthesized a ZSM-5@silicalite-1 core–shell structured zeolite.55 By precisely adjusting the shell thickness to minimize the external acidity while ensuring smooth diffusion of intermediates and products, they improved the BTX selectivity from 13.2% to 43.6%, with the para-xylene selectivity increasing from 7.6% to 25.3% over the Cr2O3/ZSM-5 bifunctional catalyst. Guo et al. compared the kinetics of the methylation reaction occurring at acid sites located at the intersection of two sets of 10-MR channels and those positioned inside the straight channels within the ZSM-5 framework by DFT calculation.43 Compared with the acid sites within the straight channels, those at the channel intersections provide a suitably larger reaction space and less steric hindrance, therefore favoring the formation of trimethylbenzene while inhibiting its further methylation. Based on this finding, they designed a ZnCrOx/H-ZSM-5 catalyst and realized the direct and highly selective conversion of CO2 to trimethylbenzene and ethylene. At a 17.5% CO2 conversion, the aromatic selectivity reaches 64.6%, 57.4% of which is trimethylbenzene.
 | ||
| Fig. 10 (A) Utilization of ZSM-5 with chain-like and hollow morphology in the CO2-to-aromatics reaction. Reprinted with permission from Wang et al. Copyright (2021), Elsevier B.V.53 (B) and (C) Catalytic performance of the core–shell structured ZSM-5@S-1 in the CO2-to-aromatics reaction. Reprinted with permission from Wang et al. Copyright (2019), American Chemical Society.55 (D) NH3-TPD profiles, (E) 1H NMR spectra and (F) isomerization of meta-xylene of ZSM-5 co-modified with MgO and an inert shell. Reprinted with permission from Qu et al. Copyright (2023), The Royal Society of Chemistry.61 | ||
3. Direct conversion of CO2 to aromatics through the modified Fischer–Tropsch pathway
3.1. Reaction mechanism
In the modified Fischer–Tropsch pathway, CO2 is first transformed into CO by the RWGS reaction over the metal-based catalyst, which then undergoes the Fischer–Tropsch reaction, yielding olefins as intermediates. The aromatization of olefins then occurs over the zeolite catalyst. These reactions follow eqn (3)–(5):4Reverse water–gas shift reaction (RWGS):
| CO2 + H2 → CO + H2O | (3) |
Fischer–Tropsch synthesis (FTS):
| nCO + 2nH2 → CnH2n + nH2O | (4) |
Aromatization of olefins:
| –(CH2)n– → C6+nH6+2n | (5) |
| Mn = (1 − α) αn−1 | (6) |
 | ||
| Fig. 11 (A) Reaction pathways for CO2-to-aromatics via the modified Fischer–Tropsch synthesis route. (B) Chain-growth probability α. (C) Regulating the carbon chain length distribution of products by chain-growth probability α. Reprinted and reproduced with permission from Wang et al. Copyright (2021), Elsevier Inc.,4 and Zhou et al. Copyright (2019), The Royal Society of Chemistry.80 | ||
wherein α is determined by the rates of chain propagation (rp) and chain termination (rt), as shown in eqn (7):
| α = rp/(rp + rt) | (7) |
Based on the ASF model, the carbon chain length distribution of the product can be modulated within a certain range by adjusting the chain-growth probability α of the catalyst (Fig. 11C).80 However, there exist inherent theoretical limits. For example, the selectivity to C2–C4 hydrocarbons can hardly exceed 58%, and basically no aromatic hydrocarbons are generated.83,84 Upon integration with zeolites, not only the carbon chain length distribution can be altered, but aromatics can also be formed through reactions of C–C cleavage of heavy hydrocarbons, olefin oligomerization, hydrogen transfer or dehydrogenation, and cyclization (Fig. 4).
3.2. Metal-based materials/zeolite bifunctional catalysts
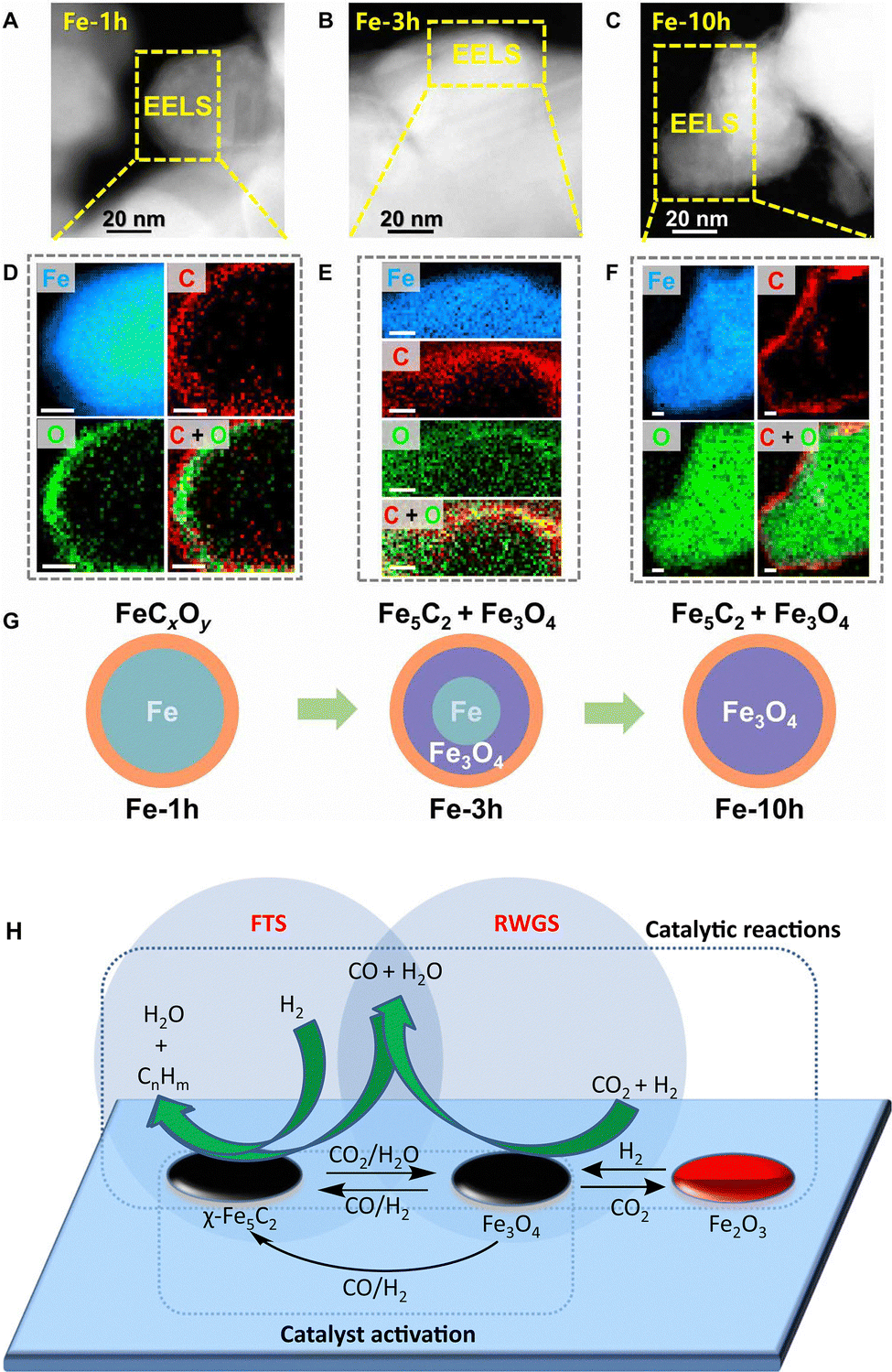 | ||
| Fig. 12 (A)–(C) High-angle annular dark-field-scanning transmission electron microscopy (HAADF-STEM) images, (D)–(F) EELS elemental distribution of Fe-xh catalysts, and (G) schematic diagrams of the evolved microstructure of the Fe-based catalyst under the reaction atmosphere of Fischer–Tropsch synthesis. Scale bar, 10 nm. Reprinted with permission from Zhu et al. Copyright (2022), The Authors.85 (H) RWGS-FTS tandem reaction path over the Fe3O4 and χ-Fe5C2 sites. Reprinted with permission from Yao et al. Copyright (2020), The Authors.86 | ||
| Catalyst | Temperature (°C) | Pressure (MPa) | Feedstock | Space velocity (mL g−1 h−1) | CO2 conv. (%) | CO Sel. (%) | Aro. Sel. (%) | Aro. STY (mmol C g−1 h−1) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a C–C coupling inhibited on the carburization-resistant LaFeO3 perovskite. | |||||||||
| Na/Fe/HZSM-5 | 320 | 1 | H2/CO2 = 3 | 2400 | 29.4 | 23.1 | 54.3 | 3.29 | 88 |
| Na–Fe/ZSM-5 | 320 | 3 | H2/CO2/Ar = 71.8/24.3/3.9 | — | 33.3 | 13.3 | 50.2 | — | 89 |
| Na–Fe@C/Z-5-ellipsoid | 320 | 3 | H2/CO2/Ar = 71.8/24.3/3.9 | 9000 | 34.3 | ∼12 | 49.6 | ∼14.62 | 90 |
| NaFe/ZSM-5 | 320 | 3 | H2/CO2 = 2 | 4000 | 27.7 | 16 | 45 | 6.23 | 91 |
| Fe2O3@KO2/ZSM-5 | 375 | 3 | H2/CO2 = 3 | 5000 | 49.8 | 12.8 | 24.9 | 6.03 | 92 |
| CuFeO2/0.15M-HZSM-5 | 320 | 3 | H2/CO2 = 3 | 8100 | 52.8 | 7.3 | 69.7 | 30.84 | 37 |
| 6.25Cu–Fe2O3/HZSM-5-c | 320 | 3 | H2/CO2/N2 = 72/24/4 | 1000 | 57.3 | 3.51 | 56.61 | 3.35 | 93 |
| 6.25Cu–Fe2O3/HZSM-5-pt | 320 | 3 | H2/CO2/N2 = 72/24/4 | 1000 | 55.38 | 4.41 | 61.94 | 3.51 | 93 |
| FeMnOx/HZSM-5 | 320 | 3 | H2/CO2/N2 = 72/24/4 | 1000 | 44.49 | 11.81 | 64.24 | 2.70 | 94 |
| 2.3Na–Cu–Fe2O3/HR-Z5-S | 320 | 3 | H2/CO2/N2 = 72/24/4 | 1000 | 33.26 | 16.03 | 68.8 | 2.06 | 95 |
| 3-NFC-5(NaCuFe)/HZSM-5 | 320 | 3 | H2/CO2/N2 = 72/24/4 | 1500 | 42.11 | ∼10 | 50.68 | ∼3.09 | 96 |
| NFC-DM (NaCuFe)/0.5 M-HZSM-5 | 320 | 3 | H2/CO2/N2 = 72/24/4 | 1500 | 46.2 | 6.5 | 56.1 | 3.89 | 97 |
| NaFeMn/core–shell HZSM-5@Si | 320 | 3 | H2/CO2/N2 = 72/24/4 | 3000 | 45 | 11 | 64 | 8.24 | 98 |
| NaFeMn/ZSM-5 | 320 | 3 | H2/CO2/N2 = 72/24/4 | 3000 | 44.49 | 11.81 | 56.65 | 7.14 | 99 |
| ZnFeOx–4.25Na/S-HZSM-5 | 320 | 3 | H2/CO2/N2 = 73/24/3 | 1000 | 41.2 | 6.9 | 75.6 | 3.11 | 49 |
| NaZnFe/ZSM-5 | 320 | 3 | H2/CO2/N2 = 72/24/4 | 1000 | 42.08 | <10 | 63.68 | <2.58 | 100 |
| Na–FeAlOx/Zn–HZSM-5@SiO2 | 370 | 3.5 | H2/CO2 = 3 | 4000 | 45.2 | — | 38.7 | — | 101 |
| LaFeO3/ZSM-5a | 350 | 3 | H2/CO2 = 3 | 1000 | 61.2 | 12 | 85.8 | 5.16 | 102 |
Adjusting the electronic and adsorption properties by alkali metal modification. The adsorption of CO2 on the Fe-based catalyst involves electron transfer from the catalyst to CO2. The electron-donating capabilities of alkali metals strengthen the Fe–C chemical bond between CO2 and the active site, thereby promoting the adsorption of CO2 molecules, while the adsorption of the electron-donating molecules, such as H2 and olefin, is weakened. Xu et al. studied the modification of the Fe-based catalysts by Na. In the Na/Fe system, Na acts as an electron donor and creates an environment with a high C*/H* ratio on the catalyst surface, which favors the formation and desorption of olefin molecules. Utilizing the bifunctional catalyst consisting of Na/Fe and ZSM-5, the aromatic selectivity reaches approximately 50%, accounting for 94% of the C5+ product.88 Wang et al. synthesized highly dispersed Fe3O4 nanoparticles encapsulated in a graphene-like carbon layer by the pyrolysis of Fe-based metal–organic frameworks (Fe-MOFs), taking advantage of the periodic structured frameworks.89 The Na–Fe@C catalyst was synthesized by further impregnation with Na2CO3, which facilitates the formation of olefins due to the high accessibility of active sites and the precise construction of the catalytic interface (Fig. 13A–D). In addition to Na, K is also adopted to modify Fe-base Fischer–Tropsch synthesis catalysts. Ramirez et al. integrated KO2-doped Fe2O3 with ZSM-5 and obtained a 61.4% aromatic fraction in the C5+ liquid product in the CO2-to-aromatics reaction.92
 | ||
| Fig. 13 (A) Preparation of Na–Fe@C by the pyrolysis of Fe-based metal–organic frameworks (Fe-MOFs). (B) TEM and (C) high resolution TEM (HRTEM) images of the used Na-Fe@C catalyst. (D) STEM image of the used Na-Fe@C catalyst and the corresponding elemental mapping of Fe, C, and Na elements. Reprinted with permission from Wang et al. Copyright (2020), Elsevier B.V.89 HRTEM images of Na modified ZnFeOx (E) after calcination and (F) after reaction. Reprinted with permission from Cui et al. Copyright (2019), American Chemical Society.49 | ||
Transition metal modification. Song et al. revealed that the modification of a Fe-based catalyst by Cu not only enhances the RWGS activity, but also promotes the stepwise reduction of Fe2O3 to Fe3O4 and subsequently to α-Fe and facilitates the formation of surface oxygen vacancies in the CO atmosphere. The synergy between Cu and Fe favors the activation and dissociation of H2, CO2 and CO, endowing the Cu–Fe2O3/HZSM-5 bifunctional catalyst with a superior performance in the CO2 hydrogenation reaction.93 Specifically, it achieves a high CO2 conversion of 56.61%, along with a low CO selectivity of 3.51%, and an aromatic selectivity as high as 61.9%. Cheng et al. discovered the reconstruction of defafossite-CuFeO2 under the reduction and reaction conditions of CO2 hydrogenation, which led to the formation of Cu, Fe3O4 and Fe5C2 phases.37 Among these active phases, Cu plays a pivotal role in promoting the reduction of Fe and enhancing CO2 adsorption, as well as facilitating olefin desorption. The integration of defafossite-CuFeO2 with hierarchical ZSM-5 achieves a high space-time yield of aromatics up to 431.8 mgCH2 gcat−1 h−1, while the total selectivity to CO and methane is as low as 12%.
The co-modification by alkali metals and transition metals. The co-modification of transition metals, including Zn, Cu, and Mn, with alkali metals can further improve the catalytic performance. Cui et al. designed a bifunctional catalyst comprising a Na-modified ZnFeOx spinel and hierarchical nanocrystalline HZSM-5 zeolite.49 During pretreatment, the ZnFeOx phase undergoes reduction to Fe3O4, FeO, and α-Fe, which subsequently transform into Fe3O4 and Fe5C2 under the reaction atmosphere (Fig. 13E and F). Therefore, the coupling of RWGS and FTS is realized. The spinel structure exhibits effective inhibition of nanoparticle sintering. Additionally, the electron-donating properties of Na not only promote CO2 adsorption and CO conversion, but also inhibit the production of CH4. It can also modulate the reducibility of ZnFeOx and optimize the active site composition of Fe3O4 and Fe5C2. The catalyst achieves a CO2 conversion of 41.2% and an aromatic selectivity of 75.6%, with the total selectivity to CH4 and CO below 20%. Yang et al. and Chen et al. independently investigated the synergistic effects of Cu and Na co-modification.95,96 They revealed that Na not only promotes the adsorption of CO2, but also acts as an RWGS active site and enhances the formation of the COOH* intermediates and subsequent CO production. Additionally, Na boosts the carburization process and the formation of Fe carbide species. The adsorption and activation of H2, along with the H-spillover effect imparted by Cu, enhances the reduction of ferrite species and the formation of oxygen vacancies. This, in turn, promotes the formation of the COOH* intermediate and the non-dissociative adsorption of CO. Additionally, the hydrogen activation and H-spillover effect of Cu can be partially attenuated by Na, which prevents the deep hydrogenation of intermediates and products. Similar co-modification of Na and Mn has also been reported for the NaMn–Fe catalyst.94,98,99 Mn increases the tolerance to the poisoning of zeolite by Na and inhibits the coking deactivation by promoting the oxidation of carbon deposition species with CO2, thus significantly prolonging the catalyst lifetime.99 By adjusting the spatial arrangement between Na and FeMnOx, the electronic properties of Fe species can be altered, as well as the distribution of the Fe3O4 and Fe5C2 species, thus remarkably influencing the catalytic performance.94 The modification of Fe-based FTS catalysts with alkali metals or transition metals enhances the aromatic selectivity, which however cannot exceed 55% and 70%, respectively (Table 2). The co-modification employing both alkali metals and transition metals exhibits greater potential, achieving a significant enhancement in aromatic selectivity up to 75.6%.
Carburization-resistant LaFeO3 perovskite inhibiting uncontrolled C–C coupling. The uncontrollable carbon chain growth on the Fe carbide sites is the determining factor for the low aromatic selectivity of the Fe-based/zeolite catalyst employed in the modified Fischer–Tropsch synthesis pathway. Regarding this, Tian et al. designed a LaFeO3 perovskite catalyst. The significant gap between the required migration Fe–Fe distance (2.6–2.8 Å) during phase transition and the actual Fe–Fe distance in the Fe-based perovskite structure (>3.7 Å and >3.90 Å for LaFeO3) makes the carburization of Fe species challenging in such a structure. Furthermore, the A-site cation (La) acts as the pillar anchoring the perovskite [FeO6] framework. Moreover, the unique empty 4f orbitals of La further enhance the stability of the [FeO6] framework through a crystal field effect, raising the energy required and suppressing the formation of Fe5C2. Consequently, the hydrogenation activity of Fe oxides is fully utilized while eliminating the undesired carbon chain growth activity originating from the Fe carbide species, leading to a breakthrough in the catalytic performance of LaFeO3/ZSM-5 in the CO2-to-aromatics reaction (Fig. 14).102 Owing to the absence of Fe carbide sites and consequently the limited C–C coupling ability, pure LaFeO3 primarily produces methane in the CO2 hydrogenation reaction, exhibiting a higher activity compared to Fe2O3 or ZnFe2O4. Upon coupling with ZSM-5, LaFeO3/ZSM-5 achieves an aromatic selectivity of up to 85.8% at a CO2 conversion of 61.2%, with HCOOH/H2CO species formed in the RWGS reaction on the Fe oxide as the key intermediates, evidenced by theoretical calculations and experimental results. In conclusion, by designing the LaFeO3 perovskite as a carburization-resistant Fe-based oxide capable of inhibiting carbon chain growth, isolating CO2 hydrogenation from C–C coupling in the tandem reaction system, and introducing the driving force by the zeolite to redirect the reaction pathway from methane-terminated to aromatic-terminated, a breakthrough in the aromatic selectivity of the Fe-based/zeolite catalyst is finally accomplished.
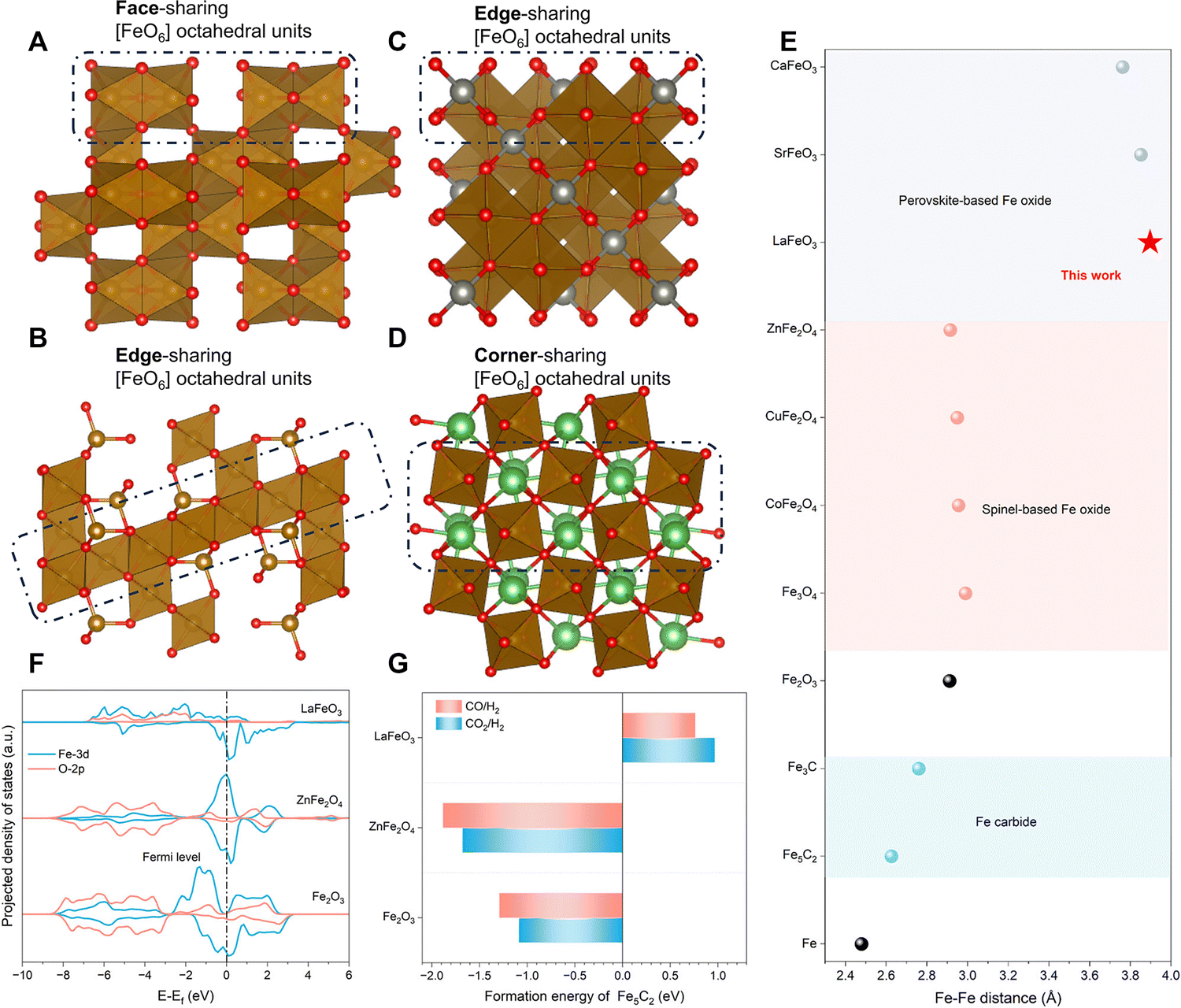 | ||
| Fig. 14 The design of the LaFeO3 perovskite as a carburization-resistant Fe-based oxide. Atomic structural model of (A) Fe2O3, (B) Fe3O4, (C) ZnFe2O4, and (D) LaFeO3, showing different connection modes of [FeO6] octahedra. Spheres in red, brown, silver, and green represent O, Fe, Zn, and La atoms, respectively. (E) Fe–Fe distances of Fe, Fe carbides, and various Fe oxides. (F) Projected density of states (PDOSs) of Fe2O3, LaFeO3, and ZnFe2O4. (G) Calculated formation energies of per mole Fe5C2 through the reduction of Fe2O3, ZnFe2O4 and LaFeO3 by typical reactive atmospheres. Reprinted with permission from Tian et al. Copyright (2024), The Authors.102 | ||
The morphology and pore structure. Compared with the methanol-mediated route, the modified Fischer–Tropsch route suffers from more significant coking deactivation, attributed to the considerable long-chain products generated by the Fischer–Tropsch synthesis reaction. Strategies aimed at improving diffusion efficiency through optimizing the morphology and pore structure of zeolite have been proposed.37,49,89,95,100 Cui et al. synthesized a nanocrystalline ZSM-5 (Fig. 15A and B), which is characterized by a hierarchical pore structure and suitable acidity, thereby facilitating the diffusion of olefins and aromatics, and consequently enhancing catalytic activity while inhibiting coke deposition.49 Wang et al. processed the parent ZSM-5 crystals using NaOH solutions of varying concentrations to create hollow-structured ZSM-5.89 Utilizing the local desilication etching induced by the radial gradient of framework aluminum, desilication is suppressed on the Al-rich surface while being promoted in the Al-poor bulk. The sample exposed to 0.2 M NaOH solution, denoted as H-ZSM-5-0.2 M, demonstrates a hollow structure comprising a thin shell of approximately 20 nm (Fig. 15C and D), featuring mesopores with a diameter of approximately 10 nm. This unique structure enhances mass transfer during the catalytic reaction and improves the reaction efficiency compared with the original ZSM-5. Jiang et al. incorporated polyethylene glycol (PEG) as a porogen during the synthesis of ZSM-5 leveraging the encapsulation effect of PEG within the crystallization system.100 They realized the construction of intracrystalline mesopores with a diameter of 4–10 nm and the weakening of strong acid sites, thereby promoting the generation and diffusion of aromatics.
 | ||
| Fig. 15 (A) and (B) The nanocrystalline ZSM-5 with hierarchical pore structure. Reprinted with permission from Cui et al. Copyright (2019), American Chemical Society.49 (C) and (D) hollow-structured ZSM-5 for the CO2-to-aromatics reaction. Reprinted with permission from Wang et al. Copyright (2020), Elsevier B.V.89 | ||
The properties and positioning of acid sites. Wei et al. explored the influence of Brønsted acidity on the formation of aromatics and coke deposition in the CO2-to-aromatics reaction, by combining a series of ZSM-5 zeolites with diverse Brønsted acidities with a NaFe catalyst.91 The study demonstrates that an increase in Brønsted acid density is beneficial for the aromatization of olefins, whereas an excessive number of Brønsted acid sites may accelerate coke deposition and catalyst deactivation. The impregnation of (NH4)2HPO3 with an appropriate amount of phosphorus can effectively reduce the Brønsted acid density and improve the aromatic selectivity.94 Regarding the side reactions, such as alkylation and isomerization, occurring on the external acid sites, various studies have reported the passivation of these sites by chemical liquid deposition of SiO2 (Fig. 16A)49,88,91 and coating with a silicalite-1 shell.37,98 For example, after the silanization treatment, the NaFe/ZSM-5 catalyst achieves a 75% proportion of light aromatics in overall aromatics and with a 72% para-xylene/xylene (PX/X) fraction.91 Similarly, the PX/X fraction reaches 75% and 66.9% in the Na–ZnFeOx/ZSM-549 and CuFeO2/ZSM-537 catalytic systems, respectively. Gu et al. synthesized ZSM-5 zeolites with varying aluminum positioning and acid properties by altering the raw materials and synthesis methods and subsequently applied them in the CO2-to-aromatics reaction in combination with Na–Fe@C.90 Consequently, the relationship between aluminum positioning and product distribution was elucidated through detailed characterization and theoretical calculation. The aluminum species located within the straight and sinusoidal channels tend to catalyze the cracking or isomerization reactions of the intermediates due to steric hindrance, while those situated at the intersection of 10-MR pore channels facilitate the aromatic formation due to the low energy barrier of the aromatization reaction (Fig. 16B and C).
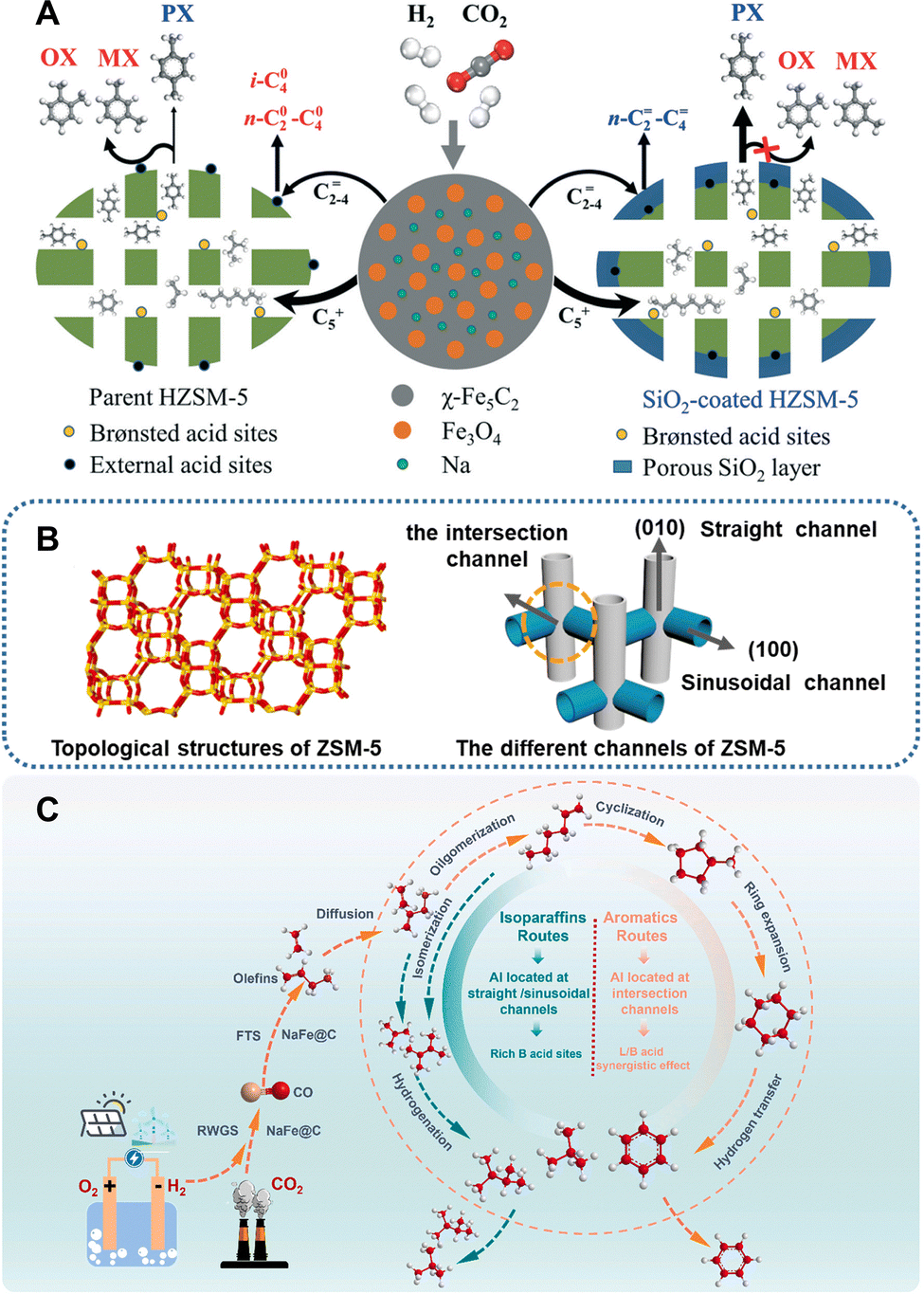 | ||
| Fig. 16 (A) Scheme of the xylene isomerization reaction on ZSM-5 with and without the modification of SiO2. Reprinted with permission from Xu et al. Copyright (2019), The Royal Society of Chemistry.88 (B) Topological structure and the corresponding channels of ZSM-5, and (C) the reaction pathway and product distribution at different aluminum positionings. Reprinted with permission from Gu et al. Copyright (2024), Elsevier B.V.90 | ||
4. Several novel coupling routes of CO2-to-aromatics
4.1. CO2 hydrogenation coupled with CO hydrogenation
RWGS is one of the principal side reactions in the CO2-to-aromatics reaction system. Despite the possibility of recycling the by-product CO for further conversion, its generation significantly reduces the carbon conversion efficiency in a single pass. Given that RWGS is a reversible reaction between CO2 and CO, the introduction of CO into the feedstock will shift the reaction equilibrium according to the Le Chatelier principle. Moreover, taking advantage of the similarity in the activation and conversion of CO2 and CO, the coupling of CO2 hydrogenation and CO hydrogenation reactions has the potential to improve the overall reaction efficiency.In an investigation focusing on the methanol-mediated pathway, Ni et al. compared the reaction performance of ZnAlOx/ZSM-5 at different CO/CO2 feed ratios. The introduction of CO has little effect on the aromatic selectivity, but it markedly reduces the CO selectivity to a minimum of 12.6% at a CO/CO2 ratio of 1.8/1 (Fig. 17A).41 According to Wang et al., the Cr2O3/ZSM-5 bifunctional catalyst exhibits a CO selectivity of 41.2% when fed with pure CO2 and H2. Upon adding 5.42% CO to the feedstock, the CO selectivity decreases drastically to 11.4%, indicating a higher conversion efficiency of CO2 to form high-value organic products (Fig. 17B).55 Similar results have also been reported for the modified Fischer–Tropsch pathway. For example, Ramirez et al. introduced an equivalent amount of CO into the CO2 hydrogenation system (a CO/CO2 ratio of 1) catalyzed by Fe2O3@KO2/ZSM-5.92 At the constant H2/(CO + CO2) ratio of 3, the COx conversion increases from 48.9% in the pure CO2 system to 58.4% in the CO/CO2 mixed system, along with the increased aromatic selectivity from 24.9% to 28.4%.
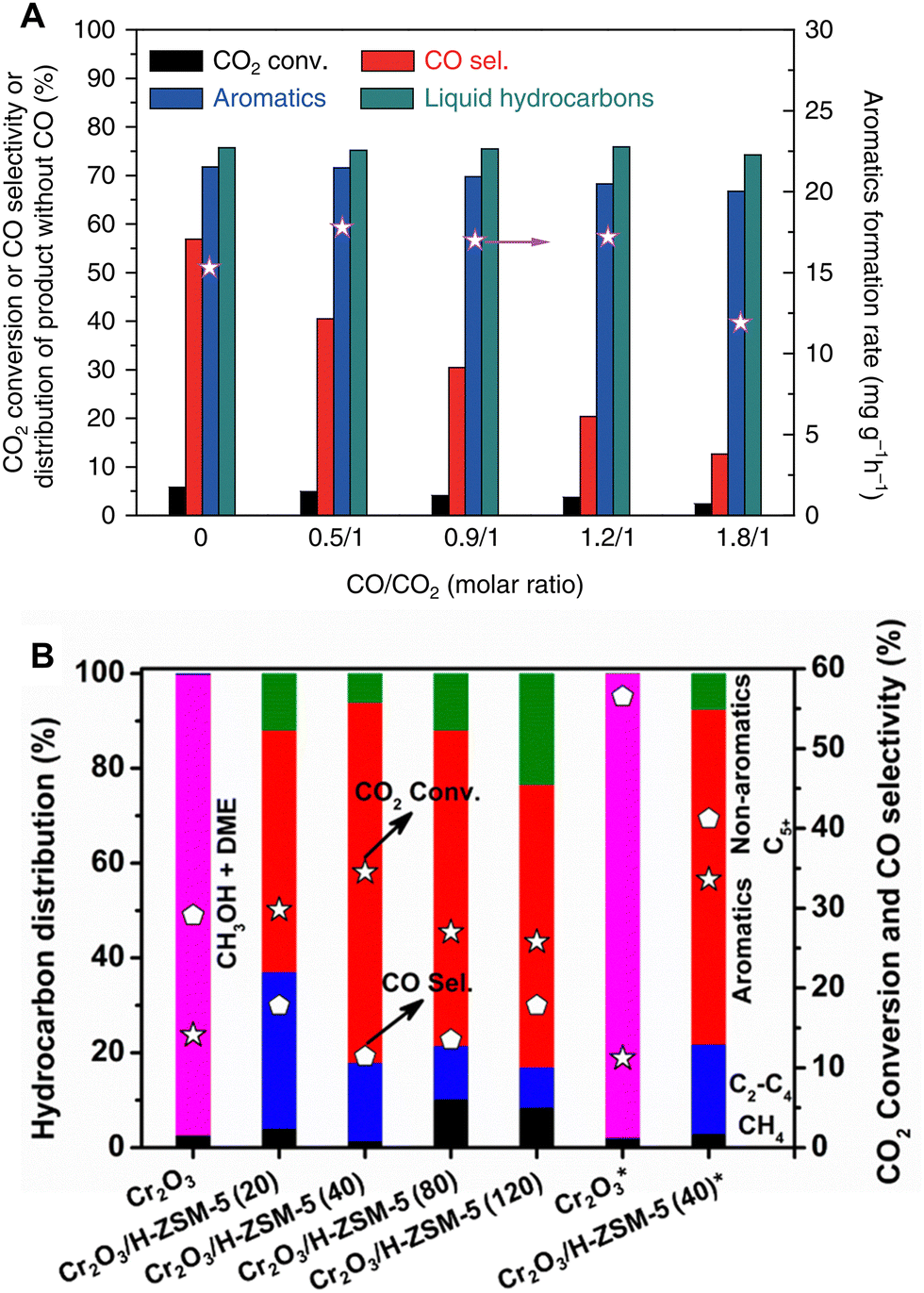 | ||
| Fig. 17 The coupling of CO2 hydrogenation with CO hydrogenation. (A) 667 mg of ZnAlOx&H-ZSM-5, 320 °C, 3.0 MPa, H2/CO2/Ar = 3/1/0.2, 66.7 mL min−1, CO = 0–26.6 mL min−1. Reprinted with permission from Ni et al. Copyright (2018), The Authors.41 (B) 0.25 g of Cr2O3 or 0.5 g of Cr2O3/H-ZSM-5, 350 °C, 3 MPa, H2/CO2 = 3 (5.42 vol% CO), 10 mL min−1, * refers to the results without CO. Reprinted with permission from Wang et al. Copyright (2019), American Chemical Society.55 | ||
4.2. CO2 hydrogenation coupled with aromatic alkylation
Zuo et al. employed the ZnZrOx/ZSM-5 catalyst for the coupling reaction of CO2 hydrogenation and toluene methylation.103 In the conventional toluene methylation reaction using methanol as the methylation reagent, the conversion of methanol into surface methoxy species is the rate-determining step. By coupling the toluene methylation reaction with the CO2 hydrogenation reaction, CO2 is first hydrogenated on the ZnZrOx surface via the formate path, generating methoxy species that subsequently migrate to the pore structure of ZSM-5 and undergo Friedel–Crafts methylation with toluene as the methylation agent (Fig. 18A). Compared with the methylation of toluene by methanol, the activation energy decreases from 108.5 ± 3.6 kJ mol−1 to 83.2 ± 1.5 kJ mol−1 upon coupling with the CO2 hydrogenation reaction. Furthermore, the para-selectivity of xylene increases from 32.7% to 70.8% due to the dynamic equilibrium between the generation and consumption of methoxy species, thereby preventing side reactions such as deep methylation and methanol to hydrocarbon (MTH) conversion resulting from the accumulation of methoxy species. To align the temperature window of the CO2 hydrogenation and methylation reactions, this team modified ZnZrOx with Cu to enhance its activity and decrease the reaction temperature. The CuZnZrOx/ZSM-5 catalyst facilitates the selective synthesis of durene by coupling the CO2 hydrogenation reaction with the methylation of trimethylbenzene.104 Cao et al. further developed a ZnZrOx/Al2O3/ZSM-5 triple composite catalyst based on ZnZrOx/ZSM-5. In this catalytic system, methanol generated from CO2 hydrogenation over ZnZrOx is dehydrated at the Lewis acid site of Al2O3 to form dimethyl ether, which then reacts with benzene to produce toluene and xylene.105 Miao et al. also achieved the coupling of CO2 hydrogenation with toluene methylation in the ZnCrOx/ZSM-5 catalytic system.106 Through optimal combination (particle mixing), the moderate proximity between the catalytic components promotes the methylation of toluene while inhibiting deep methylation, thereby improving the xylene selectivity. By further modification of ZSM-5 with P and silica, an 82.8% para-xylene fraction in xylene and a 75.8% light olefin fraction in aliphatic hydrocarbons are achieved at a CO2 conversion of 20.9% and toluene conversion of 10.6%. | ||
| Fig. 18 (A) Comparison of the performance of toluene methylation with methanol or by coupling the CO2 hydrogenation reaction. Reprinted with permission from Zuo et al. Copyright (2020), The Authors.103 (B) The proposed mechanism of n-butane aromatization coupled with CO2 over ZSM-5. Route A: formation of aromatics; Route B: formation of CO. The calculated free energy barriers of all the elementary reactions (at 450 °C) are given in kJ mol−1. Reprinted with permission from Wei et al. Copyright (2023), Dalian Institute of Chemical Physics, the Chinese Academy of Science.107 | ||
In the aforementioned studies, the methylation of aromatics with CO2 as the methoxy source has been realized by coupling the reactions of CO2 hydrogenation and aromatic methylation. Nonetheless, as the methylation reaction is prior to the side chain growth reaction, the substituents primarily consist of methyl groups, posing challenges in producing compounds with long-chain substituents, such as ethylbenzene or propylene. With respect to this, Zuo et al. established a tandem coupling reaction route consisting of “CO2 hydrogenation to light olefins” and “alkylation of benzene with light olefins”.108 In a dual-bed catalytic system comprising ZnZrOx/SAPO-34 and ZSM-5, CO2 is initially converted into ethylene or propylene through the upper bed of ZnZrOx/SAPO-34, and subsequently, the ethylene/propylene diffuses to the lower bed of ZSM-5 and reacts with benzene to form ethylbenzene or propylene. Phosphorus modification was employed to tune the medium-strong acidity of ZSM-5, thereby inhibiting the deep hydrogenation of olefins and optimizing the distribution of aromatics. The product distribution gradually shifts from ethylbenzene to propylene with the increase of phosphorus content. Consequently, the sequential construction of side chain groups and their substitution onto benzene is efficiently coupled, achieving selectivities of 83.5% and 64.7% for ethylbenzene and propylene, respectively, within the overall aromatics.
4.3. CO2 hydrogenation coupled with alkane aromatization
Introducing CO2 into the aromatization of hydrocarbons is another alternative coupling reaction pathway, in which CO2 is used as the carbon source for aromatic synthesis. Bu et al. developed a catalyst for the coupling of CO2 and propane aromatization.109 By incorporating Ga into the MFI framework to create medium-strength Brønsted acid sites, the dehydrogenation of propane is favored, while the cleavage of C–C bonds is suppressed. Simultaneously, Cu was introduced to obtain a suitable Lewis-Brønsted acid distribution, thus enhancing the adsorption and conversion of CO2 and facilitating the consumption of hydrogen species generated during alkane aromatization. The resultant Cu/Ga-MFI catalyst exhibits a 93.6% propane conversion and 73% aromatic selectivity.Wei et al. developed the coupling strategy involving CO2 hydrogenation and alkane aromatization, which is applicable for a broader spectrum of alkanes.107 By introducing CO2 into the ZSM-5-catalyzed aromatization of n-butane, n-pentane, and n-hexane, the production of methane, ethane, and propane is inhibited, resulting in aromatic selectivity exceeding 80%. This unusually high aromatic selectivity is beyond the theoretical limit (∼50%) for alkane aromatization via the hydrogen transfer pathway in the absence of CO2, suggesting that the CO2 molecules are involved in the reaction. The underlying mechanism was clarified by GC-MS analysis of the dissolved carbon species within consumed zeolite, in situ characterization, and density functional theory (DFT) calculations. In the coupling reaction system, carbonium ions generated from alkane cracking are inserted by CO2 to form alkyl carbonate species, which are further cyclized into methyl-substituted lactones (MLTOs) and then converted into methyl-substituted cycloalkenones (MCEOs). The MLTOs and MCEOs are identified as the key intermediates in this reaction, with 25% of CO2 incorporated into the aromatic products (Fig. 18B).
Research has also been conducted on the catalytic conversion of polyolefin and CO2. Chen et al. proposed a novel catalytic reaction pathway utilizing Cu–Fe3O4 and Zn/ZSM-5, which enables the simultaneous conversion of polyethylene and CO2 into aromatics and CO.110 In this reaction, CO2 is converted into CO through the RWGS reaction, which is hydrogen consuming, thereby suppressing the hydrogen transfer reaction, preventing the resulting formation of alkanes, and promoting the selective formation of aromatics with a maximum selectivity of 64.0%.
5. Rational design of multi-functional catalysts and optimizing the coupling catalytic route for CO2-to-aromatics reactions
The direct conversion of CO2 into aromatics can be realized through diverse coupling catalytic routes. Taking the methanol-mediated pathway and the modified Fischer–Tropsch synthesis route as an example, CO2 undergoes direct conversion to methanol over the metal-based catalyst or is transformed into methanol or olefins via the RWGS reaction, followed by hydrogenation or chain growth. The aromatic products with specific distribution are generated through the aromatization and alkylation of methanol or olefins over the zeolite-based catalyst. By introducing CO, alkanes, or aromatics at different reaction steps, new coupling routes are constructed, which generally adhere to the similar reaction path (Fig. 19). All aforementioned coupling catalytic strategies rely on the matching and synergy between the metal-based catalysts (metal, metal oxide, or metal carbide) and zeolites.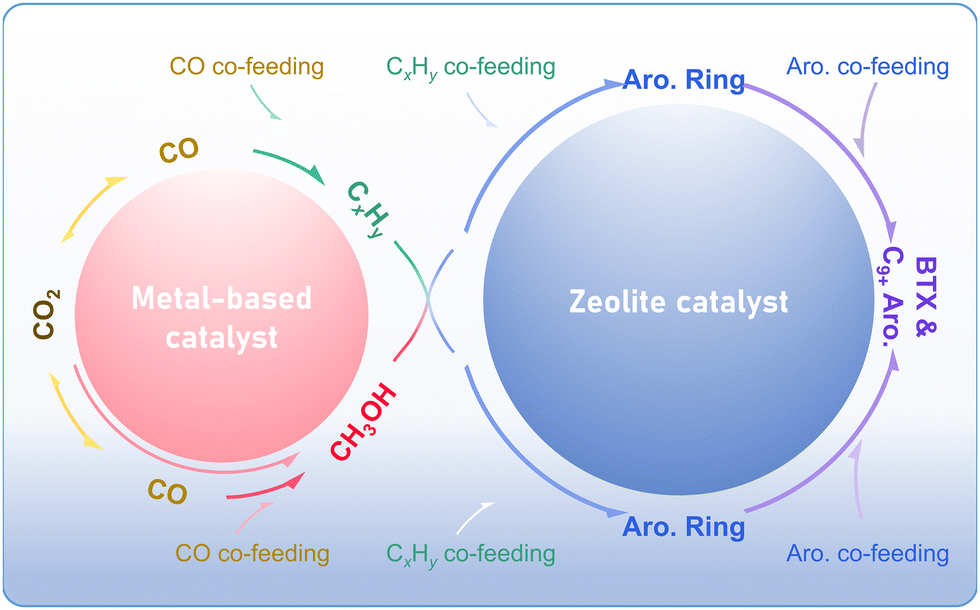 | ||
| Fig. 19 The coupling catalytic routes for CO2 conversion to aromatics over multi-functional catalysts. | ||
The activation and hydrogenation of CO2 is the primary reaction step in various coupling systems. As illustrated in Fig. 19, the RWGS reaction occurs across different systems, resulting in a considerable CO selectivity in the final product despite subsequent CO transformation. Notably, the CO–CO2 interconversion is common over metal-based catalysts. For instance, in the CO-to-aromatics reaction via the methanol-mediate pathway, CO2 emerges as the dominant byproduct due to the Boudouard or water-gas shift (WGS) reactions, with 40–50% selectivity.8 Consequently, CO and CO2 hydrogenation to aromatics exhibits comparable reaction mechanisms and catalytic system compositions. For example, the active elements are both selected from Zn, In, Cr, Zr, etc., for the methanol-mediated route, or Fe for the modified Fischer–Tropsch synthesis route. However, the CO and CO2 conversion reactions exhibit fundamental distinctions, primarily originating from the difference in C–O bond characteristics. The corresponding metal-based catalysts are different in composition, for example, the Zn–Zr binary oxide exhibits an optimal Zn/Zr ratio of 1/200 in the CO-to-aromatics system,30 compared to the higher ratio in the range of 1/8–1/2 observed in the CO2-to-aromatics reaction. In addition, the significant generation of H2O in the CO2 hydrogenation system imposes higher requirements on the water resistance of the metal-based catalysts. Based on the similarities and existing research findings on the CO- and CO2-to-aromatics reactions, developing metal-based catalysts with dual CO/CO2 hydrogenation activity represents a critical strategy to enhance the catalytic efficiency of COx-to-aromatics reactions.
Within the multi-functional catalytic system, the zeolite component catalyzes the subsequent transformation of bridging intermediates, which are generated from the metal-based catalyst. Two typical coupling reaction systems, namely the methanol-mediated route and the modified Fischer–Tropsch synthesis pathway, generate methanol and olefins as intermediates, respectively, thereby imposing different demands on the zeolite component. Specifically, the zeolite-catalyzed aromatization and alkylation processes lead to a specific distribution profile of aromatic hydrocarbons in the final product mixture. From the perspective of aromatization, the conversion of both methanol and olefins into aromatics requires the role of Brønsted acid sites, but with different strengths and densities. The ZSM-5 zeolite employed in the modified Fischer–Tropsch synthesis route exhibits a relatively lower Si/Al ratio of 10–80, whereas the methanol-mediate pathway typically demands a higher Si/Al ratio ranging from 40 to 150, with extreme cases up to 600. Furthermore, constructing the synergy between Brønsted and Lewis acidic sites and controlling the framework aluminum distribution within the pore structures are also critical strategies for tuning aromatization processes in these catalytic systems. From the perspective of alkylation, the aromatic distribution can be controlled through morphological engineering of zeolites particularly b-axis length modulation and external acidity modification. While the coupling reaction of CO2 hydrogenation with aromatic alkylation usually does not involve the formation of new aromatic rings, the shape-selectivity of zeolites remains the determining factor for the distribution of aromatic products.
Beyond the design of metal- and zeolite-based catalytic sites, the matching between these two distinct types of sites is also critical for constructing multi-functional catalysts for the coupling catalytic routes. A series of coupling effects are involved, which need to be systematically optimized and precisely controlled through the rational design of catalysts.
5.1. Coupling effects
The one-step conversion of CO2 to aromatics is realized through the coupling of diverse reaction routes, linked by the formation, transfer and conversion of intermediate species. A series of coupling effects have been revealed from the aspects of thermodynamics, kinetics and even reaction mechanisms.Taking the methanol-mediated route as an example, this route necessitates a high temperature above 400 °C due to the substantial activation energy required for the methanol to aromatic (MTA) reaction. Nonetheless, as a strong exothermic reaction, the hydrogenation of CO2 to methanol is thermodynamically unfavorable at such high temperature. By coupling the MTA and CO2 hydrogenation reactions, the conversion of CO2 to aromatics becomes more thermodynamically feasible (Fig. 20).60 Additionally, according to the Le Chatelier principle, the immediate consumption of methanol through the MTA reaction provides driving force for the CO2 conversion, which shifts the reaction equilibrium in the direction of aromatic formation.80 Li et al. compared the activity of ZnZrO and ZnZrO/ZSM-5 in the CO2 hydrogenation reaction.60 ZnZrO/ZSM-5 demonstrates significantly higher CO2 conversion and space–time yield of organic products than ZnZrO, validating that the consecutive reaction of CO2 to form aromatics is not a simple combination of reaction steps on two kinds of catalytic components, but rather a well-coupled tandem process.
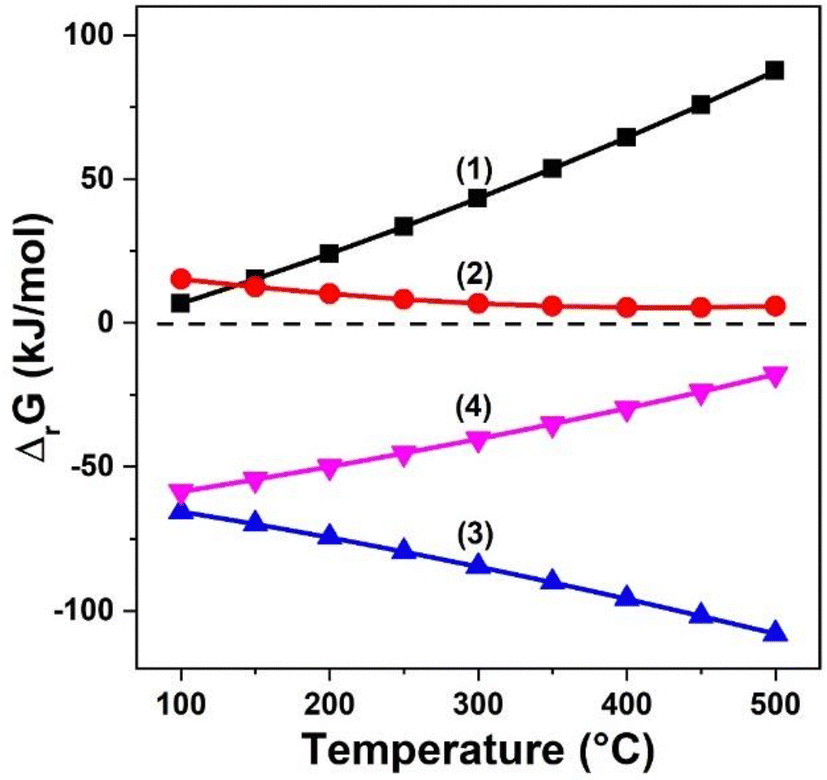 | ||
| Fig. 20 Calculation of the Gibbs free energy change of: (1) CO2 hydrogenation to methanol, (2) RWGS, (3) methanol to xylene, and (4) CO2 hydrogenation to xylene. Reprinted with permission from Li et al. Copyright (2019), Elsevier Inc.60 | ||
Wang et al. compared the performance of HZSM-5 and Cr2O3/H-ZSM-5 in the MTA and CO2-to-aromatics reactions and proposed the reinforcement effect of CO2 on aromatization in the tandem reaction system.54 Two distinct pathways compete for the conversion of olefins in zeolites, namely the deep hydrogenation leading to alkanes and the dehydro-cyclization resulting in aromatics. In the presence of Cr2O3 and CO2, the hydrogen species produced from the dehydrogenation of olefins can be consumed in-situ by reacting with CO2 under the catalysis of Cr2O3. This effectively inhibits the deep hydrogenation of olefins and enhances the formation of aromatics. Similar coupling effects have also been revealed in the ZnZrOx/ZSM-5 catalytic system, wherein the aromatization is promoted by the metal oxide-assisted CO2 dehydrogenation. Additionally, in the modified Fischer–Tropsch route, a variety of catalysts, including Na–Fe@C/ZSM-5,89 Cu–Fe/ZSM-5,93 and CuFeO2/ZSM-5,37 have exhibited the coupling effect of the CO2-assisted dehydrogenation and aromatization. For example, Song et al. proposed a “H Recycling” mechanism, in which hydrogen species resulting from the aromatization reaction diffuse out of the zeolite and are promptly consumed by reacting with CO2 over the metal oxide, thereby promoting both the CO2 hydrogenation and aromatization reactions.93
5.2. Impact of intimacy or distance on the spatial coordination of catalytic components
The coupling of two catalytic components depends not only on the precise matching of bifunctional activities, but also on the spatial coordination of catalytic sites, which determines the transfer of intermediates between different catalytic sites. Weisz et al. investigated the effect of proximity between bifunctional catalytic components in a series of reactions, such as alkane isomerization and hydrocracking.111 In reactions involving stable intermediates, a shorter distance between catalytic sites facilitates the transfer of intermediates, and therefore the catalytic performance shows the characteristic of “the closer, the better”.Using the CO2-to-aromatics reaction system via the methanol-mediated route, Wang et al., Li et al., and Zhou et al. individually evaluated the catalytic performance by employing different combination manners of catalytic components, ranging from powder mixing at the nanometer scale to granule mixing at the micrometer scale and extending to dual-bed packing at the centimeter scale.55,60,62 Enhanced catalytic activity and aromatic selectivity are observed at a smaller mixing scale (Fig. 21A). Regarding the modified Fischer–Tropsch route, Song et al. also compared the catalytic performance of Cu–Fe/ZSM-5 with different coupling manners, revealing that the short diffusion path between the two components in the granule mixing manner accelerates the transfer and consumption of olefins and therefore promotes the thermodynamic equilibrium shift of the CO2 conversion reaction.93 However, further shortening the distance through powder mixing resulted in a decrease in catalytic activity while an increase in methane selectivity (Fig. 21B). Similarly, in other catalytic systems, such as Na–Fe@C/ZSM-5,89 Na–Cu–Fe2O3/ZSM-5,95 and NaZnFe/ZSM-5,100 the optimal activity and aromatic selectivity were also achieved at a moderate coupling scale of micrometers through granule mixing.
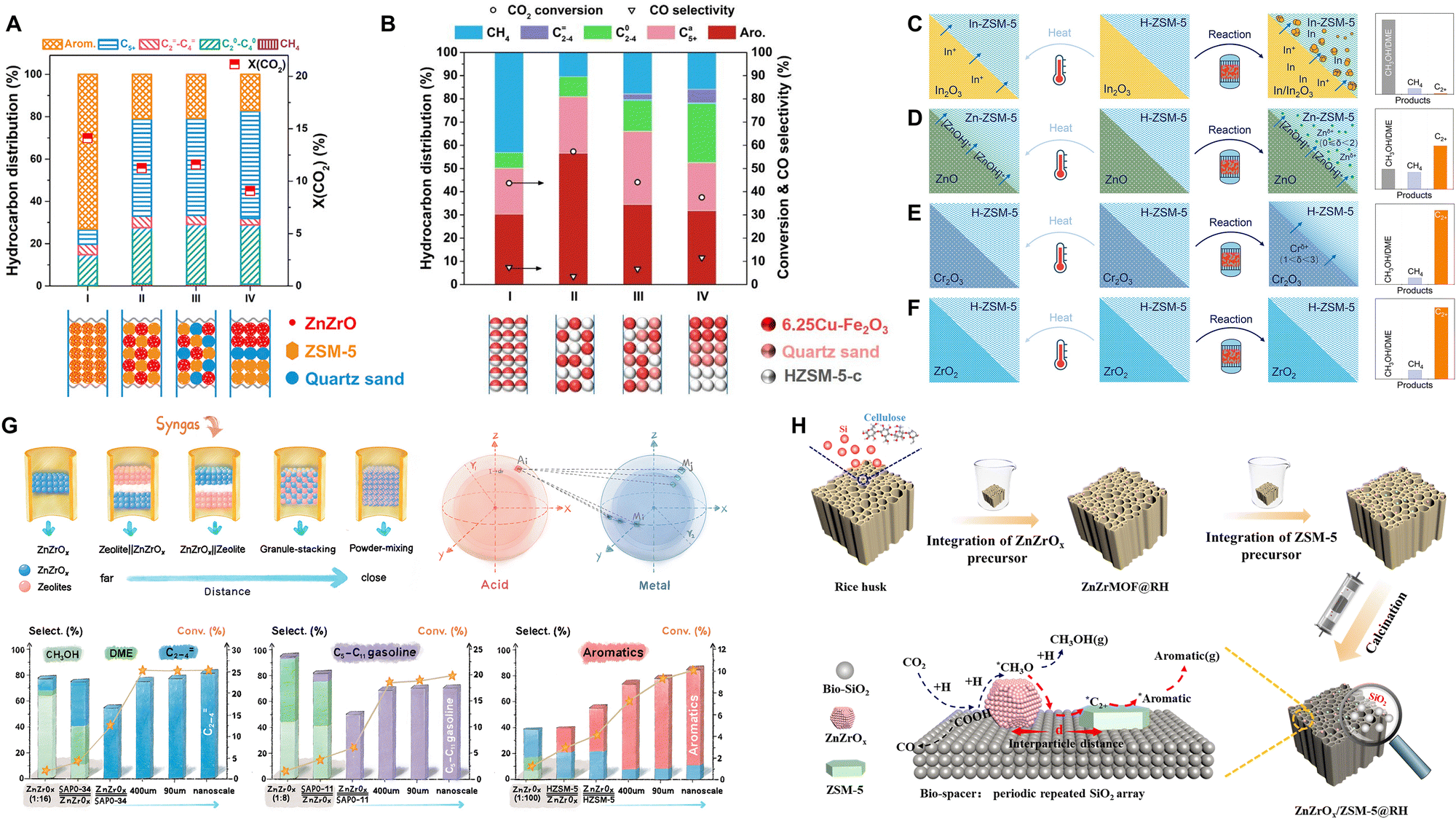 | ||
| Fig. 21 Effect of the coupling distance scale of bifunctional catalytic components in the CO2-to-aromatics reaction via (A) the methanol-mediated route and (B) the modified Fischer–Tropsch route. Reaction conditions: (A) ZnZrO/ZSM-5, 320 °C, 4.0 MPa, H2/CO2/Ar = 72/24/4, 1200 mL g−1 h−1; (B) 6.25Cu–Fe2O3/HZSM-5-c, 320 °C, 3.0 MPa, H2/CO2/N2 =72/24/4, 1000 mL g−1 h−1. Reprinted with permission from Li et al. Copyright (2019), Elsevier Inc.,60 and Song et al. Copyright (2020), American Chemical Society.93 The migration of metal species from metal oxides to zeolites driven by high-temperature treatment and reaction and its effect on the CO2 hydrogenation reaction: (C) In2O3/H-ZSM-5, (D) ZnO/H-ZSM-5, (E) Cr2O3/H-ZSM-5 and (F) ZrO2/H-ZSM-5. Reprinted with permission from Wang et al. Copyright (2021), Wiley-VCH GmbH.112 (G) Dependence of the distance between metal oxides and zeolites for reactions following the simple tandem or synergistic tandem mechanism. Reprinted with permission from Li et al. Copyright (2022), American Chemical Society.113 (H) Anchoring the metal oxide and zeolite components with the rice husk derived-SiO2. Reprinted with permission from Li et al. Copyright (2023), Elsevier B.V.114 | ||
5.3. Constructing the spatio-temporal coupling and directional spatial pathway for tandem reactions
Coupling catalysis is a strategy for achieving a multi-step reaction in a single process through the integration and synergy of multi-functional catalysts. Therefore, in tandem reactions such as CO2-to-aromatics, coupling catalysis involves spatio-temporal effects, manifesting as a temporal sequence of reactions across a spatial distribution of catalytic sites, with the transfer of intermediate species serving as the key bridge. Diffusion is governed by two crucial parameters: distance and direction. As mentioned hereinbefore, numerous studies have focused on regulating the diffusion distance of intermediate species by modulating the coupling intimacy between catalytic components, which ultimately influences the catalytic performance. However, the diffusion direction has received limited attention. The Xie and Wang group has reported advances in the direct conversion of CO/CO2 to aromatics based on the coupling catalysis of metal oxides and zeolites.79,116–118 Focusing on the oriented diffusion of intermediate species within the anisotropic pore structure of zeolites, they unveiled the spatial pathway for the diffusion and transformation of intermediates. Additionally, the directional spatial pathway was constructed by designing an orient-distributed metal oxide/zeolite bifunctional catalyst.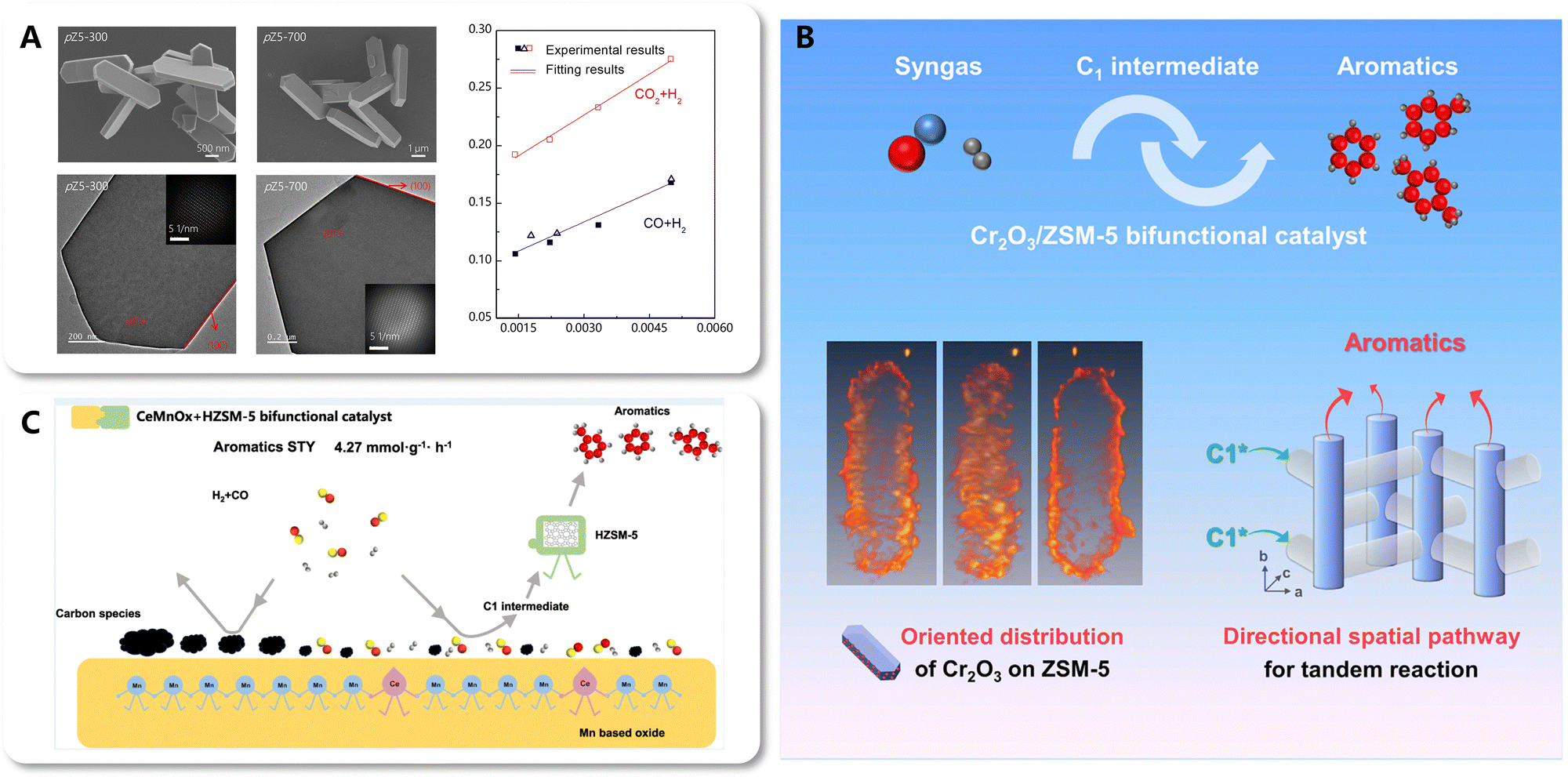 | ||
| Fig. 22 (A) The correlation between aromatic distribution and key morphology parameters. Reprinted and reproduced with permission from Liu et al. Copyright (2020), American Chemical Society.79 (B) directional spatial pathway based on orient-distributed metal oxide/zeolite catalysts. Reprinted with permission from Liu et al. Copyright (2021), Elsevier Inc.117 (C) Surface self-cleaning effect of CeMnOx boosting the efficiency of the spatial pathway for the tandem reaction. Reprinted with permission from Zhang et al. Copyright (2024), American Chemical Society.118 | ||
6. Conclusions and prospects
Aromatics are important products of the traditional petrochemical industry, possessing a substantial market demand. The development of the technologies for direct conversion of CO2 into aromatics aligns with the low-carbon transition of the chemical industry. Utilizing the strategy of coupling catalysis, two primary reaction pathways, specifically the methanol-mediated pathway and the modified Fischer–Tropsch pathway, have been intensively studied. The corresponding multi-functional catalysts, consisting of metal-based materials (metals, metal oxides or metal carbides) and zeolites, have achieved significant advancements, enabling the regulation and enhancement of CO2 conversion and aromatic selectivity through the matching and synergy between catalytic components. Furthermore, several novel coupling pathways have been developed by integrating CO2 hydrogenation with reactions such as CO hydrogenation, aromatic alkylation, or alkane aromatization. Nonetheless, further development of technologies for direct aromatic production from CO2 encounters three main challenges. First, the trade-off between CO2 conversion and aromatic selectivity: the consumption of “green hydrogen” accounts for the major cost of the direct production of aromatics from CO2, which is closely related to the product distribution. Precise selectivity control is therefore pivotal in enhancing the techno-economic viability of this process. Due to the complex reaction network, the methanol-mediated pathway exhibits high selectivity but relatively low activity, while the modified Fischer–Tropsch pathway is active yet demonstrates low selectivity to aromatics. The breakthrough of catalytic efficiency requires to overcome this trade-off effect. Second, the flexible and precise regulation of aromatic distribution: in the presence of the active intermediates, severe side reactions such as alkylation may arise and shift the aromatic distribution towards the heavy end, deviating from the market demand structure. Thus, it is essential to explore the flexible and precise regulation of aromatic distribution without compromising overall aromatic selectivity. Third, the scale-up engineering and industrial applications: in addition to the continuous deepening of understanding of catalytic mechanisms and structure–activity relationships, the iterative upgrading, scale-up engineering, and subsequent industrial applications have gained prominence, due to the increasing market demand and urgency for alternative aromatic production routes.The key lies in the clarification, construction, and optimization of the coupling reaction pathways. Future research should focus on the following aspects: (1) enhancing the understanding of reaction mechanisms, completing the reaction networks, elucidating the evolution of reaction species, in-depth characterization of catalytic activity sites, and subsequently exploring the quantitative influence of catalyst structural properties on tandem reactions; (2) developing environmentally friendly, efficient, and stable coupling catalytic systems through rational design and precise control of the location, distance, and orientation of the catalytic sites, thereby matching the adsorption and diffusion behaviors of intermediate species and boosting the tandem reaction efficiency through the directional spatial pathway; (3) expanding the coupling catalysis strategy and exploring novel coupling reaction pathways. In addition to the pathways discussed in this review, Wu et al. recently reported an FeMn&ZnCr&Z5@SiO2 catalyst for the conversion of CO in the presence of a low concentration of CO2.119 The enhancement in the space–time yield of PX is accomplished by coupling the methanol-mediated and Fischer–Tropsch pathways, representing a further exploration of the application of the coupling catalysis strategy in the aromatic production technology, using COx as the carbon source; (4) scale-up engineering issues including macro-scale fabrication of multi-functional catalysts, process design dealing with intense thermal effects and complicated product separation, along with exploration of technology integration involving CO2 capture and renewable hydrogen, ultimately aiming to provide novel technical solutions for efficient utilization of carbon resources and the advancement to a net-zero chemical industry. With the continuous development and expansion of the coupling strategy to drive the improvement of catalytic efficiency and product value, along with progress in renewable hydrogen production and carbon capture technology, the realm of CO2 conversion to aromatics will demonstrate renewed vigor and vitality.
Data availability
The data that support the results of our work, in Fig. 22A, are available from the corresponding authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the National Natural Science Foundation of China (22293025, 22293023 and U22B6011).References
- IEA ( 2024), CO2 Emissions in 2023, IEA, Paris https://www.iea.org/reports/co2-emissions-in-2023, Licence: CC BY 4.0.
- Z. Liu, Z. Deng, S. Davis and P. Ciais, Nat. Rev. Earth Environ., 2023, 4, 205–206 CrossRef PubMed.
- Y. Liu, D. Deng and X. Bao, Chem, 2020, 6(10), 2497–2514 CAS.
- D. Wang, Z. Xie, M. D. Porosoff and J. G. Chen, Chem, 2021, 7(9), 2277–2311 CAS.
- K. Cheng, Y. Li, J. Kang, Q. Zhang and Y. Wang, Acc. Chem. Res., 2024, 57(5), 714–725 CrossRef CAS PubMed.
- K. B. Tan, G. Zhan, D. Sun, J. Huang and Q. Li, J. Mater. Chem. A, 2021, 9, 5197–5231 RSC.
- G. Tian, X. Liang, H. Xiong, C. Zhang and F. Wei, EES Catal., 2023, 1, 677–686 RSC.
- K. B. Tan, K. Xu, D. Cai, J. Huang and G. Zhan, Chem. Eng. J., 2023, 463, 142262 CrossRef CAS.
- X. Lu, K. B. Tan, J. Zhao and G. Zhan, Chem. Catal., 2025, 5(3), 101264 CrossRef CAS.
- W. Wang, C. Zeng and N. Tsubaki, Green Carbon, 2023, 1(2), 133–145 CrossRef.
- W. Dai, L. Yang, G. Wu, N. Guan and L. Li, in Heterogeneous Catalysis for Sustainable Energy, ed. L. Li and J. S. J. Hargreaves, WILEY-VCH GmbH, Weinheim, 2022, ch. 10, pp. 351–388 Search PubMed.
- I. Yarulina, A. D. Chowdhury, F. Meirer, B. M. Weckhuysen and J. Gascon, Nat. Catal., 2018, 1(6), 398–411 CrossRef CAS.
- I. M. Dahl and S. Kolboe, J. Catal., 1996, 161(1), 304–309 CrossRef CAS.
- I. M. Dahl and S. Kolboe, J. Catal., 1994, 149(2), 458–464 CrossRef CAS.
- M. Bjørgen, S. Svelle, F. Joensen, J. Nerlov, S. Kolboe, F. Bonino, L. Palumbo and S. Bordiga, J. Catal., 2007, 249(2), 195–207 CrossRef.
- M. W. Erichsen, S. Svelle and U. Olsbye, Catal. Today, 2013, 215, 216–223 CrossRef.
- U. Olsbye, S. Svelle, M. Bjørgen, P. Beato, T. V. W. Janssens, F. Joensen, S. Bordiga and K. P. Lillerud, Angew. Chem., Int. Ed., 2012, 51(24), 5810–5831 CrossRef CAS PubMed.
- T. Sun, W. Chen, S. Xu, A. Zheng, X. Wu, S. Zeng, N. Wang, X. Meng, Y. Wei and Z. Liu, Chem, 2021, 7(9), 2415–2428 CAS.
- C. M. Wang, Y. D. Wang and Z. K. Xie, J. Catal., 2013, 301, 8–19 CrossRef CAS.
- W. Zhang, M. Zhang, S. Xu, S. Gao, Y. Wei and Z. Liu, ACS Catal., 2020, 10, 4510–4516 CrossRef CAS.
- A. Sassi, M. A. Wildman, H. J. Ahn, P. Prasad, J. B. Nicholas and J. F. Haw, J. Phys. Chem. B, 2002, 106(9), 2294–2303 CrossRef CAS.
- M. Bjørgen, U. Olsbye and S. Kolboe, J. Catal., 2003, 215(1), 30–44 CrossRef.
- M. Bjørgen, U. Olsbye, D. Petersen and S. Kolboe, J. Catal., 2004, 221(1), 1–10 CrossRef.
- B. Arstad and S. Kolboe, Catal. Lett., 2001, 71(3–4), 209–212 CrossRef CAS.
- B. Arstad and S. Kolboe, J. Am. Chem. Soc., 2001, 123(33), 8137–8138 CrossRef CAS PubMed.
- W. Song, J. F. Haw, J. B. Nicholas and C. S. Heneghan, J. Am. Chem. Soc., 2000, 122(43), 10726–10727 CrossRef CAS.
- S. Ilias and A. Bhan, ACS Catal., 2013, 3(1), 18–31 CrossRef CAS.
- P. Gao, J. Xu, G. Qi, C. Wang, Q. Wang, Y. Zhao, Y. Zhang, N. Feng, X. Zhao, J. Li and F. Deng, ACS Catal., 2018, 8(10), 9809–9820 CrossRef CAS.
- M. Conte, J. A. Lopez-Sanchez, Q. He, D. J. Morgan, Y. Ryabenkova, J. K. Bartley, A. F. Carley, S. H. Taylor, C. J. Kiely, K. Khalid and G. J. Hutchings, Catal. Sci. Technol., 2012, 2(1), 105–112 RSC.
- K. Cheng, W. Zhou, J. Kang, S. He, S. Shi, Q. Zhang, Y. Pan, W. Wen and Y. Wang, Chem, 2017, 3(2), 334–347 CAS.
- X. Yang, X. Su, B. Liang, Y. Zhang, H. Duan, J. Ma, Y. Huang and T. Zhang, Catal. Sci. Technol., 2018, 8(17), 4338–4348 RSC.
- D. Liu, L. Cao, G. Zhang, L. Zhao, J. Gao and C. Xu, Fuel Process. Technol., 2021, 216, 106770 CrossRef CAS.
- D. B. Lukyanov, N. S. Gnep and M. R. Guisnet, Ind. Eng. Chem. Res., 1995, 34, 516–523 CrossRef CAS.
- G. Caeiro, R. H. Carvalho, X. Wang, M. A. N. D. A. Lemos, F. Lemos, M. Guisnet and F. Ramôa Ribeiro, J. Mol. Catal. A:Chem., 2006, 255(1–2), 131–158 CrossRef CAS.
- S. Kasipandi and J. W. Bae, Adv. Mater., 2019, 31(34), e1803390 CrossRef PubMed.
- V. U. S. Rao and R. J. Gormley, Catal. Today, 1990, 6(3), 207–234 CrossRef.
- Y. Cheng, Y. Chen, S. Zhang, X. Wu, C. Chen, X. Shi, M. Qing, J. Li, C.-L. Liu and W.-S. Dong, Green Chem., 2023, 25(9), 3570–3584 RSC.
- W. Zhou, S. Shi, Y. Wang, L. Zhang, Y. Wang, G. Zhang, X. Min, K. Cheng, Q. Zhang, J. Kang and Y. Wang, ChemCatChem, 2019, 11(6), 1681–1688 CrossRef CAS.
- M. T. Arslan, B. A. Qureshi, S. Z. A. Gilani, D. Cai, Y. Ma, M. Usman, X. Chen, Y. Wang and F. Wei, ACS Catal., 2019, 9(3), 2203–2212 CrossRef CAS.
- P. Gao, S. Li, X. Bu, S. Dang, Z. Liu, H. Wang, L. Zhong, M. Qiu, C. Yang, J. Cai, W. Wei and Y. Sun, Nat. Chem., 2017, 9, 1019–1024 CrossRef CAS PubMed.
- Y. Ni, Z. Chen, Y. Fu, Y. Liu, W. Zhu and Z. Liu, Nat. Commun., 2018, 9(1), 3457 CrossRef PubMed.
- Y. Xu, D. Liu and X. Liu, Appl. Catal., A, 2018, 552, 168–183 CrossRef CAS.
- S. Guo, S. Fan, H. Wang, S. Wang, Z. Qin, M. Dong, W. Fan and J. Wang, ACS Catal., 2024, 14(1), 271–282 CrossRef CAS.
- L. Yang, C. Wang, W. Dai, G. Wu, N. Guan and L. Li, Fundam. Res., 2022, 2(2), 184–192 CrossRef CAS PubMed.
- M. T. Arslan, G. Tian, B. Ali, C. Zhang, H. Xiong, Z. Li, L. Luo, X. Chen and F. Wei, ACS Catal., 2022, 12(3), 2023–2033 CrossRef CAS.
- Y. Xu, J. Liu, J. Wang, G. Ma, J. Lin, Y. Yang, Y. Li, C. Zhang and M. Ding, ACS Catal., 2019, 9(6), 5147–5156 CrossRef CAS.
- Y. Xu, J. Wang, G. Ma, J. Lin and M. Ding, ACS Sustainable Chem. Eng., 2019, 7(21), 18125–18132 CrossRef CAS.
- B. Zhao, P. Zhai, P. Wang, J. Li, T. Li, M. Peng, M. Zhao, G. Hu, Y. Yang, Y.-W. Li, Q. Zhang, W. Fan and D. Ma, Chem, 2017, 3(2), 323–333 CAS.
- X. Cui, P. Gao, S. Li, C. Yang, Z. Liu, H. Wang, L. Zhong and Y. Sun, ACS Catal., 2019, 9(5), 3866–3876 CrossRef CAS.
- T. Li, T. Shoinkhorova, J. Gascon and J. Ruiz-Martínez, ACS Catal., 2021, 11(13), 7780–7819 CrossRef CAS.
- C. Wang, M. Hu, Y. Chu, X. Zhou, Q. Wang, G. Qi, S. Li, J. Xu and F. Deng, Angew. Chem., Int. Ed., 2020, 59(18), 7198–7202 CrossRef CAS PubMed.
- A. Hwang and A. Bhan, Acc. Chem. Res., 2019, 52(9), 2647–2656 CrossRef CAS PubMed.
- T. Wang, C. Yang, P. Gao, S. Zhou, S. Li, H. Wang and Y. Sun, Appl. Catal., B, 2021, 286, 119929 CrossRef CAS.
- Y. Wang, W. Gao, S. Kazumi, H. Li, G. Yang and N. Tsubaki, Chem. - Eur. J., 2019, 25(20), 5149–5153 CrossRef CAS PubMed.
- Y. Wang, L. Tan, M. Tan, P. Zhang, Y. Fang, Y. Yoneyama, G. Yang and N. Tsubaki, ACS Catal., 2019, 9(2), 895–901 CrossRef CAS.
- L. Zhang, W. Gao, F. Wang, C. Wang, J. Liang, X. Guo, Y. He, G. Yang and N. Tsubaki, Appl. Catal., B, 2023, 328, 122535 CrossRef CAS.
- K. Cheng, B. Gu, X. Liu, J. Kang, Q. Zhang and Y. Wang, Angew. Chem., Int. Ed., 2016, 55(15), 4725–4728 CrossRef CAS PubMed.
- J. Zhang, M. Zhang, S. Chen, X. Wang, Z. Zhou, Y. Wu, T. Zhang, G. Yang, Y. Han and Y. Tan, Chem. Commun., 2019, 55, 973–976 RSC.
- X. Zhang, A. Zhang, X. Jiang, J. Zhu, J. Liu, J. Li, G. Zhang, C. Song and X. Guo, J. CO2 Util., 2019, 29, 140–145 CrossRef CAS.
- Z. Li, Y. Qu, J. Wang, H. Liu, M. Li, S. Miao and C. Li, Joule, 2019, 3(2), 570–583 CrossRef CAS.
- Y. Qu, Z. Li, H. Hu, S. Chen, J. Wang and C. Li, Chem. Commun., 2023, 59, 7607–7610 RSC.
- C. Zhou, J. Shi, W. Zhou, K. Cheng, Q. Zhang, J. Kang and Y. Wang, ACS Catal., 2020, 10(1), 302–310 CrossRef CAS.
- H. Tian, P. Gao, X. Yang, C. Jiao, F. Zha, Y. Chang and H. Chen, Chem. Eng. J., 2023, 466, 143267 CrossRef CAS.
- B. Liu, Y. Wang, Y. Xie, L. Xiao, W. Wang and W. Wu, ACS Sustainable Chem. Eng., 2023, 11(49), 17340–17354 CrossRef CAS.
- Y. Yue, J. Tian, J. Ma, S. Yang, W. Li, J. Huang, Q. Li and G. Zhan, Appl. Catal., B, 2024, 355, 124158 CrossRef CAS.
- E. Giamello, B. Fubini, M. Bertoldi, G. Busca and A. Vaccari, J. Chem. Soc., Faraday Trans., 1989, 85(2), 237–249 RSC.
- M. Bertoldi, B. Fubini, E. Giamello, G. Busca, F. Trifirò and A. Vaccari, J. Chem. Soc., Faraday Trans., 1988, 84(5), 1405–1421 RSC.
- G. D. Piero, F. Trifiro and A. Vaccari, J. Chem. Soc., Chem. Commun., 1984, 10, 656–658 RSC.
- L. Tan, G. Yang, Y. Yoneyama, Y. Kou, Y. Tan, T. Vitidsant and N. Tsubaki, Appl. Catal., A, 2015, 505, 141–149 CrossRef CAS.
- S. Tian, S. Wang, Y. Wu, J. Gao, Y. Bai, P. Wang, H. Xie, Y. Han and Y. Tan, J. Mol. Catal. A:Chem., 2015, 404–405, 139–147 CrossRef CAS.
- Q. Zhang, X. Li and K. Fujimoto, Appl. Catal., A, 2006, 309(1), 28–32 CrossRef CAS.
- H. Song, D. Laudenschleger, J. J. Carey, H. Ruland, M. Nolan and M. Muhler, ACS Catal., 2017, 7(11), 7610–7622 CrossRef CAS.
- M. C. J. Bradford, M. V. Konduru and D. X. Fuentes, Fuel Process. Technol., 2003, 83(1–3), 11–25 CrossRef CAS.
- J. Wang, G. Li, Z. Li, C. Tang, Z. Feng, H. An, H. Liu, T. Liu and C. Li, Sci. Adv., 2017, 3, e1701290 CrossRef PubMed.
- M. V. Frash and R. A. van Santen, Phys. Chem. Chem. Phys., 2000, 2(5), 1085–1089 RSC.
- U. Lohse, B. Parlitz and V. Patzelovi, J. Chem. Phys., 1989, 93(9), 3677–3683 CrossRef CAS.
- N. O. Gonzales, A. T. Bell and A. K. Chakraborty, J. Phys. Chem. B, 1997, 101(48), 10058–10064 CrossRef CAS.
- P. Zhang, L. Tan, G. Yang and N. Tsubaki, Chem. Sci., 2017, 8(12), 7941–7946 RSC.
- C. Liu, J. Su, S. Liu, H. Zhou, X. Yuan, Y. Ye, Y. Wang, W. Jiao, L. Zhang, Y. Lu, Y. Wang, H. He and Z. Xie, ACS Catal., 2020, 10(24), 15227–15237 CrossRef CAS.
- W. Zhou, K. Cheng, J. Kang, C. Zhou, V. Subramanian, Q. Zhang and Y. Wang, Chem. Soc. Rev., 2019, 48(12), 3193–3228 RSC.
- C. Yang, H. Zhao, Y. Hou and D. Ma, J. Am. Chem. Soc., 2012, 134(38), 15814–15821 CrossRef CAS PubMed.
- T. H. Pham, Y. Qi, J. Yang, X. Duan, G. Qian, X. Zhou, D. Chen and W. Yuan, ACS Catal., 2015, 5(4), 2203–2208 CrossRef CAS.
- M. E. Dry, J. Chem. Technol. Biotechnol., 2001, 77, 43–50 CrossRef.
- F. Jiao, J. Li, X. Pan, J. Xiao, H. Li, H. Ma, M. Wei, Y. Pan, Z. Zhou, M. Li, S. Miao, J. Li, Y. Zhu, D. Xiao, T. He, J. Yang, F. Qi, Q. Fu and X. Bao, Science, 2016, 351(6277), 1065–1068 CrossRef CAS PubMed.
- J. Zhu, P. Wang, X. Zhang, G. Zhang, R. Li, W. Li, T. P. Senftle, W. Liu, J. Wang, Y. Wang, A. Zhang, Q. Fu, C. Song and X. Guo, Sci. Adv., 2022, 8(5), eabm3629 CrossRef CAS PubMed.
- B. Yao, T. Xiao, O. A. Makgae, X. Jie, S. Gonzalez-Cortes, S. Guan, A. I. Kirkland, J. R. Dilworth, H. A. Al-Megren, S. M. Alshihri, P. J. Dobson, G. P. Owen, J. M. Thomas and P. P. Edwards, Nat. Commun., 2020, 11(1), 6395 CrossRef CAS PubMed.
- J. Wei, Q. Ge, R. Yao, Z. Wen, C. Fang, L. Guo, H. Xu and J. Sun, Nat. Commun., 2017, 8(1), 15174 CrossRef PubMed.
- Y. Xu, C. Shi, B. Liu, T. Wang, J. Zheng, W. Li, D. Liu and X. Liu, Catal. Sci. Technol., 2019, 9(3), 593–610 RSC.
- Y. Wang, S. Kazumi, W. Gao, X. Gao, H. Li, X. Guo, Y. Yoneyama, G. Yang and N. Tsubaki, Appl. Catal., B, 2020, 269, 118792 CrossRef CAS.
- Y. Gu, J. Liang, Y. Wang, K. Huo, M. Li, W. Wang, R. He, S. Yasuda, X. Gao, G. Yang, M. Wu and N. Tsubaki, Appl. Catal., B, 2024, 349, 123842 CrossRef CAS.
- J. Wei, R. Yao, Q. Ge, D. Xu, C. Fang, J. Zhang, H. Xu and J. Sun, Appl. Catal., B, 2021, 283, 119648 CrossRef CAS.
- A. Ramirez, A. D. Chowdhury, A. Dokania, P. Cnudde, M. Caglayan, I. Yarulina, E. Abou-Hamad, L. Gevers, S. Ould-Chikh, K. D. Wispelaere, V. van Speybroeck and J. Gascon, ACS Catal., 2019, 9(7), 6320–6334 CrossRef CAS.
- G. Song, M. Li, P. Yan, M. A. Nawaz and D. Liu, ACS Catal., 2020, 10(19), 11268–11279 CrossRef CAS.
- G. Song, M. Li, L. Xu, X. Yang, M. A. Nawaz, H. Yuan, Z. Zhang, X. Xu and D. Liu, Ind. Eng. Chem. Res., 2022, 61(20), 6820–6830 CrossRef CAS.
- X. Yang, G. Song, M. Li, C. Chen, Z. Wang, H. Yuan, Z. Zhang and D. Liu, Ind. Eng. Chem. Res., 2022, 61(23), 7787–7798 CrossRef CAS.
- C. Chen, G. Song, Z. Wang, J. Song, Q. Jiang, Y. Zhai and D. Liu, Appl. Catal., B, 2024, 341, 123330 CrossRef CAS.
- C. Chen, X. Li, Z. Wang, J. Song and D. Liu, Chem. Eng. J., 2024, 483, 149181 CrossRef CAS.
- G. Song, Q. Jiang, Y. Zhai and D. Liu, Chem. Eng. Sci., 2023, 280, 119037 CrossRef CAS.
- G. Song, Y. Zhai, Q. Jiang and D. Liu, Fuel, 2023, 338, 127185 CrossRef CAS.
- Q. Jiang, G. Song, Y. Zhai, C. Chen, Z. Wang, H. Yuan, Z. Zhang and D. Liu, Ind. Eng. Chem. Res., 2023, 62(23), 9188–9200 CrossRef CAS.
- M. G. Sibi, M. K. Khan, D. Verma, W. Yoon and J. Kim, Appl. Catal., B, 2022, 301, 120813 CrossRef CAS.
- G. Tian, Z. Li, C. Zhang, X. Liu, X. Fan, K. Shen, H. Meng, N. Wang, H. Xiong, M. Zhao, X. Liang, L. Luo, L. Zhang, B. Yan, X. Chen, H.-J. Peng and F. Wei, Nat. Commun., 2024, 15(1), 3037 CrossRef CAS PubMed.
- J. Zuo, W. Chen, J. Liu, X. Duan, L. Ye and Y. Yuan, Sci. Adv., 2020, 6(34), eaba5433 CrossRef CAS PubMed.
- Y. Lai, B. Hong, W. Zhou, D. Wen, Y. Xie, F. Luo, L. Ye, J. Zuo and Y. Yuan, ACS Catal., 2024, 14(15), 11780–11793 CrossRef CAS.
- R. Cao, T. Fu, Y. Liu, W. Qin, Y. Guo, C. Li, S. Huang and Z. Li, ACS Catal., 2024, 14(16), 12016–12030 CrossRef CAS.
- D. Miao, X. Pan, F. Jiao, Y. Ji, G. Hou, L. Xu and X. Bao, Catal. Sci. Technol., 2021, 11(13), 4521–4528 RSC.
- C. Wei, W. Zhang, K. Yang, X. Bai, S. Xu, J. Li and Z. Liu, Chinese J. Catal., 2023, 47, 138–149 CrossRef CAS.
- J. Zuo, C. Liu, X. Han, D. Wen, X. Liu, L. Ye, W. Zhuang and Y. Yuan, Chem. Catal., 2022, 2(5), 1223–1240 CrossRef CAS.
- K. Bu, Y. Kang, Y. Li, Y. Zhang, Y. Tang, Z. Huang, W. Shen and H. Xu, Appl. Catal., B, 2024, 343, 123528 CrossRef CAS.
- W. Chen, Y. Jiao, Y. Liu, M. Wang, F. Zhang and D. Ma, CCS Chem., 2024, 6, 1422–1429 CrossRef CAS.
- P. B. Weisz, Adv. Catal., 1962, 13, 137–190 CAS.
- Y. Wang, G. Wang, L. I. van der Waals, K. Cheng, Q. Zhang, K. P. de Jong and Y. Wang, Angew. Chem., Int. Ed., 2021, 60(32), 17735–17743 CrossRef CAS PubMed.
- Y. Li, M. Wang, S. Liu, F. Wu, Q. Zhang, S. Zhang, K. Cheng and Y. Wang, ACS Catal., 2022, 12(15), 8793–8801 CrossRef CAS.
- W. Li, G. Zhan, X. Liu, Y. Yue, K. B. Tan, J. Wang, J. Huang and Q. Li, Appl. Catal., B, 2023, 330, 122575 CrossRef CAS.
- Y. Ding, F. Jiao, X. Pan, Y. Ji, M. Li, R. Si, Y. Pan, G. Hou and X. Bao, ACS Catal., 2021, 11(15), 9729–9737 CrossRef CAS.
- C. Liu, S. Liu, H. Zhou, J. Su, W. Jiao, L. Zhang, Y. Wang, H. He and Z. Xie, Appl. Catal., A, 2019, 585, 117206 CrossRef.
- C. Liu, J. Su, Y. Xiao, J. Zhou, S. Liu, H. Zhou, Y. Ye, Y. Lu, Y. Zhang, W. Jiao, L. Zhang, Y. Wang, C. Wang, X. Zheng and Z. Xie, Chem. Catal., 2021, 1(4), 896–907 CrossRef CAS.
- L. Zhang, J. Su, C. Liu, S. Liu, H. Zhou, W. Jiao, Y. Hu, F. Xiong, Y. Lu, Y. Ye, X. Zheng, Y. Zhang, Y. Wang and H. He, ACS Catal., 2024, 14(11), 8972–8982 CrossRef CAS.
- X. Wu, C. Wang, S. Zhao, Y. Wang, T. Zhang, J. Yao, W. Gao, B. Zhang, T. Arakawa, Y. He, F. Chen, M. Tan, G. Yang and N. Tsubaki, Nat. Commun., 2024, 15(1), 8064 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |