
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced activity and stability of polymeric carbon nitride photoanodes by yttrium incorporation†
Sanjit Mondala,
Ayelet Tashakorya,
Gabriel Marka,
Shmuel Barzilaib,
Angus Pedersenc,
Michael Volokh a,
Josep Alberod,
Hermenegildo Garcíad and
Menny Shalom*a
a,
Josep Alberod,
Hermenegildo Garcíad and
Menny Shalom*a
aDepartment of Chemistry and Ilse Katz Institute for Nanoscale Science and Technology, Ben-Gurion University of the Negev, Beer-Sheva 8410501, Israel. E-mail: mennysh@bgu.ac.il
bDepartment of Chemistry Nuclear Research, Centre-Negev P.O. Box 9001, Beer-Sheva 84910, Israel
cDepartment of Chemical Engineering, Imperial College London, London SW7 2AZ, England
dInstituto Universitario de Tecnología Química CSIC-UPV, Universitat Politècnica de València, València 46022, Spain
First published on 17th April 2025
Abstract
Polymeric carbon nitride materials (CNs) show promising potential as photoanodes in water-splitting photoelectrochemical cells. However, poor catalytic activity at the electrode–water interface limits their performance and longevity, resulting in low photoactivity and unwanted self-oxidation. Here, we demonstrate a high-performance photoanode based on polymeric carbon nitride doped with yttrium clusters, achieving enhanced activity and stability with high faradaic efficiency for water oxidation. Incorporating yttrium clusters enhances light harvesting, electronic conductivity, charge separation, and hole extraction kinetics, enabling efficient water oxidation. Furthermore, the strong interaction between yttrium and the CN's nitrogen groups guides the formation of yttrium-rich one-dimensional tubular structures that interconnect two-dimensional CN sheets. The optimized photoanode delivers a photocurrent density of 275 ± 10 μA cm−2 with 90% faradaic efficiency for oxygen evolution, demonstrates stable performance for up to 10 hours, and achieves external quantum efficiencies of up to 14% in an alkaline medium.
Broader contextMetal-free polymeric carbon nitride (CN) represents a promising photoanode material for water-splitting photoelectrochemical cells in neutral to alkaline media, addressing standing challenges in renewable hydrogen production. This work introduces a new strategy for oxygen evolution co-catalyst incorporation with significant improvements in catalytic activity and long-term stability compared to traditional CN photoanode designs. By incorporating yttrium dopants into the carbon nitride matrix, the photoanode exhibits enhanced charge transfer dynamics, improved light absorption characteristics, and resistance to self-degradation. These structural modifications resulted in high faradaic efficiency for oxygen evolution and extended operational longevity. |
Introduction
Photoelectrochemical cells (PEC), which convert solar energy into hydrogen, are promising for sustainable energy production.1–6 Among the various photoanode materials explored for PEC, n-type materials from the polymeric carbon nitride family (CN) have gained significant attention due to their favorable properties, such as high stability, cost-effectiveness, tunable electronic and catalytic characteristics, eco-friendliness, and appropriate energy band positions.7–14 Despite significant advancements in developing CN photoanodes,7,15 their PEC performance is limited by low charge separation and transfer efficiency, slow water oxidation kinetics, and fast charge-carrier recombination rates.12,16–20 Additionally, a significant drawback of CN photoanodes is their tendency for partial self-oxidation during operation, leading to low faradaic efficiency (FE) and oxygen production rates.16,21 These issues and the photoactive CN layer's instability pose significant obstacles to the long-term use of CN-based water-splitting PECs. Various strategies have been developed to address these challenges, including doping with different elements, nanostructuring of the CN films, and (nanoscale) co-catalyst deposition.22,23 For instance, incorporating metal-based co-catalyst nanoparticles (e.g., Ni, Fe, Co) on the surface of CN photoanodes leads to improved charge separation and transfer efficiency and water oxidation kinetics.24In this context, loading CN with rare-earth metal clusters—such as yttrium—as co-catalysts to facilitate water oxidation offers distinct advantages. As previously shown for other semiconductors, these clusters can enhance charge transfer due to their strong interaction with nitrogen-rich surfaces and introduce active catalytic sites for water oxidation by stabilizing key reaction intermediates.25 Furthermore, Y-clusters and their oxide can contribute to the durability of photoanodes, serving as a protective layer against oxidative degradation.26,27
Herein, we present the synthesis of a yttrium-doped CN photoanode with high photoelectrochemical activity and stability during water oxidation in an alkaline medium. Through detailed structural, spectroscopic, photoelectrochemical, and theoretical investigation, we demonstrate that yttrium incorporation enhances light-harvesting, electronic conductivity within the CN framework, exciton separation, hole extraction kinetics, and water oxidation efficiency. Y incorporation introduces a unique structural feature—2D sheet-like CN structures with 1D yttrium-rich tubular CN. The optimized yttrium-loaded CN-based film achieves a photocurrent density of 275 ± 10 μA cm−2 with a 90% faradaic efficiency for oxygen evolution, stable performance for up to 10 hours, and incident photon-to-current efficiency (IPCE) values reaching 14% at 390 nm.
Experimental
Synthesis of Y-incorporated supramolecular precursor and polymeric carbon nitride films
Yttrium clusters modify a porous film of CN with embedded reduced graphene oxide over fluorine-doped tin oxide-coated glass (FTO) substrates (abbreviated simply as CNGO). The Y-cluster-modified CNGO photoactive electrodes were synthesized to act as the photoanodes in a water-splitting PEC device. Initially, melamine (10 g) and yttrium acetate (in three distinct yttrium loadings: utilizing 0.25, 0.50, or 0.75 mmol Y) were dispersed in an ethanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water mixture (1:1 volume ratio). This mixture was agitated at 300 rpm and 55 °C for 8 h in an open beaker on a hot plate. Mixing this precursor with a graphene oxide dispersion and ethylene glycol formed a viscous paste, which was then doctor-bladed onto the substrate.28,29 Subsequent calcination under a nitrogen atmosphere at 550 °C for 4 hours yielded a film of porous nanosheets interconnected with twisted nanotubes over FTO, as shown in Scheme 1 (see details in the ESI†), to serve as photoanodes. For comparison, reference CNGO films (i.e., without Y) were prepared using identical conditions without treating melamine with yttrium salts (based on our previous works;23,28 see Experimental details in the ESI†).
:water mixture (1:1 volume ratio). This mixture was agitated at 300 rpm and 55 °C for 8 h in an open beaker on a hot plate. Mixing this precursor with a graphene oxide dispersion and ethylene glycol formed a viscous paste, which was then doctor-bladed onto the substrate.28,29 Subsequent calcination under a nitrogen atmosphere at 550 °C for 4 hours yielded a film of porous nanosheets interconnected with twisted nanotubes over FTO, as shown in Scheme 1 (see details in the ESI†), to serve as photoanodes. For comparison, reference CNGO films (i.e., without Y) were prepared using identical conditions without treating melamine with yttrium salts (based on our previous works;23,28 see Experimental details in the ESI†).
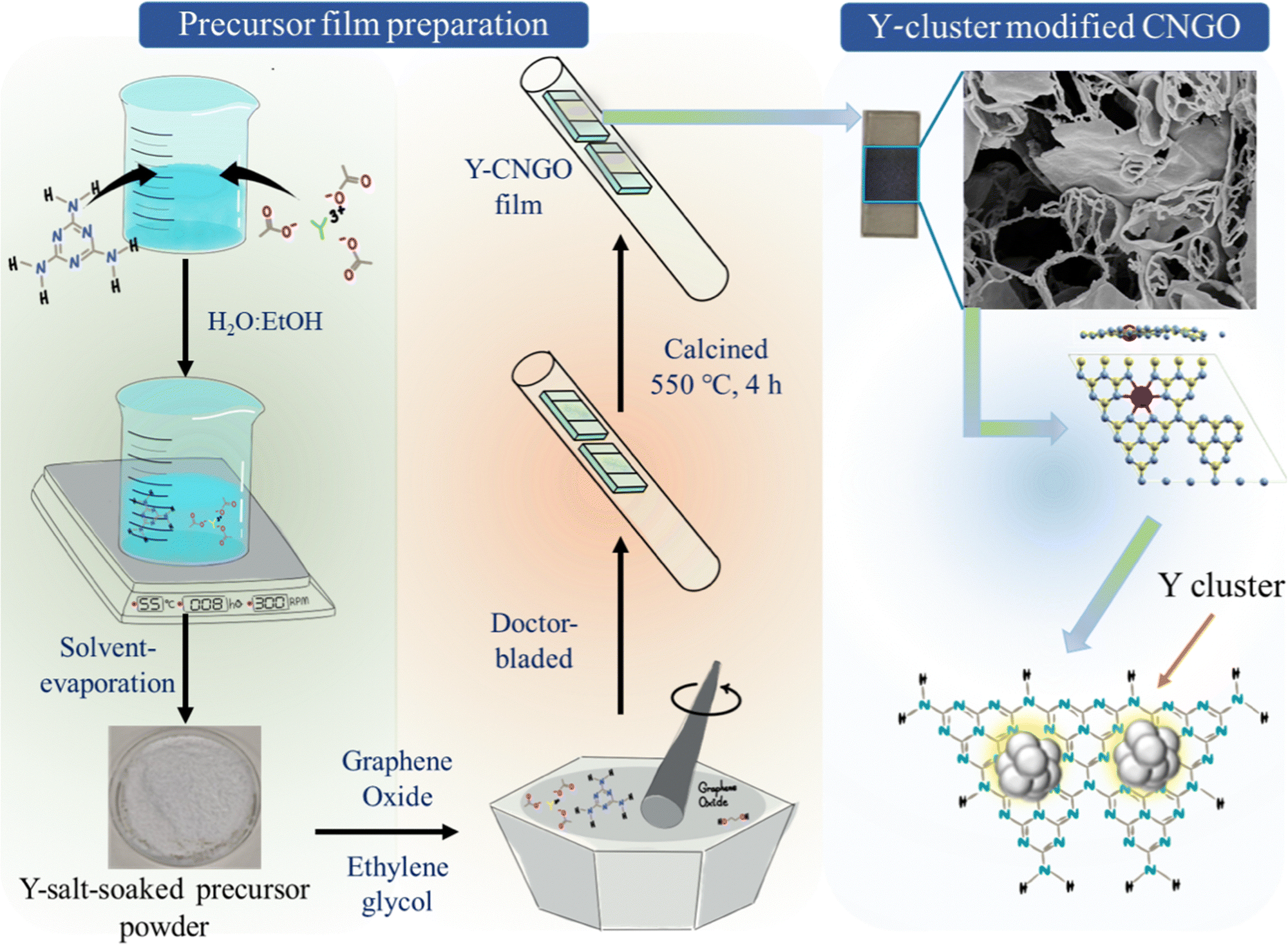 | ||
| Scheme 1 The fabrication route towards Y-cluster-modified CNGO films on FTO substrates. | ||
Results and discussion
The structural and functional characteristics of the prepared films were examined using X-ray diffraction (XRD), Fourier-transform infrared spectroscopy (FTIR), and X-ray photoelectron spectroscopy (XPS). Fig. 1a shows the XRD pattern of bare CNGO, and Y-cluster incorporated CNGO films on FTO, revealing a slight shift (0.12°) in the 2θ value of the characteristic diffraction of CN's (002) plane towards higher angles upon Y incorporation. This shift could result from distortion in the CN lattice or multiple layers in the formed CN tube within the film (as discussed later) caused by the yttrium introduction. The FTIR spectra (Fig. S1, ESI†) display typical CN stretching vibrations, with no significant changes observed upon Y incorporation. | ||
| Fig. 1 Characterization of CNGO and Y-cluster-modified CNGO films. (a) XRD. High-resolution XPS analysis: (b) N 1s, (c) Y 3d. (d) Valence band (VB) XPS (CNGO and CNGO-0.50 Y). (e) UV-vis DRS. (f) Schematic representation of the electronic band structure of CNGO and CNGO-0.50 Y films (on the normal hydrogen electrode (NHE) scale), determined using the VB position obtained from XPS measurement and the optical Eg calculation. | ||
XPS survey spectra (Fig. S2a, ESI†) confirm the successful incorporation of Y into the CNGO, showing the presence of C, N, and Y in Y-CNGO films. The high-resolution C 1s spectrum (Fig. S2b, ESI†) can be deconvoluted into three peaks centered at binding energies of 284.8, 286.4, and 288.1 eV; we attribute these peaks to C–C–H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–C, and C–(N)3, respectively.30–32 The amount of CN–C species slightly increased after Y was incorporated into the CN framework. The N 1s spectrum consists of three peaks at binding energies of 398.93, 400.21, and 401.22 eV, which are assigned to the pyridinic N, –NH, and quaternary N, respectively (Fig. 1b).33–36 The apparent shift to a lower binding energy (by 0.22 eV) of the pyridinic nitrogen after Y incorporation (compared to bare CNGO) suggests at least a partial electron cloud transfer from the incorporated Y to the CN's pyridinic N.37 The Y 3d spectrum of the CNGO-0.50 Y in Fig. 1c consists of two peaks with binding energies of 158.16 and 160.26 eV, which are attributed to the 3d5/2 and 3d3/2, respectively, of Yδ+ (positively-charged yttrium, see discussion later) bound to an electronegative atom, most probably N in this case, though O38,39 cannot be ruled out. High-resolution Y 3d spectra of the three different Y-incorporated films are provided in Fig. S2c (ESI†), showing no significant change.
N–C, and C–(N)3, respectively.30–32 The amount of CN–C species slightly increased after Y was incorporated into the CN framework. The N 1s spectrum consists of three peaks at binding energies of 398.93, 400.21, and 401.22 eV, which are assigned to the pyridinic N, –NH, and quaternary N, respectively (Fig. 1b).33–36 The apparent shift to a lower binding energy (by 0.22 eV) of the pyridinic nitrogen after Y incorporation (compared to bare CNGO) suggests at least a partial electron cloud transfer from the incorporated Y to the CN's pyridinic N.37 The Y 3d spectrum of the CNGO-0.50 Y in Fig. 1c consists of two peaks with binding energies of 158.16 and 160.26 eV, which are attributed to the 3d5/2 and 3d3/2, respectively, of Yδ+ (positively-charged yttrium, see discussion later) bound to an electronegative atom, most probably N in this case, though O38,39 cannot be ruled out. High-resolution Y 3d spectra of the three different Y-incorporated films are provided in Fig. S2c (ESI†), showing no significant change.
Valence band (VB) XPS reveals that the CNGO-0.50 Y films have a more positive VB energy (EVB) position of 1.61 V versus (vs.) normal hydrogen electrode (NHE), compared to 1.50 V for the bare CNGO (Fig. 1d and Note S1 in the ESI†), providing a better thermodynamic driving force for the oxygen evolution reaction (OER). Diffuse reflectance spectroscopy (DRS) of the films in the UV-vis region, presented in Fig. 1e, indicates a red-shift in absorption following Y incorporation at the band edge and the presence of another optical transition ca. 500 nm. The direct optical bandgaps (Eg) of the bare CNGO and CNGO-0.50 Y films are 2.86 eV and 2.82 eV, respectively (Fig. S3, ESI†). Based on the valence band XPS and Eg obtained from the Tauc plot analysis, a schematic of the band positions for the bare film vs. Y-incorporated one is presented in Fig. 1f.
The shape and morphology of bare and Y-cluster-modified CNGO were investigated using electron microscopy. Fig. 2a presents a top-view scanning electron microscopy (SEM) image of a CNGO film, which reveals porous CN nanosheets.28 Fig. 2b and c displays images of the CNGO-0.50 Y film, highlighting a porous structure with two distinct primary morphologies: 2D nanosheets and 1D tubular structures. The 2D nanosheets are interconnected by the tubular 1D structures located at their edges. The morphology of bare CNGO and CNGO films with varying yttrium loading is shown in Fig. S4–S6 (ESI†), indicating that higher yttrium incorporation leads to increased presence of the observed tubular 1D structures.
 | ||
| Fig. 2 (a) SEM image showing the morphology of a bare CNGO film. (b) and (c) Top-view SEM images of the surface of a CNGO-0.50 Y film. (d) Cross-sectional SEM images of a CNGO-0.50 Y film. (e)–(g) STEM images of a CNGO-0.50 Y sample at different magnifications. (h) and (i) HAADF-STEM images of the CNGO-0.50 Y sample; the yellow dashed circles in panel (i) highlight the Y-clusters. (j) HAADF-STEM image and the corresponding EDS mapping of a CNGO-0.50 Y sample at two different spots, focusing (1) on the tubular part or (2) on the nanosheet area, showing the distributions of C (green), N (blue), O (yellow), and Y (red). (k) SAED pattern of a CNGO-0.50 Y sample (left—low magnification TEM; right—TEM at higher magnification—showing the two distinct spots used for the SAED analysis): (1) the tubular part and (2) the nanosheet area. | ||
Several control experiments were performed to understand the origin of the tubular structures in the film. First, a graphene oxide (GO) film alone on FTO was prepared at identical calcination conditions, revealing 2D layered structures (Fig. S7a, ESI†). Similarly, a film from GO and a Y precursor (yttrium acetate) resulted in a morphology similar to that of GO alone (Fig. S7b, ESI†). In a separate experiment, a pristine CN film was prepared using only melamine (i.e., a CN precursor without adding GO), resulting in a bulk aggregated morphology on FTO (Fig. S7c, ESI†). When the doctor-bladed paste consisted of a melamine–yttrium precursor, porous nanosheets with tubular CN structures at the edges were formed (Fig. S7d, ESI†). However, the nanosheet morphology was less pronounced than in the CNGO-Y-cluster films. From these control experiments, we deduce that the tubular 1D structures originate from the interaction between melamine and the yttrium acetate during thermal polymerization.
The SEM cross-sectional image of the CNGO-0.50 Y film shows that tubular structures connect the porous nanosheets throughout the film, which is well-attached to the FTO with a thickness of approximately 50–60 μm (Fig. 2d). For comparison, cross-sectional images of a bare CNGO film over FTO are shown in Fig. S8 (ESI†). Scanning transmission electron microscopy (STEM) images in Fig. 2e–g of the CNGO-0.50 Y sample demonstrate the thin nanosheets with 1D tubular structures at their edges. High-angle annular dark-field (HAADF)-STEM images of the CNGO-0.50 Y sample (Fig. 2h and i) reveal small Y-based particles (2–4 nm), suggesting the presence of Y-clusters. Fig. 2j displays energy-dispersive X-ray spectroscopy (EDS) mapping and HAADF-STEM images of a CNGO-0.50 Y sample at two different spots, focusing on the tubular part and the nanosheet area, showing the distribution of C, N, O, and Y. The yttrium is uniformly distributed throughout the nanosheets, with a higher concentration in the tubular parts than in the nanosheets. Fig. 2k shows the selected area electron diffraction (SAED) pattern of the CNGO-0.50 Y sample at two different spots, focusing on (1) the tubular part and (2) the nanosheet area. The arc nature in the SAED pattern from the tubular area indicates multiple layers corresponding to the 002 plane (d002 = 0.32 nm) of CN, while the ring pattern from the nanosheet corresponds to a polycrystalline phase.
Photoelectrochemical water oxidation performance
The photoelectrochemical water oxidation activity of bare CNGO and CNGO modified with Y-clusters as co-catalysts (i.e., Y-CNGO) was evaluated in 0.10 M KOH aqueous electrolyte using a custom three-electrode cell at 1.23 VRHE (where VRHE represents the voltage in units of V vs. the reversible hydrogen electrode (RHE) scale) under 1-sun illumination. Fig. 3a displays the linear sweep voltammetry (LSV) curves in the dark and under illumination, illustrating a typical PEC behavior with low onset potentials of 0.35 and 0.27 VRHE, respectively, suggesting that the photocurrent initiates approximately 1 V below the thermodynamic OER potential in the dark (1.23 VRHE). The Y-CNGO film demonstrated a significant enhancement in its photoresponse compared to bare CNGO, achieving a stable photocurrent density of 275 ± 10 μA cm−2 at 1.23 VRHE, as evidenced by the chronoamperometric measurements (Fig. 3b and Fig. S9, ESI†). | ||
| Fig. 3 Photoelectrochemical characterization of bare CNGO compared to Y-CNGO photoanodes. (a) Linear sweep voltammetry (LSV) in the dark and under 1 sun illumination in 0.10 M KOH, pH ∼ 13.1. (b) Chronoamperometry (current density vs. time) of bare CNGO and different loading of Y-CNGO films in 0.10 M KOH (pH ∼ 13.1) at 1.23 V vs. RHE (VRHE) upon cycling on/off 1 sun illumination. (c) Chronoamperometry of CNGO-0.50 Y in different electrolytes (acidic, neutral, and basic) at 1.23 VRHE upon cycling on/off 1 sun illumination. (d) Oxygen (O2) production measurements data under 1 sun illumination at 1.23 VRHE in 0.10 M KOH. (e) The corresponding faradaic efficiency (FE) towards O2 production. (f) Chronoamperometric stability measurement under continuous 1 sun illumination in 0.10 M KOH at 1.23 VRHE. (g) Chronoamperometry measurements of CNGO and Y-CNGO in the presence of a hole scavenger (10% v/v TEOA) in 0.10 M KOH at 1.23 VRHE upon cycling on/off 1 sun illumination. (h) Chronoamperometric stability measurement under continuous 1 sun illumination in the presence of a hole scavenger (10% v/v TEOA in 0.10 M KOH) at 1.23 VRHE. (i) Incident photon-to-current conversion efficiency (IPCE) of bare CNGO and CNGO-0.50 Y electrodes in the 290–550 nm illumination range with and without a hole scavenger at 1.23 VRHE. | ||
Among the tested Y-CNGO films (0.25, 0.50, and 0.75 mmol Y in the precursor), particular attention is given to the CNGO-0.50 Y sample as it exhibits the highest photocurrent density, indicating optimal Y incorporation within the CN framework. Moreover, the PEC performance of CNGO-0.50 Y was also measured in acidic (0.50 M H2SO4, pH ∼ 0.3) and neutral (phosphate buffer, pH ∼ 7.0) electrolytes, yielding photocurrent densities of 230 ± 10 and 250 ± 10 μA cm−2, respectively (Fig. 3c). The good activity across a wide pH window suggests that Y-CNGO photoanodes could be used in other pH-sensitive oxidation reactions.
The O2 quantification measurements yield crucial insights, indicating that most of the measured current originates from the OER rather than from parasitic self-oxidation of the CN layer. Bare CNGO and Y-CNGO films exhibit O2 generation rates of 0.014 and 0.036 μmol cm−2 min−1, respectively (as depicted in Fig. 3d), which correlate to a high OER faradaic efficiency (FE) of 90% for the CNGO-0.50 Y film after 2 h (a 4 h water-splitting PEC experiment; Fig. 3e), whereas a bare CNGO film reaches an OER FE of only 68% under identical conditions. Moreover, H2 quantification (Fig. S10, ESI†) reveals hydrogen evolution rates of 0.025 and 0.065 μmol cm−2 min−1 for CNGO and CNGO-0.50 Y, respectively. At 2 h, the CNGO-0.50 Y photoanode exhibits a H2 FE of 91%, whereas FE for pristine CNGO is 77%. Together with the OER FE, it indicates that over 90% of the excitons are effectively separated, transported, and utilized for the desired redox reactions. Specifically, the photogenerated holes reach the photoanode–electrolyte interface, and electrons migrate through the photoanode to the Pt counter electrode, where the H2 evolution reaction occurs.
The stability assessment in alkaline electrolyte reveals a notable enhancement due to the incorporation of yttrium over a 10 h period (Fig. 3f). The CNGO-0.50 Y photoanode retained 45% of its initial activity even after continuous operation for 10 hours, attributed to the enhanced charge separation in Y-CNGO films, a significant achievement for a CN-based photoanode without a dedicated separate inorganic phase, serving as an oxidation co-catalyst (see Table S1 (ESI†) for comparison to reported CN-based water-splitting PEC performance). The bare CNGO photoanodes drop to 25% of their initial activity after 5 h, highlighting a significant enhancement in durability achieved by augmenting the CNGO blend with yttrium. Additionally, CNGO-0.50 Y demonstrated superior stability in alkaline and neutral electrolyte environments in contrast to CNGO alone (Fig. 3f and Fig. S11, ESI†).
To identify limiting factors in the PEC-OER performance, chronoamperometry measurements of the bare CNGO and CNGO-0.50 Y electrodes were performed in a 0.10 M KOH aqueous solution at 1.23 VRHE in the presence of triethanolamine (TEOA; 10% v/v), acting as an efficient hole scavenger,40,41 yielding photocurrent densities of 282 ± 10 μA cm−2 and 415 ± 10 μA cm−2, respectively (Fig. 3g). These results indicate that the primary limitation of this system lies in the challenging extraction of holes. The comparison between photocurrents in the presence of TEOA and in KOH alone allows estimation of charge extraction efficiencies (ηct) for CNGO and CNGO-0.50 Y; assuming a perfect hole extraction of this scavenger at pH ∼ 13.1, the calculated ηct are 49.9% and 66.3%, respectively, signifying that Y-loading of the CNGO photoanode improves charge transfer efficiency and thus the overall PEC performance. Additionally, the CNGO-0.50 Y films have good durability in the presence of TEOA (Fig. 3h, ∼75% of the initial current density, j0) compared to the bare CNGO.
The incident photon-to-current conversion efficiency (IPCE) across different illumination wavelengths (320–550 nm) was measured as shown in Fig. 3i. The IPCE values consistently correlate with the optical absorption spectra of the films. The IPCE values of Y-CNGO exceed those of bare CNGO (maximal values of 14.0% at 390 nm vs. 6.4% at 375 nm), suggesting the Y-cluster role in improving charge separation and transfer. Moreover, the IPCE analysis confirms that the extended absorbance of the CNGO-0.50 Y electrode in the visible range translates into photoactivity towards longer wavelengths up to ca. 525 nm. This extension is attributed to the defect states generated in the CN due to the incorporation of yttrium clusters (see the peak in the absorbance at ∼525 nm in Fig. 1e). Furthermore, the presence of TEOA noticeably increased IPCE values, reaching 14.6% for CNGO (at 375 nm) and 21.7% for CNGO-0.50 Y (at 390 nm), respectively.
Characterization after prolonged activity of CNGO-0.50 Y photoanodes was conducted to examine ensuing structural and chemical changes. For analysis, the samples before operation (‘Fresh’) are compared to samples after 30 h of operation (with TEOA) under voltage bias and illumination—a continuous stability test (‘post-mortem’). Fig. S12 (ESI†) presents SEM images of a post-mortem photoanode, disclosing minimal changes in thickness and morphology. An XPS analysis (Fig. S13, ESI†) discloses that the C 1s spectrum is identical before and after operation. However, the N 1s XPS spectra (Fig. S13c, ESI†) reveal a new peak at 402.4 eV, corresponding to N–Ox species, indicating partial oxidation of CN during the stability test (30 h period in the presence of TEOA); we believe this is the main degradation mechanism responsible for the measured loss in activity. Only slight changes are observed in the Y 3d XPS spectra (Fig. S13d, ESI†), indicative of partial oxidation. Post-operation XRD also shows only minor changes in the pattern, as shown in Fig. S13e (ESI†).
The origin of enhanced activity and stability
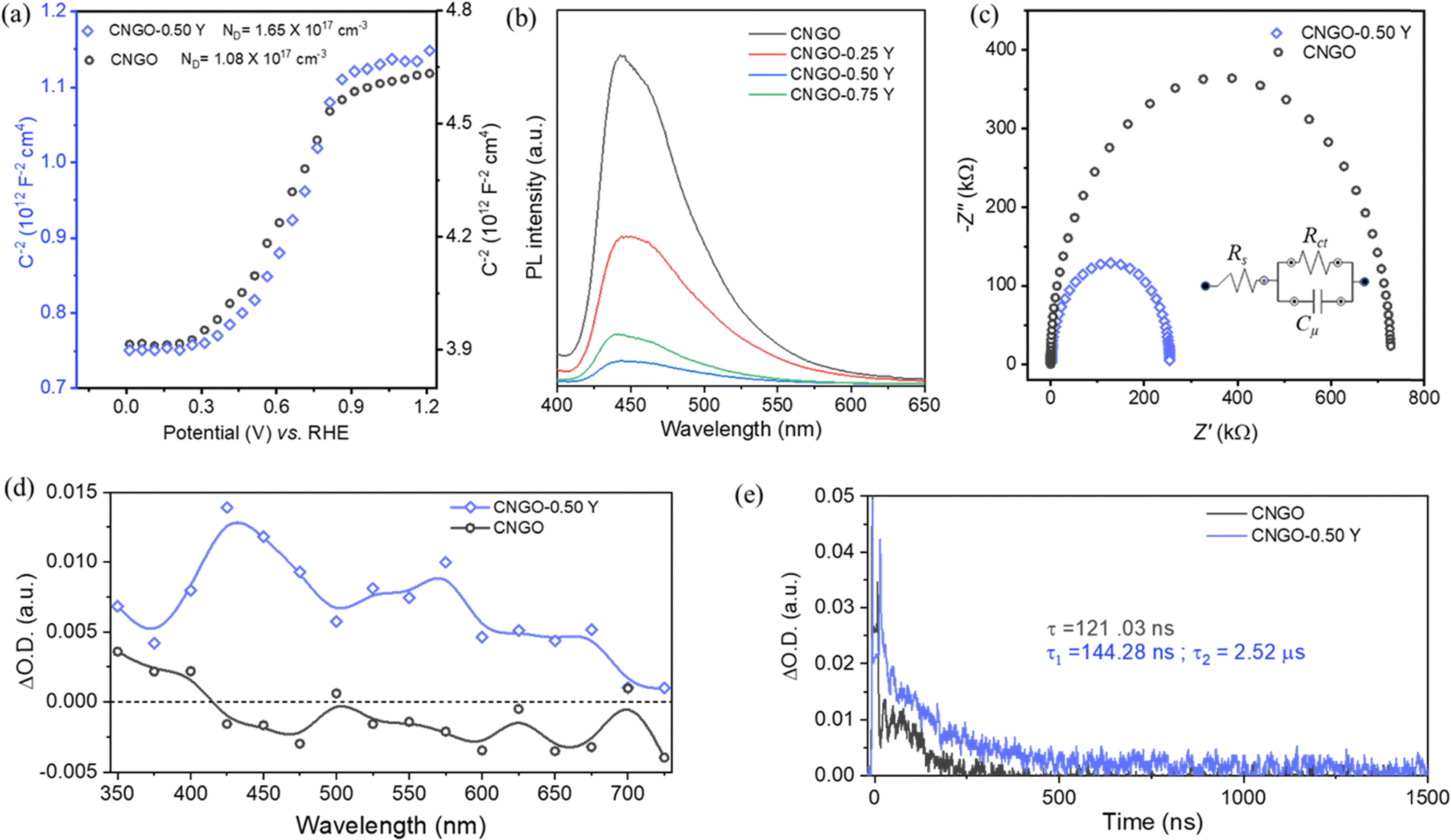 | ||
| Fig. 4 (a) Mott–Schottky analysis of CNGO and CNGO-0.50 Y films. (b) Photoluminescence (PL) emission spectra of CNGO and Y-CNGO films. (c) Nyquist plot (fitted model) of CNGO and CNGO-0.50 Y films; the inset shows the equivalent circuit used for fitting. (d) Transient absorption spectra of CNGO (black circles) and CNGO-0.50 Y (blue squares) dispersions in acetonitrile (MeCN), acquired at 250 ns. (e) Transient absorption decay of CNGO (black) and CNGO-0.50 Y (blue) dispersions in MeCN, monitored at 650 nm. The measurements were performed under N2 atmosphere upon laser excitation at 355 nm. The CNGO and CNGO-0.50 Y films dispersion in acetonitrile UV-vis absorption was adjusted at identical values at the excitation wavelength (355 nm). | ||
Electrochemical impedance spectroscopy (EIS) reveals a reduced charge transfer resistance (Rct) for CNGO-0.50 Y (254 kΩ) compared to CNGO (723 kΩ), as illustrated in Fig. 4c, upon fitting using a simple equivalent circuit (see inset). We attribute this improved charge transfer in the Y-cluster modified film to the interconnected 1D–2D morphology and the new defect states within the Y-CNGO.
Laser transient absorption spectroscopy (TAS) measurements were carried out to investigate the photo-induced charge separation and recombination processes in Y-CNGO. Fig. 4d shows the TA decay spectra of N2-saturated CNGO and CNGO-0.5 Y dispersions in acetonitrile (MeCN) upon 300 nm laser excitation. As reported before, the bare CNGO presents a positive feature in the UV region.23 Interestingly, the CNGO-0.5 Y sample exhibits an extended TA spectrum along the UV-vis spectrum, showing some prominent bands at 425, 575, and 675 nm. To assign the different features of the CNGO-0.5 Y TA spectrum, quenching experiments were carried out using an O2-saturated CNGO-0.5 Y dispersion in MeCN and a N2-saturated CNGO-0.5 Y dispersion in a methanol:MeCN (30:70, v/v) mixture (Fig. S14, ESI†). Regrettably, we could not clearly attribute these features solely to the presence of electrons or holes, and therefore, we can only ascribe them to the presence of different excited states. However, CNGO-0.5 Y presented an enhanced optical density (O.D.) compared to the CNGO sample. This is commonly related to a higher density of excited states. Therefore, we can conclude that CNGO-0.5 Y can promote improved charge separation, in good agreement with the photocurrent (Fig. 3) and PL (Fig. 4b) experiments.
Moreover, the TA decays of N2-saturated CNGO and CNGO-0.5 Y dispersions in MeCN were monitored at 400 nm upon 300 nm laser excitation (Fig. 4f). As reported before, the excited states decay recorded in the CNGO sample presents a single and fast component, in the range of ns.23 On the other hand, the excited states’ decay in CNGO-0.50 Y shows a twofold fast and slow face behavior. CNGO and CNGO-0.5 Y decays fitting to single and bi-exponential functions, (eqn (1) and (2), respectively), show a fast component in both samples of 121 and 144 ns, respectively (Table S2, ESI†). This is attributed to the internal deactivation of excited states within the CNGO.23 Notably, CNGO-0.50 Y presents an additional, extended component with a lifetime of τ2 = 2.52 μs. This supports an improved charge separation between the CNGO and the Y-clusters, as well as slower charge recombination, which is in good agreement with the photocatalytic PEC results and EIS analysis.
| F(t) = Ae−τt | (1) |
| F(t) = A1e−τ1t + A2e−τ2t | (2) |
 | ||
| Fig. 5 ToF-SIMS Y-based fragments before (fresh) and after (post-mortem) electrochemical testing of CNGO-0.50 Y in positive SIMS spectrum shown for (a) YN2C2+, (b) Y3O4+, and (c) list of identified major fragments. Fragment counts from peak areas were normalized to the total ion counts. | ||
The observed increase in normalized counts of specific post-mortem YNxCy+ fragments (e.g., YN2C2+, YNC+, and YN2C+) is similar to observations in a previous report for NiNxCy− fragments from a single atom Ni in N-doped carbon catalyst post electrochemical CO2 reduction.45 This increase in normalized counts was previously assigned to an enhancement in ionization due to a matrix effect from the KOH electrolyte used; therefore, a similar situation may have occurred here. Other YNxCy+ fragments (e.g., YNC3+, YN3C/YNC2O+, and YN2+) show decreased normalized counts, indicating that certain Y environments are unstable. Nevertheless, the persistence of YNxCy+ fragments in fresh and post-mortem samples suggests the presence of atomically dispersed Y within the N–C framework, with varying degrees of stability. Meanwhile, new peaks are observed post-mortem for other Y-based fragments (for example, YO+ and Y3O4+, see Fig. 5b and Fig. S16, ESI†), indicating that yttrium oxides form during the electrochemical test (corroborating the XPS result). It is noted that the peak assigned to Y2N6+ completely diminishes after electrochemical testing (Fig. 5c), indicating that this Y-based chemical species is unstable, which could contribute to the formation of the new observed fragments, such as Y oxides. We note that ToF-SIMS provides purely chemical information. Therefore, conclusive discussions on the bonding environment of Y cannot be made.
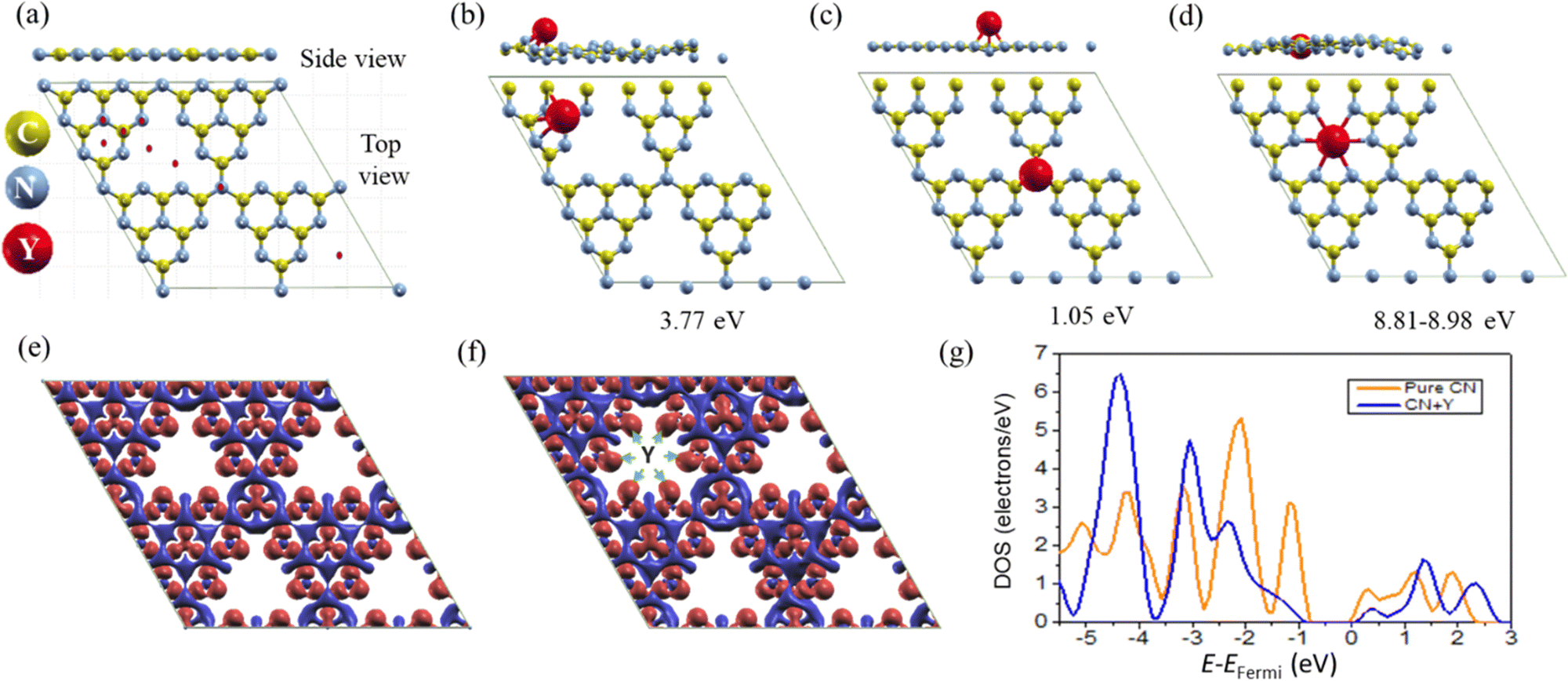 | ||
| Fig. 6 Computational (DFT) analysis of Y incorporation into CN. Side- and top-views of a relaxed CN structure, modeled using tri-s-triazine units: (a) without adsorbed atoms but with the initial positions of the Y atoms used for their relaxation, (b) and (c) intermediate adsorption sites of Y on CN, and (d) the most stable relaxed configuration. Isosurface differential electron densities of (e) pure CN layer and (f) CN + Y (i.e., CN with a Y dopant); the green inset arrows are directed to the location of the nitrogen atoms around the pore in this structure. (g) The density of states (DOS) of the 2p electron of the N atoms around the pore for pure CN (orange) and CN + Y (blue). | ||
The redistribution of electrons provides valuable insights into the nature of bonding between CN and Y. To elucidate the impact of yttrium doping on the electronic structure of CN, electron densities and density of states (DOS) were calculated for both the relaxed CN + Y structure (depicted in Fig. 6d) and pristine CN. Fig. 6e and f illustrate representative isosurface differential electron densities. The visualization reveals higher differential density for nitrogen atoms adjacent to the Y atom (shown by green arrows). Löwdin population analysis indicates that 2p electrons of N atoms, proximal to the Y dopant, exhibit an excess electron density of approximately 0.1e− per atom relative to other nitrogen atoms with similar immediate environments but lacking Y in their vicinity. Fig. 6g presents a comparative analysis of the 2p electron density of states (DOS) for nitrogen atoms in the immediate vicinity of Y and those in pristine CN. The most significant changes are observed in the energy band ranging from 1 to 4 eV below the Fermi level. In the presence of the Y atom, this band exhibits increased continuity, whereas in its absence, it appears to be segmented into three distinct regions.
The second prominent Y incorporation mechanism is Y-clusters, which are spread throughout the material, particularly between the 2D sheets of the CN. These clusters, detected in the electron microscopy analysis, undergo (partial) oxidation during OER, as evidenced by XPS and ToF-SIMS. The combination of both Y incorporation modes and the ensuing structural 1D–2D hybrid lead to a positive shift of the energy bands, new optical absorption states, higher concentration of charge carriers (Mott–Schottky analysis, TAS measurements), improved charge separation (e.g., manifesting in lower PL), and longer excited state lifetime (τ2). These analyses support the measured improved photocurrent densities during water-splitting, higher IPCE extended into the visible range, and improved FE and long-term stability.
Conclusions
In this study, we presented the incorporation of yttrium into porous CN-based films and the resulting impact on photoanodic activity and stability toward water oxidation reactions in an alkaline medium. Detailed structural, spectroscopic, photoelectrochemical, and theoretical investigations reveal that integrating yttrium clusters into the CN significantly improves light harvesting properties, electronic conductivity, charge separation, and hole extraction kinetics, enabling efficient water oxidation. The yttrium incorporation modifies the CN's electronic properties and introduces a unique structural feature that combines 1D and 2D morphologies in one platform. This design connects sheet-like 2D CN structures with 1D tubular, yttrium-rich CN components, creating a distinctive morphology of the photoactive film that manifests in improved efficiency and durability during photoelectrochemical water oxidation. The optimized CN photoanode loaded with Y-clusters exhibits a photocurrent density of 275 ± 10 μA cm−2 with 90% FE for oxygen evolution, good stability for up to 10 hours of operation (30 h with a hole scavenger), and IPCE values up to 14% in alkaline medium. This research highlights the potential of yttrium-modified CN for alkaline-to-neutral photoelectrochemical water-splitting and offers a pathway toward developing more robust and durable photocatalysts for clean energy applications.Author contributions
S. M. performed most of the experiments, analyzed the data, and wrote the initial draft of the manuscript. A. T. helped with PEC experiments. G. M. performed XPS characterization. M. V. took part in analysis, SEM imaging, and manuscript co-writing. S. B. performed the DFT calculations and wrote the related discussion. A. P. performed ToF-SIMS characterization and analysis. J. A. and H. G. conducted and analyzed the TAS measurements and reviewed the manuscript. M. S. supervised the study, co-wrote the paper, and acquired funding. All the authors discussed the results and reviewed the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Programme (Grant Agreement No. 849068). This work was partially supported by the Israel Science Foundation, Grant No. 601/21, and by the Pazy Research Foundation (Grant No. 656-2025). A. P. thanks the EPSRC Doctoral Prize Fellowship (EP/W524323/1) for funding.References
- J. M. Yu, J. Lee, Y. S. Kim, J. Song, J. Oh, S. M. Lee, M. Jeong, Y. Kim, J. H. Kwak, S. Cho, C. Yang and J.-W. Jang, Nat. Commun., 2020, 11, 5509 CrossRef PubMed.
- I. Holmes-Gentle, S. Tembhurne, C. Suter and S. Haussener, Nat. Energy, 2023, 8, 586–596 CrossRef CAS.
- R. Siavash Moakhar, S. M. Hosseini-Hosseinabad, S. Masudy-Panah, A. Seza, M. Jalali, H. Fallah-Arani, F. Dabir, S. Gholipour, Y. Abdi, M. Bagheri-Hariri, N. Riahi-Noori, Y.-F. Lim, A. Hagfeldt and M. Saliba, Adv. Mater., 2021, 33, 2007285 CrossRef CAS PubMed.
- H. C. Lee, J. H. Park, S.-I. In and J. Yang, Nanoscale, 2024, 16, 9295–9310 RSC.
- F. T. Wagner and G. A. Somorjai, J. Am. Chem. Soc., 1980, 102, 5494–5502 CrossRef CAS.
- M. Kumar, B. Meena, P. Subramanyam, D. Suryakala and C. Subrahmanyam, NPG Asia Mater., 2022, 14, 88 CrossRef CAS.
- S. F. Blaskievicz, J. L. Francisco, F. Marken and L. H. Mascaro, ACS Appl. Energy Mater., 2024, 7, 3021–3038 CrossRef CAS.
- J. Bian, L. Xi, C. Huang, K. M. Lange, R. Zhang and M. Shalom, Adv. Energy Mater., 2016, 6, 1600263 CrossRef.
- G. Peng, J. Albero, H. Garcia and M. Shalom, Angew. Chem., Int. Ed., 2018, 57, 15807–15811 CrossRef CAS PubMed.
- M. Volokh, G. Peng, J. Barrio and M. Shalom, Angew. Chem., Int. Ed., 2019, 58, 6138–6151 CrossRef CAS PubMed.
- J. Qin, J. Barrio, G. Peng, J. Tzadikov, L. Abisdris, M. Volokh and M. Shalom, Nat. Commun., 2020, 11, 4701 CrossRef PubMed.
- N. Karjule, J. Barrio, L. Xing, M. Volokh and M. Shalom, Nano Lett., 2020, 20, 4618–4624 CrossRef CAS PubMed.
- T. Shmila, S. Mondal, S. Barzilai, N. Karjule, M. Volokh and M. Shalom, Small, 2023, 19, 2303602 CrossRef CAS PubMed.
- S. Mondal, G. Mark, L. Abisdris, J. Li, T. Shmila, J. Tzadikov, M. Volokh, L. Xing and M. Shalom, Mater. Horiz., 2023, 10, 1363–1372 RSC.
- M. Volokh and M. Shalom, Ann. N. Y. Acad. Sci., 2023, 1521, 5–13 CrossRef CAS PubMed.
- A. Tashakory, S. Mondal, V. R. Battula, G. Mark, T. Shmila, M. Volokh and M. Shalom, Small Struct., 2024, 5, 2400123 CrossRef CAS.
- S. Mondal, G. Mark, A. Tashakory, M. Volokh and M. Shalom, J. Mater. Chem. A, 2024, 12, 11502 RSC.
- G. Mark, S. Mondal, M. Volokh, J. Xia and M. Shalom, Sol. RRL, 2022, 6, 2200834 CrossRef CAS.
- R. S. Roy, S. Mondal, S. Mishra, M. Banoo, L. Sahoo, A. Kumar, C. P. Vinod, A. K. De and U. K. Gautam, Appl. Catal., B, 2023, 322, 122069 CrossRef CAS.
- S. Mondal, L. Sahoo, Y. Vaishnav, S. Mishra, R. S. Roy, C. P. Vinod, A. K. De and U. K. Gautam, J. Mater. Chem. A, 2020, 8, 20581–20592 RSC.
- J. Zhang, Y. Zou, S. Eickelmann, C. Njel, T. Heil, S. Ronneberger, V. Strauss, P. H. Seeberger, A. Savateev and F. F. Loeffler, Nat. Commun., 2021, 12, 3224 CrossRef CAS PubMed.
- S. Mondal, M. Salati, M. Nicaso, J. Albero, M. Segado-Centellas, M. Volokh, C. Bo, H. García, M. Gil-Sepulcre and A. Llobet, Chem. Sci., 2024, 15, 16546–16553 RSC.
- S. Mondal, T. Naor, M. Volokh, D. Stone, J. Albero, A. Levi, A. Vakahi, H. García, U. Banin and M. Shalom, ACS Appl. Mater. Interfaces, 2024, 16, 38153–38162 CrossRef CAS PubMed.
- N. Karjule, C. Singh, J. Barrio, J. Tzadikov, I. Liberman, M. Volokh, E. Palomares, I. Hod and M. Shalom, Adv. Funct. Mater., 2021, 31, 2101724 CrossRef CAS.
- J. Kim, P.-C. Shih, K.-C. Tsao, Y.-T. Pan, X. Yin, C.-J. Sun and H. Yang, J. Am. Chem. Soc., 2017, 139, 12076–12083 CrossRef CAS PubMed.
- J. Klöwer and J.-G. Li, Mater. Corros., 1996, 47, 545–551 CrossRef.
- S. Zhang, H. Li, Z. Jiang, H. Feng, Z. Wen, J. Ren and P. Han, J. Mater. Sci. Technol., 2022, 115, 103–114 CrossRef CAS.
- G. Peng, J. Qin, M. Volokh, C. Liu and M. Shalom, J. Mater. Chem. A, 2019, 7, 11718–11723 RSC.
- G. Peng, M. Volokh, J. Tzadikov, J. Sun and M. Shalom, Adv. Energy Mater., 2018, 8, 1800566 CrossRef.
- J. Fang, H. Fan, M. Li and C. Long, J. Mater. Chem. A, 2015, 3, 13819–13826 RSC.
- Y. Yang, J. Chen, Z. Mao, N. An, D. Wang and B. D. Fahlman, RSC Adv., 2017, 7, 2333–2341 RSC.
- Y. Xu, Y. Gong, H. Ren, W. Liu, L. Niu, C. Li and X. Liu, RSC Adv., 2017, 7, 32592–32600 RSC.
- Z. Li, B. Li, X. Wu, S. A. Sheppard, S. Zhang, D. Gao, N. J. Long and Z. Zhu, Science, 2022, 376, 416–420 CrossRef CAS PubMed.
- E. Alwin, W. Nowicki, R. Wojcieszak, M. Zieliński and M. Pietrowski, Dalton Trans., 2020, 49, 12805–12813 RSC.
- H. Guo, D.-H. Si, H.-J. Zhu, Q.-X. Li, Y.-B. Huang and R. Cao, eScience, 2022, 2, 295–303 CrossRef.
- H. Liu, H. Wang, F. Zhang, J. Xue, J. Zhang and G. Zhang, Chem. Commun., 2021, 57, 927–930 RSC.
- L. Sahoo, S. Mondal, C. B. Nayana and U. K. Gautam, J. Colloid Interface Sci., 2021, 590, 175–185 CrossRef CAS PubMed.
- T. Mongstad, A. Thøgersen, A. Subrahmanyam and S. Karazhanov, Sol. Energy Mater. Sol. Cells, 2014, 128, 270–274 CrossRef CAS.
- L. Mai, N. Boysen, E. Subaşı, T. de Los Arcos, D. Rogalla, G. Grundmeier, C. Bock, H.-L. Lu and A. Devi, RSC Adv., 2018, 8, 4987–4994 RSC.
- A. Augustin, P. Ganguly, S. Shenoy, C. Chuaicham, S. C. Pillai, K. Sasaki, A. F. Lee and K. Sekar, Adv. Sustainable Syst., 2024, 8, 2400321 CrossRef CAS.
- S. T. Gebre, L. M. Kiefer, F. Guo, K. R. Yang, C. Miller, Y. Liu, C. P. Kubiak, V. S. Batista and T. Lian, J. Am. Chem. Soc., 2023, 145, 3238–3247 CrossRef CAS PubMed.
- N. S. Peighambardoust, E. Sadeghi and U. Aydemir, ACS Appl. Nano Mater., 2022, 5, 14092–14132 CrossRef CAS.
- S. Chamani, E. Sadeghi, N. S. Peighambardoust, F. Doganay, G. Yanalak, Z. Eroglu, E. Aslan, E. Asghari, O. Metin, I. H. Patir, U. Aydemir and M. Khatamian, Int. J. Hydrogen Energy, 2022, 47, 32940–32954 CrossRef CAS.
- F. Aubriet, C. Poleunis, J. Muller and P. Bertrand, J. Mass Spectrom., 2006, 41, 527–542 CrossRef CAS PubMed.
- S. C. Sarma, J. Barrio, A. Bagger, A. Pedersen, M. Gong, H. Luo, M. Wang, S. Favero, C.-X. Zhao, Q. Zhang, A. Kucernak, M.-M. Titirici and I. E. L. Stephens, Adv. Funct. Mater., 2023, 33, 2302468 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures, characterization, additional figures and tables. See DOI: https://doi.org/10.1039/d5ey00064e |
| This journal is © The Royal Society of Chemistry 2025 |