
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffective production of liquid/wax fuels from polyethylene plastics using Ru/Al2O3 catalysts†
Jueun Kim‡
 a,
Donghyeon Kim‡b,
Byung Gwan Parka,
Daewon Oha,
Shinjae Leea,
Jihun Kima,
Eonu Nama and
Kwangjin An*ab
a,
Donghyeon Kim‡b,
Byung Gwan Parka,
Daewon Oha,
Shinjae Leea,
Jihun Kima,
Eonu Nama and
Kwangjin An*ab
aSchool of Energy and Chemical Engineering, Ulsan National Institute of Science and Technology (UNIST), Ulsan, 44919, Korea. E-mail: kjan@unist.ac.kr
bSchool of Carbon Neutrality, Ulsan National Institute of Science and Technology (UNIST), Ulsan 44919, Republic of Korea
First published on 26th April 2025
Abstract
Hydrogenolysis provides a promising pathway for converting polyolefin plastics into valuable liquid and wax fuels. This process involves dehydrogenation, C–C bond cleavage, and hydrogenation at the active metal sites of the catalyst. Controlling the nature of these metal sites is crucial to optimize overall reaction activity. In this study, Ru catalysts supported on nanosheet-assembled Al2O3 (NA-Al2O3) were used for the hydrogenolysis of polyethylene (PE). Unlike commercial Al2O3, NA-Al2O3 promotes Ru–Al bond formation, leading to stronger metal–support interactions. Under identical Ru loadings, these enhanced interactions resulted in higher Ru dispersion and smaller Ru species on the NA-Al2O3 surface. To investigate the effect of Ru loading, a series of catalysts (xRu/NA-Al2O3, x = 0.5, 1, 5, and 8 wt% Ru) was synthesized, revealing that Ru particle size and electronic properties varied with Ru loading. Among them, the 1Ru/NA-Al2O3 catalyst, featuring optimally sized Ru species (∼0.8 nm) and a tailored electronic structure, demonstrated the highest efficiency in PE hydrogenolysis by effectively suppressing successive C–C bond cleavage. This catalyst achieved an outstanding PE conversion rate of 1.15 × 103 gconverted PE gRu−1 h−1 and a liquid/wax production rate of 9.23 x 102 gliquid/wax gRu−1 h−1, highlighting its superior performance in catalytic PE hydrogenolysis.
Broader contextHydrogenolysis is a promising method for the conversion of polyolefin plastics into valuable liquid/wax fuels. As a structure-sensitive reaction, polyolefin hydrogenolysis requires precise control over the size and structure of the active metal. In this study, we aimed to observe the structure of Ru by adjusting the type of support and Ru content, and to investigate the resulting changes in polyethylene (PE) hydrogenolysis reactivity. On nanosheet-assembled Al2O3 (NA-Al2O3), Ru formed Ru–Al bonds, allowing it to be highly dispersed in a smaller size. The 1Ru/NA-Al2O3 catalyst achieved an optimal Ru size and suitable electronic structure, showing the highest PE conversion rate (1.15 × 103 gconverted PE gRu−1 h−1) and liquid/wax production (9.23 × 102 gliquid/wax gRu−1 h−1). The lower gas emission and higher liquid/wax yield of 1Ru/NA-Al2O3 were attributed to its ability to suppress both successive and terminal C–C bond cleavage. This research contributes to the understanding of how the geometric and electronic properties of Ru and irreducible metal oxides, like Al2O3, can be utilized effectively in polyolefin hydrogenolysis, thus promoting advances in chemical recycling technologies. |
Introduction
Recently, global energy demand has steadily increased, which has intensified the need for alternative and sustainable resources.1 The comparable energy density of plastics to that of chemical fuels renders them as a potential future recyclable resource.2 Despite the environmental challenges posed by improper plastic waste disposal, chemical recycling technologies offer potential solutions.3 Among plastics, polyethylene (PE) and polypropylene (PP), primarily consisting of carbon and hydrogen, account for approximately 55% of global production, which makes them attractive candidates for conversion into liquid fuels.4Polyolefin hydrogenolysis, which enables the transformation of polyolefin plastics into high-quality liquid/wax fuels under mild conditions, has emerged as a promising method for chemical recycling. This process involves a series of reactions, including dehydrogenation, C–C bond cleavage, and hydrogenation. Initially, the polyolefins undergo dehydrogenation to form an intermediate that is adsorbed onto the metal surface in its dehydrogenated state. This intermediate then experiences C–C bond cleavage, followed by hydrogenation, which results in the desorption of alkanes from the metal surface.5 The efficiency and selectivity of this process are highly dependent on the successful execution of each reaction step. Notably, the geometric and electronic properties of the active metal in the catalyst play a critical role in modulating reaction pathways, particularly in controlling C–C bond cleavage activity and substrate adsorption.6–15
Extensive research has demonstrated that modifying the geometric and electronic properties of the active metal significantly influences the catalytic performance in polyolefin hydrogenolysis. For instance, Wu et al. observed that reducing the Pt particle size enhanced the reactivity of polyolefin hydrogenolysis owing to a shift in the rate constant of C–C bond cleavage with respect to Pt particle size.8 Similarly, Chen et al. reported that highly disordered Ru surfaces exhibited substantial hydrogen coverage in a hydrogen environment, which stabilized the less-dehydrogenated transition states in polyolefins.9 This stabilization favoured internal C–C bond cleavage while simultaneously suppressing excessive methane formation. In another study, Hu et al. demonstrated that PE hydrogenolysis could be effectively catalyzed using Ru-based catalysts featuring stable Ru0/Ruδ+ species, which contributed to enhanced catalytic performance.15
Considering the substantial variation in the geometric and electronic properties of the active metal depending on the support, it is essential to understand the interactions between the support and active metal. γ-Al2O3 has been recognized as a commercially important heterogeneous catalyst owing to its high-temperature stability and large surface area. By tailoring the diverse surface properties of Al2O3, metal–support interactions can be modulated, thereby altering the geometric and electronic characteristics of the active sites formed on the surface.16–18 In this study, we synthesized Ru supported on nanosheet-assembled Al2O3 (NA-Al2O3) for PE hydrogenolysis. The Ru–Al bond formation by the interaction between Ru and NA-Al2O3 allows for the formation of highly dispersed and small Ru nanoparticles on Al2O3, which significantly suppresses the excessive gas formation associated with successive C–C bond cleavage. As a result, the optimized 1Ru/NA-Al2O3 catalyst exhibited an outstanding PE conversion rate of 1.15 × 103 gconverted![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) PE gRu−1 h−1 and a liquid/wax production rate of 9.23 × 102 gliquid/wax gRu−1 h−1, demonstrating superior catalytic efficiency in PE hydrogenolysis.
PE gRu−1 h−1 and a liquid/wax production rate of 9.23 × 102 gliquid/wax gRu−1 h−1, demonstrating superior catalytic efficiency in PE hydrogenolysis.
Experimental
Materials
Polyethylene (Mw ∼ 4000, Mn ∼ 1700), potassium sulfate (K2SO4, ≥ 99.0%), p-xylene (99%), sodium borohydride (NaBH4, ≥ 96%), and C7–C30 saturated alkanes (certified reference material, 1000 μg mL−1 each component in hexane) were purchased from Sigma-Aldrich. Dichloromethane (CH2Cl2, HPLC Reagent, ≥99.9%) and n-decane (C10H22, 99.5%) were obtained from Samchun Chemical and Daejung, respectively. Ruthenium(III) chloride hydrate (RuCl3·xH2O, Ru 38% min), aluminum nitrate nonahydrate (Al(NO3)3·9H2O, 98%), n-octadecane (n-C18H38, 99%), and urea (CO(NH2)2, ≥98%) were sourced from Alfa Aesar. Commercial Al2O3 (com-Al2O3; Puralox SBa200) was purchased from Sasol.Synthesis of nanosheet-assembled Al2O3 (NA-Al2O3)
NA-Al2O3 was synthesized using a hydrothermal method, as described previously.19–21 A solution was prepared by dissolving 1.51 g of Al(NO3)3·9H2O and 0.70 g of K2SO4, and 0.50 g of CO(NH2)2 in 80 mL deionized (D.I.) water. This mixture was transferred to a 100 mL Teflon-lined stainless-steel autoclave and heated at 180 °C for 5 h. The resulting white precipitate was washed D.I. water and ethanol, dried at 60 °C overnight in a vacuum oven, and calcinated at 700 °C for 2 h.Preparation of xRu/NA-Al2O3 (x = 0.5, 1, 5, 8 wt% Ru)
Ru was loaded onto the NA-Al2O3 support using the deposition–precipitation method with urea hydrolysis (DPU).22,23 Target amounts of NA-Al2O3, urea, and RuCl3·xH2O solution were added in D.I. water at a [Ru]:[urea] molar ratio of 1:100, and the solution was heated to 80 °C and stirred for 2 h. After cooling to room temperature (25 °C), NaBH4 was slowly added at a [Ru]:[NaBH4] molar ratio of 1:2 for chemical reduction and stirred for another 2 h.24,25 The precipitates were washed with D.I. water and ethanol and dried at 70 °C overnight in a vacuum oven.
For the preparation of Ru-loaded commercial Al2O3 (com-Al2O3), calcination was performed at 700 °C for 2 h prior to Ru loading. Ru (1 wt%) was loaded using the same method as that used for xRu/NA-Al2O3, and the resulting catalyst was designated as 1Ru/com-Al2O3.
Characterization
The specific surface areas of the catalysts were determined from N2 adsorption–desorption isotherms using the Brunauer–Emmett–Teller (BET) method on a BELSORP-max system. The Ru content of the catalysts was measured through inductively coupled plasma-optical emission spectroscopy (ICP-OES) using a Varian 700-ES instrument after sample digestion in aqua regia. Thermogravimetric analysis (TGA) was performed on a TGA5500 (TA Instruments) at a heating rate of 10 °C min−1 in an air atmosphere. Transmission electron microscopy (TEM) images were obtained using a JEOL JEM-1400 electron microscope at an acceleration voltage of 120 kV. Scanning TEM (STEM) images with energy-dispersive X-ray spectrometry (EDS) elemental mapping were obtained using a Jeol JEM-2100F electron microscope at 200 kV, equipped with an Oxford x-Max spectrometer and a high-angle annular dark-field (HAADF) detector. Scanning electron microscopy (SEM) images were obtained using an FEI Nova NanoSEM. High-resolution X-ray diffraction (XRD) patterns were recorded on a Rigaku MAX2500V instrument with Cu-Kα radiation (λ = 0.154178 nm) between the 2θ range of 10–80°. X-ray photoelectron spectroscopy (XPS) was performed using a ThermoFisher K-Alpha system with an Al-Kα X-ray excitation source and the Ru 3p and Al 2p regions were analyzed. Prior to XPS measurements, all catalysts were reduced at 200 °C for 1 h under a 4% H2/Ar atmosphere. The reduced samples were briefly exposed to ambient air during transfer and were immediately loaded into the XPS. The Al 2p peak (74.7 eV) was calibrated to correct any offsets. The obtained spectra were deconvoluted using the CasaXPS software. Solid-state 27Al-nuclear magnetic resonance (NMR) analysis was performed using a Varian VNMRS 600 MHz NMR spectrometer. 1H, 13C NMR and two-dimensional (2D) heteronuclear single-quantum correlation (HSQC) spectroscopy were performed to identify the major hydrocarbon species. All 1D and 2D NMR analyses were conducted at 600 MHz NMR spectrometer. Temperature-programmed reduction by H2 (H2-TPR), temperature-programmed desorption of C10H22 (C10H22-TPD), and CO chemisorption analyses were carried out using a BELCAT II (MicrotracBEL) analyser equipped with a thermal conductivity detector (TCD). For H2-TPR, 50 mg of the catalyst was oxidized in 5% O2/He at 300 °C for 1 h, followed by cooling to 50 °C. The H2-TPR profile was obtained by increasing the temperature from 50 to 300 °C. For C10H22-TPD, the catalyst was pretreated in a 50 mL round-bottom flask under an Ar atmosphere at 150 °C for 30 min to remove surface-adsorbed moisture. C10H22 was then introduced, and the mixture was stirred for 24 h under Ar to facilitate adsorption onto the catalyst surface. The catalyst was recovered via centrifugation, dried in a vacuum oven at 60 °C for 24 h, and subsequently pretreated in a He atmosphere at 120 °C for 30 min before being cooled to 50 °C. The C10H22-TPD profile was recorded by increasing the temperature from 50 to 300 °C. In situ diffuse reflectance infrared Fourier transform (DRIFT) spectra were recorded using a Nicolet iS10 FT-IR spectrometer equipped with a mercury–cadmium–telluride (MCT) detector. Samples were pretreated at 300°C for 1 h under 20% O2/He flow and cooled to room temperature. The FT-IR spectra of the hydroxyl region were obtained at room temperature. Pyridine-adsorbed DRIFT spectra were recorded under the same pretreatment conditions with pyridine adsorption at 100 °C until sample saturation, followed by a He purge for 30 min at 100 °C to remove weakly adsorbed pyridine. Ru K-edge X-ray absorption fine structure (XAFS) measurements were performed at the 7D and 8C beamline in PLS-II. Similar to XPS analysis, all catalysts were reduced at 200 °C for 1 h under a 4% H2/Ar atmosphere prior to XAFS measurements. The reduced samples experienced a short exposure to ambient air during transfer before being rapidly loaded into the XAFS. The Ru K-edge signal (E0 = 22117 eV) was obtained in the fluorescence mode.
Catalytic test
Gas products (C1–C4) and hydrogen were collected after the temperature dropped below 10 °C. The gaseous products were analyzed using GC (Agilent Technologies 7820A) equipped with a flame ionization detector (FID, HP-PLOT Q column) and a TCD (Carboxen1000 column). The mass of the gaseous products was calculated from the weight of the samples before and after the reaction. The solid residue was extracted with dichloromethane, filtered, and dried in an oven at 80 °C. The filtrate contained hydrocarbons in the liquid/wax range (C5-C30+). The C5-C30 fraction was analysed using a GC-FID (Youngin Chromass ChroZen GC, DB-5HT column) and calibrated using a standard solution of normal alkanes. Branched alkanes were assumed to have the same calibration factors as their normal counterparts. p-Xylene was used as the internal standard. The yield of the wax products with carbon numbers greater than 30 (C30+) was calculated by subtracting the carbon moles (C-mol) of gas, liquid/wax (C5-30), and solid residues from the carbon moles of initial substrate. The conversion, yield, conversion rate, production rate, and PE C–C bond cleavage activity were calculated using the following equations:
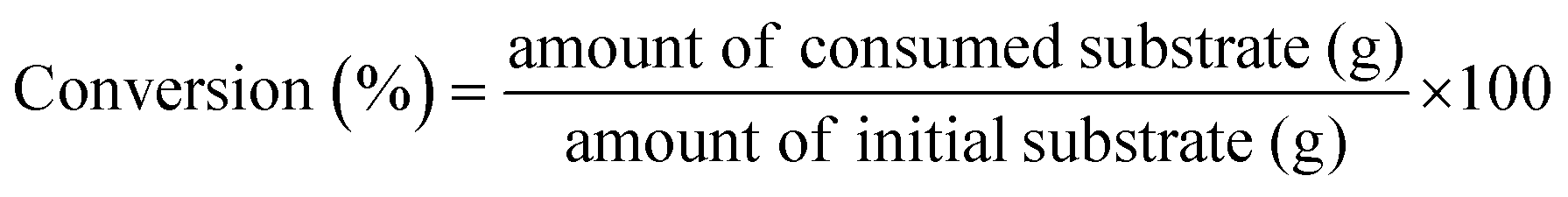 | (1) |
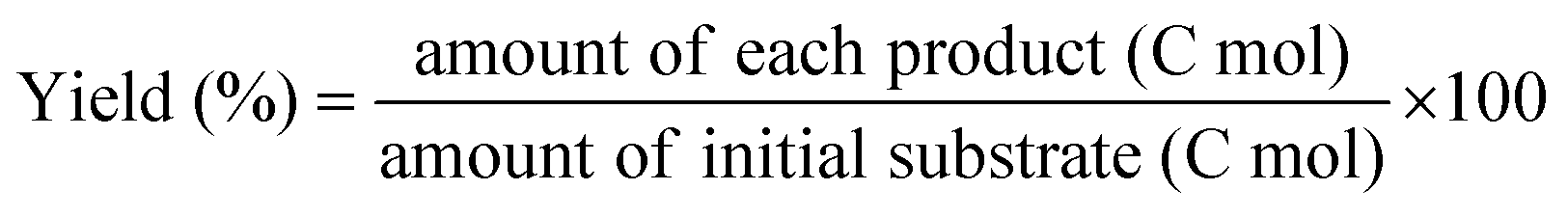 | (2) |
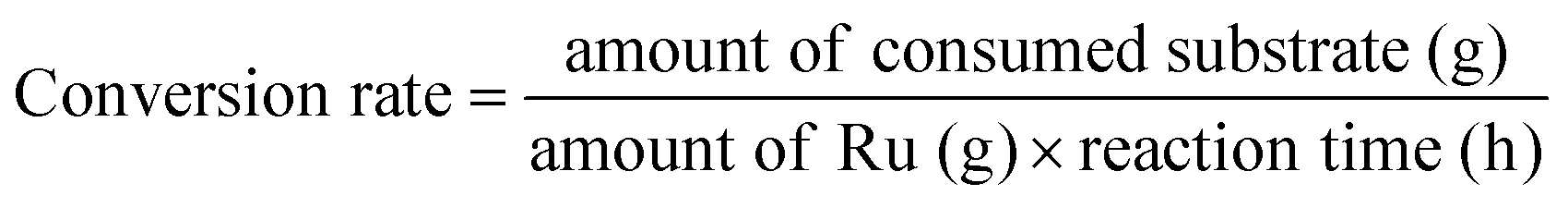 | (3) |
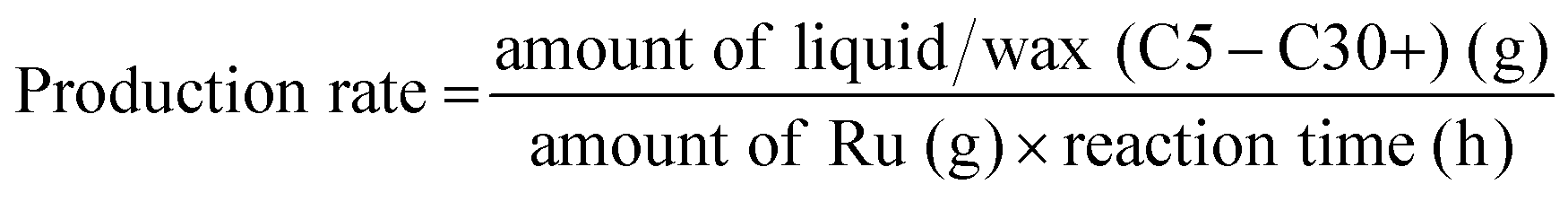 | (4) |
 | (5) |
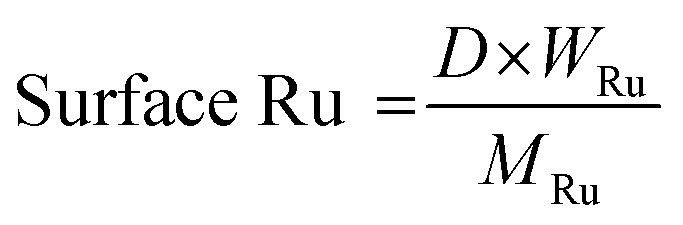 | (6) |
Regeneration of catalysts for recycling reactions
To assess catalyst reusability, the solid residue was washed with hot toluene to remove organic deposits. The spent catalyst was recovered via centrifugation and subsequently dried at 80 °C. The reusability of the catalyst was evaluated by repeating the PE hydrogenolysis under identical conditions.n-C18H38 hydrogenolysis
The n-C18H38 hydrogenolysis process and product analysis methodology followed the same procedure as described for PE hydrogenolysis. The catalytic performance in n-C18H38 hydrogenolysis was evaluated in a 45 mL Parr stainless–steel batch reactor. A reaction mixture containing 3 g n-C18H38 and an as-prepared catalyst (0.5 mg Ru metal) was placed in a glass liner equipped with a glass-coated stirrer, and the reactor was sealed. The reactor was purged three times with H2 at 50 bar, then pressured to 40 bar of H2 at room temperature, and subsequently heated to 250 °C. Upon completion of the reaction, the reactor was rapidly quenched in an ice bath.After cooling below 10 °C, gas products (C1–C4) were collected and analysed using GC-FID and TCD. Liquid-phase products were dissolved in dichloromethane (CH2Cl2) and analysed by GC-FID. The extent of C–C bond cleavage was determined using the following equation:
| C–C bond cleavage = number of C–C bonds in reacted C18H38 (mmol) − number of C–C bonds in products (mmol) | (7) |
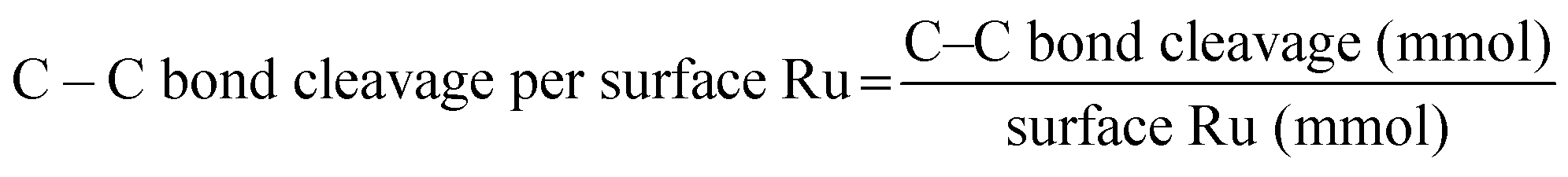 | (8) |
Results and discussion
Catalyst synthesis and characterization
NA-Al2O3 was synthesized using a previously reported hydrothermal method.19–21 The synthesis process involved urea hydrolysis and polycondensation of Al3+ and SO4−, which formed aluminum oxide hydroxide (AlOOH) with a hierarchical structure (Fig. 1(a) and Fig. S1, ESI†). After calcination at 700 °C for 2 h, a topotactic phase transformation occurred, converting AlOOH into γ-phase Al2O3, while preserving its hierarchical structure (Fig. 1(b) and Fig. S2, ESI†).26,27 Owing to its γ-phase characteristics, the resulting NA-Al2O3 exhibited a large specific surface area (SBET = 160 m2 g−1) and high thermal stability (Table S1 and Fig. S3a, ESI†). Commercially available Al2O3, com-Al2O3 calcinated under similar conditions also showed comparable surface areas (Table S1 and Fig. S3b, ESI†). The 3D hierarchical structure of NA-Al2O3 provides stability against phase transformations at elevated temperatures and enhances mass transfer, which enables the reactant molecules to easily access the active metal sites.19,28,29 Ru was loaded onto the NA-Al2O3 support through the DPU method. During this process, the Ru precursor was deposited as a hydroxide on the support and then reduced, followed by a chemical reduction using NaBH4. The catalysts exhibited similar surface areas regardless of the loaded Ru content (Table S1, ESI†). HAADF-STEM revealed that the average diameters of Ru nanoparticles were approximately 0.47 ± 0.13, 0.80 ± 0.17, 1.60 ± 0.89, 2.32 ± 1.14, and 1.17 ± 0.66 nm for 0.5Ru/NA-Al2O3, 1Ru/NA-Al2O3, 5Ru/NA-Al2O3, 8Ru/NA-Al2O3, and 1Ru/com-Al2O3, respectively (Fig. 1(c)–(g) and Fig. S4, ESI†). The 0.5Ru/NA-Al2O3 catalyst contains Ru in the form of single atoms and clusters. As the Ru loading increased, the nanoparticles agglomerated, which resulted in reduced dispersion. Comparing 1 wt% Ru-loaded catalysts, Ru was uniformly sized on NA-Al2O3 than on com-Al2O3 (Fig. S4, ESI†). The XRD patterns of all the samples showed no detectable peaks corresponding to either Ru (JCPDS 06-0663) or RuO2 (JCPDS 43-1027), which indicates a high Ru dispersion (Fig. S5, ESI†). The EDS elemental mapping further confirmed the even distribution of the Ru nanoparticles across the catalyst surface (Fig. S6, ESI†). | ||
| Fig. 1 Preparation of Ru/NA-Al2O3 catalysts with various Ru contents. (a) Synthesis scheme of xRu/NA-Al2O3 (x = 0.5, 1, 5, and 8 wt% Ru). (b) TEM images of NA-Al2O3. HAADF-STEM images of (c) 0.5Ru/NA-Al2O3, (d) 1Ru/NA-Al2O3, (e) 5Ru/NA-Al2O3, (f) 8Ru/NA-Al2O3, and (g) 1Ru/com-Al2O3. A Ru single atom is indicated by an orange dotted circle in (c). | ||
Dispersion mechanism of Ru over Al2O3
γ-Al2O3 generally has a cubic defective spinel structure, where Al3+ cations occupy both tetrahedral (AIV3+) and octahedral (AVI3+) sites.30 Unlike the typical spinel structure (AB2O4), γ-Al2O3 contains only Al3+ ions, and therefore, requires cation vacancies to maintain the stoichiometric balance. The surface of γ-Al2O3 features coordinatively unsaturated pentacoordinate Al3+ (AlV3+), diverse surface hydroxyl groups, and cationic vacancies. Kwak et al. reported that AlV3+ sites created through dehydration and dehydroxylation acted as anchoring sites that enhanced the dispersion of Pt on the support.16 Wang et al. showed that the terminal hydroxyl groups on the (100) surface acted as anchoring sites for Ag.17 Penkova et al. demonstrated that cationic vacancies on Al2O3 could accommodate Ni2+ and Mg2+, which formed NiAl2O4 and MgAl2O4 spinels, respectively.31 Therefore, understanding these surface properties of Al2O3 is critical to modulate the dispersion and structure of the active metal species.Solid-state 27Al-NMR spectroscopy was used to probe the coordination environment of the Al3+ cations. The spectra showed signals corresponding to AIVI3+, AIV3+, and AIIV3+ sites at 10, 34, and 70 ppm, respectively (Fig. 2(a) and (b)).16 NA-Al2O3 exhibited a slightly higher proportion of AlV3+ ions, compared to com-Al2O3. However, owing to the low intensity of these AlV3+ sites, it can be inferred that Ru may also be anchored at other surface sites. This disparity in the NMR results was attributed to the structural differences between NA-Al2O3 and com-Al2O3, which influence the Ru deposition on Al2O3.
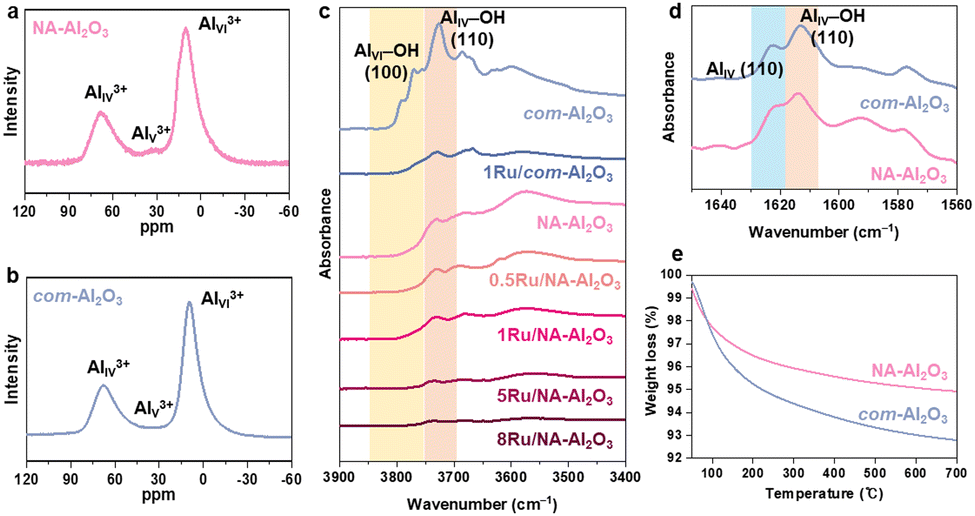 | ||
| Fig. 2 Characterization of Ru/NA-Al2O3 catalysts. Solid-state 27Al-NMR spectra of (a) NA-Al2O3 and (b) com-Al2O3. (c) In situ DRIFT spectra in OH stretching regions of NA-Al2O3, com-Al2O3, xRu/NA-Al2O3 and 1Ru/com-Al2O3 at 25 °C. (d) DRIFT (normalized to the intensity at 1613 cm−1) adsorbed in pyridine at 100 °C. (e) TGA analysis of NA-Al2O3 and com-Al2O3. | ||
To further elucidate the surface characteristics of NA-Al2O3, in situ DRIFT measurements were performed (Fig. 2(c) and Fig. S7a, ESI†). The DRIFT spectra revealed the presence of various surface hydroxyl groups in the 3900 to 3400 cm−1 region, mainly categorized as terminal hydroxyl (3790–3720 cm−1), bridged hydroxyl (3690 cm−1), and tri-bridged hydroxyl groups and H bonded (3590 cm−1).32–35 In NA-Al2O3, no peaks corresponding to AlVI–OH (100) at 3770 cm−1 and 3790 cm−1 were observed and only the peak associated with AlIV–OH (110) at 3730 cm−1 was detected.34 In contrast, com-Al2O3 exhibited both the AlVI–OH (100) and AlIV–OH (110) peaks. The intensity of the 3770 cm−1 peak correlates with the (100) facet ratio of Al2O3.36 This suggests that NA-Al2O3 had a much lower (100) facet ratio because it predominantly exposed the (110) facet owing to its nanosheet structure.19 After Ru loading, a reduction in the hydroxyl groups was observed in both the Al2O3 samples, which indicates that the terminal hydroxyl groups on the (110) facet of NA-Al2O3 and on both the (100) and (110) facets of com-Al2O3 were involved in Ru anchoring. 1Ru/com-Al2O3 exhibited a broader Ru size distribution than 1Ru/NA-Al2O3, probably because Ru was deposited on both the (100) and (110) facets of com-Al2O3 (Fig. S4, ESI†).
The role of hydroxyl groups in metal deposition is facet-dependent.18,37–39 Yang et al. argued that hydroxyl groups on the (100) facet inhibit the agglomeration of large Ru clusters, whereas those on the (110) facet promote it.18 In both NA-Al2O3 and com-Al2O3, the (110) facet appears to be involved in Ru anchoring. The differences in the Ru particle formation between the two supports can be attributed to variations in the hydroxyl density of the (110) facet. Pyridine-adsorbed DRIFT spectra, particularly in the 1625–1570 cm−1 range corresponding to Lewis acid sites (Al3+) and hydrogen-bonded hydroxyls, provided further insights into the coordination environment (Fig. 2(d) and Fig. S7b, S8, ESI†).31,32,40 Peaks at 1622 cm−1 correspond to the AlIV (110) sites, while those at 1613 cm−1 correspond to the AlIV–OH (110) sites.40 NA-Al2O3 exhibited a higher ratio of AlIV (110) to AlIV–OH (110) compared to com-Al2O3 (Fig. 2(d)), which indicates that NA-Al2O3 had fewer hydroxyl groups on its (110) facet. After Ru deposition, both the peaks diminished in intensity, which further confirmed the involvement of these sites in Ru anchoring (Fig. S8, ESI†). The relatively low hydroxyl group density on the (110) facet of NA-Al2O3 minimized the interference with Ru–Al2O3 interactions and facilitated stronger adsorption of Ru on the surface TGA showed that the weight loss above 200 °C, which was attributed to hydroxyl group desorption,41 was higher for com-Al2O3 (2.46%) than for NA-Al2O3 (1.57%) (Fig. 2(e)).
In situ DRIFT and pyridine-adsorbed DRIFT results (Fig. 2(c), (d) and Fig. S7, S8, ESI†) suggest that both the hydroxyl groups and Lewis acid sites in Al2O3 played crucial roles in Ru anchoring. However, the interaction between Ru and Al2O3 depends on the presence of anchoring sites and on their spatial distribution as well. For NA-Al2O3, the Ru deposition predominantly occurred on the (110) facet. The lower density of hydroxyl groups on (110) facet resulted in strong Ru–Al2O3 interactions, formed smaller and uniformly dispersed Ru particles, and ultimately influenced the performance of the catalyst in polyethylene (PE) hydrogenolysis.
Structural characterizations of Ru/Al2O3 catalysts
The H2-TPR analysis revealed two distinct reduction peaks (Fig. S9, ESI†): a low-temperature reduction peak at 136 °C, corresponding to bulk RuO2, and a high-temperature reduction peak at 160 °C, associated with Ru–Al2O3.42 As the Ru size increases, the intensity of the low-temperature reduction peak gradually increases, eventually becoming dominant in 5Ru/NA-Al2O3 and 8Ru/NA-Al2O3 catalysts. This trend suggests that Ru aggregation intensifies in these catalysts, leading to the formation of bulk Ru species. This phenomenon is further corroborated by HAADF-STEM images (Fig. 1(e) and (f)), which confirm the presence of aggregated Ru particles in high-loading catalysts. Additionally, when comparing catalysts with the same Ru content but different supports—1Ru/com-Al2O3 and 1Ru/NA-Al2O3—the latter exhibits a higher reduction temperature and lower intensity at lower reduction temperatures. This indicates a stronger interaction between Ru and NA-Al2O3, which enhances Ru dispersion and stability on the support.43To investigate the oxidation states of Ru in various Ru/Al2O3 catalysts, Ru K-edge X-ray absorption near edge structure (XANES) spectra were obtained (Fig. 3(a)). The white-line intensity for all the Ru catalysts was located between the reference spectra for RuO2 and the Ru foil. The 0.5Ru/NA-Al2O3 catalyst exhibited an oxidation state closer to RuO2 (Ru4+), while higher Ru loadings shifted the oxidation state towards that of metallic Ru (Ru0). Among the catalysts with 1 wt% Ru, 1Ru/NA-Al2O3 was more oxidized than 1Ru/com-Al2O3, suggesting a stronger Ru–NA-Al2O3 interaction, stabilizing Ru in a more oxidic state. These findings are further supported by the Ru 3p XPS results (Fig. S10† and Table S2, ESI†), where the Ru 3p spectra were deconvoluted into four peaks corresponding to Ru4+ 3p3/2 (463.3 eV), Ru4+ 3p1/2 (485.5 eV), Ru0 3p3/2 (461.5 eV), and Ru0 3p1/2 (483.7 eV).44,45 The variation in the Ru oxidation states can be attributed to the type of support and Ru loading, which also influence the particle size and dispersion.
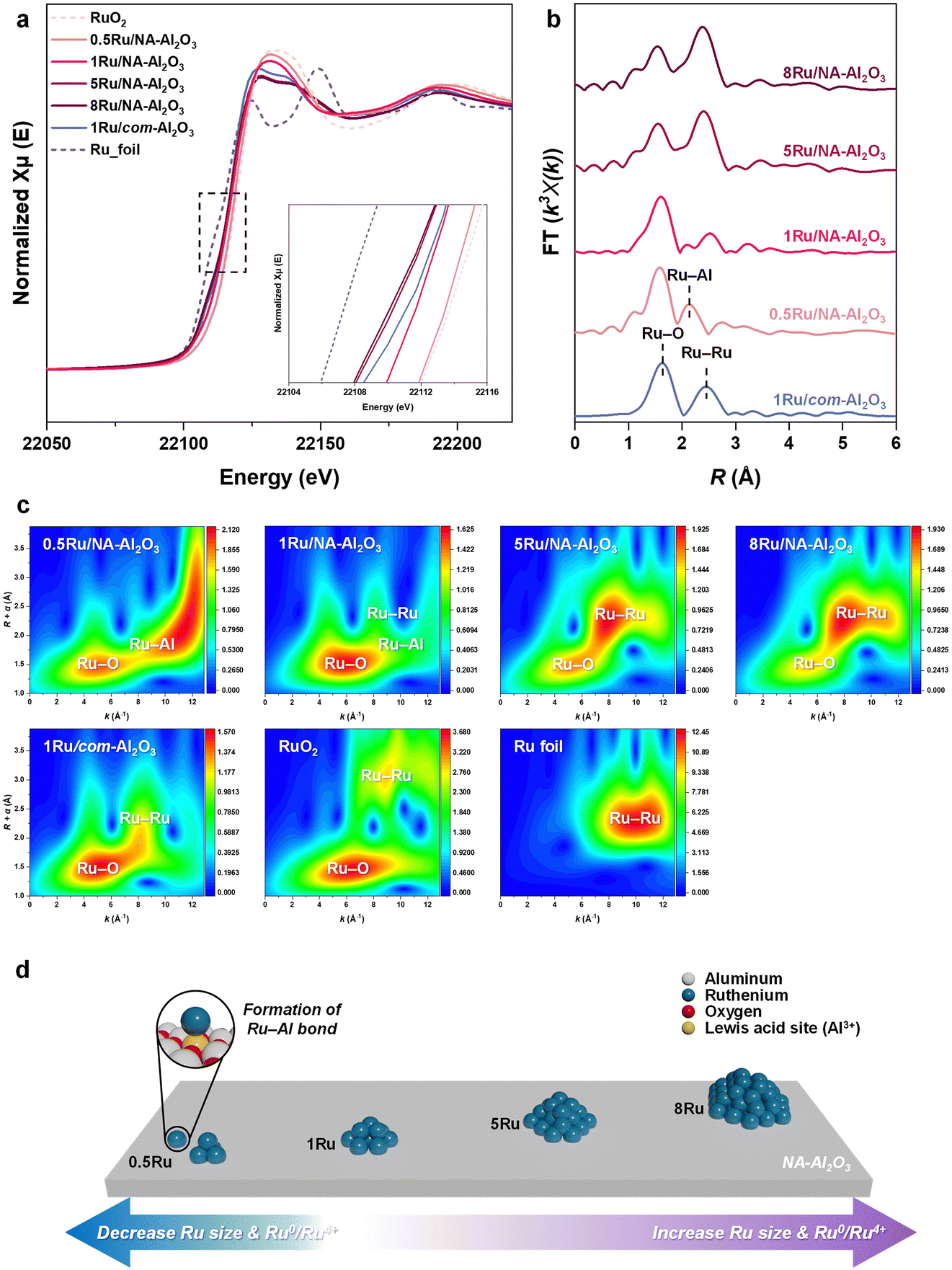 | ||
| Fig. 3 XAFS analysis of Ru/NA-Al2O3 catalysts. (a) Ru K-edge XANES spectra. (b) Ru K-edge EXAFS spectra in R-space. (c) Ru K-edge WT-EXAFS (d) schematic illustration of the changes in Ru oxidation state and structure depending on the Ru content in NA-Al2O3. | ||
The Fourier transform of the k3-weighted Ru K-edge extended X-ray absorption fine structure (EXAFS) spectra provided more detailed insights into the Ru coordination environment (Fig. 3(b)). For the more oxidic samples, including the 0.5Ru/NA-Al2O3, 1Ru/NA-Al2O3 and 1Ru/com-Al2O3 catalysts, a prominent peak corresponding to Ru–O coordination was observed. In contrast, the more metallic 5Ru/NA-Al2O3 and 8Ru/NA-Al2O3 catalysts exhibited significant peaks corresponding to both Ru–O and Ru–Ru coordination, which suggests a shift toward metallic Ru with increasing Ru content (Table S3 and Fig. S11, S12, ESI†). This progression indicates that with higher Ru loadings, the Ru species transitioned from being primarily oxidic to having a metallic character. Interestingly, the 0.5Ru/NA-Al2O3 and 1Ru/NA-Al2O3 catalysts exhibited a distinct peak corresponding to Ru–Al coordination, which appeared at a bond length longer than Ru–O coordination but shorter than metallic Ru–Ru coordination.
Wavelet transform (WT)-EXAFS analysis (Fig. 3(c)) showed that the 0.5Ru/NA-Al2O3 catalyst exhibited a very distinctive trend at high k values (8–12 Å−1), which was attributed to the formation of Ru–Al. The peak appearing at low R and k values in this region is due to Ru–Al coordination.46 Additionally, peaks were observed at higher R and k values due to Ru atoms considering that heavier atoms contribute at higher k values.47 This is different from the Ru–Ru observed in Ru foil and RuO2, indicating the formation of unique Ru species due to Ru–Al bond formation. With increasing Ru content, the Ru–Ru bonding becomes more dominant, indicating more agglomeration of Ru particles and metallic characteristics. In the 1Ru/com-Al2O3 catalyst, the WT-EXAFS peak trend related to Ru–Al formation observed in 1Ru/NA-Al2O3 was absent, and there was no Ru–Al peak in FT-EXAFS analysis either. Therefore, the Ru–Al bond is a unique feature between NA-Al2O3 and Ru, playing a crucial role in the formation of highly dispersed Ru species. The combined characterization results demonstrated that the structure and oxidation state of Ru is significantly influenced by both the support type and Ru loading (Fig. 3(d)), which is expected to have a significant impact on the catalytic performance in PE hydrogenolysis.
Polyethylene (PE) hydrogenolysis
The catalytic performance of the Ru/NA-Al2O3 series was evaluated for PE hydrogenolysis in a batch reactor (Fig. 4). At 220 °C for 4 h, 1Ru/NA-Al2O3 exhibited higher liquid/wax yields and lower solid residue compared to 1Ru/com-Al2O3, demonstrating its superior efficiency in PE hydrogenolysis (Fig. 4(b) and Table S4, ESI†). At 250 °C for 2.5 h, 1Ru/NA-Al2O3 achieved an almost complete conversion (96.2%), yielding the highest liquid/wax fraction (C5–C30+) at 78.9% (Fig. 4(b), Fig. S13 and Table S4, ESI†). The liquid/wax fraction was found to consist predominantly of n-alkanes, indicating that the reaction proceeds primarily through a hydrogenolysis pathway (Fig. S14, S15 and Table S5, ESI†).5,48,49 1Ru/NA-Al2O3 catalyst also exhibited the highest conversion rate (1.15 × 103 gconvertedPE gRu−1 h−1) and liquid/wax production rate (9.23 × 102 gLiquid/wax gRu−1 h−1), surpassing all other Ru/NA-Al2O3 catalysts (Fig. 4(c)). The liquid/wax yield (C5–C30+) followed the trend: 1Ru/NA-Al2O3 > 0.5Ru/NA-Al2O3 > 1Ru/com-Al2O3 > 5Ru/NA-Al2O3 > 8Ru/NA-Al2O3. Notably, 1Ru/NA-Al2O3 outperformed previously reported Ru-based PE hydrogenolysis catalysts (Fig. S16 and Tables S6, S7, ESI†). Post-reaction characterization of 1Ru/NA-Al2O3 after 250 °C for 2.5 h confirmed that the catalyst retained its Ru composition and particle size with minimal changes, demonstrating high structural stability (Fig. S17, ESI†). XPS analysis further revealed an increase in the Ru0/Ru4+ ratio, attributed to the reductive reaction environment (Fig. S17d, ESI†). The catalyst stability was further verified through recycling tests, where 1Ru/NA-Al2O3 maintained its activity across multiple cycles (Fig. S18, ESI†). A time-dependent analysis of PE conversion and product distribution indicated that, at approximately 96% conversion, 1Ru/com-Al2O3 exhibited higher gas production, whereas 1Ru/NA-Al2O3 favored the formation of liquid/wax (C5–C30+) products (Fig. 4(d), (e), Fig. S19 and Table S4, ESI†). These results strongly suggest that 1Ru/NA-Al2O3 is inherently more selective toward liquid/wax production, making it a superior catalyst for PE hydrogenolysis.
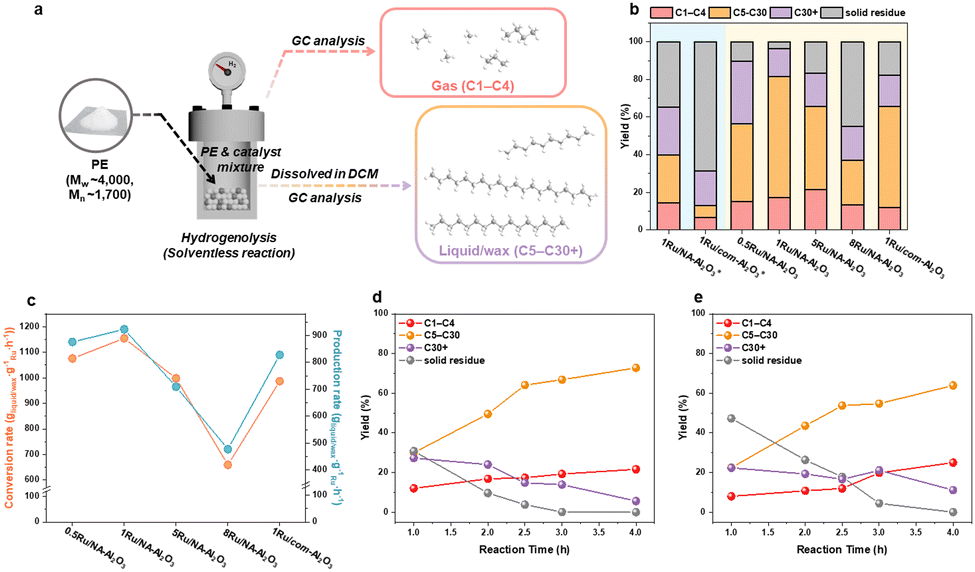 | ||
| Fig. 4 PE hydrogenolysis performances of Ru/NA-Al2O3 catalysts. (a) Schematic illustration of PE hydrogenolysis. (b) PE conversion and product yield. (c) PE conversion rate and production rate. (d), (e) Reactivity changes over time for (d) 1Ru/NA-Al2O3 and (e) 1Ru/com-Al2O3 catalysts. Standard reaction conditions: 3.0 g PE (Mw ∼4000, Mn ∼ 1700), 1 mg Ru, 250 °C, 40 bar H2, 2.5 h. * The asterisk indicates that reaction temperature and reaction time for PE hydrogenolysis were 220 °C and 4 h, respectively. | ||
Mechanistic insights from model hydrocarbon studies
Despite exhibiting a conversion rate comparable to 1Ru/com-Al2O3, 5Ru/NA-Al2O3 demonstrated a lower liquid/wax production rate (Fig. 4(c)). A detailed product distribution analysis revealed that while the total non-solid yield (gas, liquid, wax) of 5Ru/NA-Al2O3 and 1Ru/com-Al2O3 were similar, 5Ru/NA-Al2O3 produced significantly more gas. This increase in gas formation is primarily attributed to an enhanced occurrence of successive and terminal C–C bond cleavage events.5,9,10 A quantitative evaluation of C–C bond cleavage activity was conducted using hydrogen consumption measurements, based on the principle that one H2 molecule is consumed per C–C bond cleavage event.50 The results revealed a strong deviation between overall conversion trends and C–C bond cleavage activity (Fig. S20, ESI†). Specifically, C–C bond cleavage activity followed the trend: 0.5Ru/NA-Al2O3 < 1Ru/NA-Al2O3 < 8Ru/NA-Al2O3 < 1Ru/com-Al2O3 < 5Ru/NA-Al2O3, peaking at 5Ru/NA-Al2O3, before decreasing in 8Ru/NA-Al2O3. This trend suggests that 5Ru/NA-Al2O3 promotes excessive successive C–C bond cleavage, leading to higher gas formation and hydrogen consumption.To gain deeper mechanistic insights into C–C bond cleavage behavior, we conducted hydrogenolysis of n-C18H38 as a model reaction (Fig. 5(a) and Fig. S21, ESI†). The C–C bond cleavage frequency and product distribution were evaluated under low conversion conditions (20–30%), allowing a direct comparison with PE hydrogenolysis trends. Despite differences in absolute conversion efficiency, the C–C bond cleavage activity exhibited a similar trend, decreasing in the order: 5Ru/NA-Al2O3 > 8Ru/NA-Al2O3 > 1Ru/com-Al2O3 > 1Ru/NA-Al2O3 > 0.5Ru/NA-Al2O3 (Fig. 5(a)).
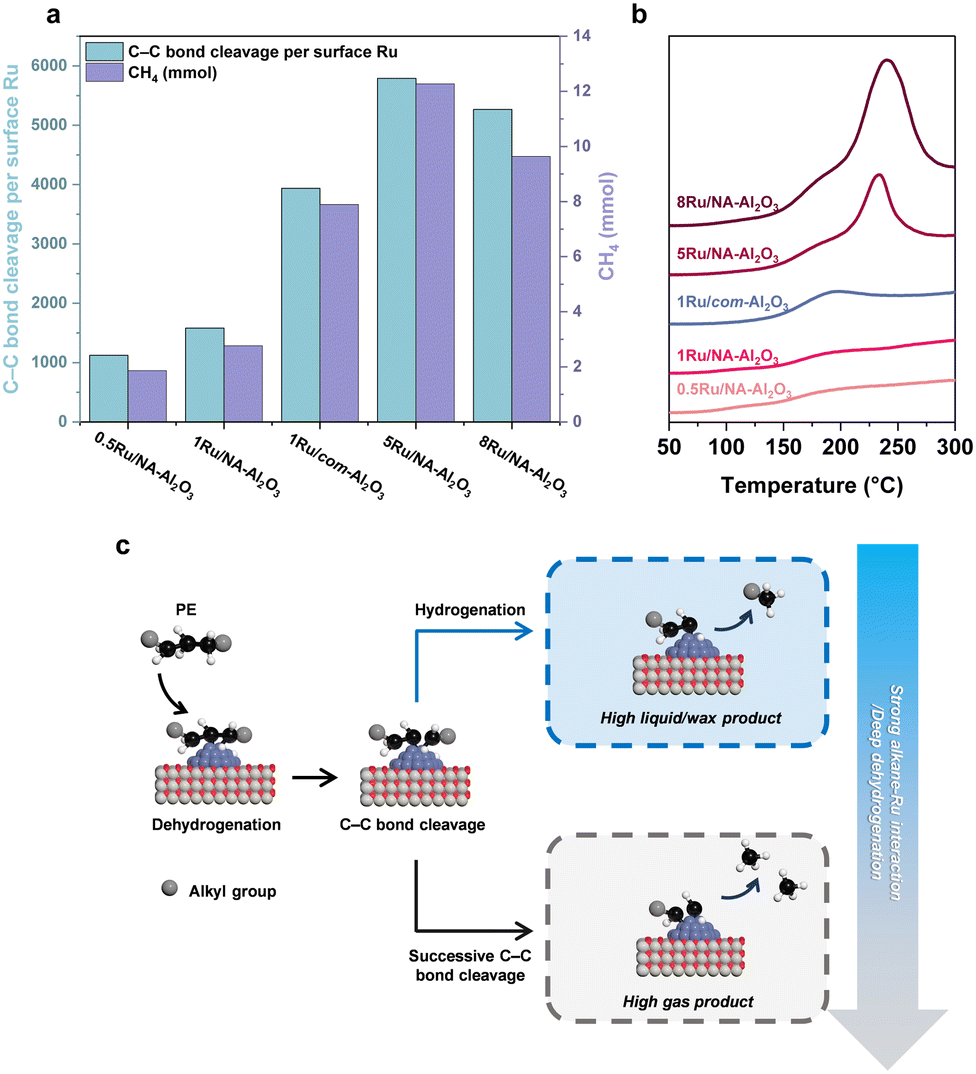 | ||
| Fig. 5 (a) C–C bond cleavage per surface Ru and methane production at similar conversions (20–30%) in C18H38 hydrogenolysis. Reaction conditions: 3.0 g C18H38, 0.5 mg Ru, 250 °C, 40 bar H2. The reaction times were 30 min for 0.5Ru/NA-Al2O3, 20 min for 1Ru/NA-Al2O3 and 1Ru/com-Al2O3, and 10 min for 5Ru/NA-Al2O3 and 8Ru/NA-Al2O3. (b) C10H22-TPD of the as-prepared Ru catalysts. (c) Schematic illustration of PE hydrogenolysis. | ||
Similar to PE hydrogenolysis, 5Ru/NA-Al2O3 exhibited the highest C–C bond cleavage activity, while 8Ru/NA-Al2O3 showed reduced activity and lower methane formation, likely due to a reduction in C–C bond cleavage activity caused by an increase in Ru particle size beyond a certain threshold.6 In contrast, 0.5Ru/NA-Al2O3, while exhibiting low C–C bond cleavage activity, produced a remarkably uniform product distribution with minimal methane formation (Fig. S22, ESI†). These findings underscore that higher C–C bond cleavage activity does not necessarily correlate with enhanced catalytic performance. Instead, selective control over C–C bond cleavage pathways is crucial for optimizing product distribution.
Methane formation in alkane hydrogenolysis using Ru catalysts is typically attributed to two competing pathways:
• Terminal C–C bond cleavage, leading to the production of C17H36 in n-C18H38 hydrogenolysis.
• Successive C–C bond cleavage, causing over-hydrogenolysis and excessive CH4 formation.
A progressive increase in terminal C–C bond cleavage was observed in the order: 0.5Ru/NA-Al2O3 < 1Ru/NA-Al2O3 < 1Ru/com-Al2O3 < 5Ru/NA-Al2O3 < 8Ru/NA-Al2O3 (Fig. S22, ESI†).9 However, the most significant contributor to methane formation was successive C–C bond cleavage, which drastically reduced catalytic efficiency. For instance, if 17 consecutive C–C bond cleavages occur within a single n-C18H38 molecule, 18 moles of CH4 are produced, with an effective conversion of only 1 mole of n-C18H38. Conversely, 17 independent C–C bond cleavage events yield 17 moles of useful hydrocarbons, highlighting the importance of selectivity control. The strong correlation between methane formation and successive C–C bond cleavage suggests that 5Ru/NA-Al2O3and 8Ru/NA-Al2O3 exhibit excessive Ru-alkane interactions, leading to undesirable product distributions.
Factors contributing to successive C–C bond cleavage
Successive C–C bond cleavage in alkane hydrogenolysis using Ru catalysts can be attributed to two key factors: (i) variations in the interaction strength between Ru and alkanes and (ii) deep dehydrogenation (Fig. 5(c)).5,10,51 To investigate the first factor, C10H22-TPD analysis was performed (Fig. 5(b)). The measurements were conducted below 300 °C to avoid alkane pyrolysis.52,53 The desorption profiles were analyzed to determine alkane adsorption behavior. The intensity of desorption signals increased with higher Ru content, indicating that alkane adsorption preferentially occurs on Ru sites. Two distinct desorption peaks were observed: one at 196 °C, which was present across all catalyst groups, and another at higher temperatures (233 °C and 240 °C) for 5Ru/NA-Al2O3 and 8Ru/NA-Al2O3, respectively. These results suggest that Ru–alkane interactions are stronger in 5Ru/NA-Al2O3 and 8Ru/NA-Al2O3, whereas the interaction is comparatively weaker in 0.5Ru/NA-Al2O3, 1Ru/NA-Al2O3, and 1Ru/com-Al2O3.54 This trend is further corroborated by TEM and H2-TPR (Fig. 1(e), (f) and Fig. S9, ESI†), which confirm that 5Ru/NA-Al2O3 and 8Ru/NA-Al2O3 contain bulk Ru species. The formation of bulk Ru particles increases the proportion of terrace sites, thereby enabling multiple interactions with alkanes and strengthening Ru–alkane adsorption.10,55,56 This strong interaction impedes alkane desorption after initial C–C bond cleavage, leading to successive cleavage events. As observed in n-C18H38 hydrogenolysis, these catalysts exhibited high C–C bond cleavage activity but poor product selectivity, resulting in elevated methane formation. Similarly, in PE hydrogenolysis, 5Ru/NA-Al2O3 and 8Ru/NA-Al2O3 promoted excessive gas formation relative to conversion, indicating their low catalytic efficiency for selective liquid/wax production. In contrast, 0.5Ru/NA-Al2O3, 1Ru/NA-Al2O3 and 1Ru/com-Al2O3 exhibited comparable alkane interaction strengths, suggesting that factors beyond Ru–alkane interactions influence C–C bond cleavage behavior. One such factor is deep dehydrogenation, which can serve as an alternative mechanism for successive C–C bond cleavage.5,51,57The extent of dehydrogenation in PE hydrogenolysis is governed by C–H bond activation, which is directly influenced by the electronic state of Ru.9,13,15 The activation of C–H bonds occurs via electron back-donation from Ru sites into the σ* orbitals of the C–H bond.58 XPS and XANES analyses (Fig. 3(a) and Fig. S10, ESI†) revealed that the Ru0/Ru4+ ratio follows the order: 0.5Ru/NA-Al2O3 < 1Ru/NA-Al2O3 < 1Ru/com-Al2O3 < 5Ru/NA-Al2O3 < 8Ru/NA-Al2O3. Furthermore, EXAFS and H2-TPR analyses (Fig. 3(b) and Fig. S9, ESI†) indicate that Ru–Al bond formation in NA-Al2O3-supported catalysts enhances metal–support interactions, leading to higher Ru oxidation states for 1Ru/NA-Al2O3 relative to 1Ru/com-Al2O3 at equivalent Ru loadings. As the Ru0 fraction increases, C–H bond activation intensifies. While moderate C–H activation is essential for initiating dehydrogenation, excessive activation leads to deep dehydrogenation, which facilitates uncontrolled successive C–C bond cleavage.
In the case of highly oxidized 0.5Ru/NA-Al2O3, successive C–C bond cleavage was significantly suppressed compared to other catalysts. However, this advantage came at the expense of lower overall C–C bond cleavage activity, limiting its hydrogenolysis efficiency. Conversely, 1Ru/com-Al2O3, while exhibiting higher C–C bond cleavage activity, also displayed more pronounced successive C–C bond cleavage, leading to uncontrolled fragmentation. Given these findings, 1Ru/NA-Al2O3 emerges as the most effective catalyst, as it maintains an optimal Ru0/Ru4+ balance, thereby achieving high catalytic activity while simultaneously regulating successive C–C bond cleavage. This selective control over C–C bond cleavage pathways is critical for maximizing the production of liquid/wax hydrocarbons while minimizing undesirable gas formation.
Conclusions
The hydrogenolysis of polyolefin plastics, particularly PE, into valuable liquid/wax fuels has emerged as a promising strategy to address both rising energy demands and plastic waste challenges. This study systematically investigated the interactions between Ru and various Al2O3 supports, along with the geometric and electronic effects of Ru loading, to elucidate their influence on catalytic reactivity in PE hydrogenolysis.Comprehensive catalyst characterization demonstrated that optimal Ru loading on NA-Al2O3 facilitates Ru–Al bond formation, resulting in smaller, highly dispersed, and more oxidic Ru species compared to com-Al2O3. The 1Ru/NA-Al2O3 catalyst, featuring optimally sized Ru particles (∼0.8 nm) and a finely tuned electronic structure, effectively suppressed successive C–C bond cleavage, leading to enhanced selectivity toward liquid/wax hydrocarbons. This catalyst achieved an exceptional PE conversion rate of 1.15 × 103 gconverted PE gRu−1 h−1 and a liquid/wax production rate of 9.23 × 102 gLiquid/wax gRu−1 h−1, significantly outperforming other Ru/Al2O3 catalysts. This outcome underscores the critical role of metal–support interactions and precise control over geometric and electronic properties in optimizing the hydrogenolysis process. Overall, this work contributes to the broader effort to develop efficient catalytic strategies for polyolefin recycling, offering a sustainable approach to mitigating plastic waste while simultaneously generating valuable energy resources.
Author contributions
J. K. and D. K. performed experiments, characterized catalysts, and analyzed data. B. P., D. O., S. L, J. K. and E. N. prepared the materials and contributed to characterization. K. A. supervised the study and edited the manuscript. All authors discussed the results and commented on the manuscript.Data availability
All data generated or analyzed during this study are included in this published article and its ESI† files.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by Basic Science Research Program (2021R1A2C2006713) and the Engineering Research Center of Excellence Program (2020R1A5A1019631) of the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT, Ministry of Education (MOE).References
- O. O. Yolcan, Innov. Green Dev., 2023, 2, 100070 CrossRef.
- B. Baytekin, H. T. Baytekin and B. A. Grzybowski, Energy Environ. Sci., 2013, 6, 3467–3482 RSC.
- OECD, Global plastics outlook: Economic drivers, environmental impacts and policy options, OECD Publishing, 2022 Search PubMed.
- US Department of Energy, Distribution of plastic production worldwide in 2018, by type, https://www.statista.com/statistics/968808/distribution-of-global-plastic-production-by-type/, (accessed May 29, 2024).
- C. Wang, T. Xie, P. A. Kots, B. C. Vance, K. Yu, P. Kumar, J. Fu, S. Liu, G. Tsilomelekis, E. A. Stach, W. Zheng and D. G. Vlachos, JACS Au, 2021, 1, 1422–1434 CrossRef CAS PubMed.
- J. A. Sun, P. A. Kots, Z. R. Hinton, N. S. Marinkovic, L. Ma, S. N. Ehrlich, W. Zheng, T. H. Epps III, L. T. Korley and D. G. Vlachos, ACS Catal., 2024, 14, 3228–3240 CrossRef CAS.
- G. Celik, R. M. Kennedy, R. A. Hackler, M. Ferrandon, A. Tennakoon, S. Patnaik, A. M. LaPointe, S. C. Ammal, A. Heyden, F. A. Perras, M. Pruski, S. L. Scott, K. R. Poeppelmeier, A. D. Sadow and M. Delferro, ACS Cent. Sci., 2019, 5, 1795–1803 CrossRef CAS PubMed.
- X. Wu, A. Tennakoon, R. Yappert, M. Esveld, M. S. Ferrandon, R. A. Hackler, A. M. LaPointe, A. Heyden, M. Delferro, B. Peters, A. D. Sadow and W. Huang, J. Am. Chem. Soc., 2022, 144, 5323–5334 CrossRef CAS PubMed.
- L. Chen, L. C. Meyer, L. Kovarik, D. Meira, X. I. Pereira-Hernandez, H. Shi, K. Khivantsev, O. Y. Gutierrez and J. Szanyi, ACS Catal., 2022, 12, 4618–4627 CrossRef CAS.
- M. Tamura, S. Miyaoka, Y. Nakaji, M. Tanji, S. Kumagai, Y. Nakagawa, T. Yoshioka and K. Tomishige, Appl. Catal., B, 2022, 318, 121870 CrossRef CAS.
- S. D. Jaydev, A. J. Martín and J. Pérez-Ramírez, ChemSusChem, 2021, 14, 5179–5185 CrossRef CAS PubMed.
- M. Chu, X. Wang, X. Wang, X. Lou, C. Zhang, M. Cao, L. Wang, Y. Li, S. Liu, T.-K. Sham, Q. Zhang and J. Chen, Research, 2023, 6, 0032 CrossRef CAS PubMed.
- H. Ji, X. Wang, X. Wei, Y. Peng, S. Zhang, S. Song and H. Zhang, Small, 2023, 19, 2300903 CrossRef CAS PubMed.
- S. S. Borkar, R. Helmer, S. Panicker and M. Shetty, ACS Sustainable Chem. Eng., 2023, 11, 10142–10157 CrossRef CAS.
- P. Hu, C. Zhang, M. Chu, X. Wang, L. Wang, Y. Li, T. Yan, L. Zhang, Z. Ding, M. Cao, P. Xu, Y. Li, Y. Cui, Q. Zhang, J. Chen and L. Chi, J. Am. Chem. Soc., 2024, 146, 7076–7087 CrossRef CAS PubMed.
- J. H. Kwak, J. Hu, D. Mei, C.-W. Yi, D. H. Kim, C. H. Peden, L. F. Allard and J. Szanyi, Science, 2009, 325, 1670–1673 CrossRef CAS PubMed.
- F. Wang, J. Ma, S. Xin, Q. Wang, J. Xu, C. Zhang, H. He and X. Cheng Zeng, Nat. Commun., 2020, 11, 529 CrossRef CAS PubMed.
- J. Yang, H. Wang, X. Zhao, Y. Li and W. Fan, RSC Adv., 2016, 6, 40459–40473 RSC.
- X. Yang, Q. Li, E. Lu, Z. Wang, X. Gong, Z. Yu, Y. Guo, L. Wang, Y. Guo, W. Zhan, J. Zhang and S. Dai, Nat. Commun., 2019, 10, 1611 CrossRef PubMed.
- W. Liu, S. Yang, Q. Zhang, T. He, Y. Luo, J. Tao, D. Wu and H. Peng, Appl. Catal., B, 2021, 292, 120171 CrossRef CAS.
- W. Cai, J. Yu, S. Gu and M. Jaroniec, Cryst. Growth Des., 2010, 10, 3977–3982 CrossRef CAS.
- L. A. Calzada, S. E. Collins, C. W. Han, V. Ortalan and R. Zanella, Appl. Catal., B, 2017, 207, 79–92 CrossRef CAS.
- K. P. de Jong, Synthesis of solid catalysts, John Wiley & Sons, 2009 Search PubMed.
- J. Lee, B. G. Park, K. Sung, H. Lee, J. Kim, E. Nam, J. W. Han and K. An, ACS Catal., 2023, 13, 13691–13703 CrossRef CAS.
- B. G. Park, H. Lee, J. Lee, E. Nam, J.-S. Bae and K. An, Catal. Today, 2024, 425, 114339 CrossRef CAS.
- X. Krokidis, P. Raybaud, A.-E. Gobichon, B. Rebours, P. Euzen and H. Toulhoat, J. Phys. Chem. B, 2001, 105, 5121–5130 CrossRef CAS.
- G. Busca, Catal. Today, 2014, 226, 2–13 CrossRef CAS.
- S. Zhang, L. Tang, J. Yu, W. Zhan, L. Wang, Y. Guo and Y. Guo, ACS Appl. Mater. Interfaces, 2021, 13, 58605–58618 CrossRef CAS PubMed.
- J. Wu, G. Zhao, M. Song, H. Wang, Y. Wei, X. Chen, G. Wang and Z. Yan, Fuel, 2022, 329, 125381 CrossRef CAS.
- B. Lippens and J. De Boer, Acta Crystallogr., 1964, 17, 1312–1321 CrossRef CAS.
- A. Penkova, L. F. Bobadilla, F. Romero-Sarria, M. A. Centeno and J. A. Odriozola, Appl. Surf. Sci., 2014, 317, 241–251 CrossRef CAS.
- C. Morterra and G. Magnacca, Catal. Today, 1996, 27, 497–532 CrossRef CAS.
- M. Digne, P. Sautet, P. Raybaud, P. Euzen and H. Toulhoat, J. Catal., 2002, 211, 1–5 CrossRef CAS.
- K. Khivantsev, N. R. Jaegers, J. H. Kwak, J. Szanyi and L. Kovarik, Angew. Chem., Int. Ed., 2021, 133, 17663–17671 CrossRef.
- H. Knözinger and P. Ratnasamy, Catal. Rev.: Sci. Eng., 1978, 17, 31–70 CrossRef.
- J. Lee, E. J. Jang, H. Y. Jeong and J. H. Kwak, Appl. Catal., A, 2018, 556, 121–128 CrossRef CAS.
- J. Yang, X. Zhao, S. Bu and W. Fan, J. Phys. Chem. C, 2018, 122, 17287–17300 CrossRef CAS.
- Z. Liu, Y. Wang, J. Li and R. Zhang, RSC Adv., 2014, 4, 13280–13292 RSC.
- J. Li, R. Zhang and B. Wang, Appl. Surf. Sci., 2013, 270, 728–736 CrossRef CAS.
- M. Digne, P. Sautet, P. Raybaud, P. Euzen and H. Toulhoat, J. Catal., 2004, 226, 54–68 CrossRef CAS.
- Y. Fan, F. Wang, R. Li, C. Liu and Q. Fu, ACS Catal., 2023, 13, 2277–2285 CrossRef CAS.
- T. W. Kim, H.-J. Chun, Y. Jo, D. Kim, H. Ko, S. H. Kim, S. K. Kim and Y.-W. Suh, J. Catal., 2023, 428, 115178 CrossRef CAS.
- J. Zhou, Z. Gao, G. Xiang, T. Zhai, Z. Liu, W. Zhao, X. Liang and L. Wang, Nat. Commun., 2022, 13, 327 CrossRef CAS PubMed.
- H. Wang, X. Li, Q. Ruan and J. Tang, Nanoscale, 2020, 12, 12329–12335 RSC.
- L. Ge, M. Qiu, Y. Zhu, S. Yang, W. Li, W. Li, Z. Jiang and X. Chen, Appl. Catal., B, 2022, 319, 121958 CrossRef CAS.
- X. Liu, C. Xing, F. Yang, Z. Liu, Y. Wang, T. Dong, L. Zhao, H. Liu and W. Zhou, Adv. Energy Mater., 2022, 12, 2201009 CrossRef CAS.
- M. G. Siebecker and D. L. Sparks, J. Phys. Chem. A, 2017, 121, 6992–6999 CrossRef CAS PubMed.
- T. Kwon, B. Ahn, K. H. Kang, W. Won and I. Ro, Nat. Commun., 2024, 15, 10239 CrossRef CAS PubMed.
- L. Chen, J. B. Moreira, L. C. Meyer and J. Szanyi, Appl. Catal., B, 2023, 335, 122897 CrossRef CAS.
- S. Chen, A. Tennakoon, K.-E. You, A. L. Paterson, R. Yappert, S. Alayoglu, L. Fang, X. Wu, T. Y. Zhao, M. P. Lapak, M. Saravanan, R. A. Hackler, Y.-Y. Wang, L. Qi, M. Delferro, T. Li, B. Lee, B. Peters, K. R. Poeppelmeier, S. C. Ammal, C. R. Bowers, F. A. Perras, A. Heyden, A. D. Sadow and W. Huang, Nat. Catal., 2023, 6, 161–173 CrossRef CAS.
- C. Wang, K. Yu, B. Sheludko, T. Xie, P. A. Kots, B. C. Vance, P. Kumar, E. A. Stach, W. Zheng and D. G. Vlachos, Appl. Catal., B, 2022, 319, 121899 CrossRef CAS.
- J. Tuo and Y. Qian, Energy Fuels, 2004, 18, 1485–1493 CrossRef CAS.
- K. R. Paserba and A. J. Gellman, J. Chem. Phys., 2001, 115, 6737–6751 CrossRef CAS.
- H. Meng, Y. Yang, T. Shen, Z. Yin, J. Zhang, H. Yan and M. Wei, ACS Catal., 2023, 13, 9234–9244 CrossRef CAS.
- M. Zare, P. A. Kots, Z. R. Hinton, T. H. Epps, L. T. Korley, S. Caratzoulas and D. G. Vlachos, Appl. Catal., B, 2024, 351, 123969 CrossRef CAS.
- M. Zare, P. A. Kots, S. Caratzoulas and D. G. Vlachos, Chem. Sci., 2023, 14, 1966–1977 RSC.
- D. W. Flaherty, D. D. Hibbitts and E. Iglesia, J. Am. Chem. Soc., 2014, 136, 9664–9676 CrossRef CAS PubMed.
- J. Y. Saillard and R. Hoffmann, J. Am. Chem. Soc., 1984, 106, 2006–2026 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Reaction results and additional characterization; SEM, XRD, N2 adsorption–desorption isotherms, HAADF-STEM, EDS mapping, Pyridine-adsorbed DRIFT, EXAFS, XPS TEM, and data. See DOI: https://doi.org/10.1039/d5ey00070j |
| ‡ J. K. and D. K. contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |