
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstructing four-in-one catalysts to realize ultralow voltage hydrogen production at ampere-level current densities†
Minghui Xinga,
Mengting Hana,
Guoqing Xua,
Zhiping Liua,
Qinglan Zhao b,
Minhua Shaob,
Jimmy Yuncd,
Peng Wange and
Dapeng Cao*a
b,
Minhua Shaob,
Jimmy Yuncd,
Peng Wange and
Dapeng Cao*a
aState Key Laboratory of Organic–Inorganic Composites, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: caodp@buct.edu.cn
bDepartment of Chemical and Biological Engineering, Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, China
cQingdao International Academician Park Research Institute, Qingdao 266000, China
dSchool of Chemical Engineering, The University of New South Wales, Sydney, NSW 2052, Australia
eQihang New Energy Technology Co. Ltd, China Railway Rolling Stock Corporation, Beijing 100039, China
First published on 18th April 2025
Abstract
An anion exchange membrane water electrolyzer (AEMWE) is emerging as key technology for hydrogen production. However, its widespread application requires further reduction of cost and improvement of efficiencies. Here, we synthesize a four-in-one catalyst (VSA-CoNx) to achieve high-efficiency coupling hydrogen production by combining with the hydrazine oxidation reaction (HzOR) and the urea oxidation reaction (UOR). The as-synthesized VSA-CoNx exhibits excellent performance in all the four reactions of HzOR, UOR and hydrogen/oxygen evolution reactions (HER/OER). The HER–HzOR coupling system only requires an ultra-low voltage of 0.21 V to deliver an ampere-level current density (1 A cm−2), while the conventional HER–OER AEMWE needs nearly an input of 1.88 V. Remarkably, this HER–HzOR coupling system largely reduces the energy expenditure of the AEMWE by approximately 90%, which hits a record in the low energy cost for all water electrolysis systems known to date. Given the energy consumption of the traditional AEMWE of approximately 4.56 kW h Nm−3 of H2 at a current density of 1 A cm−2, the HER–HzOR AEM electrolyzer only requires 0.51 kW h Nm−3 of H2. This HER–HzOR coupling system not only significantly lowers the energy expenditure of large-scale H2 production but also addresses the hydrazine-associated environmental pollution.
Broader contextTo construct a sustainably developing society, hydrogen, as a promising clean energy carrier, stands out due to its high energy density and zero-carbon emissions. An anion exchange membrane water electrolyzer (AEMWE) to produce hydrogen has been considered as one of the most promising hydrogen production technologies. However, its high energy costs have severely limited the extensive applications. Using small molecule oxidation as an alternative to the oxygen evolution reaction (OER) may be a useful solution to reduce the energy expenditure of large-scale H2 production. In this work, we develop a four-in-one catalyst, which can catalyze four reactions, to achieve high-efficiency coupling hydrogen production by combining with the hydrazine oxidation reaction (HzOR) and the hydrogen evolution reaction (HER). The HER–HzOR coupling system only requires an ultralow voltage of 0.21 V to deliver an ampere-level current density (1 A cm−2), while the conventional HER–OER AEMWE needs nearly an input of 1.88 V, which largely reduces the energy expenditure of the AEMWE by approximately 90%, hitting a record in the low energy cost for all water electrolysis systems known to date. Simultaneously, this HER–HzOR coupling system also alleviates the hydrazine-associated environmental pollution. It is expected that this work provides a useful coupling strategy to reduce the energy expenditure of large-scale H2 production. |
1. Introduction
Building a sustainably developing society heavily relies on effective utilization of clean energy sources such as solar, wind, and hydrogen. As a promising energy carrier, hydrogen energy stands out due to its high energy density and zero carbon emissions.1 Harnessing renewable energies for water electrolyzers (WEs) has been increasingly considered as a highly promising approach to H2 production.2 The cathodic hydrogen evolution reaction (HER) and the anodic oxygen evolution reaction (OER) are usually coupled in a WE cell.3 Compared with the HER, the four-electron transfer OER is a relatively slow process with sluggish kinetics.4 The high theoretical overpotential (1.23V vs. reversible hydrogen electrode (RHE)) of the OER severely hinders its practical applications,5 which requires high energy cost in the overall water splitting (OWS) reaction.6 Previous studies mainly focused on optimizing the catalysts for the HER and/or OER to achieve high-performance OWS.7 To reduce the energy cost of OWS, the electrochemical hydrogen-production coupled with an alternative oxidation (EHCO) reaction may be a brand-new route for green H2 production.In the EHCO route, coupling the HER and thermodynamically favorable small molecule oxidation with relatively low theoretical potentials to replace the OER can not only reduce the energy consumption of the overall reaction, but also simultaneously yield high-value-added products or facilitate wastewater treatment in the anode.8 In fact, a series of organic molecular oxidation reactions, such as those involving methanol,9 ethanol,10 urea,11 5-hydroxymethylfurfural,12 and hydrazine,13 have been used to replace the OER. Among them, urea and hydrazine, as the common substances in wastewater, are harmful to the environment and human beings. Compared with the OER (1.23 V vs. RHE), the urea and hydrazine oxidation reactions (UOR/HzOR) can be performed at much lower standard potentials (0.37 V vs. RHE for UOR, and −0.33 V vs. RHE for HzOR). Therefore, the anodic UOR or HzOR coupled with the cathodic HER can not only effectively reduce the energy consumption of H2 production, but also provide an opportunity to purify wastewater rich in urea or hydrazine, demonstrating significant potential for practical applications.11d,14 In this scenario, developing high-performance multi-functional catalysts is extremely important, which not only targets desirable reactions with high selectivity, but also significantly streamlines the design and fabrication process of catalytic electrodes.15
Recently, various catalysts, including noble metals14c,15a,16 and non-noble metals,17 have been developed to improve efficiencies of H2 production. Heteroatom doping engineering has been widely used as an effective modification strategy of catalyst development, due to its synergistic effects in optimizing the electronic structure, regulating the adsorption of intermediates, and promoting electron and mass transfers.18 Previous studies have shown that the incorporation of W–O groups with strong adsorption capacity into CoP can serve as H2O dissociation and N2H4 dehydration sites, effectively promoting the coupling of the HER with the HzOR.13a Similarly, Mo-doping was reported to optimize the electronic structure and achieve thermoneutral ΔGH* for the HER, which therefore raises dehydrogenation kinetics from *N2H4 to *NHNH2 for the HzOR.19 However, few studies have been reported at ampere-level current density for large-scale H2 production. Actually, developing multifunctional catalysts with excellent HER, OER, UOR and HzOR activities at ampere-level current density is crucial for H2 production via an EHCO route.
Herein, we synthesize a single-atom V-doped CoNx four-in-one-functional catalyst (VSA-CoNx), where V doping can not only optimize the surface structure of catalysts, but also promote the generation of active substances and inhibit the accumulation of high-valent metal species in the high-voltage reaction process. The HER–UOR coupling system based on VSA-CoNx as bifunctional catalysts with anion exchange membrane (AEM) only requires an input of 1.64 V to deliver a current density of 1 A cm−2, while the HER–HzOR AEM coupling system even lowers the potential to 0.21 V under the same conditions, hitting a record in the low energy consumption for all water electrolysis systems known to date. This work offers a new route of devising multifunctional catalysts to achieve green H2 production at ampere-level current densities by the EHCO approach, paving the way for a sustainable future in green hydrogen development.
2. Results and discussion
2.1. Synthesis and structure characterization
The catalyst VSA-CoNx was synthesized through a hydrothermal and nitridation procedure (Fig. 1a). First, the treated nickel foam (NF) was put into a homogeneous solution containing vanadium (V) and cobalt (Co) metal salts, and the precursor V-Co(OH)2 was obtained by a hydrothermal method. Then, the final single-atom V-doped CoNx sample (VSA-CoNx) can be obtained by calcination in a nitrogen atmosphere. For comparison, the CoNx catalyst was also prepared via the same method, but without V salt in the aqueous solution.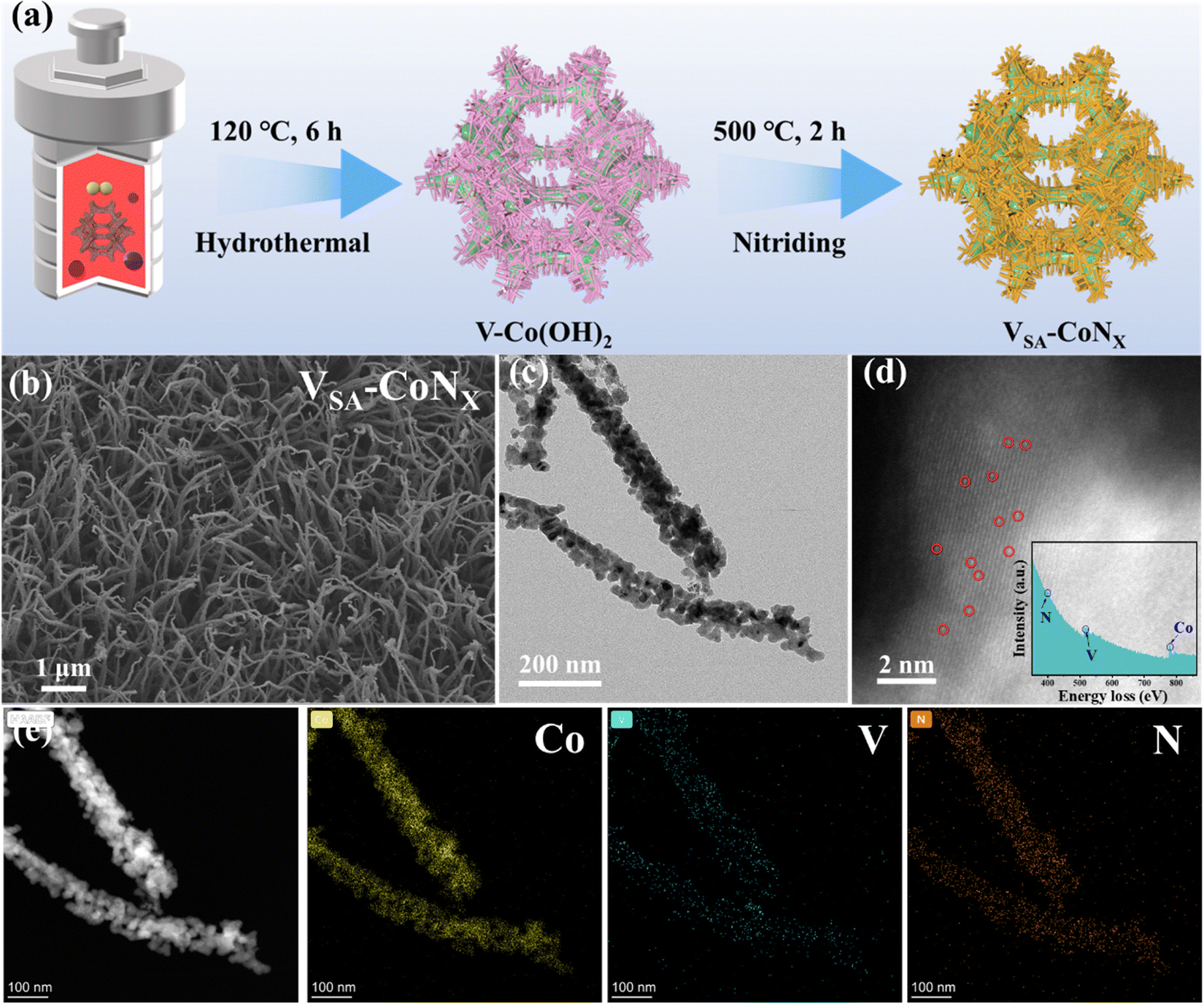 | ||
| Fig. 1 Synthesis illustration and morphological characterization of VSA-CoNx. (a) Synthesis route. (b) SEM, (c) TEM, and (d) HAADF-STEM images (the inset in (d) is the EELS analysis). (e) STEM-EDS mappings. | ||
Compared to the two samples of Co(OH)2 and CoNx without V-doping, V-doping can effectively reduce the aggregation of the VSA-CoNx and achieve better dispersion as shown in Fig. 1b and Fig. S1–S4 (ESI†). Moreover, the surface hydrophilicity (aerophobicity) of the VSA-CoNx was changed by V-doping (Fig. S5, ESI†), where VSA-CoNx has a smaller liquid contact angle and a relatively larger bubble contact angle than CoNx. The change can not only maximize the active site of the VSA-CoNx catalyst in the reaction process, but also effectively prevent the formation of large bubbles resulting in bubble blockage. Fig. 1c and Fig. S6 (ESI†) confirm the existence of the CoNx phase in VSA-CoNx, and the lattice fringe with 0.213 nm matches with the (200) plane of CoNx. HAADF-STEM and electron energy loss spectroscopy (EELS) also proved the existence of elements N, V and Co, where V is atomically dispersed in the catalyst (Fig. 1d). Furthermore, the TEM-EDS mapping indicates a homogeneous distribution of Co, V, and N throughout the nanorod (Fig. 1e). In addition, the XPS results of VSA-CoNx and CoNx (Fig. S7, ESI†) indicate that V-doping does not produce a strong electron interaction on the main catalyst phase, which suggests that the electron interaction between V and CoNx is not responsible for the enhancement of catalyst activity.
The X-ray diffraction (XRD) pattern (Fig. 2a) shows that both samples match well with the standard card JCPDS#15-0806, which indicates that no additional crystalline phases were formed in the V-doping process. However, upon closer examination of the enlarged areas in Fig. 2b, it is observed that the diffraction peaks of VSA-CoNx have shifted to lower angles relative to CoNx. This shift becomes more obvious with an increase in the X-ray diffraction angle, suggesting a lattice expansion, as dictated by the Bragg diffraction formula. This observation indicates that the incorporation of V into the CoNx structure has led to an increase in the interplanar spacing within the lattice.6b,20 The electronic structure and coordination environment of the catalyst were also analyzed by X-ray absorption spectroscopy (XAS). Fig. 2c depicts the Co K-edge XANES spectra of the Co foil, CoO, CoNx and VSA-CoNx. The absorption edge of the Co K-edge of CoNx and VSA-CoNx is between Co foil and CoO, suggesting that the state of Co is situated between 0 and +2. The over-lapped Co K-edge XANES spectra of CoNx and VSA-CoNx also verify that the electronic environments of Co sites were not significantly influenced by the incorporation of V, which is in accordance with the results of Co 2p3/2 XPS.21 The XANES spectra at the V K-edge (Fig. 2d) further provide comparable insights, revealing that the valence state of V in VSA-CoNx lies between +3 and +4 when compared with the standard samples (V foil, V2O3, VO2, and V2O5). Furthermore, the Fourier transform (FT) of the k3-weighted EXAFS oscillations in the R space for both Co and V in VSA-CoNx, as shown in Fig. 2e and f, elucidates the local coordination environment around these elements. The predominant peaks at approximately 1.46 and 1.47 Å are attributed to the first shell coordination of Co–N and V–N/O, respectively. Notably, a significant peak at around 2.15 Å is observed in the VSA-CoNx sample, which supports the presence of a Co–V bond. This peak aligns with the one at approximately 2.16 Å in the R-space of the V K-edge. Moreover, the Co–N bond length in VSA-CoNx increased significantly from 1.40 to 1.46 Å, compared to the CoNx sample without V-doping, which further proves that due to the incorporation of metal V, more unsaturated metal sites were formed, resulting in a lattice expansion of the CoNx phase.22 Strain engineering can often be used as a very effective method to improve the performance of catalytic materials.23 The EXAFS fitting was also carried out to identify the coordination structure of the Co atom in VSA-CoNx (Fig. S8, ESI†), and the fitting parameters are shown in Table S6. The EXAFS fitting curves of the other samples (Co foil, CoO and CoNx) are shown in Fig. S9 and S10 (ESI†). Compared with CoNx, the atomic distance of the Co–N bond in VSA-CoNx increases from 1.97 to 2.02 Å due to the V-doping. The average coordination number of Co–Co in CoNx samples was 4.5. The Co–Co coordination number in VSA-CoNx decreased to 4.1, while Co–V bonds appeared with a coordination number of 0.8. We conducted ICP tests on the catalytic materials. As shown in Table S7 (ESI†), the ratio of metals Co to V is approximately 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, which is consistent with the parameters fitted by using EXAFS. All these results suggest that V atoms may partially replace Co atoms in the CoNx and exists in the single-atom form (Fig. S11, ESI†).
:1, which is consistent with the parameters fitted by using EXAFS. All these results suggest that V atoms may partially replace Co atoms in the CoNx and exists in the single-atom form (Fig. S11, ESI†).
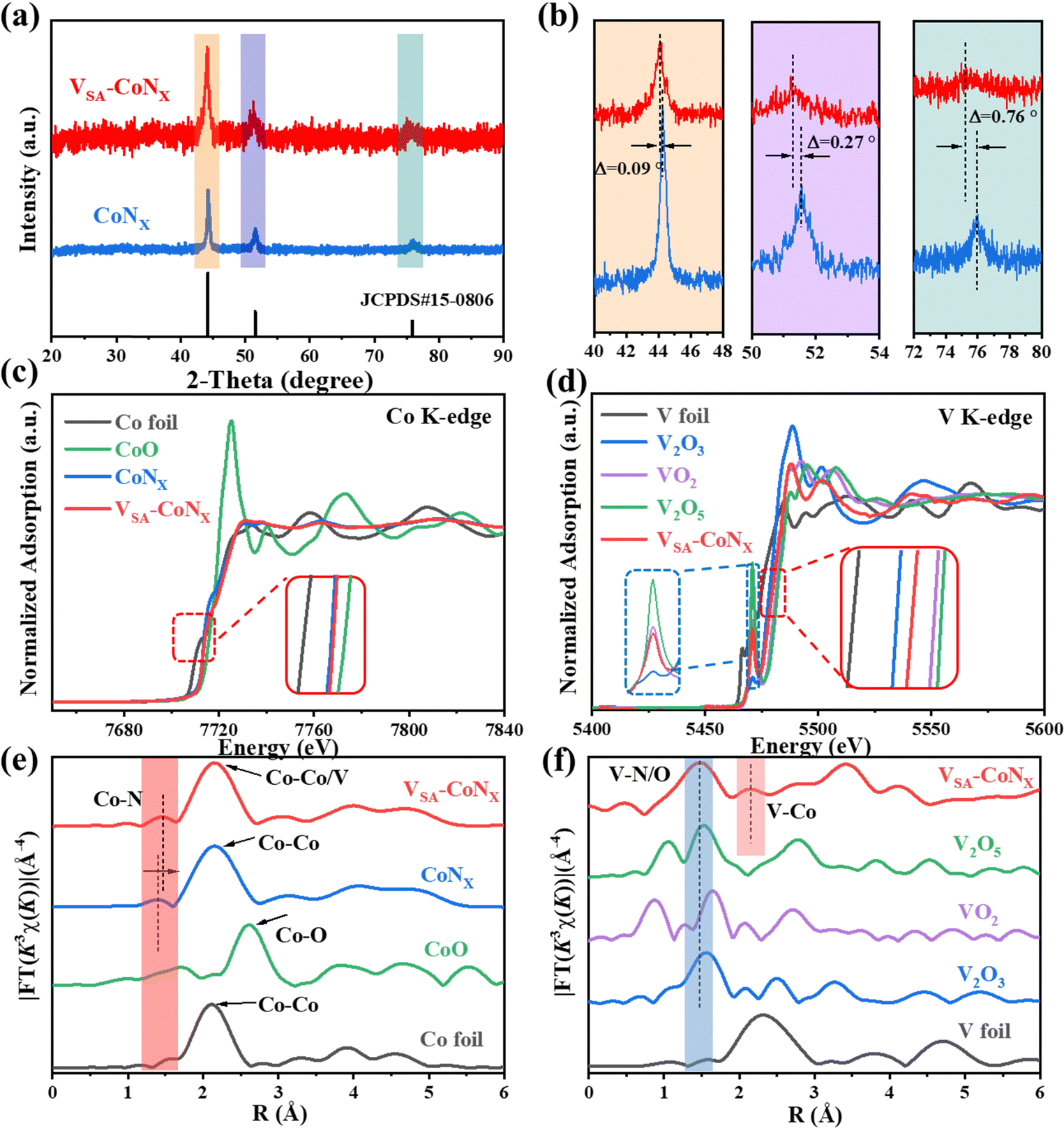 | ||
| Fig. 2 XRD characterization and coordination environment of samples. (a) XRD patterns of VSA-CoNx and CoNx. (b) The XRD of magnified regions with different colors. (c) Co K-edge and (d) V K-edge XANES spectra, and (e) and (f) the corresponding Fourier transforms of EXAFS spectra of different samples. | ||
2.2. HER–OER and HER–UOR performance
The HER and OER performances of all the samples were tested in a 1 M KOH solution, where the applied potentials relative to the reversible hydrogen electrode (RHE) were converted using the equation: ERHE = EHg/HgO + 0.925 V (Fig. S12, ESI†). Fig. 3a and Fig. S13 (ESI†) show that VSA-CoNx only needed overpotentials of ∼135 mV at a current density of 100 mA cm−2 for the HER and 1.565 V to yield a current density of 50 mA cm−2 for the OER (Fig. 3c), which exceeded the commercial Pt/C catalyst (η100 = 148 mV) for the HER and commercial RuO2 (η50 = 1.580 V) for the OER. The Tafel slope also proves its superior chemical kinetics (Fig. S14 and S15, ESI†). Fig. S16 and S17 (ESI†) show the negligible change in LSV before and after the stability test, which confirmed its high stability. Moreover, characterization of VSA-CoNx after the HER stability test indicates that it well maintained the morphology (Fig. S18, ESI†) and electronic structure (Fig. S19, ESI†). It is noted that, unlike in the HER, after the OER long-term stability test, the original nanowires of the catalyst surface exhibit an obvious nanosheet region (Fig. S20, ESI†). According to previous reports, the region of the nanosheet should be the in situ formed high-valence Co species (CoOOH) during the high-voltage stability test. The lattice constants corresponding to the CoOOH and the XPS tests also confirmed this observation (Fig. S21, ESI†).24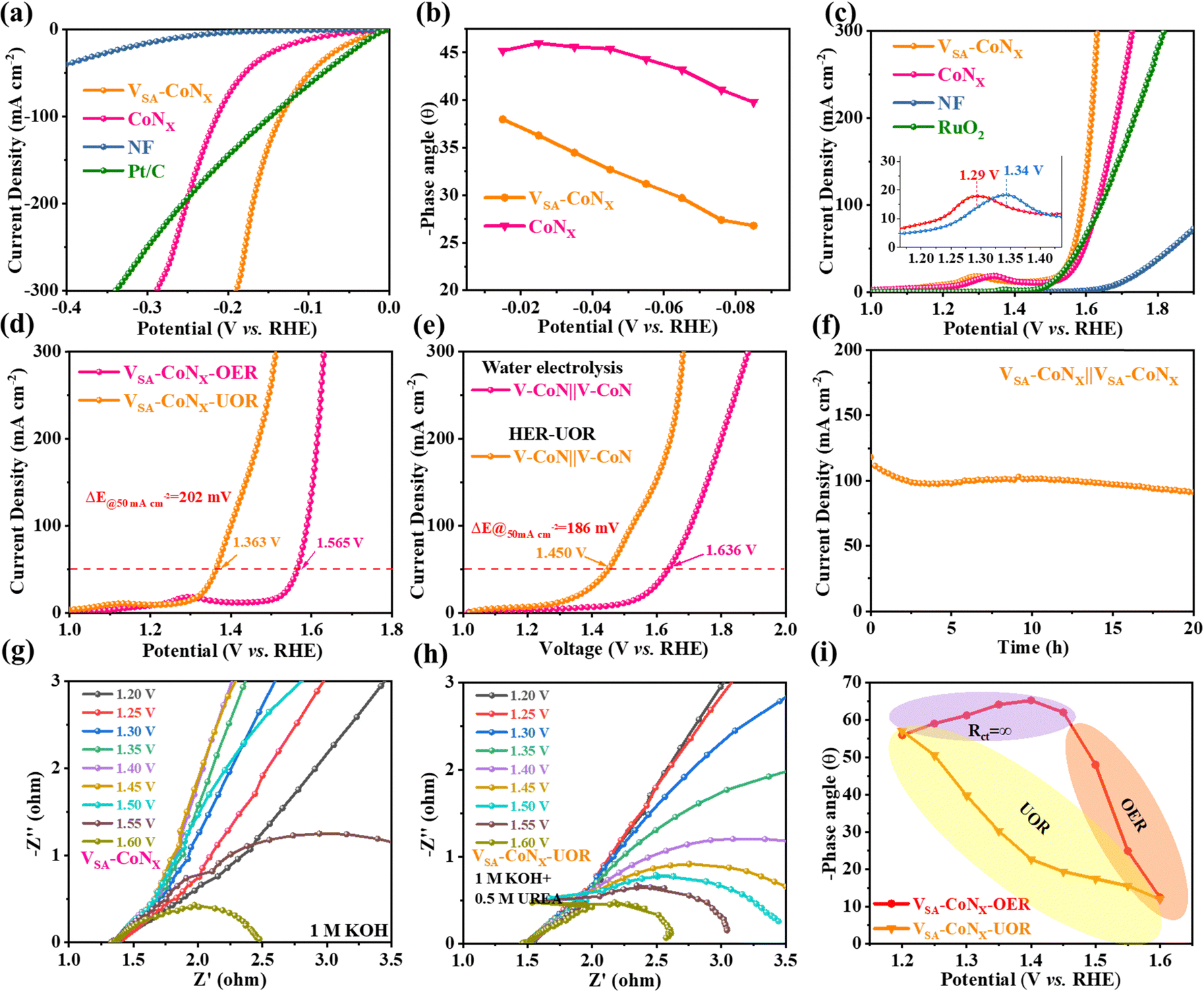 | ||
| Fig. 3 Electrocatalytic performance of the electrodes for water electrolysis and urea electrolysis. (a) The HER polarization curves and (b) phase peak angles of VSA-CoNx and CoNx. (c) The OER polarization curves. (d) Polarization curves of VSA-CoNx in the 1 M KOH electrolyte in the presence and absence of urea. (e) The LSV curves of VSA-CoNx‖VSA-CoNx for the water (HER–OER) and urea electrolysis (HER–UOR). (f) The stability test of VSA-CoNx‖VSA-CoNx for the HER–UOR. Nyquist plots of at different voltages of VSA-CoNx for the (g) OER and (h) UOR. (i) Phase peak angles of VSA-CoNx for the OER and UOR. | ||
Electrochemical impedance spectroscopy (EIS) further demonstrates that VSA-CoNx has the faster catalytic process (Fig. S22, S23 and Table S1, ESI†). Furthermore, EIS measurements were conducted at various voltages to probe the charge transfer resistance (Rct) and the electrocatalytic kinetics involved in the HER. When the applied voltages were ranging from −0.015 V to −0.085 V (Fig. S24a and b, ESI†), VSA-CoNx presented significantly lower mass transfer impedance. The Bode phase plots in Fig. S24c and d (ESI†) illustrate the phase angle as a function of frequency, which provides insights into the reaction dynamics at the electrolyte–catalyst interface and within the catalyst. The peaks in the phase angle at low and high frequencies are associated with the charge transfer at the electrolyte–catalyst interface and the electron conduction through the catalyst's inner layer, respectively.25 In the low-frequency region, the noticeably smaller phase peak angle observed for VSA-CoNx (Fig. 3b) indicates that the incorporation of V into the CoNx structure enhanced the charge transfer at the electrolyte–catalyst interface. This enhancement contributes to faster HER kinetics by facilitating the transfer of electrons between the electrolyte and the catalyst, which is crucial for the efficiency of the HER process.
Considering the excellent performance of the HER and OER of VSA-CoNx, we assembled a two-electrode electrolyzer by using VSA-CoNx as a bifunctional catalyst for the anode and cathode. As shown in Fig. S25 (ESI†), it needs 1.636 V to provide a current density of 50 mA cm−2. Moreover, the current density (50 mA cm−2) negligibly attenuates during the stability test over 200 h (Fig. S26, ESI†). Compared with CoNx without V-doping, VSA-CoNx shows much better catalytic performance. Notably, the HER/OER and OWS performance based on the VSA-CoNx catalyst are standing out among the state-of-the-art catalysts (Table S2, ESI†).
Different from the OER process (1.23 V vs. RHE), the electrochemical oxidation process of urea requires only 0.37 V.26 Inspired by the above excellent HER/OER performance of VSA-CoNx, we further explored its UOR performance. Fig. 3d and Fig. S27 and S28 (ESI†) show that the UOR performance of the VSA-CoNx in the 1 M KOH electrolyte containing 0.5 M urea. Impressively, the VSA-CoNx catalyst only needs a potential of 1.363 V to deliver a current density of 50 mA cm−2 in the UOR. Compared to the OER in the KOH electrolyte without urea, the overpotential of VSA-CoNx presents a 202 mV decrease at 50 mA cm−2 after the addition of urea. In order to verify the feasibility of using the UOR as a substitute of the OER, a two-electrode HER–UOR coupling system was constructed with KOH solution containing 0.5 M urea as the anode. The cell voltage of the HER–UOR system at 50 mA cm−2 is only 1.450 V (Fig. 3e), which is much lower than that of 1.636 V for OWS. Moreover, the potential of HER–UOR coupling system is apparently lower than OWS in all current densities (Fig. S29, ESI†), and also lower than many other electrocatalysts in HER–UOR systems (Table S3, ESI†). The time–current curve in Fig. 3f at about 100 mA cm−2 also demonstrates its excellent stability. Similar to OER, high-valence Co species appeared on the catalyst surface after UOR stability testing (Fig. S30, ESI†), and XPS results (Fig. S31, ESI†) also demonstrated the conversion of Co and the sacrifice of surface V.
To unveil the mechanism of activity enhancement for VSA-CoNx in different electrolytes, the EIS at different potentials in different electrolytes was also measured to understand the relationship between interfacial dynamics, electron transfer and mass transfer in the processes of OER and UOR.27 It is clear from Nyquist that the Rct is infinite when the voltage is below 1.4 V without the addition of urea (Fig. 3g), which indicates that no reaction occurred in the lower voltage range. With the increase of voltage, the corresponding phase angle in KOH even tended to increase (Fig. 3i and Fig. S32a, ESI†). In contrast, the phase angle of Bode phase plots in the urea-containing electrolyte decreased continuously as the potential increases (Fig. 3h–f and Fig. S32, ESI†), indicating that the catalyst can react in all the range of applied voltages. This intrinsically fast kinetics also explains why the UOR performed better than the OER.28
In order to further investigate the structural changes of the prepared catalysts at high voltages, a series of in situ Raman tests were performed. Fig. 4a shows the time-dependent in situ Raman diffraction peaks of the catalyst at a fixed voltage, and the original catalyst (t = 0 min) exhibits the characteristic peaks of the initial CoNx and CoOx.29 When a constant voltage was applied, VSA-CoNx yields new Raman peaks corresponding to Co–O (H) at approximately 499 and 573 cm−1. The contour map was gradually formed (Fig. 4a1–d1) with the intensity increase of corresponding peaks, and the Raman peak corresponding to VOx also appeared in 867 cm−1 and was stabilized over time.30 Unlike VSA-CoNx, although CoNx had a similar diffraction peak (Co–O (H)) around 499 cm−1 (Fig. 4b), its peak intensity did not become stronger over time, even if the time was prolonged to 15 min. The above analysis shows that the two as-prepared catalysts (CoNx and VSA-CoNx) will undergo a pre-oxidation process in the alkaline electrolyte, and the V-doping can effectively promote the pre-oxidation of the catalyst surface, which is beneficial for the following reaction.
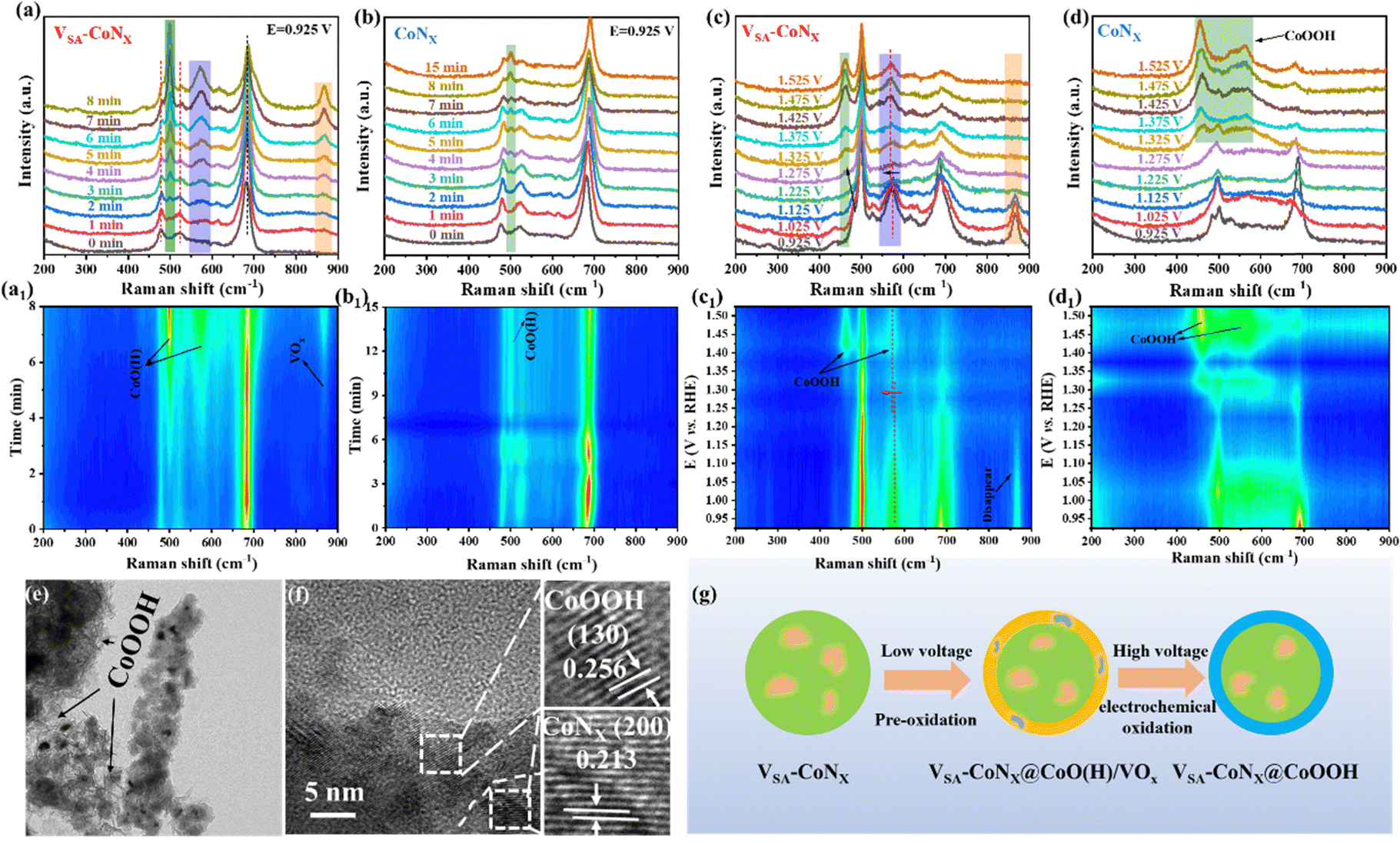 | ||
| Fig. 4 Time-dependent in situ Raman spectra of (a) VSA-CoNx and (b) CoNx at a constant voltage. Potential-dependent in situ Raman spectra of (c) VSA-CoNx and (d) CoNx. (a1)–(d1) The corresponding contour maps. (e) TEM and (f) HRTEM images of VSA-CoNx after the UOR stability test. (g) Schematic representation of active species in VSA-CoNx under different voltages. | ||
After the pre-oxidation process, different voltages were applied to investigate the phase transition of catalysts in the real process. Fig. 4c shows that the Raman peaks of VSA-CoNx at 479 and 573 cm−1 shifted to 461 and 568 cm−1, respectively, when the voltage was increased to 1.275 V, indicating the formation of a higher valence Co species (CoOOH) on the surface during the reaction. It is consistent with the previous study where the in situ generated high-valence Co species at high voltages were regarded as the real active substances.31 Concurrently, the Raman peak at 867 cm−1 gradually disappeared with the increase of the voltage, which also explains the disappearance of the surface V after the stability test under high voltages (Fig. S21 and S31, ESI†). Fig. 4d shows the Raman spectra of CoNx at different voltages. Clearly, a diffraction peak corresponding to the high-valence Co species appeared at 1.325 V. The peak intensity (CoOOH) of CoNx increased while the peak at 499 cm−1 disappeared with increasing voltage, indicating that CoNx underwent a continuous oxidation under high voltage, and the surface was all converted into high-valence Co species. However, for VSA-CoNx (Fig. 4c), with the increase of voltage, the intensity of the corresponding peak of CoOOH kept constant, meaning that V-doping can effectively inhibit the over-oxidation of Co at the high voltage. The TME and HRTEM images of VSA-CoNx after the UOR (Fig. 4e and f) show the apparent coexistence of CoOOH and CoNx. Therefore, V-doping promotes the production of active substances and inhibits the accumulation of high-valency species (Fig. 4g), which makes it promising to achieve excellent catalytic activity and stability at the high voltage.
2.4. HER–HzOR performance
In addition to urea, hydrazine has also been widely used as industrial raw materials. As a common type of pollutants, it is significant to develop an effective strategy to degrade hydrazine sewage for environmental protection.32 Compared with the urea oxidation, the hydrazine oxidation (HzOR) can be performed at an even lower standard voltage (−0.33 V vs. RHE), which means that the HER–HzOR system may further lower energy consumption of H2 production. Since the products of the HzOR are nitrogen and water, it is a greenhouse gas emission-free reaction. Therefore, the HzOR is an ideal alternative for the anodic reaction to couple with the HER. Impressively, VSA-CoNx showed a surprisingly low potential (η50 = −0.033 V), and also presented the lower mass transfer resistance to prove the origin of its excellent performance (Fig. S33, ESI†). Compared with the traditional anodic OER, the voltage-drop reached 1.598 V at 50 mA cm−2 (Fig. 5a), which was larger than 1.5 V in the wide current ranges (Fig. 5b), indicating its extraordinary promise to replace the OER. Therefore, a two-electrode HER–HzOR system with VSA-CoNx as bifunctional catalysts was constructed, and it only required 0.064 V to reach 50 mA cm−2 (Fig. 5c), which was significantly lower than 1.636 V at OWS and 1.450 V at the HER–UOR system. In addition, only 0.302 V was required at 300 mA cm−2, which shows an extremely low energy cost for practical green H2 production (Fig. S34 and Table S4, ESI†). The stability of the two-electrode electrolyzer was also tested and is shown in Fig. 5d. During the stability test, the current density continued to decrease, but it is noted that once the electrolyte was refreshed, and the current density was restored immediately. This indicates that the decay of current density was due to the decrease of the hydrazine concentration in the electrolyte. Similar to the HER, the morphology (Fig. S35, ESI†) and electronic structure (Fig. S36, ESI†) of the catalyst did not change obviously after the stability test. Since the waste water containing hydrazine is very harmful and carcinogenic to the environment, the degradation rate of hydrazine by the HER–HzOR system was also studied by UV-vis spectroscopy. As expected, in the HER–HzOR system, the degradation rate of hydrazine in the anode electrode can reach over 95% at 2 h by two cycles, and the hydrazine concentration can be reduced to 6 ppb in 3 h, lower than the 10 ppb required by the U. S. Environmental Protection Agency (EPA) (Fig. 5e and Fig. S37, S38, ESI†).15a This HER–HzOR coupling system is promising to the largely lower energy expenditure of H2 production while simultaneously treating wastewater containing hydrazine.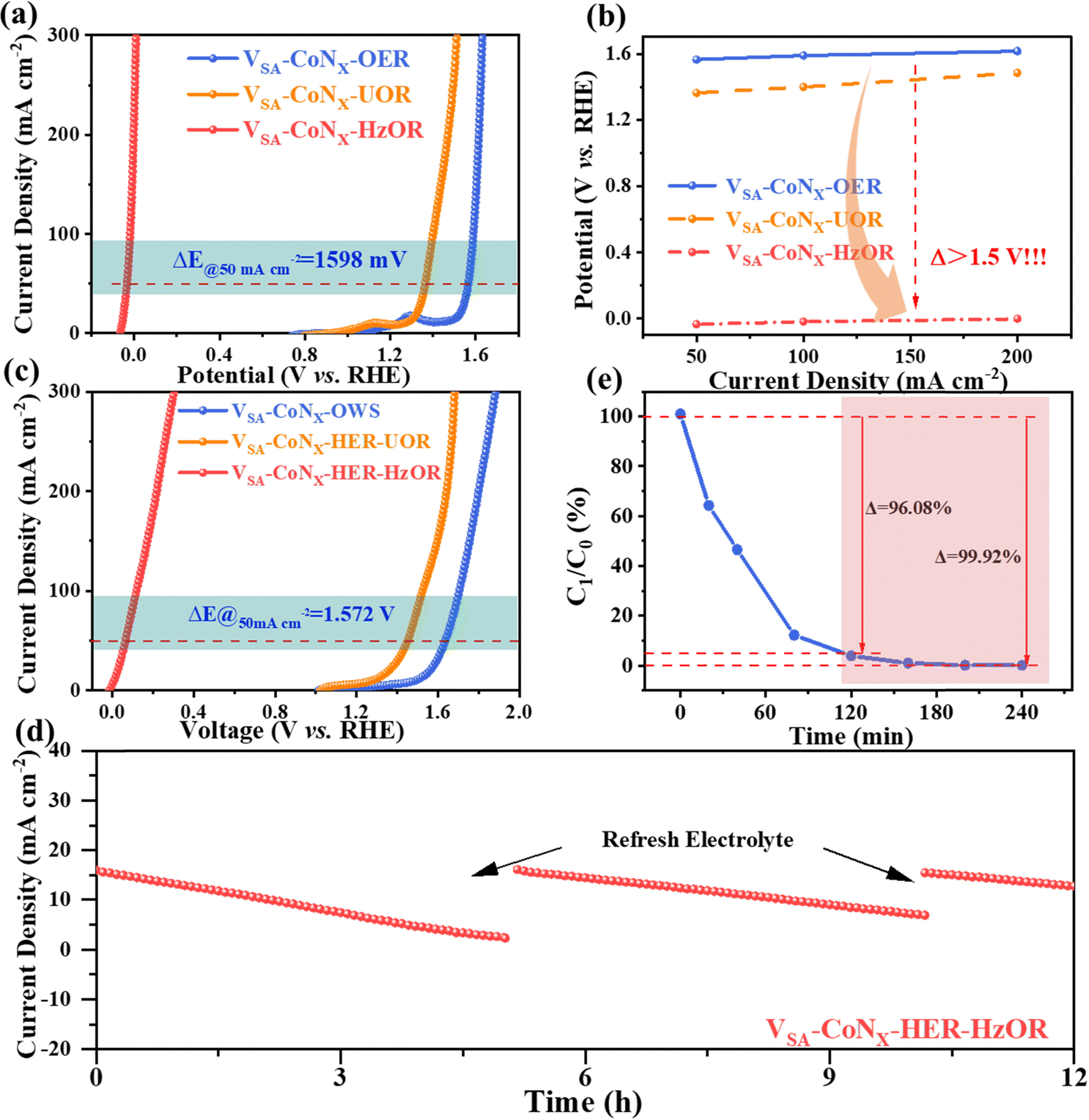 | ||
| Fig. 5 Electrocatalytic performance of the electrodes for hydrazine electrolysis. (a) Polarization curves and (b) driving potentials at different current densities of VSA-CoNx for the OER, UOR and HzOR. (c) Polarization curves of VSA-CoNx for the OWS, HER–UOR and HER–HzOR. (d) The stability test of VSA-CoNx‖VSA-CoNx for the HER–HzOR system. (e) Degradation of the hydrazine electrolyte. | ||
2.5. HER–OER (UOR/HzOR)-AEMWE performance
As mentioned above, VSA-CoNx has shown excellent catalytic performance in the HER, OER, UOR and HzOR as multifunctional catalysts. However, the H-type electrolyzer at the laboratory stage is still far from the industrial level. Therefore, to further test the commercial prospect of the catalyst material, AEMWEs using VSA-CoNx as bifunctional catalysts were also constructed (Fig. 6a). A current density of 1 A cm−2 was reached with an input voltage of 1.88 V on the AEMWE for OWS, which is superior to many reported catalysts (Fig. 6b and Table S3, ESI†). To test the adaptability of the AEMWE for intermittent wind and solar powers, we also simulated this type of electricity fluctuation by changing the input voltage and the input current in a short time to study its response. Clearly, the AEMWE with VSA-CoNx as bifunctional catalysts exhibited excellent response to the change of voltage or current (Fig. S39, ESI†), which offers the possibility for the use of renewable electricity.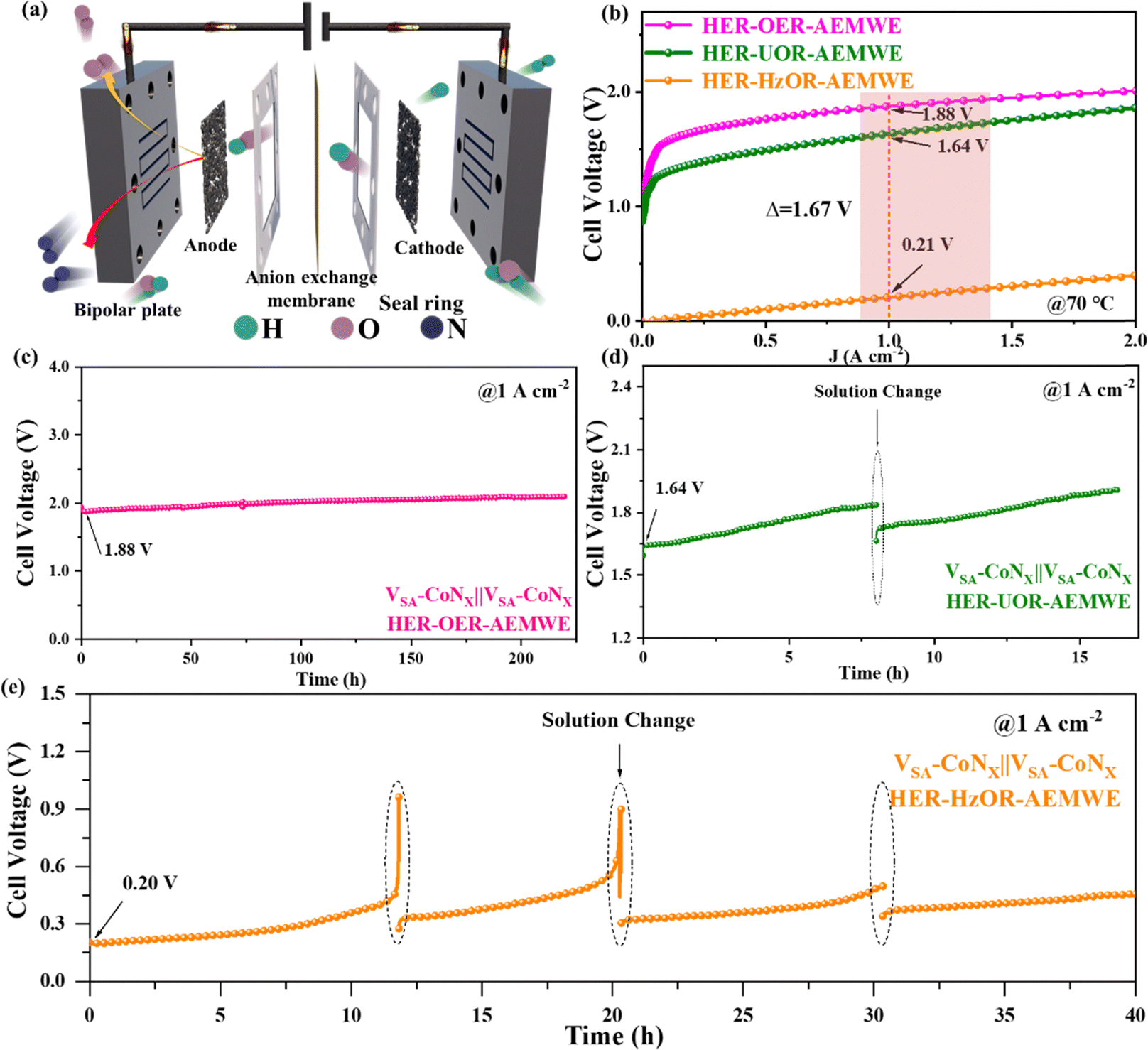 | ||
| Fig. 6 (a) Schematic illustration of the two-electrode MEA flow electrolyzer based on samples. (b) LSV curves for the HER–OER-AWMWE, HER–UOR-AEMWE and HER–HzOR-AEMWE. The stability test of VSA-CoNx‖VSA-CoNx for the (c) HER–OER-AWMWE, (d) HER–UOR-AEMWE and (e) HER–HzOR-AEMWE. | ||
Encouraged by above excellent performance, we further constructed the HER–UOR AEMWE cell, and it needs a lower input voltage of 1.64 V to deliver a current density of 1 A cm−2, which exhibits apparently lowered energy consumption for H2 production, compared with the AEMWE for OWS. To further reduce the energy cost, the AEMWE system with the HzOR replacing the OER was constructed, which only required a voltage of 0.21 V to reach a current density of 1 A cm−2, resulting in a voltage-drop of 1.67 V from the conventional AEMWE. This HER–HzOR AEM electrolyzer shows an unprecedented ultralow voltage to deliver a current density of 1 A cm−2, setting a new record among all AEM water electrolysis systems reported to date (Table S5, ESI†). Compared with conventional AEMWE devices (Fig. S40, ESI†), the HER–UOR and HER–HzOR systems only consume about 87.23% and 11.17% energy for the same amount of H2 generation. An evaluation of energy consumption revealed that at 1 A cm−2, if the energy consumption of the traditional AEMWE is about 4.56 kW h Nm−3 of H2, it is 3.95 kW h Nm−3 of H2 (equivalent to 43.954 kW h kg−1 of H2) for the HER–UOR AEM system and only 0.51 kW h Nm−3 of H2 (equivalent to 5.628 kW h kg−1 of H2) for the HER–HzOR AEM system,33 which is apparent lower than that of commercial PEM electrolyzers, about 4.5 to 5.0 kW h Nm−3 of H2 (50.00–55.56 kW h kg−1 of H2).34 The ultralow energy cost of the HER–HzOR AEM system for producing hydrogen provides a broad prospect for practical applications.35
The stability of the traditional AEMWE cell was further tested (Fig. 6c), which maintained over 200 h at 1 A cm−2, providing a promising potential for traditional water electrolysis. The HER–UOR system also shows excellent stability (Fig. 6d), where the voltage increase was caused by the decrease of the urea concentration. When the urea electrolyte was refreshed, the initial voltage was also recovered, accompanying with a slight increase. Similarly, the HER–HzOR system also exhibited the same behavior (Fig. 6e), which was due to the decrease of the hydrazine concentration. Table S7 (ESI†) also shows that the weight of the catalyst remained almost the same before and after the reaction, and the ratio of the two metals, Co and V, did not change significantly. This further confirms that the degradation of performance observed during the stability test was ascribed to the change in the hydrazine concentration, rather than the dissolution of the catalyst itself. Owing to the excellent multifunctional properties of VSA-CoNx for the HER, OER, UOR and HzOR, the AEMWE systems based on our catalysts under multi-operating conditions (including HER–OER, HER–UOR and HER–HzOR) presented outstanding performance for H2 production. In particular, the HER–HzOR system only consumed about 11% energy of the traditional AEMWE for producing the same amount of hydrogen, which hits a record of ultralow energy cost at a current density of 1 A cm−2 among all AEM water electrolysis systems reported so far.
3. Conclusions
In summary, a four-in-one catalyst of VSA-CoNx has been developed, and the VSA-CoNx can efficiently catalyze four reactions HER, OER, UOR and HzOR. By using the VSA-CoNx catalyst as both an anode and a cathode, the assembled AEMWE systems can achieve coupling H2 production under multi-operating conditions, including HER–OER, HER–UOR and HER–HzOR conditions. In particular, the HER–UOR and HER–HzOR AEM coupling systems only require 1.64 and 0.21 V to achieve an ampere-level current density (1 A cm−2), while the conventional HER–OER AEMWE system needs 1.88 V. The energy consumption of the HER–HzOR ECHO system is just about 11% of the conventional AEMWE system, which hits a record in the low energy expenditure for all water electrolysis systems known to date. Simultaneously, the HER–HzOR ECHO system can efficiently degrade hydrazine over 95% in 2 h and achieve the treatment of wastewater containing hydrazine. At a current density of 1 A cm−2, if the energy consumption of the conventional AEMWE is about 4.56 kW h Nm−3 of H2, it is 3.95 kW h Nm−3 of H2 (equivalent to 43.954 kW h kg−1 of H2) for the HER–UOR AEM electrolyzer and 0.51 kW h Nm−3 of H2 (equivalent to 5.628 kW h kg−1 of H2) for the HER–HzOR AEM electrolyzer. In short, this study offers a new perspective of achieving ultralow-cost H2 production by coupling alternative oxidation reactions under multiple operating conditions.Author contributions
D. C. conceived and designed the project. M. X. performed all the experiments and analyzed the results. M. H., G. X. performed a part of characterization. Q. Z. and M. S. analyzed the electrochemical data. Z. L., J. Y. and P. W. gave the supervision. M. X. and D. C. wrote the manuscript. All authors have reviewed, discussed and approved the results and conclusions of this article.Data availability
The data supporting this article have been included as a part of the ESI.†Conflicts of interest
The authors declare no competing interest.Acknowledgements
This work is supported by the Open Research Fund of Suzhou Laboratory (SZLAB-1308-2024-ZD009), The Science and Technology Innovation Program of Hunan Province (2023RC4002) and the Outstanding Talent Fund from Xiangtan University.References
- (a) S. Huang, F. Lin, S. Wang, X. Zeng, H. Ling, X. Hu, Z. Shen and D. Cao, Adv. Mater., 2024, 36, 2407974 CrossRef CAS PubMed; (b) Z. Xiao, P. Sun, Z. Qiao, K. Qiao, H. Xu, S. Wang and D. Cao, Chem. Eng. J., 2022, 446, 137112 CrossRef CAS.
- (a) M. Xing, S. Zhu, X. Zeng, S. Wang, Z. Liu and D. Cao, Adv. Energy Mater., 2023, 13, 2302376 CrossRef CAS; (b) S. Zhu, M. Xing, Z. Lu, Z. Qiao, S. Wang, Q. Zhao, M. Shao, J. Yun and D. Cao, Sci. China Mater., 2024, 67, 1891 CrossRef CAS; (c) J. Wang, F. Xu, H. Jin, Y. Chen and Y. Wang, Adv. Mater., 2017, 29, 1605838 CrossRef PubMed.
- (a) Z. Niu, Z. Lu, Z. Qiao, S. Wang, X. Cao, X. Chen, J. Yun, L. Zheng and D. Cao, Adv. Mater., 2024, 36, 2310690 CrossRef CAS PubMed; (b) X. Ding, R. Jiang, J. Wu, M. Xing, Z. Qiao, X. Zeng, S. Wang and D. Cao, Adv. Funct. Mater., 2023, 33, 2306786 CrossRef CAS.
- P. Sun, Z. Qiao, X. Dong, R. Jiang, Z. Hu, J. Yun and D. Cao, J. Am. Chem. Soc., 2024, 146, 15515 CrossRef CAS PubMed.
- (a) H. Sun, X. Xu, L. Fei, W. Zhou and Z. Shao, Adv. Energy Mater., 2024, 14, 2401242 CrossRef CAS; (b) Z. Qiao, R. Jiang, H. Xu, D. Cao and X. Zeng, Angew. Chem., Int. Ed., 2024, 63, e202407812 CrossRef CAS PubMed; (c) H. Xu, D. Cheng, D. Cao and X. Zeng, Nat. Catal., 2024, 7, 207 CrossRef CAS.
- (a) Q. Huang, G.-J. Xia, B. Huang, D. Xie, J. Wang, D. Wen, D. Lin, C. Xu, L. Gao, Z. Wu, J. Wu, F. Xie, W. Guo and R. Zou, Energy Environ. Sci., 2024, 17, 5260 RSC; (b) M. Xing, Z. Qiao, Z. Niu, S. Wang, Z. Liu and D. Cao, ACS Appl. Mater. Interfaces, 2023, 15, 40428 CrossRef CAS PubMed; (c) M. Xing, Z. Qiao, S. Zhu, G. Xu, J. Yun and D. Cao, Adv. Funct. Mater., 2024, 34, 2409559 CrossRef CAS.
- (a) C. Hu, L. Zhang and J. Gong, Energy Environ. Sci., 2019, 12, 2620 RSC; (b) F. Zeng, C. Mebrahtu, L. Liao, A. K. Beine and R. Palkovits, J. Energy Chem., 2022, 69, 301 CrossRef CAS.
- (a) P. Wang, J. Zheng, X. Xu, Y. Q. Zhang, Q. F. Shi, Y. Wan, S. Ramakrishna, J. Zhang, L. Zhu, T. Yokoshima, Y. Yamauchi and Y. Z. Long, Adv. Mater., 2024, 36, 2404806 CrossRef CAS PubMed; (b) Y. Song, K. Ji, H. Duan and M. Shao, Exploration, 2021, 1, 20210050 CrossRef PubMed; (c) X. Zhang, S. Feizpoor, M. Humayun and C. Wang, Chem. Catal., 2024, 4, 100840 CrossRef CAS.
- M. Yang, T. Feng, Y. Chen, J. Liu, X. Zhao and B. Yang, Appl. Catal., B, 2020, 267, 118657 CrossRef CAS.
- (a) M. Shao, F. Ning, J. Zhao, M. Wei, D. G. Evans and X. Duan, Adv. Funct. Mater., 2013, 23, 3513 CrossRef CAS; (b) H. Zhou, Z. Li, S. M. Xu, L. Lu, M. Xu, K. Ji, R. Ge, Y. Yan, L. Ma, X. Kong, L. Zheng and H. Duan, Angew. Chem., Int. Ed., 2021, 60, 8976 CrossRef CAS PubMed.
- (a) M. Liu, W. Zou, S. Qiu, N. Su, J. Cong and L. Hou, Adv. Funct. Mater., 2023, 34, 2310155 CrossRef; (b) Z. Wang, W. Liu, Y. Hu, M. Guan, L. Xu, H. Li, J. Bao and H. Li, Appl. Catal., B, 2020, 272, 118959 CrossRef CAS; (c) D. Li, W. Wan, Z. Wang, H. Wu, S. Wu, T. Jiang, G. Cai, C. Jiang and F. Ren, Adv. Energy Mater., 2022, 12, 2201913 CrossRef CAS; (d) J. Zhang, J. Feng, J. Zhu, L. Kang, L. Liu, F. Guo, J. Li, K. Li, J. Chen, W. Zong, M. Liu, R. Chen, I. P. Parkin, L. Mai and G. He, Angew. Chem., Int. Ed., 2024, 63, e202407038 CrossRef CAS PubMed.
- (a) Y. Song, W. Xie, Y. Song, H. Li, S. Li, S. Jiang, J. Y. Lee and M. Shao, Appl. Catal., B, 2022, 312, 121400 CrossRef CAS; (b) Y. Gao, L. Ge, H. Xu, K. Davey, Y. Zheng and S.-Z. Qiao, ACS Catal., 2023, 13, 11204 CrossRef CAS.
- (a) G. Meng, Z. Chang, L. Zhu, C. Chen, Y. Chen, H. Tian, W. Luo, W. Sun, X. Cui and J. Shi, Nano-Micro Lett., 2023, 15, 212 CrossRef CAS PubMed; (b) Z. Wang, L. Xu, F. Huang, L. Qu, J. Li, K. A. Owusu, Z. Liu, Z. Lin, B. Xiang, X. Liu, K. Zhao, X. Liao, W. Yang, Y. B. Cheng and L. Mai, Adv. Energy Mater., 2019, 9, 1900390 CrossRef; (c) C. Feng, M. Lv, J. Shao, H. Wu, W. Zhou, S. Qi, C. Deng, X. Chai, H. Yang, Q. Hu and C. He, Adv. Mater., 2023, 35, 2305598 CrossRef CAS PubMed.
- (a) Z.-Y. Yu, C.-C. Lang, M.-R. Gao, Y. Chen, Q.-Q. Fu, Y. Duan and S.-H. Yu, Energy Environ. Sci., 2018, 11, 1890 RSC; (b) Z. Xiao, Y. Qian, T. Tan, H. Lu, C. Liu, B. Wang, Q. Zhang, M. T. Sarwar, R. Gao, A. Tang and H. Yang, J. Mater. Chem. A, 2023, 11, 259 RSC; (c) Y. Zhu, Y. Chen, Y. Feng, X. Meng, J. Xia and G. Zhang, Adv. Mater., 2024, 36, 2401694 CrossRef CAS PubMed; (d) Y. Hu, T. Chao, Y. Li, P. Liu, T. Zhao, G. Yu, C. Chen, X. Liang, H. Jin, S. Niu, W. Chen, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2023, 62, e202308800 CrossRef CAS PubMed.
- (a) Y. Huang, X. Zhang, L. Li, M. Humayun, H. Zhang, X. Xu, S. P. Anthony, Z. Chen, J. Zeng, D. V. Shtansky, K. Huo, H. Song, C. Wang and W. Zhang, Adv. Funct. Mater., 2024, 2401011 CrossRef; (b) Q. Qin, H. Jang, P. Li, B. Yuan, X. Liu and J. Cho, Adv. Energy Mater., 2018, 9, 1803312 CrossRef.
- H. Sun, L. Li, H.-C. Chen, D. Duan, M. Humayun, Y. Qiu, X. Zhang, X. Ao, Y. Wu, Y. Pang, K. Huo, C. Wang and Y. Xiong, Sci. Bull., 2022, 67, 1763 CrossRef CAS PubMed.
- (a) X. Gao, X. Bai, P. Wang, Y. Jiao, K. Davey, Y. Zheng and S.-Z. Qiao, Nat. Commun., 2023, 14, 5842 CrossRef CAS PubMed; (b) Y. Liu, J. Zhang, Y. Li, Q. Qian, Z. Li, Y. Zhu and G. Zhang, Nat. Commun., 2020, 11, 1853 CrossRef CAS PubMed.
- (a) J. Han, H. Wang, Y. Wang, H. Zhang, J. Li, Y. Xia, J. Zhou, Z. Wang, M. Luo, Y. Wang, N. Wang, E. Cortés, Z. Wang, A. Vomiero, Z. F. Huang, H. Ren, X. Yuan, S. Chen, D. Feng, X. Sun, Y. Liu and H. Liang, Angew. Chem., Int. Ed., 2024, 63, e202405839 CrossRef CAS PubMed; (b) X. Zhang, L. Tong, X. Shi, Z. Li, Z. Xiao, Y. Liu, T. Zhang and S. Lin, J. Colloid Interface Sci., 2024, 668, 502 CrossRef CAS PubMed.
- J. Chi, L. Guo, J. Mao, T. Cui, J. Zhu, Y. Xia, J. Lai and L. Wang, Adv. Funct. Mater., 2023, 33, 2300625 CrossRef CAS.
- Y. Wang, Y. Jiao, H. Yan, G. Yang, C. Tian, A. Wu, Y. Liu and H. Fu, Angew. Chem., Int. Ed., 2022, 61, 202116233 CrossRef PubMed.
- H. Liao, G. Ni, P. Tan, Y. Liu, K. Chen, G. Wang, M. Liu and J. Pan, Appl. Catal., B, 2022, 317, 121713 CrossRef CAS.
- L. Zhou, C. Zhang, Y. Zhang, Z. Li and M. Shao, Adv. Funct. Mater., 2021, 31, 2009743 CrossRef CAS.
- (a) Y. Wang, Y. Jiao, H. Yan, G. Yang, C. Tian, A. Wu, Y. Liu and H. Fu, Angew. Chem., Int. Ed., 2022, 61, 202116233 CrossRef PubMed; (b) M. Xing, Z. Qiao, Z. Niu, S. Wang, Z. Liu and D. Cao, ACS Appl. Mater. Interfaces, 2023, 15, 40428 CrossRef CAS PubMed.
- X. Guo, H. Zhang, W. Xia, M. Ma, D. Cao and D. Cheng, Adv. Funct. Mater., 2024, 34, 2316539 CrossRef CAS.
- L. Li, G. Zhang, J. Xu, H. He, B. Wang, Z. Yang and S. Yang, Adv. Funct. Mater., 2023, 33, 2213304 CrossRef CAS.
- T. Yu, Q. Xu, J. Chen, G. Qian, X. Zhuo, H. Yang and S. Yin, Chem. Eng. J., 2022, 449, 137791 CrossRef CAS.
- W. Luo, H. Tian, Q. Li, G. Meng, Z. Chang, C. Chen, R. Shen, X. Yu, L. Zhu, F. Kong, X. Cui and J. Shi, Adv. Funct. Mater., 2023, 34, 2306995 CrossRef.
- X. Zheng, J. Yang, P. Li, Z. Jiang, P. Zhu, Q. Wang, J. Wu, E. Zhang, W. Sun, S. Dou, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2023, 62, e202217449 CrossRef CAS PubMed.
- J. Song, D. Yu, X. Wu, D. Xie, Y. Sun, P. Vishniakov, F. Hu, L. Li, C. Li, M. Y. Maximov, K. M. El-Khatib and S. Peng, Chem. Eng. J., 2022, 437, 135281 CrossRef CAS.
- (a) A. Moysiadou, S. Lee, C.-S. Hsu, H. M. Chen and X. Hu, J. Am. Chem. Soc., 2020, 142, 11901 CrossRef CAS PubMed; (b) V. M. Dzhagan, M. Y. Valakh, O. F. Isaieva, V. O. Yukhymchuk, O. A. Stadnik, O. Y. Gudymenko, P. M. Lytvyn, O. A. Kulbachynskyi, V. S. Yefanov, B. M. Romanyuk and V. P. Melnik, Opt. Mater., 2024, 148, 114894 CrossRef CAS.
- H. Yin, L. Qian, H. Xiao, L. Zhang, X. Li, X. Zhou, S. Yuan, C. Li, Q. Lu, F. Pan, L. Zeng, J. Guo and S. Li, Chem. Eng. J., 2024, 490, 151699 CrossRef CAS.
- X. Wang, H. Hu, X. Yan, Z. Zhang and M. Yang, Angew. Chem., Int. Ed., 2024, 63, e202401364 CrossRef CAS PubMed.
- Y. Wu, C. Guo, R. Yao, K. Zhang, J. Li and G. Liu, Adv. Funct. Mater., 2024, 2410193 CrossRef CAS.
- R. Zeng, Q. Gao, L. Xiao, W. Wang, Y. Gu, H. Huang, Y. Tan, D. Tang and S. Guo, J. Am. Chem. Soc., 2024, 146, 10023 CrossRef CAS PubMed.
- S. Wang, T. Shen, C. Yang, G. Luo and D. Wang, ACS Catal., 2023, 13, 8670 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ey00117j |
| This journal is © The Royal Society of Chemistry 2025 |