
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComparison of extraction methods to obtain high-purity protein concentrate from green microalgae Nannochloropsis oceanica
Thi Phuong Linh
Le
*a,
Jayani
Samarathunga
a,
Max
Gabard
b,
Katrina
Strazdins
b,
Jeroen
Rens
b and
Benu
Adhikari
 *a
*a
aSchool of Science, RMIT University, Melbourne, VIC 3083, Australia. E-mail: s3952016@student.rmit.edu.au; benu.adhikari@rmit.edu.au
bBega Group, Melbourne, VIC 3008, Australia
First published on 6th February 2025
Abstract
Nannochloropsis oceanica is a promising and sustainable source of high-value protein. This study compared various extraction methods to implement an optimal protocol for obtaining high-purity protein concentrate. These methods included non-defatting protocols using ultrasound and high-pressure homogenization, as well as solvent-based defatting with chloroform, ethanol, isopropanol, and acetone. The most effective protocol involved acetone defatting, followed by alkaline extraction at pH 12.0 and acid precipitation at pH 2.0, obtaining 86% protein content by dry weight. This protocol outperformed non-defatting methods, which achieved only 42% protein content by dry weight. The higher effectiveness of the acetone-defatting process was attributed to its capacity to break down cell walls and remove lipids and pigments, including chlorophyll a. These findings establish that acetone defatting is an effective method for obtaining high-purity protein concentrate from N. oceanica and could support further research and applications in protein extraction from this and other microalgal species.
Sustainability spotlightThis study contributes to the United Nations Sustainable Development Goals (SDGs), particularly SDG 2 (Zero Hunger) and SDG 12 (Responsible Consumption and Production), by advancing the sustainable extraction of high-purity protein from microalgal resources, specifically Nannochloropsis species. The proposed method achieves a high protein content (86%), is easy to implement, and environmentally benign. As a sustainable protein source, microalgae require minimal energy and nutrients while producing protein-rich biomass. By presenting an efficient extraction and purification process, this research promotes the diversification of protein supply chains, minimizes the environmental impact of protein production, and helps establish microalgae as a resource-efficient solution for meeting the growing global demand for high-value, protein-rich foods. |
1. Introduction
The world's human population reached 8.0 billion in 2022 and is expected to reach 10 billion by 2053.1 A massive expansion of mainstream agricultural and food industry resources is required to meet the protein needs of this population. However, such expansion cannot be easily realized due to associated sustainability constraints and negative impact to the environment. In addition, there is an increasing demand for protein ingredients and protein-rich foods. Consequently, food scientists, industry, and human society are exploring alternative protein sources to meet the demand for protein without compromising sustainability. The United Nations (UN) Sustainable Development Goal (SDG)2 has rightly emphasized reducing protein malnutrition through developing sustainable food systems.3 Protein derived from mainstream sources such as animals (in particular) and terrestrial plants is unlikely to meet the increased protein demand, and also they require more land, high energy, and water input.4 Thus, there is a need to consider alternative sources of protein that are less costly (in terms of energy, water and nutrients) and also require less land.5 Microalgae are a well-known source of proteins and have the potential to be used in many food products. Microalgae have a low carbon footprint and are able to absorb 10–50 times more CO2 than other natural plants.6 In addition, they grow fast, can be densely grown in ponds and reactors and require less input in terms of energy, water and nutrients.7Some microalgal species contain up to 70% of their dry biomass protein.7,8 They also contain sufficient essential amino acids comparable to plant proteins.9,10 Despite the potential health benefits of microalgae, particularly in preventing age-related metabolic issues,11 their use in human diets is limited. Nannochloropsis spp. is known for its robust growth and high levels of essential nutrients like protein (30–43% in dry weight (DW)), lipids (28–45% in DW) and polyunsaturated fatty acids, carbohydrates (15–35% in DW), antioxidants, and vitamins,12 represent promising candidates for future dietary inclusion. Nannochloropsis spp. has been known as a high docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA)-containing strain in Australia. According to Food and Agriculture Organization (FAO) recommendations, it contains sufficient amounts of essential amino acids and can meet the daily requirement of 29 to 40% of the proteins or 13–15 mg of these amino acids per kilogram of body weight.13,14 However, the extracted protein from microalgae has drawbacks such as high dewatering cost, complex extraction, and purification. The sensory issues associated with the fishy smell and pigment also restrict their application in food.15
Nannochloropsis oceanica belongs to single-celled eukaryotes. Their cell is protected by a thick multi-bilayered cell wall,16 primarily composed of algaenan. Algaenan is a complex, highly resistant carbohydrate that is difficult to break down due to its chemical structure, which includes long-chain aliphatic hydrocarbons linked together by ether bonds.17 This cell wall structure and characteristics are a barrier to solvent penetration, making extracting intracellular compounds difficult when conventional extraction methods are used. Therefore, the cell wall of microalgae must be disrupted by a suitable combination of physical (e.g., mechanical, thermal, and thermo-mechanical processing), chemical (e.g., organic solvents, osmotic shock, and acid-alkali treatment), or biological (e.g., enzymatic) pre-treatments to make the extraction process more efficient. Recent research efforts on Nannochloropsis oceanica have primarily aimed at improving cell disruption efficiency to optimize lipid yield and quality.18–22 In contrast, efforts to extract and characterize other components, such as proteins, have been limited, with far fewer studies dedicated to protein isolation.
Therefore, this study aimed to optimize and compare protein extraction methods to obtain concentrates with greater than 80% (w/w) protein from Nannochloropsis oceanica. Various mechanical cell disruption and solvent-based defatting methods were applied to increase protein purities. The effects of these methods on the release of intracellular compounds, as well as the purity of the extracted protein, were evaluated through compositional and morphological analyses.
2. Materials and methods
2.1. Materials
The freeze-dried Nannochloropsis oceania (N. oceanica) biomass (4.3% ± 0.0 moisture content) was received from Qponics Limited (Queensland, Australia). Ethanol, isopropanol, and acetone were purchased from ChemSupply Australia Pty. Ltd (Victoria, Australia). All other chemicals used in this study were purchased from Sigma-Aldrich Australia (New South Wales, Australia) and were used as received.2.2. Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×g for 30 min at ambient temperature.
000×g for 30 min at ambient temperature.
The isoelectric point of the extracted protein was determined by recording zeta potential as a function of pH. A dynamic light scattering (Zetasizer ZS-90, Malvern Instruments, Worcestershire, UK) was used for these measurements at 25 °C. The supernatants (obtained after centrifugation) of each sample were transferred into folded capillary cells for the zeta potential measurements. The zeta potentials were calculated using the Smoluchowski model for all measurements. Every supernatant was measured in triplicate with five readings per measurement.23 The isoelectric point was determined as the pH value where the net charge or zeta potential was zero or close to zero.24
2.2.2.1. Non-defatting of procedure. The freeze-dried biomass was dissolved in Milli-Q at a ratio of 1
:20 (biomass:water) and stirred at 700 rpm for an hour at room temperature to obtain complete hydration. This biomass suspension was used in pre-treatments and protein extraction as mentioned below.
2.2.2.1.1 Procedure I: alkali solubilization and acid precipitation. The biomass suspension was adjusted to pH 12.0 (obtained from part 2.2.1) by 1.0 M NaOH and agitated at 700 rpm for 5 h at ambient temperature (25 ± 2 °C). Subsequently, the extract was centrifuged at 10
000×g for 30 min (Sorvall LYNX 6000, Thermo Fisher Scientific, USA). The supernatant was collected, and its pH was adjusted to 2.0 which was found to be the iso-electronic point of N. oceanica protein (Section 2.2.1), by using 1.0 M HCl to precipitate the protein. The extracted protein was stored at 4 °C overnight to allow complete protein precipitation. This precipitated protein was centrifuged at 10000×g for 30 min, redispersed in Milli-Q water, and then neutralized using 1.0 M NaOH. The neutralized protein mass was freeze-dried at 0.1 mbar and −40 °C (VaCo 10, Zirbus Technology GmbH, Germany). The freeze-dried protein sample was vacuum sealed and stored at −20 °C till further use.
2.2.2.1.2 Procedure II: ultrasound-assisted extraction. Sonication was applied to the biomass suspension as a pretreatment to improve protein extraction. A laboratory ultrasonicator Q500 (Qsonica, Newtown, CT, USA) equipped with a 1/2-inch tip probe at 60% of amplitude for cell disruption. Sonication was applied for 5 min with a pulse mode of 5 seconds ON and 5 seconds OFF at 500 W maximum power and 20 kHz frequency. The sonication was carried out in an ice bath to minimize temperature increase and avoid possible undesired reactions.25 The ultrasound-treated biomass suspension was subjected to alkali extraction–acid precipitation procedure as described in Procedure I above.
2.2.2.1.3 Procedure III: high-pressure homogenization-assisted extraction. High-pressure homogenization was applied as a pretreatment to improve protein extraction. The biomass suspension was homogenized using a laboratory two-stage high-pressure homogenizer (Panda PLUS 2000, GEA Niro Soavi Americas, Bedford, USA). Three passes were applied at a fixed homogenizing pressure of 150 MPa with a volumetric flow rate of 9 L h−1. The temperature during this homogenization process varied from 22–30 °C in the first pass and thus was cooled down to 22 °C before undertaking the next pass.26 The high-pressure-homogenized biomass suspension was subjected to alkaline extraction and acid precipitation procedure described in Procedure I above.
2.2.2.2. Defatting procedure. The freeze-dried biomass was defatted in different solvents at a ratio of 1
:5 (biomass:solvent) and stirred at 500 rpm for 2 h at room temperature (20–25 °C). Chloroform:methanol (2:1, v/v) (Procedure IV), ethanol 50% (Procedure V), isopropanol 60% (Procedure VI), and acetone 98% (Procedure VII) were applied in the defatting process. The supernatants containing the lipids were removed. The defatting process was repeated twice. The defatted biomass obtained in this way was dried in a fume hood for 72 h to allow residual solvents to evaporate. Subsequently, protein from the dried defatted biomass was extracted using the alkaline extraction–acid precipitation procedure described above.
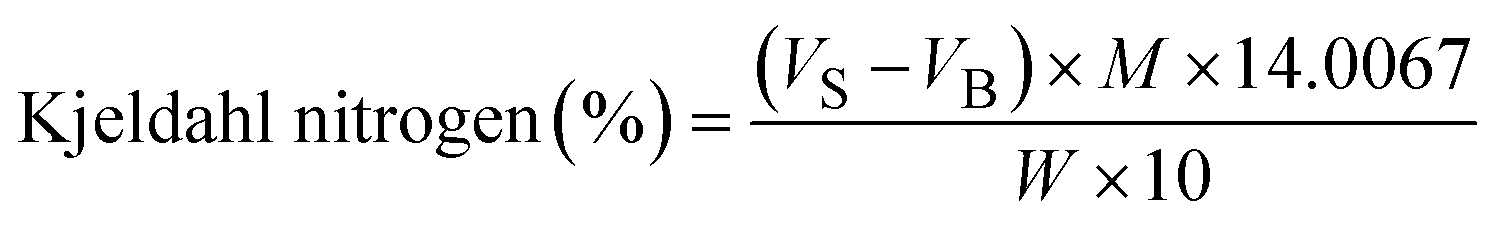
| Crude protein (%) = Kjeldal nitrogen (%) × F |
2.3. Statistical analysis
Statistical analysis was performed using IBM SPSS Statistics 28 (IBM, USA). All experimental measurements were conducted at least in triplicate. The data were reported as mean value ± standard deviation. The analysis of variance (ANOVA) was performed to determine the significant difference between any two mean values. Duncan test was implemented on the data sets using a 95% significance level (P < 0.05).3. Results and discussion
3.1. Protein content and isoelectric point
In the process of protein extraction, it is essential to know the isoelectric point of the protein being extracted as its solubility is lowest at this point and easiest to separate it from the complex mixture by precipitation/centrifugation. pH is a primary factor influencing colloidal stability as proteins exhibit pH-dependent surface charge alterations (in solution state) due to their ionisable groups. Ionic strength also modulates protein's charge and, consequently, their stability. Adjusting the pH to the protein's isoelectric point makes it least soluble and most easily precipitable out of the solution, allowing for its separation from other components. A isoelectric point (pI) of proteins is most readily determined as pH where the net charge (measured through zeta potential on the protein is zero).30To establish suitable pH values for alkali solubilization and acid precipitation the zeta potential of N. oceanica protein was determined as a function of pH (pH 2.0 to 12.0) (Fig. 1). This range was chosen for two reasons: (1) many algal proteins exhibit isoelectric point at lower pH and most proteins typically have high solubility at high alkaline condition.23
 | ||
| Fig. 1 The variation of electrostatic charge density (zeta potential) of Nannochloropsis oceanica protein as a function of pH at ambient temperature (25 ± 2 °C). | ||
As can be observed (Fig. 1), the isoelectric point of N. oceanica protein is 2.0, indicating that it could be optimally separated at this pH. It showed negative zeta potential at a pH value above 2.0, and the magnitude zeta potential values increased with the increase in pH. The highest negative value of zeta potential (−34.6 mV) was observed at pH 12.0, indicating that the protein component in N. oceanica would be optimally solubilized at this pH. In the aqueous environment, the values of the zeta potential indicate potential stability or instability of the colloidal systems. A high magnitude of zeta potential solutions of N. oceanica protein at and above pH 6.0 indicated that these solutions would be highly stable above this pH.31 The magnitude of zeta potential values of the solutions was more than 30 mV at pH 11.0 and 12.0, indicating that N. oceanica protein was readily soluble and stable and thus most readily extractable.
This work aimed to precipitate the protein to a maximum extent. When the pH was lowered, the magnitude of zeta potentials increased rapidly from −29.4 mV at pH 10.0 to −0.8 mV at pH 3.0. The zeta potential versus pH curve passed through zero zeta potential at pH 2.0, indicating that it was its isoelectric point. Thus, this pH was used for extraction (precipitation) of N. oceanica protein.
The pH values for solubilisation (pH 12.0) of N. oceanica protein determined in this study were comparable to those of Spirulina and Chlorella protein. The pH values of the best solubilisation of Spirulina protein and Spirulina protein were reported to be 8.0–11.0 and 10.0–12.0, respectively. However, the pI of N. oceanica protein was substantially lower than that of Spirulina protein (pH 3.0–4.0) and Chlorella protein (pH 4.0–5.5).32–36 Protein from Tetraselmis sp. was reported to be highly soluble above pH 6.0, and it was sparingly soluble at pH values below 4.0.37 Notably, the low pI of N. oceanica protein, compared to proteins from other algae strains, can be attributed to its high content of acidic amino acids. Aspartic acid and glutamic acid together constitute 8–12% of the total amino acids in most microalgae.8 However, N. oceanica proteins have a higher proportion of these acids, accounting for 21.6–22.5% of the total protein.10,38,39 At neutral pH, the negatively charged side chains of these amino acids contribute to a net negative charge on the protein, thus lowering the pI.40 Based on this result, pH 12.0 was chosen as the solubilization-pH and pH 2.0 was chosen as the precipitation-pH in the further extraction process.
3.2. Protein content
The protein content in the Nannochloropsis oceanica biomass was 52.8 ± 0.7% on a dry weight (DW) basis. Seven different non-defatting and defatting protein extraction procedures were carried out to determine which could produce protein content >80% in DW (w/w) in the extracted protein sample.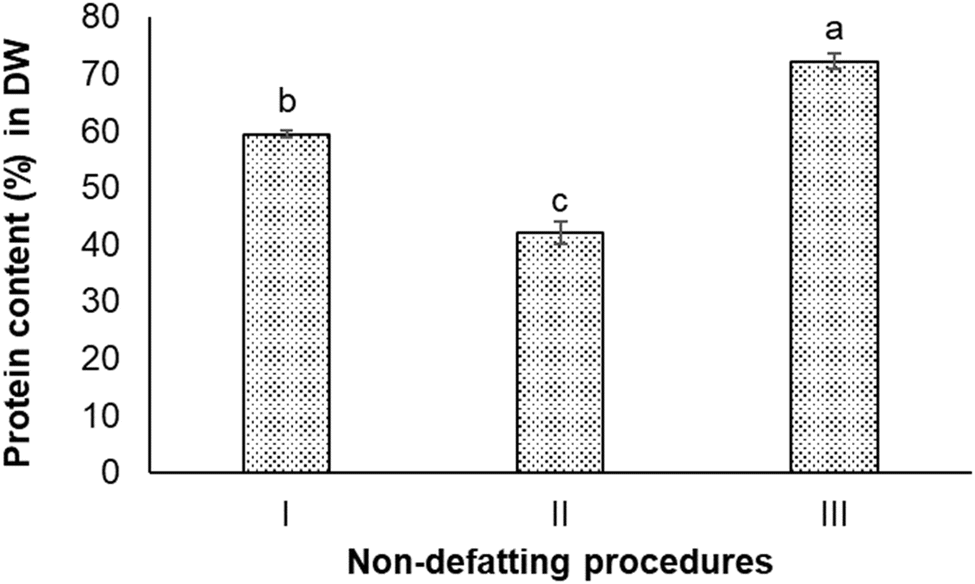 | ||
| Fig. 2 Protein contents of three extracts from three different non-defatting protein extraction procedures: (I) alkali extraction–acid precipitation, (II) ultrasound-assisted alkali extraction–acid precipitation, and (III) high-pressure homogenisation-assisted alkali extraction–acid precipitation. | ||
As can be seen, the ultrasound-treated extracts had the lowest protein content (42.0 ± 2.0%). The simplest procedure (Procedure I), which did not involve intense sonication or homogenization, achieved 59.4 ± 0.5% protein content. The homogenization-assisted extraction produced the extract with the highest protein content (72.2 ± 1.4%) among these three procedures.
In Procedure I of the alkali extraction–acid precipitation, the maximum solubilisation of N. oceanica protein occurred at pH 12.0, and the highest precipitation occurred at pH 2.0, indicating these two pH values to be optimum for extraction and precipitation (separation), respectively. At pH 12.0, the protein component in the cell wall and the cytoplasm of N. oceanica cells was solubilized to a maximum extent. The highly alkaline condition could also have disrupted the intermolecular forces between biopolymers within the cell wall and sever certain resistant covalent bonds. This could have altered or increased the cell wall's porosity and permeability,37 or caused the breakdown of cell wall components into smaller organic fragments that would dissolve in water easily.41 In addition, N. oceanica biomass is reported to contain a considerable amount of lipids (10–69% in DW), carbohydrates (5–28% in DW), polyphenols, pigments, and minerals.42 This meant intracellular components were expected to be co-released with protein through the weakened cell wall at alkaline conditions. Hydroxide ions permeate the algal cell wall and membrane to interact with intracellular lipids, resulting in the saponification of triglycerides into glycerol and fatty acid anions.43 These non-protein components are expected to precipitate together with protein under acidic environment. As a result, the protein extract obtained from the alkali extraction-isoelectric precipitation procedure did not have high protein content or high purity. The algaenan (a highly resistant biopolymer presenting in the cell walls of some green algae species) in Nannochloropsis spp. and specifically, in Nannochloropsis oceania is composed of long-chain aliphatic hydrocarbons that undergo an ether cross-linking process.16 Due to their resistance to digestion and degradation, the recalcitrant nature of these microalgal cell walls presents a significant challenge for extracting biomolecules, including proteins. Ultrasound and high-pressure homogenization have been widely used as primary physical methods to disrupt the cell wall, allowing the release of intracellular products and extracting protein.44 The ultrasound and high-pressure homogenization processes are considered to be quite effective in disrupting cell walls. High-pressure homogenization pre-treatments are known to break down biomass into smaller particle sizes and release a higher concentration of protein in the extracts compared to ultrasonication.26,45–47 This trend was also observed in this study, where high-pressure homogenization obtained extracts with higher protein concentrations than those obtained through ultrasonication. In this study, both methods did not enable extraction or protein from N. oceanica to ≥80%. The data presented in Fig. 2 shows that the N. oceanica protein extracts obtained from Procedure II (ultrasound-assisted alkali extraction–acid precipitation) extraction showed 40.6 ± 2.0% protein content. The ultrasonication is known to create high shear and cavitation effects, which are expected to induce intense mechanical stress on the cell wall,48 which would ultimately release intracellular components along with protein. The cavitation effect could promote the unfolding of proteins, which could increase the ability of proteins to bind with other molecules, such as polysaccharides, covalently.49,50 Such binding decreases the protein content or protein purity in the extract. Thus, it is also essential to remove the lipids and other compounds from biomass before extracting protein to improve the purity or protein content in the extracts.
:methanol (2:1, v/v) (Procedure IV), ethanol 50% (Procedure V), isopropanol 60% (Procedure VI), and acetone 98% (Procedure VII). All four defatting procedures obtained higher protein content in the extracts compared to the non-defatting procedure. The highest protein content (92.3 ± 1.7% in DW) was observed in the extract obtained from Procedure IV (chloroform and methanol mixture), followed by the extract obtained from Procedure VII (acetone) with 86.4 ± 3.4% protein in DW. There was a significant difference in protein content in extracts obtained from Procedures IV and VII (P < 0.05). The extracts from Procedure V and VI involving alcohols (ethanol and isopropanol) contained lower protein content compared to others (Fig. 3).
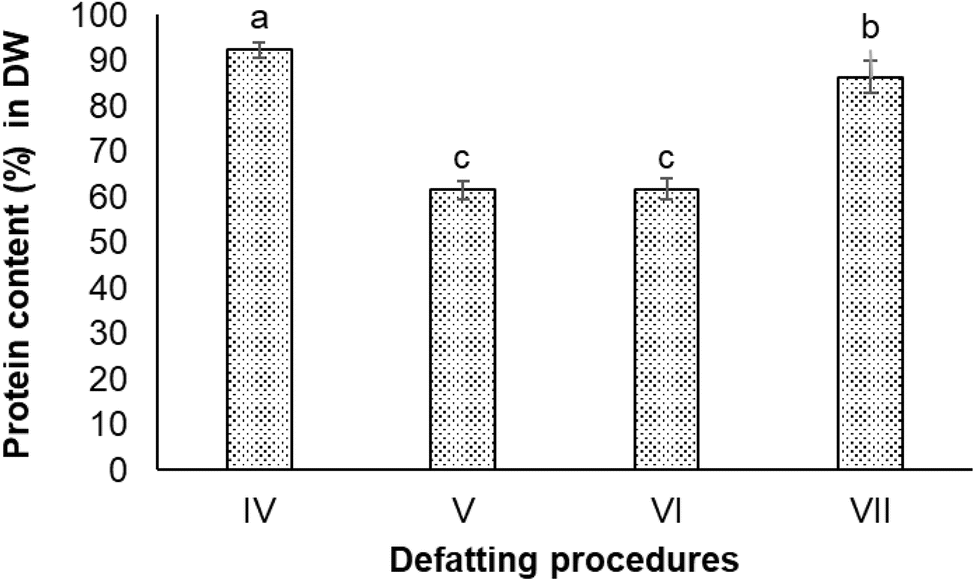 | ||
| Fig. 3 Protein contents in extracts obtained from defatting procedures which used different solvents. (IV) Chloroform:methanol (2:1); (V) ethanol 60%; (VI) isopropanol 50%; and (VII) acetone 98%. | ||
The chloroform–methanol mixed solvent is considered to be highly effective in removing lipids.51,52 Non-polar organic solvents such as chloroform can penetrate microalgal cell membranes and extract neutral lipids. These lipids are subsequently solubilized and transported across the cell membrane into the solvent. However, neutral lipids are complexed with polar lipids within the cell, forming stable associations with membrane proteins via hydrogen bonding. Thus, the non-polar solvents are insufficient to disrupt these lipid–protein complexes, necessitating polar solvents, such as methanol, for more effective lipid removal.53 Chloroform–methanol mixtures have been used for more effective lipid extraction, including hydrocarbons, carotenoids, chlorophyll, sterols, triglycerides, waxes, aldehydes, fatty acids, phospholipids, and glycolipids, which are found to abundant in N. oceanica.16 However, because of the toxicity concern, chloroform–methanol mixtures are often used only for research rather than industrial cases.
The organic solvents such as ethanol and isopropanol are also trialled as alternatives for chloroform–methanol mixtures in the process of defatting biomass.54 In the preliminary trials of lipid extraction using different concentrations (50, 60 and 70%) of ethanol and isopropanol, a promising result revealed that isopropanol 60% and 70% could obtain higher protein content of de-fatted biomass (72% in DW) compared to when chloroform:methanol solvent was used (data not reported). It could be explained that isopropanol 60–70% efficiently extracted oil from microalgae.54 Thus, this solvent was effective in the case of N. oceanica, which had a high polar lipid content. Ethanol and isopropanol could penetrate through the cell membrane and dissolve the lipids and the lipoproteins of chloroplast membranes and solubilise pigments such as chlorophyll a and b present in N. oceanica cells and other green algae.55 This is why defatted biomass of N. oceanica by ethanol and isopropanol had higher protein content. However, the protein extracts obtained from chloroform:methanol defatting treatment still had a higher protein content than those subjected to defatting by isopropanol and ethanol. The high alcohol concentration in the solvent likely contributed to protein loss and reduced overall purity of the protein extract. It might have partially unfolded the proteins and disrupted non-covalent bonds, such as hydrogen bonds and hydrophobic interactions, during lipid extraction, making the denatured proteins difficult to dissolve even at highly alkaline conditions.25 The high alcohol concentration could also have affected the solubility of other components in the samples, promoting non-specific binding between proteins and these components, leading to their co-extraction along with the protein.56
Acetone, a polar aprotic solvent, has shown to be one of the most effective candidates for defatting green algal biomass.57 The fact that it has high solvency for lipids and relatively low toxicity and flammability compared to other organic solvents makes it a better alternative. The degree of polarity of acetone is higher than that of chloroform:methanol mixtures. Hence, acetone was able to remove more of the polar lipids from N. oceanica biomass. Moreover, because the cell membrane of N. oceanica mainly consisted of polar lipids, including glycolipids, phospholipids, and betaine lipids, the use of polar solvents could increase the release of lipids from it.58,59 Chlorophyll, the primary pigment in Nannochloropsis spp., is present in high abundance.60 Organic solvent extraction is an efficient method for chlorophyll production, as it requires less energy and is more cost-effective compared to other methods. Common solvents used for pigment extraction, including chlorophyll, are polar organic liquids such as methanol, ethanol, acetone, and dimethylformamide.61 Studies have shown that 100% acetone can remove up to 70.9% of total lipids and achieve a 75.2% chlorophyll (a + b) removal rate in Scenedesmus spp. Microalgae.62 However, acetone was found to extract only about 32% of chlorophyll a from Nannochloropsis spp.61 Although previous research indicated that polar solvents such as ethanol and methanol are more suitable for chlorophyll extraction than acetone,61 its use effectively prevents the formation of degradation products during the extraction process.55 The protein content in acetone-defatted N. oceanica biomass was 86% (DW), much higher than that obtained from ethanol-defatted biomass. This indicates that acetone was more effective than ethanol at removing chlorophyll during the defatting process. However, the yield (mass of protein in the extract/mass of protein in the biomass) was 7.5 ± 0.5% across all the above-mentioned extraction procedures. This low yield was due to the removal of components covalently bound to the protein, resulting in lower recovery. Based on these results, acetone was used for defatting lipids before extracting protein (Fig. 4).
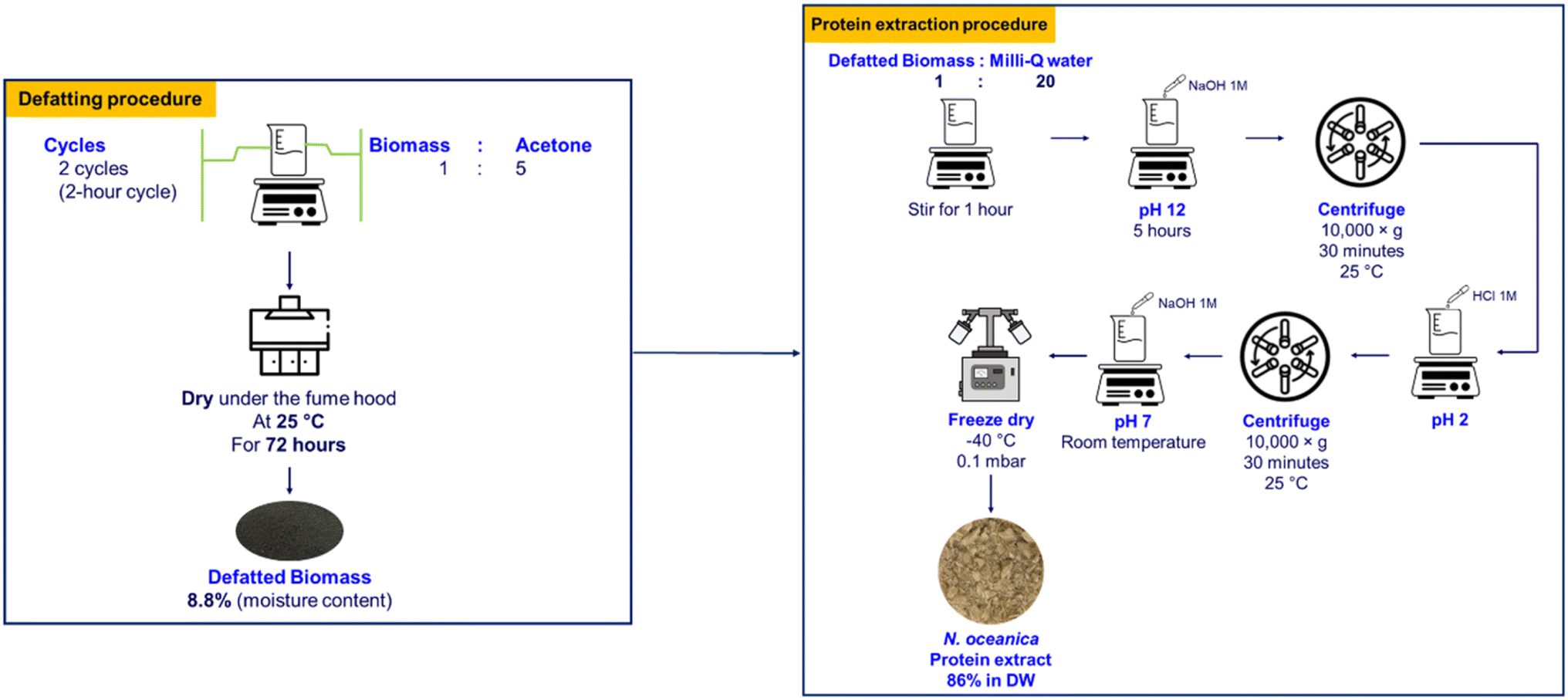 | ||
| Fig. 4 Flow diagram of the process used to extract protein from Nannochloropsis oceanica. | ||
3.3. Microscopic characteristics of Nannochloropsis oceanica surface
Fig. 5 (SEM images) shows the surface of N. oceanica cells before and after pre-treatments and protein extraction. In Fig. 5A, the cells were observed to be round shaped with slightly sprinkled and clumped on the surface. After the alkaline–acid extraction, the N. oceanica cells shrunk and became rougher (Fig. 5B) than the freshly freeze-dried microalgae cells. The surface structure of microalgae cells was more severely altered and became flat and more shrunken. This indicated that the alkaline–acid extraction process did not entirely disrupt or damage the cell wall of N. oceanica, although it was expected to alter the chemical composition of the cell wall. This would have changed the cell wall's porosity, permeability, and structure. The increased permeability of the cell wall enabled increased outward release of intracellular components through the cell wall. On the other hand, acetone caused significant damage to the N. oceanica's cell wall during the defatting procedure. The cells became flattened and deformed, with visible holes in the cell wall (Fig. 5C). The acetone disrupted the cell membranes by dissolving polar lipids such as glycolipids, phospholipids, and betaine lipids, which are the key components of the membrane.59 This process also weakened the bonds within the cell wall, leading to the release of intracellular compounds. Due to acetone's solubility with lipids and carbohydrates, these substances were easily removed during defatting. | ||
| Fig. 5 SEM images of N. oceanica cells (A) freeze-dried biomass; (B) biomass obtained after alkali solubilisation–acid precipitation; (C) biomass obtained after acetone defatting followed by alkali solubilisation–acid precipitation. | ||
3.4. NMR study of Nannochloropsis oceanica biomass, acetone defatted biomass and protein extracts
NMR spectroscopy is a valuable method for determining the chemical composition and structure of natural substances, including those in microalgae.63,64 In this study, carbon-13 (13C) NMR spectroscopy was applied to solid samples rather than liquid samples to capture both soluble and insoluble compounds. The experiments covered qualitative and quantitative analyses of Nannochloropsis oceanica components, including proteins, lipids, and carbohydrates. This study employed NMR spectroscopy to evaluate the effectiveness of the acetone-based defatting and protein extraction procedures, and to characterize both the defatted biomass and the protein extracts of N. oceanica. The non-destructive nature of NMR, along with its minimal sample preparation requirements, made it an ideal tool for this analysis.To determine the efficacy of the defatting and protein extraction processes for N. oceanica, the whole-cell biomass, acetone-defatted biomass, and protein extracts were analyzed using the NMR protocol described above. The effectiveness of the established protocol was further assessed by comparing the CP/MAS 13C NMR spectra of the three samples (Fig. 6). The chemical shifts of carbon atoms in lipids observed in 13C NMR can be categorized into four regions: aliphatic (10–35 ppm), glycerol backbone (60–72 ppm), unsaturated (olefinic) (124–134 ppm), and carbonyl or carboxyl (172–178 ppm).65 The aliphatic 13C chemical shifts of carbon atoms in proteins also appear in the region of 10–70 ppm.66 In the spectra of the defatted biomass and protein extract, the signals corresponding to lipid aliphatic carbon atoms in the upfield region were significantly less intense than in the initial biomass sample. A similar trend was observed for the unsaturated double-bond carbons (olefinic carbons, 124–134 ppm). A significant drop in signal intensity was noted in the CP/MAS 13C NMR spectrum of the defatted sample compared to the initial biomass. Additionally, the signals for these olefinic carbons appeared at very low intensity in the protein extract. In contrast, the 13C signal from amine groups (37–45 ppm), characteristic of carbons in protein molecules, showed a notable increase in intensity compared to both the initial and defatted biomass. These findings indicate that the defatting and protein extraction processes considerably removed lipids, thereby enhancing the purity of the protein extracts.
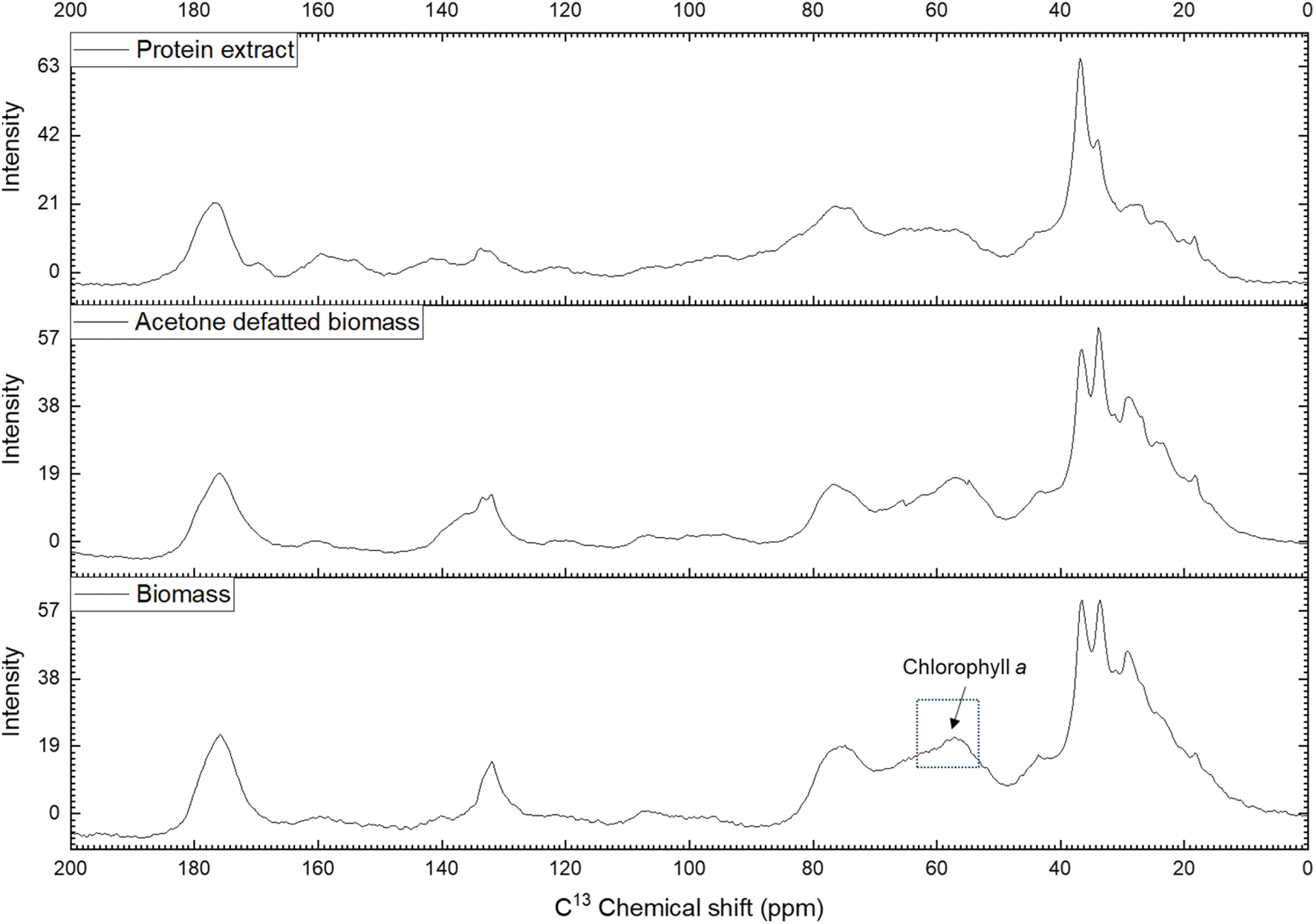 | ||
| Fig. 6 CP/MAS 13C NMR spectrum of biomass, acetone defatted biomass and protein extract of Nannochloropsis oceanica. | ||
Meanwhile, the signals corresponding to carbohydrates (60–112 ppm) remained almost unchanged between the defatted and initial biomass, suggesting that most of the carbohydrates remained within the cell wall or membranes. In contrast, the intensity of these signals decreased noticeably in the protein extract, indicating partial removal of carbohydrates during the protein extraction process. Interestingly, a decrease in signal intensity was observed in the 55–65 ppm region, with the lowest intensity in the protein extract, followed by the defatted biomass, and the highest intensity in the initial biomass. The 13C signal in the 55–65 ppm region can be attributed to the C7, C8, and C10 carbons of chlorophyll a.67 Chlorophyll a is abundant in the biomass of green microalgae, including Nannochloropsis spp. During the defatting procedure, chlorophyll a was largely dissolved and removed along with the lipids by acetone, and further separated during the protein extraction process. This resulted in a decreased 13C signal intensity for chlorophyll a and lipids in the CP/MAS 13C NMR spectra of the defatted biomass and protein extract. The removal of chlorophyll a during extraction was evident from the yellowish color of the protein extract. These findings confirm the effective removal of lipids and partial removal of carbohydrates from the N. oceanica biomass using the procedure applied in this study.
4. Conclusion
Protein was extracted from freeze-dried N. oceanica biomass using various procedures. The most effective method involved acetone defatting followed by alkaline extraction and acid precipitation, obtaining a protein concentrate with 86% protein by dry weight. This process outperformed non-defatting methods, including high-pressure homogenization and ultrasound pre-treatment, which produced concentrates with only 42% protein. The higher effectiveness of acetone defatting was attributed to its ability to disrupt the cell wall and dissolve lipids and pigments, including chlorophyll a, from the biomass. In contrast, high-pressure homogenization and ultrasound treatment were unable to sufficiently disrupt the robust cell wall of N. oceanica. Although the acetone-based procedure obtained slightly lower protein purity compared to chloroform extracts (92% protein in the extract), it is a safer option for the defatting process, as it is generally recognized as safe (GRAS). This study provides a viable approach for obtaining high-purity protein concentrate from N. oceanica with safety considerations. Given the growing global demand for sustainable protein sources, the findings contribute towards advancing research and increasing the utilization of microalgae as a valuable protein source.Data availability
The manuscript contains all data necessary to support the findings, statements, and conclusions. Spreadsheet data will be made available upon request. Any data taken from the literature, if applicable, have been appropriately acknowledged and cited.Author contributions
Thi Phuong Linh Le: conceptualization, methodology, writing – original draft. Jayani Samarathung: conceptualization, writing – review and editing. Max Gabard: conceptualization, supervision. Katrina Strazdins: conceptualization, supervision, review and editing. Jeroen Rens: conceptualization, supervision. Benu Adhikari: conceptualization, supervision, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the Bega Group for the contribution through scholarship support. We acknowledge Qponics Limited (Queensland, Australia) for providing the freeze-dried N. oceanica used in this research. We also acknowledge the technical support and access to RMIT Microscopy and Microanalysis Facility and Analytical Chemistry laboratories undertake NMR and SEM-related work.References
- United Nations, Department of Economic and Social Affairs, Population Division, 2019 Search PubMed.
- United Nations, Goal 2: End hunger, achieve food security and improved nutrition and promote sustainable agriculture, https://unstats.un.org/sdgs/report/2016/goal-02/ Search PubMed.
- UNICEF and World Health Organization, The state of food security and nutrition in the world 2017: building resilience for peace and food security, Food & Agriculture Organization of the UN (FAO), 2017 Search PubMed.
- D. Pimentel and M. Pimentel, Am. J. Clin. Nutr., 2003, 78, 660S–663S CrossRef CAS PubMed.
- M. L. Colgrave, S. Dominik, A. B. Tobin, R. Stockmann, C. Simon, C. A. Howitt, D. P. Belobrajdic, C. Paull and T. Vanhercke, J. Agric. Food Chem., 2021, 69, 15076–15083 CrossRef CAS PubMed.
- O. Helen, M. Taghi, O. KeChrist, H. Abarasi, A. Christian and T. A.-S. Zainab, Carbon Capture Sci. Technol., 2021, 1, 100007 CrossRef.
- A. K. Koyande, K. W. Chew, K. Rambabu, Y. Tao, D.-T. Chu and P.-L. Show, Food Sci. Hum. Wellness, 2019, 8, 16–24 CrossRef.
- O. K. Mosibo, G. Ferrentino and C. C. Udenigwe, Foods, 2024, 13, 733 CrossRef CAS PubMed.
- S. H. Gorissen, J. J. Crombag, J. M. Senden, W. H. Waterval, J. Bierau, L. B. Verdijk and L. J. van Loon, Amino Acids, 2018, 50, 1685–1695 CrossRef CAS PubMed.
- B. Prandi, F. Boukid, S. Van De Walle, S. Cutroneo, J. Comaposada, G. Van Royen, S. Sforza, T. Tedeschi and M. Castellari, Foods, 2023, 12, 2395 CrossRef CAS PubMed.
- J. Ampofo and L. Abbey, Foods, 2022, 11, 1744 CrossRef CAS PubMed.
- L. Zanella and F. Vianello, J. Funct. Foods, 2020, 68, 103919 CrossRef CAS.
- R. Du Preez, M. E. Majzoub, T. Thomas, S. K. Panchal and L. Brown, Nutrients, 2021, 13, 3991 CrossRef CAS PubMed.
- J. Y. Wu, R. Tso, H. S. Teo and S. Haldar, Front. Nutr., 2023, 10, 1277343 CrossRef PubMed.
- K.-Y. Show, D.-J. Lee, J.-H. Tay, T.-M. Lee and J.-S. Chang, Bioresour. Technol., 2015, 184, 258–266 CrossRef CAS PubMed.
- S. Suda, M. Atsumi and H. Miyashita, Phycologia, 2002, 41, 273–279 CrossRef.
- F. Gelin, I. Boogers, A. A. Noordeloos, J. S. S. Damste, R. Riegman and J. W. De Leeuw, Org. Geochem., 1997, 26, 659–675 CrossRef CAS.
- E. Mienis, D. Vandamme and I. Foubert, Front. Mar. Sci., 2024, 11, 1359090 CrossRef.
- S. Shivakumar, N. Serlini, S. M. Esteves, S. Miros and R. Halim, Fermentation, 2024, 10, 608 CrossRef CAS.
- S. Piyatilleke, B. Thevarajah, P. Nimarshana and T. U. Ariyadasa, Food Bioprod. Process., 2024, 255–268 CrossRef CAS.
- M. C. Quesada-Salas, G. Delfau-Bonnet, G. Willig, N. Préat, F. Allais and I. Ioannou, Processes, 2021, 9, 369 CrossRef CAS.
- K. Zhao, L. Zhang, M. Zhang, H. Tian, D. He and J. Zheng, Eur. J. Lipid Sci. Technol., 2022, 124, 2100200 CrossRef CAS.
- L. Grossmann, J. Hinrichs and J. Weiss, LWT–Food Sci. Technol., 2019, 105, 408–416 CrossRef CAS.
- A. A. Tokmakov, A. Kurotani and K.-I. Sato, Front. Mol. Biosci., 2021, 8, 775736 CrossRef CAS PubMed.
- J. A. Gerde, T. Wang, L. Yao, S. Jung, L. A. Johnson and B. Lamsal, Algal Res., 2013, 2, 145–153 CrossRef.
- N. Grimi, A. Dubois, L. Marchal, S. Jubeau, N. Lebovka and E. Vorobiev, Bioresour. Technol., 2014, 153, 254–259 CrossRef CAS PubMed.
- AOAC, Official Methods: Gaithersburg, MD, USA, 2005 Search PubMed.
- A. C. Vítor, A. E. Francisco, J. Silva, M. Pinho, S. A. Huws, J. Santos-Silva, R. J. Bessa and S. P. Alves, Sci. Rep., 2021, 11, 21878 CrossRef PubMed.
- S. Belyakov, M. Voigtmann, K. Y. Win, C. Lee, D. Lee, M. N. Antipina, R. Y. M. Teo, L. W. Khoo, Y. Kanagasundaram and C. T. Busran, ACS Food Sci. Technol., 2024, 2058–2068 CrossRef CAS.
- A. Gerzhova, M. Mondor, M. Benali and M. Aider, Food Chem., 2016, 201, 243–252 CrossRef CAS PubMed.
- C. N. Lunardi, A. J. Gomes, F. S. Rocha, J. De Tommaso and G. S. Patience, Can. J. Chem. Eng., 2021, 99, 627–639 CrossRef CAS.
- M. A. Devi, G. Subbulakshmi, K. M. Devi and L. Venkataraman, J. Agric. Food Chem., 1981, 29, 522–525 CrossRef CAS PubMed.
- N. Boukhari, A. Doumandji and A. Ferradji, Mediterr. J. Nutr. Metab, 2018, 11, 235–249 Search PubMed.
- S. Benelhadj, A. Gharsallaoui, P. Degraeve, H. Attia and D. Ghorbel, Food Chem., 2016, 194, 1056–1063 CrossRef CAS PubMed.
- Y. Ladjal-Ettoumi, L. H. Douik, M. Hamadi, J. A. A. Abdullah, Z. Cherifi, M. N. Keddar, M. Zidour and A. Nazir, Food Biophys., 2024, 19, 439–452 CrossRef.
- A. M. Pereira, C. R. Lisboa and J. A. V. Costa, Innovative Food Sci. Emerging Technol., 2018, 47, 187–194 CrossRef CAS.
- A. Schwenzfeier, P. A. Wierenga and H. Gruppen, Bioresour. Technol., 2011, 102, 9121–9127 CrossRef CAS PubMed.
- Ö. Gencer, Int. J. Pure Appl. Sci. Technol., 2024, 10, 276–282 Search PubMed.
- X. Guo, Q. Wang, Y. Wu, X. Liu and Z. Gong, Food Hydrocolloids, 2024, 110112 CrossRef CAS.
- F. H. Stephenson, in Calculations for Molecular Biology and Biotechnology, ed. F. H. Stephenson, Academic Press, Boston, 2016, 3rd edn, pp. 375–429, doi: DOI:10.1016/B978-0-12-802211-5.00011-4.
- Y. Qiu, C. Frear, S. Chen, P. Ndegwa, J. Harrison, Y. Yao and J. Ma, Renewable energy, 2020, 149, 691–700 CrossRef CAS.
- Y. Xu, J. Agric. Food Chem., 2022, 70, 11500–11509 CrossRef CAS PubMed.
- C. Wu, Y. Xiao, W. Lin, J. Li, S. Zhang, J. Zhu and J. Rong, Bioresour. Technol., 2017, 223, 312–316 CrossRef CAS PubMed.
- F. Zhao, Z. Wang and H. Huang, Processes, 2024, 12, 2059 CrossRef.
- C. Safi, A. V. Ursu, C. Laroche, B. Zebib, O. Merah, P.-Y. Pontalier and C. Vaca-Garcia, Algal Res., 2014, 3, 61–65 CrossRef.
- W. J. C. Sow and J. Du, Ultrason. Sonochem., 2024, 105, 106851 CrossRef PubMed.
- R. Zhang, N. Grimi, L. Marchal, N. Lebovka and E. Vorobiev, Algal Res., 2019, 40, 101524 CrossRef.
- M. Kurokawa, P. M. King, X. Wu, E. M. Joyce, T. J. Mason and K. Yamamoto, Ultrason. Sonochem., 2016, 31, 157–162 CrossRef CAS PubMed.
- W. Wang, Y. Feng, W. Chen, K. Adie, D. Liu and Y. Yin, Ultrason. Sonochem., 2021, 70, 105322 CrossRef CAS PubMed.
- G.-Y. Li, Q.-H. Chen, C.-R. Su, H. Wang, S. He, J. Liu, A. Nag and Y. Yuan, Innovative Food Sci. Emerging Technol., 2021, 68, 102612 CrossRef CAS.
- J. Folch, M. Lees and G. S. Stanley, J. Biol. Chem., 1957, 226, 497–509 CrossRef CAS PubMed.
- V. Matyash, G. Liebisch, T. V. Kurzchalia, A. Shevchenko and D. Schwudke, J. Lipid Res., 2008, 49, 1137–1146 CrossRef CAS PubMed.
- R. Halim, M. K. Danquah and P. A. Webley, Biotechnol. Adv., 2012, 30, 709–732 CrossRef CAS PubMed.
- L. Yao, J. A. Gerde and T. Wang, J. Am. Oil Chem. Soc., 2012, 89, 2279–2287 CrossRef CAS.
- A. Hosikian, S. Lim, R. Halim and M. K. Danquah, Int. J. Chem. Eng., 2010, 2010, 391632 Search PubMed.
- J. X. Tan, C.-C. Tan, J. Dharmawan and S. S. J. Leong, Food Bioprod. Process., 2023, 141, 73–80 CrossRef CAS.
- X. Ren, X. Zhao, F. Turcotte, J.-S. Deschênes, R. Tremblay and M. Jolicoeur, Microb. Cell Fact., 2017, 16, 1–13 CrossRef PubMed.
- G. S. Araujo, L. J. Matos, J. O. Fernandes, S. J. Cartaxo, L. R. Gonçalves, F. A. Fernandes and W. R. Farias, Ultrason. Sonochem., 2013, 20, 95–98 CrossRef CAS PubMed.
- Y. Li-Beisson, J. J. Thelen, E. Fedosejevs and J. L. Harwood, Prog. Lipid Res., 2019, 74, 31–68 CrossRef CAS PubMed.
- W. A. Faé Neto, C. R. Borges Mendes and P. C. Abreu, Aquacult. Res., 2018, 49, 2527–2535 CrossRef.
- L. Julia, K. Minsoo, C. Yong Keun and K. Donghyun, Sep. Purif. Technol., 2021, 276, 119248 CrossRef.
- T. Li, J. Xu, H. Wu, G. Wang, S. Dai, J. Fan, H. He and W. Xiang, Mar. Drugs, 2016, 14, 162 CrossRef PubMed.
- C. D. S. C. Caprara, T. K. Mathias, M. D. F. C. Santos, M. G. D'Oca, C. D. R. D'Oca, F. Roselet, P. C. Abreu and D. F. Ramos, Metabolites, 2023, 13, 202 CrossRef CAS PubMed.
- B. W. Vogler, J. Brannum, J. W. Chung, M. Seger and M. C. Posewitz, Algal Res., 2018, 36, 152–158 CrossRef.
- E. Alexandri, R. Ahmed, H. Siddiqui, M. I. Choudhary, C. G. Tsiafoulis and I. P. Gerothanassis, Molecules, 2017, 22, 1663 CrossRef PubMed.
- R. B. Pritchard and D. F. Hansen, Nat. Commun., 2019, 10, 1747 CrossRef PubMed.
- R. J. Abraham and A. Rowan, in Chlorophylls, CRC Press, 1991, pp. 797–834 Search PubMed.
| This journal is © The Royal Society of Chemistry 2025 |