 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxidation by ozone of linoleic acid monolayers at the air–water interface in multi-component films at 21 °C and 3 °C†
Ben
Woden
a,
Yizhou
Su
 b,
Maximilian W. A.
Skoda
c,
Adam
Milsom
b and
Christian
Pfrang
*bd
b,
Maximilian W. A.
Skoda
c,
Adam
Milsom
b and
Christian
Pfrang
*bd
aDepartment of Chemistry, University of Reading, Reading, UK
bSchool of Geography, Earth and Environmental Sciences, University of Birmingham, Birmingham, UK. E-mail: c.pfrang@bham.ac.uk
cISIS Neutron and Muon Source, Didcot, UK
dDepartment of Meteorology, University of Reading, Reading, UK
First published on 3rd December 2024
Abstract
Aqueous aerosols are often covered in thin films of surface-active species, such as fatty acids which are prominent components of both sea spray and cooking emissions. The focus of our study is one-molecule thin layers of linoleic acid (LOA) and their behaviours when exposed to ozone in multi-component films at the air–water interface. LOA’s two double bonds allow for ozone-initiated autoxidation, a radical self-oxidation process, as well as traditional ozonolysis. Neutron reflectometry was employed as a highly sensitive technique to follow the kinetics of these films in real time in a temperature-controlled environment. We oxidised deuterated LOA (d-LOA) as a monolayer, and in mixed two-component films with either oleic acid (h-OA) or its methyl ester, methyl oleate (h-MO), at room temperature and atmospherically more realistic temperatures of 3 ± 1 °C. We found that the temperature change did not notably affect the reaction rate (ranging from 1.9 to 2.5 × 10−10 cm2 s−1) which was similar to that of pure OA. We also measured the rate coefficient for d-OA/h-LOA to be 2.0 ± 0.4 × 10−10 cm2 s−1. Kinetic multi-layer modelling using our Multilayer-Py package was subsequently carried out for further insight. Neither the change in temperature nor the introduction of co-deposited film components alongside d-LOA consistently affected the oxidation rates, but the deviation from a single process decay behaviour (indicative of autoxidation) at 98 ppb is clearest for pure d-LOA, weaker for h-MO mixtures and weakest for h-OA mixtures. As atmospheric surfactants will be present in complex, multi-component mixtures, it is important to understand the reasons for these different behaviours even in two-component mixtures of closely related species. The rates we found were fast compared to those reported earlier. Our work demonstrates clearly that it is essential to employ atmospherically realistic ozone levels as well as multi-component mixtures especially to understand LOA behaviour at low O3 in the atmosphere. While the temperature change did not play a crucial role for the kinetics, residue formation may be affected, potentially impacting on the persistence of the organic character at the surface of aqueous droplets with a wide range of atmospheric implications.
1 Introduction
Many anthropogenic and biogenic organic compounds emitted into the atmosphere show surfactant activity and therefore can partition to the air–water interfaces that form the surfaces of atmospheric droplets.1–3 Aqueous aerosols are thus often covered in thin films of surface-active species, such as fatty acids which are prominent components of sea spray and cooking emissions. Various properties of these films and their reactivity have been probed mainly for simple fatty acids,4–8 while less work has focussed on polyunsaturated fatty acids such as linoleic acid (LOA) which is the main focus of the present study.The oxidation of these organic species to form secondary organic aerosols (SOAs) has been studied extensively in the past,9–12 and the SOA thereby produced will have climatic cloud lifetime effects. However, the partitioning of these species into monolayers at the surfaces of cloud droplets has also been observed to affect their reactivity.4–6,13 Perhaps more significant, though, may be the effects that the presence of a surface monolayer has on the cloud droplets themselves. Cloud formation, growth, evaporation, and rainout processes have a critical dependence upon surface tension as a result of the Kelvin effect.14 The presence of insoluble organic films at the air–water interface has a lowering effect on the surface tension of the resultant water aerosols, with impacts on interactions with clouds, and with cloud lifetime effects.3 The lowering of the surface tension associated with the presence of a monolayer would seem, via a weakening of the Kelvin effect, to suppress the destabilisation of small droplets and resultant suppression of nucleation that the Kelvin effect produces.
On this basis, the presence of these surfactants on aerosols could promote nucleation via reducing the surface tension of the droplets thereby nucleated. Additionally, the presence of such monolayers on the surface of existing droplets enhances their stability at lower diameters. This enhancement of nucleation and comparative stabilisation of lower diameter droplets could lead to increased cloud lifetimes.
There are also several other routes via which the presence of a monolayer film on the surface of a cloud droplet has important consequences for atmospheric chemistry. On the one hand, such films can act as a barrier to mass transport between the water droplet and the atmosphere. Many studies have shown that insoluble films at the interface are able to significantly retard both the evaporation of the water droplet itself and the transport of oxygen (O2), ozone (O3), ammonia (NH3), and many other important atmospheric species between the water droplet bulk phase and the atmospheric bulk phase.15–22 This inhibition of cross-interfacial transport could hinder the progress of atmospheric reactions inside aqueous aerosols that rely heavily on mass-transport steps across the air–water boundary.23
On the other hand, the surface film itself can act as a ‘2D solvent’ for species from the atmosphere, thus allowing the dissolution of species that would not normally dissolve in a water droplet, or altering the solvation behaviour of species that normally would dissolve in the aqueous droplet.24–28 This effect may play an important role in the transport of various species by water droplets, as it allows species that would not normally be transported by water droplets to be adsorbed at the coated interface.3 These changes in solvation behaviour at the interface could impact the rates and even nature of atmospheric reactions that occur heterogeneously at the surface of such particles. Studies of some reactions have shown a reduction in rates, such as the rate of production of nitric acid (HNO3) from dinitrogen pentoxide (N2O5) and water29,30 as a result of the presence of these coatings3 which is a key atmospheric process for the redistribution of nitrogen amongst various species in the atmosphere.
In summary, the reactivity and properties of these surfactant films deserve further study, as they likely have a vast range of effects that are of climatic and meteorological significance, be that by affecting the reactivity of the film components and associated SOA production, by weakening the Kelvin effect and thus extending the lifetime of reflective clouds, or by modifying key atmospheric chemical processes that involve adsorption to the surface of or diffusion into the bulk of water droplets.
The by far most commonly studied unsaturated fatty acid in the context of atmospheric aerosol coatings is oleic acid (OA).5,6,31–34 OA will react with highly reactive gas-phase species in the atmosphere such as O3,5,32,33,35 OH34 and NO3.6,31 OA’s single double bond limits the complexity of potential products being formed making it a fairly well-understood model system for ozone interaction with atmospheric surfactants.36 The poly-unsaturated analogue of OA, linoleic acid (LOA), represents a logical next step for investigations.
The focus of the study presented here is one-molecule thin layers of linoleic acid (LOA) and their behaviours when exposed to ozone in multi-component films at the air–water interface. LOA contains two double bonds allowing for ozone-initiated autoxidation, a radical self-oxidation process, as well as traditional ozonolysis.37,38 The core of the experiment involved studying the oxidation of custom-deuterated LOA (d-LOA) as a pure monolayer, and in mixed monolayers with oleic acid (h-OA) and methyl oleate (h-MO; the methyl ester analogue of OA) at both room temperature and a more atmospherically relevant temperature of 3 ± 1 °C. The OA/LOA system was also studied in a reverse deuteration configuration (d-OA/h-LOA) in both these temperature conditions. Exploratory data from d-LOA/stearic acid (h-SA; the saturated analogue of OA) mixed monolayers was also gathered. The custom-deuterated surfactants employed in the present study are displayed in Fig. 1.
 | ||
| Fig. 1 Custom-deuterated surfactant molecules employed in the present study: (a) deuterated linoleic acid (d-LOA); and (b) deuterated oleic acid (d-OA). | ||
Compared to OA reactivity, there has been fairly little previous work on the oxidation of LOA by ozone: He et al.39 studied a different LOA morphology and used much higher ozone concentrations than found in the atmosphere; Chu et al.37 suggested that competing LOA reaction mechanisms dominate under different ozone conditions.
2 Methodology
The experiments involved oxidation of custom-deuterated LOA (d-LOA) as a monolayer, and in mixed two-component films with hydrogenated oleic acid (h-OA) or its methyl ester, methyl oleate (h-MO), at room temperature (21 ± 1 °C) and atmospherically more realistic temperatures of 3 ± 1 °C. We also carried out initial experiments on mixed monolayers containing deuterated oleic acid (d-OA) and linoleic acid (h-LOA) to establish the possible impact of the linoleic acid co-deposition on the well-established reactivity of oleic acid towards ozone. The oxidation of the surfactant films was followed at ozone levels of less than 100 ppb to ca. 1 ppm. Neutron reflectometry (NR) was employed as a highly sensitive technique to follow the kinetics of these films in real time in a temperature-controlled environment. Below, the experimental set-up is outlined first, before briefly introducing the neutron reflectometry method followed by the multi-layer modelling analysis.2.1 Experimental set-up
The experimental set-up has been described previously (Woden et al., 2021;33 compare also Skoda et al., 2017 (ref. 40) and Woden et al., 2018 (ref. 41) for earlier variations of the set-up for study of related systems) and is only briefly outlined here. All experiments were performed on the specular neutron reflectometry instrument INTER at the ISIS Neutron and Muon Source.A custom-built aluminium gas flow cell (volume of ca. 1.5 L) was fitted with a PTFE liquid trough (inner dimensions of 238 mm × 70 mm). This reaction chamber was mounted on the sample stage and interfaced with the gas delivery system. The trough was filled with 90 mL of null-reflecting water (NRW). The height of the air–liquid interface was aligned with respect to the neutron beam using a Keyence laser displacement sensor (model no. LK-G402), which was coupled into the sample chamber via a quartz window to allow automated height adjustment during the measurements. Height adjustments over a ca. 2 hours experiment were always less than 0.15 mm for a water height of ca. 5 mm in the trough. The chamber was designed to provide a controlled and confined environment in which monolayers can be oxidised by a gas-phase oxidant while under analysis by both neutron reflectometry and infra-red reflection absorption spectroscopy (IRRAS) as described in detail in a method paper.40 For the present study, a further iteration of the sample environment development was deployed to allow for cooling of the subphase, in order to access relevant atmospheric temperature conditions (compare Woden et al., 2021).33 Because of the low relative humidity (RH) used in the work presented here, we did not observe any condensation within the reaction chamber or on the windows throughout the experiments even at near-freezing temperatures (3 ± 1 °C).
Monolayers were spread using 20–40 μL of the spreading solutions in chloroform, leaving a monolayer of the dissolved species after solvent evaporation. Dry oxygen was continuously flowing (flow rate: 1.2 L min−1) into the chamber to provide a low (<10%) RH environment and to avoid build-up of any gas-phase products. Data were recorded for several minutes before ozone (O3) was admitted into the chamber. O3 was generated by exposing the O2 flow to UV light using a commercial PenRay ozoniser (UVP Ltd, Cambridge) to ozonise the stream of O2 (99.999%; BOC Ltd) regulated by an electronic mass flow controller to achieve O3 mixing ratios in the range of 98–983 ppb; the ozoniser was calibrated offline using UV-vis absorption at 254 nm and an absorption cross-section value of 1.13 × 10−17 cm−2 (see Daumont et al., 1992).42 We were working in large excess of O3 compared to the organic monolayer, and [O3] remained approximately constant during the reaction.
2.2 Neutron reflectometry (NR)
Our neutron reflectometry (NR) methodology was described elsewhere.40 In short, specular NR experiments were conducted using the white beam INTER reflectometer at the Rutherford Appleton Laboratory in Oxfordshire, UK, employing neutron wavelengths ranging from 2.0 to 17.0 Å. The reflected intensity was recorded at an incident angle of 0.8°, with a non-polarising supermirror positioned at 0.75°, as a function of the momentum transfer, q = (4π![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ)/λ, where λ represents the wavelength and θ denotes the incident angle. The data were collected with a resolution (Δq/q) of 7%, covering a total illuminated length of 165 mm. To minimize meniscus effects, the beam width was adjusted to 50 mm. Time-resolved measurements were performed over intervals ranging from minutes to several hours, with a time resolution of 20 seconds.
sinθ)/λ, where λ represents the wavelength and θ denotes the incident angle. The data were collected with a resolution (Δq/q) of 7%, covering a total illuminated length of 165 mm. To minimize meniscus effects, the beam width was adjusted to 50 mm. Time-resolved measurements were performed over intervals ranging from minutes to several hours, with a time resolution of 20 seconds.
Neutron reflectivity (the fraction of incident neutrons reflected, R) varies as a function of the energy and reflection angle of the incident neutrons (expressed as momentum transfer, Q) and the scattering length density, SLD (ρ) and thickness (τ) of the monolayer as shown in eqn (1) (based on Lu et al., 2000):43
 | (1) |
The SLD and layer thickness can be inferred from the relationship between reflectivity and momentum transfer measured by the instrument. The two parameters are fitted over the whole Q range as a combined ρτ value, which corresponds to a surface concentration of scattering length, from which the surface concentration of the surfactant of interest can be determined if the scattering length is known (e.g. b = 315 fm for deuterated oleic acid). Reflectivity curves of R vs. Q were fitted using MOTOFIT44 to give ρτ values for each 20 s time slice. These fitted ρτ values can be converted into surface concentration (Γ) values for the surfactant following eqn (2):43
 | (2) |
The error bars for the SLD × thickness product were propagated using the calculated fitting errors for each parameter obtained from MOTOFIT.44 In order to achieve a sufficient contrast of the monolayer compared to the other phases in the experimental system, a deuterated form of oleic acid (d-OA), was used (Sigma-Aldrich at 98% atom D; 99.9%). Spreading onto an aqueous subphase will cause the acidic deuterium to be exchanged with the subphase, so we use the scattering length of oleic acid with 33 deuterium atoms (315 fm) to calculate the surface concentration from ρτ values. For custom-deuterated linoleic acid (d-LOA; Sigma-Aldrich at 98% atom D; 98%), the equivalent consideration leads to the presence of 31 deuterium atoms in the fully deuterated LOA.
2.3 Multilayer-Py modelling
Following the initial, basic kinetic analysis presented first, we also modelled the experimental data using the Multilayer-Py framework45 in order to gain further mechanistic insights.Multilayer-Py45 provides a framework for constructing kinetic multi-layer models, so that the model code is produced automatically for the user, removing potential human error in typing out the model code. This code is written in a readable format, enabling the code to be shared easily and facilitating more reproducible modelling results. This is further supported through the Jupyter notebook,46 which is a document that incorporates both Python code and markdown text and is becoming an increasingly popular way of sharing and describing scientific code. The usefulness of Multilayer-Py has been demonstrated by application to the ozonolysis of OA45 employing both KM-SUB47 and KM-GAP48 modelling approaches.
A detailed description of the KM-SUB model concept used in the present study has been presented previously47 and is not repeated here. Essentially, the model splits our experimental system into a number of layers. The diffusion of reactants between each layer and the reaction of each component within each layer are resolved. Surface chemistry and the adsorption and desorption of gaseous species are also resolved. For this specific study, we introduced four different treatments of the oxidation of LOA by ozone: (i) single step oxidation considering the gradual build-up of ozone in our reaction chamber; (ii) the same assumptions as in (i), but now also assuming an inert residue remaining; (iii) two-step oxidation with a reaction product reacting with the reactant fatty acid in the second step; and (iv) the same assumptions as in (iii), but now also assuming an inert residue remaining at the interface.
3 Results and discussion
In this section, experimental work is presented first followed by basic kinetic analysis and subsequently multi-layer modelling analysis is presented.3.1 Oleic acid ozonolysis in the presence of co-surfactant linoleic acid
We first studied two-component monolayers containing deuterated oleic acid (d-OA) and linoleic acid (h-LOA) to establish the possible impact of the linoleic acid co-deposition on the well-established reactivity of oleic acid towards ozone.32,33,36Fig. 2(a) and (b) show the ozonolysis of a d-OA/h-LOA monolayer at a variety of ozone concentrations at room temperature and 3 ± 1 °C, respectively. Fig. 2(c) shows ozonolysis under the two temperature conditions overlaid, displaying only the highest and lowest ozone concentrations for visual clarity. These figures illustrate clearly that the change in temperature did not notably affect the rate of reaction.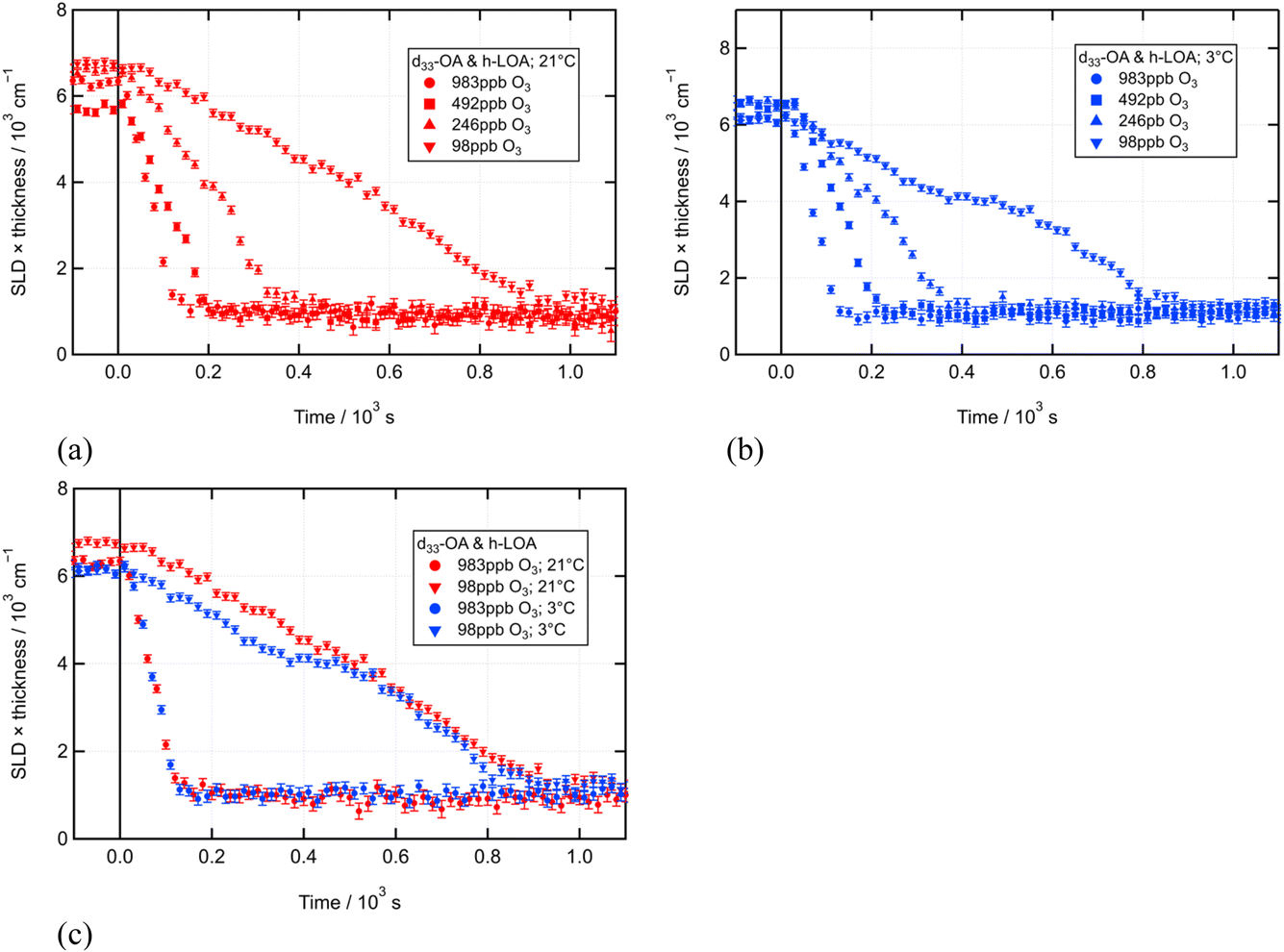 | ||
| Fig. 2 Time evolution of fitted neutron reflectometry signal during oxidation of d-OA in a mixed monolayer with h-LOA (21 μL of 0.81 g L−1 d-OA/0.59 g L−1 h-LOA; O3 introduced at t = 0 s at various [O3]) at (a) room temperature (21 ± 1 °C) and (b) near-freezing (3 ± 1 °C). (c) Re-plots the data for the highest and lowest ozone concentrations to compare directly the behaviour at the two different temperatures. | ||
Pseudo-first-order rate coefficients were fitted to these neutron reflectometry time series displayed in Fig. 2 using the stretched exponential analytical model outlined in the ESI Section S3 in Woden et al. (2021).33Fig. 3 displays a second order plot for this reaction (omitting data from the highest ozone concentration, as these reaction conditions were judged to be too fast to be fitted, given the limitation of a 20 seconds time resolution in our experimental approach). The second order rate coefficient obtained from these data is 2.0 ± 0.4 × 10−10 cm2 s−1. This is slightly lower than the values we measured for pure OA ((2.2 ± 0.4) × 10−10 cm2 s−1 at 21 ± 1 °C and (2.2 ± 0.2) × 10−10 cm2 s−1 at 2 ± 1 °C),33 but the error bars overlap significantly.
 | ||
| Fig. 3 Second order rate plot for the oxidation of d-OA in a mixed monolayer with h-LOA based on the data displayed in Fig. 2 (omitting the data for [O3] = 983 ppb). | ||
A further experiment was performed collecting neutrons over the full Q range accessible to INTER, in order to quantify any residue left behind after ozonolysis (as we have reported for low temperature ozonolysis of a pure OA monolayer; see Woden et al., 2021).33Fig. 4 shows reflectivity curves before and after oxidation at 3 ± 1 °C where a residue can clearly be identified. Fitting monolayer parameters to these reflectivity curves to quantify the absolute amount of deuterated material adsorbed at the interface determined that 13% of the material present before ozonolysis remained afterward. This is similar to the range seen for pure OA monolayers at these temperatures (up to 11.1% deuterated residue was reported in Woden et al., 2021).33Fig. 4 shows that the fit to the data collected before reaction deviates at high Q values. This is because the reflectivity background is not always Q-independent, as assumed in the MOTOFIT model and this becomes important at high Q where there is little signal compared to the background. This can be avoided by using a fixed background parameter derived from a clean air-NRW measurement, and this is the procedure used for fitting most of the data presented. This does, however, assume that the background will not change across the conditions studied (including temperature variations). Therefore, when carrying out experiments specifically to quantify post-oxidation residue under various conditions, in which distinguishing genuine residue from background signal is particularly important, this fixed-background method was avoided.
 | ||
| Fig. 4 R vs. Q reflectivity plots before and after ozonolysis of d-OA in a mixed monolayer with h-LOA (21 μL of 0.81 g L−1 d-OA/0.59 g L−1 h-LOA; [O3] = 983 ± 150 ppb; 3 ± 1 °C); null-reflecting water (NRW) background is shown for comparison. | ||
Due to time constraints during neutron beamtime, no analogous experiment could be performed at room temperature. However, the kinetic data shown in Fig. 2(a) and (c) – collected across a restricted Q range and therefore not suitable for precise absolute quantification of small amounts of material – suggests that a similar residual monolayer remained.
3.2 Linoleic acid oxidation by ozone
The primary focus of the experiments reported here was the study of the oxidation of linoleic acid (which can be achieved via ozonolysis or ozone-initiated autoxidation) as a monolayer at the air–water interface, and the effects of temperature and co-deposited film components on that oxidation process. To this end, pure d-LOA monolayers and mixed monolayers containing either h-OA or h-MO were oxidised under a variety of ozone concentrations at room temperature (21 ± 1 °C) and at 3 ± 1 °C. Fig. 5–7 display the time series of the neutron reflectometry data for these reactions. | ||
| Fig. 5 Time series of neutron reflectometry data for the oxidation of pure d-LOA monolayers (21 μL of 1.4 g L−1 d-LOA; O3 introduced at t = 0 s at various [O3]) at (a) room temperature (21 ± 1 °C) and (b) near freezing (3 ± 1 °C). | ||
 | ||
| Fig. 6 Time series of neutron reflectometry data for the oxidation of a mixed monolayer of d-LOA with h-OA (21 μL of 0.68 g L−1 d-LOA/0.75 g L−1 h-OA; O3 introduced at t = 0 s at various [O3]) at (a) room temperature (21 ± 1 °C) and (b) near freezing (3 ± 1 °C). | ||
 | ||
| Fig. 7 Time series of neutron reflectometry data for the oxidation of a mixed monolayer of d-LOA with h-MO (21 μL of 0.65 g L−1 d-LOA/1.0 g L−1 h-MO; O3 introduced at t = 0 s at various [O3]) at (a) room temperature (21 ± 1 °C) and (b) near freezing (3 ± 1 °C). | ||
Fig. 8(a) to (c) overlay the temperature conditions, displaying only the highest and lowest ozone concentrations for visual clarity.
 | ||
| Fig. 8 Time series of neutron reflectometry data contrasting the two temperature conditions for the highest and lowest [O3] for the oxidation (see Fig. 5–7 for individual plots for all [O3] studied) of (a) a pure d-LOA monolayer; (b) a mixed monolayer of d-LOA with h-OA; and (c) a mixed monolayer of d-LOA with h-MO. | ||
Fig. 9 displays a second-order plot for the LOA oxidation, and Table 1 summarises the fitted second-order rate coefficients for the three monolayer types in the two temperature conditions, as well as the rate coefficients for each monolayer type (treating temperature as irrelevant), for each temperature (treating monolayer type as irrelevant), and for all data combined. Table 1 demonstrates that neither the change in temperature nor the introduction of a co-deposited film component alongside d-LOA consistently affected the rate of reaction of LOA with ozone.
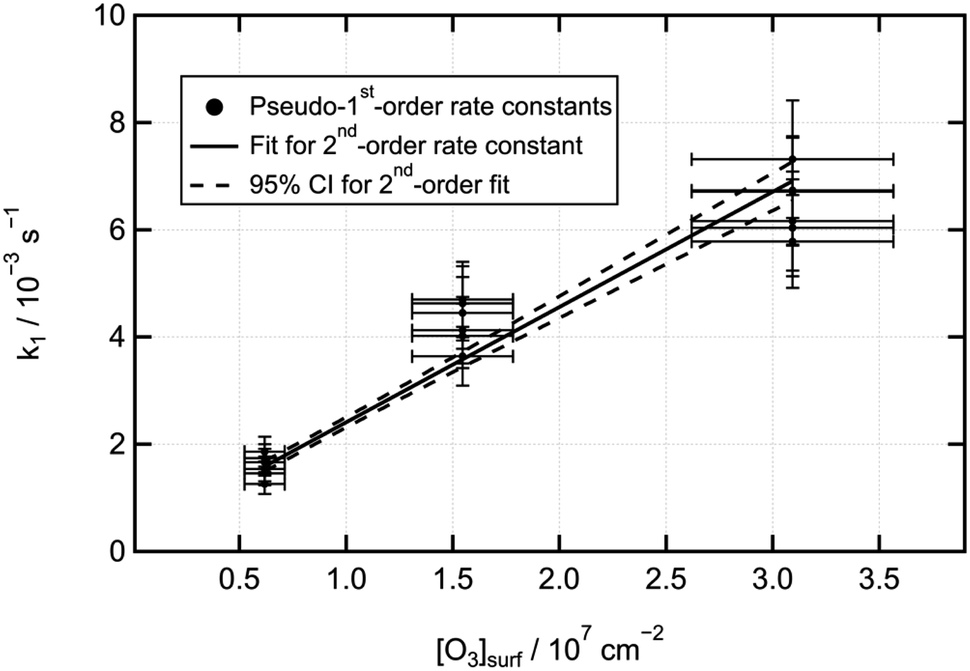 | ||
| Fig. 9 Second-order rate plot for the data displayed in Fig. 5–8. | ||
| Monolayer | Rate coefficient/10−10 cm2 s−1 | ||
|---|---|---|---|
| 21 ± 1 °C | 3 ± 1 °C | Combined | |
| d-LOA | 2.1 ± 0.7 | 2.0 ± 0.7 | 2.0 ± 0.4 |
| d-LOA/h-OA | 2.0 ± 0.7 | 2.5 ± 0.7 | 2.3 ± 0.4 |
| d-LOA/h-MO | 1.9 ± 0.7 | 2.5 ± 0.7 | 2.0 ± 0.4 |
| Combined data | 2.0 ± 0.2 | 2.3 ± 0.3 | 2.1 ± 0.2 |
The rates displayed in Table 1 are higher compared to those reported by He et al.39 However, our study uses a very different morphology (monolayer at the air–water interface) compared to that used by He et al. Furthermore, He et al. use a much higher ozone concentration than our study (ca. 10 ppm compared to ca. 100 to 1000 ppb in the present study) and Chu et al. have specifically warned that, due to the competing mechanisms for this reaction that dominate under different conditions, extrapolating from high ozone concentrations downwards is likely to be problematic.37 Other studies on this heterogenous reaction have mostly reported uptake coefficients, rather than rate coefficients, and these have varied by around an order of magnitude as reviewed by He et al.39 The use of an uptake coefficient, which is more dependent on reaction conditions and geometry than is a rate coefficient, makes these studies less useful as a guide for what to expect in our study, which uses a monolayer at the air–water interface rather than particulate phase or film-coated flow tube setups. Differences in mechanism may also be driving some of these discrepancies.
An experiment analogous to that described earlier for the d-OA/h-LOA system (compare Fig. 4) was performed in order to quantify any post-oxidation residue. Again, only the low temperature was studied due to time constraints at neutron beamtime experiments. Fig. 10(a)–(c) display reflectivity curves before and after oxidation at 3 ± 1 °C for d-LOA, d-LOA/h-OA and d-LOA/h-MO, respectively.
 | ||
| Fig. 10 R vs. Q reflectivity plots before and after reaction with O3 of (a) a pure d-LOA monolayer (21 μL of 1.4 g L−1 d-LOA; [O3] = 983 ± 150 ppb; 3 ± 1 °C); (b) d-LOA in a mixed monolayer with h-OA (21 μL of 0.68 g L−1 d-LOA/0.75 g L−1 h-OA; [O3] = 983 ± 150 ppb; 3 ± 1 °C); and (c) d-LOA in a mixed monolayer with h-MO (21 μL of 0.65 g L−1 d-LOA/1.0 g L−1 h-MO; [O3] = 983 ± 150 ppb; 3 ± 1 °C). Null-reflecting water (NRW) backgrounds are shown for comparison. | ||
For d-LOA as a pure monolayer (Fig. 10(a)), a residue is clearly observed, and fitting the monolayer parameters quantifies this as ca. 7% of the initial adsorbed deuterated material. For d-LOA in a mixed monolayer with h-OA, a residue is also observed and quantified as ca. 11% of the initial adsorbed deuterated material. The data for d-LOA in a mixed monolayer with h-MO are slightly more difficult to interpret. The fitting process used throughout this work does successfully converge and fit a curve (displayed in Fig. 10(c) as the red line) that would represent ca. 13% of the initial adsorbed deuterated material. However, a visual appraisal of Fig. 10(c) suggests that the post-oxidation reflectivity curve differs mainly from the null-reflecting water background in that it is unusually noisy at very low Q. From these noisy data alone it is very difficult to quantify the amount of residual monolayer, so further experimental confirmation would be useful. As well as repeating the low temperature residue quantification experiment for d-LOA/h-MO, future work on this system could perform analogous room temperature experiments. As for the OA ozonolysis in the presence of LOA discussed earlier, the kinetic data collected over a limited Q range (see Fig. 5–8) suggests that similar residual monolayers are present, but a full Q range characterisation would be very useful to confirm this. As further experiments were not feasible within the limitations of time-constrained beamline facility access, this was one of the key motivations for the more detailed modelling analysis presented in Section 3.4.
The mechanism for LOA oxidation is far more complex than that for OA oxidation; the former involves multiple competing pathways of ozonolysis and ozone-initiated autoxidation that result in different products. One mechanism or the other has previously been observed to dominate depending on ozone concentration and relative humidity.37 The linear increase in pseudo-first-order reaction rate with increasing ozone concentration suggests that one dominant mechanism is being observed across the range of ozone concentrations used in this work (ca. 100 to 1000 ppb). Chu et al. noted that higher ozone concentrations (above 250 ppb) inhibited the build-up of autoxidation products, as did higher relative humidity.37 All our work is carried out with ozone dissolved in a stream of dry oxygen, so the relative humidity will be very low. Based on the range of ozone concentrations we employed and the low relative humidity in our system, based on this literature we would have expected to observe mainly autoxidation except for the highest [O3] conditions. The autoxidation mechanism is complex and is not presented here as we were not conducting any product analysis to directly distinguish reaction mechanism, but rather focussing on reaction kinetics and residue formation in two-component monolayers at different temperatures (a detailed explanation of LOA autoxidation can be found in Chu et al.).37
Given the abundance of saturated surfactants in atmospheric aerosols, we also carried out exploratory experiments to test the effect of co-deposited stearic acid (SA) – the saturated analogue to OA – on the LOA monolayer oxidation. Fig. 11(a) displays the oxidation of a d-LOA/h-SA monolayers at two ozone concentrations at the two temperatures of interest. While this data set was too limited to reliably calculate a second-order rate coefficient, a visual appraisal of the data suggests that temperature once again does not exert a significant effect on the reaction rate. As far as the effect of SA on the reaction rate goes, however, this preliminary data suggest that there may be an impact. Fig. 11(b) illustrates the room temperature data overlaid with the analogous data for d-LOA/h-OA oxidation. It does appear that the reaction is faster in the presence of SA. Further work would be useful to establish whether this effect is reproducible, as there may be a degree of inter-run variability.
 | ||
| Fig. 11 (a) Time series of NR data contrasting the two temperature conditions for the highest and lowest [O3] for the oxidation of d-LOA in a mixed monolayer with h-SA (21 μL of 0.65 g L−1 d-LOA/0.80 g L−1 h-SA); O3 introduced at t = 0 s; (b) time series of neutron reflectometry data contrasting the highest and lowest [O3] for the oxidation of d-LOA at room temperature in mixed monolayers with h-SA and h-OA (21 μL of 0.68 g L−1 d-LOA/0.75 g L−1 h-OA or 0.65 g L−1 d-LOA/0.80 g L−1 h-SA; O3 introduced at t = 0 s at two [O3]); and (c) R vs. Q reflectivity plots before and after reaction with ozone of d-LOA in a mixed monolayer with h-SA (21 μL of 0.65 g L−1 d-LOA/0.80 g L−1 h-SA; [O3] = 983 ± 150 ppb; 3 ± 1 °C; air-NRW background is shown for comparison). | ||
A full Q range residue quantification experiment was performed for the d-LOA/h-SA system at 3 ± 1 °C, and the results are shown in Fig. 11(c). The post-oxidation reflectivity appears no different from the air-NRW background barring an intriguing oscillation at low Q that would merit further investigation as would the residue behaviour at 21 ± 1 °C.
3.3 Multilayer-Py modelling results
The Multilayer-Py framework45 was used with the KM-SUB model47 to gain further mechanistic insights. The model was run in four scenarios: (i) considering a single reaction process with initial ozone build-up modelled in the reaction chamber; (ii) additionally considering a residue remaining after reaction; (iii) additionally considering a secondary reaction step without residue; and (iv) considering the additional reaction step and residue formation. Model fits quickly demonstrated that both ozone build-up in the reaction chamber and residue formation need to be considered to obtain a reasonable fit, while one stage and two stage oxidation mechanisms resulted in very similar decay shapes. We thus focussed on the single-step oxidation with ozone build-up and residue formation to fit the entire experimental data set with a consistent set of assumptions with minimum complexity. Examples of the optimised model output are displayed in Fig. 12–14 (the full set of model fits for the different [O3] is provided in the ESI†). | ||
| Fig. 12 Multilayer-Py45 modelling fits to selected [O3] for pure d-LOA oxidation (fits to the full set of [O3] conditions is presented in the ESI;† the model runs presented here consider ozone build-up in the reaction chamber, a single-stage oxidation process and residue formation). The figures display the normalised decay of d-LOA as a function of time with experimental data (red and blue symbols), the results of the global optimisation using Markov chain Monte Carlo (MCMC) sampling (pink lines close to the best fit) and the optimised model fit (solid black line). (a) 983 ppb & 21 ± 1 °C; (b) 983 ppb & 3 ± 1 °C; (c) 492 ppb & 21 ± 1 °C; and (d) 492 ppb & 3 ± 1 °C. | ||
 | ||
| Fig. 13 Multilayer-Py45 modelling fits to selected [O3] for d-LOA/h-OA oxidation (fits to the full set of [O3] conditions is presented in the ESI;† the model runs presented here consider ozone build-up in the reaction chamber, a single-stage oxidation process and residue formation). The figures display the normalised decay of d-LOA as a function of time with experimental data (red and blue symbols), the results of the global optimisation using Markov chain Monte Carlo (MCMC) sampling (pink lines close to the best fit) and the optimised model fit (solid black line). (a) 983 ppb & 21 ± 1 °C; (b) 983 ppb & 3 ± 1 °C; (c) 492 ppb & 21 ± 1 °C; and (d) 492 ppb & 3 ± 1 °C. | ||
 | ||
| Fig. 14 Multilayer-Py45 modelling fits to selected [O3] for d-LOA/h-MO oxidation (fits to the full set of [O3] conditions is presented in the ESI;† the model runs presented here consider ozone build-up in the reaction chamber, a single-stage oxidation process and residue formation). The figures display the normalised decay of d-LOA as a function of time with experimental data (red and blue symbols), the results of the global optimisation using Markov chain Monte Carlo (MCMC) sampling (pink lines close to the best fit) and the optimised model fit (solid black line). (a) 983 ppb & 21 ± 1 °C; (b) 983 ppb & 3 ± 1 °C; (c) 492 ppb & 21 ± 1 °C; and (d) 492 ppb & 3 ± 1 °C. | ||
The kinetic multi-layer modelling demonstrated that model optimisation using Markov chain Monte Carlo (MCMC) sampling yields good fits for the rate coefficients obtained with standard kinetic fitting (see fits in Fig. 12–14 which are using the rate coefficients stated in Table 1).
Furthermore, Multilayer-Py allowed optimised residue fitting. The results (see Table 2) suggest that the residues for pure d-LOA (Fig. 12) are consistently lower (11–14%) than for its mixtures with h-OA (13–27%; Fig. 13) followed by the mixtures with h-MO (19–27%; Fig. 14). The modelling showed no significant difference between residues at the two different temperatures: only for the d-LOA/h-MO mixtures (see Table 2) did we find a small difference with the residue averaging at ca. 23% for 3 ± 1 °C compared to ca. 20% for 21 ± 1 °C, however no clear difference was observed for the other monolayer compositions.
| Monolayer | Relative residue/% | |||||||
|---|---|---|---|---|---|---|---|---|
| 21 °C & 983 ppb | 21 °C & 492 ppb | 21 °C & 246 ppb | 21 °C & 98 ppb | 3 °C & 983 ppb | 3 °C & 492 ppb | 3 °C & 246 ppb | 3 °C & 98 ppb | |
| d-LOA | 14.48 | 12.86 | 14.31 | 12.07 | 10.87 | 13.59 | 12.39 | 13.17 |
| d-LOA/h-OA | 19.38 | 13.90 | 16.22 | 20.21 | 16.40 | 14.31 | 12.80 | 26.79 |
| d-LOA/h-MO | 21.32 | 19.14 | 20.68 | 19.94 | 22.49 | 19.76 | 22.94 | 27.39 |
Overall, the modelling at room temperature results in slightly better fits across the different monolayer compositions at all [O3] except for the lowest concentration of 98 ppb (see Fig. S25 in the ESI†). For pure d-LOA at low [O3] (98 ppb) the decay shape consistently deviates from that compatible with a single decay process (see Fig. S25(a) & (b)†); this deviation is weaker, but still visible, for the mixtures with h-MO and less apparent for the h-OA mixtures (contrast Fig. S25(a)–(d)†); the data were too limited to quantify this effect, but this observation is consistent with previous work suggesting that the additional process of autoxidation becomes important at low O3.37
3.4 Effects of low temperatures
Overall, we found that the temperature change from 21 ± 1 to 3 ± 1 °C for the systems studied here did not affect the reaction rate, which ranges from 1.9 to 2.5 × 10−10 cm2 s−1 and is thus similar to that of pure oleic acid (2.2 × 10−10 cm2 s−1 both at 21 ± 1 and 2 ± 1 °C).33 This lack of a measurable effect on the kinetics over the comparatively small temperature range accessible in our system is consistent with our previous work – temperatures are likely to play a more important role for the kinetics when phase boundaries are crossed, which would warrant further study.Kinetic multi-layer modelling using our Multilayer-Py package allowed us to optimise the residue fitting as residue formation showed a temperature dependence in our previous work for oleic acid ozonolysis (Woden et al., 2021).33 While the modelling results presented here suggest that the residues for pure d-LOA are consistently lower (11–14%) than for its mixtures with h-OA (13–27%) and h-MO (19–27%), it showed no clear difference between residues at the two different temperatures: only for the d-LOA/h-MO mixtures did we find a small difference with the residue averaging at ca. 23% for 3 ± 1 °C compared to ca. 20% for 21 ± 1 °C, but no significant difference was observed for the other monolayer compositions across the different O3 levels.
Since the low-temperature conditions used here are atmospherically realistic, it is key to understand if a product film persists and thus needs to be considered when assessing the impact of unsaturated fatty acid partitioned to the air–water interface. The presence of stable (non-oxidisable) reaction products could also lead to a build-up of inert monolayers during the aerosol life cycle with potential implications for cloud formation. Our previous work on oleic acid ozonolysis33 showed that a residual surface film (likely formed of ozonolysis products nonanoic acid and a mixture of azelaic and 9-oxononanoic acids) was retained at the interface after ozonolysis at low temperatures, but not at room temperature. For the binary mixtures studied here, we did not find such a clear temperature dependence of residue formation. As surfactants will be present in the atmosphere in complex, multi-component mixtures, it is important to understand the reasons for these different behaviours even for closely related systems. While the temperature change did not impact on the kinetics, residue formation may be affected, thus altering the persistence of the organic character at the surface of aqueous droplets.
3.5 Future work
As LOA, OA, and MO are all closely related, unsaturated fatty acids (a fatty acid methyl ester in the case of MO) with broadly similar physical properties, it is likely that they mix well at the interface, and it is also likely that LOA and stearic acid (SA) mix poorly, as do OA and SA (see Skoda et al., 2017).40 Due to this similarity to systems already studied, offline characterisation (e.g. via Brewster angle microscopy and Wilhelmy plate tensiometry) was not performed, but such investigations would be useful, especially if less well understood atmospheric surfactants would be considered. Additional work on the mixture with SA would allow the quantification of the rate coefficient in this mixed monolayer system which would be a very useful addition since the reaction appears to be faster in the presence of SA based on our preliminary work, but this needs confirmation since there is a reasonable degree of inter-run variability.Furthermore, it would be very useful to explore further the kinetic behaviour at lower ozone concentrations (as long as reactions can still be observed within the limited timeframe of neutron beamtime experiments) given the unusual decay shapes we found especially for pure d-LOA at 98 ppb. This would also extend the range of data available for second-order fitting increasing the robustness of the fitted parameters.
Finally, it would be worth establishing the residue remaining after reaction for a wider range of conditions also including full Q range runs at room temperature.
4 Conclusions
Our experiments focussed on the oxidation of deuterated LOA (d-LOA) as a monolayer, and in mixed two-component films with either oleic acid (h-OA) or its methyl ester, methyl oleate (h-MO), at two temperatures using ozone levels of ca. 100 ppb to 1 ppm. We found that the temperature change did not affect the reaction rate. We also measured the rate coefficient for oleic acid in d-OA/h-LOA mixed monolayers to be 2.0 ± 0.4 × 10−10 cm2 s−1 and thus ca. 10% below that measured for pure d-OA earlier,33 but the uncertainties overlap significantly.Kinetic multi-layer modelling was used to: (i) confirm the rate coefficients obtained with standard kinetic fitting; (ii) illustrate that for pure d-LOA at low [O3] (98 ppb) the decay shape consistently deviates from that compatible with a single decay process; this deviation was found to be weaker, but still visible, for the mixtures with h-MO and less apparent for the h-OA mixtures (this observation is consistent with previous work suggesting that the additional process of autoxidation becomes important at low O3, but the reason for the differences between pure d-LOA and the two-component mixtures merits further investigation); and (iii) optimise the residue fitting suggesting that the residues for pure d-LOA are consistently lower (11–14%) than for its mixtures with h-OA (13–27%) followed by the mixtures with h-MO (19–27%). The latter trend in the model data is consistent with full Q range NR experiments carried out at the highest [O3] conditions, although these experiments suggest a lower residue proportion ranging from ca. 7% to 13%.
In summary, neither the change in temperature nor the introduction of co-deposited film components alongside d-LOA consistently affected the LOA oxidation rates, but the deviation from a single process decay behaviour (indicative of autoxidation) at 98 ppb is clearest for pure d-LOA, weaker for the h-MO mixtures and the weakest for h-OA mixtures.
As surfactants will be present in the atmosphere in complex, multi-component mixtures, it is important to understand the reasons for these different behaviours even in two-component mixtures of closely related species. The rates we found were fast compared to those reported earlier for a different LOA morphology under much higher ozone concentrations. Our work demonstrates clearly that it is essential to employ atmospherically realistic ozone levels as well as multi-component mixtures especially to understand LOA behaviour at low O3 in the atmosphere. While the temperature change did not play a crucial role for the kinetics, residue formation may be affected, potentially impacting on the persistence of the organic character at the surface of aqueous droplets with a wide range of atmospheric implications.
Data availability
The modelling data supporting this article have been included in the ESI.† The underlying neutron reflectometry data is available at the ISIS Neutron & Muon Source (https://doi.org/10.5286/ISIS.E.RB1910615) and the experimental work presented here is also part of chapter 7 of Ben Woden’s PhD thesis which is available in the University of Reading’s repository (https://centaur.reading.ac.uk/96396/2/19011604_WODEN_Thesis.pdf).Author contributions
BW carried out the experimental work (jointly with MWAS, CP and AM) and wrote a thesis chapter for his PhD at the University of Reading that formed the basis for this manuscript; CP was lead supervisor for BW’s PhD; CP wrote the abstract submitted for the Faraday Discussions and the first draft of this manuscript; MWAS co-supervised BW’s PhD project, supported the experimental work and data analysis at ISIS and contributed substantially to data analysis and interpretation; YS carried out the Multilayer-Py modelling analysis and created the modelling plots; AM contributed to the experiments and model analysis; all co-authors contributed to the final paper.Conflicts of interest
The authors confirm that there are no conflicts of interest to declare.Acknowledgements
BW’s PhD was funded through the SCENARIO NERC DTP and supported by ISIS Neutron and Muon Source; AM was supported through SCENARIO/CENTA NERC DTPs and subsequently through NERC (grant number NE/T00732X/1); ISIS beamtime was awarded at the INTER instrument (https://doi.org/10.5286/ISIS.E.RB1910615).References
- P. S. Gill, T. E. Graedel and C. J. Weschler, Rev. Geophys., 1983, 21, 903–920 CrossRef CAS.
- G. B. Ellison, A. F. Tuck and V. Vaida, J. Geophys. Res., 1999, 104, 11633–11641 CrossRef CAS.
- D. J. Donaldson and V. Vaida, Chem. Rev., 2006, 106, 1445–1461 CrossRef CAS PubMed.
- L. F. Voss, M. F. Bazerbashi, C. P. Beekman, C. M. Hadad and H. C. Allen, J. Geophys. Res., 2007, 112, D06209 Search PubMed.
- M. D. King, A. R. Rennie, K. C. Thompson, F. N. Fisher, C. C. Dong, R. K. Thomas, C. Pfrang and A. V. Hughes, Phys. Chem. Chem. Phys., 2009, 11, 7699–7707 RSC.
- F. Sebastiani, R. A. Campbell, K. Rastogi and C. Pfrang, Atmos. Chem. Phys., 2018, 18, 3249–3268 Search PubMed.
- W. R. Barger and W. D. Garrett, J. Geophys. Res., 1970, 75, 4561–4566 CrossRef CAS.
- E. Adams and H. Allen, Atmosphere, 2013, 4, 315–336 Search PubMed.
- M. K. Shrivastava, R. Subramanian, W. F. Rogge and A. L. Robinson, Atmos. Environ., 2007, 41, 9353–9369 CrossRef CAS.
- H.-M. Hung and C.-W. Tang, J. Phys. Chem. A, 2010, 114, 13104–13112 CrossRef CAS PubMed.
- A. K. Y. Lee and C. K. Chan, J. Phys. Chem. A, 2007, 111, 6285–6295 CrossRef CAS PubMed.
- K. E. Huff Hartz, E. A. Weitkamp, A. M. Sage, N. M. Donahue and A. L. Robinson, J. Geophys. Res., 2007, 112, D04204 CrossRef.
- C. Pfrang, F. Sebastiani, C. O. M. Lucas, M. D. King, I. D. Hoare, D. Chang and R. A. Campbell, Phys. Chem. Chem. Phys., 2014, 16, 13220–13228 RSC.
- M. H. P. Ambaum, Thermal Physics of the Atmosphere, Wiley-Blackwell, Reading, 2010 Search PubMed.
- G. T. Barnes, Colloids Surf., A, 1997, 126, 149–158 Search PubMed.
- G. L. Gaines, Insoluble Monolayers at the Liquid Gas Interface, Interscience Publishers, Geneva, 1966 Search PubMed.
- V. K. La Mer, Retardation of Evaporation by Monolayers: Transport Processes, Academic Press Inc., London, 1962 Search PubMed.
- V. K. La Mer, J. Colloid Sci., 1964, 19, 673–684 Search PubMed.
- I. Benjamin, Chem. Rev., 1996, 96, 1449–1476 CrossRef CAS PubMed.
- W. D. Garrett, J. Atmos. Sci., 1971, 28, 816–819 Search PubMed.
- A. K. Ray, B. Devakottai, A. Souyri and J. L. Huckaby, Langmuir, 1991, 7, 525–531 CrossRef CAS.
- E. K. Rideal, J. Phys. Chem., 1925, 29, 1585–1588 CrossRef CAS.
- B. Daumer, R. Niessner and D. Klockow, J. Aerosol Sci., 1992, 23, 315–325 CrossRef.
- J. B. Gilman, T. L. Eliason, A. Fast and V. Vaida, J. Colloid Interface Sci., 2004, 280, 234–243 CrossRef CAS PubMed.
- J.-H. A. Lo and W.-M. G. Lee, Chemosphere, 1996, 33, 1391–1408 CrossRef CAS.
- B. T. Mmereki, S. R. Chaudhuri and D. J. Donaldson, J. Phys. Chem. A, 2003, 107, 2264–2269 CrossRef CAS.
- B. T. Mmereki and D. J. Donaldson, Phys. Chem. Chem. Phys., 2002, 4, 4186–4191 RSC.
- M. Tomoaia-Cotisel and D. A. Cadenhead, Langmuir, 1991, 7, 964–974 CrossRef CAS.
- M. Folkers, Th. F. Mentel and A. Wahner, Geophys. Res. Lett., 2003, 30, 1644 CrossRef.
- J. A. Thornton and J. P. D. Abbatt, J. Phys. Chem. A, 2005, 109, 10004–10012 CrossRef CAS PubMed.
- F. Sebastiani, R. A. Campbell and C. Pfrang, Environ. Sci.: Atmos., 2022, 2, 1324–1337 CAS.
- M. D. King, S. H. Jones, C. O. M. Lucas, K. C. Thompson, A. R. Rennie, A. D. Ward, A. A. Marks, F. N. Fisher, C. Pfrang, A. V. Hughes and R. A. Campbell, Phys. Chem. Chem. Phys., 2020, 22, 28032–28044 RSC.
- B. Woden, M. W. A. Skoda, A. Milsom, C. Gubb, A. Maestro, J. Tellam and C. Pfrang, Atmos. Chem. Phys., 2021, 21, 1325–1340 CrossRef CAS.
- X. Zhang, K. M. Barraza, K. T. Upton and J. L. Beauchamp, Chem. Phys. Lett., 2017, 683, 76–82 CrossRef CAS.
- M. D. King, K. C. Thompson and A. D. Ward, J. Am. Chem. Soc., 2004, 126, 16710–16711 CrossRef CAS PubMed.
- J. Zahardis and G. A. Petrucci, Atmos. Chem. Phys., 2007, 7, 1237–1274 CrossRef CAS.
- Y. Chu, T. F. Cheng, M. Gen, C. K. Chan, A. K. Y. Lee and M. N. Chan, ACS Earth Space Chem., 2019, 3, 779–788 CrossRef CAS.
- U. Molteni, M. Simon, M. Heinritzi, C. R. Hoyle, A.-K. Bernhammer, F. Bianchi, M. Breitenlechner, S. Brilke, A. Dias, J. Duplissy, C. Frege, H. Gordon, C. Heyn, T. Jokinen, A. Kürten, K. Lehtipalo, V. Makhmutov, T. Petäjä, S. M. Pieber, A. P. Praplan, S. Schobesberger, G. Steiner, Y. Stozhkov, A. Tomé, J. Tröstl, A. C. Wagner, R. Wagner, C. Williamson, C. Yan, U. Baltensperger, J. Curtius, N. M. Donahue, A. Hansel, J. Kirkby, M. Kulmala, D. R. Worsnop and J. Dommen, ACS Earth Space Chem., 2019, 3, 873–883 CrossRef CAS.
- X. He, C. Leng, S. Pang and Y. Zhang, RSC Adv., 2017, 7, 3204–3213 RSC.
- M. W. A. Skoda, B. Thomas, M. Hagreen, F. Sebastiani and C. Pfrang, RSC Adv., 2017, 7, 34208–34214 RSC.
- B. Woden, M. W. A. Skoda, M. Hagreen and C. Pfrang, Atmosphere, 2018, 9, 471 CrossRef CAS.
- D. Daumont, J. Brion, J. Charbonnier and J. Malicet, J. Atmos. Chem., 1992, 15, 145–155 CrossRef CAS.
- J. R. Lu, R. K. Thomas and J. Penfold, Adv. Colloid Interface Sci., 2000, 84, 143–304 CrossRef CAS PubMed.
- A. Nelson, J. Appl. Crystallogr., 2006, 39, 273–276 CrossRef CAS.
- A. Milsom, A. Lees, A. M. Squires and C. Pfrang, Geosci. Model Dev., 2022, 15, 7139–7151 CrossRef CAS.
- J. M. Perkel, Nature, 2018, 563, 145–146 CrossRef CAS PubMed.
- M. Shiraiwa, C. Pfrang and U. Pöschl, Atmos. Chem. Phys., 2010, 10, 3673–3691 CrossRef CAS.
- M. Shiraiwa, C. Pfrang, T. Koop and U. Pöschl, Atmos. Chem. Phys., 2012, 12, 2777–2794 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4fd00167b |
| This journal is © The Royal Society of Chemistry 2025 |