 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransport of continental particulate over the Labrador Sea and entrainment are important pathways for glaciation of remote marine clouds†
Hugh Coe *ab,
Huihui Wuac,
Nicholas Marsdenab,
Michael Biggarta,
Keith N. Bowera,
Tom Choulartona,
Michael Flynna,
Martin W. Gallaghera,
Kezhen Hua,
Gary Lloydab,
Graeme J. Nottd,
Paul F. Fieldef and
Benjamin J. Murrayf
*ab,
Huihui Wuac,
Nicholas Marsdenab,
Michael Biggarta,
Keith N. Bowera,
Tom Choulartona,
Michael Flynna,
Martin W. Gallaghera,
Kezhen Hua,
Gary Lloydab,
Graeme J. Nottd,
Paul F. Fieldef and
Benjamin J. Murrayf
aDepartment of Earth and Environmental Sciences, University of Manchester, Manchester, M13 9PL, UK. E-mail: hugh.coe@manchester.ac.uk
bNational Centre for Atmospheric Sciences, University of Manchester, Manchester, M13 9PL, UK
cUniversité Paris Cité and Univ Paris Est Créteil, CNRS, LISA, 75013 Paris, France
dFAAM Airborne Laboratory, Cranfield, UK
eMet Office, FitzRoy Road, Exeter, Devon EX1 3PB, UK
fSchool of Earth and Environment, University of Leeds, Woodhouse Lane, Leeds, LS2 9JT, UK
First published on 17th January 2025
Abstract
Marine Arctic clouds greatly influence the radiative balance across the Arctic region and their effectiveness at scattering radiation changes considerably depending on cloud phase. Glaciation of these clouds relies on the presence of ice nucleating particles, which are often limited in number, so often clouds may be liquid even at temperatures well below 0 °C. As the Arctic region warms, cloud feedbacks may accelerate change or lessen absorbed solar radiation. Understanding aerosol–cloud interactions and the sources and pathways of aerosol particles across the Arctic region is central to improving our knowledge of these poorly understood processes. In this paper, aircraft observations of single particle chemical and physical properties are presented and the composition of cloud residuals in both warm and glaciated clouds are examined using a single-particle laser ablation aerosol particle mass spectrometer (LAAPToF). In cloud, the LAAPToF sampled behind a Counterflow Virtual Impactor (CVI) to detect cloud particle residuals, separated into liquid, mixed phase and ice clouds using in situ observations of the fractional ice water content. Three different air mass regimes were sampled: northerly winds in both the marine boundary layer and the lower free troposphere; westerly winds from Canada in both the marine boundary layer and the free troposphere; and periods when the boundary layer winds were northerly but the air immediately above the boundary layer was from continental Canada. When the air in the boundary layer and free troposphere was from the north, most clouds were in the liquid phase, however, considerably more glaciation was observed when the air immediately above the boundary layer clouds was from Canada regardless of the flow direction in the boundary layer. Sea salt particles dominate the observed out of cloud aerosol particle population and liquid cloud particle residuals. However, in the detected mixed phase and ice cloud particle residuals dust and bioaerosol particles were substantial in number. Since these are known to be effective ice-nucleating particles, the observations suggest that long range transport of continental air and entrainment is an important pathway for the supply of aerosol to the remote Arctic boundary layer.
Introduction
Aerosol particles play an important role in the climate system in Arctic regions since they can interact with radiation through both scattering and absorption,1 and via deposition to ice and snow surfaces which may reduce the surface albedo and enhance melting.2 Mixed phase clouds are prevalent across high latitude regions. Hygroscopic aerosols act as efficient cloud condensation nuclei and so influence cloud droplet numbers in liquid clouds. For a cloud to glaciate, ice-nucleating particles (INP) need to be present to initiate ice formation at temperatures typically seen in marine boundary layer clouds at high latitudes. Ice multiplication processes are often important in the glaciation of these clouds. The Arctic region is warming rapidly and cloud responses to these changes are a major component of the overall cloud feedback but remain very uncertain.3 Warming drives an increase in the amount of supercooled water in clouds since both the absolute humidity available for condensation and the ice nucleating (IN) efficiency are both strongly temperature dependent. However, the size of the response is also dependent on the extent of glaciation of the clouds, and hence secondary ice production, in the present day system. All of these processes are rather poorly understood and all depend on a knowledge of the aerosol particle physical and chemical properties and mixing state, which are themselves not well known.4Early characterisation of aerosols in the Arctic region focused on long range transport of pollution to the Arctic during late winter and early springtime, the so-called Arctic Haze5 when pollution from northern mid-latitudes emitted north of the polar front can be transported to the Arctic. During the summer and autumn, such pathways are very limited and in situ sources of particulate become important.6 There is therefore a strong seasonal cycle in Arctic aerosol loading.7,8 More recently, long term annual studies of aerosol composition have been carried out at a number of ground based stations across the Arctic region. An examination of long term trends and seasonal cycles in aerosol composition using samples from multiple ground based observation sites showed that transport to the Arctic during late winter and early spring has reduced but remains a strong source of aerosols in the Arctic at this time of year.9 In summertime, natural aerosols including sea salt, non-sea salt (nss) marine sulphur and dust all contribute though no long term trends were apparent. Outside the late winter-early spring period, northern Greenland and the European Arctic boundary layer aerosol is influenced heavily by marine sources, notably nss-sulphate and sea salt.10 Multi-site observations have shown that in addition to sea salt and nss-sulphate, organic aerosol is a major contributor to particulate mass.11 The work used air mass analysis to show that these components arise predominately from inner Arctic source regions. Nevertheless, understanding aerosol composition across the Arctic has to a large extent focused on the contribution to particulate mass (PM), not least since experimental logistics in the region are extremely challenging, However, since cloud processes are driven by single particle composition, and in the case of ice nucleation, often result from minor components, bulk mass analysis may not reveal the most important contributions to cloud processes.
Single particle observations of aerosol composition in the Arctic are far less common. Observations at an Alaskan coastal station showed that primary marine aerosol was commonly internally mixed with secondary sulphate and depleted in chloride during wintertime.12 Long term observations of aerosol and cloud particle residuals have been made at the Zeppelin Observatory, Svalbard over 4 years using a multi-inlet system consisting of an aerosol inlet and a counterflow virtual impactor (CVI) respectively.13 Samples were analysed for single particle composition using scanning transmission electron microscopy-energy dispersive X-ray spectroscopy (STEM-EDS). During periods of sampling warm, liquid clouds (T > 0 °C), the aerosol and cloud residual data were similar with main components identified as mineral dust, sea salt, K-bearing, sulphate, and carbonaceous. However, at temperatures below 0 °C, sea spray and mineral dust particles were dominant in the cloud residual samples and a far greater proportion of these particle types were internally mixed compared to the aerosol samples, indicating the importance of sea spray and mineral dust in mixed phase cloud processes.
Mineral dust and biological or biogenic particles have long been known to be effective INP14 even at warm temperatures (>−10 °C) as often experienced in Arctic boundary layer clouds. IN active biological particles may be emitted from the open ocean15 and so may be mixed with sea salt. They may also be transported from land based sources such as boreal forests.16 Mineral dust and biological particles have also been observed to be internally mixed17 and have been demonstrated to be effective INP18,19 at relatively high temperatures. The effectiveness of mineral dust emanating from dry, low latitude regions has been extensively studied but more recently high latitude dust sources have been identified as important in the Arctic.20 Such high latitude sources have been shown to be a potentially important type of ice nucleating particle21–24 though long range transport has also been shown to be important.25,26 Most recently, biological particles have been shown to be present in cloud residuals at concentrations consistent with high temperature ice nuclei concentrations with a seasonal summer maximum.27
Much of what is known of aerosol composition in the Arctic region has emerged from surface based studies. While such long term observations are extremely valuable, they cannot examine vertical variation in aerosol composition or be used to readily identify transport pathways of aerosol into cloudy regions via the free troposphere. Airborne studies have previously been carried out to address some of these challenges. The vertical structure in submicron aerosol composition above the polar dome during high Arctic springtime (close to 80°N) in northern Canada showed that composition varied with altitude, with submicron aerosol composition dominated by organic matter, whereas closer to the surface sulphate was the dominant species.28 The presence of submicron sea salt was also significantly enhanced at the surface. In the same study (NETCARE), transport processes affecting aerosol within the high Arctic during spring and summer were studied.29 These transport features drove dominance of pollution aerosol in later winter and spring and local sources were more important in summertime.
Single particle laser ablation mass spectrometry combined with high resolution aerosol mass spectrometry has been used to make airborne observations of aerosol composition in the Arctic region during summertime.30 Aerosol particles above the boundary layer were shown to be dominated by chemically aged particles rich in organic matter and sulphate that had been transported from lower latitudes, elevated nitrate and ammonium being associated with wildfires in northern Canada. Aerosol composition within the boundary layer was dominated by sources within the Arctic region and included sea spray and marine biogenic sources. To date there have been relatively few airborne observations of aerosol composition of cloud residual particles that may shed light on the important aerosols involved in aerosol–cloud interactions. INP concentrations have been made from aircraft in the Arctic region combined with offline filter sampling of aerosol composition. In the Alaskan Arctic spring time, INP and aerosol concentrations have been shown to be low and were dominated by sea spray and mineral dust.31 The mineral dust particles dominated the ice nucleating activity in the samples analysed. It was also shown that the INP concentrations were more consistent with soil dust that had an active biogenic component rather than it being the feldspar minerals that determined INP effectiveness. The results were consistent with long range transport rather than local production. Similarly, INP spectra have been reported in the spring-time European Arctic,26 but in this study much higher INP concentrations were found compared to the Alaskan Arctic. They attributed those INPs to long-range transported mineral dust–biogenic mixtures.
In this work, conducted as part of the UK Natural Environment Research Council (NERC) M-Phase project within the CloudSense programme, airborne observations of aerosol composition and mixing state made using a single particle mass spectrometer sampling aerosol particles when out of cloud and cloud residuals in cloud are used to examine the likely sources of aerosols above the Labrador Sea during autumn 2022. Transport pathways through the lower free troposphere and marine boundary layer are investigated and the prevalence of different aerosol components in the residuals of both warm (liquid) and glaciated marine boundary layer cloud particles are presented. The paper presents a description of the experiment and our methodology, discusses the characterisation of air masses, presents the vertical structure of aerosol through the lower atmosphere and their physical and chemical properties. Case studies of sampled cloud systems are presented and the chemical composition of residuals is detailed followed by a discussion and conclusions.
Methods
The M-Phase flight campaign was conducted over the Labrador Sea between 55°N and 60°N utilizing the UK’s BAe-146-301 Atmospheric Research Aircraft. A total of 16 scientific flights (labelled from C318 to C333) were carried out between September 27 and November 3, 2022.Each flight took off from Goose Bay, Canada (53.34°N, 60.39°W), beginning with an ascending transit profile toward the Labrador Sea. The following missions consisted of a combination of aerosol runs and/or cloud passes, depending on atmospheric conditions. Aerosol runs were performed at low altitudes and above cloud layers to characterize aerosol loadings, size distributions, and chemical composition. Cloud passes followed a saw-tooth flight pattern to characterize cloud microphysics through the depth of the boundary layer clouds. During cloud passes, a Counterflow Virtual Impactor (CVI) inlet was also employed to sample residue particles from cloud particles. The FAAM aircraft was equipped with a suite of core instruments to measure atmospheric dynamics and thermodynamic variables, including wind speed and direction, temperature (T) and humidity. This study focuses on aerosol and cloud residue measurements, related instruments are briefly described below. Fig. 1 shows the tracks of flights (C321 to C332) used in this study, where aerosol and cloud residue measurements were available. The flight dates are shown in Table 1.
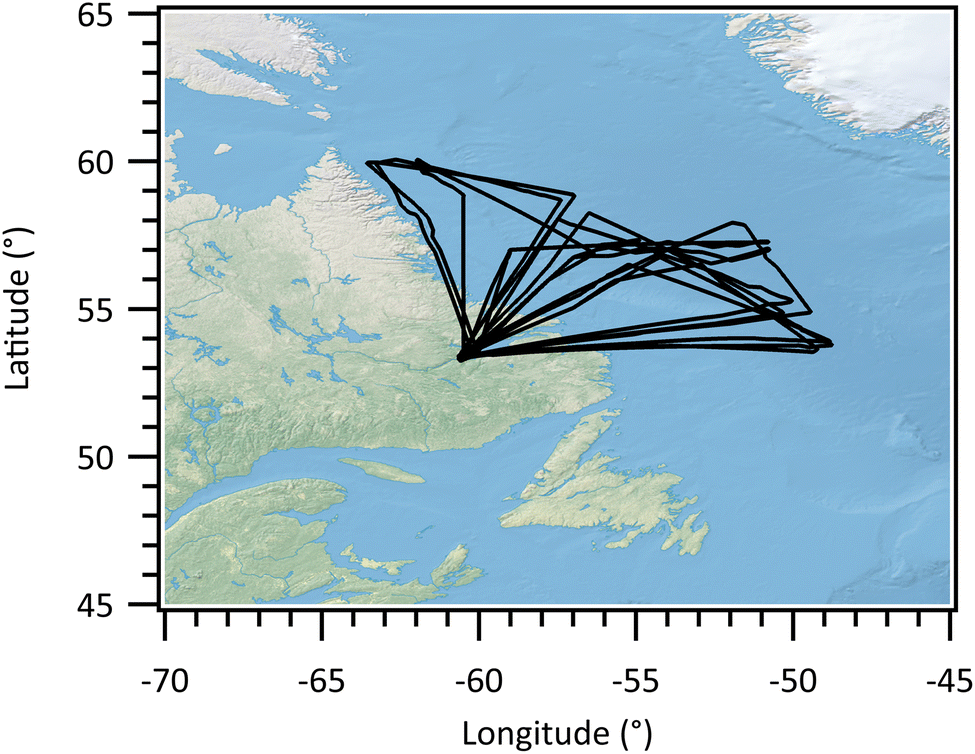 | ||
| Fig. 1 Tracks of flights (C321 to C332) used in this study. | ||
| Classification by air mass source | Flight (date) | Cloud passes T range (K) | Cloud passes LAT range (°) | Total cloud sample time (s) | Mixed-phase/ice cloud fraction |
|---|---|---|---|---|---|
| a (1) Flight C325 focused on aerosol samples, insufficient cloud data can be provided. (2) The mixed-phase/ice cloud fraction was estimated using the 1 Hz measurements of TWC, LWC and IWC from Nevzorov. If the IWC/TWC was less than 0.1 the cloud was characterised as liquid, if the IWC/TWC was greater than 0.9 then the cloud was characterised as being ice phase and at intermediate values of IWC/TWC the cloud was identified as being mixed phase. The mixed-phase/ice cloud fraction was defined as the ratio of the sample time of mixed-phase/ice cloud to the total cloud sample time. | |||||
| Canadian: long range transport was mostly from W (for both high and low levels) | C321 (23/10/2022) | 261.4–268.7 | 57.4–59.6 | 707 | 0.89 |
| C325 (28/10/2022) | n/a | n/a | n/a | n/a | |
| C326 (28/10/2022) | 254.4–266.0 | 57.0–57.3 | 2671 | 0.74 | |
| C327 (29/10/2022) | 266.1–275.1 | 56.3–57.1 | 1292 | 0.63 | |
| Northerly: long range transport was mostly from NW/N (for both high and low levels) | C322 (24/10/2022) | 262.3–269.6 | 58.8–60.0 | 1069 | 0.08 |
| C323 (24/10/2022) | 260.9–273.1 | 53.5–55.6 | 867 | 0.35 | |
| C330 (03/11/2022) | 254.8–267.9 | 56.3–60.0 | 2295 | 0.22 | |
| C331 (03/11/2022) | 256.5–271.2 | 53.9–56.4 | 2579 | 0.35 | |
| BLNFTC (mixed sources): low levels: mostly from N/NW. Higher levels: mostly from SW/W | C324 (25/10/2022) | 262.1–270.2 | 54.9–57.9 | 1254 | 0.23 |
| C328 (31/10/2022) | 257.6–272.0 | 55.2–57.7 | 1308 | 0.44 | |
| C329 (01/11/2022) | 262.5–271.6 | 53.8–56.7 | 1426 | 0.56 | |
| C332 (04/11/2022) | 260.1–267.2 | 54.4–58.1 | 2021 | 0.06 | |
Instrumentation
The aerosol size distributions were determined using a wing-mounted passive cavity aerosol spectrometer probe (PCASP), which measured aerosol number concentrations at 5 Hz resolution across 30 size bins spanning nominal diameters (Dp) from 0.1 to 3 μm. Calibration of the PCASP was performed using di-ethyl-hexylsebacate (DEHS) and polystyrene latex (PSL) spheres with known size and refractive index.32 Bin sizes were calculated based on Mie scattering theory, under the assumption that particles were spherical with a refractive index of 1.396 + 0.002i. Integration of the PCASP size distributions yielded aerosol number concentrations in the accumulation mode (Na, 0.1 < Dp < 3 μm).A Single Particle Soot Photometer (SP2) (Droplet Measurement Technologies, Boulder, CO, USA) was employed to measure the refractory black carbon (BC). It can detect BC-containing particles, with an equivalent spherical diameter in the range of 70–850 nm. As these particles pass through the laser beam, they absorb the laser and heat up. Upon reaching their boiling point, these particles incandesce and emit visible light, which is captured by two detectors. The SP2 incandescence signal, which is proportional to the mass of BC present in the particle, was calibrated using Aquadag BC standards (Aqueous Deflocculated Acheson Graphite, manufactured by Acheson Inc., USA). Details regarding the SP2, including its principles, operation, calibration and data interpretation for aircraft deployment have been previously documented.33 PCASP and SP2 measurements were corrected to standard temperature and pressure (STP) (273.15 K and 1013.25 hPa).
A Laser Ablation Aerosol Particle Time-of-Flight mass spectrometer (LAAPToF; AeroMegt GmbH) was employed to characterize single-particle chemical composition and mixing state.34,35 Briefly, aerosols enter the instrument via an aerodynamic lens inlet, which focuses the incoming particles into a narrow but divergent beam. The incoming particles then pass through an optical detection chamber, which is equipped with a custom-built fibre-coupled system that integrates a Nd:YAG diode-pumped solid-state laser with collimated laser beams at multiple wavelengths. Once a single particle is detected, an excimer laser pulse (λ = 193 nm, 8 ns pulse) is triggered for a one-step desorption and ionization of the particle. The resulting cations and anions are subsequently analyzed by a bipolar time-of-flight mass spectrometer (BTOF-MS, Tofwerks AG). In this study, the LAAPToF efficiently detected particles with aerodynamic diameters (Daero) ranging from 0.5 to 2.5 μm. The LAAPToF data were processed using Igor software (Version 1.0.0, AeroMegt GmbH) for the analysis of single-particle spectral information.
A CVI inlet36 was employed to sample the residual particles of cloud droplets during cloud passes. The CVI inlet with counterflow on allows only cloud droplets larger than a controllable minimum size to enter the inlet, subsequently collecting the cloud residues by evaporating the cloud water using dry, particle-free carrier air. During the campaign, the droplet cut-off size was approximately Daero ∼8.5 μm but was calculated as a function of time. The residual particles were then characterized using various online aerosol instruments downstream of the CVI inlet. Here, we focus on the chemical composition and mixing state of cloud residues, which were analyzed using the LAAPToF.
Cloud periods were identified using measurements from a Cloud Droplet Probe (CDP). The CDP provided measurements of cloud droplet number concentrations at a 25 Hz resolution across 30 size bins within a nominal diameter range of 2–50 μm. The operation and calibration procedures of the CDP are detailed elsewhere.37 Size calibrations were conducted pre-flight using 10 different-size glass beads with known diameters and refractive indices.32 The scattering cross-section was converted to droplet size based on the refractive index of water, 1.33 + 0i. Cloud droplet number concentration (Nd) and liquid water content (LWCCDP) were derived from the CDP’s cloud droplet spectrum using the following equations:
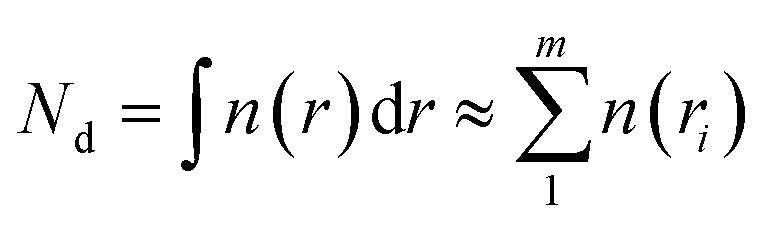
where n(ri) represents the number of cloud droplets within a specific size bin, ri represents the middle radius value for each size bin, and ρwater represents the density of liquid water. A LWC threshold of 0.01 g m−3 was used to define the presence of clouds. The CDP measurements were also corrected to STP.
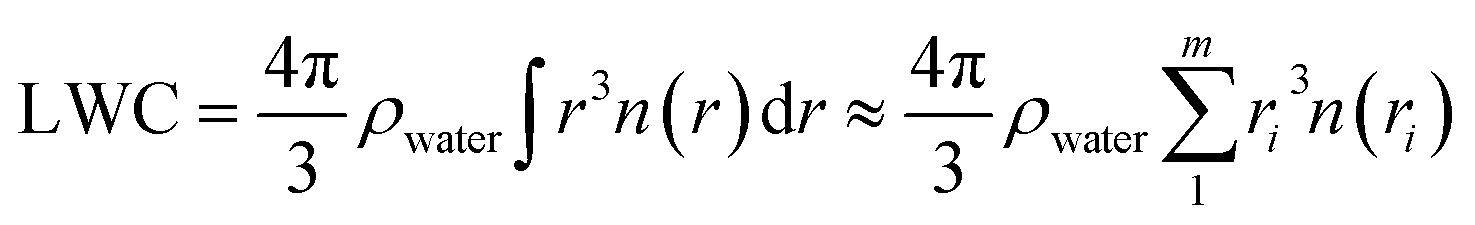
A Nevzorov probe,38 which consists of two separate sensors, was used to measure the bulk total condensed-water content (TWC) and the liquid water content (LWCNev). Nevzorov data were corrected for the baseline drift of the measured bulk water contents.39 LWCNev and ice water contents (IWC) were then derived from measurements based on the following assumptions: (1) a value of 0.11 is assumed to estimate a slight reaction of the liquid water sensor with respect to impacting ice crystals; (2) the collection efficiencies of hydrometeors are assumed to be 1; (3) the difference between the TWC and LWCNev measurement is due to ice particles. The ratio of IWC to TWC (IWC/TWCNev) was calculated to identify cloud phase state.
Back trajectory analysis
To examine the sources and transport pathways of the sampled air masses during the campaign, a back-trajectory analysis was conducted utilizing the Numerical Atmospheric Modelling Environment (NAME) developed by the UK Met Office.40 For each flight, tracer particles were released at two-minute intervals along the flight track over the Labrador Sea. The trajectories of released tracer particles were tracked backward over a five-day period, based on three-dimensional gridded (3D) meteorological fields from the UK Met Office’s global Numerical Weather Prediction (NWP) model, the Unified Model (MetUM).41 These meteorological fields were updated every three hours and featured a high horizontal resolution of 0.11° longitude by 0.1° latitude and 59 vertical levels up to approximately 29 km altitude. The longitude, latitude, and altitude of the released tracer particles were recorded along their backward trajectories.Results
Air mass characterization
During the M-Phase flight campaign, the FAAM aircraft sampled a range of different air masses. The back trajectories for each flight track are shown in Fig. 2, the colour of the trajectories shows the altitude of the air parcel at that point in the trajectory. Fig. S1† shows the same trajectories but coloured by the arrival height of the trajectory along the flight track of the aircraft to demonstrate the differing transport pathways of sampled boundary layer or free tropospheric air. Though there is some flight to flight variability in the air mass histories there are also some clear patterns that are summarised in Table 1. Flights C321, C325, C326 and C327 (Canadian) show the air to have advected from the west across continental Canada and show similar transport pathways for all aircraft locations and altitudes throughout each of the flights. These contrast with flights C322, C323, C330 and C331 (northerly). The trajectories associated with these flights are predominately more northerly for all launch points along the flight track, both within the boundary layer and the lower free troposphere. The air masses are oceanic in nature with no significant passage over land mass and are characteristic of cold air outbreaks when cold Arctic Ocean air advects southward. Low level stratiform cloud is commonly present in these conditions and transitions to cumulus cloud as the air moves to the south. The third group of trajectories includes flights C324, C328, C329, and C332. The air masses during these flights are characterised by northerly to north-westerly air arriving at the aircraft sample location when the aircraft was sampling in the boundary layer at low levels, and more westerly air masses when the aircraft was sampling in the free troposphere. This flow regime is termed Boundary Layer Northerly, Free Troposphere Canadian (BLNFTC). The aerosol properties and cloud residuals are examined based on these air mass classifications in subsequent sections.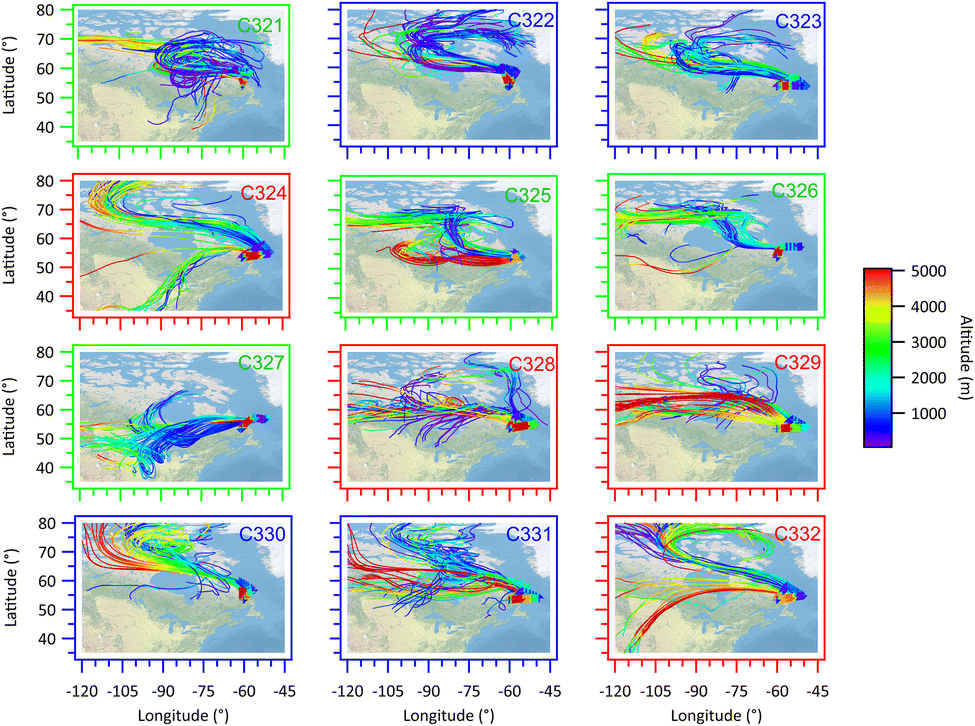 | ||
| Fig. 2 5 Day back trajectories along the flight track over the Labrador Sea for each flight. The trajectories were calculated from the aircraft position and time every 2 minutes along the flight track for each of the flights labelled in the top right hand corner of each panel. The heights of the back trajectories in metres are shown as colours given by the scale bar. The flight numbers and plot borders are coloured to identify the flow regime sampled during the flight: green – Canadian; blue – northerly; red – BLNFTC. | ||
Aerosol vertical structure
Average vertical profiles of accumulation mode aerosol number (Na), black carbon (BC) and cloud liquid water content are shown for each of the flights in Fig. 3. There is considerable variability from flight to flight. In general, the aerosol accumulation mode number concentrations are low during Canadian airmasses, typically <50 particles cm−3 in the boundary layer, though during C326 and C327, average boundary layer particles numbers were between 50 to 100 cm−3. Concentrations were between 50 and 100 cm−3 in northerly marine surface air during all flights (northerly and BLNFTC airmasses). There was little black carbon in the boundary layer and so no evidence of aged pollution or biomass burning in boundary layer air during any of the flights. Typically, the cloud top was close to 2 km for many of the flights but was slightly lower (1.5 km) on flights C323, C327 and C333 and around 1 km on C322. Liquid water contents ranged from 0.4 to 0.8 g cm−3 at cloud top. Table 1 shows that there is some variability in cloud depth, as given by cloud base and cloud top temperatures, between the different flights but no systematic difference was noted between clouds in each of the flow regimes. In the northerly regime cloud top temperatures varied between 255–262 K across the 4 flights. Cloud bases varied between 268 and 273 K. In the Canadian flow regime cloud top temperatures were between 255 and 266 K and cloud bases were 268–275 K. In BLNFTC conditions cloud tops were 257–262 K and cloud bases 267–272 K.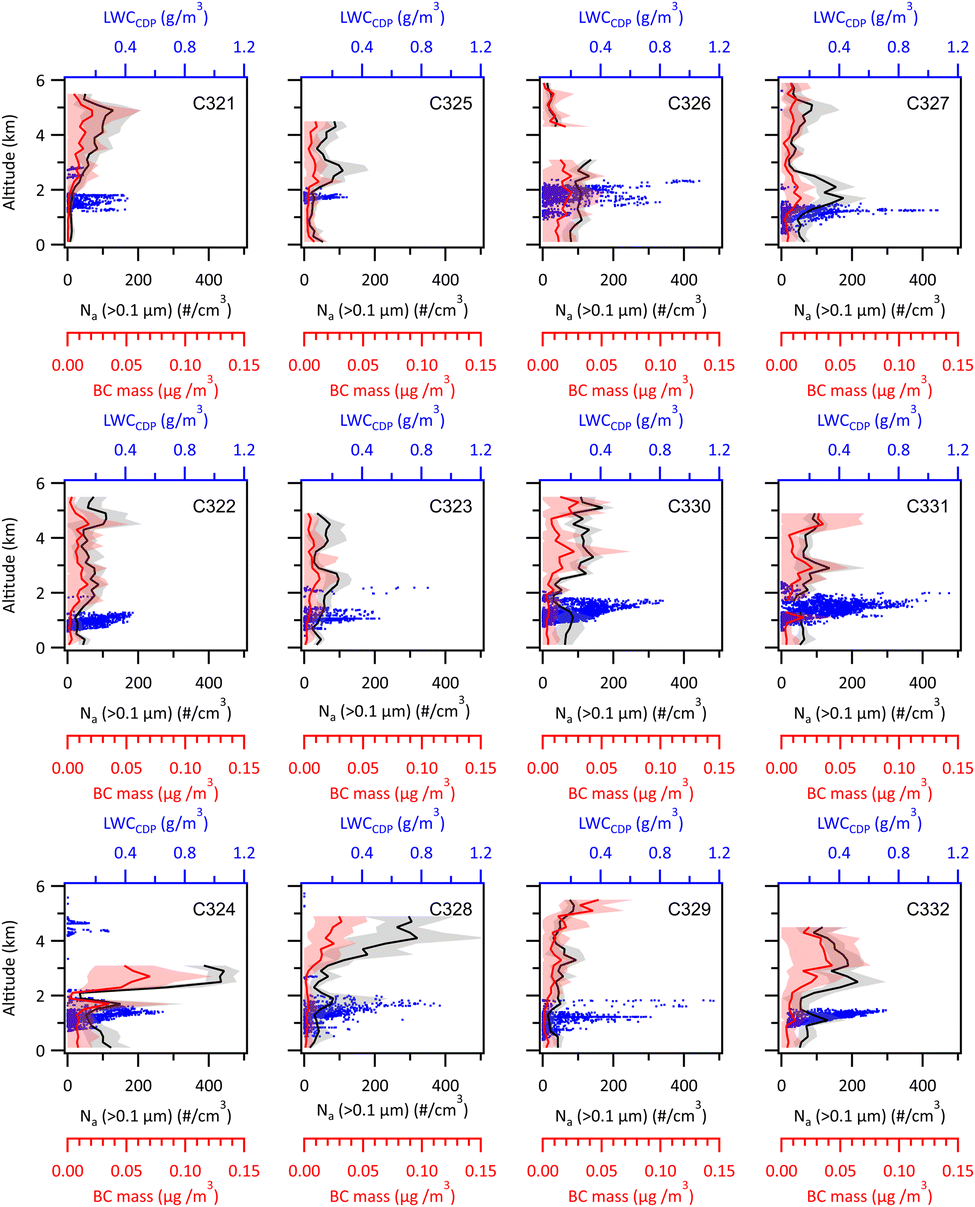 | ||
| Fig. 3 Average vertical distributions of aerosol number concentrations (Na, > 0.1 μm cm−3) from PCASP measurements and BC mass concentrations (μg m−3) from SP2 measurements in each flight. Black (PCASP) and red (BC) lines and shades represent average values and one standard deviation in every 200 m bin. The corresponding 1 Hz LWCcdp (g m−3) values are shown as blue dots. Top row includes flights in the Canadian flow regime; middle row includes flights in the northerly flow regime; bottom row includes flights in the BLNFTC regime. | ||
Aerosol above the boundary layer was variable with altitude and also from flight to flight. High altitude layers were sampled on some flights containing significant amounts of black carbon, for example aerosol layers were observed on flights C331, C332 in Canadian airflows and in most of the BLNFTC regime flights. However, rarely was there substantial evidence of elevated layers penetrating the boundary layer, the exceptions being on flights C327 and C331.
Aerosol physical and chemical properties
Average aerosol size distributions are shown as a function of altitude for each flight in Fig. 4. Since cloud was typically present between 1 and 2 km no size distributions were obtained for these altitudes. In the sub-cloud surface layer, accumulation mode aerosol number concentrations are typically lower than in the free troposphere, largely since low level long-range transport particles have been depleted through both dry and wet removal. Coarse mode number concentrations typically peak close to Dp ∼ 1 μm and are enhanced in the marine air masses from the north/north-west (northerly and BLNFTC), suggesting significant marine sea salt inputs compared to the air masses advected from the Canadian continent (Canadian). Small, accumulation mode particles (Dp < 500 nm) are enhanced by between 2 and 5 times in the free troposphere compared to the sub-cloud layer but coarse mode particles are reduced by one order of magnitude or more, demonstrating that marine particles dominate the coarse mode in the boundary layer during all flights.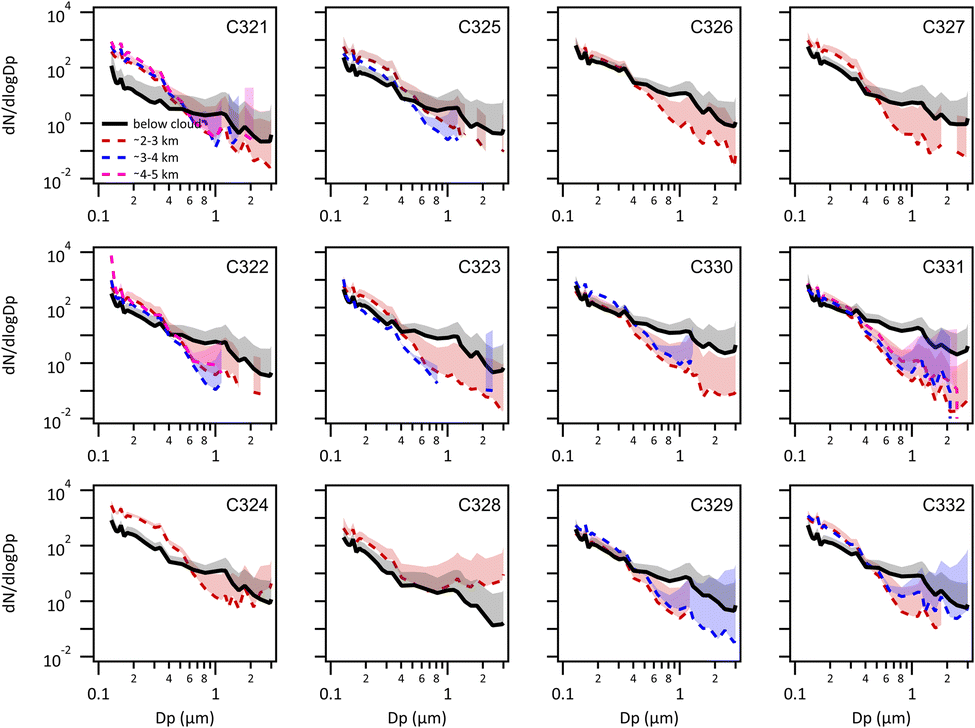 | ||
| Fig. 4 Average aerosol number size distributions (PCASP) at different sample heights in each flight. Top row includes flights in the Canadian flow regime; middle row includes flights in the northerly flow regime; bottom row includes flights in the BLNFTC regime. | ||
The chemical composition and mixing state was analysed by the LAAPToF. Both positive and negative mass spectra for each particle were categorised using a k-means clustering algorithm.17 The chemical composition and mixing state was analysed by the LAAPToF. The mass spectra for each particle were categorised using the fuzzy c-means clustering within the software LaapTof Data Analysis V1.02, AeroMegt GmbH.42 A fuzzifier of 1.2 was used. Six classes were chosen to produce particle composition classes for the aerosol data. Two similar types of fresh sea salt were merged, and two similar types of aged sea salt were merged. Nine classes were selected within the cluster analysis package when clustering the cloud residue spectra sampled through the CVI. The same two similar types of fresh sea salt were merged, and two similar types of aged sea salt were merged. Once the cluster analysis had been carried out, the results were checked manually. Since only one spectrum of bioaerosol was detected when sampling from the aerosol inlet, it was not classified by the toolkit and was identified during the post-clustering manual checking procedure.
The mean mass spectra for each cluster when sampling aerosol through the aerosol inlet are shown in Fig. 5. The numbers of particles of each class detected by the LAAPToF when sampling out of cloud aerosol particles as a function of air mass class are summarised in Table S1.† The data have been normalised by the detection efficiency of each particle type by the LAAPToF. Sea salt was the most prevalent particle type and consists only of Na+, Cl−, and associated cluster ions in both the positive and negative ion modes. The next most prevalent particle cluster was aged sea salt, which has similar Na and Cl ion combinations but in addition NO2− and NO3− peaks are present, evidence that nitric acid uptake has displaced some of the chloride in the sea salt during atmospheric transport. Some pollution particles were observed, identified by the sulphate/organic cluster; HSO4− is the dominant ion with m/z 43 providing evidence of organic material and a small amount of NO+ from nitrate. The dust class is commonly identified due to the presence of Al+, SiO3−, Ca+, CaO+ and OH− ions resulting from aluminosilicate mineralogies with nitrate, evidence of heterogeneous reactions typical of atmospheric transport. Only a single bioaerosol particle was identified when the LAAPToF was sampling from the aerosol inlet. This spectrum could be identified through the PO2− and PO3− ions and CN− and CNO− as has previously been reported.43 The lack of this particle type arises from the low detection efficiency of the LAAPToF for the larger particles typical of bioaerosol, the low sample volume and the very low coarse mode aerosol concentrations observed in Arctic marine air.
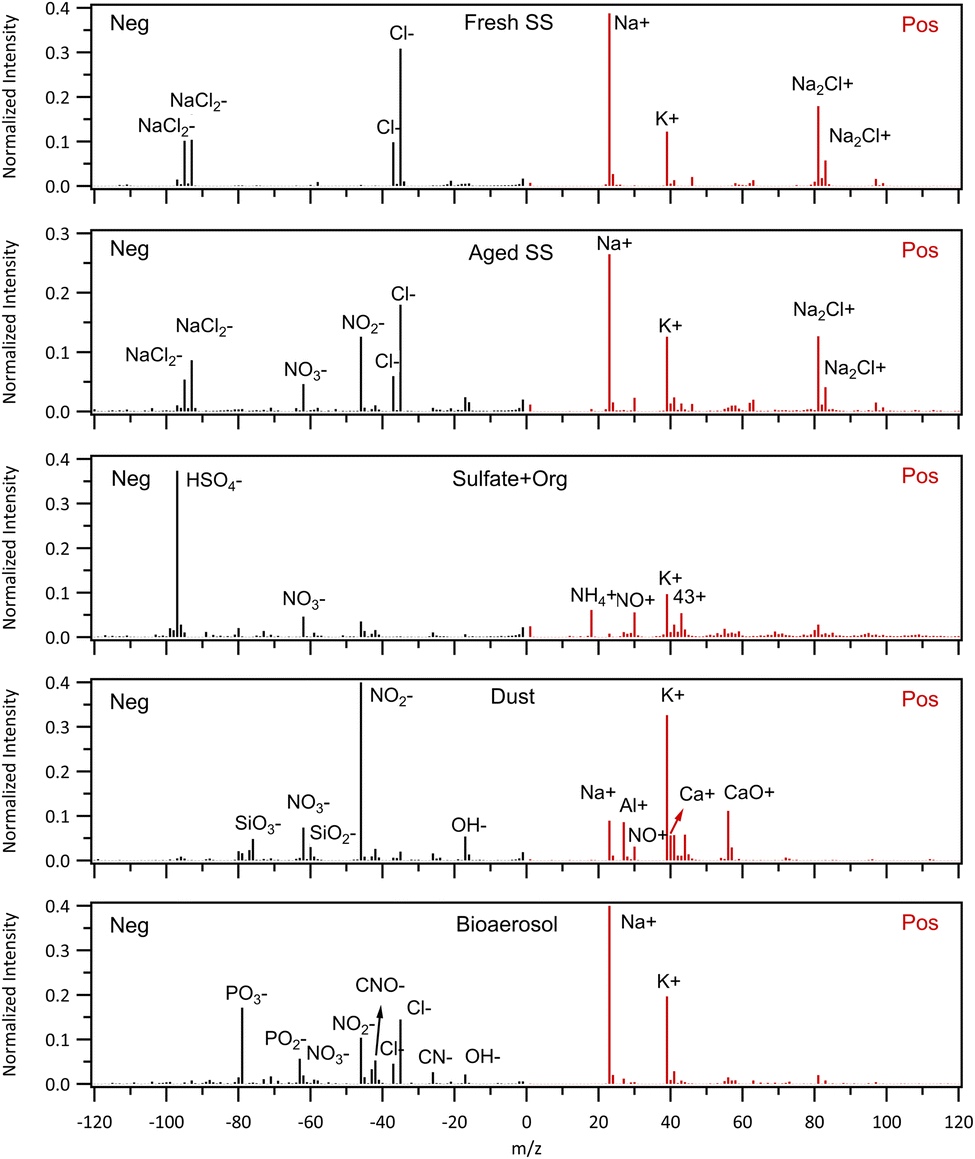 | ||
| Fig. 5 Average spectra of (from the top to bottom) fresh sea salt, aged sea salt, sulfates, dust, and bioaerosol, from aerosol sampling. It is noted that only one spectrum of bioaerosol in aerosol mode was detected. | ||
The average below cloud aerosol number (Dp between 0.5–3 μm as sampled by the PCASP) over each of the flights together with the total number of LAAPToF spectra are shown in Fig. 6a and the corresponding statistics for the above cloud layer are shown in Fig. 6b. There are clearly sufficient numbers of aerosol measured by the LAAPToF below cloud to be statistically significant and the fraction of the k-means clusters in the sub-cloud layer sampled during each flight are shown in Fig. 6a. However, there are very few particles sampled by the LAAPToF in the above cloud air and so further statistics on particle type are not possible. The sub-cloud aerosol is dominated by fresh sea salt particles with only a minor component of aged sea salt present (<10%) on only a few flights. The remaining clusters represent a very small fraction of the total particles sampled.
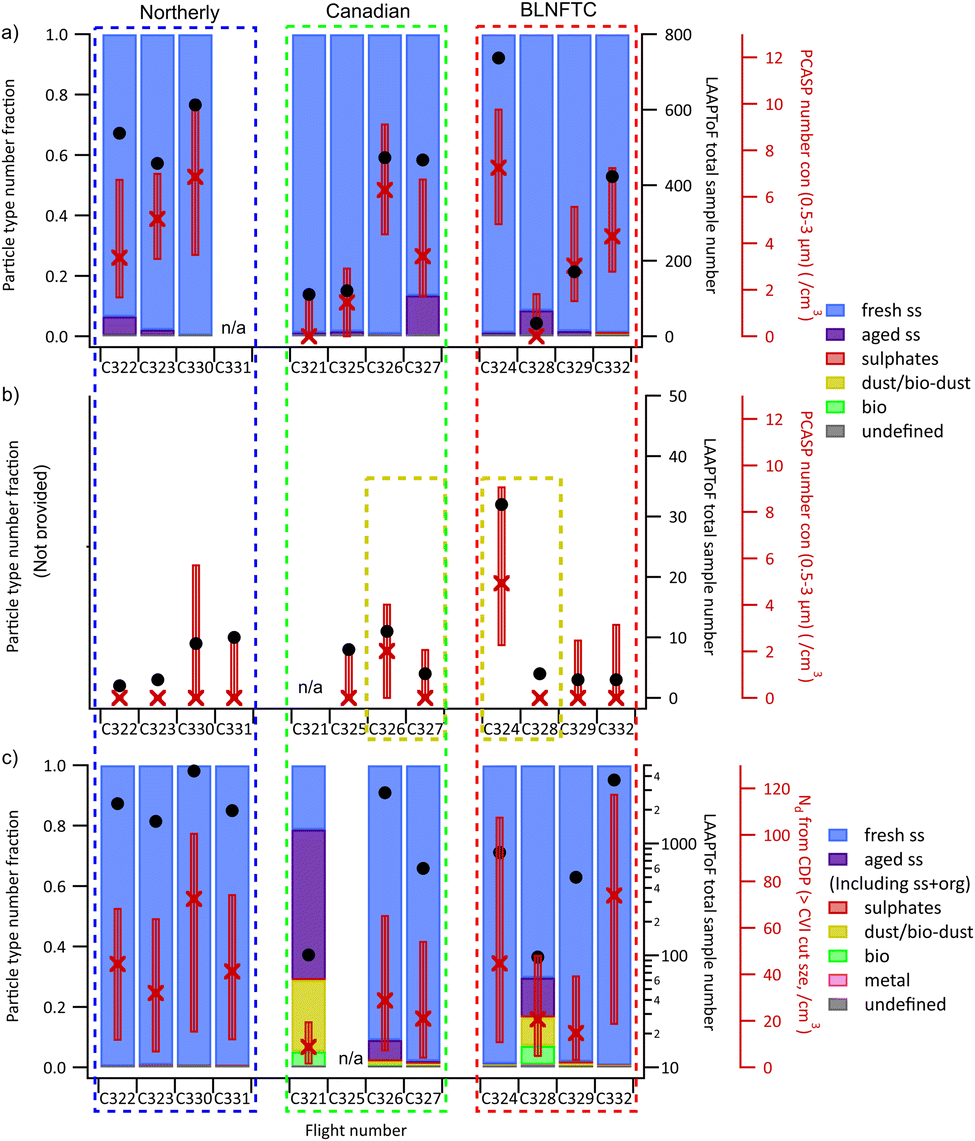 | ||
| Fig. 6 The number fractions of each detected particle type from (a) below-cloud aerosol sampling and (c) cloud residue sampling, in each flight. Flights are grouped by airmass, from left to right Canadian; northerly; BLNFTC. The number fractions of each detected particle type are not provided for (b) above-cloud sampling, due to limited sample amounts. The dashed yellow boxes indicate the flights with dust or/and bioaerosols detected during the above-cloud sampling. The corresponding total sampled particle amounts from LAAPToF are shown as black circles. The aerosol (0.5–3 μm) number concentrations from PCASP measurements are shown in (a) and (b). The cloud droplet number concentrations from CDP measurements are shown in (c). The red X markers and boxes represent median, 10% and 90% values. | ||
Cloud sampling case studies
Fig. 7 and 8 show examples of cloud sampling segments of two different flights, C321 (Canadian) and C322 (northerly) respectively. In both these flight segments the aircraft was carrying out profile climbs and descents through the cloud layer from below cloud base to above cloud top (Fig. 7a and 8a). During these types of sampling periods the LAAPToF sampled from the CVI inlet and therefore measured cloud residues. The CVI enhances particle counting statistics since the inlet is sub-isokinetic, leading to increasing enhancement factors with original particle size and this has been corrected for in the data presented here. The lower size cut of the CVI was 9 ± 1 μm during all flights. During these flights cloudbase temperatures were −2 °C to −4 °C and cloud top temperatures were close to −10 °C on most flights but on some flights colder as the clouds were deeper, notably C326 (Canadian), C330 (northerly) and C328 (BLNFTC), with cloud top temperatures of 254 K, 255 K and 258 K respectively (Table 1).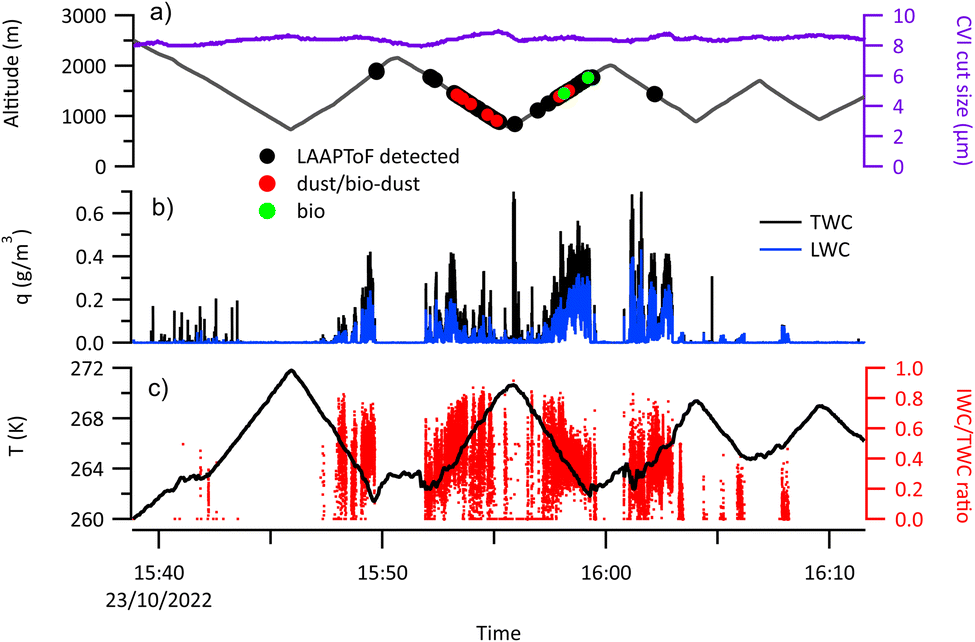 | ||
| Fig. 7 A portion of flight C321 (Canadian air mass) when the aircraft was sampling cloud using a sawtooth flight pattern and cloud residuals were being sampled using the CVI. (Panel a) Shows the flight altitude (black line) and the lowest cut size of the CVI is shown in purple. The circles superimposed on the flight altitude line show points when residual particles were detected by the LAAPToF. All particles are marked as black circles; particles identified as mixed dust-biological particles marked as red circles, and particles identified as biological in type marked as green circles. (Panel b) Shows the time series of total water content (TWC, black) and liquid water content (LWC, blue). (Panel c) The temperature (black line) and the fractional ice water content (IWC/TWC) (red dots). | ||
 | ||
| Fig. 8 A portion of flight C322 (northerly air mass) when the aircraft was sampling cloud using a sawtooth flight pattern and cloud residuals were being sampled using the CVI. (Panel a) Shows the flight altitude (black line) and the lowest cut size of the CVI is shown in purple. The circles superimposed on the flight altitude line show points when residual particles were detected by the LAAPToF. (Panel b) Shows time series of total water content (TWC, black) and liquid water content (LWC, blue). (Panel c) The temperature (black line) and the fractional ice water content (IWC/TWC) (red dots). If IWC/TWC is below 0.1 the cloud particles are predominately liquid, if it exceeds 0.9 the cloud is characterised as ice phase, intermediate values are characterised as mixed phase. | ||
Cloud state was identified using the ice water content (IWC) and total water content (TWC) measured by the Nevzorov probe (Fig. 7b and 8b). When the fractional ice water content (IWC/TWC) was less than 0.1, as shown in Fig. 7c and 8c, the cloud was characterised as liquid, if the IWC/TWC was greater than 0.9 then the cloud was characterised as being ice phase and at intermediate values of IWC/TWC the cloud was identified as being mixed phase. The cloud residual particle detections by the LAAPToF during each flight are shown in Fig. 7a and 8a by large dots superimposed on the altitude time series.
During C322 (northerly) there is very little glaciation and the IWC/TWC is largely less than 0.1 (Fig. 8b and c) indicating that the cloud was largely liquid. This was typical of low level clouds sampled during the cold air outbreak events where there was little ice phase observed and the residuals are likely dominated by those from liquid droplets. Even in the northerly case with the colder cloud top (C330), though there was more glaciation, liquid water was often greater than ice. During C321 (Canadian), there was considerably more ice present (Fig. 7b and c) at similar temperatures to the warmer northerly cases and the cloud is characterised as mixed phase. This was typical of boundary layer cloud sampled when the surface air had travelled across the Canadian continent.
Table 1 shows the duration of time spent in-cloud sampling using the CVI on each flight and also the fraction of this time the cloud was characterised as mixed phase or glaciated based on the IWC/TWC thresholds. A greater fraction of cloud sampled during the Canadian air masses were classified as either mixed phase or glaciated (63–89%), compared to the clouds sampled during northerly air masses (8–35%) across all flights. Although these fractions were less in BLNFTC airmasses (6–56%) than those in Canadian air masses, they were considerably higher than those in northerly conditions.
Chemical composition of cloud residuals
Single particle cloud residuals were measured by the LAAPToF when sampling from the CVI. A k-means clustering was performed on the cloud residual single particle spectra irrespective of the phase of the cloud particles and the main classes are shown in Fig. 9 using a similar approach to that performed on the out of cloud data (Fig. 5). The numbers of particles of each class detected by the LAAPToF when sampling via the CVI as a function of air mass class are summarised in Table S2.† The data have been normalised by the detection efficiency of each particle type by the LAAPToF. Fresh and aged sea salt particles were the most commonly observed residuals but are not shown here since the spectra are similar to those shown in Fig. 5 when sampling out of cloud. The residue dust class is broadly similar to that seen when sampling the aerosol out of cloud but the negative ion spectra commonly show additional organic nitrogen peaks (CN−, CNO−) and are also internally mixed with sea salt (Cl−, Na+, and cluster ions such as Na2Cl+). The residue bioaerosol is similar to that observed during aerosol sampling (Fig. 5), again with evidence of internal mixing with sea salt. In addition to the residue dust and residue bioaerosol classes, a mixed dust and bioaerosol residual class was observed, indicating prevalence of internal mixing of these two types, that was also mixed with sea salt. Two minor particle types were observed in the residuals that were not seen during the out of cloud sampling of the aerosol particles. A residue sea salt/organic class and a metal class. The latter of these two may well be associated with contamination from impaction on the inside of the CVI, leading to production of metal particles in the inlet as has previously been observed.44 Only 20 particles of this metal type were sampled throughout the whole campaign, mostly on flights C326 (15) and C328 (3). No metal particles were detected on flights C321, C323, C324, C327, C329, C331 and C332. When metal particles were detected, they appeared mostly intermittently and were predominantly associated with periods of mixed-phase/ice cloud sampling. Particles of all types were removed from further analysis during the periods when metal particles were continuously detected (e.g. in C326).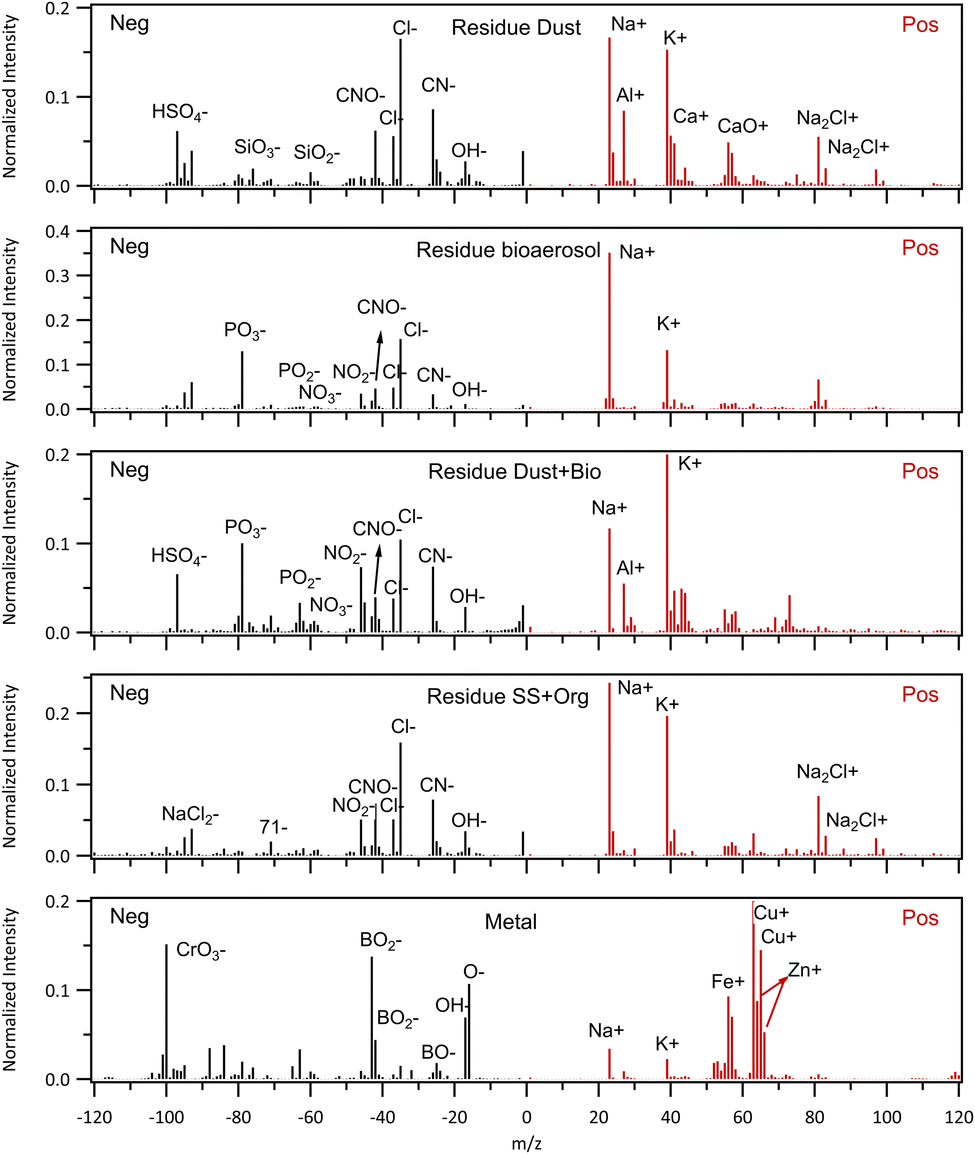 | ||
| Fig. 9 Average spectra of (from the top to bottom) dust, bioaerosol, dust mixed with bioaerosol, sea salt mixed with organic (SS + Org), and metal (pollutants from CVI inlet surface) from cloud residue sampling. Residual dust and bioaerosol were always mixed with sea salt. (SS + Org) and metal were not detected during aerosol sampling. Fresh sea salt and aged sea salt detected were also detected in residue mode, but are not shown here as the spectra are similar to those shown in Fig. 5 when sampling through the aerosol inlet. | ||
The sampled fractional contribution each residue particle class makes to the total particles observed when the LAAPToF was sampling from the CVI is shown in Fig. 6c for each flight. There is no discrimination between cloud phases but only low cloud data are shown. In most flights, the residuals are dominated by sea salt particles. The aged sea salt residue particle type is next most prevalent during cloud sampling and is the most dominant type during flight C321 and significant during flight C328. The dust, biological or bio/dust types are only significant in the flights in Canadian or BLNFTC airmasses, suggesting entrainment of free tropospheric air may well be an important source of these particles into the clouds. Unlike the data collected below cloud, only a very few sulphate particle residuals were observed. This is perhaps not unsurprising since the number of sulphate aerosol particles in the sub-cloud region were very low (Fig. 6a).
The prevalence of the different particle types sampled by the LAAPToF in liquid clouds is shown in Fig. 10 and in mixed phase and ice clouds in Fig. 11 based on the fractional ice water content thresholds described previously and the data have also been separated by air mass class. The upper and lower panels in both figures show the same data on linear and logarithmic scales respectively to highlight the relative contributions and examine the variation in the relative minor particle composition classes between flights, air masses and cloud phases. The data have been normalised by CVI sample efficiency but no normalisation has been made for cloud sample duration or cloud particle number sampled during each flight so the total numbers are not comparable between flights, though some comparison can be made from the sample duration statistics in Table 1. Rather, the relative populations of particle classes should be compared from one flight to another. The sea salt residue class dominates in every flight in both phases and all air masses.
 | ||
| Fig. 10 Number and classification of cloud residuals sampled during periods of liquid cloud, using the LAAPToF behind the CVI for each flight. The number of particles have been corrected for collection efficiency for different particle types. Numbers of particles observed in each flight are shown as a single bar; the types of particle, coloured by chemical classification, are shown as stacked bars. Panel (a) is plotted on a linear number scale and panel (b) is the same data plotted on a logarithmic scale. The flights are separated by air mass: flights in the dashed blue box were sampled in northerly air masses; flights in the dashed green box were sampled in Canadian air masses; flights in the dashed red box were sampled in the BLNFTC air masses. | ||
 | ||
| Fig. 11 Number and classification of cloud residuals sampled during periods of mixed phase or glaciated cloud, using the LAAPToF behind the CVI for each flight. The number of particles have been corrected for collection efficiency for different particle types. Numbers of particles observed in each flight are shown as a single bar; the types of particle, coloured by chemical classification, are shown as stacked bars. Panel (a) is plotted on a linear number scale and panel (b) is the same data plotted on a logarithmic scale. The flights are separated by air mass: flights in the dashed blue box were sampled in northerly air masses; flights in the dashed green box were sampled in Canadian air masses; flights in the dashed red box were sampled in the BLNFTC air masses. | ||
In northerly air masses very few classes other than sea salt or aged sea salt were observed. During liquid phase cloud sampling (Fig. 10), the dust/bioaerosol type was only observed during 3 flights and only a small number of particles were detected. A single sulphate particle was observed on flight C322. Ice and mixed phase cloud residuals during northerly air masses were also dominated by sea salt with aged sea salt as a minor component (Fig. 11). During two flights around 1% of the particles were observed to be of the dust/bioaerosol type. This contrasted with the air masses from the Canadian land mass. In liquid phase clouds, residuals were again dominated by sea salt and aged sea salt and the dust/bioaerosol type was only observed as a minor component during one flight (C326, Fig. 10). However, during periods of ice or mixed phase cloud sampling significant numbers of dust/bioaerosol and bioaerosol types were observed, comprising between 3 and 30% of the total numbers of particles sampled (Fig. 11). We have little evidence for dust or bioaerosol classes in residual particles from clouds of any phase in northerly conditions but are able to demonstrate that these particle types make a significant contribution to the residual particles when ice or mixed phase clouds were sampled in Canadian air masses.
During periods when northerly marine air masses influenced the boundary layer but lower free tropospheric air came from continental outflow (BLNFTC), the sea salt and aged sea salt particles types dominated the sampled residuals of liquid cloud droplets and bioaerosol and dust/bioaerosol types were only sampled during two flights and made only a small contribution (<1%) to the total residual particles sampled (Fig. 10). When ice or mixed phase clouds were sampled under this air mass type, the relative distribution of particle types is much more similar to that observed during Canadian airmasses, with both bioaerosol and mixed dust/bioaerosol types contributing between 5 and 30% of the total residual particles sampled during two of the four flights (Fig. 11). This suggests that free tropospheric continental outflow entrains dust and bioaerosol into cloud tops during transport over the Labrador Sea.
Since the range of cloud base and cloud top temperatures is not significant between the different cloud types these differences are more likely reflect the presence or absence of the dust and bioaerosol types in the different samples rather than different cloud temperatures driving different ice activation efficiencies.
It is worth emphasising that the CVI operates sub-isokinetically and so particles are over-sampled by over an order of magnitude in the case of larger particles. This may contribute to observations of dust/bioaerosol particles being observed by the LAAPToF in cloud while very few were observed out of cloud.
Discussion and conclusions
The vertical and spatial variation of aerosol particle composition and mixing state has been characterised across the Labrador Sea (55–60°N; 45–60°W) region during autumn 2022 using the BAe-146 research aircraft. A single particle laser ablation time of flight mass spectrometry (LAAPToF) was used to determine the chemical composition of single particles in the diameter (Daero) range 0.5 to 2.5 μm. The LAAPToF cannot quantitatively determine mass loading, the response to different chemical components differs due to differences in ionization efficiency though this can be normalised to some extent, and mass spectral intensities can vary through changes in absorbance of laser light by a particle due to shot to shot and/or matrix effects changing ion production. However, single particle laser ablation can provide valuable information on particle mixing state and composition for larger accumulation mode particles and smaller coarse mode particles that may well be important in cloud processes.The boundary layer and lower free troposphere was characterised during autumn 2022 under a range of conditions when the marine boundary layer was largely cloud filled. Three distinctly different regimes were identified. Periods of northerly outflows were characteristic of cold air outbreaks from the high Arctic. The northerly regime was characterised by northerly or north-westerly flows in both the marine boundary layer and the lower free troposphere. In the Canadian regime, winds were from the west in both the boundary layer and lower free troposphere transporting air from the Canadian continental landmass. In the third regime, marine boundary layer flows were from the north but flow in the free troposphere was from the Canadian landmass to the west (BLNFTC).
Accumulation mode aerosol number concentrations were observed to be greater in the free troposphere than the boundary layer, where concentrations were very low (few tens cm−3) due to wet and dry removal. In the free troposphere, increases in concentration of particles below 500 nm in diameter of a factor of between 2 and 5 were observed compared to the sub-cloud boundary layer. Coarse mode number concentrations were largest in northerly flows within the marine boundary layer with a mode diameter of around 1 μm.
Sea salt dominated the particle numbers observed by the LAAPToF in both the boundary layer and lower free troposphere (above cloud layer) in all air masses though sulphate, dust and bioaerosol were a minor detected number fraction in the boundary layer. Low particle concentration limited particle sample statistics above cloud in the lower free troposphere. Prior observations have shown that sulphate dominates the accumulation mode in Arctic flows and organic matter is likely to play a major role in continental outflows.4 The LAAPToF only detects particles larger than Dp ∼ 500 nm so is unlikely to observe most of the particles of these compositions since they would not be detected. In the marine Arctic, sea spray is an important source of larger particles10 and our results are consistent with these observations. Previously observed single particle aerosol composition in Arctic aerosols above Alaska23 during springtime is also consistent with our observations showing sea spray and dust particles are most prevalent in the larger accumulation mode up to around 3 μm.
We have also shown that the LAAPToF can provide insights into cloud processes by coupling to a counterflow virtual impactor that selectively samples cloud particles and dries them to allow their residuals to be sampled, while preventing unactivated aerosols from entering the sample line. Boundary layer clouds were sampled across the region in all 3 regimes. Ice and mixed phase clouds were discriminated from liquid clouds using a threshold fractional ice water content (IWC/TWC). Clouds were identified as ice if IWC/TWC > 0.9, clouds were classed as liquid if IWC/TWC < 0.1, and otherwise clouds were classed as mixed phase. Previous work has shown that when sampling ice clouds using a CVI, ice particles may collide with the inlet walls and dislodge surfaces, creating residual particles that are non-representative of the cloud.44 However, a single particle mass spectrometer has previously been coupled to a CVI successfully to show that biological particles are found in ice crystal residuals and can be effective ice nuclei.45 We observed a few short periods when a small number of metal particles were observed and removed these periods from our analyses. There are no indications of contamination through the remaining data set.
During northerly airmasses the clouds were predominately liquid and very little ice phase was detected, this was the case even in the clouds with colder tops (C330, 255 K; and C331, 256 K Table 1). Even when ice was detected in these clouds very few dust or bioaerosol classes were identified by the LAAPToF. The lack of such particles in the boundary layer indicates that, at least in the Labrador Sea region away from coastal regions during late summer/autumn, high latitude dust sources are not strong and bioaerosol either from marine or coastal locations is limited.
In Canadian outflows or in periods of northerly marine air when above-cloud air was advected from continental Canada (BLNFTC), ice was much more prevalent in boundary layer clouds and the mixed phase was common. The residuals sampled during periods of liquid cloud were dominated by sea salt particle types in all airmass regimes. This is likely a result of the minimum size cut of the LAAPToF (Dp ∼ 0.5 μm) preventing detection of most accumulation mode particles that are likely dominated by sulphate or organic matter28 but is broadly consistent with previous observations of larger submicron and smaller coarse mode particles in previous studies10,31 since sea salt is hygroscopic and mineral dust has low hygroscopicity. Only one bioaerosol particle type was detected during sub-cloud aerosol sampling throughout the experiment. Improved particle counting statistics were possible in cloud compared to out of cloud since the CVI enhances particle concentrations by upwards of a factor of 10.
When sampling clouds containing ice (either mixed phase or ice) in the Canadian or BLNFTC regimes, a very different picture emerges from the northerly regime. The largest residual aerosol type remains sea salt regardless of the air mass regime. However, there are significant differences in the fractional contribution of other particle types in the cloud residuals between periods when both the free troposphere and boundary layer experienced northerly air masses and periods when the air immediately above the boundary layer was westerly and from continental Canada, regardless of whether the boundary layer flow was northerly or westerly in nature (both Canadian and BLNFTC regimes). In the northerly regime, only a small fraction (∼1%) of the detected residual particles were classified as dust and no other particle types were detected. However in Canadian and BLNFTC regimes, between 5 and 30% of residual particles observed during most of the flights were classified as being either the dust/bioaerosol type or bioaerosol type. Since the cloud depths and cloud top temperatures show the same range in the different flow regimes it is unlikely that formation of ice at different temperatures can explain the result. We argue that such marked differences in the prevalence of ice result from the availability of transported continental aerosol, specifically INPs, from Canada.
The data show that ice clouds are more prevalent across the Labrador Sea region in the autumn when flow within the marine boundary is from continental Canada or when the air immediately above the boundary layer cloud originated from the west above the Canadian land mass even if the marine boundary layer flow was northerly from the Arctic region. This strongly suggests that there is a paucity of INPs in the Arctic region to the north of the Labrador Sea. Such low INP concentrations have previously been reported during March 2018.31 Under these conditions, warming of the Arctic will likely further reduce the likelihood of ice formation in these clouds, since primary ice particle production would be less likely.
The enhanced fraction of detected dust and bioaerosol classes detected by the LAAPToF in ice and mixed phase cloud residuals in periods when flows immediately above marine clouds originated from continental Canada demonstrates the importance of long range transport as an important aerosol pathway, as has previously been noted.4,28,29 Since bioaerosol and dust are important INPs,14,18,43 our work suggests that continental Canada is likely to be an important source of INP into these cloud systems and during cold air outbreaks, entrainment of continental free tropospheric air could be a major route for initial glaciation of the clouds. Previous work has shown that Greenland is an important source of dust and biological INP to the Arctic during springtime.46 There is no evidence for increases in aerosol of these types in northerly air streams in our work, though given the lack of INP under these conditions, this source could be important but exist at concentrations below our detectable limit.
How such a pathway changes the prevalence of ice phase clouds in a future warmer Arctic region is complex. Clearly, warmer conditions reduce the effectiveness of primary ice formation since the number of active INP is strongly temperature dependent. However, if transport and entrainment supplies sufficient INP and these are active at warmer temperatures in the temperature regimes where secondary ice processes (SIP) are efficient, then INP supply and SIP may synergise so that some clouds will contain more ice in a warmer world, influencing cloud reflectivity and impacting spatial precipitation patterns.
Our work only identifies the dominant pathways for aerosol transport and its interaction with cloud during a month long study period in the Labrador Sea region in the autumn. It is recommended that similar studies examine other regions and seasons to establish the importance of these different processes since the extent to which clouds glaciate across the Arctic region is important and the underlying processes driving the phase of clouds is central to how clouds across the region may respond to a rapidly warming environment. Such observations should ideally be coupled with a more extensive characterisation of smaller particles to gain a fuller interpretation of the role of accumulation particles in future studies.
Data availability
Airborne measurements are available from the Centre for Environmental Data Analysis via Facility for Airborne Atmospheric Measurements; Natural Environment Research Council; Met Office (2022): M-Phase: Airborne atmospheric measurements from core instrument suite on board the BAe-146 aircraft. NERC EDS Centre for Environmental Data Analysis. Last accessed Jan 2025 (http://catalogue.ceda.ac.uk/uuid/affe775e8d8890a4556aec5bc4e0b45c/). The data used in this manuscript are available from H. Wu, & H. Coe, (2025), M-PHASE Project: Airborne atmospheric measurements on board the BAE-146 aircraft [Data set]. Zenodo https://doi.org/10.5281/zenodo.14900461.Author contributions
BM, TWC, MWG and HC designed the research; HW, NM, MF, KNB, GJN, BJM performed field experiments; BJM was overall PI of the M-Phase project and together with the M-Phase team planned the flights and served as mission scientist on a number of flights. HW, NM, KH, GJN, MB, PF prepared datasets of LAAPToF, SP2, PCASP; CDP; Nevzorov; HW analysed datasets; HC supervised the research; and HC and HW wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research has been supported by the Natural Environment Research Council through M-Phase project, part of the CloudSense programme (grant no. NE/T00648X/1, NE/T006463/1). This project has also received funding from Horizon Europe programme under Grant Agreement No. 101137680 via project CERTAINTY (Cloud-aERosol inTeractions & their impActs IN The earth sYstem). Airborne data was obtained using the BAe-146-301 Atmospheric Research Aircraft [ARA] flown by Airtask Ltd and managed by FAAM Airborne Laboratory, jointly operated by UKRI and the University of Leeds. We thank Dr Steve Abel for processing the Nevzorov data.References
- J. Schmale, P. Zieger and A. M. L. Ekman, Nat. Clim. Change, 2021, 11, 95 CrossRef.
- T. Aoki, S. Matoba, S. Yamaguchi, T. Tanikawa, M. Niwano, K. Kuchiki, K. Adachi, J. Uetake, H. Motoyama and M. Hori, Bull. Glaciol. Res., 2014, 32, 21 CrossRef.
- T. Storelvmo, I. Tan and A. V. Korolev, Curr. Clim. Change Rep., 2015, 1, 288 CrossRef.
- M. D. Willis, W. R. Leaitch and J. P. Abbatt, Rev. Geophys., 2018, 56, 621 CrossRef.
- L. A. Barrie, Atmos. Environ., 1986, 20, 643 CrossRef CAS.
- S. Sharma, L. A. Barrie, E. Magnusson, G. Brattström, W. R. Leaitch, A. Steffen and S. Landsberger, J. Geophys. Res.:Atmos., 2019, 124, 14133 CrossRef CAS.
- E. Asmi, V. Kondratyev, D. Brus, T. Laurila, H. Lihavainen, J. Backman, V. Vakkari, M. Aurela, J. Hatakka, Y. Viisanen, T. Uttal, V. Ivakhov and A. Makshtas, Atmos. Chem. Phys., 2016, 16(3), 1271 CrossRef CAS.
- E. Freud, R. Krejci, P. Tunved, R. Leaitch, Q. T. Nguyen, A. Massling, H. Skov and L. Barrie, Atmos. Chem. Phys., 2017, 17, 8101 CrossRef CAS.
- J. Schmale, S. Sharma, S. Decesari, J. Pernov, A. Massling, H.-C. Hansson, K. von Salzen, H. Skov, E. Andrews, P. K. Quinn, L. M. Upchurch, K. Eleftheriadis, R. Traversi, S. Gilardoni, M. Mazzola, J. Laing and P. Hopke, Atmos. Chem. Phys., 2022, 22, 3067 CrossRef CAS.
- K. Adachi, Y. Tobo, M. Koike, G. Freitas, P. Zieger and R. Krejci, Atmos. Chem. Phys., 2022, 22, 14421 CrossRef CAS.
- V. Moschos, J. K. Schmale, W. Aas, S. Becagli, G. Calzolai, K. Eleftheriadis, C. E. Moffett, J. Schnelle-Kreis, M. Severi, S. Sharma, H. Skov, M. Vestenius, W. Zhang, H. Hakola, H. Hellen, L. Huang, J. L. Jaffrezo, A. Massling, J. Nojgaard, T. Petaja, O. Popovicheva, R. J. Sheesley, R. Traversi, K. E. Yttri, A. S. H. Prevot, U. Baltensperger and I. El Haddad, Environ. Res. Lett., 2022, 17, 034032 CrossRef CAS.
- R. M. Kirpes, A. L. Bondy, D. Bonanno, R. C. Moffet, B. Wang, A. Laskin, A. P. Ault and K. A. Pratt, Atmos. Chem. Phys., 2018, 18(6), 3937 CrossRef CAS.
- L. Karlsson, R. Krejci, M. Koike, K. Ebell and P. Zieger, Atmos. Chem. Phys., 2021, 21, 8933 CrossRef CAS.
- Z. A. Kanji, L. A. Ladino, H. Wex, Y. Boose, M. Burkert-Kohn, D. J. Cziczo and M. Krämer, Meteorol. Monogr., 2017, 58, 1.1–1.33 CrossRef.
- J. Kojoj, G. P. Freitas, M. Muilwijk, M. A. Granskog, T. Naakka, A. M. L. Ekman, B. Heutte, J. Schmale, A. Da Silva, R. Lapere, L. Marelle, J. L. Thomas, C. Melsheimer, B. J. Murray and P. Zieger, Tellus B, 2024, 76(1), 47 CrossRef.
- J. Schneider, K. Höhler, P. Heikkilä, J. Keskinen, B. Bertozzi, P. Bogert, T. Schorr, N. S. Umo, F. Vogel, Z. Brasseur, Y. Wu, S. Hakala, J. Duplissy, D. Moisseev, M. Kulmala, M. P. Adams, B. J. Murray, K. Korhonen, L. Hao, E. S. Thomson, D. Castarède, T. Leisner, T. Petäjä and O. Möhler, Atmos. Chem. Phys., 2021, 21, 3899 CrossRef CAS.
- N. A. Marsden, H. Coe, J. Allan, P. Williams and D. Liu, Atmos. Chem. Phys., 2019, 19(4), 2259 CrossRef CAS.
- S. Augustin-Bauditz, H. Wex, C. Denjean, S. Hartmann, J. Schneider, S. Schmidt, M. Ebert and F. Stratmann, Atmos. Chem. Phys., 2016, 16, 5531 CrossRef CAS.
- D. O’Sullivan, B. J. Murray, T. L. Malkin, T. F. Whale, N. S. Umo, J. D. Atkinson, H. C. Price, K. J. Baustian, J. Browse and M. E. Webb, Atmos. Chem. Phys., 2014, 14, 1853 CrossRef.
- J. E. Bullard, M. Baddock, T. Bradwell, J. Crusius, E. Darlington, D. Gaiero, S. Gassó, G. Gisladottir, R. Hodgkins, R. McCulloch, C. McKenna-Neuman, T. Mockford, H. Stewart and T. Thorsteinsson, Rev. Geophys., 2016, 54, 447 CrossRef.
- Y. Tobo, K. Adachi, P. DeMott, T. C. J. Hill, D. S. Hamilton, N. M. Mahowald, N. Nagatsuka, S. Ohata, J. Uetake, Y. Kondo and M. Koike, Nat. Geosci., 2019, 12, 253 CrossRef CAS.
- S. L. Barr, B. Wyld, J. B. McQuaid, R. R. Neely III and B. J. Murray, Sci. Adv., 2023, 9, eadg3708 CrossRef CAS PubMed.
- A. Sanchez-Marroquin, O. Arnalds, K. J. Baustian-Dorsi, J. Browse, P. Dagsson-Waldhauserova, A. D. Harrison, E. C. Maters, K. J. Pringle, J. Vergara-Temprado, I. T. Burke, J. B. McQuaid, K. S. Carslaw and B. J. Murray, Sci. Adv., 2020, 6, eaba8137 CrossRef CAS PubMed.
- Y. Xi, C. Xu, A. Downey, R. Stevens, J. O. Bachelder, J. King, P. L. Hayes and A. K. Bertram, Environ. Sci.: Atmos., 2022, 2, 714 CAS.
- R. J. Herbert, A. Sanchez-Marroquin, D. P. Grosvenor, K. J. Pringle, S. R. Arnold, B. J. Murray and K. S. Carslaw, Atmos. Chem. Phys., 2025, 25, 291 CrossRef CAS.
- E. N. Raif, S. L. Barr, M. D. Tarn, J. B. McQuaid, M. I. Daily, S. J. Abel, P. A. Barrett, K. N. Bower, P. R. Field, K. S. Carslaw and B. J. Murray, Atmos. Chem. Phys., 2024, 24, 14045 CrossRef CAS.
- G. Pereira Freitas, B. Kopec, K. Adachi, R. Krejci, D. Heslin-Rees, K. E. Yttri, A. Hubbard, J. M. Welker and P. Zieger, Atmos. Chem. Phys., 2024, 24, 5479 CrossRef.
- M. D. Willis, H. Bozem, D. Kunkel, A. K. Lee, H. Schulz, J. Burkart, A. A. Aliabadi, A. Herber, W. R. Leaitch and J. P. Abbatt, Atmos. Chem. Phys., 2019, 19, 57 CrossRef CAS.
- H. Bozem, P. M. Hoor, D. Kunkel, F. Köllner, J. Schneider, A. Herber, H. Schulz, W. R. Leaitch, A. A. Aliabadi, M. D. Willis, J. Burkart and J. P. Abbatt, Atmos. Chem. Phys., 2019, 19, 15049 CrossRef CAS.
- F. Köllner, J. Schneider, M. D. Willis, H. Schulz, D. Kunkel, H. Bozem, P. Hoor, T. Klimach, F. Helleis, J. Burkart, W. R. Leaitch, A. A. Aliabadi, J. P. D. Abbatt, A. B. Herber and S. Borrmann, Atmos. Chem. Phys., 2021, 21, 6509 CrossRef.
- A. Sanchez-Marroquin, S. L. Barr, I. T. Burke, J. B. McQuaid and B. J. Murray, Atmos. Chem. Phys., 2023, 23, 13819 CrossRef CAS.
- P. D. Rosenberg, A. R. Dean, P. I. Williams, J. R. Dorsey, A. Minikin, M. Pickering and A. Petzold, Atmos. Meas. Tech., 2012, 5, 1147 CrossRef.
- D. Liu, M. Flynn, M. Gysel, A. Targino, I. Crawford, K. Bower, T. Choularton, Z. Jurányi, M. Steinbacher, C. Hüglin, J. Curtius, M. Kampus, A. Petzold, E. Weingartner, U. Baltensperger and H. Coe, Atmos. Chem. Phys., 2010, 10, 7389 CrossRef CAS.
- N. Marsden, M. J. Flynn, J. W. Taylor, J. D. Allan and H. Coe, Atmos. Meas. Tech., 2016, 9, 6051 CrossRef CAS.
- N. A. Marsden, M. J. Flynn, J. D. Allan and H. Coe, Atmos. Meas. Tech., 2018, 11, 195 CrossRef CAS.
- T. Shingler, S. Dey, A. Sorooshian, F. J. Brechtel, Z. Wang, A. Metcalf, M. Coggon, J. Mülmenstädt, L. M. Russell, H. H. Jonsson and J. H. Seinfeld, Atmos. Meas. Tech., 2012, 5, 1259 CrossRef.
- S. Lance, C. A. Brock, D. Rogers and J. A. Gordon, Atmos. Meas. Tech., 2010, 3, 1683 CrossRef.
- A. V. Korolev, E. Emery and K. Creelman, J. Atmos. Ocean. Technol., 2013, 30, 690 CrossRef.
- S. J. Abel, R. J. Cotton, P. A. Barrett and A. K. Vance, Atmos. Meas. Tech., 2014, 7, 3007 CrossRef.
- A. Jones, D. Thomson, M. Hort and B. Devenish, Air Pollut. Model. Appl. XVII, 2007, 17, 580 Search PubMed.
- A. Brown, S. Milton, M. Cullen, B. Golding, J. Mitchell and A. Shelly, Bull. Am. Meteorol. Soc., 2012, 93, 1865 CrossRef.
- P. Reitz, S. R. Zorn, S. H. Trimborn and A. M. Trimborn, J. Aerosol Sci., 2016, 98, 1 CrossRef CAS.
- M. A. Zawadowicz, K. D. Froyd, D. M. Murphy and D. J. Cziczo, Atmos. Chem. Phys., 2017, 17, 7193 CrossRef CAS.
- D. M. Murphy, D. J. Cziczo, P. K. Hudson, D. S. Thomson, J. C. Wilson, T. Kojima and P. R. Buseck, Aerosol Sci. Technol., 2004, 38, 401 CrossRef.
- K. A. Pratt, P. J. DeMott, J. R. French, Z. Wang, D. L. Westphal, A. J. Heymsfield, C. H. Twohy, A. J. Prenni and K. A. Prather, Nat. Geosci., 2009, 2, 398 CrossRef CAS.
- K. C. H. Sze, H. Wex, M. Hartmann, H. Skov, A. Massling, D. Villanueva and F. Stratmann, Atmos. Chem. Phys., 2023, 23, 4741 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5fd00005j |
| This journal is © The Royal Society of Chemistry 2025 |