 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Silica biomineralization in plants alters the structure of lignin†
Srinath Palakurthy a,
Michael Elbaumb and
Rivka Elbaum*a
a,
Michael Elbaumb and
Rivka Elbaum*a
aThe Robert H. Smith Institute of Plant Sciences and Genetics in Agriculture, The Hebrew University of Jerusalem, 7610001 Rehovot, Israel. E-mail: rivka.elbaum@mail.huji.ac.il
bWeizmann Institute of Science, 7610001 Rehovot, Israel
First published on 17th February 2025
Abstract
Biomineralization of silica is a major process in plants, which may contribute 3–10% of tissue dry weight. For reasons that remain unclear, plants containing silica are less sensitive to abiotic and biotic stress. In particular, the mechanisms of silica deposition and stress amelioration are still not fully understood. Silica resides mostly in the extracellular volume (the apoplast) which is made of the lignocellulosic cell wall. In a previous work we showed that synthetic lignin catalyses the formation of silica nanoparticles at RC-OSi(OH)3 positions. Since the phenolic O-4 position is the most reactive during lignin polymerization, the binding sites form at the expense of β-O-4 lignin backbone bonds. Therefore, synthetic lignin becomes more branched when polymerized in the presence of silicic acid, as compared to lignin polymerized without silicic acid. To study lignin–silica relationships in the plant, we extracted lignin from stems of wild type sorghum and compared it to lignin extracted from mutants exhibiting high and low silica contents. The thermal stability of both non-extracted biomass and extracted lignin was measured using thermogravimetric analysis (TGA). High-silica biomass was thermally less stable than low-silica biomass, suggesting lower content of ether (β-O-4) linkages. This interpretation was supported by gas chromatography-mass spectroscopy (GC-MS). Fourier transform infrared (FTIR) and X-ray photoelectron spectra (XPS) indicated lignin with C–O–Si modifications in all genotypes and further showed silicic acid binding to lignin phenolics and carbonyl moieties. Our results show that lignin extracted from genotypes with native-silicon levels have higher affinity to silicic acid, catalysing silica deposition through Si–O-4 (Si–phenoxyl) bonds, and suggest that the presence of silicic acid during in vivo lignin polymerization reduces β-O-4 ether linkages.
1. Introduction
Biomineralization, the process by which organisms produce minerals, is indeed found in a wide variety of organisms, including plants, fungi, bacteria, and animals. The self-amplifying nature of biomineralization is particularly fascinating, as it involves a feedback loop in which small initial mineral particles or structures gradually grow into larger and more complex structural supports (e.g., shells, skeletons, bone and teeth). This synergistic response helps organisms to maximize their survival, contributing to both growth and survival in a dynamic and changing environment. Mineral formation by organisms is usually carried out in an aqueous environment at atmospheric pressure and temperature, and involves organic biomacromolecules that self-organize into highly ordered structures under regulation of the organisms. These macromolecules act as templates that control the morphology and molecular/atomic structure of mineralised phases. These hybrid structures of organic and inorganic materials have unique functions not seen in single-component materials. Therefore, understanding the biomineralization process can promote novel methods for the production of green functional materials to meet the needs of modern society.1–3Plants growing in silica-rich soil take-up soluble silicic acid from the soil solution and transform it into solid amorphous silica. Silicic acid moves with water in the extracellular space, called the apoplast, which is made of lignocellulosic cell walls and voids. With the help of the transpiration stream and dedicated protein transporters,4 silicic acid is distributed in the plant body and gets deposited mostly at the epidermis of leaves and stems (Fig. 1).6 The organic environment of cell walls, including abundant proteins, polysaccharides (cellulose and hemicellulose), and phenolic (lignin) compounds, is likely to play an ultimate role in the formation of silica.7 A few reports suggest that silicon (Si, as silicic acid or silica) affects the composition of plant cell walls in vivo8–10 and in vitro, by binding to phenolic components in polymerization reactions.11 Reciprocally, silica formation was linked with lignin formation in vitro12–14 and in vivo.15,16 However, the molecular details explaining lignin-assisted biosilicification in plants are incomplete.
 | ||
| Fig. 1 Cell wall model, demonstrating silica deposited into the cell wall matrix. (a) Sorghum plant planted in silicate (red) soil which contains silicic acid available for root intake. The bulk of silicic acid resides in the apoplast, i.e. the extracellular space, and moves with the water transpiration stream to the shoot. (b) Silica deposits (red) in the cell wall, onto a lignin (orange)–hemicellulose (blue) matrix that binds cellulose microfibrils (green). (c) Lignin model structure, made of three canonical monomer units, H, G, and S, bound via β-O-4 backbone connection and other phenylpropanoids via a random radical-driven dehydrogenation. β-O-4 are the most abundant linkages in natural lignins.5 | ||
The simplified in vitro system of peroxidase-catalyzed polymerization of lignin model compounds shows that silica is precipitated by polymerizing lignin, but not by lignin monomers, and that silica prevents the formation of large lignin fragments.11,12 We have shown that silicic acid binding at the phenoxyl radical/quinone methide moieties of lignin reduces alkyl-aryl ether (β-O-4) backbone linkages in the final lignin. Synthetic lignin catalyses silica deposition through covalent Si–O–C bonding, which leads to the growth of 2–5 nm silica particles.17
In plants, lignin is secreted into a cellulose–hemicellulose structure in secondary cell walls. Pure lignin typically accounts for 15–30% of lignocellulosic biomass. It is covalently linked to hemicellulose, usually by ester bonding, and thereby crosslinks polysaccharides, providing mechanical strength and rigidity to the cell wall (Fig. 1).18–20 Delignification, the process of extracting lignin during pulping pretreatment procedures, disrupts the glycosidic bonds in polysaccharides. As a result, hydrolysable linkages in lignin may break as well.21 Lignocellulosic biorefinery technologies have developed a process using acetic and formic acid-based organosolv fractionation of lignin from biomass without degradation or extensive modification.22,23 This method adds carboxylic acid groups to extracted lignin molecules through esterification (acetate and formate) that are cleavable upon hydrolysis.24,25
Sorghum is a highly productive grain staple crop, tolerant to drought and salinity.26 Sorghum biomass is also a research target for developing second-generation biofuels that do not compete with food production.27 The high silicon accumulation in sorghum may interfere with its dual role as food and fodder/biofuel source.28 Further, sorghum is a model plant for studying silica deposition.29 As such, a sorghum mutant carrying a defective gene for silicon root transporter – Sblsi1, was isolated, which absorbs about 200 times less silica as compared to the wild type (WT) plant.30
In the present work, the in planta lignin–silica relationship was studied by analysing the lignin of native- and low-silicon (Sblsi1) sorghum genotypes. We compare the effects of Si uptake in the background of both wild type (WT) and a lignin mutant presenting extra aldehyde at the expense of hydroxyl lignin groups (brown mid-rib 6, bmr6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 31). The pyrolysis behaviour of unextracted biomass and of acid-based organosolv isolated lignin, indicated a higher content of native aryl-alkyl ether (β-O-4) linkages in low-silicon genotypes. Lignin extracted from high silicon genotypes had higher catalytic activity in silicic acid polymerization, as compared to lignin extracted from low-silicon genotypes. We identified Si–O–C bonds that formed during lignin synthesis in planta and suggest that these positions catalyse formation of SiO2 nanoparticles.
31). The pyrolysis behaviour of unextracted biomass and of acid-based organosolv isolated lignin, indicated a higher content of native aryl-alkyl ether (β-O-4) linkages in low-silicon genotypes. Lignin extracted from high silicon genotypes had higher catalytic activity in silicic acid polymerization, as compared to lignin extracted from low-silicon genotypes. We identified Si–O–C bonds that formed during lignin synthesis in planta and suggest that these positions catalyse formation of SiO2 nanoparticles.
2. Materials and methods
2.1 Materials and reagents
Formic acid (FA, reagent grade, ≥95%, Sigma-Aldrich) and glacial acetic acid (AA, Reagent Plus, ≥99%, Sigma-Aldrich) were used in lignin extractions. Dipotassium hydrogen orthophosphate (K2HPO4) and potassium dihydrogen phosphate (KH2PO4) were used to prepare 0.1 M potassium phosphate buffer solution at pH 7.4. 1 M. Silicic acid was produced by adding 150 μL tetramethyl orthosilicate (TMOS, Sigma-Aldrich) to 850 μL 1 mM hydrochloric acid (HCl, ACS reagent, 37%, Sigma-Aldrich), and mixing for 30 min. Boron trifluoride diethyl etherate (>46.5% BF3, Sigma-Aldrich), ethanethiol (99+%, Thermo Scientific), 1,4-dioxane (99.8%, Sigma-Aldrich), bisphenol-A (≥99%, Sigma-Aldrich), sodium bicarbonate (NaHCO3, 99.5%, Merck), and ethyl acetate (99.9%, Sigma-Aldrich) were used in thioacidolysis.2.2 Plant materials
Sorghum bicolor (L.) Moench, line BTx623 (wild type, WT), low-silicon mutant (Sblsi1), with about 1:200 silica content in relation to WT plants,30 and brown midrib (bmr6) containing altered lignin composition,31 were grown in a green house. In addition, we produced a cross between bmr6 and Sblsil (bmr6×lsi1), F1 hybrid, and propagated it via self-fertilization to F2 inbred lines. Selection of the F2 line carrying both bmr6 and Sblsi1 mutations was done based on PCR amplification of the mutated genes (ESI SI1†). Plants mutated at both Sblsi1 and bmr6 were grown in parallel to the other genotypes. No visual variation in growing parameters was detected between the four genotypes. Stems of approximately 3 month-old plants were collected, cut into less than half-centimetre pieces, and thoroughly washed under running tap water and then distilled water. The washed pulp was dried in an oven at 70 °C for 3 days. The final dried samples were stored for later use.
2.3 Determination of silica content in biomass
Approximately 2 grams of chopped and dried sorghum stems were placed in a muffle furnace using porcelain crucibles and heated at 600 °C for 12 h. The ash was collected and treated with 1 M HCl and washed once with distilled water. The final acid insoluble ash (AIA) was weighed, AIA was considered to be mainly silica, and its percent per dry weight biomass was calculated.322.4 Isolation of lignin
The lignin fraction was extracted by mixing 1 g (dry matter) of chopped sorghum stems in a volume of 10 mL acetic acid/formic acid/water medium (AA/FA/H2O: 55/30/15 volume ratio).23 It was pre-soaked for 30 min at 50 °C on a heating plate. After soaking, the mixture was kept at 100 °C for 2.5 h with continuous stirring. The mixture was allowed to cool down to room temperature and then filtered with a vacuum filter funnel assembled with a filter disk (11 μm pore size; Whatman filter paper). Acids were evaporated from the filtrate fraction containing the extract liquor until they reached around 60% volume by vacuum evaporation (320 mbar). Distilled water was added to the concentrated extract liquors while stirring, to increase the solution pH from a baseline value below 1 to precipitate the lignin at around pH 2. We noted that the water/concentrated liquor ratio to achieve optimal lignin precipitation was 2/1 for WT and bmr6 and 4/1 for lsi1 and bmr6×lsi1. The precipitates were recovered by centrifugation and washed once with phosphate buffer (pH 7.4) and two times with distilled water.2.5 Lignin–silicic acid precipitation process
Isolated never-dried lignin particles (approximately equal to the dried weight of 3 mg) were dispersed in 3.8 mL of 0.1 M phosphate buffer solution at pH 7.4. Silicic acid at final concentrations of 2.5 and 5 mM was added to the solution and agitated at 200 rpm for 3 days in the dark. Samples were centrifuged at 9000 rpm and the precipitate was washed 3 times with double distilled water, and dried under vacuum at room temperature.2.6 Characterization
10 (%T)). The relative change in the integrated absorption area of the deconvoluted absorption bands was calculated as the difference between the normalized (peak area/total area of fitted range) peak area.Energy-dispersive X-ray spectroscopy measurements were acquired in a Talos 200-X microscope (Thermo-Fisher Scientific) equipped with a QUANTAX FlatQUAD spectrometer (Bruker). As above, the measurements were performed under cryogenic conditions. Data were analysed using the embedded Velox software.
3. Results and discussion
3.1 Variations in biomass composition of four sorghum genotypes
To understand the effect of silicic acid and silica on lignin structures, we grew sorghum wild type (WT) plants, mutants in lignin biosynthesis (brown midrib 6, bmr631), and mutants in silicic acid intake (low silicon 1, lsi130). We also produced double mutant plants by crossing a mutant in bmr6 with a mutant in lsi1 (bmr6×lsi1, see SI1† for details). Silica content was measured in the stems (Table 1). As expected, a much lower percentage of silica was observed in the biomass of both low-silicon genotypes (lsi1 and bmr6×lsi1, herein BM-LowSi) as compared to the genotype with native-silicon intake (WT and bmr6, herein BM-HighSi). Furthermore, a slightly higher percentage of silica was observed in both lignin mutants (bmr6 and bmr6×lsi1) as compared respectively to plants with native lignin (WT and lsi1), in accordance with published results.16
| Sorghum genotype | Silica (% weight per biomass dry weight) | Thermal gravimetric analysis (TGA) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Biomass | Lignin | ||||||||||
| Stage (i) (40–170 °C) | Stage (ii) (170–480 °C) | Stage (iii) (480–900 °C) | Stage (ii) (170–480 °C) | Residue (%) | |||||||
| Weight loss (%) | Peak (°C) | Weight loss (%) | Peak (°C) | Weight loss (%) | Peak (°C) | Weight loss (%) | Peak 1 (°C) | Peak 2 (°C) | |||
| WT | 2.0 ± 0.2 | 3.4 | 154 | 56.7 | 316 | 21.1 | ∼600 | 55.2 | 256 | 353 | 23.3 |
| lsi1 | 0.04 ± 0.02 | 5.8 | 160 | 59.9 | 343 | 10.7 | ∼600 | 57.5 | 269 | 363 | 28.1 |
| bmr6 | 2.3 ± 0.2 | 5.6 | 154 | 55.7 | 320 | 11.8 | ∼600 | 57.2 | 254 | 338 | 23.3 |
| bmr6×lsi1 | 0.3 ± 0.1 | 8.7 | 154 | 53.2 | 331 | 8.5 | ∼600 | 59.2 | 263 | 342 | 25.8 |
The thermal gravimetric analysis (TGA) of stems from flowering plants presented a three-stage decomposition process, with distinct weight loss rates, as seen by differential thermal gravimetry (DTG) (Fig. 2a, b and Table 1): (i) moisture loss (40–170 °C); (ii) lignin rapid devolatilization (170–480 °C) and polysaccharide decomposition (220–480 °C) including hemicellulose (220–310 °C) and cellulose (300–480 °C); and (iii) char formation (>480 °C), where the carbonaceous lignin reduced to graphite.36 A shift of the polysaccharide decomposition (stage (ii)) to higher temperatures was detected in the BM-LowSi as compared to BM-HighSi (Table 1). This could indicate increased crosslinking in the low-silicon cell walls, pointing to changes in lignin structure (see Fig. 1).37 Furthermore, the mass which was lost during char formation (stage (iii), >480 °C) was lower in BM-LowSi as compared to BM-HighSi (Table 1). This could result from the possible role of silica in facilitating reduction and evaporation of lignin radicals.17
 | ||
| Fig. 2 Thermal decomposition of biomass (a and b) and isolated lignin (c and d) from stems of four sorghum genotypes. Biomass thermal degradation (continuous lines) and degradation rates (dotted lines) of (a) WT (black) compared to low silicon lsi1 (red), and (b) lignin mutant bmr6 (black) compared to low silicon lignin mutant bmr6×lsi1 (red). Thermal degradation and degradation rates of lignin extracted from stems of (c) WT (black) and lsi1 (red), and (d) bmr6 (black) and bmr6×lsi1 (red). Degradation was divided into stages (i)–(iii), and % weight of each stage was calculated (Table 1). | ||
To further confirm the increase in crosslinking and other possible changes in lignin structure in BM-LowSi (herein Lig-LowSi) in comparison to lignin in BM-HighSi (herein Lig-HighSi), lignin monomers were isolated via thioacidolysis of the biomass, and β-O-4 cleavage derived monomers were quantified by GC/MS (Fig. S1†). Thioacidolysis relies on the selective cleavage of β-O-4 ether linkages to produce thioethylated H, G and S monomers (see inset of Fig. 3).38,39 G and S-derived monomers were the major products in all genotypes, while H-derived monomers were detected at trace levels. The yield of monomers and S/G ratio derived from lignin mutants bmr6 and bmr6×lsi1 were lower compared to WT and lsi1 genotypes (Fig. 3), in agreement with published analyses of lignin composition in the bmr6 sorghum genotype.31 All three monomer yields obtained from BM-LowSi were higher than those obtained from BM-HighSi, similar to published data.10 This could be interpreted as an increase in the lignin fraction in the BM-LowSi relative to BM-HighSi. However, our TGA results do not show such trends (Fig. 2a, b and Table 1). Therefore, we relate this variation simply to increased β-O-4 linkages in the BM-LowSi as compared to BM-HighSi, similar to synthetic lignin produced in vitro.17
 | ||
| Fig. 3 Distribution of major lignin units in stems of sorghum genotypes quantified by GC-MS following thioacidolysis. (a) Abundance of monomer units calculated by measuring peak area of ions relative to an internal standard. Arrows demonstrate a trend of increased monomer release in BM-LowSi (dark shadow) as compared to BM-HighSi (light shadow) in native (green) and mutated (red) lignin genotypes. (b) Calculated monomer S/G ratio. Color-codes are similar to panel (a). Inset: representative chemical reaction of the formation of thioethylated H, G and S monomers by the cleavage of β-O-4 ethers in lignin. | ||
3.2 Variation in structure of extracted lignin
To highlight the effects of silicic acid or silica on lignin structure, lignin was isolated from the stem’s biomass by acid based organosolv extraction. Scanning electron micrographs (SEM) of isolated lignins showed spherical lignin particles of 10 to 50 nm in diameter that aggregated into a porous mesh (Fig. 4). Lignin extracted from bmr6 mutants (bmr6 and bmr6×lsi1) aggregated into a finer mesh, as compared to the lignin extracted from WT and lsi1 plants, indicating some variations in the self-assembly of the mutated polymer. The Lig-LowSi formed a more open network, as compared to Lig-HighSi. All lignin samples had residual Si, as detected by energy dispersive X-ray spectroscopy (EDX) elemental analysis. However, the average Si content was higher in Lig-HighSi than in Lig-LowSi (tables in Fig. 4). Interestingly, the fraction of oxygen was higher, and carbon was lower, in Lig-HighSi as compared to Lig-LowSi, suggesting that the functional groups were more oxidized in Lig-HighSi than in Lig-LowSi. | ||
| Fig. 4 Scanning electron micrographs of lignin extracted from the four sorghum genotypes, as marked on the images. Insets: tabulated mean element content ± standard deviations of at least five EDX spectral measurements. | ||
Pyrolysis of the extracted lignin occurred over a wide temperature range, from about 200 °C to 800 °C, with fast weight loss rate in parallel to the polysaccharide decomposition (stage (ii), 170–480 °C) (Fig. 2c, d and Table 1). Stage (ii) thermal decomposition of the isolated lignin, was divided into two maxima, at 254–269 °C and 338–350 °C. The first decomposition peak was associated with cleavage of aryl-alkyl ether (β-O-4) linkages to produce phenols and aromatic hydrocarbons.40,41 This peak was bigger and shifted to higher temperatures in Lig-LowSi as compared to Lig-HighSi. Possibly, Lig-LowSi contained a higher fraction of ether bonds that broke to produce a higher concentration of volatile molecules, as compared to Lig-HighSi. The second peak was attributed to lignin side chain decomposition such as carboxylic acid and carbonyl group cleavage and oxidation, and dehydroxylation and hydroxyl cracking in lignin hydrogen bond networks.40,41 This peak varied in lignin extracted from the different genotypes, possibly indicating variation in the distribution of side chain functional group content.
Fourier transform infrared (FTIR) spectroscopy of the isolated lignins indicated structural differences between the samples. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch of conjugated aldehyde carbonyls at 1655 cm−1 was more abundant in mutated lignin extracted from bmr6 and bmr6×lsi1, while non conjugated carbonyls of ketone and esters at 1710 cm−1 were abundant in lignin extracted from WT and lsi1. The intensities of absorptions assigned to S units at 834 cm−1 (C–H deformation) and 1330 cm−1 (C–O stretch) relative to the aromatic skeletal vibration at 1514 cm−1 were lower in mutated lignin (bmr6 and bmr6×lsi1) compared to WT and lsi1 lignin (Fig. 5 and Table 2). These results indicate that bmr6 sorghum mutants have reduced S:G ratio, consistent with GC-MS results (Fig. 3) and the literature showing significantly reduced S-units and production of more aldehyde groups during biosynthesis in bmr6 sorghum mutants.31,48
O stretch of conjugated aldehyde carbonyls at 1655 cm−1 was more abundant in mutated lignin extracted from bmr6 and bmr6×lsi1, while non conjugated carbonyls of ketone and esters at 1710 cm−1 were abundant in lignin extracted from WT and lsi1. The intensities of absorptions assigned to S units at 834 cm−1 (C–H deformation) and 1330 cm−1 (C–O stretch) relative to the aromatic skeletal vibration at 1514 cm−1 were lower in mutated lignin (bmr6 and bmr6×lsi1) compared to WT and lsi1 lignin (Fig. 5 and Table 2). These results indicate that bmr6 sorghum mutants have reduced S:G ratio, consistent with GC-MS results (Fig. 3) and the literature showing significantly reduced S-units and production of more aldehyde groups during biosynthesis in bmr6 sorghum mutants.31,48
 | ||
| Fig. 5 FTIR transmission spectra of isolated lignin. Lignin extracted from wild type (WT, black), lsi1 mutant (lsi1, red), bmr6 mutant (bmr6, blue), and a double mutant in lsi1 and bmr6 (lsi×bmr6, green) sorghum genotypes, showing characteristic functional groups between 400 and 4000 cm−1 (a), and zoomed-in between 800 and 1800 cm−1 (b), as marked by a rectangle in panel (a). Structural differences in lignin, as a result of the bmr6 mutation, are indicated by black vertical dotted lines. Conjugated carbonyl groups associated with silica–lignin interactions as a result of the lsi1 mutation are indicated by the red vertical dotted line. Arrows indicate an increase in the absorption of these carbonyls in lignins of low-silicon as compared to native-silicon genotypes. See Table 2 for band assignments. | ||
| Observed bands (cm−1) | Assignment | Source |
|---|---|---|
| 3390–3435 | O–H stretching | 42 and 43 |
| 3005 | C–H stretch of OCH3 | 42 and 43 |
| 2935 | Symmetric C–H stretch of OCH3 and antisymmetric stretch of CH2OH | 42 and 43 |
| 2848 | Symmetric C–H stretch of OCH3 | 42 and 43 |
| 1710 | CO stretch of non-conjugated carbonyls |
42 and 43 |
| 1654 | CO stretch of conjugated carbonyls |
42 and 43 |
| 1630 | O–H bending | 42 and 43 |
| 1605 | Aromatic ring stretch | 42 and 43 |
| 1514 | Aromatic ring stretch | 42 and 43 |
| 1463 | C–H bending of OCH3 and CH2 | 42 and 43 |
| 1423 | Aromatic ring stretching with in plane C–H deformation | 42 and 43 |
| 1369 | Aliphatic C–H stretch in CH3; not in OCH3; phenolic OH | 42 and 43 |
| 1330 | C–O stretch of S ring; ring stretch of asymmetric-tetrasubstituted rings | 42 and 43 |
| 1275 | C–O stretch of G ring; CO stretch |
42 and 43 |
| 1265 | C–O stretch of G ring; CO stretch |
42 and 43 |
| 1240 | Si–phenoxy | 44 |
| 1217 | C–C, C–O, CO stretch; G condensed > G etherified |
42, 43 and 45 |
| 1204 | C–C stretch | 42, 43 and 45 |
| 1168 | CO stretch in ester group of HGS lignin |
42, 43 and 45 |
| 1158 | Si–O–C asymmetric stretching or C–O–Si cage link structure | 44 and 46 |
| 1125 | Aromatic C–H deformation of G units | 42 and 43 |
| 1116 | Si–O–C asymmetric stretching or C–O–Si open link structure | 46 |
| 1085 | C–O deformation in secondary alcohols and aliphatic ethers | 42 and 43 |
| 1063 | Si–O–C asymmetric stretching or C–O–Si ring link structure | 44 and 46 |
| 1050 | Si–O–Si asymmetric stretching of open chain siloxanes | 44 and 46 |
| 1034 | C–O deformation primary alcohols; C–O stretch of methoxy groups | 42 and 43 |
| 1015 | Si–O–Si asymmetric stretching of cyclic siloxanes | 46 |
| 986 | HCCH out-of-plane deformation |
42 and 43 |
| 975 | Si–phenoxy | 44 |
| 832 | C–H bending of S units | 42 and 43 |
| 523 | Aromatic ring C–H deformation | 42 and 43 |
| 469 | Si–O–Si bending vibrations | 47 |
Plant silicon intake also affected the lignin FTIR signature. The strong absorption band at 3100–3600 cm−1, attributed to the stretching vibrations of OH groups, was broader for Lig-HighSi as compared to Lig-LowSi (Fig. 5a). This could be attributed to a high concentration of hydroxyl moieties in Lig-HighSi on the expense of β-O-4 linkages, in agreement with the TGA and GC-MS results (Fig. 2 and 3). In accordance, the peak at 1168 cm−1 was bigger in Lig-LowSi compared to the corresponding Lig-HighSi (red arrows in Fig. 5b). This peak could be assigned to conjugated carbonyl moieties (CO located at α position) and commonly observed in H–G–S type lignin.45,49
XPS of C 1s showed the presence of four species of carbon atoms with distinct binding energies (Fig. 6a). The C1 peak at 285 eV corresponds to non-oxidized carbon (C–H, C–C, CC); the C2 peak at 286.5 eV corresponds to carbon bound to one oxygen through a single bond (C–OH, C–O–C); the C3 peak at 288.2 eV corresponds to carbon bound to oxygen with two bonds (CO), attributed to carbonyls; and the C4 peak at 289.2 eV corresponds to carbon with three bonds to oxygen (O–CO), attributed to ester and carboxylic acid groups.50,51
 | ||
| Fig. 6 High resolution XPS C 1s and Si 2p spectra of the four sorghum genotype lignins and lignin–silicic acid (lignin+Si) precipitates: (a) C 1s spectra of lignin extracted from WT, lsi1, bmr6 and bmr6×lsi1 sorghum genotypes. C1 (C–C, CH, red), C2, (C–OH, green) C3 (O–C–O, CO, blue), and C4 (O–CO, brown) peaks are deconvolved. (b) C 1s spectra of lignin+Si from WT, lsi1, bmr6 and bmr6×lsi1 sorghum genotypes precipitated with 2.5 mM silicic acid solution. Reduction in C3 peaks (blue) with addition of silicic acid indicate reduction in CO groups that possibly reacted to give Si–O–C. (c) Si 2p spectra of lignin extracted from WT, lsi1, bmr6 and bmr6×lsi1 sorghum genotypes. Lignin+Si samples showed similar Si 2p spectra with higher signal-to-noise ratio (Fig. S2†). Curves with filled area are deconvoluted peaks, the black-symbol curves represent the measured intensity, and the red curves represent the cumulative fit. | ||
The atomic ratio of carbon to oxygen (O/C) quantified by XPS was higher in Lig-HighSi than in Lig-LowSi (Table 3), in agreement with SEM-EDX results (Fig. 4). This may indicate a higher content of phenolic hydroxyls (C–OH) at the expense of β-O-4 ether bonds (C–O–C) in Lig-HighSi relative to Lig-LowSi, in accordance with the FTIR indication of increased hydroxyls in Lig-HighSi relative to Lig-LowSi (Fig. 5a). The lower fraction of C3 in Lig-HighSi indicates that it may contain less carbonyls (CO) than Lig-LowSi (Fig. 6a and Table 3). Interestingly, we noted that the liquor extracted from BM-HighSi was less acidic than the liquor extracted from BM-LowSi (see Materials and methods, Section 2.4). This supports our analysis that Lig-HighSi contains a high fraction of phenolic hydroxyl groups (pKa ∼10) and low fraction of carbonyl groups (pKa ∼4.4).52
| Samples | Percentage of carbon species (%) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| O/C | C1 (C–C, C–H, CC) |
C2 (C–O) | C3 (CO, O–C–O) |
C4 (O–CO) |
||||||
| 0 mM Si | 2.5 mM Si | 0 mM Si | 2.5 mM Si | 0 mM Si | 2.5 mM Si | 0 mM Si | 2.5 mM Si | 0 mM Si | 2.5 mM Si | |
| WT | 0.29 | 0.3 | 48 | 48 | 40 | 45 | 9 | 4 | 3 | 4 |
| lsi1 | 0.24 | 0.3 | 42 | 41 | 37 | 49 | 19 | 7 | 2 | 4 |
| bmr6 | 0.26 | 0.3 | 51 | 52 | 40 | 42 | 6 | 3 | 3 | 3 |
| bmr6×lsi1 | 0.22 | 0.3 | 53 | 56 | 33 | 37 | 12 | 4 | 2 | 3 |
3.3 Identification of Si–O–C bonds and lignin potential to polymerize silicic acid
To study the chemistry between lignin and silica and to highlight possible Si–O–C bond formation, we reacted lignins with silicic acid solution (2.5, 5 mM) in phosphate buffer, herein lignin+Si. The solubility concentration of silicic acid in water is 1.7 mM,53 above which, polymerisation to oligomeric silicic acid occurs. Therefore, we expected only minute amounts of silica polymerization. The C 1s XPS spectra of lignin+Si precipitates shows the reduction of C3 (CO, carbonyls) and the increase in C2 (C–O, hydroxyls and ethers), compared to lignins that precipitated without silicic acid (Fig. 6a, b and Table 3). This suggests that the surface carbonyl moieties on the lignin reacted with silicic acid to give Si–O–C bonds. High resolution Si 2p spectra of lignin+Si were similar to lignin samples before silicic acid addition, however, with increased signal-to-noise ratio (Fig. 6c and S2†). Si–O–C bonds were detected in all samples, including lignin and lignin+Si, by the two major peaks at 102.1 eV (Si 2p3/2) and 102.7 eV (Si 2p1/2) (Fig. 6c), consistent with Si–O–C linkage.54 In addition, two minor signals at 103.5 eV (Si 2p3/2) and 104.1 eV (Si 2p1/2) indicated Si–O–Si and Si–OH of silica (SiO2).55 Our results suggest that with the addition of silicic acid, surface Si–O–C and Si–O–Si bonds increased by a similar factor, while Si–O–C occupied most of the CO positions in Lig-LowSi, reducing surface carbonyls significantly.
FTIR spectra of lignin+Si in comparison to the corresponding lignin exhibited an interesting variation in the band at 469 cm−1, assigned to Si–O–Si bending modes. Under reaction with a marginally saturated silicic acid solution of 2.5 mM, lignin+Si spectra of only Lig-HighSi and not Lig-LowSi showed this peak. When silicic acid concentration was 5 mM it appeared in all lignin+Si samples, but its intensity was significantly higher in Lig-HighSi as compared to Lig-LowSi (Fig. 7). Further, pyrolysis residue during TGA of lignin+Si precipitates was found to be significantly higher in Lig-HighSi compared to Lig-LowSi (Fig. S3†). Our results indicated that Lig-HighSi has higher catalytic activity in polymerizing silicic acid. This could possibly occur through Si–phenoxyl bonds as Lig-HighSi contains higher free phenolics at the expense of β-O-4 linkages (Fig. 2 and 3) compared to Lig-LowSi.17
 | ||
| Fig. 7 FTIR transmission spectra of lignin and lignin+Si precipitants. Lignins extracted from biomass of (a) WT, (b) lsi1, (c) bmr6 and (d) bmr6×lsi1 plants (black lines) were reacted with silicic acid at 2.5 mM (red) and 5 mM (green) to form lignin+Si precipitates. Peaks and shoulder peaks of Si–O–Si and Si–O–C vibration modes are marked in blue text. Lignin major functional groups, marked in black text, overlapped with silica related peaks. Traces of Si–O–Si absorption band (469 cm−1) appear when lignin reacted with 2.5 mM silicic acid only in WT and bmr6. This band is also stronger in lignins extracted from the biomass of WT and bmr6 as compared to lsi1 and bmr6×lsi1 at 5 mM silicic acid. | ||
Supporting this, lignin extracted using the alkali pretreatment procedure in glass beakers showed a strong 469 cm−1 band only when extracted from BM-HighSi and not from BM-LowSi (ESI SI2 and Fig. S4†). This suggests that the Lig-HighSi could nucleate SiO2 from silicic acid released from the glass beaker under alkaline pH, through binding silicic acid to the abundant phenolic hydroxyls and the formation of Si–phenoxyl bonds.
With the addition of silicic acid, we detected an increase in a shoulder at 975 cm−1 and a band at 1240 cm−1, exclusively assigned to Si–O–phenoxyl44 (Fig. 7 and Table 2). Further minor variations in the spectra could be attributed to Si–O–Si asymmetric stretching in cyclic siloxanes at 1015 cm−1, open chain siloxanes at 1050 cm−1, Si–O–C asymmetric stretching in the ring-link at 1063 cm−1, open-link Si–O–C at 1116 cm−1, and cage-link Si–O–C asymmetric stretching modes at 1158 cm−1.44,46
In order to highlight these variations, we deconvoluted spectra of lignin and lignin+Si reacted with 2.5 mM silicic acid (Fig. 8). The area of the lignin bands at 1030 cm−1 assigned to C–O deformation of primary alcohols and methoxy groups, and at 1168 cm−1 assigned to conjugated CO stretching, decreased with the addition of silicic acid, suggesting these bonds react with silicic acid. Fitted peaks assigned to Si–O–C bonds at 975, 1240, 1116 and 1158 cm−1, and Si–O–Si bonds at 1050 cm−1 were observed in the spectra of all samples, and their relative integrated absorption area increased in lignin+Si precipitates. Two very small peaks, attributed to Si–O–Si asymmetric cyclic siloxane stretching (1015 cm−1), and Si–O–C ring-link modes (1063 cm−1) could be fitted only in the spectra of lignin+Si precipitates.
 | ||
| Fig. 8 Deconvolution of FTIR spectra of extracted lignin and lignin+Si reacted with 2.5 mM silicic acid. Si–O–C and Si–O–Si bonding modes are highlighted in the wavenumber range 950 to 1250 cm−1. Spectra of lignin extracted from biomass of (a) WT, (c) lsi1, (e) bmr6, and (g) bmr6×lsi1 plants were compared to lignin+Si precipitates of (b) WT, (d) lsi1, (f) bmr6, and (h) bmr6×lsi1 plants. The black line represents infrared absorption, and red dashed line represents the cumulative peak fit of lignin (coloured-dashed lines) and Si–O (full lines with filled area). | ||
Based on the fit, we calculated the difference between the intensities of selected bands in lignin+Si and lignin samples (Fig. 9). As expected from our previous analysis (Fig. 7), with silicic acid addition, the increment in the integrated absorption area of the bands at 975 and 1240 cm−1 assigned to Si–O–phenoxyl bonding, was higher in Lig-HighSi compared to Lig-LowSi (Fig. 9a). Furthermore, the relative increase in the integrated absorption area of Si–O–Si asymmetric stretching in cyclic siloxanes (at 1016 cm−1) and open chain siloxanes (at 1050 cm−1) was greater in Lig-HighSi compared to Lig-LowSi (Fig. 9b).
 | ||
| Fig. 9 Relative changes in the integrated absorption of lignin functional groups and Si–O–Si and Si–O–C modes in lignins from the different sorghum genotypes as a result of reaction with silicic acid. (a) Changes in bands at 975 and 1240 cm−1 assigned to the Si–O–C bonding modes in which Si is bonded to the phenoxyl group. (b) Changes in bands at 1016 cm−1, assigned to Si–O–Si cyclic siloxanes, and at 1050 cm−1, assigned Si–O–Si open chain siloxanes. (c) Changes in bands at 1116 and 1158 cm−1 assigned to open-link and cage-link Si–O–C bonds respectively. (d) Bands at 1168 cm−1, assigned to conjugated CO, and at 1030 cm−1, assigned to C–O deformation in primary alcohols and methoxy groups, reduced with addition of silicic acid. | ||
Peaks related to the open-link Si–O–C bonds at 1116 cm−1 and cage-link Si–O–C bonds at 1158 cm−1 increased with the addition of silicic acid more in Lig-LowSi compared to Lig-HighSi (Fig. 9c). In parallel, a similar decrement was observed in the relative absorption of conjugated CO stretching at 1168 cm−1, and less so in C–O deformations at 1030 cm−1 (Fig. 9d). Taking into account the XPS results (Fig. 6a and b), the higher formation rate of Si–O–C with Lig-LowSi could be due to the availability of surface carbonyl groups to readily react with silicic acid and form Si–O–C bonds.
3.4 Silica nanoparticle distribution in lignin of high-silicon genotypes
Tomography using scanning electron transmission microscopy (STEM) was performed in order to examine the distribution of silica within the agglomeration of lignin particles. Specimens were prepared using lignin extracted from bmr6 and bmr6×lsi1 plants, both exposed to silicic acid solution of 2.5 mM concentration. Fig. 10 shows 60 nm thick virtual sections from the top, middle, and bottom of the specimens in a semi-transparent volume display mode (in addition, depth sectioning movies are provided in the ESI: video clips S1 and S2†). The two specimens appear as similar agglomerates of near-spherical particles some tens of nm in diameter forming a film of overall thickness 300–350 nm. Particles are clearly visible at the top and bottom of the clusters. They appear to have fused together near the central plane, but this may reflect a loss of resolution for boundaries and small channels within the bulk of the material. Notably, small dense particles of about 6–10 nm diameter appear near the mid-section of the bmr6 specimen, but are absent in the bmr6×lsi1. Given the evidence from SEM-EDS, indicating a Si:C ratio of 3%, the small particles would not appear to account for all the silicon. Other silicon fractions could be bound as single or oligomeric silicic acid units dispersed within the lignin matrix. In order to investigate this, the same grids used for tomography were examined by the more sensitive STEM-EDS. A significant concentration of Si was found in the bmr6 specimen, distributed throughout the lignin. Si was detected in the bmr6×lsi1 specimen only at a level very close to background. These results establish strong evidence that lignin of the high silicon genotype, which has fewer β-O-4 linkages and free phenolics, effectively catalyse silicic acid to incorporate silica within the lignin matrix.
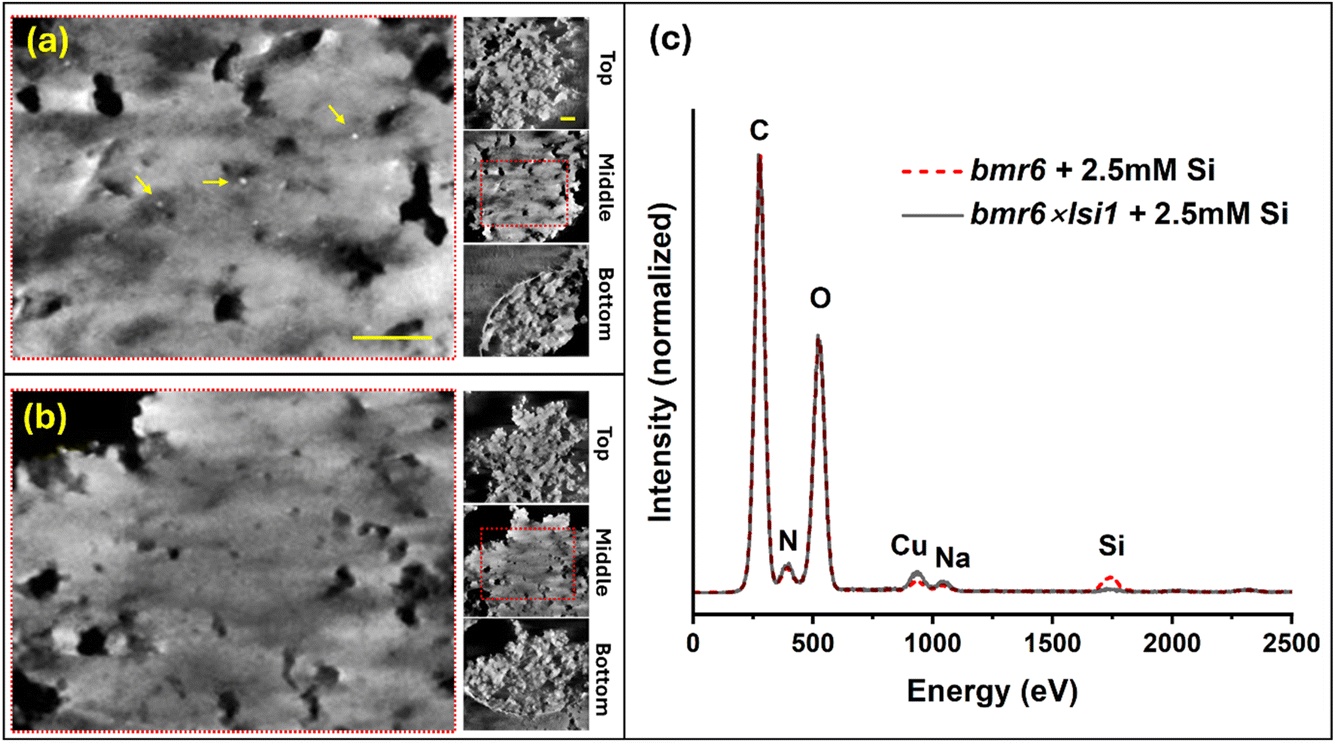 | ||
| Fig. 10 Electron tomography of agglomerates of lignin extracted from bmr6 and bmr6×lsi1 plants, exposed to silicic acid at a concentration of 2.5 mM. (a) Thick virtual section reconstructed from the mid-plane of the bmr6 specimen; note the dense nanoparticles indicated by yellow arrows, which we identify as silica deposits due to the strong electron scattering. Insets to the right show the top, middle, and bottom sections to indicate the texture of small, clustered particles of lignin at the top and bottom. The bright circular arc in the bottom section is the edge of a hole (2 μm diameter) in the specimen support film. (b) Virtual sections of the bmr6×lsi1 specimen, organized as above. Bright nanoparticles were not observed. Scale bar in panel (a) is 200 nm, common to (a) and (b). Scale bar in the top inset is 200 nm, common to all insets. (c) EDX spectra recorded from the same grids, showing higher concentration of Si in the lignin extracted from the bmr6 plant than the bmr6×lsi1 plant. | ||
4. Conclusions
The presented results indicate that Si–O–C bonds form primarily on lignin phenolic hydroxyls and conjugated carbonyl moieties either in the plant or during extraction. Lignin extracted from the biomass of plants with native-silicon intake showed a higher catalytic activity, polymerizing SiO2 with higher content of Si–phenoxyl bonds, as compared to SiO2 forming on lignin in low-silicon mutants. In synthetic lignin, the presence of phenoxy radicals and/or quinone methides of short lignin fragments have high affinity towards silica, and form cyclic siloxanes with Si–phenoxyl bonds instead of an extended Si–O–Si network.17 The low content of β-O-4 ether linkages between monolignol units and high content of Si–O–Si formation with Lig-HighSi suggest that free phenolics (O-4) catalyse silicic acid condensation. In the bmr6 genotype we measured higher SiO2 content as compared to the WT genotype (Table 1 and Fig. 7), in agreement with our published data.16 This could be explained by the increase in aliphatic carbonyl moieties forming under bmr6 mutation, which enhance the catalytic activity of lignin in silicic acid condensation.Interestingly, lignin and silica are not colocalized in the plant. Most lignin is polymerized in the vascular bundles, in xylem tracheary elements and fibre cells. In contrast, most silica is deposited at the epidermis, in silica cells and hairs and forming a double layer with the cuticle. Nonetheless, our work indicates significant variations in the lignin structure as a result of silicic acid intake. The presented data indicate that silicic acid, when present in the apoplast, affects the radical dehydrogenation and polymerization of monolignols by capping the O-4 phenoxyl position. These positions are available in the polymerized lignin for H-bonding to other polymers and molecules and for binding cations. Such variations may explain some of the beneficial effects of silica in plants exposed to heavy metals.56
The presence of silicic acid during in vivo lignin polymerization apparently leads to the aryl-silyl ether (Si–O-4) bonds between silicic acid and monolignol phenoxyl radicals/quinone methides. This may lead to abundant phenoxyl Si–O–C bonds that effectively catalyse the polymerization of silicic acid into SiO2 nanoparticles. In contrast, Lig-LowSi may be produced with abundant aryl-alkyl ether (β-O-4) bonds between monomers that would increase the cell wall density and the extension of conjugated carbonyls. This could explain the appearance of dense lignified cell walls with red-shifted autofluorescence in roots grown under low silicon conditions.37 The common paradigm asserts that silica reduces biomass digestibility in parallel to lignin.57,58 This work highlights a more complex relationship between the two materials as silica actually changes lignin, and vice versa. Extending this research will show whether the modified lignin has implications on the biological function of the tissue and valorisation of biomass.
Data availability
The STEM and STEM-EDX data that support the findings of this study are available under at https://doi.org/10.5281/zenodo.14686717.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Nerya Zexer for producing double mutant plants by crossing bmr6 and lsi1 sorghum plants, Shula Blum for isolating the homozygote bmr6×lsi1 plants, and Lothar Houben for assistance with STEM-EDS. S. P. is thankful for a scholarship from Lady Davis and Golda Meir. This work was funded in part by the Israel Science Foundation grant 958/21. Contribution of ME was supported by the European Union (ERC, CryoSTEM, 101055413; Views and opinions expressed are however those of the authors only and do not necessarily reflect those of the European Union or the European Research Council. Neither the European Union nor the granting authority can be held responsible for them). ME is incumbent of the Sam and Ayala Zacks Professorial Chair in Chemistry.References
- L. A. Estroff, Introduction: Biomineralization, Chem. Rev., 2008, 108, 4329–4331 Search PubMed.
- R. Wood, Exploring the drivers of early biomineralization, Emerging Top. Life Sci., 2018, 2, 201–212 CrossRef CAS PubMed.
- P. U. P. A. Gilbert, K. D. Bergmann, N. Boekelheide, S. Tambutté, T. Mass, F. Marin, J. F. Adkins, J. Erez, B. Gilbert, V. Knutson, M. Cantine, J. Ortega Hernández and A. H. Knoll, Biomineralization: Integrating mechanism and evolutionary history, Sci. Adv., 2022, 8, 1–16 Search PubMed.
- N. Mitani-Ueno, N. Yamaji, S. Huang, Y. Yoshioka, T. Miyaji and J. F. Ma, Nat. Commun., 2023, 14, 6522 CrossRef CAS PubMed.
- J. D. Gargulak, S. E. Lebo and T. J. McNally, Kirk-Othmer Encyclopedia of Chemical Technology, Wiley, 2015, pp. 1–26 Search PubMed.
- S. Kumar, M. Soukup and R. Elbaum, Front. Plant Sci., 2017, 8, 438 Search PubMed.
- N. Zexer, S. Kumar and R. Elbaum, Ann. Bot., 2023, 131, 897–908 Search PubMed.
- Z. Lukačová, R. Švubová, J. Kohanová and A. Lux, Plant Growth Regul., 2013, 70, 89–103 CrossRef.
- A. T. Fleck, T. Nye, C. Repenning, F. Stahl, M. Zahn and M. K. Schenk, J. Exp. Bot., 2011, 62, 2001–2011 CrossRef CAS PubMed.
- R. R. Rivai, K. Yamazaki, M. Kobayashi, Y. Tobimatsu, T. Tokunaga, T. Fujiwara and T. Umezawa, Plant Cell Physiol., 2024, 65, 1983–1992 CrossRef CAS PubMed.
- K. Radotić, D. Djikanović, A. Kalauzi, G. Tanasijević, V. Maksimović and J. Dragišić Maksimović, Int. J. Biol. Macromol., 2022, 198, 168–174 CrossRef PubMed.
- J. Fang, H. Wang, Y. Chen and F. Zhang, Prog. Nat. Sci., 2003, 13, 501–504 CAS.
- J. yu Fang and X. long Ma, J. Zhejiang Univ., Sci., B, 2006, 7, 267–271 CrossRef PubMed.
- M. Soukup, V. M. Rodriguez Zancajo, J. Kneipp and R. Elbaum, J. Exp. Bot., 2020, 71, 6807–6817 CrossRef CAS PubMed.
- C. Zhang, L. Wang, W. Zhang and F. Zhang, Plant Soil, 2013, 372, 137–149 CrossRef CAS.
- N. Zexer and R. Elbaum, J. Exp. Bot., 2022, 73, 1450–1463 CrossRef CAS PubMed.
- S. Palakurthy, L. Houben, M. Elbaum and R. Elbaum, Biomacromolecules, 2024, 25, 3409–3419 CrossRef CAS PubMed.
- D. Delmer, R. A. Dixon, K. Keegstra and D. Mohnen, Plant Cell, 2024, 36, 1257–1311 CrossRef PubMed.
- J. H. Coomey, R. Sibout and S. P. Hazen, New Phytol., 2020, 227, 1649–1667 CrossRef CAS PubMed.
- L. Salmén, Cellulose, 2022, 29, 1349–1355 CrossRef.
- J. Gierer, Wood Sci. Technol., 1985, 19, 289–312 CrossRef CAS.
- J. Snelders, E. Dornez, B. Benjelloun-Mlayah, W. J. J. Huijgen, P. J. de Wild, R. J. A. Gosselink, J. Gerritsma and C. M. Courtin, Bioresour. Technol., 2014, 156, 275–282 CrossRef CAS PubMed.
- H. Labauze, N. Cachet and B. Benjelloun-Mlayah, Ind. Crops Prod., 2022, 187, 115328 CrossRef CAS.
- F. Abdelkafi, H. Ammar, B. Rousseau, M. Tessier, R. El Gharbi and A. Fradet, Biomacromolecules, 2011, 12, 3895–3902 CrossRef CAS PubMed.
- Q. Zheng, L. Chai, B. Du, W. Li, L. H. Fu and X. Chen, Polymers, 2023, 15, 1867 CrossRef CAS PubMed.
- I. Tari, G. Laskay, Z. Takács and P. Poór, J. Agron. Crop Sci., 2013, 199, 264–274 CrossRef CAS.
- W. L. Rooney, J. Blumenthal, B. Bean and J. E. Mullet, Biofuels, Bioprod. Biorefin., 2007, 1, 147–157 CrossRef CAS.
- S. Głazowska, L. Baldwin, J. Mravec, C. Bukh, T. H. Hansen, M. M. Jensen, J. U. Fangel, W. G. T. Willats, M. Glasius, C. Felby and J. K. Schjoerring, Biotechnol. Biofuels, 2018, 11(171), 1–18 Search PubMed.
- A. G. Sangster and D. W. Parry, Ann. Bot., 1976, 40, 361–371 CrossRef.
- O. Markovich, S. Kumar, D. Cohen, S. Addadi, E. Fridman and R. Elbaum, Silicon, 2019, 11, 2385–2391 CrossRef CAS.
- E. D. Scully, T. Gries, D. L. Funnell-Harris, Z. Xin, F. A. Kovacs, W. Vermerris and S. E. Sattler, J. Integr. Plant Biol., 2016, 58, 136–149 CrossRef CAS PubMed.
- Z. Peleg, Y. Saranga, T. Fahima, A. Aharoni and R. Elbaum, Physiol. Plant., 2010, 140, 10–20 CrossRef CAS PubMed.
- A. E. Harman-Ware, C. Foster, R. M. Happs, C. Doeppke, K. Meunier, J. Gehan, F. Yue, F. Lu and M. F. Davis, Biotechnol. J., 2016, 11, 1268–1273 CrossRef CAS PubMed.
- S. Seifer, L. Houben and M. Elbaum, Microsc. Microanal., 2021, 27, 1476–1487 CrossRef CAS PubMed.
- D. N. Mastronarde and S. R. Held, J. Struct. Biol., 2017, 197, 102–113 CrossRef PubMed.
- T. Emiola-Sadiq, L. Zhang and A. K. Dalai, ACS Omega, 2021, 6, 22233–22247 CrossRef CAS PubMed.
- N. Zexer and R. Elbaum, J. Exp. Bot., 2020, 71, 6818–6829 CrossRef CAS PubMed.
- J. Ralph and J. H. Grabber, Holzforschung, 1996, 50, 425–428 CrossRef CAS.
- C. Lapierre, B. Pollet, B. Monties and C. Rolando, Holzforschung, 1991, 45, 61–68 CrossRef CAS.
- T. Faravelli, A. Frassoldati, G. Migliavacca and E. Ranzi, Biomass Bioenergy, 2010, 34, 290–301 CrossRef CAS.
- X. Lu and X. Gu, Biotechnol. Biofuels, 2022, 15, 106 CrossRef CAS PubMed.
- R. J. Sammons, D. P. Harper, N. Labbé, J. J. Bozell, T. Elder and T. G. Rials, BioResources, 2013, 8, 2752–2767 CrossRef.
- P. Bock, P. Nousiainen, T. Elder, M. Blaukopf, H. Amer, R. Zirbs, A. Potthast and N. Gierlinger, J. Raman Spectrosc., 2020, 51, 422–431 CrossRef CAS PubMed.
- L. J. Bellamy, The Infra-red Spectra of Complex Molecules, Chapman and Hall, London, 1975, pp. 374–384 Search PubMed.
- T. You and F. Xu, Applications of Molecular Spectroscopy to Current Research in the Chemical and Biological Sciences, InTech, 2016, pp. 235–260, DOI:10.5772/64581.
- G. L. L. Barry Arkles, Silicon Compounds – Silanes and Silicones, Gelest, Morrisville, USA, 3rd edn, 2013 Search PubMed.
- R. Al-Oweini and H. El-Rassy, J. Mol. Struct., 2009, 919, 140–145 Search PubMed.
- S. E. Sattler, D. L. Funnell-Harris and J. F. Pedersen, J. Agric. Food Chem., 2010, 58, 3611–3616 CrossRef CAS PubMed.
- H. L. Hergert and E. F. Kurth, J. Org. Chem., 1953, 18, 521–529 CrossRef CAS.
- J. Wang, K. Yao, A. L. Korich, S. Li, S. Ma, H. J. Ploehn, P. M. Iovine, C. Wang, F. Chu and C. Tang, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3728–3738 CrossRef CAS.
- C. M. Popescu, C. M. Tibirna and C. Vasile, Appl. Surf. Sci., 2009, 256, 1355–1360 CrossRef CAS.
- M. Ragnar, C. T. Lindgren and N. O. Nilvebrant, J. Wood Chem. Technol., 2000, 20, 277–305 CrossRef CAS.
- R. K. Iler, The Chemistry of Silica : Solubility, Polymerization, Colloid and Surface Properties, and Biochemistry, Wiley, New York, 7th edn, 1979 Search PubMed.
- A. Avila, I. Montero, L. Galán, J. M. Ripalda and R. Levy, J. Appl. Phys., 2001, 89, 212–216 CrossRef CAS.
- G. Dakroub, T. Duguet, J. Esvan, C. Lacaze-Dufaure, S. Roualdes and V. Rouessac, Surf. Interfaces, 2021, 25, 101256 CrossRef CAS.
- B. Bokor, C. S. Santos, D. Kostoláni, J. Machado, M. N. da Silva, S. M. P. Carvalho, M. Vaculík and M. W. Vasconcelos, J. Hazard. Mater., 2021, 416, 126193 CrossRef CAS PubMed.
- P. J. Van Soest and L. H. P. Jones, J. Dairy Sci., 1968, 51, 1644–1648 CrossRef.
- M. P. Silva, C. Whitehead, R. L. Ordonio, T. C. Fernando, M. P. B. Castillo, J. L. Ordonio, T. Larson, D. J. Upton, S. E. Hartley and L. D. Gomez, Biomass Bioenergy, 2024, 182, 107099 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Production and isolation of a sorghum line carrying both bmr6 and lsi1 mutations (SI1), lignin extracted using alkali pretreatment process (SI2), a representative GC-MS chromatogram of trimethyl-silyl (TMS) derivatives of sorghum genotype thioacidolysis products (Fig. S1), high resolution XPS Si 2p spectra of lignin+Si from WT, lsi1, bmr6 and bmr6×lsi1 sorghum genotype at silicic acid concentration of 2.5 mM (Fig. S2), thermal decomposition behaviour of lignins reacted with different concentrations of silicic acid in phosphate buffer solution (Fig. S3), FTIR transmission spectra of lignin extracted using alkali pretreatment process (Fig. S4), tomographic sections of the high silicon specimen (bmr6 + 2.5 mM Si), scale 1.5 nm per pixel (Movie 1), tomographic sections of the low silicon specimen (bmr6×lsi1 + 2.5 mM Si), scale 1.5 nm per pixel (Movie 2). See DOI: https://doi.org/10.1039/d5fd00011d |
| This journal is © The Royal Society of Chemistry 2025 |