 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSilylated peptides as building blocks for material synthesis using sol–gel polymerization†
Mehmet B. Karakaplan ab,
Vinay S. Tiwaria,
Omer Agazania,
Cécile Echalierb,
Gilles Subrab and
Meital Reches*a
ab,
Vinay S. Tiwaria,
Omer Agazania,
Cécile Echalierb,
Gilles Subrab and
Meital Reches*a
aInstitute of Chemistry and The Center for Nanoscience and Nanotechnology, The Hebrew University of Jerusalem, Jerusalem 91904, Israel. E-mail: meital.reches@mail.huji.ac.il
bIBMM, Université de Montpellier, CNRS, ENSCM, 34293 Montpellier, France
First published on 19th February 2025
Abstract
The bottom-up approach exploits simple building blocks to generate new materials with desired physical and chemical characteristics. Here, we combine two bottom-up routes that occur under mild conditions, self-assembly and sol–gel synthesis, to program the shape and structure of materials. While self-assembly occurs through non-covalent interactions, sol–gel synthesis involves forming covalent bonds. As a proof of concept, we chose the self-assembled peptide Phe-Phe and its fluorinated analogue Phe(4-F)-Phe(4-F) to template the sol–gel process. These peptides were silylated to allow their self-mineralization. Scanning electron microscopy and atomic force microscope analysis revealed the formation of rod-shaped structures for the silylated Phe-Phe while spherical particles were formed by its fluorinated analogue. The size of the particles ranges from nano to micron scale. Fourier transform infrared spectrometry suggested the presence of parallel β-sheet secondary structure and siloxane bond formation that can stabilize these structures. Overall this approach can be adopted for other self-assembled peptides for generating new materials using a bottom-up approach.
Introduction
Precise control of material properties is essential for their successful application in diverse fields such as medicine,1 biomedical devices,2 energy storage,3 coatings,4 and environmental technologies.5 This entails the usage of a material synthesis strategy that can provide control over the smallest building blocks.6 The bottom-up strategy is one of the best methods for the design and fabrication of new materials from the atomic to the micron scale. This strategy encompasses many approaches, such as self-assembly, polymerization, sol–gel, biosynthesis, and electrochemical deposition. This variety in fabrication methods provides materials with miscellaneous characteristics.7Among these approaches, self-assembly has attracted great attention. The self-assembly process is driven by non-covalent interactions such as van der Waals, π–π, and electrostatic interactions.8 Self-assembly can result in the spontaneous formation of highly ordered structures at the nano and micron-scale.9 This process can occur under mild conditions. Using different building blocks and altering the environmental conditions allows generating diverse architectures.10
Among these building blocks, peptides especially draw significant interest in creating self-assembled supramolecular structures due to their molecular recognition properties.11–13 By designing various peptide sequences, it is possible to obtain versatile assemblies.14–19 Self-assembling peptides are also excellent templates to initiate inorganic growth and mimic biomineralization.20–22
Another method to generate novel materials is by sol–gel synthesis. The sol–gel reaction starts with the hydrolysis of alkoxysilanes (Si–OR) into hydroxysilanes (Si–OH) and proceeds with chemoselective condensation to form a siloxane (Si–O–Si) network of covalent bonds. One of the advantages of sol–gel synthesis is the ability to tailor the morphology, shape, and size of the materials by adjusting the solvent composition, pH, and precursor concentration.23 Another way to control the structure of the materials is by structure-directing agents such as cetyltrimethylammonium bromide (CTAB).24 Like self-assembly, sol–gel synthesis can be performed at room temperature.25 The combination of these two fabrication methods can generate versatile materials with different characteristics.
Here, we combine self-assembly and sol–gel synthesis for making novel materials using a one-pot method. As a proof of principle, we used the dipeptide Phe-Phe, a well-known self-recognition element, and its fluorinated analogue Phe(4-F)-Phe(4-F). 3-Isocyanatopropyltriethoxysilane (ICPTES) was used to silylate the peptides on their free amino groups (N-terminal). The silylated peptides were used as a precursor for material synthesis in aqueous acidic conditions (pH = 1.5). These conditions favour the hydrolysis of ethoxysilanes into the hydroxysilanes but prevent the spontaneous formation of siloxane bonds at low concentrations. However, the molecular recognition among the dipeptides drives the structural organization of the system by bringing the silanol groups into proximity and favouring their condensation. Based on this mechanism, the silane groups on peptides can “freeze” the structure covalently and provide stability. This simple concept can pave the way for designing materials that are driven by self-assembly but stabilized by covalent bonds.
Experimental section
Material
All solvents and reagents were used as supplied. Solvents used for high-performance liquid chromatography (HPLC) were HPLC grade. N,N′-Dimethylformamide (DMF), acetonitrile (ACN), trifluoroacetic acid (TFA), tetrahydrofuran (THF), diethyl ether, and diisopropylethylamine (DIEA) were purchased from Bio-Lab (Jerusalem, Israel). Triple distilled water (TDW) was obtained through a Milli-Q water filtering system (Millipore). 3-(Triethoxysilyl)propyl isocyanate (ICPTES) was obtained from Thermo Fisher Scientific (Lancashire, UK). Copper grids (pure carbon film 400 mesh) were purchased from Ted Pella Inc. (Redding, CA).Silylation of peptides
One equiv. of peptide 1 or 2 was completely dissolved in 1 mL DMF at a concentration of 0.2 M. DIEA (4.5 equiv.) was added to the reaction and stirred for 5 min. Lastly, ICPTES (2 equiv.) was added to the reaction mixture and stirred for 2 hours under argon. At the end of the reaction, the silylated peptides (compound 3 and compound 4) were precipitated by adding 30 mL of diethyl ether and centrifuged. The pellet was resuspended in fresh diethyl ether and centrifuged thrice in total. The pellet was dried under vacuum overnight and kept at +4 °C with argon for further use (Scheme 1).20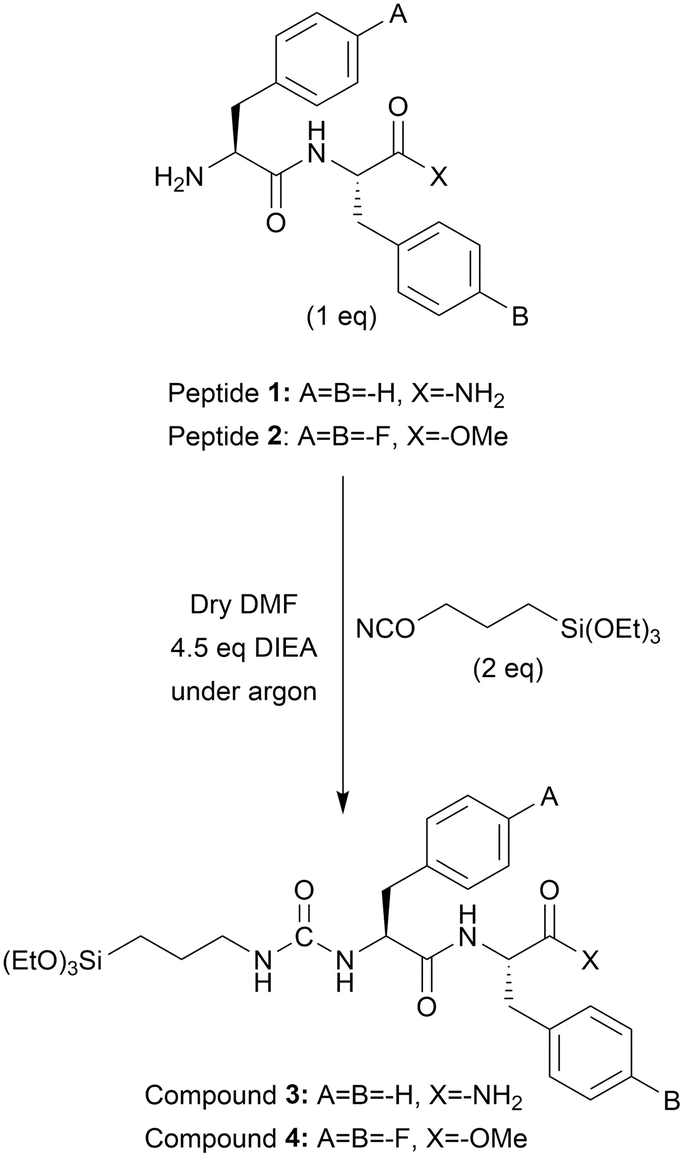 | ||
| Scheme 1 Synthesis of compound 3 and compound 4. | ||
Material synthesis
Compounds 3 and 4 were separately dissolved in a mixture of TDW (HCl, pH 1.5)/THF (1/2, v/v) at two different concentrations, 1.78 mM and 17.80 mM, resulting in four distinct reactions. After a short sonication, the reactions were stirred for 24 hours at room temperature. The solvent mixtures were evaporated overnight in a fume hood to induce the condensation reaction. The obtained white products were washed 3 times with water (HCl, pH 1.5) and dispersed in TDW for further use. The concentration of 17.80 mM was selected based on our findings from a previous study.20 The initially published concentration (38.5 mM) led to rapid gelation and condensation for compounds 3 and 4. To obtain dispersed particles, the concentration was lowered by half to 17.80 mM. Furthermore, to investigate the effect of significantly lower concentration on material architecture and size, 10 times diluted concentration (1.78 mM) was employed for material synthesis.High-performance liquid chromatography (HPLC)
Samples (peptide 1, peptide 2, compound 3, and compound 4) for HPLC were dissolved in ACN/TDW (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50, v/v) including 0.1% TFA at 2 mg mL−1 concentration. The analysis was performed using a Waters Alliance reverse-phase analytical HPLC with an XSelect C18 column (3.5 μm 130 Å, 4.6 × 150 mm). UV detection was performed at 220 nm and 254 nm. A linear gradient (5% to 95%) of ACN (with 0.1% TFA) in TDW (with 0.1% TFA) was used to elute the peptides and silylated peptides at a flow rate of 1 mL min−1. Peptide 2 was eluted with a linear gradient (30% to 95%) of ACN (with 0.1% TFA) in TDW (with 0.1% TFA).
:50, v/v) including 0.1% TFA at 2 mg mL−1 concentration. The analysis was performed using a Waters Alliance reverse-phase analytical HPLC with an XSelect C18 column (3.5 μm 130 Å, 4.6 × 150 mm). UV detection was performed at 220 nm and 254 nm. A linear gradient (5% to 95%) of ACN (with 0.1% TFA) in TDW (with 0.1% TFA) was used to elute the peptides and silylated peptides at a flow rate of 1 mL min−1. Peptide 2 was eluted with a linear gradient (30% to 95%) of ACN (with 0.1% TFA) in TDW (with 0.1% TFA).
Mass spectrometry (MS)
The mass analysis (for peptide 1, peptide 2, compound 3, and compound 4) was performed by electron spray ionization mass spectrometry using a SCIEX Triple Quad 3500 (Framingham, MA, USA). The results were analysed via SCIEX Analyst software.Scanning transmission electron microscopy (STEM)
A drop of 10 μL from the material dispersions in TDW (1 mg mL−1) was placed on a clean silicon substrate or copper grids and dried under vacuum. STEM images were recorded using an analytical high-resolution scanning electron microscope Apreo 2S (Thermo Fisher Scientific, OR, USA) with 2–20 kV acceleration voltage, current of 0.1–0.4 nA, and 4–10 mm working distance.Atomic force microscopy (AFM)
The material dispersions were drop-casted on a clean silicon wafer. All the AFM images were taken at AC mode with a Si3N2 tip with a spring constant of 3 N m−1 by a JPK NanoWizard3® (Berlin, Germany).Dynamic light scattering (DLS)
The hydrodynamic diameter of the materials was measured by a Zetasizer Nano ZS DLS instrument (Malvern Instruments, UK). The material suspensions were prepared in TDW at a concentration of 1 mg mL−1. For accurate measurements, they were sonicated for 20 min and vortexed for 1 min after adding 10 μL of Tween 80. The measurements were carried out three times and standard deviations were included.Attenuated total reflectance (ATR) Fourier transform infrared (FTIR) spectrometry
White powder samples (N3H, N3L, N4H, and N4L) were analysed via an ATR-FTIR spectrometer (Alpha II, Bruker Optics GmbH). The measurements were taken at 1 cm−1 resolution and 10 scans.X-ray diffraction (XRD) analysis
The phases of the materials (N3H, N3L, N4H, and N4L) were identified by powder XRD (X-ray diffractometer – D8 Advance, Bruker) (2θ = 10–50°, at a step size of 0.02 deg s−1) using Cu Kα (λ = 0.15406 nm) and a solid state NaI dynamic scintillation detector. The data was analysed via FullProf Suite software.Results and discussion
Peptide 1 and peptide 2 were synthesized via liquid-phase peptide synthesis (LPPS). See Scheme S1 in the ESI.† Peptide 1 (H-Phe-Phe-NH2) was synthesized with a purity of 99% according to the HPLC analysis (Fig. S1A in the ESI†). The molecular weight was confirmed with MS (m/z = 312, (M + H)+ = 312.3) (Fig. S1B in the ESI†). Silylation of peptide 1 yielded compound 3. Compound 3 was analysed using HPLC and MS (m/z = 475.19, (M + H)+ = 475) to confirm its identity (Fig. S3 in the ESI†). After dissolving compound 3 at the concentrations of 1.78 mM or 17.80 mM in TDW (HCl, pH 1.5)/THF (1/2, v/v) solvent mixture, the reactions were stirred for 24 hours. Then, the solvents were evaporated for an additional 24 hours. See Fig. 1. After solvent evaporation, the obtained white solid materials were washed with TDW (HCl, pH 1.5) three times. The obtained white solid materials were denoted as N3L for 1.78 mM and N3H for 17.80 mM. At 1.78 mM, the reaction remained clear for 24 hours suggesting that the sol–gel condensation did not proceed. Indeed, at pH 1.5, the hydrolysis of triethoxysilane is fast whereas the condensation is unfavourable. In this case, condensation was induced by the evaporation of the solvents.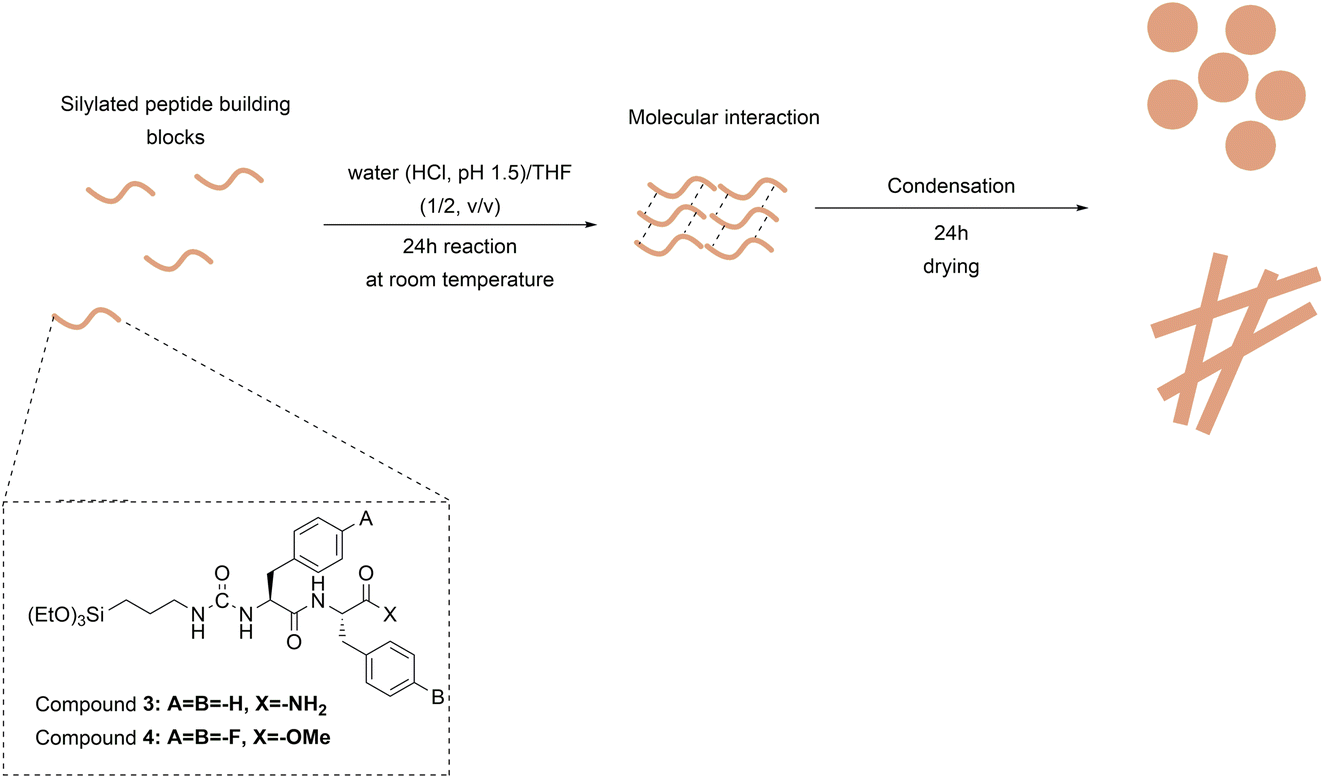 | ||
| Fig. 1 Schematic representation of material synthesis from silylated peptide building blocks. The proposed mechanism involves the hydrolysis of the silylated peptides followed by their condensation driven by self-assembly. | ||
The formation of N3H displays the same principle as N3L. Differently, N3H showed a slight turbidity, compared to N3L, a few hours after the initiation of the reaction. This can be explained by the fast self-assembly and condensation due to the high concentration of compound 3. Moreover, when the concentration was above 17.80 mM, the precursors rapidly condensed and generated a gel-like structure and therefore the concentration was adjusted to obtain dispersions rather than gels. As shown in Fig. 2A and G, SEM analysis revealed that N3H and N3L are rod-shaped materials with a range of sizes. In Table 1, DLS analysis demonstrated that the polydispersity index (PDI) for N3H is high. This means that N3H has a broad size particle distribution from nano to micron scale. Although the average size for N3H was 582 ± 19 nm, low-magnification SEM images showed the presence of micron-scale particles (Fig. S5A in the ESI†). N3L had a relatively larger average size than N3H. However, the PDI value was smaller than N3H, which means it has a narrower size distribution.
 | ||
| Fig. 2 Structural characterization of the materials formed by combining self-assembly and sol–gel process. (A) SEM, (B) STEM and (C) AFM representative images of the materials formed by N3H, (D) SEM, (E) STEM and (F) AFM of N4H, (G) SEM, (H) STEM, and (I) AFM representative images of the materials formed by N3L, and (J) SEM, (K) STEM and (L) AFM representative images of the materials formed by N4L. | ||
| Material | Z-Average (nm) | PDI (AU) |
|---|---|---|
| N3H | 582 ± 19 | 0.444 |
| N3L | 843 ± 41 | 0.252 |
| N4H | 612 ± 68 | 0.695 |
| N4L | 768 ± 41 | 0.301 |
Peptide 2 (H-Phe(4-F)-Phe(4-F)-OMe) was synthesized with a purity of 98% (Fig. S2A in the ESI†) and analysed by MS (m/z = 363, (M + H)+ = 362.7) (Fig. S2B in the ESI†). Following the synthesis, peptide 2 was reacted with 2 equiv. of ICPTES to yield compound 4. Compound 4 was characterized by HPLC (Fig. S4A in the ESI†) and its purity was 86%. Once the identity of compound 4 was confirmed with MS (m/z = 525.17), (M + H = 525) (Fig. S4B in the ESI†), it was used for the material synthesis by combining self-assembly and sol–gel process. Two different concentrations of compound 4 were dissolved in a mixture of TDW (HCl, pH 1.5)/THF (1/2, v/v), at 1.78 mM and 17.80 mM. At 1.78 mM, the reaction remained clear for 24 hours suggesting that the sol–gel reaction did not proceed as in the case of N3L. After solvent evaporation, N4L was recovered as a solid precipitate and analysed using STEM. Fig. 2D and J show the formation of ovoid-shaped materials by N4H and N4L. These structures by N4L and N4H are in the nano and micron scale. The particles formed by N4H showed a wide range of size distribution to the extent that SEM images of N4H demonstrated particles from the nanoscale to microscale (Fig. S5B in the ESI†). On the other hand, similar to N3L, N4L has a narrower size distribution with 0.301 PDI.
In nanomaterial synthesis, it is generally expected that as the concentration of the precursor increases, larger particles are obtained.26,27 Although the average size (obtained by DLS) of higher reaction concentrations (N3H and N4H) is relatively smaller than lower reaction concentrations (N3L and N4L), we could not detect a large difference in size using STEM analysis (Fig. 2B, E, H, and K). This can be due to the high polydispersity of the particles that resulted in misleading values by DLS.28 However, it can be noted from the PDI values that lower reaction concentration can provide better size uniformity (Table 1). These results were confirmed by the AFM for N4H and N4L. The nano-scale spheres of N4H aggregated into micron-level agglomerates (Fig. 2F), while N4L created separated and uniform spheres (Fig. 2L). AFM morphology analysis of N3H (Fig. 2C) and N3L (Fig. 2I) were consistent with STEM and TEM images.
ATR-FTIR spectrometry was used to analyse the secondary structures and siloxane bonds. As expected, N3H and N3L have very similar spectra (Fig. 3A). This suggests that the different reaction concentrations did not affect the chemical structure of these materials. The characteristic peak for siloxanes falls within the range between 1000 and 1200 cm−1.29 The band at 1028 cm−1 is associated with linear siloxane bonds.30 Besides, Si–O–Si asymmetric stretching was observed at 1056 cm−1 which is seen in the strained geometry.31 The peaks at 1056 and 1080 cm−1 also represent the Si–O–C vibrations.20,30 In addition, amide II (N–H) at 1560 cm−1 arises primarily from N–H bending.32 The peak at 1632 cm−1 is attributed to amide I (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) in the material33 and also suggests a β-sheet structure.34,35 The distance between amide II and amide I (Δν) is a good indication of the strength of the hydrogen bonds. The lower Δν value demonstrates the stronger hydrogen bonds. In a study that is reported by Jebors et al., Δν is around 90 which addresses the strong hydrogen bonding.20 For N3H and N3L, the Δν value was 72 suggesting the formation of a secondary structure of a parallel β-sheet.36
O) in the material33 and also suggests a β-sheet structure.34,35 The distance between amide II and amide I (Δν) is a good indication of the strength of the hydrogen bonds. The lower Δν value demonstrates the stronger hydrogen bonds. In a study that is reported by Jebors et al., Δν is around 90 which addresses the strong hydrogen bonding.20 For N3H and N3L, the Δν value was 72 suggesting the formation of a secondary structure of a parallel β-sheet.36
 | ||
| Fig. 3 FTIR spectra of (A) N3H (blue) and N3L (red), and (B) N4H (green) and N4L (black). | ||
For N4H and N4L (Fig. 3B), we observed a sharp peak that represents the siloxane bond at 1150 cm−1 and another sharp peak that represents the silica network at 825 cm−1.37,38 Additionally, we observed a band at 1737 cm−1 that represents methyl ester peptide 2. Since peptide 2 is composed of a fluorinated analogue we observe a sharp peak at 1220 cm−1 for the C–F bond on the benzene ring. Differently, N4H and N4L showed a peak at 3305 cm−1 that was attributed to free –OH of hydroxysilanes. Amide II and amide I were visible at 1561 cm−1 and 1633 cm−1, respectively. Same as N3H and N3L, Δν for these materials is 72 which shows strong hydrogen bonds. This also suggests a parallel β-sheet secondary structure in the amide I band at 1630 cm−1 like N3L.36 Antiparallel β-sheets give a small peak at 1694 cm−1 which was not observed for any of the other materials.39 This would be compatible with a mechanism in which parallel β-sheet formation brought into proximity the N-terminals of compounds 3 and 4 allowing them to condense. Additionally, all materials showed common peaks such as ∼2930 and ∼2850 cm−1 for N–H stretching, ∼1450 cm−1 for N–H deformations of urea, and ∼1555 cm−1 for CC from phenyls.
XRD analysis was performed to analyse the phases of the materials. All materials (N3H, N3L, N4H, and N4L) showed a broad diffraction peak centred at 20.4°, typical for amorphous silica.40 See Fig. 4. Differently, we observed diffraction peaks at 20°, 22.5°, and 26.7° for N3L. Although N3L exhibited an amorphous phase, its diffraction peaks at 20°, 22.5°, and 26.7° corresponded to those observed in diphenylalanine peptide assemblies, as reported in a study by Ji et al.41 N4H and N4L showed similar patterns in XRD analysis and they were amorphous.
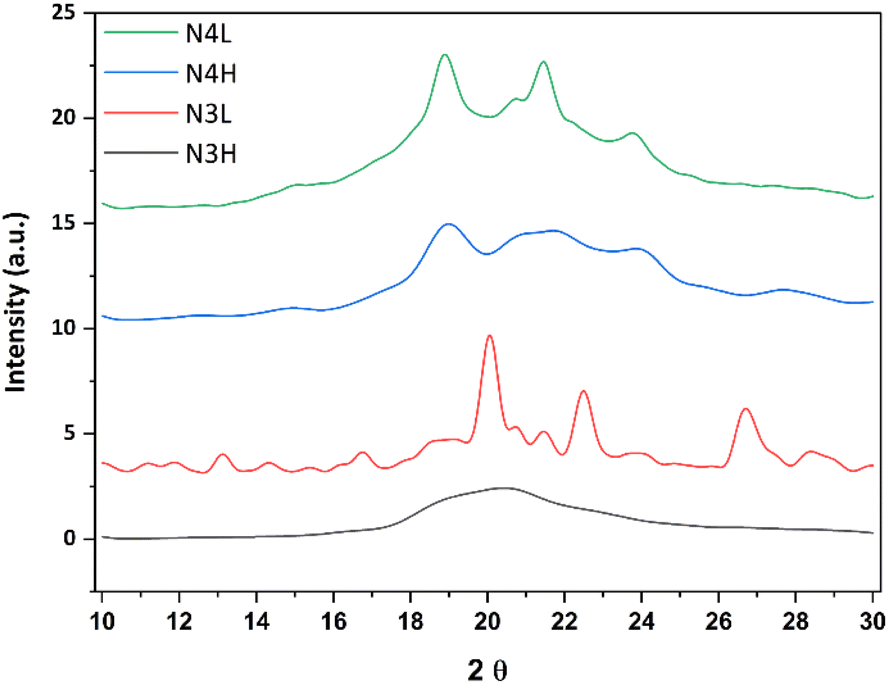 | ||
| Fig. 4 XRD patterns of N3H (black), N3L (red), N4H (blue), and N4L (green). | ||
Conclusions
Peptides provide simple building blocks for generating new materials through a bottom-up approach termed self-assembly. This process yields various architectures by non-covalent interactions formed under mild conditions. These architectures can assemble only under specific conditions and their stability can be disrupted when the conditions change. Here, we demonstrated the combination of self-assembly with another bottom-up approach, sol–gel synthesis. Sol–gel synthesis includes the formation of covalent siloxane bonds that can stabilise the resulting assemblies. We demonstrated the combination of self-assembly and sol–gel synthesis utilising the peptide diphenylalanine and its fluorinated analogue. These two peptides were silylated and used to template the growth of a sol–gel condensation reaction. Rod-shaped and spherical structures were formed by this approach. Their size ranges from the nano to the micron scale. FT-IR spectra of the materials indicated the presence of parallel β-sheet secondary structures and siloxane bond formations. Overall, the combination of these two bottom-up approaches, self-assembly and sol–gel, can provide a novel method for generating stable materials with miscellaneous characteristics in terms of shape, size, and morphology.Data availability
The raw data of this publication are available on the Dataverse website at the link: https://dataverse.unimi.it/dataverse/HUJI-UM-MBK-1. The details of experimental procedures are provided in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project received funding from the European Union’s research and innovation programme under the Marie Sklodowska-Curie grant agreement No. 101072645.References
- M. Vallet-Regí, M. Colilla and B. González, Chem. Soc. Rev., 2011, 40, 596–607 RSC.
- W. Park, H. Shin, B. Choi, W.-K. Rhim, K. Na and D. Keun Han, Prog. Mater. Sci., 2020, 114, 100686 CrossRef CAS.
- X. Zhao, Z. Li, Q. Guo, X. Yang and G. Nie, J. Alloys Compd., 2021, 855, 157480 CrossRef CAS.
- I. Zvonkina and M. Soucek, Curr. Opin. Chem. Eng., 2016, 11, 123–127 CrossRef.
- M. Ş. A. Eren, H. Arslanoğlu and H. Çiftçi, J. Environ. Chem. Eng., 2020, 8, 104247 CrossRef CAS.
- U. Ulusoy, Minerals, 2023, 13, 91 CrossRef CAS.
- L. V. Srinivasan and S. S. Rana, Discover Appl. Sci., 2024, 6, 371 CrossRef CAS.
- J. Wang, K. Liu, R. Xing and X. Yan, Chem. Soc. Rev., 2016, 45, 5589–5604 RSC.
- R. Chang, C. Yuan, P. Zhou, R. Xing and X. Yan, Acc. Chem. Res., 2024, 57, 289–301 CrossRef CAS PubMed.
- C. Yuan, Q. Li, R. Xing, J. Li and X. Yan, Chem, 2023, 9, 2425–2445 CAS.
- F. Fan, X. Chen, J. Lin, M. Lin, L. Li, Y. Gu, Y. Chai, H. Zhang, X. Chen and Q. Li, Adv. Funct. Mater., 2024, 34, 2470125 CrossRef.
- R. Xing, C. Yuan, W. Fan, X. Ren and X. Yan, Sci. Adv., 2023, 9, eadd8105 CrossRef CAS PubMed.
- C. Yuan, W. Fan, P. Zhou, R. Xing, S. Cao and X. Yan, Nat. Nanotechnol., 2024, 19, 1840–1848 CrossRef CAS PubMed.
- D. Mandal, A. Nasrolahi Shirazi and K. Parang, Org. Biomol. Chem., 2014, 12, 3544–3561 RSC.
- G. Ghosh, R. Barman, A. Mukherjee, U. Ghosh, S. Ghosh and G. Fernández, Angew. Chem., Int. Ed., 2022, 61, e202113403 CrossRef CAS PubMed.
- S. Yuran, Y. Razvag and M. Reches, ACS Nano, 2012, 6, 9559–9566 CrossRef CAS PubMed.
- Z. Jin, Y. Li, K. Li, J. Zhou, J. Yeung, C. Ling, W. Yim, T. He, Y. Cheng, M. Xu, M. N. Creyer, Y. Chang, P. Fajtová, M. Retout, B. Qi, S. Li, A. J. O’Donoghue and J. V. Jokerst, Angew. Chem., Int. Ed., 2023, 62, e202214394 CrossRef CAS PubMed.
- S. Maity, S. Nir, T. Zada and M. Reches, Chem. Commun., 2014, 50, 11154–11157 RSC.
- M. Kaganovich, K. Shlosman, E. Goldman, M. Benchis, T. Eitan, R. Shemesh, A. Gamliel and M. Reches, Microbe Killer Polymeric Films Made by Melt-Compounding and Compression of Peptide Assemblies and Polyethylene, Research Square, 2022, preprint, DOI:10.21203/rs.3.rs-1710346/v1.
- S. Jebors, L. Valot, C. Echalier, B. Legrand, R. Mikhaleff, A. Van Der Lee, R. Arenal, P. Dumy, M. Amblard, J. Martinez, A. Mehdi and G. Subra, Mater. Horiz., 2019, 6, 2040–2046 RSC.
- M. C. Mañas-Torres, G. B. Ramírez-Rodríguez, J. I. García-Peiro, B. Parra-Torrejón, J. M. Cuerva, M. T. Lopez-Lopez, L. Álvarez De Cienfuegos and J. M. Delgado-López, Inorg. Chem. Front., 2022, 9, 743–752 RSC.
- S. Jebors, S. Cecillon, C. Faye, C. Enjalbal, M. Amblard, A. Mehdi, G. Subra and J. Martinez, J. Mater. Chem. B, 2013, 1, 6510 RSC.
- M. Catauro and S. V. Ciprioti, Materials, 2021, 14, 1788 CrossRef CAS PubMed.
- D. Desai, D. S. Karaman, N. Prabhakar, S. Tadayon, A. Duchanoy, D. M. Toivola, S. Rajput, T. Näreoja and J. M. Rosenholm, Open Mater. Sci., 2014, 1 DOI:10.2478/mesbi-2014-0001.
- D. Bokov, A. Turki Jalil, S. Chupradit, W. Suksatan, M. Javed Ansari, I. H. Shewael, G. H. Valiev and E. Kianfar, Adv. Mater. Sci. Eng., 2021, 2021, 5102014 CrossRef.
- S. Stopic, F. Wenz, T.-V. Husovic and B. Friedrich, Metals, 2021, 11, 463 CrossRef CAS.
- M.-Z. Yu, J.-Z. Lin and T.-L. Chan, Chem. Eng. Sci., 2008, 63, 2317–2329 CrossRef CAS.
- E. Tomaszewska, K. Soliwoda, K. Kadziola, B. Tkacz-Szczesna, G. Celichowski, M. Cichomski, W. Szmaja and J. Grobelny, J. Nanomater., 2013, 2013, 313081 CrossRef.
- T. Hayeri and V. Mannari, J. Coat. Technol. Res., 2025, 22, 225–237 CrossRef.
- M. Masmoudi, C. Rahal, M. Abdelmouleh and R. Abdelhedi, Appl. Surf. Sci., 2013, 286, 71–77 CrossRef CAS.
- H. Mori, Y. Miyamura and T. Endo, Langmuir, 2007, 23, 9014–9023 CrossRef CAS PubMed.
- N. Ardila, F. Daigle, M.-C. Heuzey and A. Ajji, Molecules, 2017, 22, 100 CrossRef PubMed.
- Z. Ma, Y. Hong, D. M. Nelson, J. E. Pichamuthu, C. E. Leeson and W. R. Wagner, Biomacromolecules, 2011, 12, 3265–3274 CrossRef CAS PubMed.
- G. Zandomeneghi, M. R. H. Krebs, M. G. McCammon and M. Fändrich, Protein Sci., 2004, 13, 3314–3321 CrossRef CAS PubMed.
- J. P. Lomont, J. S. Ostrander, J.-J. Ho, M. K. Petti and M. T. Zanni, J. Phys. Chem. B, 2017, 121, 8935–8945 CrossRef CAS PubMed.
- L. P. DeFlores, Z. Ganim, R. A. Nicodemus and A. Tokmakoff, J. Am. Chem. Soc., 2009, 131, 3385–3391 CrossRef CAS PubMed.
- A. Issa and A. Luyt, Polymers, 2019, 11, 537 CrossRef PubMed.
- Y.-S. Li and A. Ba, Spectrochim. Acta, Part A, 2008, 70, 1013–1019 CrossRef PubMed.
- K. Zhaliazka and D. Kurouski, ACS Chem. Neurosci., 2022, 13, 2813–2820 CrossRef CAS PubMed.
- P. Lu and Y.-L. Hsieh, Powder Technol., 2012, 225, 149–155 CrossRef CAS.
- W. Ji, Y. Tang, P. Makam, Y. Yao, R. Jiao, K. Cai, G. Wei and E. Gazit, J. Am. Chem. Soc., 2021, 143, 17633–17645 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5fd00014a |
| This journal is © The Royal Society of Chemistry 2025 |