 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploiting nanoprobe X-ray techniques for imaging of biomineralisation; chemical, structural and in situ opportunities
Jessica M. Walker a,
Miguel A. Gomez-Gonzaleza,
Johannes Ihlib and
Julia E. Parker*a
a,
Miguel A. Gomez-Gonzaleza,
Johannes Ihlib and
Julia E. Parker*a
aDiamond Light Source, Diamond House, Harwell Science and Innovation Campus, Didcot, Oxfordshire OX11 0DE, UK. E-mail: julia.parker@diamond.ac.uk
bDepartment of Materials, University of Oxford, Parks Road, Oxford, OX1 3PH, UK
First published on 1st May 2025
Abstract
Advances in X-ray nanoprobe beamlines at synchrotrons across the world present exciting opportunities for rich multimodal imaging of biomineral structures and their formation processes. The combination of techniques provides a sensitive probe of both chemistry and structure, making X-ray nanoprobes an important tool for investigating crystallite growth and orientations, interfaces, and assembly of building blocks into hierarchical structures. A discussion of these capabilities is presented with reference to recent examples using a range of nanoprobe imaging techniques for investigating enamel structure, as well as coccolith properties. Key opportunities for the use of X-ray nanoprobes lie in exploiting the penetrating power and coherence properties of synchrotron X-rays in order to image in situ processes or apply coherent diffractive imaging techniques to obtain higher resolutions. To this end initial results demonstrating the observation of calcium phosphate mineralisation, in a liquid environment, using nano-X-ray fluorescence mapping are presented, and the role of X-ray dose and beam induced effects is considered. Finally novel results from tomographic ptychography imaging of Mytilus edulis mussel shell calcite prisms are discussed, where the segmentation of the phase density into organic and mineral content gives insights into the mechanisms underlying mineral prism formation and the role of the organic matrix in biomineralisation.
1. Introduction
Biominerals are inorganic materials synthesised by living organisms, composed of a wide range minerals such as calcium carbonate, hydroxyapatite and silica. From the shells of marine molluscs to the bones of vertebrates, biominerals demonstrate remarkable diversity in composition and morphology, serving essential structural, protective, and functional roles.1,2 The composite nature of biominerals, comprising both mineral crystalline phase and organic matrix, imparts unique properties, such as the mechanical strength and fracture toughness of mussel shell when compared to synthetic equivalents.3–5Organisms are able to exert precise control over the mineralisation process, controlling the crystal phase and polymorph formed (even switching between polymorphs in a single structure), and the resulting structure and morphologies. It is proposed that this control occurs through; regulating of the chemical environment around cells; mediating ions and precursor availability; controlling pH levels; confinement of processes; and the presence of organic matrices which can act as nucleation and ion binding sites.6,7 Observations of biomineralisation processes, and the identification of stable amorphous precursor phases in biominerals, has contributed to a plethora of discussions about non-classical crystallisation pathways, encompassing prenucleation clusters, orientated attachment and mesocrystallinity.7,8 Hence, there continues to be much interest in imaging mineralisation processes, across multiple lengths and timescales, in order to gain further insights into the underlying mechanisms.
This fine control over mineralisation is often exhibited as exquisite structures and forms, from coccolithophore calcite formation to the spiral of the narwhal tusk, with highly organised hierarchical structures.9 This hierarchical structure contributes to the superior mechanical properties and functionality of biominerals. Understanding the organisation of biominerals, across the different structural levels, is therefore important for a thorough understanding of formation processes, properties, and development of biomimetic approaches for materials development.10
The structural complexity of biominerals presents significant challenges for characterisation. Understanding both structure and formation of these complex materials requires a range of characterisation and imaging techniques across multiple length scales and modalities. Here X-ray imaging can be used to complement other microscopies (such as light, Raman and electron microscopy) and provide a non-destructive means of probing the structure and composition of biominerals.
With focussed X-ray beam sizes of 25–100 nm, combined with the increased spatial resolution achievable through coherent diffractive imaging techniques such as ptychography, the experimental envelope of X-ray nanoprobes bridges the resolutions and fields of view between transmission electron microscopies and microscale techniques, whilst the penetrating power of hard X-rays increases the availability of options for in situ imaging studies. X-ray nanoprobes allow a combination of techniques including nano-X-ray fluorescence (n-XRF), spectroscopy, nano-X-ray diffraction (n-XRD), and advanced phase imaging, which can be applied to biomineralisation studies, revealing details of nanoscale elemental compositions, structures and morphologies.11
A key advantage of using X-rays is the ability to penetrate ‘thicker’ sample environments in order to image in situ processes.12–14 Of particular interest for biomineralisation is the ability to observe crystallisation processes and kinetics. Here, the design challenges of in situ liquid cells are discussed, and initial results demonstrating the observation of calcium phosphate mineralisation in a liquid environment, using n-XRF mapping are presented. These results illustrate the potential advantages and role of in situ nanoprobe techniques for mineralisation studies and highlight key aspects for future development and study. These include the role played by X-ray dose and beam induced effects, an important consideration when designing experimental approaches and interpreting results which has not yet been widely studied for X-ray nanoprobe in situ experiments.
The final factor in exploiting X-ray nanoprobes for biomineral imaging lies in the direction of applying coherent imaging techniques. Tomographic ptychography, a scanning coherent diffractive imaging technique, enables quantitative 3D visualisation of electron density, making it highly suited for exploring the impact of organic–mineral interfaces on structure and properties. Novel results from tomographic ptychography imaging of a Mytilus edulis mussel shell calcite prism are discussed, where the nanoscale segmentation of the phase density into organic and mineral content gives insights into the mechanisms underlying mineral prism formation and the role of the organic matrix in biomineralisation.
2. Hard X-ray nanoprobe imaging
X-ray nanoprobe beamlines are found at most synchrotrons worldwide,15–21 often forming a central role in facilities' upgrade programs as they are well positioned to exploit the increased brilliance and coherence of a fourth generation light source. In the last 10–15 years advances in X-ray optics have led to ever decreasing beam sizes, and hence higher image resolutions. Beamlines use highly specialised optics (including zone plates, Kirkpatrick–Baez mirrors, multilayer Laue lens) where focusing to sub 10 nm X-ray beam sizes have been demonstrated,22 and 10–50 nm are routinely achieved23–25 at operational beamlines, which, combined with high stability environments and precision scanning stages, make a unique probe for examining elemental, chemical and structural information of materials.Nanoprobes are multi-modal probes, allowing simultaneous imaging using a combination of different techniques, at the same resolution, on the same sample. X-ray fluorescence (XRF) enables the mapping of elemental distributions in a sample, providing information on material compositions and heterogeneity. Phase contrast imaging techniques (such as differential phase contrast imaging, DPC)26 gives information on the sample morphology and density. This is often crucial in biological samples to provide a context for the elemental XRF maps, where lighter elements (including carbon, nitrogen and oxygen) are not observed. Thus a combination of DPC and XRF imaging can provide simultaneous imaging of the cellular structures and the distribution of metal nanoparticles or metallo–organic drugs, for example when investigating treated cancer cells.27,28 X-ray diffraction (XRD) probes the crystalline, atomic structure of the materials, and can be used to fingerprint the presence of different phases, such as mineral distributions in samples or, with a more detailed analysis, map lattice strain variations.
By repeating n-XRF maps at different energies the technique can be extended to give X-ray Absorption Near Edge Structure (XANES) spectroscopy maps (nano-XANES). 2D n-XRF maps are collected on a region at different energies through an elemental absorption edge. The elemental maps extracted from the XRF spectra at each energy are aligned and stacked to give a 3D image stack where each pixel (x, y) has a third coordinate, energy. The nano-XANES can be analysed, typically through principal component and cluster analysis, to group the spectra based on spectral similarities followed by fitting to reference data, to extract amounts and locations of distinct species. Repeating scans for different energies increases substantially the time and X-ray dose required to image a sample, limiting the use of this technique for in situ or biological samples. The X-ray dose and time can be reduced by the application of a variety of approaches, including sparse energy sampling29 or undersampling the raster scans.30 Implementation of these options begins to open the possibility of applying nano-XANES approaches to beam and time sensitive experiments.
The coherent nature of nanoprobe X-rays allows the application and development of coherent diffraction imaging techniques such as ptychography.31 In ptychography, far field diffraction patterns are collected at a series of overlapping scan positions. An iterative phase retrieval algorithm is used to recover the phase and amplitude of the scattered wave, reconstructing both the phase and adsorption image of the sample. Importantly, the image resolution is not limited by the size of the X-ray beam, offering the possibility of achieving higher resolution images than probe-limited techniques such as DPC.
These techniques can be extended to three dimensions through tomography – collecting maps or projections at a series of different angles as the sample is rotated and reconstructing to form a 3D volume. Current challenges for the implementation of tomographic techniques at nanoprobes centre around sample preparation and speed of acquisition. The demands of sample preparation, to produce a 10–20 μm pillar, extracted from a specific region in a larger sample and mounted on a pin, require specialised approaches, such as FIB-SEM, and dedicated time and expertise. Smaller samples are currently required in order to keep the sample in the focus of the beam and reduce the inherent scanning time limitations. In most cases the limitation on scanning speed is not a flux but rather a hardware, controls and detector limitation. However, the desire to extend to larger volume imaging means an increase in scanning speeds is a current development push.32 Overall, combining information from different X-ray modalities, through for example correlative ptychographic tomography and n-XRF, or n-XRF and n-XRD, is a powerful approach for imaging a wide range of materials, from biological cells,33 to catalyst34 and battery35 particles, and historic paint samples.36
2.1 I14 – Diamond's hard X-ray nanoprobe
Beamline I14 is the hard X-ray nanoprobe beamline at Diamond Light Source.16 Fig. 1(a) outlines the beamline components; the beamline is 185 m long from the source (a U22 undulator) to the sample position with the major optical components inside the main synchrotron ring building. The first mirror horizontally collimates the beam onto the secondary source aperture, vertically the source is imaged directly. The beam is monochromated by a Si (111) horizontal bounce double crystal monochromator, to tune the X-ray energy within the range 5–20 keV.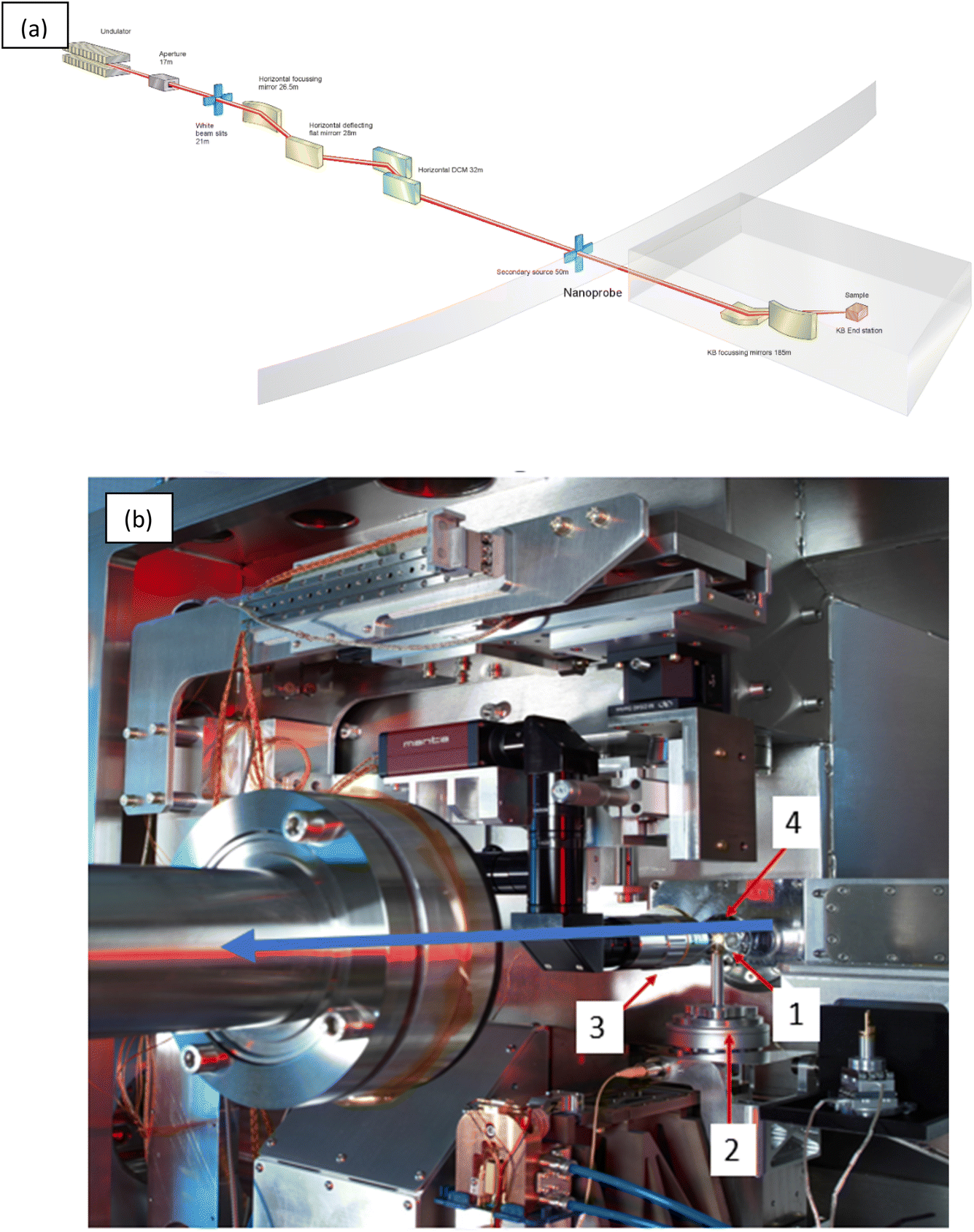 | ||
| Fig. 1 (a) A schematic of the I14 hard X-ray nanoprobe beamline layout showing the major components. (b) Inside the I14 hard X-ray nanoprobe experimental hutch showing the endstation, the blue arrow indicates the X-ray beam direction. 1–4 show the position of the (1) sample mounting position, (2) scanning and rotation stages, (3) sample alignment microscope, and (4) four-element silicon drift detector (XRF detector). | ||
The experimental hutch is located in a purpose built external building. At the endstation, Fig. 1(b), the X-ray beam is focused to 50 nm at the sample position (1) using a pair of in vacuum nano focusing KB mirrors. The sample, typically on a silicon nitride membrane window, or TEM grid, is mounted onto a magnetic holder and placed in the sample position (1). The sample spigot is attached via a kinematic mount to the scanning stages (2). The kinematic mounting system, combined with the long working distance focus of the KB optic, and in air environment, provides a flexible space for incorporating bespoke sample mounting solutions and a variety of in situ environments and facilitating easy changeover of experimental setups.
The scanning stage is a novel stage design concept, a Delta Robot.37 The delta robot uses three orthogonal voice coils, which actuate parallelogram flexures for a 3 mm travel range in x, y and z directions. The design enables high speed scanning at nm precision over mm travel ranges, with space and load capacity for flexible setups and sample environments. On top of the delta robot, underneath the kinematic mount, is a rotation stage to enable tomography measurements.
Once mounted a sample is aligned to the beam focus and region of interest found using the retractable sample alignment microscope (3). XRF data are collected in backscatter geometry using a four-element silicon drift detector (4). Downstream of the sample scattered or transmitted X-ray can be collected for DPC, ptychography or XRD analysis using silicon pixelated photon counting detectors; a quad merlin (Quantum Detectors), Excalibur 3M or an Eiger 500K (Dectris). These detectors are mounted on independent translation rails on a movable granite table to facilitate alignment and changeover between detector distances and setup – e.g. for ease of switching between DPC and XRD data collections in one experiment. Setup up of scanning regions (maps), and viewing of live data collections, is facilitated through an intuitive interface to the beamline controls system using a mapping GUI in the data acquisition system.
The capability of X-ray nanoprobe beamlines to collect correlative data across different modalities, across three dimensional and in situ experiments, and developments in faster scanning, present challenges in data processing and analysis. I14 has therefore placed a great deal of emphasis on ease of experiment and automated data processing pipelines, to enable live processing on Diamond's high performance computing cluster during a scan of; elemental maps from XRF spectra; processing of DPC data to give a quantitative phase image; stacking and alignment of XANES data; integration of 2D XRD images to 1D patterns and automated submission of ptychography reconstructions. This is complemented by offline processing tools using a web GUI for workflow submissions.
2.2 Hard X-ray nanoprobe imaging of biomineralization at I14
These capabilities, combined with the beamline's flexibility, make I14 a powerful tool for the imaging of biominerals. This strength can be further exploited by combining with complementary techniques, such as electron microscopy for a multi length scale imaging approach.X-ray nanoprobe measurements at I14 were combined with soft X-ray ptychography, electron microscopy and larger scale X-ray tomography to investigate the chemical and structural properties of carious enamel.38,39 The hierarchical structure of human dental enamel has orientated hydroxyapatite (Hap) crystals arranged into rod and interrod regions where the nanoscale arrangement contributes to the mechanical strength of the enamel.40 Demineralization during caries formation leads to chemical and structural changes which reduce the stiffness and strength of the enamel. Besnard et al.39 investigated these changes in FIB-lamellae extracted from locations in tooth enamel corresponding to normal and demineralised areas. Using a I14, n-XRF mapping revealed calcium chemical gradients across the rod and interrod areas corresponding to different demineralization rates. The chemical information was correlated with crystal orientations extracted from n-XRD measurements and structural changes observed in DPC. Higher resolution images of the same sample using soft X-ray tomography (at Diamond's I08 beamline) allowed alignment of the n-XRF maps to start to reveal details of crystallite orientation gradients within the enamel.
Coccolithophores play an important role in global biogeochemical cycles.41 They produce a calcium carbonate exoskeleton, composed of coccoliths. Coccoliths are arrays of nanoscale crystals that form intracellularly, a key example of the fine control biomineralising organisms exhibit over the chemistry and morphology of their mineral phases. Understanding their formation necessitates information on elemental distribution and speciation at a nanoscale level. n-XRF tomography of a coccolith (the lopadoliths of Scyphosphaera apsteinii) at I14 revealed an uneven distribution of strontium,42 with stripes of different concentrations, as shown in Fig. 2. This is in contrast with current Sr fractionation models which predict an even distribution. The n-XRF tomography was complemented by nano-XANES analysis of the same sample to show that Sr resides in a Ca site in the calcite lattice in both high and low Sr stripes, confirming a central assumption of current Sr fractionation models.
 | ||
| Fig. 2 3D volume renderings of the reconstructed 3D XRF data for (a) calcium and (b) strontium signals individually and (c) combined (calcium signal in red, strontium signal in green), showing both the full image of the lopadolith and a cut through along the growth direction. The corresponding reconstruction slices are also shown (d). Reproduced from ref. 42 with permission from the Royal Society of Chemistry. | ||
Nano-XANES at I14 has also been used, in combination with DPC and nano-XRF, to investigate magnetosome formation at the single-cell level in magnetotactic bacteria (MTB).43 MTB synthesize single-domain magnetic nanoparticles composed of magnetite (Fe3O4) or greigite (Fe3S4) within organelles known as magnetosomes, yet the mechanisms of biomineralization are still unclear. Recent work suggests a large fraction of intracellular iron, at least partly composed of ferrous species, is not incorporated into magnetosomes.44 Wild-type M. gryphiswaldense MSR-1 bacteria and a genetic variant lacking ferritin proteins were studied under varied iron concentrations and at different stages of magnetosome formation. DPC and nano-XRF mapping was first used to reconstruct both organic and inorganic components of bacterial cells to identify intracellular regions. Fe K-edge nano-XANES was then used to differentiate magnetosome particles from other intracellular iron species and determine their relative amounts, Fig. 3, confirming a significant presence of intracellular iron species distinct from magnetite during biomineralization.
 | ||
| Fig. 3 Nano-XANES data for MTB with complete magnetosome chains. (A and F) WT-high-iron and WT-low-iron nano-XANES regions (white box), (B and G) PCA maps (red signal), (C and H) maps of cluster centers, (D and I) normalized offset Fe K-edge XANES spectra (inset with normalized spectra retaining edge jump values), and (E and J) a summarized composite of LC fitting results. Reproduced from ref. 43 under creative commons CC BY 4.0 licence. | ||
3. A liquid cell in situ study of calcium phosphate precipitation
The I14 endstation was designed to have flexibility to mount a variety of different in situ sample environments, to be able to incorporate thicker or larger sample environments; hard X-rays are able to penetrate much thicker materials than electrons or soft X-rays. However, the thickness of a sample or in situ cell has an impact on the achievable resolution of the technique, as the beam is scattered through more sample material or the sample thickness exceeds the depth of field of the X-ray focus. The design of a sample environment must also be carefully considered so as not to import drift or vibrations to the measurement, the design parameters being similar to those of environments developed for TEM in situ studies. Given the complementarity of nanoprobe and TEM techniques and the desire for correlative approaches, it is possible to incorporate TEM sample environments into X-ray nanoprobes.12–14 I14 have developed mounts to hold commercial TEM MEMS chip devices, allowing the easy use of existing systems for both gas and liquid flow. A schematic of liquid cell chips made up of 2 silicon nitride windows to form a sandwich structure is shown in Fig. 4(a). To avoid window bulging and thick liquid layers preventing transmission of electrons, TEM liquid cell chips have small (50 × 50 μm) window areas and thickness spacings of 200 nm or less. When considering crystallisation experiments this presents a confined environment which may influence system kinetics through limited diffusion of ions or on morphologies of crystals formed. In this work we were able to relax these confinement effects to some extent by using chips with larger windows and a much thicker (8 μm) spacing. Assembled chips are mounted in a holder, either the removeable tip piece of a TEM holder (DENS systems) or a beamline mount (Protochips), on a custom kinematic mounted plate on the I14 stages, Fig. 4(b). | ||
| Fig. 4 (a) Sketch of MEMS liquid nanochip assembly showing the (left) bottom and top chip silicon nitride windows (perpendicular to each other), when assembled and mounted on the beamline the window area can be observed in the sample microscope (right). The windows are 50 μm in width. (b) Assembled chip mounted in a liquid cell on I14 endstation. | ||
3.1 Materials and methods
A calcium phosphate (CaP) mineralisation solution was prepared by mixing equal volumes of a Ca solution (1.7 mM CaCl2 in 125 mM NaCl, 50 mM Tris, pH 7.40) and phosphate solution (9.5 mM Na2HPO4 in 125 mM NaCl, 50 mM Tris, pH 7.40). The same solution conditions were used by Wang et al.45 All reagents were purchased from Sigma-Aldrich and dissolved in deionised water. After mixing, the CaP solution was immediately loaded into a 1 ml glass syringe and a syringe pump used to flow the solution through the assembled liquid cell. The Ca-Kα fluorescence signal was monitored using n-XRF and once signal was observed in the cell window area flow was stopped and n-XRF mapping commenced.Repeat n-XRF maps were collected on beamline I14, Diamond Light Source16 using a 50 nm focused X-ray beam at an energy of 12 keV. XRF spectra were collected using a 4 element silicon drift detector in backscatter geometry. Ca elemental maps were produced by integrating the intensity in the XRF spectra over a 200 eV window around the Ca-Kα fluorescence peak (3.6 keV). The P signal can be mapped from the same spectra, however due to the lower X-ray energy and the absorption by the liquid and air path the signal is much weaker and so is not presented here, although the distributions match those of Ca.
3.2 In situ crystallisation results and discussion
Fig. 5 shows a time series of Ca maps taken over 3 hours. After 1 hour, the CaP appears to have formed an extended network of small nanoscale particles (it is possible these are clusters of ions or amorphous precursors, or crystallites). Over a further 1–2 hours these can be seen to transform and growth of denser larger particles occurs. After 3 hours, additional smaller crystals can be seen to form at the bottom left of the field of view. Integrating the total Ca signal intensity in the two regions outlined in Fig. 6(a) for an area where these denser particles formed and one where it did not, show that the intensity in Region 1 increased sharply 100 minutes after mixing, see Fig. 6(b). The intensity in Region 2 does not change, suggesting that the CaP in these regions is not being redissolved in order to precipitate the larger structure formation observed in Region 1; diffusions of ions over larger distances must be occurring. | ||
| Fig. 5 Ca-Kα fluorescence maps extracted from n-XRF spectra at different time points during in situ CaP crystallisation in a liquid cell. Scale bar = 500 nm. | ||
 | ||
| Fig. 6 (a) Ca-Kα XRF map indicating the location of Region 1 (white box) and Region 2 (red box) (b) the total Ca signal in Region 1, Region 2 and the whole mapped area as a function of time. | ||
The total Ca signal intensity in the entire mapped area decreased over time and we note that after 24 hours, the formed crystals in the field of view dissolved, and other larger crystals were found nearby when scanning nearby areas – raising the question of the role of beam effects in the dissolution process. Existing models of X-ray dose effects on liquids suggest that X-ray exposure can contribute to radiolysis of the solution, and generate radicals which change the local solution pH. Over time – and in the absence of flow – dispersion of these species is diffusion limited, so effects will accumulate.
These initial results can be compared to a TEM in situ liquid cell study of CaP crystallisation.45 As the same solution concentrations were used, similar kinetics might therefore be expected in the absence of beam induced effects and any confinement effects due to the different cell geometries. Wang et al. observed branched morphologies after 1 hour, these appear to be very similar in nature to the structures observed here.45 It is clear however that in situ TEM offers improved resolution, both spatial and temporal, above these initial nanoprobe measurements for imaging biomineral formation mechanisms.46–49 However, we suggest that they can be complementary in a multiscale approach; access to larger fields of view, as well as the chemical information that can be extracted, is important for examining at the mesoscale the assembly into hierarchically ordered structures. The greater flexibility offered in terms of sample environments opens interesting avenues in terms of customised microfluidic cells for controlling crystallisation, and bridging the gap between TEM and microscale microscopy techniques.
In order to progress from these initial results an improved understanding, and mitigation, of beam effects is required, building on ongoing modelling for electron dose effects including consideration of scanning paths, damage models and propagation. Improvements in speed can also be achieved by various routes, for example by continued application and development of sparse sampling approaches.29,30
4. A ptychographic tomography study of mussel shell calcite prisms
Mussel shell has a structural toughness which serves to protect them from prey. Shells consist of three layers, from the inner nacreous layer, the prismatic layer and the outer periostracum organic layer. The structural toughness arises from a combination of the different microstructures of the layers and their thickness.50,51 In the prismatic layer, calcite prisms, surrounded by an interprismatic organic matrix, are orientated at an angle to the shell surface. Here we investigate the internal structure of a calcite prism, using ptychography, in order to determine the relationship between the mineral phase and organic component.4.1 Materials and methods
Sections of Mytilus edulis shell, approximately 10 × 10 mm, selected from regions where no, or only a very thin, nacreous layer was observed, were washed in deionised water and the outer organic periostracum removed. Sections were soaked in 4% NaOCl solution for 96 hours in order to dissolve the interprismatic organic matrix and separate individual calcite prisms. Once separated the solution was filtered and the isolated prisms washed with DI water. Prisms were imaged using a SEM (Hitachi Tabletop TM 1000). Isolated prisms were selected using optical microscopy and mounted on a cSAXS OMNY tomography pin.52Ptychographic X-ray tomography experiments were carried out at the cSAXS beamline of the Swiss Light Source.53 The photon energy was 6.2 keV. A Fresnel zone plate 220 μm in diameter with an outermost zone width of 60 nm was coherently illuminated, and the sample placed 1.2 mm downstream of the zone plate focal point. The sample was mounted in the in vacuum cryo-ONMY scanning instrument at −180 °C and scanned through the X-ray beam with a Fermat spiral covering 20 × 10 μm with a step size of 1.3 μm. Far-field coherent diffraction patterns were acquired with exposure times of 0.1 s per point using a Pilatus detector with 172 μm pixel size at a sample-detector distance of 7.342 m. A flight tube between sample and detector was used to reduce air scatter and absorption effects. 1000 angular tomography projections were acquired.
Ptychographic reconstructions were obtained using the difference map algorithm,54 with maximum-likelihood refinement.55 Tomographic reconstruction of the phase images was carried out following alignment using a modified filtered back-projection algorithm as described by Guizar-Sicairos.55
The X-ray dose imparted to a prism sample during tomogram acquisition was estimated to be on the order of 108–109 Gy. The estimated dose is based on the average area flux density of each scan and the assumed mass density of the specimen, assuming a composition of calcite and chitin.
The half-period spatial resolution of ptychographic tomograms was estimated by Fourier shell correlation (FSC). The threshold criteria for the FSC was the ½ bit criteria. From this analysis the resolution is estimated to be 75 nm.
4.2 Ptychographic tomography results and discussion
SEM images of calcite prisms extracted from Mytilus edulis shell are shown in Fig. 7. It can be seen that there exist what appear to be elongated isolated crystals, of needle-like shape. This is in agreement with previous results which demonstrate the shell prismatic layer consists of an array of crystallites, oriented along their long axis, within an organic matrix. Individual prisms are of the order 40 μm long and 5 μm diameter. Larger aggregates can also be found, presumably where the treatment to remove interprismatic organics has not fully removed the organic component. Isolated prisms, similar to those shown in Fig. 7, were selected for imaging using ptychographic tomography. | ||
| Fig. 7 SEM image of calcite prisms from M. edulis after dissolution of interprismatic organic matrix. Scale bar 10 um. | ||
Images from the ptychographic tomography reconstruction of the Mytilus edulis calcite prism are shown in Fig. 8(a)–(d). What was assumed to be a single prism here when mounting is in fact multiple smaller prisms, with their longitudinal axis co-aligned with the long axis of the larger needle-shaped prism being imaged. In contrast to micro- X-ray CT results examining the grain growth of Pinna nobilis calcite prisms,56 where each prism was observed to be a regular polygonal shape, these individual smaller prisms are much more irregular, Fig. 8(c) and (d). This may be due to interspecies variation (Pinna nobilis prisms are much larger than those of Mytilus edulis), the region of the shell from which the prisms were extracted (e.g. fresh growth vs. fully formed crystals) or the differing resolutions and length scales accessible via the different imaging techniques.
 | ||
| Fig. 8 Segmentation of the 3D volume reconstruction to show (a) and (b) the outermost 100 nm, (c) and (d) the rest of the prism, (e) the histogram of electron density for the prism and outermost 100 nm, and (f) the variation in electron density with distance from the centre of a prism. (g) and (h) Volume segmentation of the voxels where the organic content was calculated to be 55–75%. | ||
The 3D volume was segmented to separate the intensity based on the outermost 100 nm of each prism (Fig. 8(a) and (b)) compared to the internal prism structure. This reveals the intricate web of organic matrix around each individual prism, in agreement with electron microscopy studies that have imaged these prismatic envelopes in Pinna nobilis and Atrina rigida prismatic layers.57,58 Here we are able to observe them in 3D without prior dissolution of the mineral component, this enables visualisation of the close link between organic and mineral components at the outer regions of the prisms.
An advantage of ptychography is that the measured intensity corresponds to a quantitative electron density, this is shown for the internal prism and outermost 100 nm in the histogram in Fig. 8(e). Comparing to the known electron densities for chitin and calcite suggest the outermost layer is a good match to the electron density of chitin. The mineral phase of the prisms shows however a lower electron density than calcite, which is presumably due to differences in crystallinity and packing density, hydration and the presence of water as well as organics in the crystal structure.59
The electron density can be plotted with distance from the centre of a prism, as an average across the prisms imaged, Fig. 8(d). In addition to the higher organic content at the outer 100 nm, it can be seen that the inner 300 nm core of the prisms has a much lower electron density. The organic content of each voxel can be estimated using the known electron densities; segmentation based on 55–75 vol% organic is shown in Fig. 8(e) and (f). The presence of an organic rich core running through the centre of each prism can be clearly seen.
The observations of an organic web-like prismatic envelope, and a central organic core is suggestive of a formation mechanism in which calcite nucleation occurs within a dense organic fluid precursor phase, with the crystal growing to fill the space defined by the prismatic envelope, resulting in a calcite prism with high incorporated organic content. This is in broad agreement with existing results observing the organic web and a dense intra-crystalline network of chitin within Atrina rigida calcite prisms.58,60
5. Conclusions
The advancements in X-ray nanoprobe beamlines have opened new frontiers for multimodal imaging of biomineral structures, offering insights into their formation and hierarchical organisation. By leveraging the combination of X-ray fluorescence, diffraction, and ptychographic techniques, researchers can probe both the chemical composition and structural arrangement of biominerals at the nanoscale. A particularly promising direction lies in the in situ imaging of mineralisation processes, where the penetrating power of synchrotron X-rays enable real-time observations. Initial experiments using n-XRF mapping to monitor calcium phosphate mineralisation in a liquid environment demonstrate the feasibility of tracking biomineralisation dynamics with high spatial resolution. However, careful consideration of X-ray dose effects remains critical to minimize beam-induced artifacts and further developments to improve speeds and optimise scanning pathways are required.Tomographic ptychography has provided novel insights into the formation mechanisms of Mytilus edulis calcite prisms, revealing the distinct contributions of organic and mineral components in biomineralization. These findings underscore the potential role of X-ray nanoprobes in biomaterials research, paving the way for future studies that exploit higher resolution imaging and in situ methodologies to deepen our understanding of biomineral formation processes.
Data availability
Data is available from the corresponding author on request.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We would like to thank Dave Mahoney and Mark Hooper for their design and technical efforts to enable the in situ cell mounting and integration. Gea van de Kerkhof is acknowledged for her essential contributions to the liquid cell development work and experimental procedures. Diamond Light Source is acknowledged for beamtime on I14 under proposal mg30893 for the in situ liquid cell studies. For the ptychography tomography Aaron Parsons is thanked for advice and support with the planning and experiment and the assistance of the cSAXS team at the Swiss Light Source is gratefully acknowledged, in particular Klaus Wakonig for experiment and data reconstruction, and Mirko Holler and Ana Diaz for OMNY setup.References
- H. A. Lowenstam and S. Weiner, On Biomineralization, Oxford University Press, New York, 1989 Search PubMed.
- S. Mann, Biomineralization, Principles and Concepts in Bioinorganic Materials Chemistry, Oxford University Press, 2001 Search PubMed.
- A. P. Jackson, J. F. Vincent and R. M. Turner, The mechanical design of nacre, Proc. Roy. Soc. Lond. B Biol. Sci., 1988, 234(1277), 415–440 CrossRef.
- J. D. Currey, The effect of drying on the strength of mollusc shells, J. Zool., 1979, 188, 301–308 CrossRef.
- H. Gao, B. Ji, I. L. Jäger, E. Arzt and P. Fratzl, Materials become insensitive to flaws at nanoscale: Lessons from nature, Proc. Natl. Acad. Sci. U. S. A., 2003, 100(10), 5597–5600 CrossRef CAS PubMed.
- K. Kahil, S. Weiner, L. Addadi and A. Gal, Ion Pathways in Biomineralization: Perspectives on Uptake, Transport, and Deposition of Calcium, Carbonate, and Phosphate, J. Am. Chem. Soc., 2021, 143(50), 21100–21112 CrossRef CAS PubMed.
- P. U. P. A. Gilbert, et al., Biomineralization: Integrating mechanism and evolutionary history, Sci. Adv., 2022, 8, eabl9653 CrossRef CAS PubMed.
- M. Jehannin, A. Rao and H. Cölfen, New Horizons of Nonclassical Crystallization, J. Am. Chem. Soc., 2019, 141(26), 10120–10136 CrossRef CAS PubMed.
- J. Seto, Y. Ma, S. A. Davis, F. Meldrum, A. Gourrier, Y. Y. Kim, U. Schilde, M. Sztucki, M. Burghammer, S. Maltsev, C. Jäger and H. Cölfen, Structure-property relationships of a biological mesocrystal in the adult sea urchin spine, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(10), 3699–3704 CrossRef CAS PubMed.
- P. Fratzl, Biomimetic materials research: what can we really learn from nature's structural materials?, J. R. Soc. Interface, 2007, 4(15), 637–642 CrossRef CAS PubMed.
- K. Medjoubi, K. Benzerara, J. Debrie, E. Tang, D. Bazin, E. Letavernier, K. Desjardins and A. Somogyi, State-of-the-art multimodal scanning hard X-ray imaging and tomography sheds light at multiple length-scales on biomineralization related processes, Front. Environ. Chem., 2024, 5, 1339829 CrossRef.
- J. E. Parker, M. Gomez-Gonzalez, Y. Van Lishout, H. Islam, D. Duran Martin, D. Ozkaya, P. D. Quinn and M. E. Schuster, A cell design for correlative hard X-ray nanoprobe and electron microscopy studies of catalysts under in situ conditions, J. Synchrotron Radiat., 2022, 29, 431 CrossRef CAS PubMed.
- G. T. van de Kerkhof, J. M. Walker, S. Agrawal, S. M. Clarke, M. H. Sk, D. J. Craske and J. E. Parker, An in situ liquid environment for synchrotron hard X-ray nanoprobe microscopy, Mater. High Temp., 2023, 40(4), 371–375 CrossRef.
- S. Das, M. Kahnt, Y. Valen, T. Bergh, S. Blomberg, M. Lyubomirskiy, C. Schroer, H. Venvik and T. Sheppard, Restructuring of Ag catalysts for methanol to formaldehyde conversion studied using in situ X-ray ptychography and electron microscopy, Catal. Sci. Technol., 2024, 14, 5885–5898 RSC.
- J. C. da Silva, A. Pacureanu, Y. Yang, F. Fus, M. Hubert, L. Bloch, M. Salome, S. Bohic and P. Cloetens, High-energy cryo x-ray nano-imaging at the ID16A beamline of ESRF, X-Ray Nanoimaging: Instruments and Methods III, Proc. SPIE, 2017, vol. 10389 Search PubMed.
- P. D. Quinn, L. Alianelli, M. Gomez-Gonzalez, D. Mahoney, F. Cacho-Nerin, A. Peach and J. E. Parker, The Hard X-ray Nanoprobe beamline at Diamond Light Source, J. Synchrotron Radiat., 2021, 28, 1006 CrossRef CAS PubMed.
- R. P. Winarski, M. V. Holt, V. Rose, P. Fuesz, D. Carbaugh, C. Benson and D. Shu, et al., A hard x-ray nanoprobe beamline for nanoscale microscopy, J. Synchrotron Radiat., 2012, 19, 1056–1060 CrossRef CAS PubMed.
- A. Somogyi, C. M. Kewish, M. Ribbens, T. Moreno, F. Polack, G. Baranton, K. Desjardins and J. P. Samama, Status of the nanoscopium scanning hard x-ray nanoprobe beamline of Synchrotron Soleil, J. Phys.: Conf. Ser., 2013, 463, 012027 CrossRef.
- S. Chen, J. Deng, Y. Yuan, C. Flachenecker, R. Mak, B. Homberger and Q. Jin, et al., The bionanoprobe: hard x-ray fluorescence nanoprobe with cryogenic capabilities, J. Synchrotron Radiat., 2014, 21, 66–75 CrossRef CAS PubMed.
- E. Nazaretski, H. Yan, K. Lauer, N. Bouet, X. Huang, W. Xu, J. Zhou, D. Shu, Y. Hwu and Y. S. Chu, Design and performance of an X-ray scanning microscope at the Hard X-ray Nanoprobe beamline of NSLS-II, J. Synchrotron Radiat., 2017, 24, 1113–1119 CrossRef CAS PubMed.
- U. Johansson, D. Carbone, S. Kalbfleisch, A. Bjorling, M. Kahnt, S. Sala, T. Stankevic, M. Liebi, A. Rodriguez Fernandez, B. Bring, D. Paterson, K. Thanell, P. Bell, D. Erb, C. Weninger, Z. Matej, L. Roslund, K. Ãhnberg, B. Norsk Jensen, H. Tarawneh, A. Mikkelsen and U. Vogt, NanoMAX: the hard X-ray nanoprobe beamline at the MAX IV Laboratory, J. Synchrotron Radiat., 2021, 28, 1935–1947 CrossRef CAS PubMed.
- S. Bajt, M. Prasciolu and H. Fleckenstein, et al., X-ray focusing with efficient high-NA multilayer Laue lenses, Light Sci. Appl., 2018, 7, 17162 CrossRef CAS PubMed.
- J. Patommel, S. Klare, R. Hoppe, S. Ritter, D. Samberg, F. Wittwer, A. Jahn, K. Richter, C. Wenzel, J. W. Bartha, M. Scholz, F. Seiboth, U. Boesenberg, G. Falkenberg and C. G. Schroer, Focusing hard x rays beyond the critical angle of total reflection by adiabatically focusing lenses, Appl. Phys. Lett., 2017, 110(10), 101103 CrossRef.
- H. Mimura, S. Handa, T. Kimura, H. Yumoto, D. Yamakawa, H. Yokoyama, S. Matsuyama, K. Inagaki, K. Yamamura and Y. Sano, et al., Nat. Phys., 2010, 6, 122 Search PubMed.
- S. P. Kruger, H. Neubauer, M. Bartels, S. Kalbfleisch, K. Giewekemeyer, P. J. Wilbrandt, M. Sprung and T. Salditt, Sub-10nm beam confinement by X-ray waveguides: design, fabrication and characterization of optical properties, J. Synchrotron Radiat., 2012, 19, 227–236 CrossRef CAS PubMed.
- P. D. Quinn, F. Cacho-Nerin, M. A. Gomez-Gonzalez, J. E. Parker, T. Poon and J. M. Walker, Differential phase contrast for quantitative imaging and spectro-microscopy at a nanoprobe beamline, J. Synchrotron Radiat., 2023, 30, 200–207 CrossRef PubMed.
- E. M. Bolitho, C. Sanchez-Cano and H. Shi, et al., Single-Cell Chemistry of Photoactivatable Platinum Anticancer Complexes, J. Am. Chem. Soc., 2021, 143(48), 20224–20240 CrossRef CAS PubMed.
- F. Fus, Y. Yang, H. Z. S. Lee, S. Top, M. Carriere, A. Bouron, A. Pacureanu, J. C. da Silva, M. Salmain, A. Vessières, P. Cloetens, G. Jaouen and S. Bohic, Angew. Chem., Int. Ed., 2019, 58, 3461 CrossRef CAS PubMed.
- P. D. Quinn, M. S. Landman, T. Davis, M. Freitag, S. Gazzola and S. Dolgov, Optimal Sparse Energy Sampling for X-ray Spectro-Microscopy: Reducing the X-ray Dose and Experiment Time Using Model Order Reduction, Chem. Biomed. Imaging, 2024, 2(4), 283–292 CrossRef CAS PubMed.
- T. Oliver, S. Gazzola, S. Dolgov and P. Quinn, Undersampling raster scans in spectromicroscopy for a reduced dose and faster measurements, Opt. Express, 2022, 30, 43237–43254 CrossRef PubMed.
- F. Pfeiffer, X-ray ptychography, Nat. Photon., 2018, 12, 9–17 CrossRef CAS.
- D. Batey, C. Rau and S. Cipiccia, High-speed X-ray ptychographic tomography, Sci. Rep., 2022, 12, 7846 CrossRef PubMed.
- J. Deng, et al., Correlative 3D x-ray fluorescence and ptychographic tomography of frozen-hydrated green algae, Sci. Adv., 2018, 4, eaau4548 CrossRef CAS PubMed.
- K. W. Bossers, R. Valadian, S. Zanoni, R. Smeets, N. Friederichs, J. Garrevoet, F. Meirer and B. M. Weckhuysen, Correlated X-ray Ptychography and Fluorescence Nano-Tomography on the Fragmentation Behavior of an Individual Catalyst Particle during the Early Stages of Olefin Polymerization, J. Am. Chem. Soc., 2020, 142(8), 3691–3695 CrossRef CAS PubMed.
- T. M. M. Heenan, A. Wade, C. Tan, J. E. Parker, D. Matras, A. S. Leach, J. B. Robinson, A. Llewellyn, A. Dimitrijevic, R. Jervis, P. D. Quinn, D. J. L. Brett and P. R. Shearing, Identifying the Origins of Microstructural Defects Such as Cracking within Ni-Rich NMC811 Cathode Particles for Lithium-Ion Batteries, Adv. Energy Mater., 2020, 10, 2002655 CrossRef CAS.
- T. H. B. Fréderique, et al., Correlated x-ray fluorescence and ptychographic nano-tomography on Rembrandt's The Night Watch reveals unknown lead “layer”, Sci. Adv., 2023, 9, eadj9394 CrossRef PubMed.
- J. Kelly, A. Male, N. Rubies, D. Mahoney, J. M. Walker, M. A. Gomez-Gonzalez, G. Wilkin, J. E. Parker and P. D. Quinn, The Delta Robot—A long travel nano-positioning stage for scanning x-ray microscopy, Rev. Sci. Instrum., 2022, 93(4), 043712 CrossRef CAS PubMed.
- C. Besnard, M. Ali, S. Sasidharan, P. Buček, J. M. Walker, J. E. Parker, M. C. Spink, R. A. Harper, S. Marathe, K. Wanelik, T. E. J. Moxham, E. Salvati, K. Ignatyev, M. M. Kłosowski, R. M. Shelton, G. Landini and A. M. Korsunsky, Multi-resolution Correlative Ultrastructural and Chemical Analysis of Carious Enamel by Scanning Microscopy and Tomographic ImagingACS, ACS Appl. Mater. Interfaces, 2023, 15(31), 37259–37273 CrossRef CAS PubMed.
- C. Besnard, M. Ali, S. Sasidharan, P. Buček, J. M. Walker, J. E. Parker, T. E. J. Moxham, B. Daurer, B. Kaulich, M. Kazemian, R. M. Shelton, G. Landini and A. M. Korsunsky, Nanoscale correlative X-ray spectroscopy and ptychography of carious dental enamel, Mater. Des., 2022, 224, 111272 CrossRef CAS.
- J. Wilmers and S. Bargmann, Acta Biomater., 2020, 107, 1 CrossRef CAS PubMed.
- W. M. Balch, The Ecology, Biogeochemistry, and Optical Properties of Coccolithophores, Ann. Rev. Mar. Sci., 2018, 10, 71–98 CrossRef PubMed.
- J. M. Walker, H. J. M. Greene, Y. Moazzam, P. D. Quinn, J. E. Parker and G. Langer, Environ. Sci.: Processes Impacts, 2024, 26, 966–974 Search PubMed.
- D. M. Chevrier, E. Cerdá-Doñate, Y. Park, F. Cacho-Nerin, M. Gomez-Gonzalez, R. Uebe and D. Faivre, Synchrotron-Based Nano-X-Ray Absorption Near-Edge Structure Revealing Intracellular Heterogeneity of Iron Species in Magnetotactic Bacteria, Small Sci., 2022, 2, 2100089 CrossRef CAS PubMed.
- M. Amor, F. P. Mathon, C. L. Monteil, V. Busigny and C. T. Lefevre, Iron-biomineralizing organelle in magnetotactic bacteria: function, synthesis and preservation in ancient rock samples, Environ. Microbiol., 2020, 22, 3611–3632 CrossRef CAS PubMed.
- X. Wang, J. Yang and C. Andrei, et al., Biomineralization of calcium phosphate revealed by in situ liquid-phase electron microscopy, Commun. Chem., 2018, 1, 80 Search PubMed.
- K. He, et al., Revealing nanoscale mineralization pathways of hydroxyapatite using in situ liquid cell transmission electron microscopy, Sci. Adv., 2020, 6, eaaz7524 CrossRef CAS PubMed.
- L.-A. DiCecco, T. Tang, E. D. Sone and K. Grandfield, Exploring Biomineralization Processes Using In Situ Liquid Transmission Electron Microscopy: A Review, Small, 2025, 21, 2407539 Search PubMed.
- L.-A. DiCecco, R. Gao, J. L. Gray, D. F. Kelly, E. D. Sone and K. Grandfield, Liquid Transmission Electron Microscopy for Probing Collagen Biomineralization, Nano Lett., 2023, 23(21), 9760–9768 Search PubMed.
- B. Jin, Z. Liu, C. Shao, J. Chen, L. Liu, R. Tang and J. J. De Yoreo, Phase Transformation Mechanism of Amorphous Calcium Phosphate to Hydroxyapatite Investigated by Liquid-Cell Transmission Electron Microscopy, Cryst. Growth Des., 2021, 21(9), 5126–5134 Search PubMed.
- J. D. Currey, Form and Function, Academic Press, 1988, vol. 11, pp. 183–210 Search PubMed.
- B. Chen, X. Peng, J. G. Wang and X. Wu, Laminated microstructure of Bivalva shell and research of biomimetic ceramic/polymer composite, Ceram. Int., 2004, 30(7), 2011–2014 CrossRef CAS.
- M. Holler, J. Raabe, R. Wepf, S. H. Shahmoradian, A. Diaz, B. Sarafimov, T. Lachat, H. Walther and M. Vitins, OMNY PIN—A versatile sample holder for tomographic measurements at room and cryogenic temperatures, Rev. Sci. Instrum., 2017, 88(11), 113701 CrossRef CAS PubMed.
- M. Holler, et al., An instrument for 3D x-ray nano-imaging, Rev. Sci. Instrum., 2012, 83, 073703 CrossRef CAS PubMed.
- P. Thibault, M. Dierolf, O. Bunk, A. Menzel and F. Pfeiffer, Probe retrieval in ptychographic coherent diffractive imaging, Ultramicroscopy, 2009, 109(4), 338–343 CrossRef CAS PubMed.
- P. Thibault and M. Guizar-Sicairos, Maximum-likelihood refinement for coherent diffractive imaging, New J. Phys., 2012, 14, 063004 CrossRef.
- B. Bayerlein, P. Zaslansky and Y. Dauphin, et al., Self-similar mesostructure evolution of the growing mollusc shell reminiscent of thermodynamically driven grain growth, Nat. Mater., 2014, 13, 1102–1107 CrossRef CAS PubMed.
- C. Jean-Pierre, O. Belhadj, S. Borensztajn, M. Gèze, S. Trigos-Santos, P. Prado and Y. Dauphin, Prism substructures in the shell of Pinna nobilis (Linnaeus, 1758), Mollusca – Evidence for a three-dimensional pulsed-growth model, Heliyon, 2020, 6(7), e04513 CrossRef PubMed.
- F. Nudelman, H. H. Chen and H. A. Goldberg, Steve Weiner and Lia Addadi, Lessons from biomineralization: comparing the growth strategies of mollusc shell prismatic and nacreous layers in Atrina rigida, Faraday Discuss., 2007, 136, 9–25 RSC.
- A. S. Schenk and Y.-Y. Kim, Unraveling the internal microstructure of biogenic and bioinspired calcite single crystals, MRS Bull., 2015, 40(6), 499–508, DOI:10.1557/mrs.2015.100.
- A. Gerardo Checa, A. B. Rodríguez-Navarro and F. J. Esteban-Delgado, The nature and formation of calcitic columnar prismatic shell layers in pteriomorphian bivalves, Biomaterials, 2005, 26(32), 6404–6414 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2025 |