
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChallenges and opportunities in catalytic hydrogenolysis of oxygenated plastics waste: polyesters, polycarbonates, and epoxy resins†
Harisekhar
Mitta
 a,
Lingfeng
Li
a,
Mohammadhossein
Havaei
a,
Dambarudhar
Parida
bc,
Elias
Feghali
bd,
Kathy
Elst
b,
Annelore
Aerts
b,
Karolien
Vanbroekhoven
b and
Kevin M.
Van Geem
*a
a,
Lingfeng
Li
a,
Mohammadhossein
Havaei
a,
Dambarudhar
Parida
bc,
Elias
Feghali
bd,
Kathy
Elst
b,
Annelore
Aerts
b,
Karolien
Vanbroekhoven
b and
Kevin M.
Van Geem
*a
aLaboratory for Chemical Technology (LCT), Department of Materials, Textiles and Chemical Engineering, Ghent University, Technologiepark125, Zwijnaarde, B-9052 Gent, Belgium. E-mail: Kevin.VanGeem@UGent.be
bSustainable Polymer Technologies (SPOT) Team, Flemish Institute for Technological Research (VITO), Boeretang 200, Mol, Belgium
cCellulose & Wood Materials Group, Swiss Federal Laboratories for Materials Science and Technology (Empa), Ueberlandstrasse 129, 8600 Dübendorf, Switzerland
dChemical Engineering Program, Notre Dame University-Louaize, Zouk Mosbeh 1211, Lebanon
First published on 25th November 2024
Abstract
This review comprehensively explores various homogeneous and heterogeneous catalytic systems for the hydrogenolysis of oxygenated plastic waste (OXPs), presenting an adaptable solution to plastic pollution and generating valuable feedstock. Research demonstrates enhanced hydrogenolysis efficiency with reduced energy consumption, yielding alcohols, alkanes, alkenes, and aromatics. The effectiveness of depolymerization and the product distribution are influenced by factors such as solvents, ligands, metals, catalyst support, and reaction conditions. Scaling up these processes remains challenging, highlighting the need for non-toxic, highly active catalysts. Promising homogeneous catalysts, such as Ru(triphos-Xyl), and heterogeneous catalysts, such as Ru/Nb2O5, show potential in OXP depolymerization but face cost-related scalability issues. Homogeneous catalysts encounter commercialization obstacles due to harsh reaction conditions and difficulties in product separation, whereas heterogeneous catalysts like Ru/Nb2O5 provide effectiveness and stability with easier product separation. Nonetheless, challenges in scaling up, cost reduction, and catalyst reusability persist. Achieving economic viability is crucial for the commercialization of OXP hydrogenolysis and the reduction of plastic waste. The review emphasizes the shortage of depolymerization facilities for polyesters like poly(ethylene terephthalate) (PET), poly(bisphenol A carbonate) (BPA-PC), and epoxy resins (EP). It addresses recycling process challenges, focusing on sorting and supply chain issues, and identifies specific difficulties in recycling BPA-PC, PET, and EP materials, proposing chemical recycling as a viable solution to improve economic competitiveness and environmental sustainability.
1. Introduction
Oxygenated plastic waste (OXPs) are a group of polyesters containing oxygen atoms in their backbone, including poly(ethylene terephthalate) (PET), poly(bisphenol A carbonate) (BPA-PC), epoxy resins (EP), and poly(lactic acid) (PLA).1–4 These plastics account for over 10% of annual waste, with significant amounts of PET, BPA-PC, EP, and PLA being consumed each year.3,5–7 This raises concerns about the potential leaching of hazardous substances into the environment.8–10 Efficient recycling processes are crucial for a circular plastic economy to reduce waste and greenhouse gas emissions.11–13 Chemical recycling can convert OXPs into quality monomers for premium product creation without using fossil fuels.14–19 Biomass-derived compounds can replace BPA, but sustainable polymer production is still important.18,20 Bio-derived polyesters such as polyethylene furanoate (PEF), polybutylene succinate (PBS), and PLA are eco-friendly alternatives to petroleum-based plastics, driving the growth of the bioplastics market. Global bioplastic production is projected to reach 2.43 million tons by 2024, with a significant portion being non-biodegradable.21–23Various commercial methods have been developed for depolymerizing PET waste into monomers, including solvolysis and enzymatic depolymerization24–47 (Fig. 1). Companies like DePoly, Tyton BioSciences, and Gr3n are using hydrolysis on a large scale48–52 (Table 1), while Rewind® PET, IONIQA, and VolCat are running pilot-scale processes for glycolysis.53–57 Carbios has shown the commercial potential of biocatalytic PET depolymerization.58–60 Recent advancements in the depolymerization of BPA-PC through hydrolysis and alcoholysis, often using metal catalysts, have been made (Fig. 2).5,61–72 In addition, aminolysis has been found to be a promising method for depolymerization into BPA, creating stronger amine bonds and generating recyclable monomers.61,73–79 On the industrial level, Covestro has developed a new method for breaking down BPA-PC into its monomers with a pilot project in Leverkusen, Germany. Efforts are ongoing to refine and advance this process,80 with more technologies expected to enter the market soon. As for epoxy resins, most of the research focuses on selective bond cleavage at low temperatures using specific catalysts or depolymerizing agents (Fig. 3).81–87 Organic amine solvents effectively selectively break carbonyl carbon bonds in epoxy resins, making aminolysis a promising method for recycling anhydride-cured epoxy.77,88
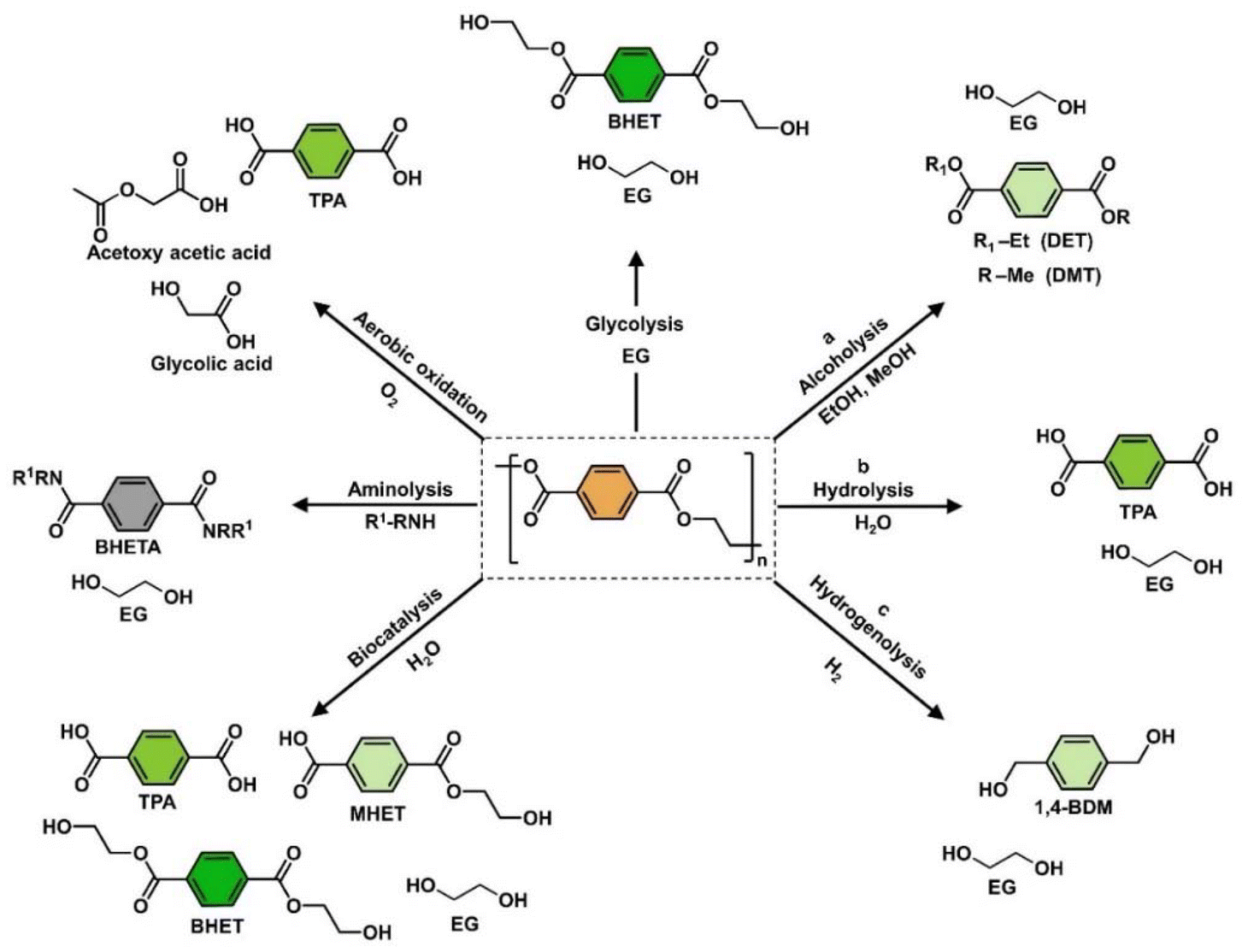 | ||
| Fig. 1 Representation of various chemical methods for converting PET (orange) into different compounds, including TPA (green), 1,4-BDM, DMT, DET and MHET (light green), BHET (dark green), and BHETA (black). | ||
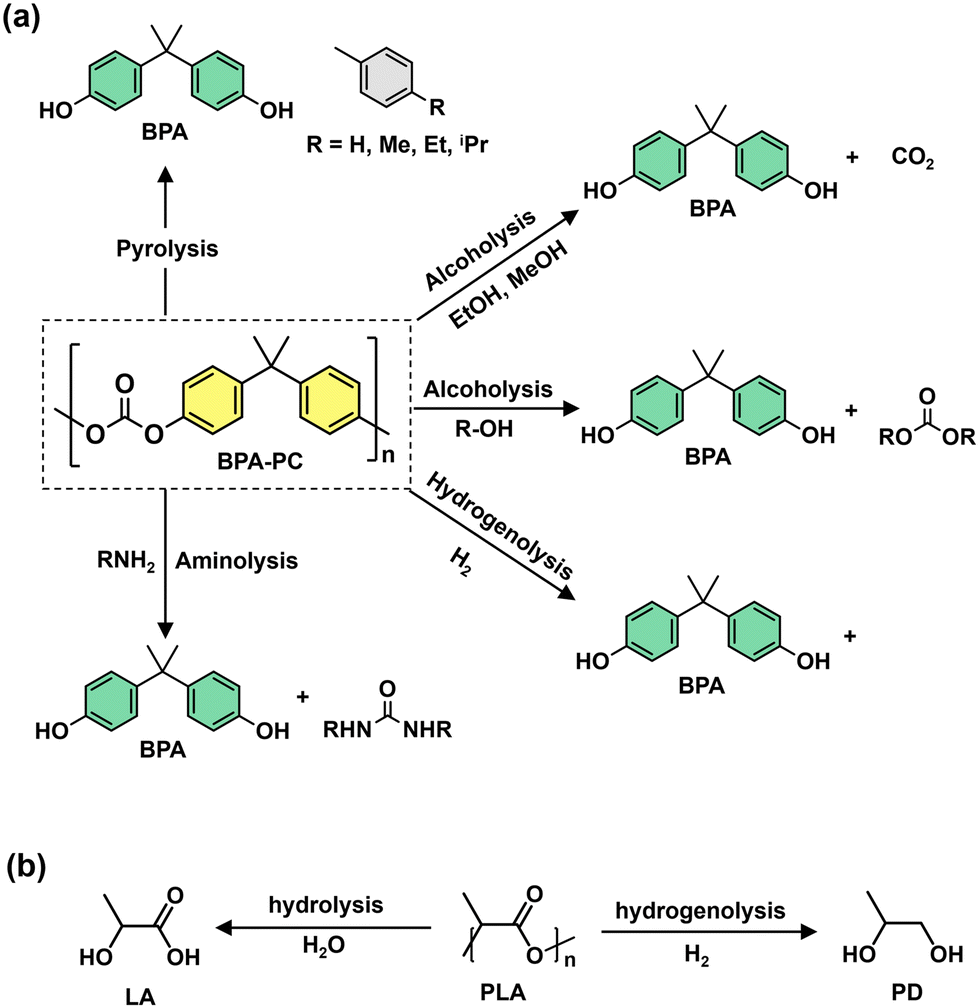 | ||
| Fig. 2 (a) Chemical recycling of BPA-PC (yellow) waste into various compounds. (b) Hydrogenolysis and hydrolysis of PLA waste yield various compounds. | ||
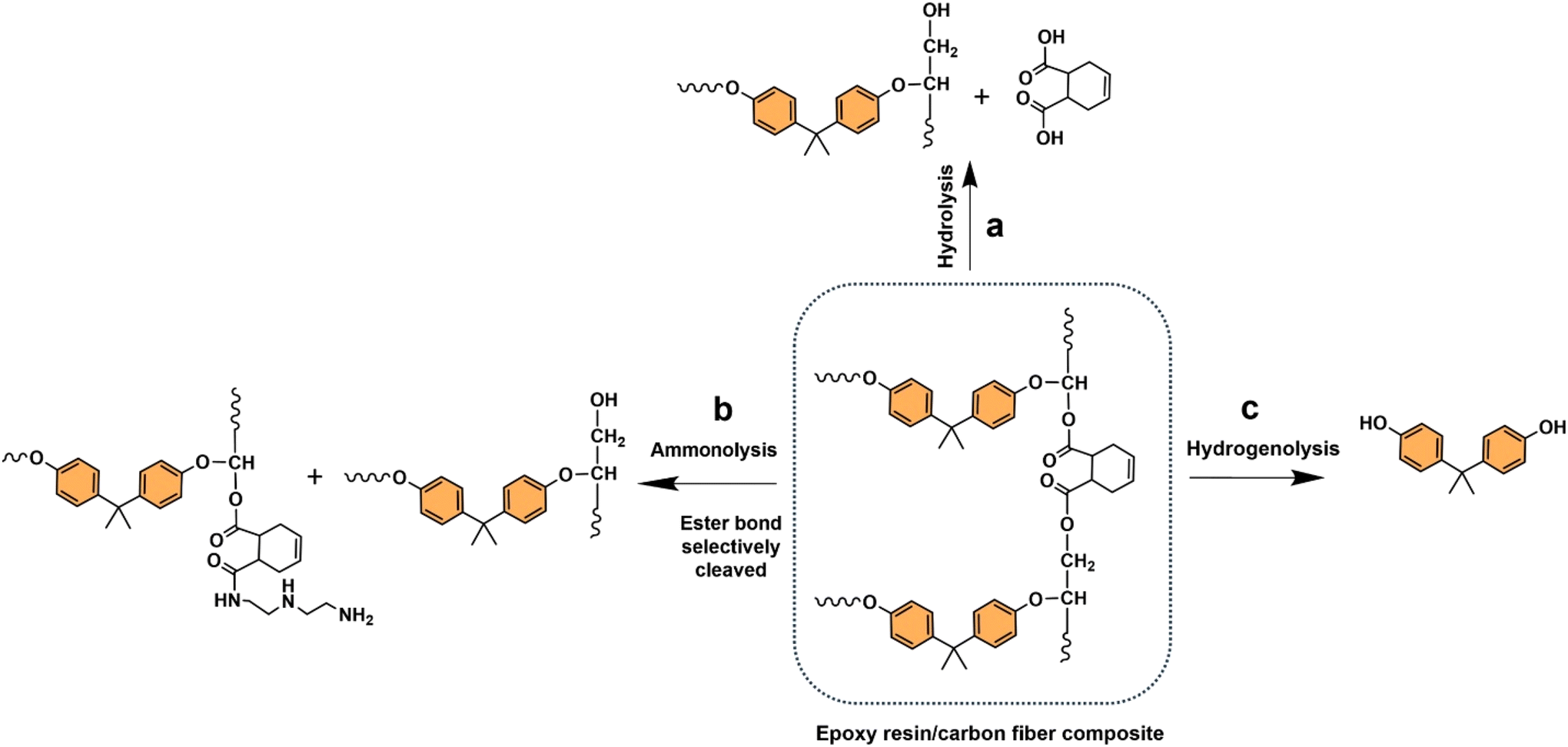 | ||
| Fig. 3 (a) Chemical recycling of epoxy resin (orange) waste into various compounds: (a) hydrolysis, (b) ammonolysis, and (c) hydrogenolysis. | ||
| Name of project | Location | Scale (per year) | Method | Feedstocks | Products | Collaborations | Ref. |
|---|---|---|---|---|---|---|---|
| DePoly | Switzerland (Start-up) | 50 to 500 tons | Basic hydrolysis + TiO2 or metal oxide at RT | PET and mixed plastics | TPA, EG, DMT | BASF Venture Capital and Wingman Ventures | 39 and 49 |
| Tyton BioSciences (Circ) | US, (commercialization) | 100 million tons | Basic hydrolysis at high T & P | PET fiber | TPA, EG | Ellen MacArthur Foundation | 50 and 51 |
| Gr3n | Spain (pilot plant) | 40.000 tons | Microwave-assisted hydrolysis | PET fiber and resin | TPA, EG | Intecsa Industrial | 52 |
| Infinite loop's | Ulsan, South Korea (pilot plant) | 84 million tons | Methanolysis, inorganic alkoxide catalyst | PET resin or fiber | DMT, MEG | SK Global Chemical | 45 |
| Québec, Canada (pilot plant) | 70 million tons | Inovista and Suez | |||||
| Spartanburg, USA (pilot plant) | 40 million tons | Indorama ventures | |||||
| Eastman Chemical | Tennessee, USA (commercialization) | 100.000 metric tons | Methanolysis, zinc acetate at high T | PET scraps | EG, DMT, EG | Polyester renewal technology | 46 and 47 |
| Rewind® PET | Kitakyushu-city, Japan (pilot plant) | 1000 tons | Glycolysis, metal hydroxide catalyst | PET bottles or fibers | BHET | Axens, IFPEN and JEPLAN | 53 |
| Ioniqa | Brightlands Chemelot Campus, Netherlands (industrial scale) | 10 kilotons | Glycolysis, imidazole base iron oxide nanoparticle | PET packaging polyesters | BHET | EUT and KTS | 55 and 56 |
| VolCat | Frankfurt Research Center, Germany (industrial scale) | 20 metric tons | Glycolysis, volatile organic base, volatile catalyst | PET packaging polyesters | BHET | Technip Energies, IBM, and Under Armour | 57 |
| Carbios | Lyon, France (industrial scale) | 64 million tons | Enzymatic catalysis-hydrolysis | PET fibers | TPA, EG | TechnipFMC | 58–60 |
Various methods are used to upcycle plastic waste, including pyrolysis, hydrocracking, solvolysis, aminolysis, and hydrogenolysis.89,90 The depolymerization of plastic is a key focus in materials science research, aiming to address challenges in commercial-scale production through optimization studies. Review articles play a crucial role in summarizing advancements in this field for researchers. Recent research has shown potential in selective catalytic hydrogenative depolymerization for plastic upcycling, with a focus on developing efficient catalysts in both homogeneous and heterogeneous forms. Hydrogenolysis, a promising process for chemical recycling, is being investigated with transition metal catalysts in laboratory trials. Successful scale-up of hydrogenolysis could revolutionize plastic recycling. Advances in the hydrogenative depolymerization of polyesters, emphasizing the cleavage of Csp3–O bonds and subsequent hydrogenation to form C–H and O–H bonds, have been reported.79,91–97
Some protocols use expensive and hazardous transition metal-based catalysts to break ethereal C–O bonds. Recent studies have demonstrated effective depolymerization of polyester plastic waste using homogeneous catalysts like Ru, Rh, and Ir, along with reductants such as H2, silane, or amino borane, or under external reductant-free conditions.5,25,86,98,99 Further research is needed to develop cost-effective methods for polyester hydrogenolysis, including output optimization, scaling up technology, and ensuring safety measures. Recent studies have aimed to enhance the hydrogenolysis activity of plastic waste using heterogeneous catalysts, such as solid acid catalysts.100,101 These catalysts offer advantages in terms of easier synthesis, recovery, recycling, and commercialization infrastructure. Jing et al. demonstrated the catalytic conversion of PET plastics back to BTX by unlocking hidden hydrogen in the ethylene glycol part over Ru/Nb2O5, involving C–O/C–C cleavage through parallel hydrogenolysis and decarboxylation.90 Solid acid catalysts like aluminosilicate zeolites (e.g., ZSM-5, beta, Y zeolite) combined with metals (Pt, Pd, Ru) and Mn-based catalysts have been used for the C–O/C–C cleavage of polyesters to cycloalkanes.102 Ru-based bimetallic solid acid catalysts have shown higher selectivity for cyclic hydrocarbons, with researchers developing Ru-based bimetallic catalysts to enhance catalytic activity and reduce costs.103 Chemical recycling of other polyesters like PLA and PBT for engineering thermoplastic applications is also being explored. Depolymerizing PLA to the lactide monomer faces challenges due to side reactions.104–107 Specific catalysts (e.g., ZnO) used in the hydrogenolysis of PLA can effectively depolymerize, producing propylene glycol.108 Due to the absence of carbonyl groups, epoxy resins with crosslinked networks pose difficulties in depolymerization. Recent studies suggested that metals like Rh, Ru, and Ni are effective for the hydrogenolysis of BPA-type epoxy resins, hydrogenating aromatic rings rather than cleaving C–O bonds under mild conditions.109–112 Carbon-based materials like graphite and activated carbon are commonly used as catalyst supports due to their high surface area and dispersibility.113–125
This review delves into converting OXP waste into chemicals using hydrogen gas with various catalysts, focusing on the hydrogenolysis of plastic waste to minimize operational disruptions and promote sustainability. The goal is to identify effective catalysts for commercializing C–O/C–C-based products for next-generation catalytic technologies. The review contributes to the development of sustainable heterogeneous catalysts for chemical production from plastic waste, with a focus on bio-derived polymeric materials and catalytic reductive depolymerization. It discusses the use of selected transition metal catalysts, such as Ru, Pt, Ni, Co, Mo, Rh, and Ir for the catalytic hydrogenolysis of polyester waste.61,94,100,108,126–161 The research findings suggest that ruthenium and nickel-based catalyst systems show high potential and activity for these reactions. They offer a promising approach to producing commercial-scale hydrocarbon fuels and valuable chemicals from waste plastics with reduced environmental impact.
2. Hydrogenolysis
Catalytic hydrogenolysis has already proved to be a key method for breaking down polymers like lignin into valuable monomers or dimers using metal catalysts such as cobalt, nickel, and molybdenum.162–169 Selective cleavage of carbon–oxygen bonds is crucial for converting lignin into valuable chemicals, such as phenolic compounds. Current research aims to enhance productivity by achieving a more useful product distribution through the selective cleavage of C–O bonds. Various catalysts have been tested for lignin model compound hydrogenolysis, offering alternative methods for converting waste polymers into valuable compounds.163,166,167,169 Hydrogenolysis of oxygenated plastic waste (OXPs) can recycle and produce valuable molecules such as BPA, 1,4-BDM, phenol, and methanol. Challenges like C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond saturation, hydrogen adsorption, and hydrogen spillover need to be addressed for optimal hydrogenolysis processes. Further research is needed to optimize OXP conversion into marketable products.108,134,170–174
O bond saturation, hydrogen adsorption, and hydrogen spillover need to be addressed for optimal hydrogenolysis processes. Further research is needed to optimize OXP conversion into marketable products.108,134,170–174
Chemical recycling of OXPs involves the depolymerisation of the polymer into monomers such as BPA, 1,4-BDM, aromatics, 1,2-PD, and LA through depolymerization processes (shown in Fig. 1–3). OXP hydrogenolysis techniques are used on a lab scale, with researchers working to optimize the process for higher yields and lower energy consumption while considering environmental factors. The E factor, which accounts for solvent recycling, can evaluate the environmental impact of depolymerization studies (eqn (1) & (2)).175–178 The environmental energy impact factor ξ combines energy consumption and waste generation to assess sustainability (eqn (4)).177 Calculating the E-factor and energy economy coefficient (ε) for hydrogenolysis processes helps assess environmental impact and energy consumption. Y represents the monomer yield, while T and t refer to reaction temperature and time, respectively. Lower ξ values indicate higher efficiency with reduced waste generation and energy consumption. Processes with low E and ξ values and high ε values are considered the best, enabling fair comparisons among different catalyst systems for polyester hydrogenolysis and paving the way for industrial-scale oxygenate polyester depolymerization.
 | (1) |
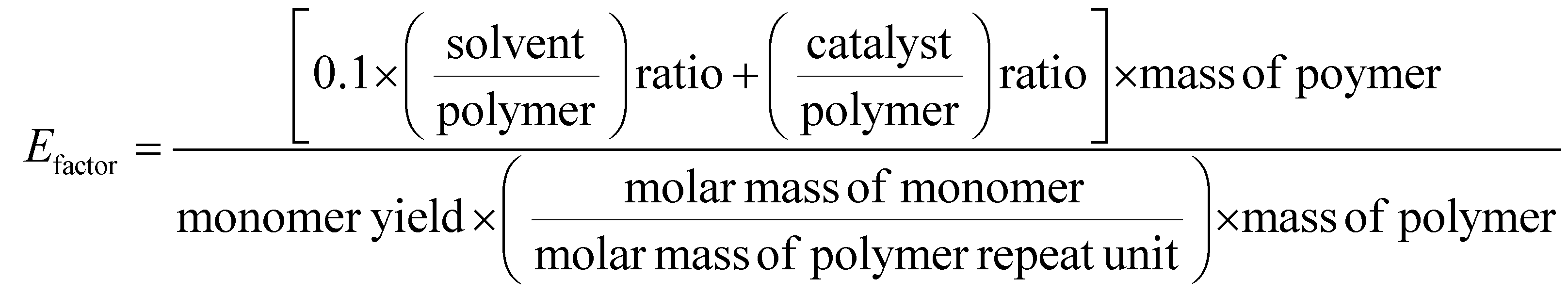 | (2) |
 | (3) |
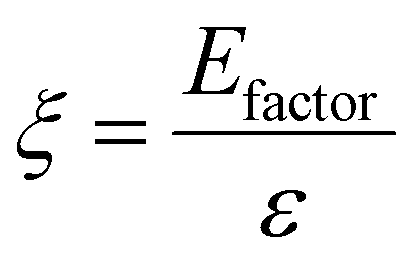 | (4) |
3. Homogeneous catalysts
3.1. Hydrogenolysis of polyesters and epoxy resins
Homogeneous catalysis plays a crucial role in efficiently breaking down polyesters through hydrogenolysis, allowing for selective transformations under specific conditions. For instance, Pd-based catalyst systems can effectively hydrogenate α- and β-unsaturated carbonyl compounds.179 Lanthanum(III) tris(amide) catalyst enables the reductive depolymerization of polyesters with hydroboranes, yielding alcohols and diols with high selectivity at low catalyst loadings (Fig. 4b).180 Ruthenium complexes achieve over 95% CO2 conversion in methanol hydrogenation.181 Hf(OTf)4 and Pd/C efficiently convert polyesters to terephthalic acid (TPA) in a solvent-free process under 1 atm H2 (Fig. 4c). Homogenous catalysts and in situ hydrogenation-based catalysts play a crucial role in cleaving C–O bonds to produce alcohols, TPA, and aromatics, along with ethane, EG, and ethylene as by-products. For more detailed information, refer to published articles.182–185 In liquid-phase reactions, H2, silanes (R3Si-H), and hydroborate (R2B-H) can function as hydrogen donors186–196 (Fig. 4). Liu et al. demonstrated the catalyst-free depolymerization of PCL to silylated monoesters and iodide derivatives using iodosilanes at temperatures of 100–150 °C for 5–6 hours, marking the first use of iodosilanes in this context.197 Homogeneous catalysis offers high activity and selectivity with minimal catalyst usage but faces challenges with catalyst robustness and environmental impact. Solutions like membrane separation and phase-transfer catalyst design can address these issues, promoting economic and environmental sustainability. Ongoing research aims to optimize catalyst recovery and expand practical applications of homogeneous catalysis. The use of homogeneous molecular catalysts for the hydrogenolysis of OXPs under severe reaction conditions is summarized, with a focus on developing catalysts that can operate under milder conditions and control product distribution by selectively cleaving C–O or C–C bonds.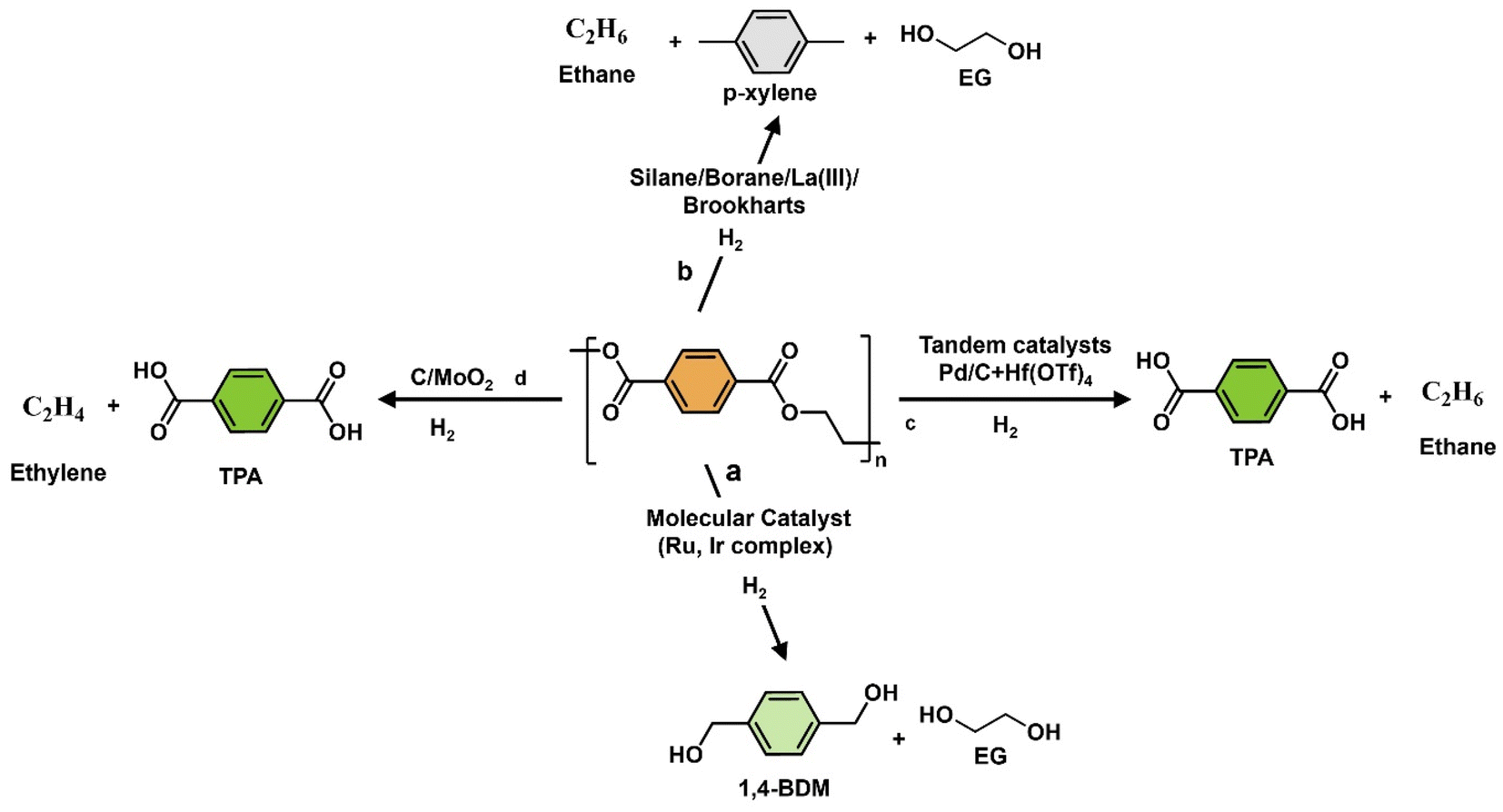 | ||
| Fig. 4 Possible reductive catalytic pathways for the depolymerization of PET.155,183–185 | ||
Recent advances in tailor-made molecular complexes with appropriate bases such as KOt-Bu, KOH, and KH have been used for the depolymerization of PET to diols.85,198–202 The reductive depolymerization of esters to valuable alcohols is an attractive strategy for recycling PET. In 2005, Zhang's group developed the first homogeneous organometallic catalytic systems (Ru(II) PNN and PNP ligands) for the hydrogenolysis of esters to alcohols as shown in Fig. 5.203 These catalysts were investigated at 150 °C, 50–70 bar H2, and a runtime of 24–48 hours using DMSO/1,4-dioxane/THF/mesitylene as solvents. A 1,4-BDM selectivity ranging between 75–80% and a PET depolymerization rate of 85–90% were achieved (Fig. 4a). However, high H2 pressures, long reaction times (16–48 hours), expensive solvents, air-sensitive Ru catalysts, and potentially toxic ligands are major drawbacks of such processes. In this case, the donor atoms cooperate with the metal center in ruthenium pincer complexes with a finely tuned ligand environment (NNP, NCN, and PNS), enabling heterolytic hydrogen activation and achieving C–O, C–N, and finally C–C bond cleavage.75–78,130,149,203,204
 | ||
| Fig. 5 A schematic diagram of homogeneous catalytic systems for the hydrogenolysis of PET. | ||
In 2014, áMc Ilrath et al. introduced a Milstein catalyst (Fig. 5) with KOt-Bu as the base for efficient hydrogenolysis of 0.19 g of PET waste into 1,4-BDM and EG as products instead of phthalate. They achieved a remarkable conversion rate of >99% and a selective monomer yield over of 77% using 0.003 g of catalyst, 0.014 g of KOt-Bu base, and 4 g of anisole![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :THF as a solvent, at 160 °C for 24 hours under 54.4 bar H2.200 The process demonstrates high selectivity, controlled depolymerization, and no observed self-recombination reactions (Table 2, entry 1). However, it has drawbacks such as high costs for solvents and noble metals, challenges in scaling up, non-reusable and air-sensitive Ru catalysts, potentially toxic ligands, and limited robustness under harsh conditions (160 °C and 54.4 bar H2). On the other hand, the advantages include high monomer yield and lower co-products. Table 2 and Fig. S1† show that the Milstein catalyst has a high ε coefficient of 4.296 × 10−6 °C−1 min−1 and a low ξ value of 259895 °C min (Table 2 and Fig. S1†). These results were achieved due to a combination of monomer yield (1,4-BDM, 75%), low catalyst requirements (0.0007 g), and a shorter reaction time of 24 hours compared with the Ru(triphos) catalyst (48 hours). In 2015, Fuentes et al. developed four Ru(II) pincer catalysts with tridentate amino phosphine-based ligands for PET hydrogenolysis. The most effective catalyst was the phosphine-amino-imide-derived Ru variant. The 1.36 g PET hydrogenolysis reaction was conducted with 0.27 g of KOt-Bu, and 0.22 g of catalyst in a 6 g THF–anisole mixture reaction conducted at a low temperature of 110 °C and 50 bar H2.205 After 48 hours, the PET depolymerization yielded a slightly low selectivity of 73% for 1,4-BDM (Table 2, entries 2 and 3), and EG as a product (N.D). The Ru(II) pincer catalyst has drawbacks such as reduced selectivity, higher cost, and sustainability issues in harsh conditions (110 °C and 54 bar H2). Challenges include product separation, scaling up the process, and catalyst recovery. Compared with heterogeneous catalysts, the Ru(II) pincer catalyst is less versatile and sustainable. Table 2 and Fig. S1† demonstrate that the Ru(II) pincer catalyst has a low ε coefficient value (2.3042 × 10−6 °C−1 min−1) resulting from a lower yield (73%) and longer reaction time (48 hours). The E factor for the Milstein catalyst is slightly lower (1.0) than for the Ru(ii) pincer catalyst due to higher solvent-to-polymer ratio (3.7) and catalyst-to-polymer ratio (0.028) The Milstein catalyst is more efficient for the hydrogenolysis of PET compared with the Ru(II) pincer catalyst due to a higher ε coefficient (4.29688 × 10−6 °C−1 min−1).
:THF as a solvent, at 160 °C for 24 hours under 54.4 bar H2.200 The process demonstrates high selectivity, controlled depolymerization, and no observed self-recombination reactions (Table 2, entry 1). However, it has drawbacks such as high costs for solvents and noble metals, challenges in scaling up, non-reusable and air-sensitive Ru catalysts, potentially toxic ligands, and limited robustness under harsh conditions (160 °C and 54.4 bar H2). On the other hand, the advantages include high monomer yield and lower co-products. Table 2 and Fig. S1† show that the Milstein catalyst has a high ε coefficient of 4.296 × 10−6 °C−1 min−1 and a low ξ value of 259895 °C min (Table 2 and Fig. S1†). These results were achieved due to a combination of monomer yield (1,4-BDM, 75%), low catalyst requirements (0.0007 g), and a shorter reaction time of 24 hours compared with the Ru(triphos) catalyst (48 hours). In 2015, Fuentes et al. developed four Ru(II) pincer catalysts with tridentate amino phosphine-based ligands for PET hydrogenolysis. The most effective catalyst was the phosphine-amino-imide-derived Ru variant. The 1.36 g PET hydrogenolysis reaction was conducted with 0.27 g of KOt-Bu, and 0.22 g of catalyst in a 6 g THF–anisole mixture reaction conducted at a low temperature of 110 °C and 50 bar H2.205 After 48 hours, the PET depolymerization yielded a slightly low selectivity of 73% for 1,4-BDM (Table 2, entries 2 and 3), and EG as a product (N.D). The Ru(II) pincer catalyst has drawbacks such as reduced selectivity, higher cost, and sustainability issues in harsh conditions (110 °C and 54 bar H2). Challenges include product separation, scaling up the process, and catalyst recovery. Compared with heterogeneous catalysts, the Ru(II) pincer catalyst is less versatile and sustainable. Table 2 and Fig. S1† demonstrate that the Ru(II) pincer catalyst has a low ε coefficient value (2.3042 × 10−6 °C−1 min−1) resulting from a lower yield (73%) and longer reaction time (48 hours). The E factor for the Milstein catalyst is slightly lower (1.0) than for the Ru(ii) pincer catalyst due to higher solvent-to-polymer ratio (3.7) and catalyst-to-polymer ratio (0.028) The Milstein catalyst is more efficient for the hydrogenolysis of PET compared with the Ru(II) pincer catalyst due to a higher ε coefficient (4.29688 × 10−6 °C−1 min−1).
| Entry | Polymer | Solvent | Solvent/polymer ratio | Catalyst | Cat./polymer ratio | Time (hour) | Temp. (°C) | Monomer yield (%) | E factor (a.u.) | ε (°C−1 min−1) | ξ (°C min) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | PET | Anisole:THF |
6.68 | Milstein catalyst KOt-Bu | 0.00074 | 24 | 160 | 75 | 1.16 | 4.29688 × 10−6 | 259895 |
200 |
| 1,4-BDM | ||||||||||||
| 2 | PET | Anisole:THF |
3.74 | Ru(II) pincer KOt-Bu | 0.028 | 48 | 110 | 73 | 1.0 | 2.30429 × 10−6 | 395890 |
205 |
| 1,4-BDM | ||||||||||||
| 3 | PET | Dioxane | 14.6 | Ru(triphos-Xyl) (TMM) HNTf2 | 0.0029 | 16 | 140 | 99 | 2.44 | 7.36607 × 10−6 | 331909 |
182 |
| 1,4-BDM | ||||||||||||
| 4 | PBT | 12.6 | 99 | 2.37 | 7.36607 × 10−6 | 322333 |
||||||
| 1,4-BDM | ||||||||||||
| 5 | PET | DMSO–mesitylene | 4.7 | Pd/C Hf(OTf)4 | 0.004 | 24 | 265 | 97 | 0.68 | 2.54193 × 10−6 | 268585 |
185 |
| TPA | ||||||||||||
| 6 | PET | DMSO–mesitylene | 4.7 | 0.0041 | 96 | 0.68 | 2.54193 × 10−6 | 274209 |
||||
| TPA | ||||||||||||
| 7 | PET | DMSO–mesitylene | 4.7 | 0.0041 | 95 | 0.69 | 2.51572 × 10−6 | 280012 |
||||
| TPA | ||||||||||||
| 8 | PET | DMSO–mesitylene | 4.7 | 0.0041 | 98 | 0.67 | 2.48952 × 10−6 | 263131 |
||||
| TPA | ||||||||||||
| Ethane | ||||||||||||
| 9 | PBT | DMSO–mesitylene | 24.0 | 0.0009 | 99 | 3.75 | 2.56813 × 10−6 | 1445831 |
||||
| TPA | ||||||||||||
| Butane | ||||||||||||
| 10 | PBT | DMSO–mesitylene | 24.4 | 0.0009 | 93 | 3.89 | 2.59434 × 10−6 | 1596701 |
||||
| TPA |
In 2018, the Westhues research group introduced two Ru molecular catalysts specifically designed for the selective conversion of PET/PBT into diols. These catalysts, Ru(triphos)TMM and Ru(triphos-Xyl)TMM are distinguished by their unique ligands: TMM, triphos-Xyl, and triphos (Fig. 5). The refined triphos-Xyl-anchored Ru catalyst achieved a full conversion rate of PET and 99% selectivity for 1,4-BDM and EG (1%) as minor by-products under specific conditions: 0.2 g polymer, 0.01 g catalyst, 0.002 g bistriflimide (HNTf2) co-catalyst, 140 °C, 100 bar H2 pressure, 16 hours reaction time, using 1,4-dioxane or 1,2-PD solvents.182,206 The catalyst was used to depolymerize various types of polyester waste, including PET and PBT (Table 2, entries 3 and 4). In 2016, the same research group successfully utilized Ru-triphos-based catalysts for lactam hydrogenation, targeting methyl benzoate and caprolactam. They used 0.001 g methanesulfonic acid as a co-catalyst and isopropyl as a solvent at 200 °C and 100 bar H2 pressure for 16 hours.206 In 2018, a study using Ru(triphos)tmm catalyst achieved 42% conversion of PET to 1,4-BDM (yield 64%) and EG. Acid-activated catalyst favored ether as a side product formation, reducing selectivity. In PBT hydrogenolysis, full conversion occurred with only 22% diols. NMR studies showed catalyst dimer formation and acid release promoting etherification. To address side reactions, a modified catalyst Ru(triphos-xyl)TMM was introduced, showing higher activity and stability. This modified catalyst in polyester hydrogenolysis achieved excellent selectivity towards diol species without any side reactions or self-recombination reactions. It can efficiently process a variety of commercial products with additives, ensuring high polyester conversion and product selectivity. However, the process uses complex ligands, reagents and robustness towards harsh catalytic conditions at high H2 pressures (54–100 atm) and requires long reaction times (16–48 hours). The Ru(triphos)TMM catalyst exhibited the highest ε value (7.3660 × 10−6 °C−1 min−1) at 140 °C for 16 hours, resulting in a 99% product yield, making it an efficient option for polyester hydrogenolysis. The E factor was low at 2.0–2.4 (Table 2 and Fig. S1†). Despite its efficiency, challenges such as scale-up and sustainability need to be addressed.
In 2021, Kratish et al. developed a solvent-free tandem catalyst system combining homogenous (Hf(OTf)4) and heterogenous (Pd/C) catalysts. The reaction converted 1 g of commercial and post-consumer PET, such as water bottles, shirts, and pillow stuffing, into TPA with a yield of ≥95% (Table 2, entries 5–8). Ethane was the major co-product during depolymerization conducted at 265 °C for 24 hours under 1 bar of hydrogen using 0.03 g of Pd/C catalyst and 0.121 g of Hf(OTf)4. A solvent mixture of 5.63 g DMSO:mesitylene was used.185 The same group applied 0.1 g of PBT hydrogenolysis, resulting in ≥93% of TPA yield with small amounts of butane (Table 2, entries 9 and 10). The operating conditions were the same, except for the catalyst and solvent, which included 0.003 g of Pd/C and 0.01 g of Hf(OTf)4 and 2.63 g of d6-DMSO:mesitylene. The tandem catalyst system effectively breaks down polyester without requiring a solvent or base. This catalyst is versatile, sustainable, and selective in C–O bond-forming reactions. It is robust under reaction conditions and prevents self-recombination reactions, allowing for controlled reactions. The system combines a recyclable homogeneous catalyst (Hf(OTf)4) with a heterogeneous hydrogenation catalyst (Pd/C) under 1 atm of H2. However, a drawback is the need for a high reaction temperature close to the polymer's melting point and a longer reaction time.
Comparison of tandem Pd/C + Hf(OTf)4 with other Ru-based catalysts like Milstein catalyst, Ru pincer, and Ru(triphos-Xyl) showed that it has the lowest E factor (0.68)and ξ (268 585 °C min) (Table 2, entry 5) values in PET hydrogenolysis due to a high monomer yield (97%), lower solvent requirement, and no reagent consumption. This catalyst exhibited a low ε coefficient value of 2.54193 × 10−6 °C−1 min−1 attributed to the high temperature (265 °C) and long reaction time (24 hours). In contrast, the Ru(triphos)TMM catalyst had the highest ε value (7.3660 × 10−6 °C−1 min−1) due to lower temperatures (140 °C), 16 hours reaction time, and higher product yield (99%), making it efficient for polyester hydrogenolysis despite a slightly higher E factor (2.0–2.4) (Table 2 and Fig. S1† for further details). Overall, Pd/C + Hf(OTf)4 catalysts are considered the best tandem catalysts for polyester hydrogenolysis, supporting catalytic chemical recycling of diverse plastics on a large scale and providing insights for addressing commercial plastic impurities and scaling up challenges in the future.
3.2. Hydrogenolysis of polycarbonates (BPA-PC/PPC)
Catalysts that break down PET are likely to also break down BPA-PC due to their similar functional groups. Pincer catalysts and other customized variants are efficient in reductive depolymerization, using external hydrogen. In 2012, Han and his group demonstrated that 0.015 g of PNP-Ru(II) catalyst efficiently converted 2.69 g of aliphatic polycarbonates like polypropylene carbonate (PPC) into 1,2-propanediol (1,2-PD) and methanol.207 The reaction achieved a 99% yield of each product within 24 hours at 140 °C under 50 bars of H2 (Table 3, entry 1). The author did not observe any self-recombination reaction or by-product formation that affected the 1,2-PD under controlled conditions, resulting in the recovery of pure monomer. The reaction used a low KOt-Bu of 0.003 g and 22.2 g of THF as a solvent. The recycling of waste polycarbonate shows promise with low E (1.04) and ξ (211782 °C min) values (details in Table 3 and Fig. S2†) due to a low solvent-to-polymer ratio (7.2) and low catalyst to polymer ratio (0.0006). The system also exhibited a high ε coefficient of 4.910 × 10−6 °C−1 min−1, attributed to a maximum monomer yield of 99% at a low temperature of 140 °C. However, challenges such as excessive solvent usage, high hydrogen pressure, single-use catalysts, expensive ligands and metals, and scale-up issues must be addressed.| Entry | Polymer | Solvent | Solvent/polymer ratio | Catalyst | Cat./polymer ratio | Time (hour) | Temp. (°C) | Monomer yield (%) | E factor (a.u.) | ε (°C−1 min−1) | ξ (°C min) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | PPC | THF | 7.2 | PNP-Ru(II) complex KOt-Bu | 0.0006 | 24 | 140 | 99 | 1.0 | 4,91071 × 10−6 | 211782 |
207 |
| 1,2-PD | ||||||||||||
| 2 | PPC | THF:anisole |
13.2 | Milstein catalyst KOt-Bu | 0.003 | 24 | 160 | 99 | 1.8 | 4,2960 × 10−6 | 420007 |
200 |
| 1,2-PD | ||||||||||||
| 3 | PEC | THF:anisole |
13.4 | 0.03 | 48 | 160 | 91 | 2.4 | 19740 × 10−6 |
1229526 |
||
| EG | ||||||||||||
| 4 | PPC | Dioxane | 40.4 | Mn-NNP complex KOt-Bu | 0.0024 | 16 | 140 | 91 | 5.9 | 67700 × 10−6 |
881909 |
209 |
| 1,2-PD | ||||||||||||
| 5 | PPC | Toluene | 4.7 | Mn PNN complex. KH | 0.02 | 30 | 110 | 68 | 1.0 | 3,4340 × 10−6 | 119264 |
210 |
| 1,2-PD | ||||||||||||
| 6 | PPC | THF | 3.6 | Fe-PNP pincer complex. KOt-Bu | 0.03 | 30 | 140 | 65 | 0.8 | 2,5719 × 10−6 | 313713 |
211 |
| 1,2-PD | ||||||||||||
| 7 | BPA-PC | 1,4-Dioxane | 2.5 | Ru-trios complex. NHTf2 | 0.002 | 16 | 140 | 99 | 0.3 | 7.3667 × 10−6 | 39564 |
182 |
| BPA | ||||||||||||
| 8 | BPA-PC | THF | 53.6 | Ru-MACHO-BH complex | 0.002 | 6 | 140 | 96 | 6.2 | 1.9047 × 10−5 | 326781 |
212 |
| BPA | ||||||||||||
| 9 | 24.5 | 0.0004 | 16 | 80 | 81 | 3.4 | 1.0546 × 10−5 | 320359 |
||||
| BPA | ||||||||||||
| 10 | BPA-PC | THF | 4.3 | Milstein catalyst KOt-Bu | 0.01 | 24 | 140 | 99 | 0.5 | 4.91071 × 10−6 | 101482 |
213 |
| BPA | ||||||||||||
| 11 | BPA-PC | THF | 4.2 | 0.007 | 93 | 0.5 | 4.6131 × 10−6 | 111757 |
||||
| BPA | ||||||||||||
| 12 | BPA-PC | THF | 16.8 | Fe-PNP pincer complex | 0.0017 | 24 | 120 | 81 | 2.3 | 4.6875 × 10−6 | 492978 |
214 |
| BPA | ||||||||||||
| 13 | PPC | THF | 34.5 | 0.0021 | 77 | 6.0 | 4.45602 × 10−6 | 1353294 |
||||
| 1,2-PD | ||||||||||||
| 14 | BPA-PC | Dibutyl ether | 6.8 | Co(III) complex. KOt-Bu | 0.0023 | 24 | 160 | 83 | 0.9 | 3.60243 × 10−6 | 257778 |
215 |
| BPA | ||||||||||||
| 15 | PPC | Dibutyl ether | 76 | 1.2 | 3.29861 × 10−6 | 370464 |
||||||
| 1,2-PD | ||||||||||||
| 16 | BPA-PC | THF–MeOH | 16.8 | Zn(II) complex | 0.0021 | 1 | 50 | 95 | 1.9 | 3.16667 × 10−4 | 6241 | 216 |
| BPA | ||||||||||||
| 17 | BPA-epoxide | THF–toluene | 92.45 | Ru(triphos-TMM) | 0.00024 | 16 | 160 | 83% | 18.5 | 5.4035 × 10−6 | 3439597 |
220 |
| BPA | ||||||||||||
| 18 | Carbon fiber | THF–toluene | 7.26 | Ru(triphos-TMM) | 0.0064 | 72 | 160 | 57% carbon fiber | 1.72 | 1.2 × 10−6 | 1676551 |
220 |
| 13% BPA |
In 2014, áMc Ilrath and Zhang and colleagues developed Ru(II) PNN (Milstein) homogenous catalysts for controlled depolymerization hydrogenolysis of PPC and poly(ethylene carbonate) (PEC).200,208 They optimized the reaction parameters to 160 °C and 54.4 bar of H2 for 24 hours with 0.326 g of PPC/PEC, 0.015 g of catalyst, a slightly high KOt-Bu of 0.007 g, 5.63 g of a (50:50) mixture of anisole, and THF as the solvent. Both amine and pyridine were found to be equally effective in the hydrogenolysis of polycarbonates. PPC was converted into 1,2-PD and methanol with a yield of 99%, while PEC was transformed into ethylene glycol (EG) and methanol with a yield of 91% (Table 3, entries 2 and 3). The study showed high product yields with reduced solvent volume, but scaling up the processes is challenging due to high reagent consumption, temperature requirements, and purification needs. The system had slightly higher E (2.4 a.u.) & ξ (1.2 × 106 °C min) values compared with PNP-Ru(II) (Table 3 and Fig. S2†). This suggests that the depolymerization method has a slightly low ε coefficient (4.296 × 10−6 °C−1 min−1) due to the high temperature (160 °C) and long time (24 hours), making it less efficient than PNP Ru(II).207
In 2018, Zuber and his group developed non-precious Mn-NNP pincer catalysts (Fig. 6) for the hydrogenolysis of PPC.209 The reaction was carried out using 0.1 g of polymer loading and 0.01 g catalyst, at 140 °C and 50 bar H2 over 16 hours in 4.13 g of 1,4-dioxane as the solvent with 0.0056 g of KOt-Bu as the base. The reaction yielded 91% of 1,2-PD and a boosted methanol yield of 84% respectively (Table 3, entry 4). This study introduces a novel catalytic system utilizing a stable and low-catalyst-loading Mn-PNN pincer for the selective hydrogenation of polycarbonate plastics. The process is highly efficient, producing by-products under mild conditions. These results have the potential to pave the way for the creation of less toxic base-metal catalysts. However, there are some drawbacks in scaling up reactions, which can increase costs and pose challenges in catalyst recovery, product separation, and the need for high hydrogen pressure. In 2018, A. Kumar and his team used slightly a higher amount of 0.036 g of Mn-PNN pincer catalyst (structure shown in Fig. 6) to convert 0.312 g of PPC into a 68% yield of 1,2-PD, with 59% methanol yield and 30% propylene carbonate yield as co-products. The reaction occurred at 110 °C over 30 hours, with 50 bar H2, using 0.006 g of KH as a strong base and 1.78 g of toluene as the solvent (Table 3, entry 5).210 The main advantage of this catalyst is its earth-abundant complex, but it has several drawbacks, i.e., the Mn-PNN pincer/KH catalyst did not perform better than the Mn-PNN pincer/KOt-Bu, and it produced two by-products. Zuber and Kumar found that using the Mn-PNP pincer catalyst for the hydrogenolysis of PPC/PEC led to a decrease in monomer alcohol yield from 91% to 68%. This decrease was likely due to factors such as low reaction temperature (110 °C), extended reaction time (30 hours), and minimal solvent usage. Consequently, the E factor, ξ values, and ε values decreased from 5.97 to 1.0, 881909 to 119264 °C min, and 6.7 × 10−6 to 3.4 × 10−6 °C−1 min−1, respectively (Table 3 and Fig. S2†). Despite these changes, the hydrogenolysis of aliphatic polycarbonate using this catalyst is efficient and environmentally friendly, making it a promising area for further research.
 | ||
| Fig. 6 A schematic representation of various homogeneous catalysts for the hydrogenolysis of polycarbonates (BPA-PCs) to diols. | ||
In 2019, Liu's group introduced the earth-abundant Fe-PNP pincer catalyst for converting PPC to 1,2-PD and methanol (Fig. 6).211 By optimizing the polymer loading to 0.2 g, catalyst to 0.03 g, and KOt-Bu to 0.006 g, the reaction was conducted at 140 °C for 30 hours, in 0.8 g of THF. Additionally, 3.1 g of isopropanol was clearly used as the hydrogen source, resulting in yields of 65% for 1,2-PD and 43% for methanol (Table 3, entry 6). This catalytic protocol demonstrates the use of poly(propylene carbonate) to produce valuable fuels like propylene glycol and methanol, which serve as essential building blocks for various chemicals. The protocol utilizes an iron-based catalyst, offering a more sustainable alternative to other catalyst systems. Isopropanol or ethanol, sourced from renewable resources, are employed as hydrogen donors, eliminating the necessity to handle flammable hydrogen gas. This protocol has a lower environmental impact with reduced waste generation compared with the catalysts reported by Zuber and Kumar (Mn(II) NNP, Mn(II)PNN), with an E factor of 0.80 and ξ value of 3.1 × 104 °C min (details in Table 3 and Fig. S2†). However, it is less efficient as it requires a longer reaction time at 140 °C and results in a lower monomer yield of approximately 67%.
Also in 2018, Westhues and colleagues evaluated triphos-based Ru catalysts (Fig. 6) for the hydrogenolysis of polycarbonate (BPA-PC). Their method involved 0.87 g of BPA-PC, 0.01 g of catalyst, and 0.003 g of HNHf2 co-catalyst and occurred at 140 °C and 100 bar H2 over 6 hours, using 0.88 g of 1,4-dioxane as the solvent. Under optimized reaction conditions it is fully converted into a 99% yield of both BPA and methanol in 6 hours. No side reactions were observed.182 Challenges include high hydrogen pressure, product separation, and catalyst recovery. The use of a co-catalyst instead of a base improved the monomer BPA yield to 99% in 16 hours at low temperature (140 °C). However, the ε coefficient (7.3 × 10−6 °C−1 min−1) is the lowest, along with the E factor (0.3 a.u.) and ξ (39564 °C min), due to a low solvent to polymer ratio (2.5) resulting in maximum yield compared with the RuMACHO and Milstein catalysts (Table 3 and Fig. S3†). BPA-PC recycling poses economic challenges but generates less waste with minimal environmental impact. In 2019, Kindler and his group conducted experiments on the hydrogenolysis of BPA-PC using a ruthenium-MACHO-BH catalyst with a tridentate PNP ligand (Fig. 6).212 The study revealed that achieving a significant monomer BPA-PC yield of 96% from 0.017 g of BPA-PC required high catalyst loadings (0.002 g), base (0.04 g), high temperatures (140 °C), and long reaction times (6 hours). The catalyst's efficiency was demonstrated on 1.72 g of BPA-PC with a lower catalyst loading (0.02 g), base (0.04 g), and shorter reaction time (16 hours) at 80 °C and 45 bar H2, resulting in an 81% isolated BPA yield and methanol as the only product. No self-recombination reactions were observed due to the controlled depolymerization process. The ruthenium-MACHO-BH catalyst showed lower activity compared with the Milstein catalyst, indicating its efficiency in the depolymerization process.213 Challenges include the difficulty in recovering the catalyst, product separation, and cost-effectiveness for scale-up due to the use of expensive ligands and noble metals. The ruthenium-MACHO-BH catalyst is tolerant and robust under harsh catalytic conditions. The sample exhibited a high ε value of 1.9 × 10−5 °C−1 min−1, attributed to a rapid monomer yield of 96% in just 6 hours at 140 °C. Additionally, it had a high E factor of 6.2 and a ξ value of 326781 °C min, likely due to the high solvent to polymer ratio of 53 (Table 3 and Fig. S3†).
In the same year, Alberti's team successfully depolymerized 0.34 g of BPA-PC using 0.03 g Milstein catalysts. The hydrogenolysis process was carried out at 140 °C with 45 bar H2, using 1.7 g of THF as the solvent and 0.04 g of KOt-Bu. This resulted in complete depolymerization of BPA to monomer with a 99% yield within 24 hours.213 Even with a reduced Milstein catalyst concentration of 0.015 g, the BPA yield remained high at 93% under optimized reaction conditions (Table 3, entries 10 and 11). Bisphenol A was isolated with an 86% yield, indicating no need to purify the end-of-life DVD before depolymerization. This result is comparable to Klankermayer's work,182 achieving a 37% yield at a lower hydrogen pressure of 10 bar, but with higher catalyst loading and a longer reaction time. The reusability, cost, and sustainability of ruthenium catalysts are still a concern. Milstein catalysts have a E factor (0.50 a.u.) and exhibit low values of ξ (1.0 × 104 °C min) due to a low solvent to polymer ratio (4.2). Although they yield a high monomer yield, the ε coefficient is low (4.613 × 10−6 °C−1 min−1) because of long reaction times (24 hours) at a high temperature of 140 °C. This results in less environmental impact but lower ε values (Table 3 and Fig. S3†).
In 2020, Alberti and his team introduced a non-noble Fe-PNP pincer catalyst (Fig. 6) for hydrogenolysis of BPA-PC and PPC yielding high diol amounts under mild condtions.214 For example, using 0.03 g of catalyst, 1 g of BPA-PC in 17.7 g of THF at 120 °C and 45 bar H2 for 24 hours resulted in an 81% BPA yield. Similarly, 1 g of PPC produced 1,2-PD with a 77% yield using 0.08 g of catalyst and 35.6 g of THF under the same reaction conditions (Table 3, entries 12 and 13). Compared to a Mn-based catalyst, the Fe-based system showed higher diol yields in shorter reaction times without the need for additional base.210 Additionally, the Fe-based system operated at lower temperature and pressure compared to the work of Krall's group.200 A non-noble Fe-PNP pincer catalyst efficiently hydrogenolyses PPC in THF with a catalyst loading ratio of 34.5. However, it has a high E factor (6.03 a.u.) and ξ value (1.3 × 106 °C min) due to the high solvent to polymer ratio used. The low ε value (4.4 × 10−6 °C−1 min−1) is attributed to the excessive reaction time, resulting in a lower monomer yield (80%). This process is not efficient for BPA hydrogenolysis due to high energy consumption (Table 3 and Fig. S3†).
In 2021, Dahiya's group introduced a new Cp*Co(III) catalyst (Fig. 6) for converting 5 g of BPA-PC/PPC into BPA and 1,2-PD. The reaction used 0.1 g of catalyst, 0.884 g of KOt-Bu base, and 38.5 g of n-Bu2O as solvent, at 160 °C, 60 bar H2 for 24 hours. The catalyst demonstrated high efficacy, yielding 83% of BPA and 76% of 1,2-PD under specific conditions215 (Table 3, entries 14 and 15). This cost-effective catalyst is ideal for large-scale production as it is non-noble and works well with isopropanol as a transfer hydrogenating reagent. It reduces reagent consumption with a low solvent to polymer ratio of 6.8, resulting in a better monomer yield of 83% compared to Fe pincer catalysts. This leads to a lower environmental impact factor of 0.9–1.2 a.u. and ξ of 3.7 × 104 °C min (Table 3 and Fig. S3†). However, drawbacks include cobalt's toxicity, the need for high hydrogen pressure, and the requirement for a base in the reaction.
In 2022, Payne and colleagues developed Zn(II) catalysts, Zn(S)2 and Zn(S)Et, based on half-salen (S) ligands (Fig. 6). They achieved a BPA yield of 85–90% by transforming 0.25 g of BPA-PC using 0.01 g of catalyst at room temperature in 4.4 g of 2-Me-THF as the solvent (Table 3, entry 16).216 These catalysts efficiently degraded commercial polyesters and polycarbonates to produce valuable chemicals like green solvents and building blocks. Zn(S)2 and Zn(S)Et showed rapid depolymerization of BPA-PC at room temperature in 2-Me-THF with high activity. The product distribution varied with the reaction conditions. This research offers new possibilities for polymer design and recycling in the plastics industry, with ongoing efforts to optimize these catalysts for chemical recycling applications. Table 3 and Fig. S4† demonstrate that the Zn(II) catalyst has a low ξ of 6241 °C min and a high ε coefficient of 3.16 × 10-4 °C−1 min−1. This indicates that the Zn(II) catalyst can achieve a high monomer yield in just 1 hour using the same amount of catalyst compared to the Co(III) catalyst which required 24 hours. These findings suggest that Zn(II) is a promising candidate for the industrial production of BPA from BPA-PC waste (Table 3 and Fig. S4†). The catalyst's recyclability also simplifies the purification process. The transition from noble to non-noble metals in chemical waste plastic recycling signifies a shift towards more efficient, cost-effective, and environmentally friendly solutions. Recent research and development have focused on utilizing various catalysts and reducing agents to improve depolymerization techniques. Future challenges in this field will revolve around improving catalyst affordability and enhancing depolymerization efficiency.217–219
Ahrens and his group conducted experiments using chemical recycling methods for thermoset epoxy resins and composites, including wind turbine blade shells. They successfully recovered 83% BPA from BPA-based epoxides using 0.043 g of waste epoxy resins, 0.001 g of Ru-trios catalyst, and 4.5 g of THF/toluene mixture at 160 °C for 16 hours.220 Carbon fibre-based epoxides were recycled using 0.187 g of waste, 0.011 g of catalyst, and 1.53 g of THF/toluene mixture at 160 °C for 72 hours, resulting in a 57% carbon fibre yield, 13% BPA, and residual acetone and phenol fractions (details in Table 3, entries 17 and 18). The selectivity in cleaving phenols over alkyl alcohols is attributed to the difficulty in cleaving alkyl alcohols. Ruthenium plays a key role in facilitating dehydrogenation and oxidative addition processes, activating C–O bonds. The reduction of Ru to a Ru intermediate helps in releasing phenol, acetone, and phenolics, resulting in the complete depolymerization of the substrate. The author did not observe any self-recombination reactions during the process. The high purity of the recovered BPA enables potential reuse in epoxy resin and polyester production chains, as well as in high-quality glass and carbon fibers. However, challenges such as the catalyst's toxic nature, scale-up difficulties, sustainability, and catalyst recycling need to be addressed. Recycling BPA-based epoxides leads to more waste and a higher environmental impact compared with depolymerizing carbon fibre-based epoxides. This is because BPA-based epoxides have higher E (18.5) and ξ (3.4 × 106 °C min) values due to excessive solvent consumption (92.4) with 83% BPA recovered. In contrast, depolymerization of carbon fibre-based epoxides results in a lower E factor (1.7) and ξ values (1.1 × 10−6 °C−1 min−1) due to a lower solvent-to-polymer ratio (7.26). The ε value is also lower (1.2 × 10−6 °C−1 min−1) at high temperatures (160 °C) with longer reaction times (16–17 hours) (Table 3 and Fig. S3†). However, industrial feasibility is not viable based on the green chemistry matrix parameter.
3.3. In situ hydrogen donor through hydrosilylation and hydroboration
In recent years, silanes (R3Si-H) and hydroboranes (R2B-H) have emerged as promising candidates for reducing OXPs and unsaturated organic compounds. This is because the slightly polar and weak Si–H and B–H bonds are more easily activated than the strong nonpolar H–H bond (bond dissociation energy of 92 kcal mol−1 in SiH4, 99 kcal mol−1 in BH3, and 104 kcal mol−1 in H2).221–223 These reductants can improve the ability to produce high-quality products with high efficiency. They can be used in metal-free catalytic systems under mild reaction conditions, such as a low temperature of ≤120 °C and pressure of ≤1 bar. Few groups have reported the successful selective hydrogenation of C–O bonds in the presence of organometallic catalytic systems using silanes/hydroboranes as the reducing agents.112,187 Hydrosilylation of carbonyl functions, which is both kinetically and thermodynamically helpful, can also be taken advantage of by hydrogen transfer to the carbonyl group.3.4. In situ hydrogenolysis of polyesters
Hydrosilylation methods can be used to convert polyesters into monomers, while reductive depolymerization breaks C–O or C–C bonds directly. Various hydrosilylation approaches with homogeneous catalyst systems, such as Ir(III), La(III), and Zr(IV) catalysts, have been developed. Selective deconstruction of polycarbonates and polyesters into valuable alcohols or carboxylic acids has been achieved with high yields. Hydrogenation of polymers requires harsh conditions (54 atm H2, 160 °C). Hydrosilylation of carbonyl functions is kinetically and thermodynamically favored.155,159,160,187,197In 2015, Feghali demonstrated the efficient breakdown of various polyesters using hydrosilanes and metal-free catalysts. The catalysts B(C6F5)3 and [Ph3C+,B(C6F5)4−] were paired with affordable reductants like triethylsilane (Et3SiH), tetramethyldisiloxane (TMDS), and polymethylhydrosiloxane (PMHS) under ambient conditions. The reaction involved 0.096 g of PET, 0.005 g of TPFP-B, 0.125 g of Et3SiH, and 2.23 g of CH2Cl2 solvent, run at room temperature for 3 hours.184 Impressive yields of 1,4-BDM-Si and EG-Si were achieved at 91% and 72%, respectively. The authors overlooked self-recombination reactions, enabling controlled reactions via reagent concentrations and conditions. Hydrolysis with TBAF 3H2O resulted in the quantitative extraction of both 1,4-BDM and EG. In another reaction, using 0.096 g of PET and 0.005 g of TPFP-B with 0.183 g of Et3SiH resulted in a 49% yield of p-xylene (Table 6, entry 2). When 0.611 g of PMHS or 0.201 g of TMDS were used instead of Et3SiH under the same conditions, yields of p-xylene ranged from 75% to 82% (Table 4, entries 3 and 4). This sustainable synthesis, devoid of noble metals, from plastic wastes is a significant advancement, though scalability and toxicity remain challenges.112,174,184 The protocol has a high E factor (3 to 9) and a ξ value of 2.022 × 10−4 °C−1 min−1 compared with homogeneous catalysts with base or co-catalysts. This is because of the high solvent-to-polymer ratio (≤28), resulting in a slightly higher environmental impact. However, the protocol demonstrates a high ε value (15225 °C min) due to the high monomer yield at a low temperature (25 °C) and short reaction time (3 hours) (Table 4 and Fig. S4†). Alternative methods using cost-effective, environmentally friendly, and widely available metal catalysts are needed for efficient plastic waste hydrogenolysis.
| Entry | Polymer | Solvent | Solvent/polymer ratio | Catalyst | Reagent | Cat./polymer ratio | Time (hour) | Temp. (°C) | Monomer yield (%) | E factor (a.u.) | ε (°C−1 min−1) | ξ (°C min) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | PET | CH2Cl2 | 23.5 | TPFP-B | Et3SiH 0.125 g | 0.002 | 3 | RT | 91 | 3.1 | 2.02112 × 10−4 | 15225 |
184 |
| 1,4-BDM-Si | |||||||||||||
| 2 | CH2Cl2 | 24.1 | Et3SiH 0.183 g | 16 | 49 | 9.4 | 2,04106 × 10−5 | 460157 |
|||||
| p-Xylene | |||||||||||||
| 3 | CH2Cl2 | 28.6 | PMHS 0.611 g | 75 | 7.2 | 3.12051 × 10−5 | 232781 |
||||||
| p-Xylene | |||||||||||||
| 4 | CH2Cl2 | 24.3 | TMDS 0.201 g | 82 | 5.6 | 3.41661 × 10−5 | 165616 |
||||||
| p-Xylene | |||||||||||||
| 5 | PET | C6D5Cl | 16.8 | Brookhart's | Et3SiH 0.201 g | 0.004 | 72 | 70 | 63 | 3.2 | 2.08333 × 10−6 | 1525717 |
155 |
| 1,4-BDM-Si | |||||||||||||
| 6 | PET | Toluene | 51.1 | Mo-dioxo complex | PhSiH3 0.162 g | 0.05 | 96 | 160 | 65 | 15.1 | 7.05295 × 10−7 | 21389339 |
112 |
| p-Xylene | |||||||||||||
| 7 | PET | Toluene | 50.0 | 63 | 15.2 | 6.83594 × 10−7 | 22299617 |
||||||
| p-Xylene | |||||||||||||
| 8 | PBT | Toluene | 44.4 | 86 | 11.1 | 9.3316 × 10−7 | 11958289 |
||||||
| p-Xylene | |||||||||||||
| 9 | PET | C6H5Cl | 49 | Zn (OAc)2 | PhSiH3 0.162 g | 0.002 | 96 | 160 | 31 | 30.1 | 3.36372 × 10−7 | 89402530 |
156 |
| p-Xylene | |||||||||||||
| 10 | PET | C6H5Cl | 48.7 | (EtO)2MeSiH 0.162 g | 61 | 15.2 | 6.61892 × 10−7 | 22972283 |
|||||
| p-Xylene | |||||||||||||
| 11 | PET | C6H5Cl | PhSiH3 0.162 g | 59 | 15.7 | 6.40191 × 10−7 | 24556124 |
||||||
| p-Xylene | |||||||||||||
| 12 | PBT | C6H5Cl | 42.0 | PhSiH3 | 67 | 13.5 | 7.26997 × 10−7 | 18634770 |
|||||
| p-Xylene | |||||||||||||
| 13 | C6H5Cl | 42.3 | (OEt)2MeSiH | 15 | 60.2 | 1.6276 × 10−7 | 370137914 |
||||||
| p-Xylene | |||||||||||||
| 14 | PBT | C6H5Cl | 42.0 | PhSiH3 | 52 | 17.3 | 5.64236 × 10−7 | 30663376 |
|||||
| p-xylene |
In 2018, Monsigny and his colleagues developed a pincer complex of iridium(III) with boranes [B(C6F5)4] (POCOP = 1,3-(tBu2PO)2C6H3) for the depolymerization of aliphatic and aromatic polyesters PET, PPC, BPA-PC, and PLA wastes. 0.04 g of PET was converted into silylated 1,4-benzenedimethanol (BDM-Si), EG-Si, ethane, and water using 0.2 g of Et3SiH and 0.003 g of an iridium catalyst at 70 °C for 72 hours in 0.48 g of C6D5Cl as a solvent. The yields obtained were 63% and 48% (Table 4, entry 5). Further heating at 90 °C for 2 weeks resulted in the formation of para-xylene and ethane.155 These silyl ethers can be utilized in Ullman's coupling reactions to produce ethers or can be hydrolyzed to obtain useful alcohols. Hydrolyzing BDM-Si from PET yields 1,4-benzenedimethanol (BDM), a valuable building block for pesticides, perfumes, or dyes. The iridium(III) pincer catalyst is effective for low catalyst loading hydrogenolysis of OXPs, but its high cost hinders scalability and reusability, and poses challenges for monomer separation. Toxic solvent waste (dichloromethane and chlorobenzene) poses environmental risks. Green chemistry metrics favor the metal-free silylation process.112,184 However, the use of iridium(III) with boranes exhibited a low environmental impact (E factor: 3.2 a.u. and ξ = 2.1 × 10−6 °C−1 min−1) due to a lower solvent to polymer ratio (16.8), higher catalyst usage, and lower ε coefficient (1.5257 × 106 °C min) resulting from an enhanced reaction temperature (70 °C) and extended reaction time (72 hours) (Table 4 and Fig. S4†). However, it is not an efficient process for industrial use.
In 2020, Nunes and his group studied silane-based molybdenum-dioxo complex (MoO2Cl2) (Fig. 6), showing its profound efficacy in hydrosilylation across an array of functional groups, particularly ester groups.112 Remarkably, this complex proved adept for the hydrogenolysis of polyesters, encompassing 0.048 g of PET being converted into silylated p-xylene and ethylene glycol as co-products using 0.2 g of PhSiH3 and 0.112 g of a Mo-dioxo catalyst in 2.2 g of toluene at 160 °C for 96 hours. The yields obtained were 65% and 63% (Table 4, entries 6–8) despite an extended reaction time and the same temperature. In a similar experiment, 0.5 g of PBT was converted into p-xylene and butanediol as co-products with a yield of 86% using 0.112 g of a Mo-dioxo catalyst, 0.2 g of PhSiH3, and 2.2 g of toluene at 160 °C for 96 hours. However, the Mo complex/silane-based catalyst may not be suitable for industrial-scale plastic recycling due to high waste generation, a high solvent to polymer ratio of 50, a low product yield (65–80%), and an excess reaction time of 96 hours. The high E factor of 15 a.u. and ξ value of 2.22996 × 107 °C min, along with a low ε coefficient value of 6.835 × 10−6 °C−1 min−1, were observed (Table 4 and Fig. S4†).112
In 2021, Fernandes and colleagues unveiled a practical approach using the affordable and ubiquitously available salt, Zn(OAc)2·2H2O.156 The study investigated the depolymerization of 0.048 g PET waste using 0.005 g Zn (OAc)2·2H2O as a catalyst. Initially, the reaction was attempted in dioxane with 0.162 g PhSiH3 as the reducing agent but was unsuccessful due to low PET solubility. Changing the solvent to 2.2 g chlorobenzene and increasing the temperature to 160 °C resulted in a 65% yield of p-xylene and a 43% yield of ethylene glycol after 96 hours. Reducing the catalyst loading to 0.0025 g decreased the p-xylene yield to 39%. Using a different reducing agent, 0.162 g (EtO)2MeSiH, under the same conditions in 2.2 g of chlorobenzene yielded a 55% p-xylene yield. The method was also applied to PET shirts from domestic waste, resulting in 59–61% p-xylene and 36–41% ethylene glycol yields (Table 4, entries 9–14). This approach offers a sustainable method for producing p-xylene from plastic waste, a valuable compound with various industrial applications. Additionally, the depolymerization of 0.055 g PBT using 0.153 g (EtO)2MeSiH as the reducing agent and 0.005 g Zn (OAc)2·2H2O in same amount of chlorobenzene at 160 °C for 4 days yielded a 15% p-xylene yield along with a mixture of intermediates (THF, butane). In contrast, using 0.153 g of PhSiH3 instead of (EtO)2MeSiH under the same conditions resulted in a mixture of p-xylene (67%) and THF (70%).156 This catalyst is efficient for recycling plastic waste on a gram scale, but has drawbacks such as low monomer yield, high reaction temperature, and long reaction time. Table 4 and Fig. S4† show that the Zn (OAc)2·2H2O catalysts have a high environmental factor values of 13.5 and 60.2, with an energy environmental impact of 3.7 × 107 °C min, leading to increased waste generation and environmental impact.
3.5. In situ hydrogenolysis of polycarbonates
In 2015 Feghali and his group also successfully silylated H-BPA-PC and CH3-BPA-PC without using metal.184 Hydrosilylation of PC-BPA is faster and less selective than the reduction of polyesters. TMDS is a strong reductant in the depolymerization of BPA-PC. 0.0961 g of BPA-PC, 0.244 g of Et3SiH, and 0.0051 g of TPFP-B in 2.23 g of CH2Cl2 as a solvent at room temperature for 1 hour resulted in an 82% yield of H-BPA-Si and a 28% yield of CH3-BPA-Si. The highest yield of H-BPA-Si (98%) was achieved using 0.295 g of TMDS under the same conditions using 2.2 g of CH2Cl2 solvent. In addition, 0.944 g of PMHS resulted in gel formation despite the use of benzene. Instead of B(C6F5)3, they used 0075 g of [PH3C]+ [B(C6F5)4]− as a catalyst along with 0.244 g of Et3SiH, and 0.0961 g of BPA-PC for hydrogenolysis to produce H-BPA-Si and CH3-BPA-Si with yields of 47% and 26% at room temperature after 16-hours (Table 5, entries 1–4). The in situ hydrogenolysis of polycarbonates using the TPFP-B catalyst with silane as a reducing agent was successful at low temperatures and short reaction times. The strong reducing agent TMDS yielded higher product yields, resulting in a higher ε coefficient (6.53 × 10−4 °C−1 min−1) and a low E factor of 2.89 compared with other silanes (Table 5 and Fig. S5†) due to the short reaction time at room temperature along with the high solvent to polymer ratio.| Entry | Polymer | Solvent | Solvent/polymer ratio | Catalyst | Cat./polymer ratio | Reagent | Time (hour) | Temp. (°C) | Monomer yield (%) | E factor (a.u.) | ε (°C−1 min−1) | ξ (°C min) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | BPA-PC Me-BPA-PC | CH2Cl2 | 24.5 | TPFP-B | 0.0020 | Et3SiH (0.244 g) | 1 | RT | 82 | 3.4 | 5.4666 × 10−4 | 6202 | 184 |
| BPA-Si | |||||||||||||
| 2 | CH2Cl2 | 25.3 | TMDS 0.295 g | 98 | 2.8 | 6.5333 × 10−4 | 4425 | ||||||
| BPA-Si | |||||||||||||
| 3 | CH2Cl2 | 32.1 | PMHS (0.947 g) | 0 | 0 | 0 | 0 | ||||||
| Gel formation | |||||||||||||
| 4 | CH2Cl2 | 26.2 | [PH3C]+, [B(C6F5)4]− | Et3SiH (0.244 g) | 16 | RT | 47 | 6.2 | 1.9855 × 10−5 | 318422 |
|||
| BPA-Si | |||||||||||||
| 5 | PPC | C6D5Cl | 22.7 | Brookhart's iridium(III) | 0.002 | Et3SiH (0.116 g) | 3 | 65 | 99 | 3.1 | 8.4615 × 10−4 | 36771 |
155 |
| 1,2-PD-Si | |||||||||||||
| 6 | BPA-PC | C6D5Cl | 15.1 | 0.002 | 6 | 88 | 1.9 | 3.7606 × 10−4 | 51696 |
||||
| BPA-Si | |||||||||||||
| 7 | C6D5Cl | 12.3 | 0.003 | TMDS/NaOH | 12 | 65 | 83 | 3.2 | 1,2808 × 10−5 | 251364 |
|||
| BPA | |||||||||||||
| 8 | BPA-Si | THF | 9.2 | — | 0.27 | [Bu4N]+ [F]−3H2O (1.08 g) | 1 | RT | 88 | 2.6 | 5.8667 × 10−4 | 4446 | |
| BPA | |||||||||||||
| 9 | PPC | C7D8 | 12.3 | MgBu2 | 0.01 | HBpin | 3 | 65 | 91 | 1.8 | 2.000 × 10−4 | 9061 | 224 |
| 1.2-PD boronate | |||||||||||||
| 10 | PPC | C7D8 | 2.6 | Mg2(Xyl Nacnac) | 0.1 | HBpin | 6 | RT | 96 | 0.5 | 1.011 × 10−4 | 4678 | 226 |
| 1.2-PD boronate | |||||||||||||
| 11 | BPA-PC | C6D6 | 10.9 | Lanthanum(III) tris(amide) | 0.0045 | HBpin | 24 | 100 | 92 | 1.34 | 6.322 × 10−6 | 211047 |
180 |
| BPA-B |
In 2018, Monsigny and his team developed a method to increase the production yield of aliphatic and aromatic polycarbonates by reducing the homogenous catalyst amount through hydrosilylation. They tested Brookhart's iridium(III) catalyst as an alternative to B(C6F5)3 and found that it enabled successful hydrosilylation with lower catalyst loadings, albeit at higher temperatures and longer reaction times.155 In the initial experiment, reductive depolymerization of 0.02 g of PPC using hydrosilylation conditions with catalyst (0.01 g), Et3SiH (0.134 g), and C6H5Cl (0.33 g) at 65 °C for 3 hours yielded MeOSiEt3 and disilylether PG-Si in quantitative yield (99%) (Table 5, entries 5–8). In a second experiment, depolymerizing 0.0284 g of PC-BPA under similar conditions with the same amount of catalyst (0.01 g), Et3SiH (0.116 g), and chlorobenzene (0.33 g) for 6 hours resulted in BPA-Si in high yield (88%), along with CH4 gases and methyl bisphenol derivatives as Friedel–Crafts-like products, while no self-recombination reaction was observed. Hydrolysis of the hydrosilylation products from PPC and PC-BPA yielded the corresponding alcohols, propylene glycol, and bisphenol A (BPA), which are the original polymer's monomers. A third experiment involved the hydrolysis of silylative polycarbonates of PC-BPA (0.456 g) using 1.08 g of TBAF in 3.5 g of THF at room temperature for 2 hours, resulting in the recovery of the original monomers, such as bisphenol A (BPA), in 88% yield. Additionally, hydrogenolysis of 0.254 g PC-BPA with the same catalyst, 0.134 g of TMDS, and 3.33 g C6H5Cl at 65 °C for 12 hours led to gel formation without Friedel–Crafts products. Subsequent hydrolysis of the crude mixture with 1 ml of NaOH in water/methanol at room temperature for 2 hours yielded BPA of 83%. The catalyst effectively regenerates original monomers or valuable chemicals during plastic depolymerization with low catalyst loadings, but its high cost hinders scale-up. The energy-intensive production of hydrosilanes and silicon by-products necessitates the exploring of alternative reduction methods. The hydrosilylation strategy using Brookhart's iridium(III) catalyst shows lower waste generation (E = 2 to 3) and environmental impact (ξ = 51696 °C min) compared with TPFP-based hydrogenolysis184 (Table 5 and Fig. S5†). It also offers higher energy efficiency (ε = 3.76 × 10−5 °C−1 min−1) due to a high product yield, less catalyst and solvent consumption, and time. However, it may not be feasible for industrial use due to its lack of sustainability.
Szewczyk et al. utilized a readily accessible ligand-free Mg(II)Bu2 catalyst (0.014 g), 0.1 g of PPC, and 0.396 g of pinacolborane to efficiently convert PPC into borate ester 1,2-PD.224 The reaction was carried out at 65 °C under atmospheric pressure for 3 hours in 0.988 g of C7D8 solvent, resulting in high yields (91%) and methanol borate as co-products (Table 5, entry 9). This method reduces catalyst usage, shortens reaction times, and yields impressive results by using boranes as reducing agents instead of high-pressure hydrogenations. Leitner and colleagues developed a transition metal-catalyzed reduction of organic carbonates using pinacolborane (HBpin), which efficiently converts polycarbonates into valuable alcohols with low catalyst loadings.225 However, there is a need for transition metal-free protocols using earth alkaline metals. This procedure offers mild reaction conditions, fast reaction times, low catalyst loading, and a broad scope, competing favorably with transition metal-catalyzed protocols. While not entirely environmentally friendly, they have slightly higher environmental factors (E = 1.9; ξ = 9061.1 °C min) and economic processes (ε = 2.0 × 10−4 °C−1 min−1) compared to Brookhart's iridium(iii) catalyst (Table 5 and Fig. S5†).
In 2020, Cao and his group developed a solvent-free low-valent magnesium(I) catalyst [((XylNacnac)Mg)2] (0.02 g) for the efficient hydroboration of PPC (0.05 g) using HBpin (0.166 g) as a reducing agent. The hydroboration products can also be hydrolyzed to form desired diols. The reaction yielded 96% borate ester 1,2-PD and MeO-B at room temperature, and atmospheric pressure over a 6 hour period (Table 5, entry 10).226 The reactions between [((XylNacnac)Mg)2] and polycarbonate in 1:1 or 1:2 molar ratios were controlled but did not produce new intermediates. Using [((XylNacnac)Mg)2] in the hydroboration depolymerization of PPC resulted in a 96% yield of borate ester 1,2-PD. The protocol was more efficient than manganese and divalent Mg(II)Bu2 catalyzed hydroboration of organic carbonates. It can also serve as an effective pre-catalyst for the catalytic hydroboration of various esters under mild conditions. Challenges such as catalyst recovery, scale-up, and stability need to be addressed. Table 4 and Fig. S5,† demonstrate the approach has a low environmental impact with an E factor of 0.5 and ξ value of 4678 °C min due to reduced solvent consumption and high monomer yield (96%). While a slightly lower ε coefficient 1.011 × 10−4 °C−1 min−1 was observed at room temperature after 6 hours, it is still more environmentally friendly compared with hydrosilylation/hydroboration-based homogeneous catalysts, leading to reduced waste generation.
In a recent study, Kobylarsk and his group reported that a commercially available La(III) tris(amide) (LaN*3) catalyst system with HBpin as a reducing agent can successfully depolymerize PPC and /BPA-PC into diols under mild conditions. Hydroboranes were used to depolymerize carbonylated polymers as a hydride source. Reaction with LaN*3 and HBpin at room temperature for 24 hours resulted in the decomposition of PPC into PG-B and MeO-B (70% yield), while at 100 °C, PC-BPA was transformed into BPA-B (78%) and MeO-B (83%). LaN*3 (0.034 g) and HBpin (0.117 g) reacted at 100 °C for 24 hours, decomposing 0.034 g of PC-BPA into BPA-B (78%) and MeO-B (83%) in the presence of 0.285 g of C6D6 as solvent (Table 5, entry 11). No over-reduced by-products were observed, even with excess HBpin (6.6 equiv.).180 In comparison, using B(C6F5)3 (0.005 g) with TMDS (0.295 g) as the organo-catalyst for PC-BPA depolymerization, silylated bisphenol-A was obtained with a 98% yield in just 1 hour without heating. Challenges include scaling up the process, catalyst recovery, and the slightly high environmental impact (1.34) with high ξ values (211047 °C min) compared with Mg(I) catalyst due to the high solvent to polymer ratio, high temperature (100 °C) with long reaction times (24 hours).184 Waste generation is slightly high, and the process is not considered sustainable or robust under the current conditions (Table 5 and Fig. S5†).
4. Development of heterogeneous catalysts for hydrogenolysis of OXPs
4.1. Heterogeneous catalytic hydrogenolysis overview
The homogeneous hydrogenolysis of OXPs into diols is currently carried out using molecular catalysts and expensive solvents (anisole, 1,4-dioxane, and THF). These processes require high H2 pressures, long reaction times (16–48 hours), expensive ligands, and air-sensitive catalysts. Recycling catalysts in these processes is extremely difficult.183,227 To overcome these challenges, heterogeneous catalysis was investigated. An ideal heterogeneous catalyst for hydrogenolysis must be easy to separate, thermally stable, resistant to moisture and air, nontoxic, selective, indefinitely recyclable, as well as capable of running in solvent-free environments.228 For these reasons, researchers are focusing on studying heterogeneous catalysts to achieve the desired outcome. Heterogeneous catalysts typically comprise an active metal or metal oxide, a promoter, and a support.229–231 Active species in heterogeneous catalysts are responsible for catalytic activity and typically consist of one or more compounds, each contributing unique functional properties or interacting with one another at their interfaces to produce synergistic effects.232 Promoters are added to catalyst compositions to improve their physicochemical properties and tune their activity, selectivity, and lifetime.233The support in heterogeneous catalysis plays a crucial role as a carrier or medium for delivering catalytic substances to aid in chemical reactions ensuring improved metal dispersion and catalyst lifetime.234 The preparation method of the catalyst is a key factor, as each method has its benefits and drawbacks, and the method chosen is based on the specific benefits.90,235–246Table 6 depicts the process parameters that influence the production of the desired material.238
| Parameters | Affected properties |
|---|---|
| pH | Phases, particle sizes, and textural properties |
| Anion (surfactants) | Morphology and textural properties |
| Aging | Purity, crystallinity, and textural properties |
| Additives | Textural properties |
| Precipitating agent | Phase and homogeneity |
| Solvents | Crystallinity and textural properties |
| Mixing consequence | Precipitate composition and homogeneity |
| Temperature | Phase, particle sizes, and textural properties |
Biomass and plastics share similar chemical bonds like C–C, C–O, and C–N. Plastics like PE, PP, PVC, PS, and PF contain C–C bonds like lignin and fatty acids.247 Cellulose, hemicellulose, chitin, and phenolic monomers have C–O linkages. Ester linkages are common in triglycerides and polyesters like PET, and BPA-PC, while C–N linkages are found in proteins, polyamides, and PU. Heterogeneous catalysts have been effective in converting biomass into valuable products by breaking down high-energy bonds in biomass molecules to produce monomers and other valuable products. These catalysts can also adjust selectivity by changing catalyst acidity and textural properties.239,240 This review focuses on the hydrogenolysis of OXP, specifically PET and BPA-PC, using heterogeneous catalysts and external hydrogen sources to convert plastic waste into monomers. It explores the selective cleavage of C–O/C–C bonds depending on catalyst type, properties, and operating conditions. Recent advancements in heterogeneous catalysts are discussed, along with their impact on OXP hydrogenolysis and consideration of green chemistry principles. A key challenge is identifying a catalyst capable of efficiently catalyzing OXP hydrogenolysis to produce valuable compounds like aromatics, oxygenates, jet fuel range compounds, and cycloalkanes (Fig. 7). To date, no catalytic system has achieved this goal successfully.
 | ||
| Fig. 7 Hydrogenolysis of PET (orange) and BPA-PC (yellow) to produce valuable compounds. | ||
4.2. Hydrogenolysis of PET/PBT/PLA/EP
In 2018, Uekert and his group developed CdS/CdOx quantum dots and carbon nitride/nickel phosphide (CNx/Ni2P) heterogeneous catalyst used for the production of clean H2 fuel from 0.1 g of PET, with 0.1 g of catalyst at 25 °C for 120–144 hours under KOH/NaOH solution. They also obtained organic byproducts such as formate, acetate, and pyruvate (Fig. 8a and Table 7).241,242 | ||
| Fig. 8 (a) PET photoreformation into clean H2 fuel. (b) Synthesis of gasoline and jet fuel (yellow) range compounds from PET using Pt/C and Ru–Cu/SiO2. | ||
| Entry | Polymer | Solvent | Solvent/polymer ratio | Catalyst | Cat./polymer ratio | Time (hour) | Temp. (°C) | Monomer yield (%) | E factor (a.u.) | ε (°C−1 min−1) | ξ (°C min) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | PET (water bottle) | 0 | — | CdS/CdOx | — | 4 | 25 | H2 | — | — | — | 241 |
| 2.17 ± 0.38% | ||||||||||||
| H2/gsub | ||||||||||||
| 2 | 0 | — | CNx/Ni2P | — | 25 | 25 | H2 | — | — | — | 242 | |
| 34.3 ± 3.1 H2/gsub | ||||||||||||
| 3 | 0 | — | Pt/C + 2.5Ru–2.5Cu/SiO2 | — | 7 | 370 | 73 | — | — | — | 243 | |
| Aromatics | ||||||||||||
| 4 | Commercial PET | H2O | 100 | 2Ru/Nb2O5 | 0.01 | 16 | 320 | 84 | 22.6 | 2.73438 × 10−6 | 8288397 |
90 |
| Aromatics | ||||||||||||
| 5 | H2O | 2Ru/TiO2 | 72 | 26.4 | 2.34375 × 10−6 | 11281430 |
||||||
| Aromatics | ||||||||||||
| 6 | H2O | 2Ru/HZSM-5 | 32 | 59.4 | 1.04167 × 10−6 | 57112242 |
||||||
| Aromatics | ||||||||||||
| 7 | H2O | 2Ru/ZrO2 | 73 | 26.0 | 2.3763 × 10−6 | 10974467 |
||||||
| Aromatics | ||||||||||||
| 8 | PET | DMSO-d6 | 23.1 | Mo-dioxo/C | 0.043 | 24 | 260 | 87 | 3.7 | 2.323 × 10−6 | 1600936 |
183 |
| TPA | ||||||||||||
| 9 | PET | H2O | 75.3 | 2Ru/Nb2O5 | 0.01 | 12 | 280 | 91.3 | 15.7 | 5.76389 × 10−6 | 2727120 |
244 |
| Aromatics | ||||||||||||
| 10 | MeOH | 197.8 | Cu–Na/SiO2 | 0.004 | 6 | 210 | 99 | 37.7 | 1.30952 × 10−5 | 2878166 |
246 | |
| p-Xylene | ||||||||||||
| 11 | PBT | MeOH | 197.8 | Cu–Na/SiO2 | 0.004 | 6 | 210 | 99 | 46.6 | 1.30952 × 10−5 | 3255699 |
246 |
| p-Xylene | ||||||||||||
| 12 | PET | Dodecane | 10.0 | 5Co/TiO2 | 0.09 | 24 | 320 | 89 | 2.3 | 1.93142 × 10−6 | 1206914 |
245 |
| Aromatics | ||||||||||||
| 13 | PLA | H2O | 504 | ZnO | 0.005 | 24 | 130 | 99 | 63.6 | 528846 × 10−6 |
522249 |
108 |
| Lactic acid | ||||||||||||
| 14 | PET | H2O | 150 | 0.006 | 24 | 180 | 99 | 20.1 | 3,81944 × 10−6 | 432112 |
||
| TPA | ||||||||||||
| 15 | PET | H2O | 73.8 | Ru/TiO2 | 0.1 | 30 | 200 | 99 | 0.4 | 2.00075 × 10−6 | 131668 |
248 |
| Aromatics | ||||||||||||
| 16 | PBT | H2O | 85 | 97 | 0.3 | 2.11175 × 10−6 | 129819 |
|||||
| Aromatics | ||||||||||||
| 17 | BPA-based epoxides | — | 0.5 | Ni complex | 0.04 | 24 | 200 | 66 | 0.2 | 2.29167 × 10−6 | 71191 |
249 |
| BPA | ||||||||||||
| 18 | BPA-PC | Octane | 267.8 | 2Ru/Nb2O5 | 0.01 | 16 | 320 | 83 | 49.4 | 2.7020 × 10−6 | 18312776 |
90 |
| Aromatics | ||||||||||||
| 19 | IPA | 17.0 | RANEY®@Ni/H-USY | 0.01 | 1 | 190 | 80 | 2.5 | 7.0502 × 10−5 | 35343 |
250 | |
| C6–C15 oxygenates | ||||||||||||
| 20 | BPA-PC | C6H12 | 36.5 | Ni/HZSM-5 | 0.01 | 16 | 190 | 81 | 5.2 | 5.423 × 10−6 | 964047 |
251 |
| C15 cycloalkane | ||||||||||||
| 21 | BPA-PC | AcOH | 3.3 | Pd/C, La(OTf)3 | 0.004 | 8 | 140 | 92 | 0.32 | 1.458 × 10−5 | 21893 |
99 |
| Jet fuel range | ||||||||||||
| 22 | BPA-PC | C6H12 | 30.7 | Pt/C, H-bèta zeolite | 0.02 | 4 | 140 | 81 | 4.6 | 2.410 × 10−5 | 191631 |
252 |
| C15 cycloalkane | ||||||||||||
| 23 | BPA-PC | H2O | 39.3 | 5%Rh/H-bèta zeolite | 0.005 | 12 | 200 | 86.9 | 5.5 | 6.04167 × 10−6 | 906675 |
101 |
| C15 cycloalkane | ||||||||||||
| 24 | BPA-PC | Decane | 35.8 | Ni–Ru/H-bèta zeolite | 0.01 | 12 | 180 | 90 | 1.3 | 7.0216 × 10−6 | 193682 |
100 |
| Cycloalkanes | ||||||||||||
| 25 | PBS | C6D6 | 65.6 | Si–Mn | 0.02 | 24 | 160 | 91 | 13.5 | 4.07986 × 10−6 | 3200484 |
91 |
| Butanediol-B | ||||||||||||
| 26 | PCL | Toluene | 89.9 | Si–Mn | 0.02 | 24 | 160 | 88 | 15.0 | 3.29861 × 10−6 | 4552598 |
|
| 1,4-BDM-B | ||||||||||||
| 27 | BPA-PC | Toluene | 45.7 | Si–Mn | 0.02 | 24 | 160 | 91 | 6.2 | 3.60243 × 10−6 | 1711696 |
|
| BPA-B |
Tang and his colleagues developed a bifunctional stable heterogenous catalyst of 0.1 g Pt/C + Ru-Cu/SiO2 catalyst for the transesterification of 0.1 g PET into DMT with a yield of 97.3% at 370 °C under 40 bar H2 (Fig. 8b). DMT can be separated automatically from methanol at low temperatures (25 °C) and then converted to DMCD through solvent-free hydrogenation over Pt/C catalysts. DMCD was then hydrodeoxygenated to produce cycloalkanes (C7–C8) and aromatics (C7–C8) with selectivity of 60.1% and 12.6%, respectively (Table 7). Ru-Cu/SiO2 catalyst showed promise for hydrodeoxygenation. Researchers suggested using DMCD in combination with oxygenates derived from biomass to produce jet fuel with the desired cycloalkane and aromatic contents.243 The Ru–Cu/SiO2 catalyst offers advantages such as robustness and lack of deactivation under the tested conditions for the solvent-free HDO of DMCD. However, a drawback is the need for high temperatures and pressure, as well as the toxicity of methanol. Precious metals are commonly used in catalysis, but their scarcity and high cost raise sustainability concerns. This has led to the development of non-metal catalysts as an alternative.
In 2020, Kratish and his group reported an air- and moisture-stable carbon-supported single-site molybdenum-dioxo heterogenous catalyst (Fig. 9a). The study investigated PET depolymerization/hydrogenolysis using a C/MoO2 catalyst at temperatures close to PET's melting point (260 °C). The reactions yielded 87% TPA, ethylene, and trace acetaldehyde (<5%) from PET powder (40:1 ester:Mo ratio) under 1 atm H2 for 24 h, with 2.38 g of d6-DMSO solvent. Lowering the catalyst loading to 100:1 ester:Mo and extending the reaction time to 96 h resulted in a 85% TPA yield. A yield of 86% TPA was achieved from waste PET, showing minimal impact of additives on the catalytic process (Table 7, entry 8). Without C/MoO2, only a 9% TPA yield was obtained, possibly due to partial PET thermolysis. The authors did not observe any self-recombination reactions under operating reaction conditions. Therefore, it is a promising catalyst for chemical recycling of polyesters due to its selectivity, robustness, abundance, non-toxicity, and recyclability. Challenges in this process include scaling up, operating at high temperatures, and dealing with extended reaction times. The high E factor of 4 and ξ value of 1 600 936 °C min are due to high solvent to polymer (23.1) ratios and catalyst loading, resulting in high energy consumption (low ε values, 2.3 × 10−6 °C−1 min−1) under harsh conditions (260 °C) and prolonged reaction times (24 hours)183 (Table 7 and Fig. S6†).
 | ||
| Fig. 9 (a) Hydrogenolysis of PET produces TPA and EG using C/MoO2 as the catalyst. (b) Ru/Nb2O5 catalyst used for PET hydrogenolysis to aromatics (light black), (c) the hydrogenolysis of PET (orange) to aromatics uses a Ru/Nb2O5 catalyst with EG as an in situ hydrogen source. (d) PET hydrogenolysis produces aromatics using a Co/TiO2 catalyst. | ||
In 2021, Jing and his group reported on various heterogenous catalytic systems for the selective cleavage of PET interunit C–O and C–C to aromatics (Fig. 9b). The systems included Ru/Nb2O5, Pd/Nb2O5, Pt/Nb2O5, Ru/TiO2, Ru/ZrO2, and Ru/HZSM-5. The 0.1 g of Ru/Nb2O5 catalyst showed an excellent performance with a 95.2% conversion of 0.1 g of PET and a selectivity to total aromatics yield of 83.6%, including p-xylene (45%), benzene (14%), and toluene (41%) in a single-step reaction. This new method provides a highly efficient way to produce aromatics from waste plastics. Aromatic separation can be easily achieved through distillation (Table 7, entries 4–7). It was evaluated at 320 °C and 5 bars of H2 for 16 hours in a water solvent. Among all supported Ru catalysts, Ru/Nb2O5 exhibited better activity due to ultra-small Ru species on Nb2O5 that prevent aromatic rings from hydrogenating, unlike on the other supports. Furthermore, adding NbOx species for C–O bond activation and Brønsted acid sites for C–C bond cleavage achieves the desired reactivity towards aromatic in plastics depolymerization.90 The process offers advantages such as easy product separation, water as the only solvent, and chemical robustness compared with homogeneous catalysts. However, it has drawbacks such as lower productivity due to solid–solid contact between plastics and catalysts affecting catalytic performance in breaking C–C and C–O bonds. While cost-effective compared with non-noble catalysts, it may compromise sustainability, and scaling up can be challenging. The Ru/HZSM-5 catalysts had the highest E factor of 60 and ξ values of 5.711 × 106 °C min due to a high solvent to polymer ratio (100) and low monomer yield (32%) with a low ε value of 1.0 × 10−6 °C−1 min−1 compared with Ru/TiO2 (2.34375 × 10−6), Ru/ZrO2 (2.3763 × 10−6), and Ru/Nb2O5 (2.73438 × 10−6 °C−1 min−1) catalysts. Overall, the series of Ru-based catalysts exhibited high E factor and ξ values with low ε values compared with other heterogeneous catalysts like RANEY® Ni (E = 2.5 and ε = 7.0502 × 10−5 °C−1 min−1), Ni-HZSM-5 (E = 5.2 and ε = 5.423 × 10−6 °C−1 min−), and Pd/C (E = 0.3 and ε = 1.458 × 10−5 °C−1 min−), resulting in high waste generation and limited industrial process potential (Table 7 and Fig. S6†).
In 2021, Lu and colleagues successfully converted common PET plastics into BTX using a tandem-catalysis strategy. They utilized the hidden hydrogen in ethylene glycol to achieve this conversion (Fig. 9c). The study involved testing the direct conversion of PET plastics, such as 0.2 g Coca-Cola bottles, polyester film, and clothes, using 0.2 g of Ru-based Nb2O5 catalyst at 220 °C with 20 bar nitrogen for 12 hours. Coca-Cola bottles and polyester film resulted in BTX with total yields of 88.7% and 93.3%, respectively, demonstrating the system's effectiveness. Initially, polyester clothes yielded 18.9% BTX, but with increased temperature (280 °C) and time (16 hours), the yield rose to 81.6% (Table 7, entry 9).244 This study highlights the versatility of Ru/Nb2O5-catalyzed H2-free conversion for various PET plastics, contributing to a circular economy for PET materials and promoting sustainability through catalysis. The Ru/Nb2O5 catalyst offers several advantages, including a high concentration of Ru δ+ species resulting from the interaction between Ru and NbxO, which helps prevent undesired decarboxylation. The catalyst also demonstrates strong hydrogenolysis capability, and easy separation of catalyst and products, and can operate at low pressure and temperature without requiring external hydrogen gas. The system used for the hydrogenolysis of polyesters has a low E factor (15.7) and high product yield, polymer loading, and ε coefficient (5.76 × 10−6 °C−1 min−1) compared with Ru/TiO2, Ru/ZrO2, and Ru/HZSM-5 (Table 7 and Fig. S6†). These four heterogeneous catalysts are stable, reusable, and less sustainable due to the use of noble metals. However, they are not feasible, generate high waste, consume high energy, and are costly.
In 2021, Hongkailers and his group successfully converted 0.3 g of PET into aromatics with a 78.9% yield of in 3 g of octane as a solvent using a 0.3 g of Co/TiO2 catalyst (Fig. 9d). The reaction was carried out at 320 °C and 30 bar H2, and with run times ranging from 4 to 24 hours.245 The total product yield after 4 hours was 15.6%, which increased to 56.6% and 89.7% after 8 and 24 hours, respectively (Table 7, entry 12). The main products after 8 hours were alkylbenzoic acids, alkylbenzoates, and coupling products, while after 24 hours, aromatics like xylene and toluene were predominant. This shift suggests that arenes are formed at the expense of other products. The increase in total product yields from 8 to 24 hours indicates the presence of oligomers in the early stages of the reaction. The catalyst's main limitation is its instability during the reaction, caused by the phase transformation of the TiO2 support and the loss of metallic Co in the form of CoTiO3. The E factor (2.3) and ξ (1.2 × 106 °C min) values were lower compared with Ru/Nb2O5 (Table 7 and Fig. S6†) due to a low solvent-to-polymer ratio (10) and catalyst to polymer ratio (0.09). The process had a low ε value of 1.9314 × 10−6 °C−1 min−1 due to the high temperature (320 °C) and long reaction time (24 hours), leading to higher energy for a reasonable monomer yield (90%). However, it was an efficient process as compared with Ru/Nb2O5 and C/MoO catalysts. This is unstable under the current conditions, non-reusable, and unsustainable because of toxic metals.
In 2022, Gao and his colleagues developed non-noble Cu–Na/SiO2-HT catalysts for converting polyesters (PET/PBT) (0.12 g) into p-xylene with a yield of 96.4% and EG as a by-product. They used 23.76 g of methanol as a solvent and hydrogen donor at 210 °C, 34–38 bar N2 for 6 hours (Fig. 10a). Similar results were obtained with 0.12 g of PBT under the same conditions. PBT at 210 °C yielded 99% p-xylene and 1,4-butanediol in methanol, releasing 28 bar gases (60% H2) (Table 7, entries 10 and 11). The PET is degraded into p-xylene using a tandem process that includes PET methanolysis and selective HDO, with in situ H2 production from methanol decomposition.246 CuNa/SiO2 is a cost-effective method for converting waste PET and PBT into valuable energy without requiring external hydrogen. The system uses in situ hydrogen production from methanol dehydrogenation, PET methanolysis, and DMT hydrodeoxygenation to xylene on a CuNa/SiO2 catalyst with a high Cu+/Cu0 ratio. This study evaluates different depolymerization conditions for polyesters based on three green chemistry metrics. The results show that the base metal catalysts achieved the highest ε coefficient (1.303 × 10−5 °C−1 min−1) with a product yield of 99%, a reaction temperature of 210 °C, and a reaction time of 6 hours. The process had a high ε coefficient (2.8781 × 105 °C−1 min−1) but a short duration (6 hours) at 210 °C, resulting in the highest monomer yield compared with Co/TiO2 and Ru/Nb2O5 catalyst. However, it also had a high E factor (37.7) and ξ value (2878166 °C min) due to a high amount of solvent to polymer ratio (197) used, making it impractical for industrial scale-up (Table 7 and Fig. S6†). CuNa–SiO2 is a sustainable catalyst with in situ hydrogen generation and non-noble metal components. It is reusable but has lower stability.
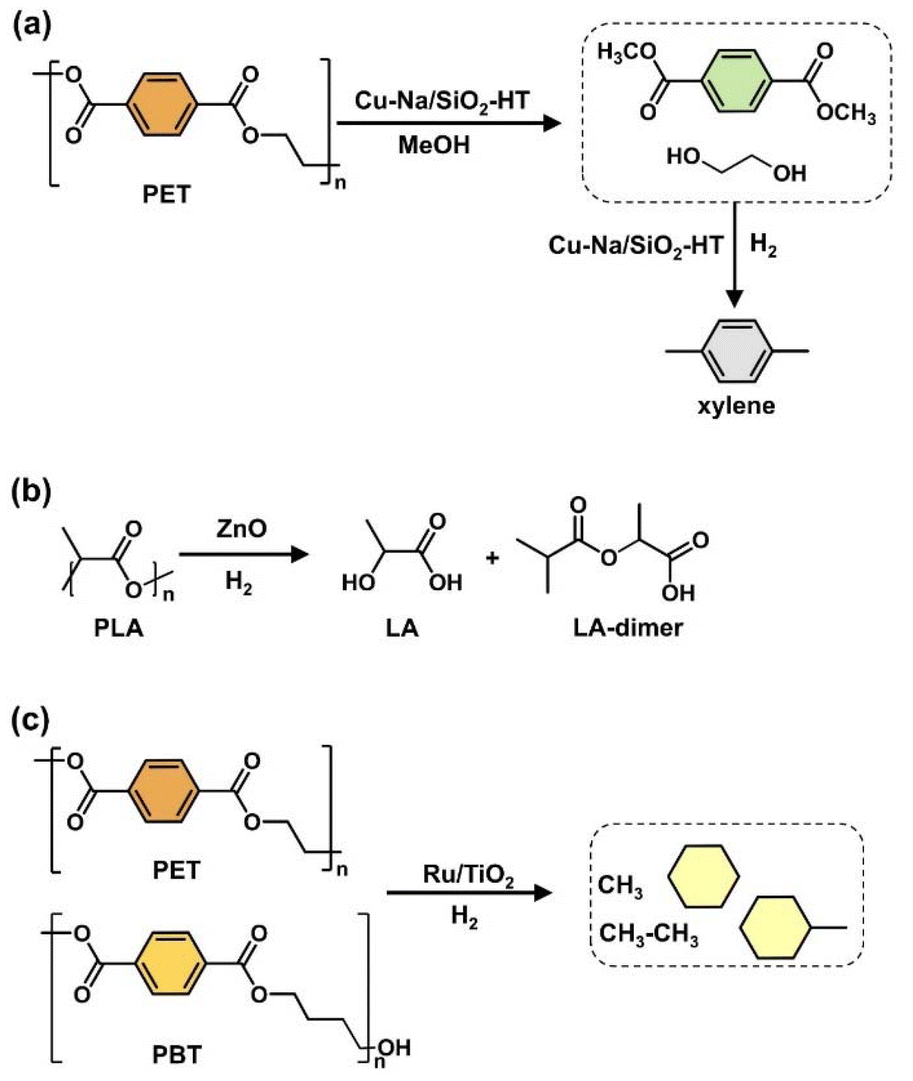 | ||
| Fig. 10 shows different hydrogenolysis reactions using various catalysts: (a) Cu–Na/SiO2-HT catalyst for the conversion of PET to p-xylene (light black), (b) ZnO catalyst for the hydrogenolysis of PLA to LA, and (c) Ru/TiO2 catalyst for the hydrogenolysis of PET/PBT (orange) to alkanes. | ||
In a recent study, Pierluigi Barbaro and colleagues successfully used a ZnO catalyst to fully depolymerize polyesters (PLA and PET) into lactic acid (LA) and TPA/EG with up to 100% selectivity. They achieved this by converting 0.02 g of PLA at 130 °C, with 0.052 g of catalyst and 10 g of water (Fig. 10b) for 24 hours. For 0.1 g of PET, they used 0.1 g of catalyst and 15 g of water, and conducted the reaction at 180 °C for 24 hours under atmospheric pressure (Table 7, entries 13 and 14).108 This method offers benefits such as using water as a solvent, no need for noble metals, and operating at 180 °C under atmospheric pressure. The catalyst is recoverable and reusable, cost-effective, and easily accessible, making the process sustainable and robust under optimized conditions. The study evaluates the depolymerization of polyesters using green chemistry metrics. The ZnO catalyst achieved a 99% product yield for PLA but had high E factor (63.6) and ξ (522249 °C min) values, requiring 24 hours and a high solvent to polymer ratio (504). PET depolymerization with the same catalyst resulted in lower waste compared with PLA (64 to 20) due to a lower solvent to polymer ratio (150). However, the catalytic process generated high waste and had a significant environmental impact, making it an inefficient process (Table 7 and Fig. S6†).
In a separate study, Rongxiang Li and his group utilized a Ru/TiO2 heterogeneous catalyst for the hydrogenolysis of PET/PBT into alkanes (Fig. 10c). The study tested the Ru/TiO2 catalyst for converting polyesters (PET/PBT) into alkanes. The reaction used 0.192 g of PET/PBT (0.221 g), 0.06 g of catalyst, and a small amount of water (0.2 g) at 200 °C and 60 bar hydrogen for 30 hours. The optimized conditions resulted in PET converting to alkanes, primarily cyclohexane (73%) and methane (88.6%), with a total carbon yield of 99.6%. PBT yielded 73% cyclohexane, along with methane (91.3%) and ethane (78.9%) under the same reaction conditions (Table 7, entries 15 and 16). The strong interaction between the TiO2 support and Ru nanoparticles enables electron flow from TiO2 to Ru, allowing Ru/TiO2 to catalyse polyester hydrolysis and hydrogenation simultaneously.248 Advantages include using only water as a reagent, mild operating temperatures, recoverable and reusable catalyst, cost-effectiveness, easy accessibility and sustainability. This system has a low environmental impact factor (0.35) and energy environmental impact (1.31668 × 104 °C min) during polyester hydrogenolysis, with a high ε coefficient (2.00075 × 10−6 °C−1 min−1). This efficient process minimizes waste generation and energy consumption compared with other heterogenous catalysts (Table 7 and Fig. S6†).
4.3. Hydrogenolysis of BPA-PC
Yumeng Liao and team recovered 66% BPA from 0.139 g of BPA-based epoxide using a 0.014 g Ni-based complex hydrogenolysis method at 200 °C for 24 hours under H2 (1 atm) solvent-free conditions (Table 7, entry 17). The white suspension turned into a dark green solution, indicating successful epoxy resin conversion. While Ni(bis(1,5-cyclooctadiene)nickel)2/PPh3 did not lead to significant BPA formation, the use of 1,2-bis(dicyclohexylphosphino)ethane (dcype) as a ligand was most effective due to its hemilabile nature. The Ni complex catalyst is effective for low catalyst loading hydrogenolysis of epoxide waste, but its high cost hinders scalability and reusability, and poses challenges for achieving high monomer yield. No toxic solvents and non-noble metal were used in the production process, making it environmentally friendly. The catalyst used is sustainable. This system has the lowest environmental impact factor (0.16) and energy environmental impact (7.1119 × 103 °C min) with a high ε coefficient (2.29167 × 10−6) due to the absence of solvents. However, a high temperature (200 °C) and long reaction times (36 hours) in BPA-based epoxy hydrogenolysis may be required (details in Table 7 and Fig. S7†), which minimizes waste generation and energy consumption during the process. The process not efficient because it consumes high energy compared with other heterogeneous catalyst systems.249In 2021, Jing and colleagues also successfully applied 0.006 g of Ru/Nb2O5 catalyst for the selective cleavage and depolymerization of 0.015 g of PC-BPA to aromatics at an elevated temperature of 320 °C and 5 bar H2 for 16 hours under 2.8 g of octane. The total aromatics yield was 83.3%, with high selectivity to benzene (78%) and cumene (21%) (Table 7, entry 18).90 The catalyst successfully cleaved ester and C–C linkages, demonstrating its effectiveness. Advantages include robustness catalyst and easy product separation, but challenges include scaling up the process and cost-effectiveness. The study also identified three green chemistry metrics for PET hydrogenolysis, with this catalyst showing high E factor (49.4) and ξ (1.8312 × 106 °C min), and low ε values (2.7018 × 10−6 °C−1 min−1) (Table 7 and Fig. S7†) due to the low yield, high temperature (320 °C), excess reaction time (16 hours), and high solvent to polymer ratio (267.8). The process shows that chemical depolymerization of PET is not efficient.
In 2021, Wang and his group used RANEY® Ni catalyst and USY as co-catalyst to hydrodeoxygenate BPA-PC into high-density aviation fuel using isopropanol as the solvent and hydrogen donor (Fig. 11a). The catalyst system (0.4 g) achieved complete conversion of 4 g of BPA-PC into a mixture of C6–C15 oxygenates and cyclic hydrocarbons as co-products at 190 °C and 30 bar H2 for 1 hour under 35.7 g of isopropanol as solvent, with a reported total carbon yield of 80% for C6–C15 aromatics and co-by-product cycloalkanes (Table 7, entry 19). The RANEY® Ni + USY catalyst effectively converts BPA-PC waste into cyclic hydrocarbons, with a 80% total carbon yield for C6–C15 hydrocarbons. No self-recombination reactions were detected. This process could potentially produce 3.68 tons of C6–C15 cyclic hydrocarbons from 6 million tons of BPA-PC waste.250 This process's key benefits include using a sustainable catalyst with non-noble metals, robustness under various conditions, easy recovery and reuse multiple times, low cost, and minimal environmental impact. The environmental impact factor is 2.5, and the energy environmental impact is 35343 °C min during BPA-PC hydrogenolysis. This is attributed to a lower solvent-to-polymer ratio (17) (Table 7), resulting in a high ε coefficient (7.0502 × 10−5 °C−1 min−1) and a shorter reaction time (1 hour) with an 80% yield at low temperature (190 °C) (details in Table 7 and Fig. S7†). This sustainable process minimizes waste generation, does not require external hydrogen, and is more efficient than other heterogeneous catalysts.
 | ||
| Fig. 11 The catalytic hydrogenolysis of BPA-PC into various products oxygenates (pink), aromatic compounds (light black), and cycloalkanes (yellow) using different catalysts; (a) shows the results using Raney® Ni or Co catalyst, while (b) shows the results using Ni/HZSM-5 catalyst, (c) shows the conversion of BPA-PC (red) to cycloalkanes using various heterogeneous catalysts, and (d) shows the results of the BPA-PC conversion into cycloalkanes using Ru–Ni/H-beta zeolite. | ||
In 2023, Liu and his coworkers successfully converted 0.3 g of BPA-PC waste into cycloalkanes with a 99.3% yield using 0.1 g of Ni/HZSM-5 catalysts under mild reaction conditions (190 °C, 4 MPa H2 for 16 hours, and 11.2 g of cyclopentane used as solvent) (Table 7, entry 20)251 (Fig. 11b). At 10% Ni loading, the yield of C15 dicycloalkane was 37.7%, with a total cycloalkane yield of 87.5% (Table 7, entry 20). Increasing Ni loading to 20% further enhanced the yields of C15 dicycloalkane and cycloalkane co-products. The 20Ni/HZSM-5(200) catalyst favored the production of C15 dicycloalkanes as the main products, with aromatics and oxygenates as intermediates. The total yield of cycloalkanes was 99.3%, with 81.2% being C15 dicycloalkane as co-product. The catalyst efficiently converted phenolic compounds to cycloalkanes. With the proper metal–acid balance, the 20Ni/HZSM-5(Si/Al-200) catalyst showed good activity toward C15 dicycloalkane. The catalyst can be reused up to 6 times without losing activity and efficiently converts various polycarbonate wastes (CDs, DVDs, PC sheets) into cycloalkanes with a yield of up to 98%. The Ni/HZSM-5 catalyst also offers cost savings, easy recovery, and improved product yield, making it a sustainable and robust option. Table 7 and Fig. S6† demonstrate that HZSM-5-based Ni catalysts led to a lower ε value (5.423 × 10−6 °C−1 min−1) due to a longer reaction time (16 hours) compared with RANEY® nickel catalysts in the conversion of BPA-PC. Despite high polymer and catalyst amounts, a 99% cycloalkane yield was achieved, with a slightly elevated E-factor (5.2) and ξ value (964047 °C min) (Table 7 and Fig. S7†). This comparison highlights the waste generation and environmental impact of the BPA-PC depolymerization process using HZSM-5-based Ni catalysts, which is slightly less efficient than using RANEY® nickel (Fig. S7†).
In the same year, Luo and his group successfully converted 2.5 g of BPA-PC into a C15 dicycloalkane with a 98% yield at 140 °C, 60 bar hydrogen pressure, using 7.9 g of AcOH, and 0.35 g of a tandem catalyst of Pd/C and La(OTf)3 (Fig. 11c). They achieved a 95–98% yield of 2,2-dicyclohexylpropane for 8 hours of reaction time (Table 7, entry 21).99 The reaction process involved the activation of polycarbonates by La(OTf)3, followed by hydrogenation catalyzed by Pd/C to produce hydrogenated polycarbonates (H-PC). The synergistic effect of Pd/C and La(OTf)3 was crucial for the depolymerization of polycarbonates. The solvent and catalyst were successfully recovered, and the Pd/C catalyst demonstrated good stability (robustness) and cost-effectiveness compared with noble metal catalysts, making it a sustainable option. The reusability of the solvent and La(OTf)3 was tested, with the yield remaining stable after multiple cycles, demonstrating the efficiency of the method. Its catalyst showed the lowest environmental impact factor (0.32) and energy environmental impact (21893 °C min) values in BPA-PC hydrogenolysis. This was due to a low solvent to polymer ratio compared with other catalysts (Ni/HZSM-5, Ru/Nb2O5), resulting in a high ε coefficient (1.458 × 10−5 °C−1 min−1) and a high yield of 98% (Table 7 and Fig. S7†). This process is efficient, minimizes waste generation, and has less environmental impact compared with Ni/HZSM-5.
Tang and his group also reported a one-step method that converted 1 g of BPA-PC into 2,2-dicyclohexylpropane with an 81% yield using 0.35 g of Pt/C and H-USY at 140 °C for 4 hours, 30 bar hydrogen pressure, and 31.1 g of cyclohexane as a solvent (Table 7, entry 22). This process can be quickly separated through liquid separation with low energy consumption, as the alkane is not soluble in water.252 The process has advantages such as mild temperature, reusable catalyst, and easy monomer recovery. However, it also has disadvantages like harsh hydrogen pressure and excessive solvent usage. The catalyst system showed a high ε coefficient value (2.4104 × 10−5 °C−1 min−1) due to the low temperature and reaction time, resulting in an 81% monomer yield. However, the E factor (4.6) and ξ (191631 °C min) values were slightly higher in BPA-PC depolymerization compared with RANEY® Ni and Pd/C, likely due to a high solvent to polymer ratio and lower monomer yield. This indicates a higher level of waste generation and environmental impact, as well as lower efficiency. The group achieved an 87% yield in the direct conversion of 1 g of BPA-PC to C15 cycloalkanes at 200 °C, 35 bar hydrogen, for 12 hours. They used 40 g of water as a solvent and 0.2 g of Rh on H–Y zeolite catalyst (Fig. 11d) (Table 7, entry 23).101 Both studies confirmed the effectiveness of the bi-catalytic system for transforming BPA-PC. However, mono-catalytic systems are preferred for their ease of production, recovery, and sustainable as well as simpler commercialization infrastructures.101,252 The catalyst exhibited a slightly lower ε coefficient value (6.0416 × 10−6 °C−1 min−1) compared with Pt/C and H-USY, likely due to operating at high temperatures and a longer reaction time of 12 hours. The E factor (5.5) and ξ values (906675 °C min) were slightly higher, attributed to the high solvent to polymer ratio, despite using water as a solvent (details in Table 7 and Fig. S7†).
Manal and his team used a bi-functional Ru–Ni/H-beta catalyst to convert BPA-PC into C15 cycloalkanes with over 90% selectivity, achieving complete conversion to jet fuel range under mild reaction conditions (Fig. 11d). The reaction conditions were as follows: 2 g of BPA-PC, 0.3 g of catalyst, 21.9 g of decane as a solvent, 180 °C temperature, 30 bar H2 pressure and 12 hours reaction time (Table 7, entry 24).100 The study found that polycarbonate breaks down into bisphenol A (BPA) through hydrogenolysis, with BPA further transforming into cyclic hydrocarbons. Methane (CH4) was also detected, likely from the methanation of carbon monoxide (CO). Alloying Ni with Ru improved activity and selectivity. The study also looked at recyclability, scalability, and structure–activity evaluation for PC waste. The Ru–Ni/H-beta catalyst demonstrated a high ε coefficient value of 7.0216 × 10−6 °C−1 min−1, outperforming Rh/C + H-USY and Ni/HZSM-5 catalysts at low temperatures over a 12 hour reaction period. It achieved a 91% monomer yield with a low E factor of 1.3 and ξ values of 193682 °C min, attributed to a low solvent to polymer ratio and consistent catalyst amount (details in Table 7 and Fig. S7†).
Lourenço and her team recently published an article on the efficient depolymerization of polyesters like PBS, PCL, and BPA-PC using a commercially available heterogeneous catalysts (Mn(II) ethyl/butyl phosphonate silica) (Si–Mn). They achieved at least 14 catalytic cycles for the depolymerization of 0.043 g of PBS with a 93% yield at 110 °C for 24 hours using 2.6 g of toluene as a solvent, 0.05 g of catalyst, and 0.2 g of HBpin. The same catalyst also successful hydrogenolysed 0.031 g of PCL and 0.061 g of BPA-PC, under the same catalyst and reducing agent amount along with reaction conditions with yields of 76% and 83%, respectively91 under the same reaction conditions (Table 7, entries 25–27). The catalyst effectively breaks down polyester and polycarbonate plastic waste using manganese compounds as catalysts. It combines a common metal with mild reducing agents to produce valuable compounds in high yields. The Si–Mn catalyst also performs well, enabling multiple cycles with excellent yields in the depolymerization of PBS/BPA-PC. For more information on the hydrogenolysis process of converting mixed plastic waste into valuable compounds, please refer to the following articles and reviews.113,253–257 The Si/Mn catalyst shows promise for the chemical recycling of polyester due to its selectivity, abundance, and sustainability. Challenges like scale-up, high temperature, and long reaction times must be overcome. In the hydrogenolysis of BPA-PC, the Si/Mn catalyst exhibited a low E factor of 6.2 and a ξ value of 1711696 °C min, attributed to a low solvent-to-polymer ratio and the same catalyst loading, resulting in reduced waste generation (details in Table 7 and Fig. S7†). Despite achieving an 83% yield at low temperature, the Si/Mn catalyst showed slightly lower efficiency (ε of 2.3 × 10−6 °C−1 min−1) due to prolonged reaction times. Compared with other catalyst systems like RANEY® Ni, Pd/C, Ni–Ru, and Pt/C, the Si/Mn catalyst was found to be less efficient for BPA-PC hydrogenolysis.
5. Conclusion and perspective
This work reviews various catalytic systems, both homogeneous and heterogeneous, that have been studied for the hydrogenolysis of OXPs. This technology offers versatile solutions to combat plastic pollution while producing valuable feedstock for recycling and upcycling. Studies have shown improved activity with reduced energy input, resulting in lower operating temperatures and higher yields of alcohols and aromatic compounds. The resulting liquids contain alcohols (1,4-dimethanolbenzene, bisphenol A, 1,2-propylene diol, methanol, ethylene glycol) and aromatics including benzene, toluene, and xylene along with products such as methane, carbon monoxide, and carbon dioxide. This review evaluates various depolymerization conditions for OXP polymers using three green chemistry metrics: energy economy coefficient (ε), environment factor (E), and their combined effect (ξ). These metrics allow for a quantitative comparison of studies to assess the effectiveness of chemical depolymerization for hydrogenolysis of the OXPs and other polymer materials.Reaction variables, such as solvents, ligands, metals, catalyst support (acidic/basic nature), and reaction conditions, can significantly influence the depolymerization of OXPs and the product distribution, aiding in the protonation in plastics to promote hydrogenolysis. Metal sites are essential for activating C–O bonds, while acidic or basic sites are crucial for activating C–C bonds in hydrogenolysis. Operating parameters, like temperature, partial pressure, and the ratio of plastics-to-catalyst, base and reducing agent concentrations greatly impact the conversion process. Optimized conditions lead to increased product yields, enhancing the conversion to alcohol/aromatics. Despite numerous studies on both homogeneous and heterogeneous catalysts, challenges remain in scaling up. Active catalysts that do not rely on toxic and expensive multi-step ligands are needed for depolymerization, while also avoiding the use of harmful solvents. Catalysts devoid of hydrogen or ligands, such as silane/borane-based catalysts, have shown high performance under mild reactions conditions. However, catalyst deactivation during the reaction limits reusability, and their unstable nature under environmental conditions and high costs are significant drawbacks. Processes that operate with high efficiency and selectivity under mild conditions are required. Successful homogeneous catalysts include Ru(triphos-Xyl), the Milstein catalyst, Zn(II) half-salen, [Ph3C]+[B(C6F5)4]−, Ru-MACHO-BH, and ((Xyl Nacnac)Mg)2. However, using such catalysts on an industrial scale is unlikely due to the excessive cost of the components. The most successful results have been achieved with homogeneous catalyst systems, demonstrating high efficiency and versatility across various applications. This approach involves breaking down various types of plastic waste, such as polyesters, polycarbonates, and epoxy resin. Apart from finding a cost-effective method for converting plastic waste into valuable compounds, the use of catalysts without complex ligands offers an advantage from a scale-up perspective. This differs from methods employing ruthenium, iridium, or manganese catalysts. While ruthenium catalysts require high temperatures and pressures, iridium catalysts and silanes exhibit low selectivity in the hydrogenolysis process.
Considerable progress has been made in breaking down plastic waste using an available heterogeneous catalyst, and in-house synthesized catalysts like Ru/Nb2O5, Pt/C, RANEY® Ni, Zn, and Mn have proved effective and stable, simplifying product separation and eliminating the need for toxic and expansive ligands. Despite high conversion rates and product yields, further cost reduction is necessary to facilitate industrial application. Emphasizing economic viability can accelerate the commercialization of these technologies. Depolymerizing plastic waste with heterogeneous catalysts holds promise for producing valuable chemicals and fuels, contributing to a new circular economic model by reducing non-biodegradable polymers in oceans and landfills. This study demonstrates the hydrogenolysis of various polyester and polycarbonate plastic waste using cost-effective manganese compounds as catalysts. Both homogeneous and heterogeneous catalysts containing non-noble metals have been utilized, resulting in the sustainable production of valuable compounds with high yields.
Chemical recycling technology offers a solution to the limitation of mechanical recycling. However, there is currently no technology available for the hydrogenolysis of polycarbonate, polyesters and epoxy resin, as indicated by the literature review. The catalytic hydrogenolysis process is being developed at a commercial level and shows promise for producing high-purity monomers that can be easily separated from the reaction mixture, making it a promising process for chemical recycling. Large-scale facilities for depolymerizing polycarbonates and epoxy resins are currently lacking. Table 8 presented that a SWOT analysis was conducted to assess the internal and external factors affecting the hydrogenolysis of oxygenated plastic waste as a potential future technology. Among the four factors considered, “opportunities” emerged as the most significant, followed by “strengths”, “threats”, and “weaknesses”. The potential of catalytic hydrogenolysis as a versatile process is highlighted by the fact that its range of opportunities exceeds the sum of all other factors. The top five opportunities identified include prioritizing eco-design principles to facilitate the hydrogenolysis of oxygenated plastic wastes, government support for tax privileges for green and alternative fuel (O1), developing sustainable energy sources and identifying new business prospects (O2), catalytic hydrogenolysis targets of international authorities in 2030 and 2050 (S5), and recent advancements in catalysis and materials science that can lead to more efficient and selective catalysts for polyester hydrogenolysis (O3). On the other hand, factors such as insufficient infrastructure, complex production processes, and a lack of experience, especially with current technology (W4), safety concerns related to hydrogen, and deforestation are also among the prioritized weaknesses and threats.
| Action | Strengths | Weaknesses | Opportunities | Threats |
|---|---|---|---|---|
| Hydrogenolysis | Utilizing heterogeneous catalysis to enhance energy efficiency and reduce production costs for the hydrogenolysis of oxygenated plastic waste. | The high initial investment and ongoing production costs, particularly for developing units and implementing efficient catalysts, may present a challenge to the economic feasibility of the process. | By prioritizing eco-design principles, we can facilitate the hydrogenolysis of oxygenated plastic wastes. The government supports tax incentives for green and alternative fuels. | Health problems can occur during energy production, especially those related to respiration and deforestation |
| Resource efficiency | Hydrogenolysis is a versatile approach that can be applied to both circular and linear economic models. | Safety concerns related to hydrogen (H2) are significant due to its incompatibility with certain locations and production methods. | Developing sustainable energy sources and identifying new business opportunities. | New technologies for recycling polyester or alternative materials could threaten the widespread use of heterogeneous catalytic hydrogenolysis. |
| Process optimization | To improve reaction efficiency, we can adjust residence time, catalyst-to-polymer mass ratio, and temperature. Heterogeneous catalysis helps develop selective catalysts, reducing by-product formation. | The process may require harsh reaction conditions, limiting its applicability to certain types of polyester. | Recent advancements in catalysis and materials science can create more efficient and selective catalysts for polyester hydrogenolysis. | The initial investment and operating costs for developing and implementing effective catalysts may pose economic challenges. |
| Productivity | Heterogeneous catalysts can be used multiple times, reducing the overall cost of the process, and minimizing waste generation. | Contaminated plastics can poison the catalyst, so removing impurities from feedstock is challenging. | Techno-economic assessments are necessary to gain comprehensive insights into the economic feasibility and challenges associated with the hydrogenolysis pathways. | The economic feasibility of the project may be questioned due to the first investment and operating costs, particularly in the development and implementation of an effective catalyst. |
| Technology implementation | Catalytic hydrogenolysis targets of international authorities in 2030 and 2050 | The challenges include insufficient infrastructure, complex production processes, and a lack of experience, especially with current technology. | Create employment opportunities for the local population and contribute to economic growth. | The public's perception of the safety and effectiveness of the process could influence its acceptance and adoption. |
By focusing on eco-design principles and addressing these challenges, the conversion of oxygenated plastic waste through hydrogenolysis can be promoted effectively. Tables 2–5 and 7 and ESI in Fig. S1–S7† clearly illustrate the effectiveness of different catalyst systems in the depolymerization of polyesters. The highest ε value was observed in the homogeneous Zn II complex catalyst at 3.166 × 10−4 °C−1 min−1, resulting in a 95% yield of BPA monomer at 50 °C for 1 hour (Fig. S3†). The solvent-to-polymer ratio of 16.8 led to a low E factor of 1.9 and the smallest ξ value of 6241 °C min compared with other homogeneous catalyst systems. In heterogeneous catalysis, RANEY® nickel also showed a high ε coefficient of 7.0502 × 10−5 °C−1 min−1, a low E factor of 2.5 and a ξ value of 35343 °C min due to a shorter reaction time (1 hour) and solvent to polymer ratio of 17, at 190 °C with an 80% monomer yield (Fig. S7†). Tandem catalysis (Pd/C + La(OTf)3) exhibited a high ε coefficient of 1.458 × 10−5 °C−1 min−1, a low E factor of 0.3 and a ξ value of 21893 °C min (Fig. S7†) due to a low solvent to BPA-PC ratio of 3.3 with a monomer yield of 92% for a short reaction time of 8 hours at 140 °C. Heterogeneous catalysts such as RANEY® Ni, Pd/C, Ru/TiO2, and Mg(II) are efficient in reductively depolymerizing polyester waste, minimizing waste generation. These catalysts are preferred over homogeneous ones due to their efficiency, sustainability, scalability, and ease of product separation along with fewer solvents consumed. The hydrogenolysis pathway using heterogeneous catalysts is environmentally and economically viable, although it may yield lower monomer quantities than homogeneous systems. This review explores various catalyst systems to optimize monomer selectivity and minimize side reactions, highlighting the importance of catalysts, co-catalysts, reducing agent concentrations, and reaction conditions in determining reaction pathways. The green chemistry matrix in each catalyst system offers valuable insights into the catalysts’ role in chemical reactions, driving progress in the field.
Future research should focus on testing the technology on real waste streams, streamlining permit acquisition processes, developing efficient sorting techniques, conducting techno-economic assessments, and promoting eco-design principles in plastic products for sustainable circularity. Transforming waste plastics into diols enables recycling within the circular economy vision. Detailed studies have shown the catalyst's tolerance toward various polymer additives and the transformation and separation of polymer mixtures. The method's effectiveness and impact can be expanded by envisioning a catalytic process with “green hydrogen” from water electrolysis based on renewable energy. Additionally, the catalytic combination of recycled diols with carbon dioxide allows the sustainable synthesis of linear and cyclic acetals only from waste carbon resources. Understanding catalytic active sites and reaction mechanisms is crucial for future research. Investigating the relative activity of different metal components for hydrogenation reactions and utilizing microkinetic modelling for catalyst design is important. Addressing compositional and molecular sizes of plastics and improving contact between polymers and catalytic sites are key challenges. Handling impurities in raw feedstocks, such as alkali metals and heavy metals, also requires attention. These processes require careful consideration of various factors such as feedstock composition, reaction conditions, catalyst stability, and product selectivity. By optimizing these parameters, researchers can enhance the efficiency and sustainability of plastic conversion processes, ultimately contributing to developing a more circular economy. This research offers valuable insights into the role of catalysts in chemical reactions, advancing knowledge in the field.
Abbreviations
| AA | Acetoxyacetic acid |
| 1,4-BDM | 1,4-Benzenedimethanol |
| BG | 1,4-Butylene glycol |
| BPA-PC | Bisphenol A polycarbonate |
| BHET | Bis(2-hydroxyethyl)terephthalate |
| DMC | Dimethyl carbonate |
| DMT | Dimethyl terephthalate |
| DMCD | Dimethyl cyclohexane-1,4 dicarboxylate |
| EG | Ethylene glycol |
| EP | Epoxy resin |
| Half-salen (S) | 2,2′-Ethylene bis(nitrilomethylidene)diphenol |
| HDO | Hydrodeoxygenation |
| HFIP | 1,1,1,3,3,3-Hexafluoro-2-propanol |
| HNTf2 | Bis(trifluoromethanesulfonyl)imide |
| GA | Glycolic acid |
| K app | Rate constant |
| Nacnac | 1,3-Diketimines (anionic bidentate ligands) |
| 2-NCA | 2-Naphthalenecarboxylic acid |
| OXPs | Oxygenated plastic wastes |
| HBpin | Pinacolborane |
| PBT | Polybutylene terephthalate |
| PCL | Polycaprolactone |
| 1,2-PD | 1,2-Propylene diol |
| PDO | Polydioxanone |
| PE | Polyethylene |
| PECHD | Polyethylene 1,4-cyclohexane-dicarboxylate |
| PET | poly(ethylene terephthalate) |
| PF | Phenol formaldehyde resins |
| PLA | Poly(lactic acid) |
| PMHS | Polymethylhydrosiloxane |
| PPC | Poly(propylene carbonate) |
| PGEE | Propylene glycol ethyl ether |
| PS | Polystyrene |
| PVC | Polyvinylchloride |
| PNN | 2-((Di-tert-butylphosphinomethyl)-6-diethylaminomethyl)pyridine |
| PNP | 2,6-Bis((diisopropylphosphino)methyl)pyridine |
| PU | Polyurethane |
| RT | Room temperature (°C) |
| TBAF | Tetra-n-butylammonium fluoride |
| TPFP-B | Tris(pentafluorophenyl)borane |
| TMDS | Tetramethyldisiloxane |
| TMM | Trimethylenemethane |
| TPA | Terephthalic acid |
| Triphos-Xyl | 1,1,1-Tris(3,5-dimethylphenylphosphinomethyl)ethane |
| Triphos | 1,1,1-Tris(diphenylphosphinomethyl)ethane |
| Xyl | Xylidine (2,6-dimethylaniline) |
Data availability
No original research findings, software, or code were included, and no new data were generated or analyzed in this review. Existing data from publications that support this article have been compiled.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to gratefully acknowledge the financial support of the Flemish Government and Flanders Innovation & Entrepreneurship (VLAIO) through the Moonshot project CYCLOPS HBC.2021.0584.References
- N. R. J. Hynes, R. Sankaranarayanan and J. A. J. Sujana, J. Cleaner Prod., 2021, 292, 125317 CrossRef.
- Global polycarbonate market outlook, https://www.expertmarketresearch.com/reports/polycarbonate-market (accessed 2023).
- Epoxy resin market analysis, https://www.chemanalyst.com/industry-report/epoxyresin-market.
- L. D. Ellis, N. A. Rorrer, K. P. Sullivan, M. Otto, J. E. McGeehan, Y. Román-Leshkov, N. Wierckx and G. T. Beckham, Nat. Catal., 2021, 4, 539–556 CrossRef CAS.
- J. G. Kim, Polym. Chem., 2020, 11, 4830–4849 RSC.
- E. Rezvani Ghomi, F. Khosravi, A. Saedi Ardahaei, Y. Dai, R. E. Neisiany, F. Foroughi, M. Wu, O. Das and S. Ramakrishna, Polymers, 2021, 13, 1854 CrossRef PubMed.
- X. Bai, D. R. Aireddy, A. Roy and K. Ding, Angew. Chem., Int. Ed., 2023, 62, e202309949 CrossRef CAS PubMed.
- C. Z. Yang, S. I. Yaniger, V. C. Jordan, D. J. Klein and G. D. Bittner, Environ. Health Perspect., 2011, 119, 989–996 CrossRef CAS.
- S. Biedermann, P. Tschudin and K. Grob, Anal. Bioanal. Chem., 2010, 398, 571–576 CrossRef CAS PubMed.
- J. Liu, L. Zhang, G. Lu, R. Jiang and Z. Yan, Ecotoxicol. Environ. Saf., 2020111481 Search PubMed.
- S. Cruz and M. Zanin, J. Appl. Polym. Sci., 2006, 99, 2117–2123 CrossRef CAS.
- A. Kassab, D. Al Nabhani, P. Mohanty, C. Pannier and G. Y. Ayoub, Polymers, 2023, 15, 3881 CrossRef CAS PubMed.
- Z. O. G. Schyns and M. P. Shaver, Macromol. Rapid Commun., 2021, 42, 2000415 CrossRef CAS.
- P. T. Benavides, J. B. Dunn, J. Han, M. Biddy and J. Markham, ACS Sustainable Chem. Eng., 2018, 6, 9725–9733 CrossRef CAS.
- G. Bishop, D. Styles and P. N. Lens, Resour., Conserv. Recycl., 2021, 168, 105451 CrossRef CAS.
- D. Parida, A. Aerts, K. Vanbroekhoven, M. Van Dael, H. Mitta, L. Li, W. Eevers, K. M. Van Geem, E. Feghali and K. Elst, Prog. Polym. Sci., 2024, 149, 101783 CrossRef CAS.
- M. Comí, B. Van Ballaer, J. Gracia-Vitoria, D. Parida, A. Aerts, K. Vanbroekhoven and R. Vendamme, ACS Sustainable Chem. Eng., 2024, 12, 9279–9289 CrossRef.
- F. Liguori, C. Moreno-Marrodan and P. Barbaro, Chem. Soc. Rev., 2020, 49, 6329–6363 RSC.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y.-X. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803–814 CrossRef CAS PubMed.
- D. Chen, K. Kannan, H. Tan, Z. Zheng, Y.-L. Feng, Y. Wu and M. Widelka, Environ. Sci. Technol., 2016, 50, 5438–5453 CrossRef CAS.
- Y. Dai and X. Zhang, Polym. Chem., 2017, 8, 7429–7437 RSC.
- J.-G. Rosenboom, R. Langer and G. Traverso, Nat. Rev. Mater., 2022, 7, 117–137 CrossRef.
- L. M. Nico Becker and A. Siebert-Raths, Biopolymers facts and statistics 2022, 2023 Search PubMed.
- N. George and T. Kurian, Ind. Eng. Chem. Res., 2014, 53, 14185–14198 CrossRef CAS.
- R. Chen, S. Deng, T. Cui, S. Duan, Q. Jia and L. Zhang, Prog. Rubber, Plast. Recycl. Technol., 2023, 40(1), 77–97 CrossRef.
- K. Dutt and R. Soni, Polym. Sci., Ser. B, 2013, 55, 430–452 CrossRef CAS.
- B. Raj, J. Rahul, P. K. Singh, V. V. K. Rao, J. Kumar, N. Dwivedi, P. Kumar, D. Singh and K. Strzałkowski, Sustainability, 2023, 15, 12269 CrossRef CAS.
- H. Abedsoltan, Polym. Eng. Sci., 2023, 2651–2674 CrossRef CAS.
- F. Cao, L. Wang, R. Zheng, L. Guo, Y. Chen and X. Qian, RSC Adv., 2022, 12, 31564–31576 RSC.
- H. Jones, F. Saffar, V. Koutsos and D. Ray, Energies, 2021, 14, 7306 CrossRef CAS.
- J. Campanelli, D. Cooper and M. Kamal, J. Appl. Polym. Sci., 1994, 53, 985–991 CrossRef CAS.
- G. Güçlü, T. Yalçınyuva, S. Özgümüş and M. Orbay, Thermochim. Acta, 2003, 404, 193–205 CrossRef.
- T. Yoshioka, T. Sato and A. Okuwaki, J. Appl. Polym. Sci., 1994, 52, 1353–1355 CrossRef CAS.
- S. Zhang, M. Li, Z. Zuo and Z. Niu, Green Chem., 2023, 25, 6949–6970 RSC.
- V. A. Kosmidis, D. S. Achilias and G. P. Karayannidis, Macromol. Mater. Eng., 2001, 286, 640–647 CrossRef CAS.
- L. Zhang, J. Gao, J. Zou and F. Yi, J. Appl. Polym. Sci., 2013, 130, 2790–2795 CrossRef CAS.
- D. Stanica-Ezeanu and D. Matei, Sci. Rep., 2021, 11, 4431 CrossRef CAS PubMed.
- A. B. Raheem, Z. Z. Noor, A. Hassan, M. K. Abd Hamid, S. A. Samsudin and A. H. Sabeen, J. Cleaner Prod., 2019, 225, 1052–1064 CrossRef CAS.
- J. Xin, Q. Zhang, J. Huang, R. Huang, Q. Z. Jaffery, D. Yan, Q. Zhou, J. Xu and X. Lu, J. Environ. Manage., 2021, 296, 113267 CrossRef CAS PubMed.
- F. R. Veregue, C. T. Pereira da Silva, M. P. Moises, J. G. Meneguin, M. R. Guilherme, P. A. Arroyo, S. L. Favaro, E. Radovanovic, E. M. Girotto and A. W. Rinaldi, ACS Sustainable Chem. Eng., 2018, 6, 12017–12024 CrossRef CAS.
- A. Bohre, P. R. Jadhao, K. Tripathi, K. K. Pant, B. Likozar and B. Saha, ChemSusChem, 2023, 16, e202300142 CrossRef CAS PubMed.
- J. W. Gamble, A. N. A. Andrius and R. B. DeBruin, U.S. Pat., 529853029, 1994 Search PubMed.
- M. Han, in Recycling of Polyethylene Terephthalate Bottles, Elsevier, 2019, pp. 85–108 Search PubMed.
- D. D. Pham and J. Cho, Green Chem., 2021, 23, 511–525 RSC.
- Making Cutting Edge Science an Everyday Reality, https://www.loopindustries.com/en/about (accessed 01/08/22).
- J. Paben , Plast. Recycl. Updat, 2021.
- A. A. Naujokas and K. M. Ryan, U.S. Pat., 505152824, 1991 Search PubMed.
- Technology: Simple Sustainable Recycling, https://www.depoly.ch (accessed accessed 01/08/22).
- S. L. Anderson, C. P. Ireland, B. Smit and K. Stylianou, Degradation of plastic materials into terephthalic acid (tpa), ethylene glycol and/or other monomers that form the plastic materials, 2020 Search PubMed.
- F. G. Barla, T. Showalter, H.-C. Su, J. Jones and I. Bobe, U.S. Pat., 1050159910, 2019 Search PubMed.
- Threading Together the Future of Circular Fashion, https://circ.earth/, (accessed Feb 02, 2022).
- Moving from Linear to Circular, https://gr3n-recycling.com/technology.html/, (accessed 01/08/22).
- BRING Technology, https://www.jeplan.co.jp/en/technology/, (accessed 01/08/22).
- P. Heikkilä, et al., Technologies and Model for Sustainable Textile Recycling, 2024 Search PubMed.
- Ioniqa takes the first 10-kiloton PET upcycling factory into operation, https://ioniqa.com/ioniqa-takes-first-10-kilotonpet-upcycling-factory-into-operation/, (accessed 01/08/22).
- Technip Energies, IBM and Under Armour form joint venture to advance the possibilities of plastics recycling technology, https://www.technipenergies.com/media/news/technip-energies-ibm-under-armour-form-joint-ventureadvance-possibilities-plastics-recycling/, (accessed 01/08/22).
- IFP Energies Nouvelles, https://www.ifpenergiesnouvelles.fr/innovation-et-industrie/nos-expertises/climat-et-environnement/recyclage-desplastiques/nos-solutions, 2023.
- Enzymatic Recycling: Removing the Constraints of Current Processes, https://www.carbios.com/en/enzymaticrecycling/, (accessed 01/08/22).
- Our Partners, https://www.carbios.com/en/partnering/, (accessed 01/08/22).
- Technip announces the successful launch of the Carbios industrial demonstration plant, https://www.hydrocarbonprocessing.com/news/2021/11/technip-announces-successful-launch-ofcarbios-industrial-demonstration-plant/, (accessed 01/19/2022).
- H. Li, H. A. Aguirre-Villegas, R. D. Allen, X. Bai, C. H. Benson, G. T. Beckham, S. L. Bradshaw, J. L. Brown, R. C. Brown and V. S. Cecon, Green Chem., 2022, 24, 8899–9002 RSC.
- S. H. Kim and S. H. Hong, ACS Catal., 2014, 4, 3630–3636 CrossRef CAS.
- K. Onida, M. Fayad, S. Norsic, O. Boyron and N. Duguet, Green Chem., 2023, 25, 4282–4291 RSC.
- A. Gluth, Z. Xu, L. S. Fifield and B. Yang, Renewable Sustainable Energy Rev., 2022, 170, 112966 CrossRef CAS.
- M. Watanabe, Y. Matsuo, T. Matsushita, H. Inomata, T. Miyake and K. Hironaka, Polym. Degrad. Stab., 2009, 94, 2157–2162 CrossRef CAS.
- H. Weingärtner and E. U. Franck, Angew. Chem., Int. Ed., 2005, 44, 2672–2692 CrossRef PubMed.
- G. Grause, N. Tsukada, W. J. Hall, T. Kameda, P. T. Williams and T. Yoshioka, Polym. J., 2010, 42, 438–442 CrossRef CAS.
- G. Grause, K. Sugawara, T. Mizoguchi and T. Yoshioka, Polym. Degrad. Stab., 2009, 94, 1119–1124 CrossRef CAS.
- G. Grause, R. Kärrbrant, T. Kameda and T. Yoshioka, Ind. Eng. Chem. Res., 2014, 53, 4215–4223 CrossRef CAS.
- G. O. Jones, A. Yuen, R. J. Wojtecki, J. L. Hedrick and J. M. Garcia, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 7722–7726 CrossRef CAS.
- Y. Huang, S. Liu and Z. Pan, Polym. Degrad. Stab., 2011, 96, 1405–1410 CrossRef CAS.
- E. Quaranta, Appl. Catal., B, 2017, 206, 233–241 CrossRef CAS.
- T. M. Shaikh, A. Sudalai, K. Cho and C. Jo, Eur. J. Org. Chem., 2024, e202301308 CrossRef CAS.
- M. Hong and E. Y.-X. Chen, Green Chem., 2017, 19, 3692–3706 RSC.
- E. Quaranta, A. Dibenedetto, F. Nocito and P. Fini, J. Hazard. Mater., 2021, 403, 123957 CrossRef CAS.
- C.-H. Wu, L.-Y. Chen, R.-J. Jeng and S. A. Dai, ACS Sustainable Chem. Eng., 2018, 6, 8964–8975 CrossRef CAS.
- Q. Zhao, L. An, C. Li, L. Zhang, J. Jiang and Y. Li, Compos. Sci. Technol., 2022, 224, 109461 CrossRef CAS.
- L. Shao, Y.-C. Chang, B. Zhao, X. Yan, B. J. Bliss, M.-E. Fei, C. Yu and J. Zhang, Chem. Eng. J., 2023, 464, 142735 CrossRef CAS.
- Y. Liao, S.-F. Koelewijn, G. Van den Bossche, J. Van Aelst, S. Van den Bosch, T. Renders, K. Navare, T. Nicolaï, K. Van Aelst and M. Maesen, Science, 2020, 367, 1385–1390 CrossRef CAS PubMed.
- Chemical recycling of polycarbonates reaches a major milestone, covestro.com.
- Y. Bai, Z. Wang and L. Feng, Mater. Des., 2010, 31, 999–1002 CrossRef CAS.
- I. Okajima, M. Hiramatsu and T. Sako, Adv. Mater. Res., 2011, 222, 243–246 CAS.
- I. Okajima and T. Sako, J. Mater. Cycles Waste Manage., 2017, 19, 15–20 CrossRef CAS.
- I. Okajima, K. Watanabe and T. Sako, J. Adv. Res. Phys., 2012, 3, 021211 Search PubMed.
- R. Piñero-Hernanz, C. Dodds, J. Hyde, J. García-Serna, M. Poliakoff, E. Lester, M. J. Cocero, S. Kingman, S. Pickering and K. H. Wong, Composites, Part A, 2008, 39, 454–461 CrossRef.
- X. Kuang, Y. Zhou, Q. Shi, T. Wang and H. J. Qi, ACS Sustainable Chem. Eng., 2018, 6, 9189–9197 CrossRef CAS.
- T. Liu, X. Guo, W. Liu, C. Hao, L. Wang, W. C. Hiscox, C. Liu, C. Jin, J. Xin and J. Zhang, Green Chem., 2017, 19, 4364–4372 RSC.
- X. Zhao, X.-L. Wang, F. Tian, W.-L. An, S. Xu and Y.-Z. Wang, Green Chem., 2019, 21, 2487–2493 RSC.
- X. Wei, J. Yu, J. Du and L. Sun, Energy Fuels, 2021, 35, 5026–5038 CrossRef CAS.
- Y. Jing, Y. Wang, S. Furukawa, J. Xia, C. Sun, M. J. Hülsey, H. Wang, Y. Guo, X. Liu and N. Yan, Angew. Chem., Int. Ed., 2021, 60, 5527–5535 CrossRef CAS PubMed.
- D. L. Lourenço, D. F. Oliveira and A. C. Fernandes, Adv. Sustainable Syst., 2024, 2300381 CrossRef.
- T. A. Branco and A. C. Fernandes, Adv. Sustainable Syst., 2023, 7, 2300217 CrossRef CAS.
- Z. Zhao, Z. Li, X. Zhang, T. Li, Y. Li, X. Chen and K. Wang, Environ. Pollut., 2022, 313, 120154 CrossRef CAS.
- M. Okamoto, J. I. Sugiyama and T. Yamamoto, J. Appl. Polym. Sci., 2008, 110, 3902–3907 CrossRef CAS.
- Y. Liu and X. B. Lu, J. Polym. Sci., 2022, 60, 3256–3268 CrossRef CAS.
- E. A. Gilbert, M. L. Polo, J. M. Maffi, J. F. Guastavino, S. E. Vaillard and D. A. Estenoz, J. Polym. Sci., 2022, 60, 3284–3317 CrossRef CAS.
- J. Xu, K. Zhou, J. Fu, Z. Tan, L. Qin, P. Duan, Y. Xu and S. Kang, Fuel, 2023, 334, 126609 CrossRef CAS.
- Q. Kang, M. Chu, P. Xu, X. Wang, S. Wang, M. Cao, O. Ivasenko, T. K. Sham, Q. Zhang and Q. Sun, Angew. Chem., Int. Ed., 2023, 62, e202313174 CrossRef CAS PubMed.
- J.-H. Luo and J. Deng, ACS Sustainable Chem. Eng., 2023, 11, 17120–17129 CrossRef CAS.
- A. K. Manal, G. V. Shanbhag and R. Srivastava, Appl. Catal., B, 2023, 123021 CrossRef CAS.
- L. Wang, G. Li, Y. Cong, A. Wang, X. Wang, T. Zhang and N. Li, Green Chem., 2021, 23, 3693–3699 RSC.
- H. Liu, J. Zhang, J. Gu, R. Shan, H. Yuan and Y. Chen, J. Cleaner Prod., 2023, 425, 138924 CrossRef CAS.
- C. Michel and P. Gallezot, ACS Catal., 2015, 5, 4130–4132 CrossRef CAS.
- F. G. Terrade, J. van Krieken, B. J. Verkuijl and E. Bouwman, ChemSusChem, 2017, 10, 1904–1908 CrossRef CAS.
- I. A. Shuklov, H. Jiao, J. Schulze, W. Tietz, K. Kühlein and A. Börner, Tetrahedron Lett., 2011, 52, 1027–1030 CrossRef CAS.
- J. Meimoun, A. Favrelle-Huret, M. Bria, N. Merle, G. Stoclet, J. De Winter, R. Mincheva, J.-M. Raquez and P. Zinck, Polym. Degrad. Stab., 2020, 181, 109188 CrossRef CAS.
- T. Su, G.-P. Lu, K. Sun, P. Wu and C. Cai, J. Environ. Chem. Eng., 2023, 11, 111397 CrossRef CAS.
- F. Liguori, C. Moreno-Marrodán, W. Oberhauser, E. Passaglia and P. Barbaro, RSC Sustainability, 2023, 1, 1394–1403 RSC.
- B.-X. Lai, S. Bhattacharjee, Y.-H. Huang, A.-B. Duh, P.-C. Wang and C.-S. Tan, Polymers, 2020, 12, 2513 CrossRef CAS PubMed.
- A. Ahrens, A. Bonde, H. Sun, N. K. Wittig, H. C. D. Hammershøj, G. M. F. Batista, A. Sommerfeldt, S. Frølich, H. Birkedal and T. Skrydstrup, Nature, 2023, 1–8 CAS.
- W.-Y. Lu, S. Bhattacharjee, B.-X. Lai, A.-B. Duh, P.-C. Wang and C.-S. Tan, Ind. Eng. Chem. Res., 2019, 58, 16326–16337 CrossRef CAS.
- B. F. Nunes, M. C. Oliveira and A. C. Fernandes, Green Chem., 2020, 22, 2419–2425 RSC.
- T.-W. Jiang, K. S. K. Reddy, Y.-C. Chen, M.-W. Wang, H.-C. Chang, M. M. Abu-Omar and C.-H. Lin, ACS Sustainable Chem. Eng., 2022, 10, 2429–2440 CrossRef CAS.
- Y. Wang, X. Cui, H. Ge, Y. Yang, Y. Wang, C. Zhang, J. Li, T. Deng, Z. Qin and X. Hou, ACS Sustainable Chem. Eng., 2015, 3, 3332–3337 CrossRef CAS.
- K. Wakafuji, S. Iwasa, K. N. Ouchida, H. Cho, H. Dohi, E. Yamamoto, T. Kamachi and M. Tokunaga, ACS Catal., 2021, 11, 14067–14075 CrossRef CAS.
- Y. Xu, S. Dai, H. Zhang, L. Bi, J. Jiang and Y. Chen, ACS Sustainable Chem. Eng., 2021, 9, 16281–16290 CrossRef CAS.
- S. Nakamura, Y. Saegusa, H. Yanagisawa, M. Touse, T. Shirai and T. Nishikubo, Thermochim. Acta, 1991, 183, 269–277 CrossRef CAS.
- S. Nakamura and M. Arima, J. Therm. Anal. Calorim., 1993, 40, 613–619 CrossRef CAS.
- C.-M. Lin, C.-H. Chen, C.-H. Lin, W. C. Su and T.-Y. Juang, ACS Omega, 2018, 3, 4295–4305 CrossRef CAS PubMed.
- Y. Qi, Z. Weng, Y. Kou, L. Song, J. Li, J. Wang, S. Zhang, C. Liu and X. Jian, Chem. Eng. J., 2021, 406, 126881 CrossRef CAS.
- B. Krishnakumar, R. P. Sanka, W. H. Binder, V. Parthasarthy, S. Rana and N. Karak, Chem. Eng. J., 2020, 385, 123820 CrossRef CAS.
- H. Liu, H. Zhang, H. Wang, X. Huang, G. Huang and J. Wu, Chem. Eng. J., 2019, 368, 61–70 CrossRef CAS.
- Y.-Y. Liu, G.-L. Liu, Y.-D. Li, Y. Weng and J.-B. Zeng, ACS Sustainable Chem. Eng., 2021, 9, 4638–4647 CrossRef CAS.
- S. W. Shao, C. H. Chen, J. R. Chan, T. Y. Juang, M. M. Abu-Omar and C. H. Lin, Green Chem., 2020, 22, 4683–4696 RSC.
- B. Wang, S. Ma, S. Yan and J. Zhu, Green Chem., 2019, 21, 5781–5796 RSC.
- Z. Yang, X. Li, F. Xu, W. Wang, Y. Shi, Z. Zhang, W. Fang, L. Liu and S. Zhang, Green Chem., 2021, 23, 447–456 RSC.
- H. Wang, F. Xu, Z. Zhang, M. Feng, M. Jiang and S. Zhang, RSC Sustainability, 2023, 2162–2179 RSC.
- T. Abe, R. Takashima, T. Kamiya, C. P. Foong, K. Numata, D. Aoki and H. Otsuka, Green Chem., 2021, 23, 9030–9037 RSC.
- Y. Shen, Fuel Process. Technol., 2022, 236, 107437 CrossRef CAS.
- Q. Zhang, C. Hu, R. Duan, Y. Huang, X. Li, Z. Sun, X. Pang and X. Chen, Green Chem., 2022, 24, 9282–9289 RSC.
- C. E. Berdugo-Díaz, Y. S. Yun, M. T. Manetsch, J. Luo, D. G. Barton, X. Chen and D. W. Flaherty, ACS Catal., 2022, 12, 9717–9734 CrossRef.
- W. Stuyck, K. Janssens, M. Denayer, F. De Schouwer, R. Coeck, K. V. Bernaerts, J. Vekeman, F. De Proft and D. E. De Vos, Green Chem., 2022, 24, 6923–6930 RSC.
- J. Ji, T. Zhao and F. Li, Environ. Pollut., 2022, 313, 120166 CrossRef CAS.
- A. C. Fernandes, Green Chem., 2021, 23, 7330–7360 RSC.
- B. Kunwar, H. Cheng, S. R. Chandrashekaran and B. K. Sharma, Renewable Sustainable Energy Rev., 2016, 54, 421–428 CrossRef CAS.
- A. J. Martín, C. Mondelli, S. D. Jaydev and J. Perez-Ramirez, Chem, 2021, 7, 1487–1533 Search PubMed.
- J. Wei, J. Liu, W. Zeng, Z. Dong, J. Song, S. Liu and G. Liu, Catal. Sci. Technol., 2023, 1258–1280 RSC.
- J. R. Banu and V. G. Sharmila, Catal. Sci. Technol., 2023, 13, 2291–2302 RSC.
- C. Wang, H. Han, Y. Wu and D. Astruc, Coord. Chem. Rev., 2022, 458, 214422 CrossRef CAS.
- V. D. Lechuga-Islas, D. M. Sánchez-Cerrillo, S. Stumpf, R. Guerrero-Santos, U. S. Schubert and C. Guerrero-Sánchez, Polym. Chem., 2023, 14, 1893–1904 RSC.
- X. Zhang, Q. Gan, P. Zhou, Z. Chen, Z. Zhang and G.-P. Lu, Appl. Catal., B, 2024, 344, 123626 CrossRef CAS.
- B. C. Vance, P. A. Kots, C. Wang, J. E. Granite and D. G. Vlachos, Appl. Catal., B, 2023, 322, 122138 CrossRef CAS.
- C. Wang, T. Xie, P. A. Kots, B. C. Vance, K. Yu, P. Kumar, J. Fu, S. Liu, G. Tsilomelekis and E. A. Stach, JACS Au, 2021, 1, 1422–1434 CrossRef CAS.
- A. Al-Sabagh, F. Yehia, G. Eshaq, A. Rabie and A. ElMetwally, Egypt. J. Pet., 2016, 25, 53–64 CrossRef.
- S. Yoshida, K. Hiraga, T. Takehana, I. Taniguchi, H. Yamaji, Y. Maeda, K. Toyohara, K. Miyamoto, Y. Kimura and K. Oda, Science, 2016, 351, 1196–1199 CrossRef CAS PubMed.
- S. Mahalingam, B. T. Raimi-Abraham, D. Q. Craig and M. Edirisinghe, Chem. Eng. J., 2015, 280, 344–353 CrossRef CAS.
- K. Weisskopf, J. Polym. Sci., Part A: Polym. Chem., 1988, 26, 1919–1935 CrossRef CAS.
- A. B. Lende, S. Bhattacharjee and C.-S. Tan, Ind. Eng. Chem. Res., 2020, 59, 21333–21346 CrossRef CAS.
- J. C. Worch and A. P. Dove, ACS Macro Lett., 2020, 9, 1494–1506 CrossRef CAS PubMed.
- S. Al-Salem, P. Lettieri and J. Baeyens, Prog. Energy Combust. Sci., 2010, 36, 103–129 CrossRef CAS.
- Sharing insights elevates their impact, https://www.spglobal.com/commodityinsights/en/ci/products/polycarbonate-resins-chemical-economics-handbook.html/, (accessed 20–05–23).
- T. Do, E. R. Baral and J. G. Kim, Polymer, 2018, 143, 106–114 CrossRef CAS.
- S. D. Thorat, P. J. Phillips, V. Semenov and A. Gakh, J. Appl. Polym. Sci., 2003, 89, 1163–1176 CrossRef CAS.
- M. C. Delpech, F. M. Coutinho and M. E. S. Habibe, Polym. Test., 2002, 21, 155–161 CrossRef CAS.
- L. Monsigny, J.-C. Berthet and T. Cantat, ACS Sustainable Chem. Eng., 2018, 6, 10481–10488 CrossRef CAS.
- A. C. Fernandes, ChemSusChem, 2021, 14, 4228–4233 CrossRef CAS.
- M. E. Viana, A. Riul, G. M. Carvalho, A. F. Rubira and E. C. Muniz, Chem. Eng. J., 2011, 173, 210–219 CrossRef CAS.
- T. Thiounn and R. C. Smith, J. Polym. Sci., 2020, 58, 1347–1364 CrossRef CAS.
- K. V. Khopade, S. H. Chikkali and N. Barsu, Cell Rep. Phys. Sci., 2023, 101341 CrossRef CAS.
- C. Wang and O. El-Sepelgy, Curr. Opin. Green Sustain. Chem., 2021, 32, 100547 CrossRef CAS.
- F. Branda, D. Parida, R. Pauer, M. Durante, S. Gaan, G. Malucelli and A. Bifulco, Polymers, 2022, 14, 3853 CrossRef CAS PubMed.
- H. X. Huang XiaoMing, O. Gonzalez, Z. J. Zhu JiaDong, T. Korányi, M. Boot and E. Hensen, Green Chem., 2017, 19, 175–187 RSC.
- J. Hu, M. Zhao, B. Jiang, S. Wu and P. Lu, Energy Fuels, 2020, 34, 9754–9762 CrossRef CAS.
- S. Rautiainen, D. Di Francesco, S. N. Katea, G. Westin, D. N. Tungasmita and J. S. Samec, ChemSusChem, 2019, 12, 404–408 CrossRef CAS PubMed.
- M. Chui, G. Metzker, C. M. Bernt, A. T. Tran, A. C. Burtoloso and P. C. Ford, ACS Sustainable Chem. Eng., 2017, 5, 3158–3169 CrossRef CAS.
- Q. Xu, Q. Wang, L.-P. Xiao, X.-Y. Li, X. Xiao, M.-X. Li, M.-R. Lin, Y.-M. Zhao and R.-C. Sun, J. Mater. Chem. A, 2023, 11, 23809–23820 RSC.
- S. Van den Bosch, W. Schutyser, S.-F. Koelewijn, T. Renders, C. Courtin and B. Sels, Chem. Commun., 2015, 51, 13158–13161 RSC.
- G. Zichittella, A. M. Ebrahim, J. Zhu, A. E. Brenner, G. Drake, G. T. Beckham, S. R. Bare, J. E. Rorrer and Y. Román-Leshkov, JACS Au, 2022, 2, 2259 CrossRef CAS PubMed.
- X. Huang, O. M. M. Gonzalez, J. Zhu, T. I. Korányi, M. D. Boot and E. J. Hensen, Green Chem., 2017, 19, 175–187 RSC.
- J. Aubrecht, V. Pospelova, O. Kikhtyanin, M. Lhotka and D. Kubička, Catal. Today, 2022, 397, 173–181 CrossRef.
- S. Mujahed, M. S. Richter, E. Kirillov, E. Hey-Hawkins and D. Gelman, Isr. J. Chem., 2023, e202300037 CrossRef CAS.
- H. Rong, Y. Zhang, X. Ai, W. Li, F. Cao and L. Li, Inorg. Chem., 2022, 61, 14662–14672 CrossRef CAS PubMed.
- X. Wang, Z. Huo, X. Xie, N. Shanaiah and R. Tong, Chem. – Asian J., 2023, 18, e202201147 CrossRef CAS PubMed.
- K. C. Ott, Final report for the DOE chemical hydrogen storage center of excellence, Los Alamos National Laboratory, 2012 Search PubMed.
- D. Parida, A. Aerts, L. V. Perez, C. Marquez, S. Vloemans, K. Vanbroekhoven, E. Feghali and K. Elst, Chem. Eng. J., 2024, 497, 154390 CrossRef CAS.
- R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC.
- E. Barnard, J. J. R. Arias and W. Thielemans, Green Chem., 2021, 23, 3765–3789 RSC.
- A. K. Manal, G. Saini and R. Srivastava, Green Chem., 2024, 26, 3814–3831 RSC.
- M. Bartoli, L. Rosi, G. Petrucci, L. Armelao, W. Oberhauser, M. Frediani, O. Piccolo, V. D. Rathod and S. Paganelli, Catal. Commun., 2015, 69, 228–233 CrossRef CAS.
- M. Kobylarski, J.-C. Berthet and T. Cantat, Chem. Commun., 2022, 58, 2830–2833 RSC.
- N. M. Rezayee, C. A. Huff and M. S. Sanford, J. Am. Chem. Soc., 2015, 137, 1028–1031 CrossRef CAS.
- S. Westhues, J. Idel and J. Klankermayer, Sci. Adv., 2018, 4, eaat9669 CrossRef CAS PubMed.
- Y. Kratish, J. Li, S. Liu, Y. Gao and T. J. Marks, Angew. Chem., 2020, 132, 20029–20033 CrossRef.
- E. Feghali and T. Cantat, ChemSusChem, 2015, 8, 980–984 CrossRef CAS.
- Y. Kratish and T. J. Marks, Angew. Chem., Int. Ed., 2022, 61, e202112576 CrossRef CAS PubMed.
- M. Accolla, G. Santoro, P. Merino, L. Martínez, G. Tajuelo-Castilla, L. Vázquez, J. M. Sobrado, M. Agúndez, M. Jiménez-Redondo and V. J. Herrero, Astrophys. J., 2021, 906, 44 CrossRef CAS PubMed.
- M. Kobylarski, L. J. Donnelly, J.-C. Berthet and T. Cantat, Green Chem., 2022, 24, 6810–6815 RSC.
- W. T. Lee, A. P. van Muyden, F. D. Bobbink, Z. Huang and P. J. Dyson, Angew. Chem., 2019, 131, 567–570 CrossRef.
- D. L. Lourenço and A. C. Fernandes, Mol. Catal., 2023, 542, 113128 CrossRef.
- Q. Zhang, C. Hu, X. Pang and X. Chen, ChemSusChem, 2023, e202300907 Search PubMed.
- D. L. Lourenço and A. C. Fernandes, Catalysts, 2022, 12, 381 CrossRef.
- S. Suárez-Pantiga and R. Sanz, Org. Biomol. Chem., 2021, 19, 10472–10492 RSC.
- P. H. Pham, S. Barlow, S. R. Marder and O. R. Luca, Chem. Catal., 2023, 3, 100675 CrossRef CAS.
- S. Kang, W. Yuan, W. Chen, M. Du, Y. Zhang and B. Qiu, Nanotechnology, 2023, 462001 CrossRef CAS.
- A. Chaudhary and R. Srivastava, ChemistrySelect, 2023, 8, e202301709 CrossRef CAS.
- A. R. Kamali and S. Li, Appl. Energy, 2023, 334, 120692 CrossRef CAS.
- X. Liu, M. Kobylarski, J.-C. Berthet and T. Cantat, Chem. Commun., 2023, 59, 11228–11231 RSC.
- C. Gunanathan and D. Milstein, Chem. Rev., 2014, 114, 12024–12087 CrossRef CAS PubMed.
- A. Kumar, N. von Wolff, M. Rauch, Y.-Q. Zou, G. Shmul, Y. Ben-David, G. Leitus, L. Avram and D. Milstein, J. Am. Chem. Soc., 2020, 142, 14267–14275 CrossRef CAS PubMed.
- S. P. áMc Ilrath, Chem. Commun., 2014, 50, 4884–4887 RSC.
- L.-C. Hu, A. Oku and E. Yamada, Polymer, 1998, 39, 3841–3845 CrossRef CAS.
- R. Piñero, J. García and M. J. Cocero, Green Chem., 2005, 7, 380–387 RSC.
- J. Zhang, G. Leitus, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2005, 127, 10840–10841 CrossRef CAS PubMed.
- Q. Qian, G. Liu, D. Lang, C. Guo, L. Wang and R. Wu, Mater. Today Sustainability, 2022, 20, 100254 CrossRef.
- J. A. Fuentes, S. M. Smith, M. T. Scharbert, I. Carpenter, D. B. Cordes, A. M. Slawin and M. L. Clarke, Chem. – Eur. J., 2015, 21, 10851–10860 CrossRef CAS PubMed.
- S. Westhues, M. Meuresch and J. Klankermayer, Angew. Chem., Int. Ed., 2016, 55, 12841–12844 CrossRef CAS PubMed.
- Z. Han, L. Rong, J. Wu, L. Zhang, Z. Wang and K. Ding, Angew. Chem., 2012, 124, 13218–13222 CrossRef.
- J. Zhang, G. Leitus, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2006, 45, 1113–1115 CrossRef CAS PubMed.
- V. Zubar, Y. Lebedev, L. M. Azofra, L. Cavallo, O. El-Sepelgy and M. Rueping, Angew. Chem., Int. Ed., 2018, 57, 13439–13443 CrossRef CAS PubMed.
- A. Kumar, T. Janes, N. A. Espinosa-Jalapa and D. Milstein, Angew. Chem., Int. Ed., 2018, 57, 12076–12080 CrossRef CAS PubMed.
- X. Liu, J. G. de Vries and T. Werner, Green Chem., 2019, 21, 5248–5255 RSC.
- T. O. Kindler, C. Alberti, J. Sundermeier and S. Enthaler, ChemistryOpen, 2019, 8, 1410–1412 CrossRef CAS PubMed.
- C. Alberti, S. Eckelt and S. Enthaler, ChemistrySelect, 2019, 4, 12268–12271 CrossRef CAS.
- C. Alberti, E. Fedorenko and S. Enthaler, ChemistryOpen, 2020, 9, 818–821 CrossRef CAS PubMed.
- P. Dahiya, M. K. Gangwar and B. Sundararaju, ChemCatChem, 2021, 13, 934–939 CrossRef CAS.
- J. M. Payne, M. Kamran, M. G. Davidson and M. D. Jones, ChemSusChem, 2022, 15, e202200255 CrossRef CAS PubMed.
- O. Awogbemi and D. V. Von Kallon, J. Energy Inst., 2023, 106, 101154 CrossRef CAS.
- M.-Q. Zhang, M. Wang, B. Sun, C. Hu, D. Xiao and D. Ma, Chem, 2022, 8, 2912–2923 CAS.
- Y. Hu, S. Zhang, J. Xu, Y. Liu, A. Yu, J. Qian and Y. Xie, Angew. Chem., Int. Ed., 2023, 62, e202312564 CrossRef CAS PubMed.
- A. Ahrens, A. Bonde, H. Sun, N. K. Wittig, H. C. D. Hammershøj, G. M. F. Batista, A. Sommerfeldt, S. Frølich, H. Birkedal and T. Skrydstrup, Nature, 2023, 617, 730–737 CrossRef CAS PubMed.
- S. J. Grabowski, Chem. Phys. Lett., 2001, 338, 361–366 CrossRef CAS.
- P. Ríos, A. Rodríguez and S. Conejero, Chem. Sci., 2022, 13, 7392–7418 RSC.
- T. Higashi, S. Kusumoto and K. Nozaki, Chem. Rev., 2019, 119, 10393–10402 CrossRef CAS PubMed.
- M. Szewczyk, M. Magre, V. Zubar and M. Rueping, ACS Catal., 2019, 9, 11634–11639 CrossRef CAS.
- C. Erken, A. Kaithal, S. Sen, T. Weyhermüller, M. Hölscher, C. Werlé and W. Leitner, Nat. Commun., 2018, 9, 4521 CrossRef PubMed.
- X. Cao, W. Wang, K. Lu, W. Yao, F. Xue and M. Ma, Dalton Trans., 2020, 49, 2776–2780 RSC.
- T. W. Kim, D. Kim, S. H. Kim and Y.-W. Suh, ChemCatChem, 2024, e202301581 CrossRef.
- Q. Hou, M. Zhen, H. Qian, Y. Nie, X. Bai, T. Xia, M. L. U. Rehman, Q. Li and M. Ju, Cell Rep. Phys. Sci., 2021, 2, 100514 CrossRef CAS.
- G. L. Haller, J. Catal., 2003, 216, 12–22 CrossRef CAS.
- J. Limido, J. Lepage, P. Corty, E. Freund, J. Franck, Y. Jacquin, B. Juguin, C. Marcilly, G. Martino and J. Miquel, Institute Francais du Petrole, Applied Heterogeneous Catalysis Design Manufacture Use of Solid Catalysts, 1987, vol. 329.
- J. M. Thomas and W. J. Thomas, Principles and practice of heterogeneous catalysis, John Wiley & Sons, 2014 Search PubMed.
- I. Fechete and J. C. Vedrine, Nanotechnology in Catalysis: Applications in the Chemical Industry, Energy Development, and Environment Protection, 2017, pp. 57–90 Search PubMed.
- R. Ye, S. Xiao, Q. Lai, D. Wang, Y. Huang, G. Feng, R. Zhang and T. Wang, Catalysts, 2022, 12, 747 CrossRef CAS.
- J. C. Védrine, Catalysts, 2017, 7, 341 CrossRef.
- M. Boronat and P. Concepcion, Catal. Today, 2017, 285, 166–178 CrossRef CAS.
- Z. Heidarinejad, M. H. Dehghani, M. Heidari, G. Javedan, I. Ali and M. Sillanpää, Environ. Chem. Lett., 2020, 18, 393–415 CrossRef CAS.
- M. Miceli, P. Frontera, A. Macario and A. Malara, Catalysts, 2021, 11, 591 CrossRef CAS.
- C. Perego and P. Villa, Catal. Today, 1997, 34, 281–305 CrossRef CAS.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- J. Aguado, D. Serrano and J. Escola, Ind. Eng. Chem. Res., 2008, 47, 7982–7992 CrossRef CAS.
- T. Uekert, M. F. Kuehnel, D. W. Wakerley and E. Reisner, Energy Environ. Sci., 2018, 11, 2853–2857 RSC.
- T. Uekert, H. Kasap and E. Reisner, J. Am. Chem. Soc., 2019, 141, 15201–15210 CrossRef CAS PubMed.
- H. Tang, N. Li, G. Li, A. Wang, Y. Cong, G. Xu, X. Wang and T. Zhang, Green Chem., 2019, 21, 2709–2719 RSC.
- S. Lu, Y. Jing, B. Feng, Y. Guo, X. Liu and Y. Wang, ChemSusChem, 2021, 14, 4242–4250 CrossRef CAS PubMed.
- S. Hongkailers, Y. Jing, Y. Wang, N. Hinchiranan and N. Yan, ChemSusChem, 2021, 14, 4330–4339 CrossRef CAS PubMed.
- Z. Gao, B. Ma, S. Chen, J. Tian and C. Zhao, Nat. Commun., 2022, 13, 3343 CrossRef CAS PubMed.
- K. Lee, Y. Jing, Y. Wang and N. Yan, Nat. Rev. Chem., 2022, 6, 635–652 CrossRef PubMed.
- R. Li, W. Zeng, R. Zhao, Y. Zhao, Y. Wang, F. Zhang, M. Tang, Y. Wang, X. Chang and F. Wu, Nano Res., 2023, 1–7 Search PubMed.
- Y. Liao, K. Takahashi and K. Nozaki, J. Am. Chem. Soc., 2024, 146(4), 2419–2425 CrossRef CAS PubMed.
- L. Wang, F. Han, G. Li, M. Zheng, A. Wang, X. Wang, T. Zhang, Y. Cong and N. Li, Green Chem., 2021, 23, 912–919 RSC.
- J. Liu, J. Wei, X. Feng, M. Song, S. Shi, S. Liu and G. Liu, Appl. Catal., B, 2023, 338, 123050 CrossRef CAS.
- H. Tang, Y. Hu, G. Li, A. Wang, G. Xu, C. Yu, X. Wang, T. Zhang and N. Li, Green Chem., 2019, 21, 3789–3795 RSC.
- S. C. Kosloski-Oh, Z. A. Wood, Y. Manjarrez, J. P. de Los Rios and M. E. Fieser, Mater. Horiz., 2021, 8, 1084–1129 RSC.
- J. E. Rorrer, C. Troyano-Valls, G. T. Beckham and Y. Román-Leshkov, ACS Sustainable Chem. Eng., 2021, 9, 11661–11666 CrossRef CAS.
- S. D. Jaydev, M.-E. Usteri, A. J. Martín and J. Pérez-Ramírez, Chem. Catal., 2023, 3, 100564 CrossRef CAS.
- X. Si, J. Chen, Z. Wang, Y. Hu, Z. Ren, R. Lu, L. Liu, J. Zhang, L. Pan and R. Cai, J. Energy Chem., 2023, 85, 562–569 CrossRef CAS.
- S. S. Borkar, R. Helmer, S. Panicker and M. Shetty, ACS Sustainable Chem. Eng., 2023, 11, 10142–10157 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc03784g |
| This journal is © The Royal Society of Chemistry 2025 |