
Green and facile modification of mesoporous activated carbon for selective indium and gallium recovery from waste photovoltaic modules†
Wenxuan
Wang
ab,
Xinhai
Xu
*ac,
Jie
Li
a,
Tao
Liu
ab,
Hailong
Wang
ab and
Yin
Wang
 *ac
*ac
aCAS Key Laboratory of Urban Pollutant Conversion, Institute of Urban Environment, Chinese Academy of Sciences, Xiamen, 361021, China. E-mail: xhxu@iue.ac.cn; yinwang@iue.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing, 100049, China
cZhejiang Key Laboratory of Urban Environmental Processes and Pollution Control, CAS Haixi Industrial Technology Innovation Center in Beilun, Ningbo 3158, China
First published on 19th November 2024
Abstract
With the increasing deployment and subsequent retirement of photovoltaic (PV) modules, it is urgent to selectively recover critical metals involved, such as indium (In) and gallium (Ga). Activated carbon, a widely used eco-friendly adsorbent for metal ions, often requires large amounts of toxic chemicals and complicated modifications to achieve selective adsorption. Herein, a novel adsorbent with outstanding ability for In and Ga recovery, phosphoryl-functionalized waste biomass-derived mesoporous activated carbon (P-PDA@MAC), was synthesized via a green and facile one-pot method. This approach eliminates the use of toxic organic reagents and enables functionalization at ambient temperature and pressure, aligning with the principles of green chemistry. Using ethyl phenylphosphinate (EPP) as a precursor, the obtained EPP–PDA@MAC exhibited superior adsorption capacity for In3+ (125.1 mg g−1) and Ga3+ (140.7 mg g−1) and high selectivity (SFXIn = 382.4, SFXGa = 239.0) over competing ions Al3+, Zn2+, Cd2+, Cu2+, and Mg2+. Surprisingly, this adsorbent demonstrated excellent reusability, maintaining adsorption efficiencies above 85% over 9 cycles in a static system and 98% over 50 cycles in a capacitive deionization system. The superior adsorption ability of EPP–PDA@MAC was ascribed to the abundant and stable phosphoryl groups, facilitated by the adhesive polydopamine coating and covalent phosphoryl-functionalization on the high surface area of MAC. Furthermore, a comparison of the adsorption ability, green metrics, and production costs with those of commercial adsorbents underscores the significant industrial application potential of EPP–PDA@MAC. The successful extraction of 99.9% In (purity = 97.79%) and Ga (purity = 90.40%) from waste copper indium gallium selenide PV modules by EPP–PDA@MAC highlights its potential and industrial viability for the circular economy.
1. Introduction
To mitigate fossil fuel reliance and greenhouse gas emissions, renewable energy development is accelerating. Projections indicated that photovoltaic (PV) modules will comprise over half of this renewable energy, reaching millions of tons by 2030.1 Copper indium gallium selenide (CIGS) PV modules are notable for their lower semiconductor usage and high flexibility.2 However, the widespread deployment and limited lifespan (∼12 years) of CIGS modules result in substantial amounts of waste, estimated at 45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons by 2035 and a staggering 2000000 tons by 2050.3,4 Without effective treatment, these waste modules will pose serious environmental risks and lead to huge loss of the valuable components involved. Indium (In) and gallium (Ga) are rare-dispersed and technology critical metals due to their limited natural abundance and various high-tech applications.5 The In and Ga concentrations in waste CIGS modules far exceed those in mineral residues by nearly 29 and 10 times, respectively.6,7 Therefore, In and Ga recovery from waste CIGS modules is crucial to alleviate supply challenges, sustainably foster PV and other high-tech industries, and advance the circular economy.
000 tons by 2035 and a staggering 2000000 tons by 2050.3,4 Without effective treatment, these waste modules will pose serious environmental risks and lead to huge loss of the valuable components involved. Indium (In) and gallium (Ga) are rare-dispersed and technology critical metals due to their limited natural abundance and various high-tech applications.5 The In and Ga concentrations in waste CIGS modules far exceed those in mineral residues by nearly 29 and 10 times, respectively.6,7 Therefore, In and Ga recovery from waste CIGS modules is crucial to alleviate supply challenges, sustainably foster PV and other high-tech industries, and advance the circular economy.
Various methods have been developed for the recovery of critical metals In and Ga, which can be broadly categorized into pyrometallurgy, hydrometallurgy, and biotechnology.2 Pyrometallurgy, a traditional approach, is hindered by high energy consumption and toxic gas emissions, raising concerns about its environmental sustainability. Although biotechnology can offer a green approach, it requires long microbial cultivation periods and often suffers from low metal recovery efficiency, limiting its practicality for large-scale applications. In contrast, hydrometallurgy has gained prominence due to its adaptable process flows, practical applicability, and ability to achieve high metal recovery rates and purity. Hydrometallurgy encompasses several methods, such as chemical precipitation,8 solvent extraction,9 electrochemistry,10 and adsorption.11 Chemical precipitation is a straightforward method but often results in low purity of recovered metals. Electrochemical methods, despite allowing for selective recovery through strictly controlled parameters, involve complex equipment and high energy consumption. Solvent extraction is a mature, cost-effective, and flexible technique that enables the selective recovery of target metals. However, it is limited by the volatility of liquid extractants and the extensive use of organic solvents, which can lead to secondary pollution. Among these methods, adsorption stands out for its efficiency, low cost, operational flexibility, and sustainability. Modified adsorbents not only exhibit selectivity towards target metal ions but also eliminate the need for organic solvents during the adsorption process, thus offering significant potential for sustainable industrial applications.
Three typical types of adsorbents are utilized for In and/or Ga recovery based on distinct adsorption mechanisms. The first type includes composite adsorbents synthesized through the impregnation method, such as P507@MAC,12 D2EHPA@XAD-4 resin,13 and TBP@SiO2-P.14 These materials primarily achieve the selective adsorption of In3+ and Ga3+ by the impregnated liquid extractants. However, their adsorption capacity and reusability are limited by the restricted surface area of the carrier and the inadequacy of impregnation bonding. The second type involves functional group-modified adsorbents. Examples include phytic acid modified carbon fibers prepared via the hydrothermal method (413 K, 14 h),15 PO3H2-modified SiO2@GO synthesized through a multistep process (96 h) involving extensive organic reagents,16 and tannic acid-modified cellulose produced under 30 kGy irradiation and dry-ice cooling, followed by heating at 353 K for 24 h.17 These adsorbents achieved stable adsorption by incorporating functional groups rather than relying on liquid extractants. Nevertheless, their overall adsorption ability is limited, and the preparation process requires extensive toxic chemicals, complex modification steps and stringent conditions. The third type consists of advanced ion-imprinted adsorbents designed to selectively adsorb ions through specific ion-imprinted recognition sites. However, their selectivity for In and Ga remains unsatisfactory due to insufficient imprinting sites, uneven distribution, and interference from similar ions.18,19 Therefore, despite the exploration of various adsorbents, achieving exceptional capacity and selectivity simultaneously under green and facile preparation conditions remains a significant challenge.
Green chemistry plays a pivotal role in environment and materials sciences, particularly in the design of functional materials for environmental applications.20,21 When developing adsorption materials, it is essential to actively learn and adhere to the principles of green chemistry. This involves selecting non-toxic raw materials, reducing energy consumption, and minimizing waste throughout the production process. By implementing these strategies, the development of adsorbents can not only achieve superior adsorption ability for various environmental applications but also demonstrate excellent environmental friendliness and sustainability.22
Activated carbon (AC) derived from biomass waste is a typical adsorbent that is stable, eco-friendly, cost-efficient, and industrialized but lacks selective connection sites for the target metal ions.23 To the best of the authors’ knowledge, only a few AC-based adsorbents for In and Ga recovery have been reported, but they are typically synthesized via the complicated replica method, which is not only costly but also lacks selectivity and recyclability.24 Mesoporous activated carbon (MAC) shows potential because of its huge specific surface area and well-developed mesoporous network, offering sufficient reaction space for modification and adsorption. Our team previously developed an extractant–MAC composite by physical impregnation that showed great capacity and selectivity for In3+ and Ga3+.12 However, the recyclability needs further improvement due to the weak physical bonding between the extractant and MAC. In this case, functional group-modified MAC might be a promising and stable material for selective and efficient adsorption. However, the report about the green and facile synthesis of functional group-modified biomass-derived MAC for In and Ga separation has not been found. Inspired by mussels, a polydopamine (PDA) layer can be easily formed by the self-polymerization of dopamine (DA), which occurs under ambient conditions with the assistance of oxygen from the air. The PDA layer exhibits excellent adhesion on diverse substrates, controllable thickness, and abundant hydroxyl groups for further modification. Moreover, its application can be diversified through co-deposition with specific chemicals during the polymerization of DA.21,25 Therefore, utilizing PDA for MAC modification may easily achieve the tight bonding between functional groups and MAC for the selective adsorption of In and Ga. However, this concept has not been found thus far.
To impart selectivity to MAC, this study proposed a green and facile one-pot modification method at ambient temperature and pressure without using any toxic chemicals. This method employed co-deposition and the Atherton–Todd (A–T) reaction, aligning with the principles of green chemistry. Using ethyl phenylphosphinate (EPP) as the precursor and coconut shell-derived MAC as the carrier, the phosphoryl-functionalized PDA-coated MAC (EPP–PDA@MAC) was successfully synthesized. To the best of the authors’ knowledge, this is a pioneering application of such a sustainable strategy for the modification of MAC to recover critical metals. Leveraging the large surface area and phosphoryl groups of MAC, this adsorbent achieved outstanding adsorption capacity and selectivity for In and Ga. Its application in waste CIGS modules showcased near-complete recovery of In and Ga, with purities reaching 97.79% and 90.40%, respectively. Moreover, the EPP–PDA@MAC exhibited remarkable stability due to the adhesive PDA layer and robust covalent bonding between the PDA and the precursor. A comparison with commercial adsorbents highlights its potential for large-scale production. This work introduces an innovative and eco-friendly approach for the recovery of technology critical metals from waste PV modules, aiming to advance sustainable development.
2. Experiments
2.1 Materials
Six kinds of waste coconut shell-derived MAC (MAC-1 to MAC-6) were all purchased and detailed information about the manufacturers and pore structures is provided in ESI 1.† And N2 adsorption–desorption isotherms at 77 K (Micromeritics ASAP 2460, USA) were used to measure the MAC structure. Diethyl phosphite, diisopropyl phosphite, phenylphosphinic acid, and 1-methyl-2-pyrrolidone were purchased from Shanghai Acmec Biochemical Co. Ethyl phenylphosphinate was purchased from Tianjin Heowns Opde Technologies. Dimethylphosphine oxide was purchased from Shanghai D&B Biological Science and Technology Co. Dopamine hydrochloride, tris(hydroxymethyl) aminomethane hydrochloride and the other reagents were purchased from Sinopharm Chemical Reagent Co. Deionized water was used for washing solid materials and solution preparation.2.2 P-PDA@MAC and P-PDA synthesis and characterization
A mixture of deionized water (20 mL), dopamine hydrochloride (DA) (0.03–0.50 mmol), tris(hydroxymethyl) aminomethane hydrochloride (Tris) (0–0.5 mmol), and an organic phosphine-containing precursor (0–5 mmol) were sequentially introduced into a glass tube and thoroughly stirred. MAC (0.2 g) was added to the mixture after the solution pH was adjusted to 7–12. This solid–liquid mixture was shaken for 5–48 h at 293–313 K in a constant temperature air shaker. Following filtration, the solid material was washed and dried at 353 K for 6 h to yield the phosphoryl-functionalized PDA coated MAC (P-PDA@MAC). Following similar steps without adding solid MAC, the solution was directly dried after shaking to obtain a white or brown solid product (P-PDA). For example, a white solid product (EPP–PDA) was produced by using the EPP precursor. The above materials were analysed by field emission Scanning Electron Microscopy (SEM, Hitachi S-4800, Japan), Energy Dispersive Spectroscopy (EDS) coupled with SEM, 120 kV Transmission Electron Microscope (TEM, Hitachi H-7650, Japan), X-ray Photoelectron Spectroscopy (XPS, ESCALAB 250XI, USA), and Fourier Transform-Infrared spectroscopy (FTIR, Nicolet iS10, USA). Nuclear magnetic resonance spectra (NMR, Bruker Avance NEO 400 MHz, Germany) of EPP and EPP–PDA were recorded for mechanism analysis.2.3 Adsorption and desorption ability determination
To evaluate the adsorption capacity, separate solutions of indium nitrate (In(NO3)3) and gallium nitrate (Ga(NO3)3) were prepared and adjusted to specific pH by using hydrochloric acid (HNO3) and ammonia (NH3·H2O), respectively. Then, P-PDA@MAC was added to adsorb metal ions by shaking at 298 K in a constant temperature air shaker. The ion concentrations were tested by using inductively coupled plasma-optical emission spectrometry (ICP-OES, Horiba ULTIMA 2, France).The adsorption efficiency (R, %) was calculated using eqn (1):
 | (1) |
The adsorption capacity (qt, mg g−1) was calculated using eqn (2):
 | (2) |
The adsorption selectivity of adsorbents (MAC, PDA@MAC, and EPP–PDA@MAC) for In3+ and Ga3+ from mixed solutions was tested. The concentrations of all metal ions (In3+, Ga3+, Cu2+, Cd2+, Al3+, and Zn2+) were the same and the pH was in the range of 0.5 to 4. The separation factor (SFBA) representing the ability to separate A from B was calculated using eqn (3):
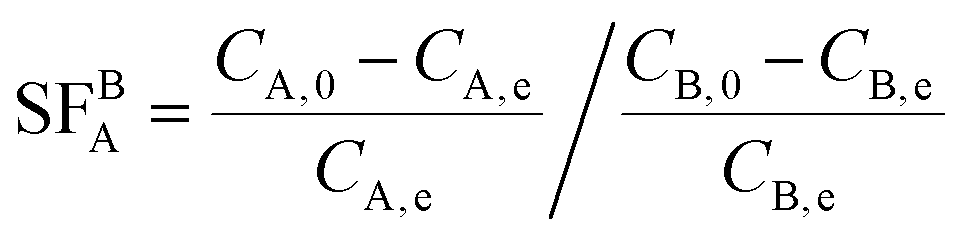 | (3) |
3. Results and discussion
3.1 P-PDA@MAC synthesis parameter optimization
The pore structure of mesoporous activated carbon (MAC) carriers significantly impacts the performance of P-PDA@MAC. Therefore, the influence of the MAC type (Fig. 1a) and its particle size (Fig. 1b) on the adsorption efficiencies for Ga and In were investigated under identical conditions. First, six types of MAC with different pore structures were ground to the same particle size, and then the corresponding P-PDA@MAC were prepared. Fig. 1a shows that the adsorption efficiency of P-PDA@MAC rapidly increased with the specific surface area of MAC. Notably, P-PDA@MAC-5 and P-PDA@MAC-6 exhibited superior adsorption efficiencies, corresponding to the larger specific surface areas of MAC-5 and MAC-6 (>1200 m2 g−1). Thus, MAC-6 was recommended as the best solid carrier. Furthermore, the particle size was investigated by grinding MAC-6 from 20–60 mesh to 220–260 mesh. As the grinding degree increased, the specific surface area of MAC gradually decreased, leading to a significant reduction in the adsorption efficiency of P-PDA@MAC (Fig. 1b, ESI 4.1†). This decline was likely attributed to the disruption of the MAC structure during the mechanical grinding process.26 The above analyses suggested that the MAC's huge surface area and appropriate pore structure offer ample space for modification and binding sites for metal ions, thereby enhancing the density of functional groups and the adsorption ability. Consequently, in the subsequent experiments, the solid-phase carrier (MAC-6, 20–60 mesh) was employed for efficient adsorption and facile adsorbent separation from solution. | ||
| Fig. 1 Effect of the (a) MAC type and pore structure, and (b) particle size on the adsorption efficiency of P-PDA@MAC. Effect of the (c) organophosphorus precursor type, (d) precursor dosage, and (e) NaOH dosage on the adsorption capacity of P-PDA@MAC. | ||
Based on the three types of organophosphorus compounds commonly used in the A–T reaction, five specific precursors were selected: diethyl phosphite (DEP) and diisopropyl phosphite (DIP), which are O,O-dialkyl phosphonates (Fig. S1, i†); phenylphosphinic acid (PPA) and ethyl phenylphosphinate (EPP), which are hypophosphonates (Fig. S1, ii†); and dimethylphosphine oxide (DMO), which is dialkyl phosphine oxides (Fig. S1, iii†). During the modification process, each precursor with the same molar mass was separately added to synthesize five types of P-PDA@MAC. The adsorption selectivity of these P-PDA@MAC was tested by isolating In3+ and Ga3+ from solutions containing In3+, Ga3+, Cu2+, Cd2+, Al3+, and Zn2+ at pH = 0.5–4 (Fig. 1c). As expected, all five types of P-PDA@MAC exhibited higher adsorption efficiencies for In3+ and Ga3+ than for other metal ions, demonstrating their adsorption selectivity. In particular, EPP–PDA@MAC, prepared using EPP as a precursor, exhibited the highest adsorption efficiencies for In3+ and Ga3+ while maintaining quite low efficiencies for other metal ions, which might be attributed to the distinct structural features of the groups attached to the phosphorus atom.27 Therefore, EPP was selected as a suitable precursor for downstream investigation of material synthesis for removing In3+ and Ga3+ with high adsorption capacity and selectivity.
The manipulation of both the DA and EPP precursor dosages had a pronounced impact on the polymerization of PDA and the phosphoryl modification process, thereby influencing the adsorption capacity of EPP–PDA@MAC. Experiments were conducted by varying the DA dosage (nDA) from 0.03 to 0.5 mmol while maintaining a Tris dosage equivalent to that of DA (nTris = nDA). As shown in Fig. 1d, the adsorption efficiency initially rose with the increasing DA dosage, but then it sharply decreased, reaching the maximum efficiency at nDA = 0.07 mmol. Moreover, the surface morphology changes (Fig. S2†) indicated that a thin and uniform PDA layer successfully formed at nDA = 0.07 mmol, effectively utilizing the large surface area of MAC and providing abundant modification spaces and binding sites for metal ions. Additionally, an investigation of the EPP precursor dosage impact demonstrated an initial increase followed by stabilization. This phenomenon was attributed to the limited bonding capacity of the PDA when the DA dosage remained constant, consequently limiting the available EPP amount for modification. Beyond the threshold dosage of EPP (1 mmol), the phosphoryl modification reached a plateau. As a result, the optimal dosages of DA and EPP (nDA = 0.07 mmol, nEEP = 1 mmol) were selected for subsequent experiments to ensure superior adsorption efficiency and minimize precursor waste.
In addition to the above parameters, the effects of the solution pH, temperature, precursor adsorption time, and polymerization time on EPP–PDA@MAC preparation were also investigated (Fig. 1e, ESI 4.3–4.4†). The solution pH during the adsorbent preparation greatly affected the degree of dopamine polymerization and the A–T reaction, consequently impacting the phosphoryl modification of EPP–PDA@MAC. Fig. 1e shows that excessive oxidation of PDA can be prevented by adjusting the solution pH to 7.3, facilitating the synergistic co-deposition and A–T reactions, resulting in the optimal adsorption ability of EPP–PDA@MAC. Moreover, temperature (298 K, 308 K, and 318 K) had a negligible impact on the adsorption ability, supporting the feasibility of conducting the preparation process at 298 K to conserve energy (Fig. S3a†). Similarly, the adsorption efficiency was not affected by the precursor adsorption time, whereas it increased followed by stabilization as the polymerization time increased, as shown in Fig. S3(b and c).† This phenomenon might be due to the lengthy air-oxidation self-polymerization reaction of dopamine.28 This finding simplified the modification process by eliminating the precursor adsorption time. Despite requiring 18 hours of polymerization, the process could be carried out at room temperature without additional conditions, highlighting the simplicity of the modification steps, facile reaction conditions, and industrial viability. Additionally, the versatility of phosphoryl modification methods has been underscored by the self-polymerization of dopamine on various carrier surfaces.
According to the SEM and EDS analyses (Fig. 2), MAC exhibited a loose and porous surface morphology. Compared with MAC, PDA@MAC displayed some uniformly distributed hemispherical particles on the surface, attributed to the production of polydopamine. Compared with PDA@MAC, the EPP@PDA-MAC demonstrated a size increase of distributed particles on the surface, suggesting that the EPP were doped into the polymer layer via a co-deposition reaction, resulting in excess DA in the form of PDA particles. More importantly, the significant increase and uniform distribution of phosphorous elements on the surface of EPP–PDA@MAC confirmed successful phosphoryl modification. TEM characterization results (Fig. 2) indicate that the thickness of the coating layer is approximately 40 nm, which is consistent with previous research findings.29 BET characterization results revealed that EPP–PDA@MAC exhibited a high specific surface area and a rich mesoporous structure, providing abundant reactive sites for interactions between grafted functional groups and metal ions (ESI 4.5†). Table S3† shows a slight decrease in both specific surface area and micropore volume compared to the original MAC, indicating that the coating layer had partially obstructed some micropores. However, the sacrifice of certain pore structures was acceptable, as the grafted functional groups were the primary source of the selective adsorption ability of EPP–PDA@MAC.
 | ||
| Fig. 2 SEM and TEM micrographs, EDS spectra, elemental mapping images of MAC, PDA@MAC, and EPP–PDA@MAC. | ||
3.2 EPP–PDA@MAC adsorption and desorption performance
Several key parameters during the adsorption and desorption processes of EPP–PDA@MAC were investigated, including the anion species, adsorbent dosage, adsorption time, adsorption selectivity, desorption phase concentration, regeneration ability, and leaching performance. Fig. S5† shows that the adsorption efficiencies of EPP–PDA@MAC for both In3+ and Ga3+ were the highest in a solution containing NO3− compared with SO42− and Cl−. Therefore, NO3− was employed for all subsequent experiments. Under the same initial In3+ and Ga3+ concentrations (0.07–0.08 mmol L−1), the influence of the adsorbent dosage (0.05–0.65 g L−1) for efficiencies and capacities was investigated. As shown in Fig. 3a, the adsorption efficiency increased substantially when the dosage was increased from 0.05 to 0.33 g L−1. However, the growth rate of adsorption efficiency slowed down when the dosage increased to a range of 0.33 to 0.65 g L−1. Meanwhile, with increasing dosage (0.05–0.65 g L−1), the adsorption capacity exhibited a consistent decrease. Consequently, the optimal balance between adsorption efficiency and capacity was achieved specifically at a dosage of 0.33 g L−1. | ||
| Fig. 3 Effect of (a) dose and (b) reaction time on the adsorption efficiency. (c) Kinetic and (d) isotherm fitting models for adsorption. (e) Adsorption efficiency for different ions in mixed solutions at pH 0.5–4. (f) Effect of nitric acid concentration on the desorption efficiency of In3+ and Ga3+. (g) Number of cycles of EPP–PDA@MAC in the static system. (h) Effect of the liquid phase composition on the adsorption ability after leaching treatment. | ||
The influence of the adsorption time on the adsorption efficiency of EPP–PDA@MAC was explored. Fig. 3b shows that adsorption efficiency for In3+ and Ga3+ rapidly increased with the adsorption time, followed by a slower increase and reaching equilibrium at 14 h and 8 h, respectively. Based on these data, the adsorption kinetics of EPP–PDA@MAC for In3+ and Ga3+ were analyzed by using six kinetic models, as shown in Fig. 3c and ESI 5.2.† The results underscored the effectiveness of the Elovich model in characterizing the In3+ and Ga3+adsorption on EPP–PDA@MAC, indicating that a chemisorption process occurred on the monomolecular layer of the nonuniform solid surface. Furthermore, the adsorption experiments at different temperatures (298 K, 308 K, and 318 K) and initial concentrations of In3+ (6–180 mg L−1) and Ga3+ (7–90 mg L−1) were conducted to analyze the adsorption isotherms by using Langmuir and Freundlich models (Fig. 3d, ESI 5.3†). Fig. 3d shows that the adsorption capacity (qe) for In3+ and Ga3+ both increased with temperature and equilibrium concentration (Ce). The high alignment of the Langmuir model (R2 > 0.993) indicated that the adsorption reaction mainly occurred in the homogeneous monolayer on the adsorbent surface (Table S5†), consistent with kinetic analysis. Additionally, the lower separation factors (RL < 1) indicated favorable adsorption processes. The modeling constant (KL) for In3+ exceeded that for Ga3+, indicating a higher adsorption energy for In3+ than for Ga3+. An elevated temperature can effectively drive the adsorption reaction, indicating a heat-absorbing process. The fitted adsorption capacities (Qm) of EPP–PDA@MAC for In3+ and Ga3+ reached 125.1 mg g−1 and 140.7 mg g−1, respectively, at 298 K.
The adsorption selectivity of an adsorbent is a critical factor to consider. To this end, a mixed solution was prepared based on the types and concentrations of primary metallic elements in waste CIGS modules, and the adsorption efficiencies of involved metal ions by MAC, PDA@MAC, and EPP–PDA@MAC were investigated under different pH conditions. Fig. 3e shows that MAC and PDA@MAC could not separately recover In3+ and Ga3+ due to the absence of specific functional groups. Instead, EPP–PDA@MAC showed significantly enhanced adsorption selectivity for both In3+ (SFXIn = 382.4, pH = 0.5) and Ga3+ (SFXGa = 239.0, pH = 2), owing to the successful phosphoryl modification. Moreover, as the pH increased, In3+ was first adsorbed followed by Ga3+, consistent with the adsorption energy order shown in the isotherm analysis results and the stability order of metal ion–ligand complexes.30 Besides, In3+ (3d10 and 4d10) has a competitive advantage over Ga3+ (3d10), as the full 4d10 orbitals are more prone to forming stable complexes with the same ligands than the 3d10 orbit, based on the ligand field theory.31
In the desorption process, nitric acid (HNO3) was utilized to maintain the anionic species in solution, and the impact of HNO3 concentration (CHNO3) was investigated. Initially, EPP–PDA@MAC-In and EPP–PDA@MAC-Ga were separately prepared through filtration, washing, and drying after In3+ and Ga3+adsorption. Subsequently, each material was desorbed by the HNO3 solution (0.1 to 8 mol L−1), respectively. After a reaction time of 3 h, the desorption efficiency was determined based on the ion concentrations in solution. Fig. 3f shows that the desorption efficiencies of the above materials increased with increasing CHNO3. This trend aligned with the cation exchange reaction, wherein H+ from the HNO3 disrupted the complexes between phosphoryl groups and ions, facilitating the desorption of ions.12 At the CHNO3 values of 5 mol L−1 and 4 mol L−1, the highest desorption efficiencies of In3+ (96.3%) and Ga3+ (97.5%) were achieved, respectively. However, avoiding excessively high concentrations of acid in practical industrial applications is imperative due to safety and environmental concerns. The data indicate that the desorption efficiencies of In3+ and Ga3+ could still reach 91.0% and 92.2% using 2 mol L−1 and 0.5 mol L−1 HNO3, respectively. Therefore, to ensure both environmental protection and efficient desorption, HNO3 concentrations of 2 mol L−1 and 0.5 mol L−1 were chosen for In3+ and Ga3+ desorption in the static system. The regeneration ability of the adsorbent plays an essential role in practical applications (Fig. 3g). The regeneration potential of EPP–PDA@MAC in a static system was investigated. First, EPP–PDA@MAC was introduced into In(NO3)3 solution (pH = 0.5) and Ga(NO3)3 solution (pH = 2). After adsorption, the mixture was subjected to filtration, followed by separate desorption using 2 mol L−1 and 0.5 mol L−1 HNO3. After nine adsorption–desorption cycles, EPP–PDA@MAC maintained high adsorption efficiencies of 85% for In3+ and 90% for Ga3+. In contrast, the adsorption efficiency of P507@MAC, synthesized by the impregnation method in our previous study, decreased to 50% for Ga3+ after the same number of cycles (ESI 9†). This significant difference demonstrates the stable connection between the functional groups and the carrier in EPP–PDA@MAC, highlighting its notable reusability.
The leaching performance of the adsorbent is crucial for determining stability. To evaluate this aspect, three liquid phase systems were employed for the leaching experiment of EPP–PDA@MAC: deionized water, HNO3 solution (pH = 0.5, simulating the highest acidity during adsorption), and 2 mol L−1 HNO3 solution (simulating the highest acidity during desorption). EPP–PDA@MAC was added to each liquid phase, subjected to ultrasonication for 6 h, followed by a 14-day soaking period. The adsorbents were then recovered to conduct adsorption experiments under identical conditions, with the fresh adsorbent used as a control. Fig. 3h shows that the adsorption efficiencies of leached adsorbents closely resembled those of the fresh adsorbent. The maximum decreases in the adsorption efficiency for In3+ and Ga3+ were only 4.7% and 4.3%, respectively, in the 2 mol L−1 HNO3 solution. These findings confirmed the strong connection between phosphoryl functional groups and MAC, highlighting the superior stabilization ability of EPP–PDA@MAC and laying a solid foundation for industrial applications.
The preparation methods, adsorption ability and desorption conditions of EPP–PDA@MAC and other adsorbents in existing studies are summarized in Table S6 (ESI 6†). Comparative analysis reveals that EPP–PDA@MAC stands out due to its straightforward one-step preparation process, which requires only mixing and shaking under mild conditions (room temperature and pressure) and avoids the use of toxic organic reagents, making it one of the most environmentally friendly options. Furthermore, EPP–PDA@MAC exhibits superior adsorption capacity, selectivity and reusability for both In3+ and Ga3+ compared to other adsorbents. During the desorption process, efficient desorption is also achieved by EPP–PDA@MAC at relatively low acid concentrations and small acid quantities, coupled with rapid desorption rates. Notably, this novel adsorbent adheres to the principles of green chemistry: less hazardous chemical syntheses (avoid using or generating substances toxic to humans and/or the environment), designing safer chemicals (products should achieve desired function while being as non-toxic as possible), safer solvents and auxiliaries, and design for energy efficiency (minimized energy requirements and conditions of ambient temperature and pressure). Additionally, EPP–PDA@MAC enables the simultaneous separation and recovery of critical metals, underscoring its comprehensive advantage in both adsorption performance and environmental sustainability over other adsorbents.12–18,32–36
3.3 Comparison between EPP–PDA@MAC and commercial adsorbent P507@Resin
Extractant P507 impregnated resin (P507@Resin) is one of the widely used commercial adsorbents, particularly for the recovery of rare metals such as In3+ and Ga3+. P507@Resin is typically synthesized using a one-step method. In this process, the extractant P507 is mixed with an organic solvent, such as dichloromethane, followed by the addition of resin. After the organic solvent is removed via rotary evaporation, P507 is impregnated into the resin, resulting in the formation of P507@Resin. Detailed information is provided in ESI 7.2.†To comprehensively compare the performance of EPP–PDA@MAC and commercial adsorbent P507@Resin, the adsorption ability, green metrics and production costs of these two adsorbents were studied. The adsorption ability of P507@Resin in the multi-metal ion system was examined under identical conditions. Fig. S7† demonstrates that while P507@Resin can selectively recover In3+ and Ga3+, its adsorption efficiency and separation factor are inferior to those of EPP–PDA@MAC, confirming the superior adsorption ability of EPP–PDA@MAC (ESI 7.1†).
Green metrics play a crucial role in evaluating the environmental impact of an adsorbent.37,38 Due to the absence of standardized data on the molecular weights of activated carbon and resin, green metrics calculations were primarily based on the input–output mass throughout the entire production process. Green metrics include reaction mass efficiency (RME), process mass intensity (PMI), mass productivity (MP), environmental impact factor (E), solvent intensity (SI), and water intensity (WI). Detailed calculations are shown in ESI 7.2.† RME, which accounts for all reactant mass, yield, and atom economy, serves as a key metric for assessing the greenness of an adsorbent. The RME calculation yielded notable results: the significantly higher RME value of EPP–PDA@MAC (61.43) compared to P507@Resin (0.55) proves that EPP–PDA@MAC is “greener” than P507@Resin. Similarly, the PMI and E evaluation indicated that EPP–PDA@MAC (84.96, 83.96) has less waste generation and more positive environmental impact than P507@Resin (138.18, 137.18). Additionally, the MP value for EPP–PDA@MAC (1.18) is higher than that for P507@Resin (0.72), indicating smaller mass waste in preparing EPP–PDA@MAC. Furthermore, the excess organic solvent used in the synthesis of P507@Resin leads to a substantial difference in SI and WI compared to the water usage in synthesizing EPP–PDA@MAC. The combined SI and WI for P507@Resin exceed for EPP–PDA@MAC, highlighting the smaller environmental impact of EPP–PDA@MAC. Overall, green metrics calculation results indicated that EPP–PDA@MAC demonstrates superior environmental friendliness compared to the commercial adsorbent P507@Resin.
The production cost of adsorbents is critical for assessing their industrial application potential. In this case, the large-scale (100 kg) production costs of EPP–PDA@MAC and P507@Resin were estimated, with the detailed information provided in ESI 7.3.† Due to the smaller usage and lower price of chemical reagents, the estimated production cost of EPP–PDA@MAC (146 RMB per kg) is significantly lower than that of P507@Resin (1598 RMB per kg). According to Jining Tangyi Chemical Co., the market price of commercial P507@Resin is 1575 RMB per kg. Considering producer profits and other associated costs, it supports the validity of the above calculated production cost. Thereby, EPP–PDA@MAC has a significant cost advantage on the industrial scale. In conclusion, based on adsorption ability, green metrics and production costs (Table 1 and Tables S7, 8†), this novel adsorbent EPP–PDA@MAC exhibits considerable advantages over the commercial adsorbent P507@Resin, underscoring its significant potential for industrial application.
| Adsorbent | Green metrics | Adsorption properties | ||||||
|---|---|---|---|---|---|---|---|---|
| RME/% | PMI/g g−1 | MP/% | E/g g−1 | SI/g g−1 | WI/g g−1 | Efficiency (R)/% | Selectivity (SF) | |
| EPP–PDA@MAC | 61.43 | 84.96 | 1.18 | 83.96 | 0 | 83.33 | R In = 73%; RGa = 86% | SFIn = 382; SFGa = 239 |
| P507@Resin | 0.55 | 138.18 | 0.72 | 137.18 | 136.36 | 45.45 | R In = 50%; RGa = 72% | SFIn = 118; SFGa = 52 |
3.4 Adsorption mechanism analysis
Adsorption isotherms and kinetics analyses were conducted for In3+ and Ga3+ to elucidate the adsorption behavior of EPP–PDA@MAC, as described in Fig. 3c and d. In addition, other characterization studies, including SEM, EDS, FTIR spectroscopy, XPS, and NMR spectroscopy, on MAC, PDA@MAC, EPP–PDA@MAC, and EPP–PDA were also performed to verify the adsorption mechanism.Firstly, the functional group conversion of MAC, PDA@MAC, EPP, EPP–PDA@MAC, EPP–PDA@MAC-Ga, and EPP–PDA@MAC-In was analyzed through FT-IR characterization (Fig. 4a). The results show that all the materials exhibited –OH and N–H stretching vibration peaks (3440 cm−1). However, the peak range in the MAC spectrum (3310–3600 cm−1) was narrower compared to that of other materials (3000–3600 cm−1), owing to the plentiful hydroxyl and amino functional groups in PDA.39 In addition, the wavenumber of the C–O–C peak in the PDA@MAC spectrum (1143 cm−1) was significantly lower than that in the MAC spectrum (1163 cm−1), which was attributed to the PDA layer.40 These changes, combined with the SEM and EDS analyses (Fig. 2), successfully confirmed the PDA coating on the MAC surface. Furthermore, EPP has been verified to exhibit characteristic peaks related to P–H groups (2351 cm−1), –P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups (1227 cm−1), and P–O–C groups (1040 cm−1).41 Because the C–O–C (1143 cm−1) peak was close to the –PO (1227 cm−1) peak, a broad peak was observed at 1183 cm−1 in the EPP–PDA@MAC spectra. Furthermore, the P–O–C peak at 1040 cm−1 was also present, indicating the successful functionalization of the –PO groups in EPP–PDA@MAC. Notably, the P–H peak at 2351 cm−1 completely disappeared in the EPP–PDA@MAC spectrum, providing evidence that the primary modification mechanism was the A–T reaction. Upon comparing the spectra of EPP–PDA@MAC and EPP–PDA@MAC-In/Ga, the most noticeable change was the significant attenuation of the –PO peak intensity, resulting in a decrease in the wavenumber of the fusion peak at 1159 cm−1 in the EPP–PDA@MAC-In spectrum. This peak even re-splits into two peaks in the EPP–PDA@MAC-Ga spectrum (1227 cm−1, 1143 cm−1), suggesting that the main adsorption mechanism is the complexation of –PO groups with metal ions. In addition, a peak related to the overlapping CC resonance vibrations within the benzene ring at 1585 cm−1 was observed. Interestingly, this peak intensity significantly increased in the EPP–PDA@MAC-In/Ga spectrum, suggesting the formation of an –PO → Me bond, which subsequently influenced the electron cloud surrounding the benzene ring.42
O groups (1227 cm−1), and P–O–C groups (1040 cm−1).41 Because the C–O–C (1143 cm−1) peak was close to the –PO (1227 cm−1) peak, a broad peak was observed at 1183 cm−1 in the EPP–PDA@MAC spectra. Furthermore, the P–O–C peak at 1040 cm−1 was also present, indicating the successful functionalization of the –PO groups in EPP–PDA@MAC. Notably, the P–H peak at 2351 cm−1 completely disappeared in the EPP–PDA@MAC spectrum, providing evidence that the primary modification mechanism was the A–T reaction. Upon comparing the spectra of EPP–PDA@MAC and EPP–PDA@MAC-In/Ga, the most noticeable change was the significant attenuation of the –PO peak intensity, resulting in a decrease in the wavenumber of the fusion peak at 1159 cm−1 in the EPP–PDA@MAC-In spectrum. This peak even re-splits into two peaks in the EPP–PDA@MAC-Ga spectrum (1227 cm−1, 1143 cm−1), suggesting that the main adsorption mechanism is the complexation of –PO groups with metal ions. In addition, a peak related to the overlapping CC resonance vibrations within the benzene ring at 1585 cm−1 was observed. Interestingly, this peak intensity significantly increased in the EPP–PDA@MAC-In/Ga spectrum, suggesting the formation of an –PO → Me bond, which subsequently influenced the electron cloud surrounding the benzene ring.42
 | ||
| Fig. 4 (a) FT-IR spectra, (b and c) XPS spectra, (d) O 1s, and (e) P 2p spectra of the MAC, PDA@MAC, EPP, EPP–PDA@MAC, and EPP–PDA@MAC-In/Ga. (f) 1H-NMR and 31P-NMR spectra of EPP and EPP–PDA. | ||
XPS characterization of MAC, PDA@MAC, EPP–PDA@MAC, EPP–PDA@MAC-Ga, and EPP–PDA@MAC-In was performed. The full spectrum analysis shown in Fig. 4(b and c) revealed the emergence of a new N 1s peak in the PDA@MAC spectrum compared to the MAC spectrum, providing evidence for the successful encapsulation of polydopamine. In addition, the appearance of P 2s and P 2p peaks in both the EPP–PDA@MAC spectrum and the EPP–PDA@MAC-In/Ga spectrum compared to the PDA@MAC spectrum confirmed the successful organophosphorus functionalization. Notably, in the EPP–PDA@MAC-In/Ga spectrum, distinct peaks corresponding to In 3d3/2, In 3d5/2, In 3d2, Ga 2p1/2, and Ga 2p3/2 were observed, and their gaps were all consistent with the reference values, substantiating the successful adsorption of metal ions.43,44 To further validate the proposed adsorption mechanism, the XPS high-resolution O 1s and P 2p spectra of the materials were analyzed, as shown in Fig. 4(d and e). In the O 1s spectrum, the C–OH peak intensity in the PDA@MAC spectrum significantly increased compared to the MAC spectrum, owing to the phenolic hydroxyl groups in polydopamine. Additionally, P–O–C and PO peaks were both observed in the EPP–PDA@MAC spectrum, confirming successful phosphoryl functionalization. After the adsorption experiment, the PO peak intensity in EPP–PDA@MAC-In/Ga both significantly decreased. Similarly, compared with that in the EPP–PDA@MAC spectrum, the PO peak density in the P 2p spectrum substantially decreased in the EPP–PDA@MAC-In/Ga spectrum. These observations provided compelling evidence that the adsorption reaction is primarily governed by the complexation of PO with metal ions.
To further verify the functionalization mechanism from MAC to EPP–PDA@MAC, 1H and 31P NMR analyses of EPP and EPP–PDA were performed (Fig. 4f). In the 31P NMR spectrum, a single P 31 signal appeared at 23.9 ppm in the spectrum of EPP. Conversely, multiple new P 31 signals were observed in the 31P NMR spectrum of EPP–PDA, indicating various chemical environments for the P atoms. Furthermore, in the 1H NMR spectrum of EPP–PDA@MAC, the H 1 signal at 8.52 ppm belonging to the P–H of EPP disappeared and several strong H 1 signals appeared due to the hydrogen atom of PDA. These results verify the successful A–T reaction between EPP and PDA during the functional process, corresponding to the FT-IR and XPS analyses described above. By integrating the areas of the aromatic protons (j–n) from EPP and –NH2 (a), aromatic protons (b, c), and –CH2– (e) from PDA in the 1H spectrum of EPP–PDA, the approximate composition of EPP–PDA was calculated (ESI 8†). The results suggested that the organic composition was the same as the monomer ratio in the feed. The weak peak attributed to phenolic hydroxyl (8.1 ppm) in the 1H NMR spectrum of EPP–PDA indicated that the A–T reaction during the modification process may have been incomplete. This implied the insufficient formation of covalent bonds between PDA and EPP. Consequently, a residual PDA layer containing hydroxyl groups may persist on the surface of EPP–PDA@MAC.
In summary, the modification mechanism of EPP–PDA@MAC involved a co-deposition reaction between the PDA and the organophosphorus precursor, as well as the A–T reaction between the hydroxyl group in PDA and the phosphoryl groups in the organophosphorus precursor. The primary adsorption mechanism of EPP–PDA@MAC was the ligand complexation reaction between the lone pair electrons on the oxygen atoms from the –PO group and the metal cations, consistent with extensive research studies on neutral organic phosphorus extractants.45 This reaction led to the formation of ligand bonds, –PO → Me, facilitating effective and selective adsorption of In and Ga by EPP–PDA@MAC. This also elucidates why EPP–PDA@MAC, derived from EPP as a precursor, exhibited the highest selectivity compared to other P-PDA@MAC. The conjugated benzyl and alkoxyl groups linked to the phosphorus atom exhibited lower electronegativity, enhancing the electron-repelling effect. As a result, the electron cloud density around the oxygen atom in PO increased, thereby improving its coordination ability with metal ions.46 Additionally, during the adsorption process, the occurrence of a chelation reaction was observed between ions and unreacted phenolic hydroxyl groups within the PDA layer. However, this reaction made a minor contribution to the adsorption mechanism, given the excellent adsorption selectivity of EPP–PDA@MAC and the weak selectivity of –OH groups. Lastly, it is imperative to consider the role of the MAC carrier in the adsorption process. Adsorption kinetics and isotherm analysis revealed that the MAC carrier served as a crucial site for adsorption reactions. Furthermore, the huge surface area and suitable pore structure of MAC could facilitate and promote the transfer and adsorption of In3+ and Ga3+, according to the pore filling theory, and prove the significant impact of the MAC structure (type and particle size) on the adsorption efficiency (Fig. 1a and b).27 The adsorption and desorption mechanism of In3+ and Ga3+ by EPP–PDA@MAC is illustrated in Fig. 5.
 | ||
| Fig. 5 Adsorption and desorption mechanism for the selective recovery of In3+ and Ga3+ by EPP–PDA@MAC from waste CIGS PV modules. | ||
3.5 Application of the EPP–PDA@MAC adsorbent
The feasibility of separating In3+ and Ga3+ from Mg2+, Al3+, Cd2+, Zn2+, and Cu2+ in waste CIGS PV modules (L2-120, Hanergy Photovoltaic Technology Co.) was evaluated. As shown in Fig. 6a, the CIGS components were crushed to remove the back plate and aluminum frame. Then, the components containing the CIGS alloy layer were subjected to leaching with nitric acid to extract metal ions. This process generated the CIGS leach solution containing valuable critical metals In3+ and Ga3+ alongside other low-value metal ions. After filtration and dilution, the solution pH was adjusted to 0.5 to facilitate the adsorption of In3+. Concentrations of In3+, Ga3+, Mg2+, Al3+, Cd2+, Zn2+, and Cu2+ in this solution were 11.79, 5.01, 0.11, 0.33, 2.31, 45.4, and 7.39 mg L−1, respectively. After the selective In3+ adsorption of EPP–PDA@MAC, the solution pH was adjusted to 2 for the further adsorption of Ga3+. Each adsorption process for In3+ and Ga3+ was repeated twice to ensure sufficient adsorption. After the adsorption process, the adsorbents (EPP–PDA@MAC-In and EPP–PDA@MAC-Ga) were subjected to washing and drying via filtration, followed by desorption using 0.5 mol L−1 and 2 mol L−1 nitric acid, respectively. The recovered In(NO3)3 and Ga(NO3)3 solutions were finally obtained. Ultimately, 99.9% of the In and Ga in the leach solution were effectively adsorbed. Furthermore, the mass ratio of In3+ in the recovered In(NO3)3 solution reached 97.79%, while the mass ratio of Ga3+ in the recovered Ga(NO3)3 solution reached 90.40% (Fig. 6b). These findings underscore the efficacy of EPP–PDA@MAC in achieving facile separation and recovery of In and Ga from real waste CIGS photovoltaic modules. | ||
| Fig. 6 (a) The application of EPP–PDA@MAC for the recovery of In3+ and Ga3+ from waste CIGS photovoltaic modules. (b) The composition of the CIGS leach solution and recovered In(NO3)3 and Ga(NO3)3 solutions. (c) Effect of the reaction time on adsorption and desorption, and (d) the number of cycles of EPP–PDA@MAC in the CDI system. | ||
Capacitive deionization (CDI) is a highly regarded technology that effectively removes ionic species through electric double-layer adsorption. Renowned for its low energy consumption and high efficiency, CDI has found extensive application in the fields of desalination, heavy metal elimination, and precious metal recovery.47 During the CDI process, a low current (voltage) is applied to the electrode to facilitate ion adsorption, followed by electrode regeneration through the reversal of the applied potential and subsequent desorption of ions. Compared to the static adsorption and desorption system, the CDI system significantly enhances the efficiency of ion adsorption and desorption, mainly due to the applied electric field. Additionally, the desorption process in CDI systems is environmentally friendly because it eliminates the need for chemical reagents. Carbon-based materials, such as AC, carbon nanotubes, carbon felt, and graphene, are commonly employed in CDI electrodes. With the advancement and industrial adoption of this technology, evaluating the performance of developed AC-based adsorbents in CDI systems has become crucial for assessing their scalability and suitability. In this study, an EPP–PDA@MAC electrode was synthesized and used in a CDI system (CD5050, Changsha Supu Lin Co.), as detailed in ESI 2.†Fig. 6c shows that an exceptional Ga3+ adsorption efficiency of 99% was achieved within 2 h, coupled with a commendable desorption efficiency of 96%. Impressively, the adsorption efficiency of EPP–PDA@MAC remained notably high at 98.05% even after 50 cycles of adsorption and desorption (Fig. 6d). This robust cycling stability further validates the suitability of this material for integration into CDI systems. Given the existence of established industrial-scale equipment utilizing CDI technology, EPP–PDA@MAC holds significant promise for widespread industrial adoption.
4. Conclusions
This study developed a novel adsorbent via a green and facile one-pot modification method, phosphoryl-functionalized polydopamine-coated mesoporous activated carbon (EPP–PDA@MAC), for the effective recovery of critical metals from waste CIGS PV modules. The successful synthesis of this material underscores the effectiveness of co-deposition and the Atherton–Todd reaction as facile and green modification methods, aligning with the principles of green chemistry. The outstanding adsorption capacity and selectivity of EPP–PDA@MAC for both In3+ and Ga3+ result from the huge surface area provided by the MAC carrier and the functional phosphoryl groups derived from the organophosphorus precursor. Notably, EPP–PDA@MAC exhibits excellent recyclability, as evidenced by its negligible loss of adsorption efficiency after leaching treatment and multiple cycles of adsorption–desorption in both static and capacitive deionization systems. Compared to commercial adsorbents, EPP–PDA@MAC offers significant advantages in terms of adsorption ability, green metrics, and production costs. The adsorption mechanism mainly involves the ligand complexation reaction between In3+/Ga3+ and the –PO groups present in EPP–PDA@MAC. Overall, the proposed adsorbent and modification method in this study presents a prospective and eco-friendly method for recovering critical metals from waste PV modules, thereby mitigating the overexploitation of natural indium and gallium resources and bolstering the sustainable development of clean energy.
Author contributions
Wenxuan Wang: conceptualization, data curation, investigation, methodology, validation, visualization, writing – original draft, and writing – review and editing. Xinhai Xu: conceptualization, methodology, resources, supervision, and writing – review and editing. Jie Li: methodology, investigation, and writing – review and editing. Tao Liu: data curation and investigation. Hailong Wang: data curation and investigation. Yin Wang: funding acquisition, project administration, resources, supervision, and writing – review and editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the Fujian Province Science and Technology Major Project (No. 2023YZ01110001), the STS Plan Supporting Project of the Chinese Academy of Sciences in Fujian Province (Grant No., 2023T3032 and 2020T3036), the Social Development Leading Key Projects of Fujian Province (Grant No., 2022Y0080), and the Natural Science Foundation of Xiamen, China (Grant No. 3502Z20227086).References
- R. Deng, P. R. Dias, M. M. Lunardi and J. Ji, Green Chem., 2021, 23, 10157–10167 RSC.
- D. Hu, B. Ma, X. Li, Y. Lv, Y. Chen and C. Wang, J. Cleaner Prod., 2022, 350, 131426 CrossRef CAS.
- D. Hu, B. Ma, X. Li, Y. Lv, W. Zhang, Y. Chen and C. Wang, Sep. Purif. Technol., 2022, 282, 120087 CrossRef CAS.
- F. Liu, T. m. Cheng, Y. Chen, K. Yueh, S. Tang, K. Wang, C. Wu, H. Tsai, Y. Yu, C. Lai, W. Chen and Y. Chueh, Sol. Energy Mater. Sol. Cells, 2022, 241, 111691 CrossRef CAS.
- A. Brewer, J. Florek and F. Kleitz, Green Chem., 2022, 24, 2752–2765 RSC.
- Y. S. Zimmermann, C. Niewersch, M. Lenz, Z. Z. Kul, P. F. Corvini, A. Schaffer and T. Wintgens, Environ. Sci. Technol., 2014, 48, 13412–13418 CrossRef CAS PubMed.
- B. A. Andersson, Prog. Photovolt.: Res. Appl., 2000, 8, 61–76 CrossRef CAS.
- Y. Lv, P. Xing, B. Ma, B. Liu, C. Wang, Y. Zhang and W. Zhang, ACS Sustainable Chem. Eng., 2019, 7, 19816–19823 CrossRef CAS.
- J. M. Edgecomb, E. E. Tereshatov, G. Zante, M. Boltoeva and C. M. Folden Iii, Green Chem., 2020, 22, 7047–7058 RSC.
- Q. Song, L. Zhang and Z. Xu, J. Hazard. Mater., 2020, 381, 120973 CrossRef CAS.
- N. R. Nicomel, L. Otero-Gonzalez, L. Arashiro, M. Garfí, I. Ferrer, P. Van Der Voort, K. Verbeken, T. Hennebel and G. Du Laing, Green Chem., 2020, 22, 1985–1995 RSC.
- W. Wang, X. Xu, D. Lai, Q. Xu, J. Li and Y. Wang, Sep. Purif. Technol., 2024, 330, 125510 CrossRef CAS.
- J.-H. Chen, Y.-H. Chang, K.-C. Hsu and J.-C. Lin, Chem. Eng. Technol., 2021, 44, 2257–2268 CrossRef CAS.
- J. J. Meng, C. L. He, J. Zhou, T. Fujita, S. Y. Ning and Y. Z. Wei, J. Appl. Polym. Sci., 2020, 138, e49732 CrossRef.
- X. Gao, Z. Cao, C. Li, J. Liu, X. Liu and L. Guo, New J. Chem., 2022, 46, 18952–18960 RSC.
- M. Li, X. Meng, K. Huang, J. Feng and S. Jiang, Hydrometallurgy, 2019, 186, 73–82 CrossRef CAS.
- J. Du, M. Zhang, Z. Dong, X. Yang and L. Zhao, Sep. Purif. Technol., 2022, 286, 120442 CrossRef CAS.
- M. Li, S. Tang, R. Liu, X. Meng, J. Feng, L. Zhou and Y. Chen, Chem. Eng. Res. Des., 2021, 168, 135–145 CrossRef CAS.
- N. Chai, L. Gao, S. Li, Y. Cao, Z. Ma, L. Li and M. Hu, J. Cleaner Prod., 2023, 387, 135861 CrossRef CAS.
- K. Lan, H. S.-H. Wang, T. Lee, C. A. de Assis, R. A. Venditti, Y. Zhu and Y. Yao, Green Chem., 2024, 26, 3466–3478 RSC.
- Y. Yuan, Y. Zhang, Z. Wang and H. Yan, Green Chem., 2024, 26, 6787–6798 RSC.
- P. M. Nowak, Green Chem., 2023, 25, 4625–4640 RSC.
- W. Jia, S. Li, J. Wang, J. T. E. Lee, C. S. K. Lin, O. Mašek, H. Zhang and X. Yuan, Green Chem., 2024, 26, 1790–1805 RSC.
- P. Sáez, I. Bernabé, J. M. Gómez, E. Díez, C. Fraile, N. García and A. Rodríguez, Separations, 2023, 10, 349 CrossRef.
- T. Zhang, J. Hu, L. Guo, Z. Gu, X. Jiang and Y. Li, Green Chem., 2024, 26, 3125–3138 RSC.
- S. M. Mak, B. T. Tey, K. Y. Cheah, W. L. Siew and K. K. Tan, J. Chem. Technol. Biotechnol., 2009, 84, 1405–1411 CrossRef CAS.
- Q. Li, W. Xu, X. Liang, B. Liu, Q. Wu, Z. Zeng, L. Li and X. Ma, Fuel, 2022, 325, 124871 CrossRef CAS.
- R. Zhou, P.-F. Ren, H.-C. Yang and Z.-K. Xu, J. Membr. Sci., 2014, 466, 18–25 CrossRef CAS.
- H. Feng, X. Zhu, R. Chen, Q. Liao, J. Liu and L. Li, Chem. Eng. J., 2016, 306, 1017–1025 CrossRef CAS.
- R. Miura, M. Tokumaru, T. Oshima and Y. Baba, Solvent Extr. Res. Dev., Jpn., 2017, 24, 123–130 CrossRef CAS.
- D. H. Nies, Metallomics, 2016, 8, 481–507 CrossRef CAS PubMed.
- C. He, Y. Yang, M. Qi, Y. Jiang, Y. Wei, T. Fujita, G. Wang, S. Ma and W. Yang, J. Environ. Chem. Eng., 2023, 11, 109372 CrossRef CAS.
- W. Li, C. Zhou, C. Li, W. Zhu, J. Shi and G. Liu, Sep. Purif. Technol., 2023, 323, 124464 CrossRef CAS.
- P. Raj, M. Patel and A. K. Karamalidis, Sep. Purif. Technol., 2023, 323, 124396 CrossRef CAS.
- M. Zhang, Q. Sun, Y. Wang, W. Shan, Z. Lou and Y. Xiong, Chem. Eng. J., 2021, 421, 129748 CrossRef CAS.
- M. M. Bhuyan, O. B. Adala, H. Okabe, Y. Hidaka and K. Hara, J. Environ. Chem. Eng., 2019, 7, 102844 CrossRef.
- F. Roschangar, R. A. Sheldon and C. H. Senanayake, Green Chem., 2015, 17, 752–768 RSC.
- X. Zhang, G. Dhawan, A. Muthengi, S. Liu, W. Wang, M. Legris and W. Zhang, Green Chem., 2017, 19, 3851–3855 RSC.
- A. Sun, Y. Zhan, W. Yang, Q. Feng, H. Dong, X. Chen and Y. Chen, J. Environ. Chem. Eng., 2022, 10, 108118 CrossRef CAS.
- Y. Duan, G. Dong, R. Wu, X. Zhao, M. Li, F. Zhang, Z. Song and H. Hao, Polym. Eng. Sci., 2022, 62, 3390–3399 CrossRef CAS.
- X. Li, H. Liang, Y. Mo and Y. Wei, Processes, 2022, 10, 1761 CrossRef CAS.
- K. Yu, R. Warsaba, H. Yazdani-Ahmadabadi, D. Lange, E. Jan and J. N. Kizhakkedathu, ACS Biomater. Sci. Eng., 2023, 9, 329–339 CrossRef CAS PubMed.
- Y. Zhang, X. Liu, Y. Wang, Z. Lou, W. Shan and Y. Xiong, J. Colloid Interface Sci., 2019, 556, 102–110 CrossRef CAS PubMed.
- X. Li, B. Ma, D. Hu, Q. Zhao, Y. Chen and C. Wang, J. Cleaner Prod., 2022, 339, 130658 CrossRef CAS.
- J. Wang, W. Xu, H. Liu, F. Yu and H. Wang, Miner. Eng., 2021, 163, 106798 CrossRef CAS.
- T. H. Siddall, Ind. Eng. Chem., 1959, 51, 41–44 CrossRef CAS.
- K. Sun, M. Tebyetekerwa, C. Wang, X. Wang, X. Zhang and X. S. Zhao, Adv. Funct. Mater., 2023, 33, 2213578 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc04204b |
| This journal is © The Royal Society of Chemistry 2025 |