DOI:
10.1039/D4GC05102E
(Paper)
Green Chem., 2025, Advance Article
Continuous flow synthesis of cyclobutenes via lithium ynolates†
Received
11th October 2024
, Accepted 30th January 2025
First published on 31st January 2025
Abstract
Batch reactions that involve the generation of highly reactive species require a cryogenic temperature, complicated manipulations by chemists, and higher amounts of reagents, resulting in energy wastage and high costs. In this study, we developed a continuous flow synthesis of functionalised cyclobutenes, where the first step was the flash generation of short-lived lithium ynolates. Lithium ynolates were generated by the reaction of α,α,α-tribromomethyl ketones and n-butyllithium at 30 °C in 2.5 s and transferred to the next reactor before decomposition. The optimal reaction time (2.5 s) and temperature (30 °C) were determined via in-line Raman spectroscopy. The one-flow process involved three steps: the generation of lithium ynolates, the [2 + 2] cycloaddition reaction with α,β-unsaturated esters, and acetylation of the resulting unstable lithium enolates. These reactions were mediated by several reactive chemical species such as lithium ynolates, ketenes, and lithium enolates. Our green, flash flow approach to generating ynolate anions does not require cryogenic conditions and is highly reproducible and scalable, making it suitable for practical applications.
Green foundation
1. We present a greener protocol to generate ynolate anions in flow. This protocol eliminates the need for cryogenic conditions and is highly reproducible and scalable. We also present a greener synthetic method of highly functionalized cyclobutenes. This three-step, one-flow process affords cyclobutenes without complicated manipulations by chemists, thus saving manpower.
2. In the optimised reaction conditions, we reduced the consumption of organolithium reagents by half. Morever, the mild reaction temperature of 30 °C and an extremely short residence time of 23.1 s, which are applied by using the flow reactor system, contributed to energy savings.
3. In the future, this flow synthesis will be applied for the process synthesis of pharmaceutically important cyclobutenes.
|
Introduction
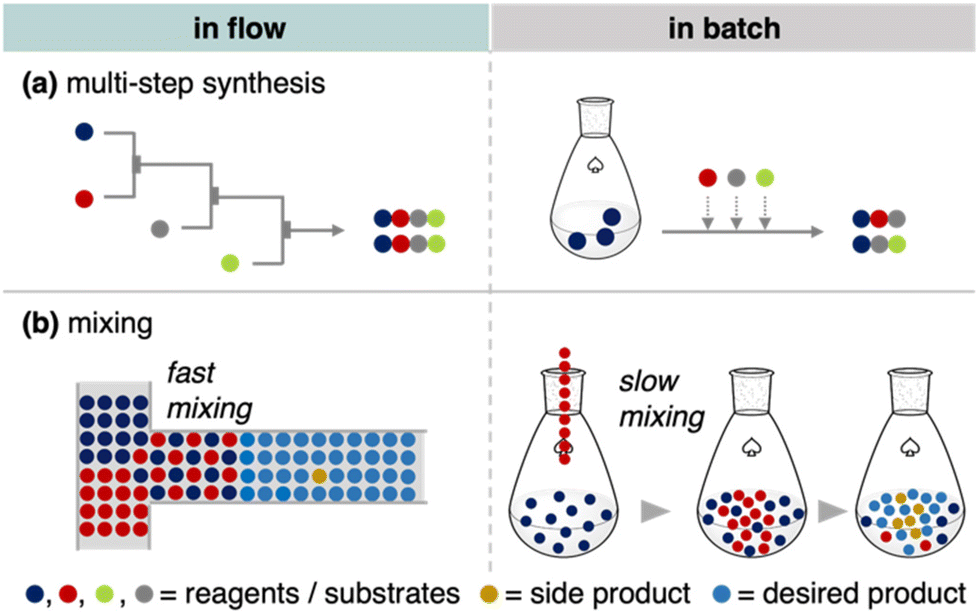
Flow reactions play a central role in modern synthetic chemistry and engineering.1,2 Flow microreactors, in particular, provide precise control over the reactivity of short-lived intermediates. In addition, continuous flow systems facilitate the integration of several reactions involving highly reactive intermediates without isolation within a few minutes.3–6 This could be attributed to the advantageous mechanical features of flow microreactors. First, the continuous stream for a precise reaction time facilitates optimal mass transfer (Fig. 1a). Flow reactions can allow a reaction time of milliseconds, which is unachievable for batch systems. Second, their small dimensions permit precise temperature control and fast mixing, facilitating the desired reactions without side reactions (Fig. 1b). Consequently, continuous flow systems have empowered fast reactions via highly reactive chemical species such as organolithium, as demonstrated in the field of flash chemistry.7 In certain cases, flow reactions eliminate the necessity of cryogenic conditions, which are typically required in batch reactions. Furthermore, continuous flow synthesis ensures increased safety, high scalability, and reproducibility, broadening the synthetic applications of short-lived species.8,9
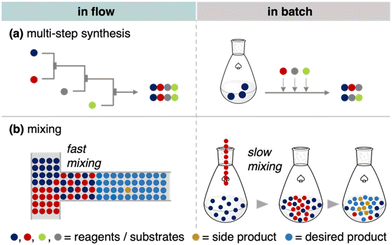 |
| | Fig. 1 Mechanical features in continuous flow and batch systems: (a) mass transfer in multi-step synthesis, (b) fast mixing in a flow system and slow mixing in a batch system. | |
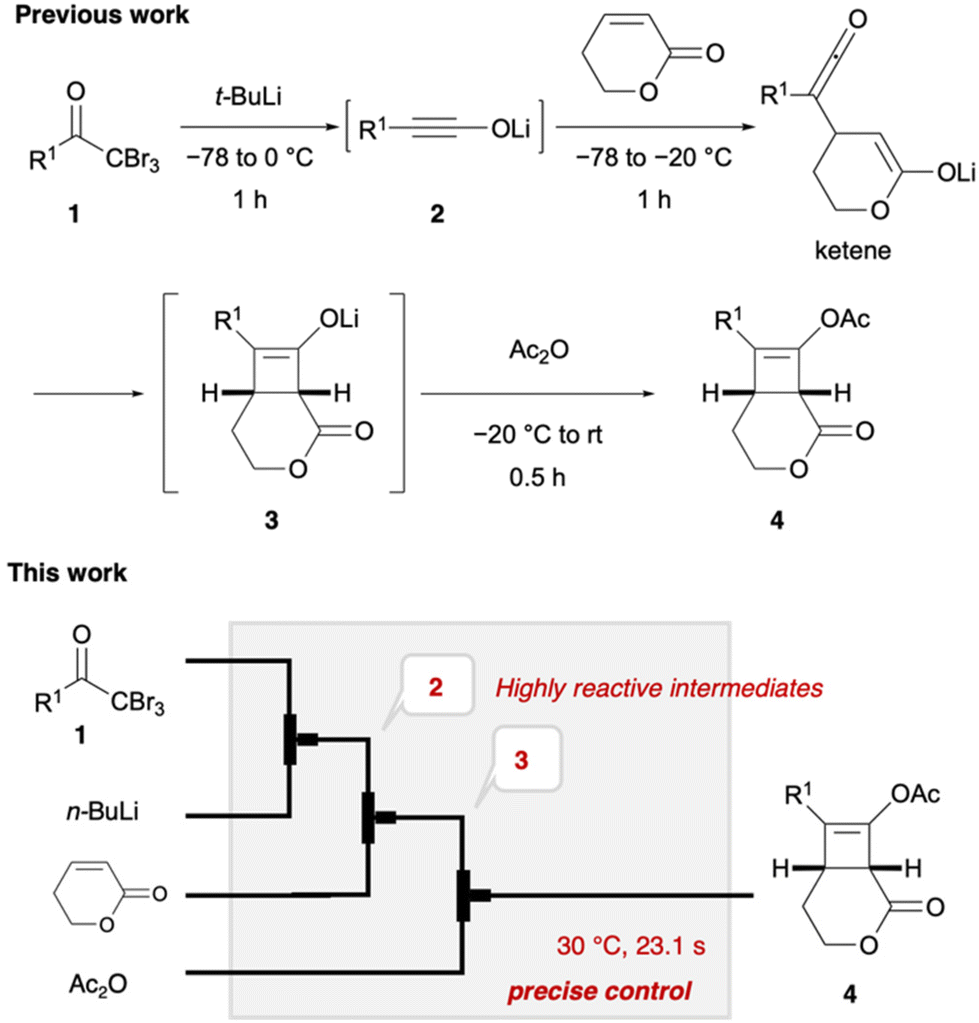
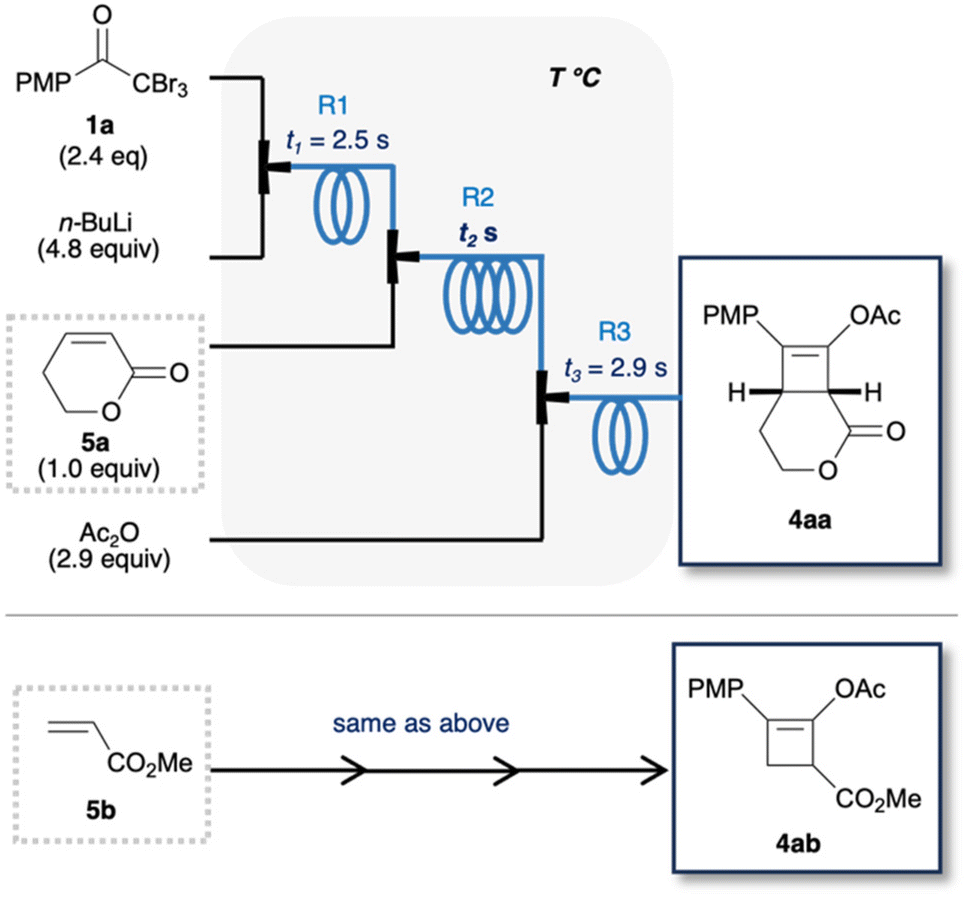
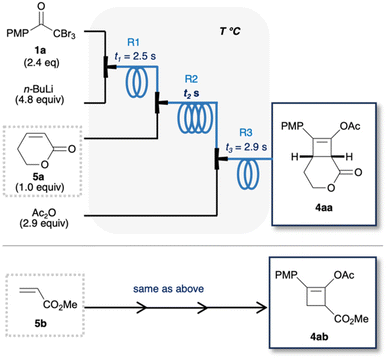
Lithium ynolates are highly reactive species.10 Except for a few isolable ynolates, most of them have not been detected and quantified.11,12 Consequently, controlling their reactivity and employing them for organic synthesis remain a significant challenge.13–15 We previously developed a one-pot synthesis of cyclobutenes 4 via the generation of lithium ynolates. The synthesis involved three reactions: (1) the generation of lithium ynolates 2 from α,α,α-tribromomethyl ketones 1 and tert-butyl lithium (t-BuLi); (2) the [2 + 2] cycloaddition of lithium ynolates 2 with α,β-unsaturated esters via reactive ketene intermediates; (3) the acetylation of the resulting lithium enolates 3 (Fig. 2).16 As alkoxide polymerises the α,β-unsaturated esters, the first step of the reaction was conducted under alkoxide-free conditions. This reaction features wide substrate scope of ynolates. However, this procedure has several drawbacks: cryogenic conditions, complicated operation, and temperature changes from −78 to 0 °C during every step. Additionally, precise temperature regulation is essential, as any deviation could lead to the rapid decomposition of two unstable intermediates, ynolates and cyclobutenes. This is a common problem in the generation and handling of lithium ynolates.17,18 Furthermore, excess consumption of t-BuLi at cryogenic temperatures hampers its application in the scalable and sustainable synthesis of cyclobutenes. Considering the advantages of flow chemistry and our previous success in flow synthesis, we envisioned that a continuous flow system would solve these drawbacks and improve the consumption of excess reagents and energy.19 Shindo's group previously utilised a microflow reactor to generate lithium ynolates from α,α-dibromo esters with precise control of the reaction temperature.20–22 However, the flash generation of lithium ynolates from α,α,α-tribromomethyl ketones has never been reported. Therefore, in this study, we developed an original flow protocol for the flash generation of lithium ynolates in a flow system and integrated it with the flow synthesis of cyclobutenes. Cyclobutenes are an important structural motif in small-molecule drug candidates. However, their green synthetic methods are limited.23,24 Our microflow synthesis is green in terms of safety, scalability, and productivity.
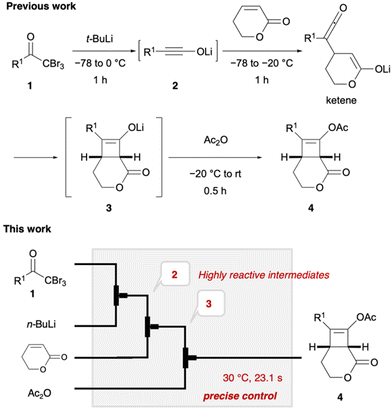 |
| | Fig. 2 Continuous synthesis of cyclobutenes in batch (previous work) and flow (this work) systems. | |
Results and discussion
Optimisation of reagents and solvents in batch for flow synthesis
We sought a less pyrophoric lithium reagent than t-BuLi for the lithium–halogen (Li–X) exchange of 1a, as t-BuLi is highly pyrophoric and may produce bubbles due to the formation of volatile isobutene.9,25 We tested other organolithium reagents for the synthesis of 4aa in batch (Table 1). Entry 1 lists the previously optimised conditions.16 The use of Et2O produces LiBr precipitate, which blocks the flow stream. Next, THF was used to prevent precipitation, which afforded 4aa in 30% yield (entry 2). The use of t-BuLi or s-BuLi in THF decreased the yield of 4aa due to side reactions of the organolithium reagents with THF (entries 2 and 3). In contrast, n-BuLi improved the yield of 4aa to 46% (entry 4). Although this yield was lower than the previously reported yield of 70%, we considered it sufficient at this stage prior to further flow optimisation.
Table 1 Optimisation of organolithium reagents and solvents in batch for flow synthesis
|
| Entry |
Solvent |
BuLi (equiv.) |
Yield of 4aa (%) |
| Reaction temperature for acetylation: −20 °C to rt. |
| 1a |
Et2O |
t-BuLi (8) |
70 |
| 2 |
THF |
t-BuLi (8) |
30 |
| 3 |
THF |
s-BuLi (4) |
13 |
| 4 |
THF |
n-BuLi (4) |
46 |
Monitoring the formation of lithium ynolates
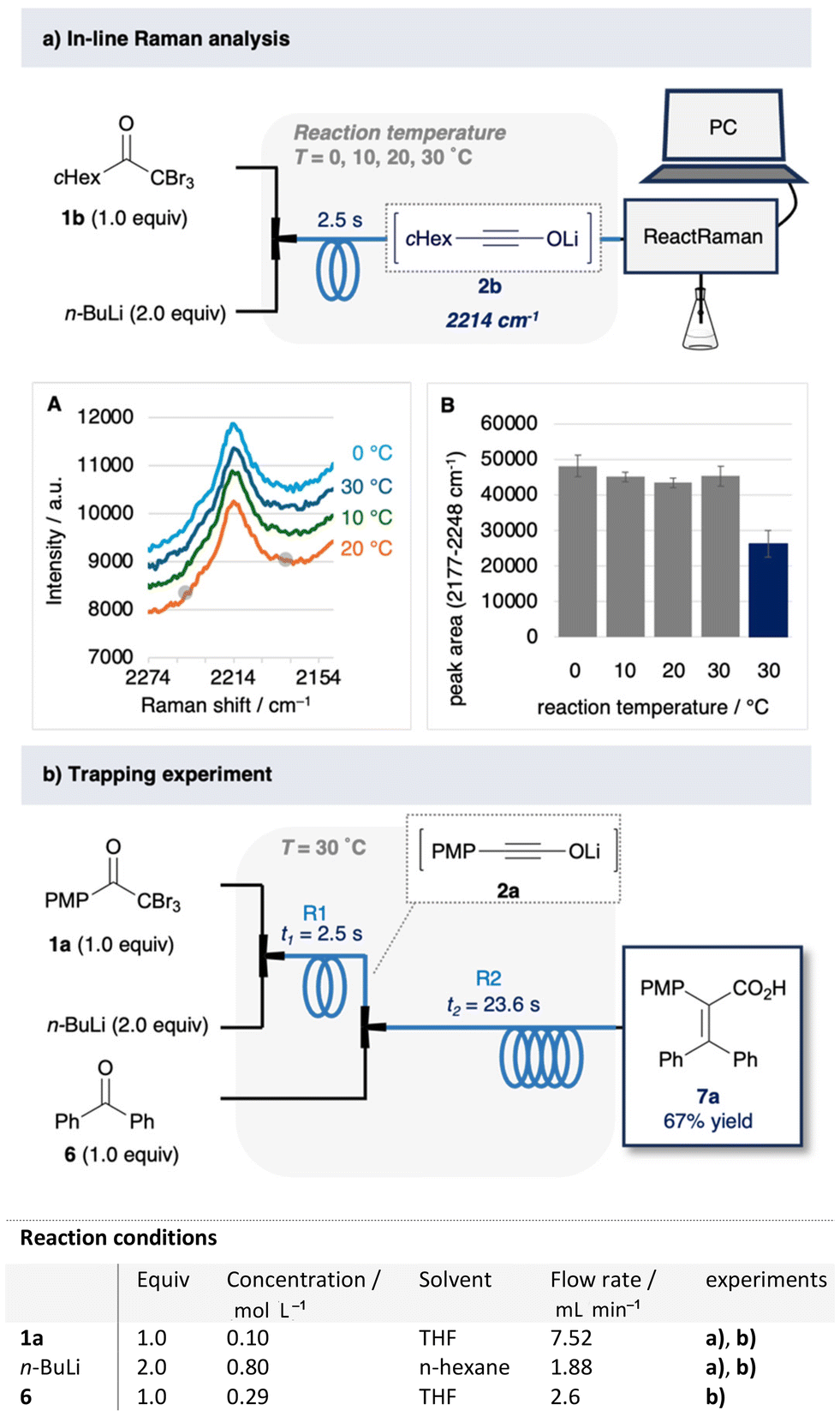
The formation of lithium ynolates was monitored in real time in a flow system (Fig. 3a).26–29 The characteristic band of the lithium ynolates at ∼2100 cm−1 can be monitored using the ReactRaman instrument.11 Hence, we integrated the ReactRaman instrument into the flow system (Fig. 3a). Lithium ynolates are formed via rapid reactions starting from double Li–X exchange.16 Therefore, to achieve rapid mixing, we employed a total flow rate of 9.4 ml min−1 and a mixer with an inner diameter of 250 μm. The reaction mixture was monitored at 30 °C after a residence time of 2.5 s. However, the monitoring experiment using 1a was hampered by the saturation of the Raman spectra. Therefore, we used α,α,α-tribromomethyl cyclohexyl ketone (1b) as the substrate. Compound 1b exhibits a characteristic Raman peak at 2214 cm−1 only when mixed with n-BuLi (Fig. S4 and Table S4†). This Raman band was identified as the alkynyl moiety of lithium ynolate 2b, which is consistent with previously reported IR stretching frequencies [ESI 4.2†]. The Raman peak intensity remained unchanged in the temperature range of 0–30 °C (Fig. 3a (A, B)). This indicates that a consistent amount of ynolate 2b was produced in this temperature range. In contrast, the prolonged residence time of 22.5 s at 30 °C reduced the Raman peak intensity (Fig. 3a and b, blue bar). Overall, these results indicate that a significant amount of ynolate was produced at room temperature at a mixing time of 2.5 s, and longer mixing times than 2.5 s may lead to ynolate decomposition. Furthermore, as a quantitative reference, we trapped the ynolate 2a with a known electrophile (Fig. 3b). Ynolate 2a reacts with benzophenone (6) to afford a stable alkene 7a.20,30 We conducted this reaction in the flow system. The generated ynolate 2a was trapped by 6 to afford alkene 7a in 67% yield. That is, 2a was obtained in 67% yield, at least in the first step.
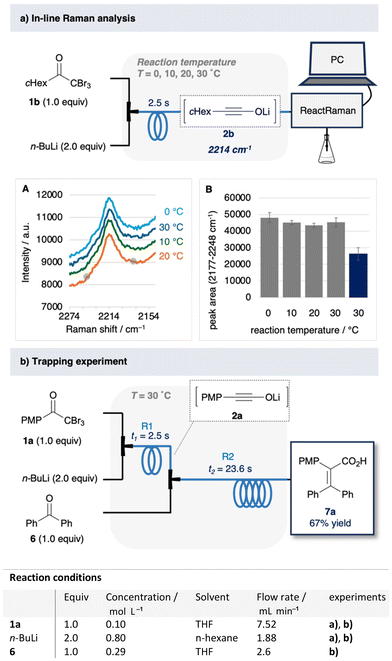 |
| | Fig. 3 (a) In-line Raman analysis of 2b. A: Observed Raman spectra, residence time = 2.5 s, B: peak area (2177–2248 cm−1), residence time = 2.5 s (grey bar), 22.5 s (blue bar). (b) Trapping experiment. | |
Optimisation of the [2 + 2] cycloaddition of ynolate 2a in flow synthesis
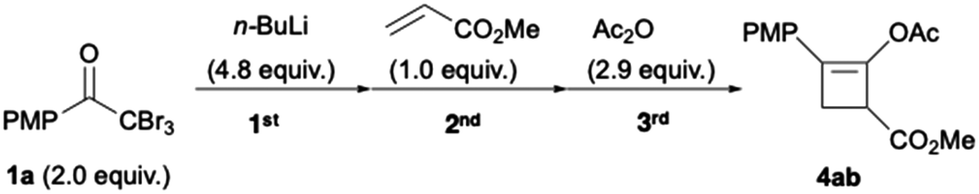
Next, we examined the continuous synthesis of cyclobutenes 4 (Table 2) in the flow system. This synthesis involves the formation of lithium ynolate 2a from 1a, the subsequent [2 + 2] cycloaddition with 5a or 5b, and acetylation. To optimise the conditions for the second step, the residence time (t1) in the first reactor loop (R1) was fixed at 2.5 s. In the second reactor loop (R2), the residence time (t2) range was set from 5.9 to 23.6 s, based on prior in-line Raman analysis that had demonstrated significant decomposition of 2b at 22.5 s (vide supra). Thus, the various t2s in R2 and the various reaction temperatures (T) in all of the reactor loops were screened in the continuous process. Screening with 5a demonstrated that a residence time between 17.7 and 23.6 s in R2 at 20–30 °C afforded the best yield of 4aa (74–75%) (Table 2a). Screening with 5b showed that a residence time of 17.7 s in R2 at 30 °C resulted in the highest yield of 4ab (63%) (Table 2b). Besides, the lower the reaction temperature, the lower the yield of 4aa or 4ab.31 Hence, the [2 + 2] cycloaddition proceeds smoothly at approximately room temperature within the time scale. Moreover, this result demonstrates the potential of flash chemistry in preventing side reactions. Although the equivalents of ynolates and residence time (t3) in the third reaction were re-examined, the initially set parameters afforded the best results (Tables S2 and S3†). Finally, a t2 of 17.7 s in R2 and 30 °C were selected as the optimised reaction conditions for generality.
Table 2 Screening of temperature and residence time (t2) to synthesise cyclobutenes 4aa and 4ab. NMR yields of (a) 4aa and (b) 4ab
|
| Reaction conditions: 1a (2.4 equiv., c1 = 0.10 mol L−1, v1 = 7.52 mL min−1, THF); n-BuLi (4.8 equiv., c2 = 0.80 mol L−1, v2 = 1.88 mL min−1, n-hexane); 5 (1.0 equiv., c3 = 0.12 mol L−1, v3 = 2.6 mL min−1, THF); Ac2O (2.9 equiv., c4 = 0.23 mol L−1, v4 = 4.0 mL min−1, THF). t1 = 2.5 s, t3 = 2.9 s. |
| (a) |
| Yield of 4aa (%) |
t2 (s) |
| 5.9 |
11.8 |
17.7 |
23.6 |
| T ( °C) |
30 |
72 |
71 |
74 (61) |
74 |
| 20 |
67 |
70 |
74 |
75 |
| 10 |
64 |
67 |
68 |
N.D. |
| (b) |
| Yield of 4ab (%) |
t2 (s) |
| 5.9 |
11.8 |
17.7 |
23.6 |
| Yields of 4 from the batch reaction are shown in the parentheses. |
| T ( °C) |
30 |
43 |
56 |
63 (70) |
62 |
| 20 |
28 |
46 |
53 |
54 |
| 10 |
22 |
30 |
41 |
47 |
Substrate scope
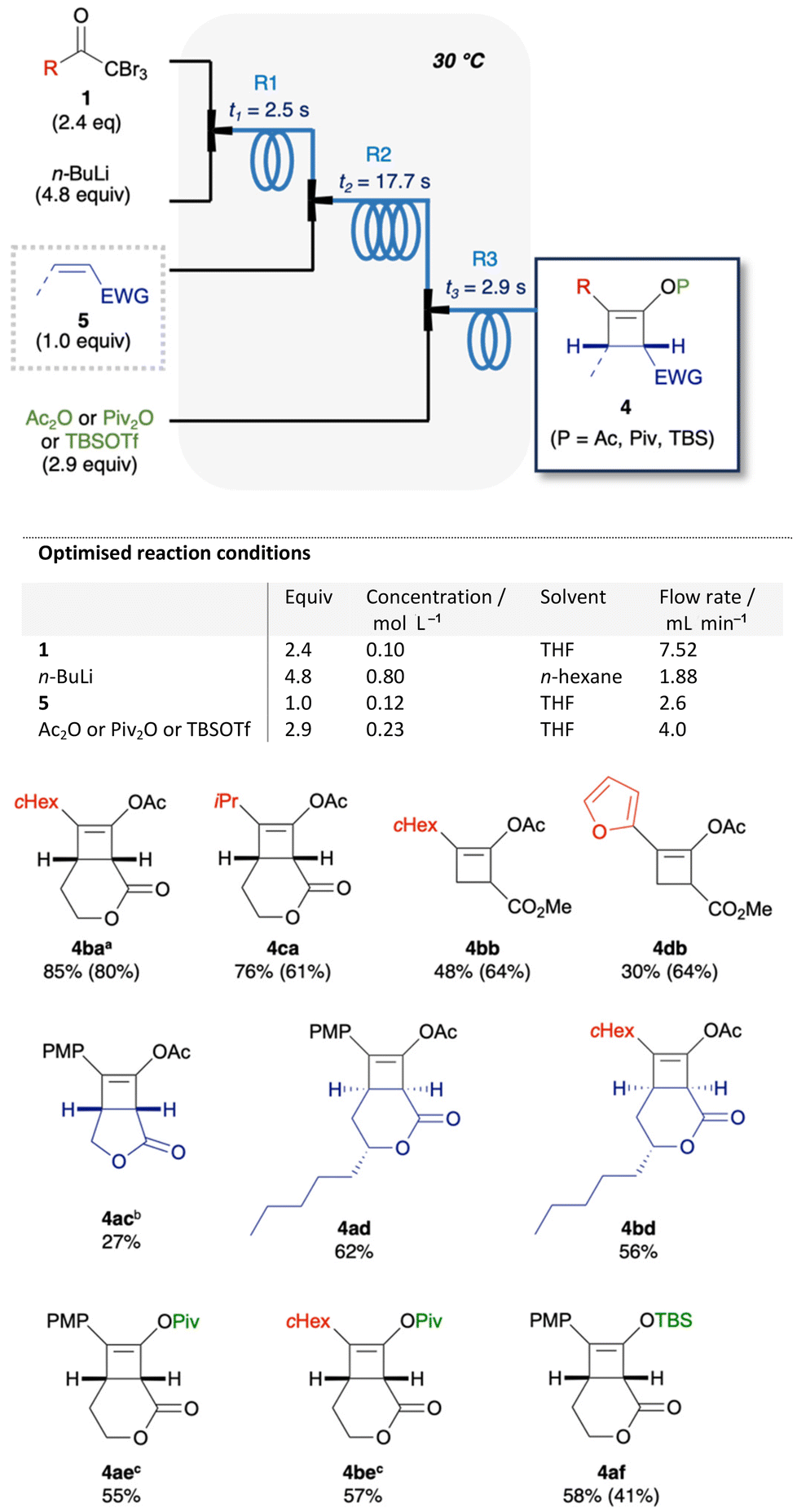
The optimised reaction conditions were applied to the flow synthesis of 10 cyclobutenes (Fig. 4). Cyclohexyl (1b) and isobutyl (1c) ketones afforded 4ba and 4ca in 85% and 76% yields, respectively. Compared to the batch process, the flow process afforded higher yields of 4 with improved reproducibility. On the other hand, the use of 5b afforded 4bb and 4db in lower yields (30–48%) than 64% yield in the batch process. It is considered that sterically less hindered 5b involved undesired side reactions in this flow system. Similarly, the use of 2,5-dihydrofuranone afforded a low yield of 4ac (27%). A plausible side reaction is considered to be the over-reaction of ketene or enolate intermediates with ynolates. The other plausible side reaction is considered to be 1,2-adition32 of ynolates with 5, because some side products from this reaction were observed in unsuccessful experiments (ESI, S10†). [2 + 2] cycloaddition with massoia lactone proceeded diastereoselectively to afford 4ad and 4bd in 62% and 56% yields, respectively. Relative stereochemistry of 4ad was confirmed by X-ray crystallography. Trapping with pivalic anhydride or t-butyldimethylsilyl trifluoromethanesulfonate (TBSOTf) afforded pivalate ester 4ae and 4be, or silyl enol ether 4af, respectively, in moderate yields (55–58%).
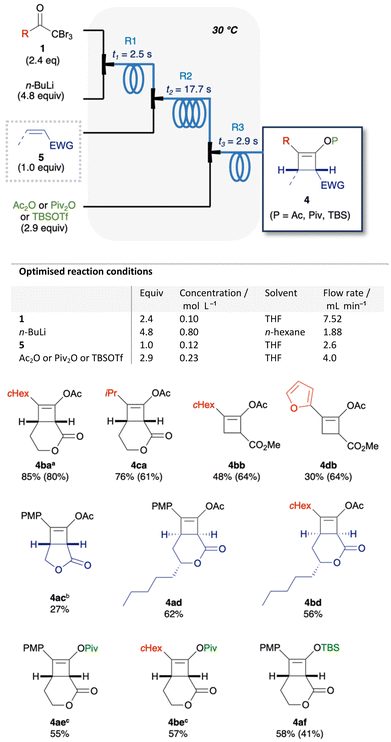 |
| | Fig. 4 Substrate scope. 1H NMR yields of cyclobutenes 4 in flow synthesis, with batch yields in the parentheses. For the flow synthesis of 4ba, 4ac, 4ae, and 4be, few parameters of the optimised reaction conditions were changed as follows; aT = 0 °C, t3 = 1.5 s. bT = 20 °C. cConcentration of 5 = 0.10 mol L−1. | |
Flow vs. batch synthesis and scale up
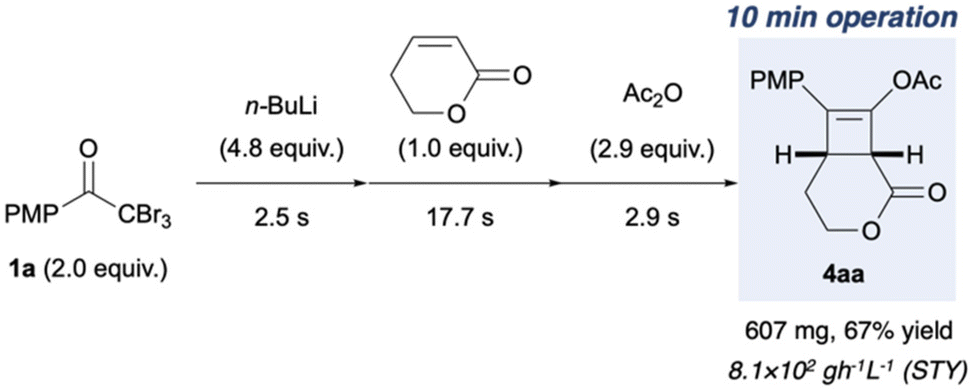
Finally, the flow conditions were compared with the corresponding batch conditions (Table 3). We used flow conditions (entry 3) for batch synthesis (entry 2), albeit with a slightly prolonged reaction time. Thus a low yield of 4ab (16%) was afforded. When the batch synthesis was conducted at −78 °C, we observed a moderate yield (70%) of 4ab, which is comparable to the flow synthesis yield (63%; entries 1 and 3). Unlike the batch systems that require cryogenic temperatures to stabilise the reactive and short-lived ynolates, the flow system did not need cryogenic temperatures. Furthermore, the space–time yields (STYs)33,34 of the flow system were over 50 times that of those in the batch system (Table 3, entries 1–3). This is attributed to the short residence time of 23.1 s over three steps in the flow system. This result is favourable for the scale up of this green synthesis. Hence, we used 3.12 mmol methyl acrylate (Fig. 5) as 0.12 M THF solution (26 mL), with a 0.1 M THF solution of 1a (75.2 mL), 0.80 M n-hexane solution of n-BuLi (18.8 mL), and 0.23 M THF solution of acetic anhydride (40 mL). A flow reaction for 10 min afforded a 67% yield of 4aa (607 mg) with a high STY of 8.1 × 102 g L−1 h−1. This demonstrates the utility of the flow protocol for the large-scale synthesis of highly functionalised cyclobutene 4aa, which is impossible in a batch system. The advantages over the corresponding batch reaction, including the avoidance of cryogenic temperatures and reduction in reaction time, were effective for a straightforward and safe scale up. Moreover, the use of n-BuLi reduced the consumption of organolithium reagents by half, which minimised wastage and lowered the synthetic cost. Our results should facilitate the development of a safe industrial process to synthesise cylobutenes.
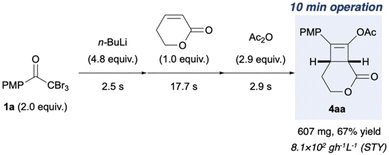 |
| | Fig. 5 Large-scale flow synthesis of 4aa. The reactions were performed under the optimised reaction conditions shown in Fig. 4, and the reaction mixture was collected for 10 min. | |
Table 3 Flow vs. batch synthesis of 4ab
|
| Entry |
Reactor |
Reaction time/temperature |
Yield of 4ab (%) |
STY (g h−1 L−1) |
| 1st |
2nd |
3rd |
| t-BuLi (8.0 equiv.) was used in a previous study instead of n-BuLi. Et2O was used instead of THF. |
| 1 |
Batcha |
1 h/−78 °C |
1 h/−78 to −20 °C |
0.5 h/−78 to −20 °C |
70 |
0.98 |
| 2 |
Batch |
60 s/30 °C |
60 s/30 °C |
60 s/30 °C |
16 |
14 |
| 3 |
Flow |
2.5 s/30 °C |
17.7 s/30 °C |
2.9 s/30 °C |
63 |
7.2 × 102 |
Conclusions
In this study, we achieved the continuous flow synthesis of cyclobutenes, starting from the flash generation of lithium ynolates. The synthesis is mediated by highly reactive species such as lithium ynolates, ketenes, and lithium enolates, which require complicated manipulations in batch. Nevertheless, we established a protocol that allows precise control of the reactivity without complicated temperature control. Non-cryogenic conditions shortened the total reaction time to 23.1 s, which prevented the decomposition of the reactive intermediates. First, we monitored the generation and degradation of lithium ynolates using in-line Raman analysis in the residence time range of 2.5–22.5 s at 30 °C to identify the best protocol for the continuous synthesis. The large-scale synthesis of cyclobutene 4aa with the optimised conditions afforded a high STY (8.1 × 102 g L−1 h−1). Unlike batch protocols, the flow protocol uses a lower amount of safer reagents, is easy to scale up, conserves energy as it does not require cryogenic temperatures, and saves time and space. The mild reaction temperature of 30 °C (nearly room temperature) and an extremely short residence time of 23.1 s contributed to energy savings. Overall, this established flow protocol is highly sustainable. Notably, this facile three-step, one-flow process affords functionalised cyclobutenes without complicated manipulations by chemists, thus saving manpower. Our results improve the chemistry of lithium ynolates and enhance their utility in green chemistry by providing a simple and sustainable protocol.
Data availability
The data supporting this article have been included in the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by JSPS KAKENHI (Grant Number 23K27295 to K. T.), MEXT KAKNHI (Grant Number JP21H05211 to K. T.) in Digi-TOS, JSPS KAKENHI (Grant Number 21K05069 and 24K09721 to Y. Y.), MEXT KAKENHI (Grant Number JP20H05740 to H. N.) in Deuterium Science, BINDS from AMED (Grant Numbers 22ama121042j0001 and 22ama121034j0001 to K. T.), and Mitsui Chemicals & Kyoto University Digital Chemistry Lab. Foundation (for K. T.). We thank Mr. Kurokawa (Graduate School of Pharmaceutical Sciences, Kyoto University) for his help with the X-ray diffraction analysis.
References
- S. G. Newman and K. F. Jensen, Green Chem., 2013, 15, 1456–1472 RSC.
- P. Natho and R. Luisi, Tetrahedron Green Chem., 2023, 2, 100015 CrossRef.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed.
- R. Porta, M. Benaglia and A. Puglisi, Org. Process Res. Dev., 2016, 20, 2–25 CrossRef CAS.
- J. Yoshida, K. Saito, T. Nokami and A. Nagaki, Synlett, 2011, 1189–1194 CrossRef CAS.
- S. T. Muller, T. Hokamp, S. Ehrmann, P. Hellier and T. Wirth, Chem. – Eur. J., 2016, 22, 11940–11942 CrossRef PubMed.
- A. Nagaki, Y. Ashikari, M. Takumi and T. Tamaki, Chem. Lett., 2021, 50, 485–492 CrossRef CAS.
- N. G. Anderson, Org. Process Res. Dev., 2012, 16, 852–869 CrossRef CAS.
- M. Power, E. Alcock and G. P. McGlacken, Org. Process Res. Dev., 2020, 24, 1814–1838 CrossRef CAS.
- M. Shindo, Synthesis, 2003, 2275–2288 CrossRef CAS.
- P. Duari, S. Mondal, M. Jorges and V. H. Gessner, Chem. Commun., 2024, 60, 9372–9375 RSC.
- J. Jubb and S. Gambarotta, J. Am. Chem. Soc., 1993, 115, 10410–10411 CrossRef CAS.
- J. Ficini, Tetrahedron, 1976, 32, 1449–1486 CrossRef CAS.
- M. Shindo, Tetrahedron, 2007, 63, 10–36 CrossRef CAS.
- M. Shindo, Chem. Soc. Rev., 1998, 27, 367–374 RSC.
-
(a) Y. Yamaoka, H. Imahori, M. Namioka, R. Nishina, Y. Kobori, M. Ueda, M. Shindo and K. Takasu, Org. Lett., 2024, 26, 1896–1901 CrossRef CAS PubMed;
(b) Y. Yamaoka, R. Nishina, K. Fujita and K. Takasu, Chem. Pharm. Bull., 2024, 72, 966–969 CrossRef CAS PubMed.
- S. Akai, S. Kitagaki, T. Naka, K. Yamamoto, Y. Tsuzuki, K. Matsumoto and Y. Kita, J. Chem. Soc., Perkin Trans. 1, 1996, 1705–1709 RSC.
- C. J. Kowalski and K. W. Fields, J. Am. Chem. Soc., 1982, 104, 321–323 CrossRef CAS.
- K. Kurahashi, Y. Takemoto and K. Takasu, ChemSusChem, 2012, 5, 270–273 CrossRef CAS PubMed.
- S. Umezu, T. Yoshiiwa, M. Tokeshi and M. Shindo, Tetrahedron Lett., 2014, 55, 1822–1825 CrossRef CAS.
- T. Yoshiiwa, S. Umezu, M. Tokeshi, Y. Baba and M. Shindo, J. Flow Chem., 2014, 4, 180–184 CrossRef.
- T. Iwata, T. Yoshinaga and M. Shindo, Synlett, 2020, 1903–1906 CAS.
- D. Didier and F. Reiners, Chem. Rec., 2021, 21, 1144–1160 CrossRef CAS PubMed.
- F. Hu, Y. Zhao, L. Che, X. Yan and Y. Xia, ChemCatChem, 2024, 16, e202301249 CrossRef CAS.
-
(a) For n-BuLi and s-BuLi: T. V. Ovaska, Encyclopedia of Reagents for Organic Synthesis, Wiley, 2001, pp. 899–914 Search PubMed;
(b) For t-BuLi: W. F. Bailey and N. Wachter-Jurcsak, Encyclopedia of Reagents
for Organic Synthesis, Wiley, 2001, 914–920 Search PubMed.
- V. Sans and L. Cronin, Chem. Soc. Rev., 2016, 45, 2032–2043 RSC.
- React IR could not be applied as the lithium ynolate 2b peak and the background peaks (1950–2350 cm−1) corresponding to the diamond IR probe overlapped.
- J. Li, H. Šimek, D. Ilioae, N. Jung, S. Bräse, H. Zappe, R. Dittmeyer and B. P. Ladewig, React. Chem. Eng., 2021, 6, 1497–1507 RSC.
- Y. Ashikari, R. Yoshioka, Y. Yonekura, D. E. Yoo, K. Okamoto and A. Nagaki, Chem. – Eur. J., 2024, 30, e202303774 CrossRef CAS PubMed.
- M. Shindo, Y. Sato and K. Shishido, Tetrahedron Lett., 1998, 39, 4857–4860 CrossRef CAS.
- This tendency was observed in our screening of the [2 + 2] cycloaddition preliminary experiment (Table S1†). The reaction temperature and residence time were screened using a freshly prepared ynolate in a batch reaction.
- M. Shindo, T. Kita, T. Kumagai, K. Matsumoto and K. Shishido, J. Am. Chem. Soc., 2006, 128, 1062–1063 CrossRef CAS PubMed.
- STY = product mol/(reactor volume × operation time).
- C. A. Hone and C. O. Kappe, Chem. Methods, 2021, 1, 454–467 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Motoki Namiokaa,
Hiroshi Naka
*a,
Motoki Namiokaa,
Hiroshi Naka