
Electrochemical dehydrogenative annulation for the synthesis of 4-oxo-oxazolines†
Yong
Yuan
 *,
Xincong
Liu
,
Feng
Zhang
,
Chunyan
Bai
,
Yuyan
Tao
,
Xiazhen
Bao
,
Dongsheng
Ji
and
Congde
Huo
*,
Xincong
Liu
,
Feng
Zhang
,
Chunyan
Bai
,
Yuyan
Tao
,
Xiazhen
Bao
,
Dongsheng
Ji
and
Congde
Huo
College of Chemistry and Chemical Engineering, Northwest Normal University, Lanzhou, Gansu 730070, China. E-mail: yuanyong@nwnu.edu.cn
First published on 2nd December 2024
Abstract
A metal- and oxidizing reagent-free electrochemical dehydrogenative annulation of enamides with O-nucleophiles has been developed, leading to a series of 4-oxo-oxazolines under environmentally friendly conditions. The present method demonstrates broad substrate scope and wide functional group tolerance. In addition to water, alcohols and carboxylic acids were also competent reaction partners. The merit of this electrochemical protocol has also been demonstrated by the gram-scale reaction and the synthesis of 2-(3,4-dimethoxyphenyl)-5-methyl-4,5-dihydrooxazol-4-ol (an antitumor nature product precursor). The mechanistic results suggest that the formation and homolytic cleavage of the N–Br bond is key to the success of this electrochemical dehydrogenative annulation reaction.
Oxazolines are ubiquitous structural units in natural products, pharmaceuticals and biologically active molecules.1 Furthermore, oxazolines are valuable synthetic building blocks that can be converted into a diverse range of more complex molecules.2 Moreover, oxazolines are also useful ligands in transition-metal-catalyzed reactions.3 Over the past decades, considerable efforts have been made to prepare this class of compounds.4 Among the numerous methods, the reaction of substituted 1,2-amino alcohols with carboxylic acids, aldehydes, or nitriles is one of the most prominent methods (Scheme 1A).5 However, reported methods often generate a large amount of waste. Moreover, these methods can only synthesize 4-carbon functional group substituted oxazolines; the reason for this may be because heteroatom-substituted 1,2-amino alcohols are not readily available. Therefore, there is a significant need for the development of new procedures to access 4-heteroatom-substituted oxazolines in an environmentally friendly and versatile fashion. Visible-light-induced dehydrogenative annulation has been demonstrated to be an enabling method to prepare 4-alkoxy- and 4-acyloxy-substituted oxazolines.6 However, this method has been suggested to be challenging for obtaining 4-hydroxy-substituted oxazolines probably due to the incompatibility of H2O with photocatalyst.
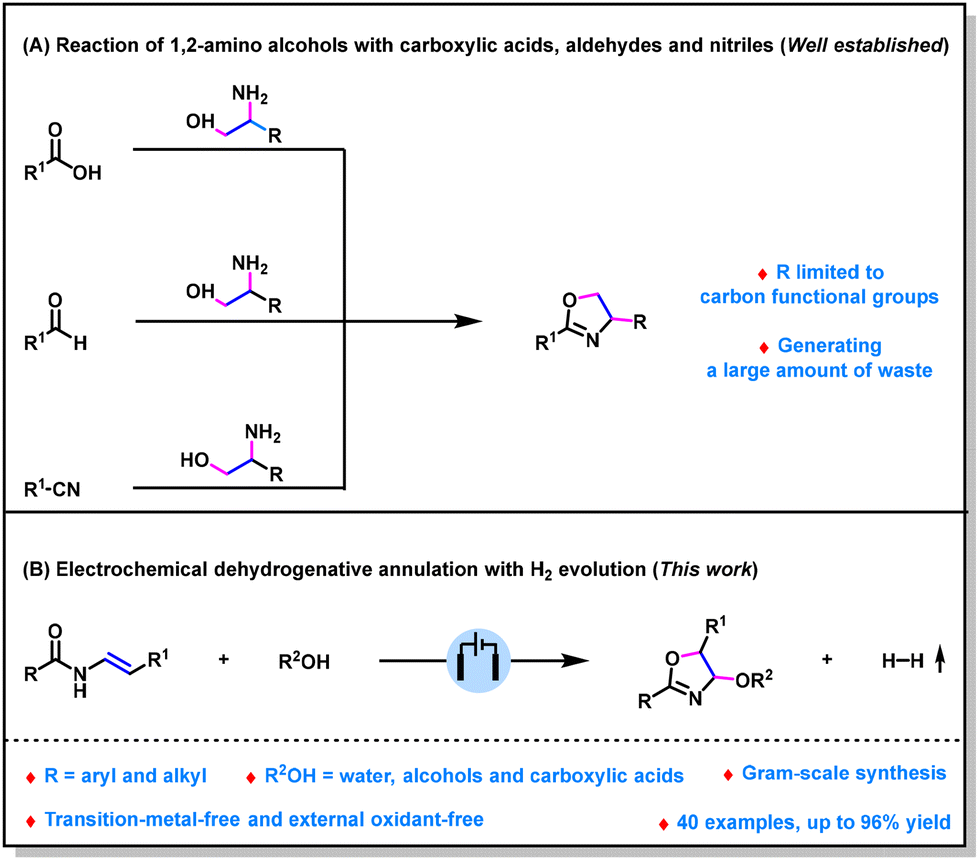 | ||
| Scheme 1 Methods for accessing 4-substituted oxazolines. | ||
Organic electrosynthesis, which uses electrons as oxidizing or reducing reagents,7 has emerged as a useful tool for organic synthesis.8 In particular, it can realize the dehydrogenative annulation with H2 evolution under environmentally friendly conditions.9,10 In this context, only very few methods have been developed for preparing 4-aryl-substituted oxazolines.11 However, to the best of our knowledge, the synthesis of 4-oxo-oxazolines through dehydrogenative annulation with H2 evolution has not been reported. Herein, as a part of our continuing research interest in organic electrosynthesis,12 we report an efficient electrochemical dehydrogenative annulation reaction of enamides with O-nucleophiles (Scheme 1B). Our method shows broad substrate scope and environmentally friendly reaction conditions, a series of 4-hydroxy-, 4-alkoxy-, and even 4-acyloxy-substituted oxazolines were obtained in up to 96% yield under metal- and oxidant-free conditions.
The electrochemical dehydrogenative annulation of N-vinylbenzamide 1a and H2O was chosen as the model reaction for optimization (Table 1). After careful exploration, the desired dehydrogenative annulation product 2a was obtained in 89% yield under a 12 mA constant current for 3.5 h in MeCN with NaBr as the mediator, nBu4NBF4 as the supporting electrolyte, carbon cloth as the anode and stainless steel plate as the cathode (Table 1, entry 1). Control experiments revealed that both electricity and NaBr were important for this dehydrogenative annulation reaction (Table 1, entries 2 and 3). Using nBu4NBr as the mediator, 83% yield of dehydrogenative annulation product 2a was obtained (Table 1, entry 4), whereas when NaCl or NaI was employed, the yield of 2a was dropped to 57% and 68% (Table 1, entries 5 and 6), respectively. Decreasing the operating current to 10 mA provided the dehydrogenative annulation product 2a in high yield (Table 1, entry 7), however, running the electrolysis under a controlled current of 14 mA for 3 h led to a decrease in the reaction efficiency (Table 1, entry 8). Performing the electrolysis with a graphite rod anode furnished 2a in 84% yield (Table 1, entry 9), whereas when the dehydrogenative annulation of N-vinylbenzamide 1a and H2O was conducted with a graphite plate anode or nickel plate cathode (Table 1, entries 10 and 11), an obvious loss of yield was observed. Using DMF as the solvent, the desired dehydrogenative annulation product 2a was afforded in high yield as well (Table 1, entry 12). In addition, when the electrolysis was conducted under an air atmosphere, the dehydrogenative annulation product 2a could still be obtained in 85% yield (Table 1, entry 13).
|
|
||
|---|---|---|
| Entry | Variation from standard conditions | Yieldb (%) |
| a Reaction conditions: undivided cell, carbon cloth as the anode, stainless steel plate as the cathode, 12 mA, 3.5 h, 1a (0.5 mmol), H2O (1.5 mL), NaBr (2.0 equiv.), nBu4NBF4 (1.0 equiv.), MeCN (10.5 mL), 70 °C, under Ar atmosphere. b Isolated yield. | ||
| 1 | None | 89 |
| 2 | No electric current | 0 |
| 3 | No NaBr | 34 |
| 4 | n Bu4NBr instead of NaBr | 83 |
| 5 | NaCl instead of NaBr | 57 |
| 6 | NaI instead of NaBr | 68 |
| 7 | 10 mA, 4.2 h | 83 |
| 8 | 14 mA, 3.0 h | 65 |
| 9 | Graphite rod anode | 84 |
| 10 | Graphite plate anode | 69 |
| 11 | Nickel plate cathode | 63 |
| 12 | DMF instead of MeCN | 85 |
| 13 | Air | 85 |

Having established the optimized reaction conditions, we set out to investigate the substrate scope and generality of this electrochemical dehydrogenative annulation reaction (Scheme 2). Firstly, a variety of enamides were examined (Scheme 2, 2a–2x). N-Vinylbenzamides bearing electron-neutral substituents (methyl, ethyl, n-butyl, t-butyl) or electron-donating groups (methoxy, ethoxy) on the phenyl ring were compatible with the reaction conditions, generating the desired dehydrogenative annulation products in good to excellent yields (Scheme 2, 2b–2i).13 Furthermore, the reaction is also compatible with the N-vinylbenzamides bearing halogen substituent (fluorine, chlorine, bromine) in the para, meta or ortho position of the phenyl ring, and the respective dehydrogenative annulation product was obtained with a high yield (Scheme 2, 2j–2n). Moreover, when 3,4-dimethoxy-N-vinylbenzamide was used, the corresponding dehydrogenative annulation product was furnished in 86% yield (Scheme 2, 2o). 2,4,6-Trimethyl-N-vinylbenzamide reacted smoothly under standard conditions to give the dehydrogenative annulation product in good yield (Scheme 2, 2p). Note that 2q, 2r, and 2s could also be synthesized in 70%–86% yield. In addition to enamides prepared from aryl chlorides (Scheme 2, 2a–2s), N-vinyl-heterarylamides were then investigated as reaction partners (Scheme 2, 2t–2w). N-heteraryl amides prepared from furan-2-carbonyl chloride (Scheme 2, 2t), thiophene-2-carbonyl chloride (Scheme 2, 2u), 6-chloronicotinoyl chloride (Scheme 2, 2v), and 6-bromonicotinoyl chloride (Scheme 2, 2w) were also amenable, providing the dehydrogenative annulation products in 57–87% yields. Notably, N-propenylphenylamide could also be used to deliver the corresponding dehydrogenative annulation product (Scheme 2, 2x).
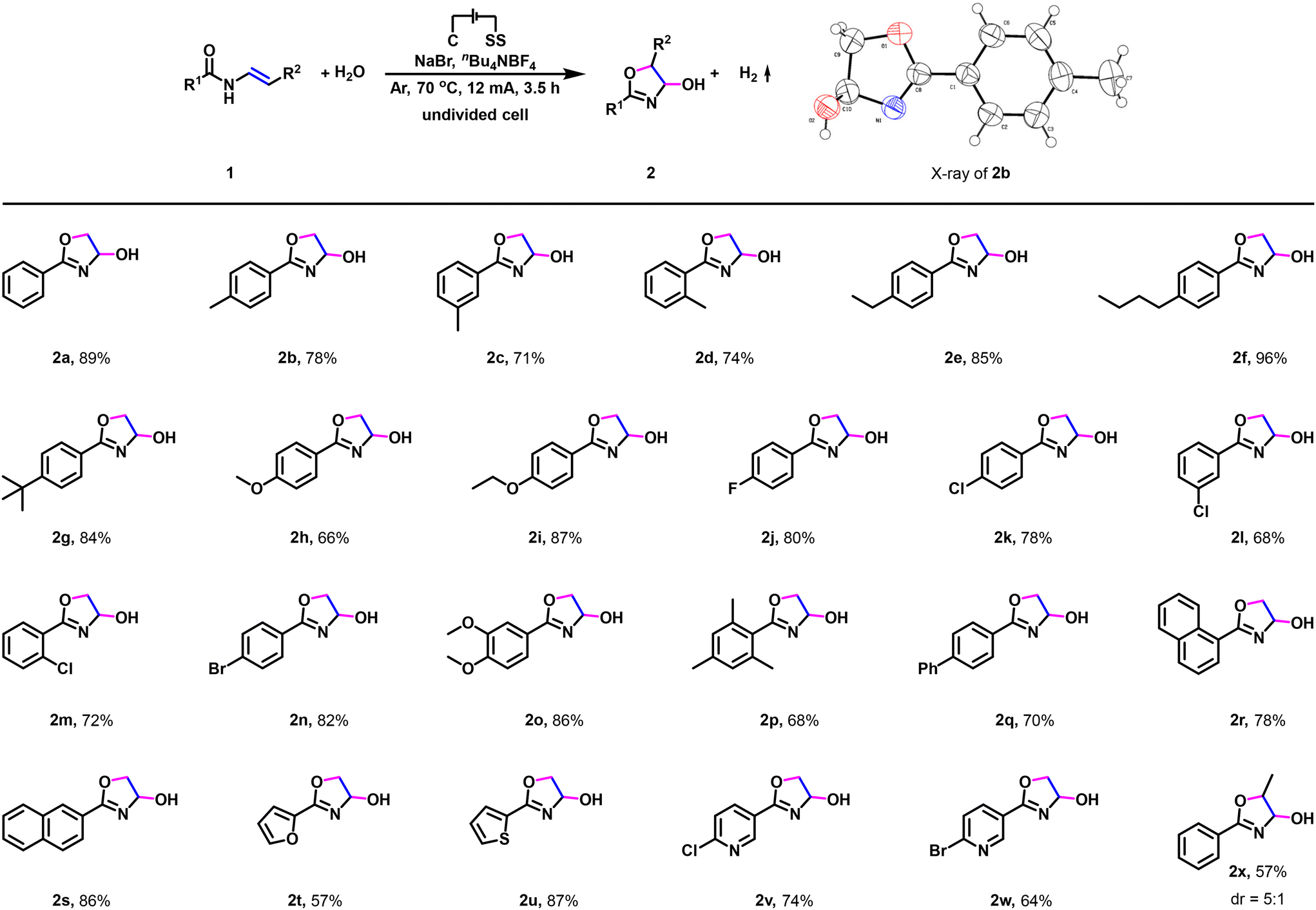 | ||
| Scheme 2 Scope of enamides. Reaction conditions: undivided cell, carbon cloth as the anode, stainless steel plate as the cathode, 12 mA, 3.5 h, 1 (0.5 mmol), H2O (1.5 mL), NaBr (2.0 equiv.), nBu4NBF4 (1.0 equiv.), MeCN (10.5 mL), 70 °C, under Ar atmosphere, isolated yield. | ||
To further establish the generality of this transformation, substrate scope for electrochemical dehydrogenative annulation with respect to O-nucleophiles was then evaluated. As shown in Scheme 3, simple primary alcohols, such as methanol, ethanol, n-butanol, isobutanol, and benzyl alcohol access to the corresponding dehydrogenative annulation products in high to excellent yields (Scheme 3, 4a–4e). Furthermore, ethylene glycol, 3-buten-1-ol, 3-butyn-1-ol, as well as citronellol were also suitable reaction partners (Scheme 3, 4f–4i), and functional groups such as hydroxy, carbon–carbon double bond, and carbon–carbon triple bond were fully tolerated in this electrochemical dehydrogenative annulation. Interestingly, the reaction was also found to be amenable to secondary alcohols. For example, isopropanol and cyclopentanol furnished the corresponding dehydrogenative annulation products 4j and 4k in 83% and 86% yields (Scheme 3, 4j and 4k), respectively. Moreover, in addition to alcohols (Scheme 3, 4a–4k), aromatic acids and fatty acids were also suitable reaction partners (Scheme 3, 4l–4o). Benzoic acid and 4-methylbenzoic acid reacted smoothly to access the corresponding dehydrogenative annulation products in 75% and 70% yields (Scheme 3, 4l–4m), respectively. By contrast, electron-deficient 4-bromobenzoic acid furnished the corresponding product in a decreased yield (Scheme 3, 4n). In addition, when palmitic acid (one of the most common fatty acids in nature) was subjected to the standard reaction conditions, the corresponding dehydrogenative annulation product was isolated in 60% yield (Scheme 3, 4o).
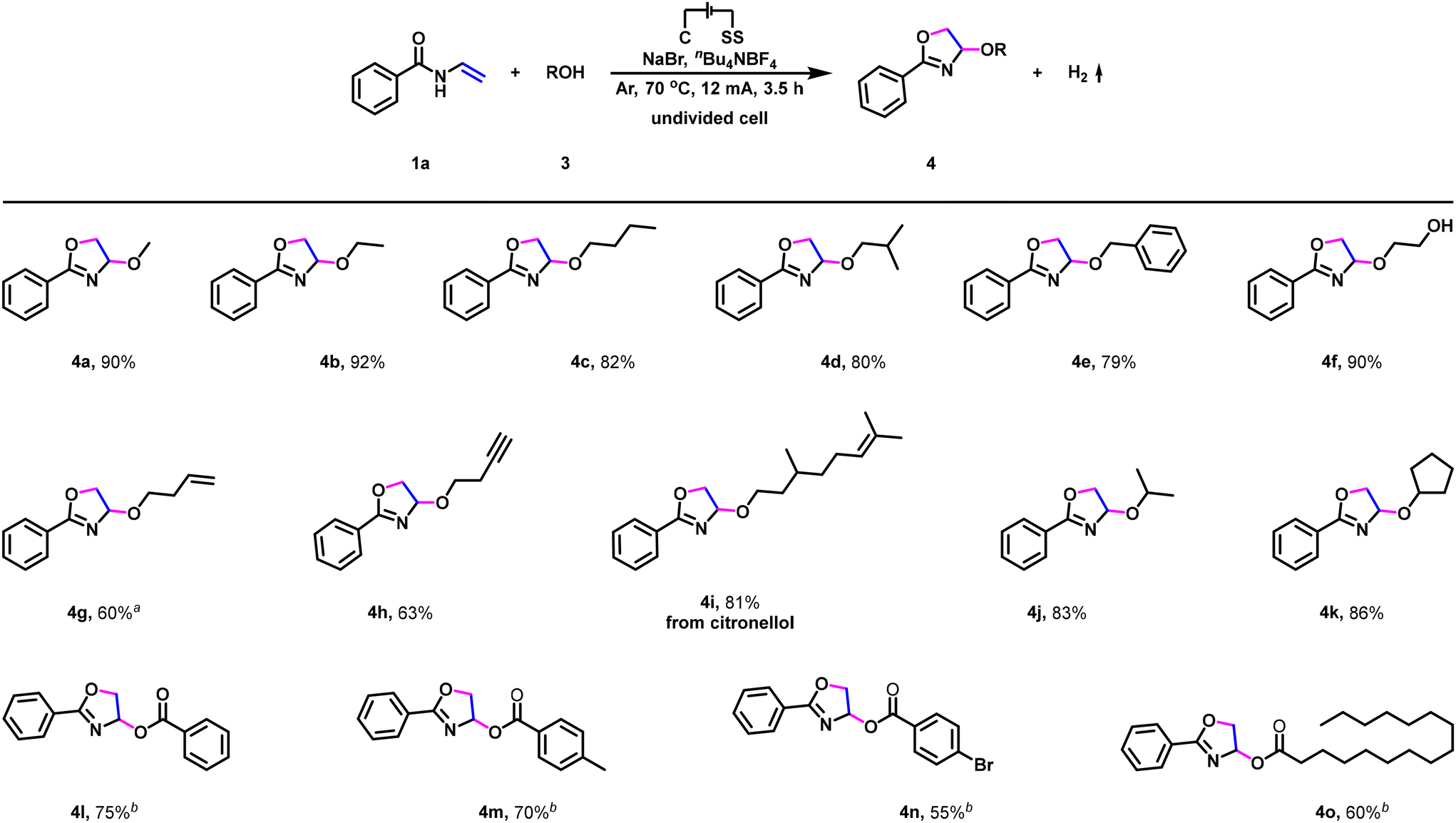 | ||
Scheme 3 Scope of O-nucleophiles. Reaction conditions: undivided cell, carbon cloth as the anode, stainless steel plate as the cathode, 12 mA, 3.5 h, 1a (0.5 mmol), 3 (0.5 mL), NaBr (2.0 equiv.), nBu4NBF4 (1.0 equiv.), MeCN (10.5 mL), 70 °C, under Ar atmosphere, isolated yield. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Using 1.0 mL of 3-buten-1-ol. bUsing 2.0 equiv. of carboxylic acids. Using 1.0 mL of 3-buten-1-ol. bUsing 2.0 equiv. of carboxylic acids. | ||
To demonstrate the synthetic application of this electrochemical approach, we performed a scale-up synthesis of 2a. To our delight, when 10 mmol 1a was subjected to the reaction, the corresponding dehydrogenative annulation product 2a could be afforded in 72% (1.17 g) yield (Scheme 4A). The potential of this electrochemical method in the formation of 4-oxo-oxazolines was further underpinned by the synthesis of 2-(3,4-dimethoxyphenyl)-5-methyl-4,5-dihydrooxazol-4-ol 2y—an antitumor nature product precursor.14 Specifically, through electrochemical dehydrogenative annulation method, 2y was obtained with 81% isolated yield (Scheme 4B).
 | ||
| Scheme 4 Synthetic applications. | ||
Trace amounts of benzamide were observed as side products in some reaction system, suggesting that intermediates C and/or D may be formed under the electrochemical conditions (Scheme 5A). To clarify whether 2a would be converted into the corresponding radical intermediate C and carbocation intermediate D, a radical trapping experiment was conducted by employing 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) as the radical scavenger. As shown in Scheme 5B, in the presence of 2.0 equiv. of TEMPO, the dehydrogenative annulation reaction was partially inhibited (only 30% yield of 2a was obtained) and the TEMPO adduct E as well as its hydrolysates F and G were all identified via HRMS, confirming the presence of radical species C. Meanwhile, the nucleophilic attack product H and its hydrolysates F and I, were also detected by HRMS (Scheme 5B). This result indicates that the intermediate D is involved in the reaction pathway as well. In addition, cyclic voltammetry (CV) experiments on 1a and NaBr were also performed to shed light on the mechanism (see the ESI† for details). The oxidation peak of NaBr was observed at 1.19 V, whereas 1a showed no oxidation peak at 0–2 V. These results indicated that the reaction might start with the anodic oxidation of bromide ion.
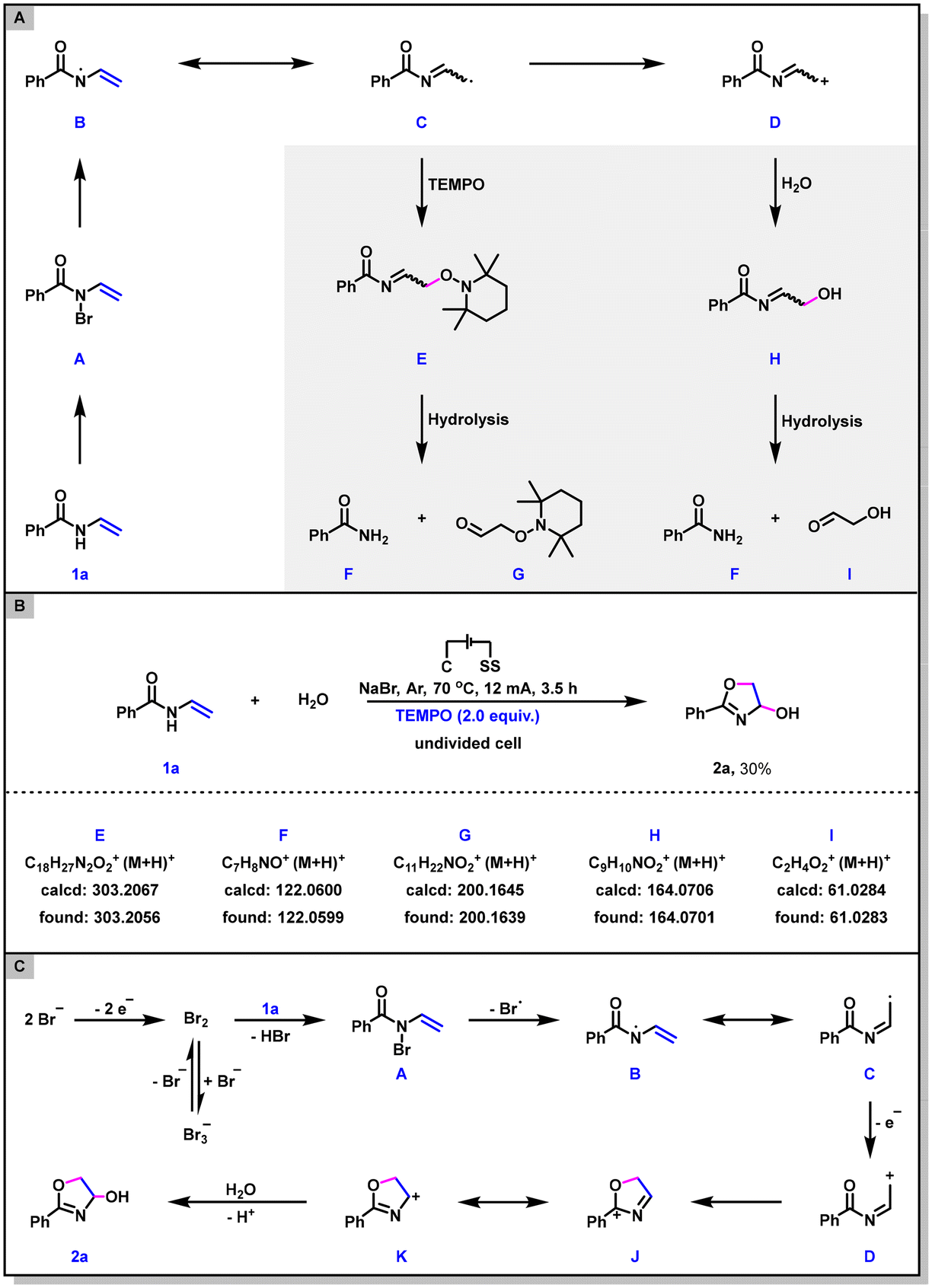 | ||
| Scheme 5 Mechanism studies and proposed mechanism. | ||
On the basis of the above experiments and previous literature,15 a plausible reaction mechanism for this electrochemical dehydrogenative annulation is presented in Scheme 5C. Firstly, anodic oxidation of the bromine ion generates molecular bromine, which then reacts with N-vinylbenzamide 1a to afford the intermediate A. Secondly, intermediate A undergoes homolytic cleavage of the N-Br bond to form the nitrogen radical intermediate B and then to furnish the carbon radical intermediate C. Thirdly, the further one-electron oxidation of radical C generates the carbocation intermediate D. Finally, intermediate D undergoes electrophilic addition, nucleophilic attack, and deprotonation to afford the dehydrogenative annulation product 2a. On the cathode, H2O is reduced to produce OH− and H2.16
In summary, we have developed an efficient electrochemical dehydrogenative annulation reaction between enamides and O-nucleophile for the synthesis of 4-oxo-oxazolines. Under mild and environmentally friendly electrochemical conditions, a series of 4-oxo-oxazolines were obtained in moderate to excellent yields. It is worth noting that this reaction features broad substrate scope; in addition to water, alcohols, aromatic acids as well as fatty acids were also competent reaction partners, providing the corresponding 4-oxo-oxazolines in moderate to excellent yields.
Data availability
Experimental details and characterization data can be found in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22401234), the Young Doctor Fund Project of Gansu Provincial Department of Education (2022QB-030), and the Northwest Normal University Young Teachers’ Research Ability Enhancement Program (NWNU-LKQN2021-02).References
- (a) H.-Z. Zhang, Z.-L. Zhao and C.-H. Zhou, Eur. J. Med. Chem., 2018, 144, 444–492 CrossRef CAS; (b) C. Minakuchi, J. Suzuki, K. Toda, M. Akamatsu and Y. Nakagawa, Bioorg. Med. Chem. Lett., 2006, 16, 4080–4084 CrossRef CAS; (c) Q. Li, K. W. Woods, A. Claiborne, I. I. S. L. Gwaltney, K. J. Barr, G. Liu, L. Gehrke, R. B. Credo, Y. H. Hui, J. Lee, R. B. Warner, P. Kovar, M. A. Nukkala, N. A. Zielinski, S. K. Tahir, M. Fitzgerald, K. H. Kim, K. Marsh, D. Frost, S.-C. Ng, S. Rosenberg and H. L. Sham, Bioorg. Med. Chem. Lett., 2002, 12, 465–469 CrossRef CAS PubMed; (d) G. Campiani, M. De Angelis, S. Armaroli, C. Fattorusso, B. Catalanotti, A. Ramunno, V. Nacci, E. Novellino, C. Grewer, D. Ionescu, T. Rauen, R. Griffiths, C. Sinclair, E. Fumagalli and T. Mennini, J. Med. Chem., 2001, 44, 2507–2510 CrossRef CAS PubMed.
- (a) K. Dong, A. Humeidi, W. Griffith, H. Arman, X. Xu and M. P. Doyle, Angew. Chem., Int. Ed., 2021, 60, 13394–13400 CrossRef CAS; (b) X. Wang, X. Yi, H. Xu and H.-X. Dai, Org. Lett., 2019, 21, 5981–5985 CrossRef CAS PubMed; (c) A. I. Meyers and E. D. Mihelich, Angew. Chem., Int. Ed. Engl., 1976, 15, 270–281 CrossRef.
- G. Yang and W. Zhang, Chem. Soc. Rev., 2018, 47, 1783–1810 RSC.
- (a) Q. Shi, Y. Huang and W. H. Liu, Precis. Chem., 2023, 1, 316–325 CrossRef CAS; (b) X.-F. Wu, H. Neumann, S. Neumann and M. Beller, Chem. – Eur. J., 2012, 18, 13619–13623 CrossRef CAS PubMed; (c) N. Luo, Z. Zhan, Z. Ban, G. Lu, J. He, F. Hu and G. Huang, Adv. Synth. Catal., 2020, 362, 3126–3130 CrossRef CAS; (d) R.-H. Liu, Q.-C. Shan, Y. Gao, T.-P. Loh and X.-H. Hu, Chin. Chem. Lett., 2021, 32, 1411–1414 CrossRef CAS; (e) H. Luo, Z. Yang, W. Lin, Y. Zheng and S. Ma, Chem. Sci., 2018, 9, 1964–1969 RSC; (f) X.-W. Liang, C. Zheng and S.-L. You, Adv. Synth. Catal., 2016, 358, 2066–2071 CrossRef CAS; (g) Á. Sinai, D. Vangel, T. Gáti, P. Bombicz and Z. Novák, Org. Lett., 2015, 17, 4136–4139 CrossRef; (h) W.-C. Gao, F. Hu, Y.-M. Huo, H.-H. Chang, X. Li and W.-L. Wei, Org. Lett., 2015, 17, 3914–3917 CrossRef CAS.
- (a) T. Ohshima, T. Iwasaki and K. Mashima, Chem. Commun., 2006, 2711–2713, 10.1039/B605066B; (b) P. Chaudhry, F. Schoenen, B. Neuenswander, G. H. Lushington and J. Aubé, J. Comb. Chem., 2007, 9, 473–476 CrossRef CAS PubMed; (c) P. J. Boissarie, Z. E. Hamilton, S. Lang, J. A. Murphy and C. J. Suckling, Org. Lett., 2011, 13, 6256–6259 CrossRef CAS; (d) M. Ishihara and H. Togo, Tetrahedron, 2007, 63, 1474–1480 CrossRef CAS; (e) T. G. Gant and A. I. Meyers, Tetrahedron, 1994, 50, 2297–2360 CrossRef CAS.
- R. Sun, X. Yang, Y. Ge, J. Song, X. Zheng, M. Yuan, R. Li, H. Chen and H. Fu, ACS Catal., 2021, 11, 11762–11773 CrossRef CAS.
- (a) K.-J. Jiao, Z.-H. Wang, C. Ma, H.-L. Liu, B. Cheng and T.-S. Mei, Chem. Catal., 2022, 2, 3019–3047 CrossRef CAS; (b) S.-H. Shi, Y. Liang and N. Jiao, Chem. Rev., 2021, 121, 485–505 CrossRef CAS; (c) L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC; (d) Y. Yuan, J. Yang and A. Lei, Chem. Soc. Rev., 2021, 50, 10058–10086 RSC; (e) C. Ma, P. Fang, Z.-R. Liu, S.-S. Xu, K. Xu, X. Cheng, A. Lei, H.-C. Xu, C. Zeng and T.-S. Mei, Sci. Bull., 2021, 66, 2412–2429 CrossRef CAS; (f) N. Chen and H.-C. Xu, Chem. Rec., 2021, 21, 2306–2319 CrossRef CAS PubMed; (g) L. Ackermann, Acc. Chem. Res., 2020, 53, 84–104 CrossRef CAS; (h) Y. Jiang, K. Xu and C. Zeng, Chem. Rev., 2018, 118, 4485–4540 CrossRef CAS PubMed; (i) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed; (j) A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS.
- (a) L. Zeng, Q. Yang, J. Wang, X. Wang, P. Wang, S. Wang, S. Lv, S. Muhammad, Y. Liu, H. Yi and A. Lei, Science, 2024, 385, 216–223 CrossRef CAS PubMed; (b) P.-Y. Chen, C. Huang, L.-H. Jie, B. Guo, S. Zhu and H.-C. Xu, J. Am. Chem. Soc., 2024, 146, 7178–7184 CrossRef CAS PubMed; (c) S. Ji, L. Zhao, B. Miao, M. Xue, T. Pan, Z. Shao, X. Zhou, A. Fu and Y. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202304434 CrossRef CAS PubMed; (d) L. Zou, X. Wang, S. Xiang, W. Zheng and Q. Lu, Angew. Chem., Int. Ed., 2023, 62, e202301026 CrossRef CAS; (e) S. Arepally, T. Kim, G. Kim, H. Yang and J. K. Park, Angew. Chem., Int. Ed., 2023, 62, e202303460 CrossRef CAS; (f) J. Lu, Y. Yao, L. Li and N. Fu, J. Am. Chem. Soc., 2023, 145, 26774–26782 CrossRef CAS PubMed; (g) M. Liu, T. Feng, Y. Wang, G. Kou, Q. Wang, Q. Wang and Y. Qiu, Nat. Commun., 2023, 14, 6467 CrossRef CAS PubMed; (h) Y. Zhao, X. Li, S. L. Homölle, B. Wang and L. Ackermann, Chem. Sci., 2024, 15, 1117–1122 RSC; (i) X. Sun and K. Zheng, Nat. Commun., 2023, 14, 6825 CrossRef CAS; (j) L. Lu, Y. Wang, W. Zhang, W. Zhang, K. A. See and S. Lin, J. Am. Chem. Soc., 2023, 145, 22298–22304 CrossRef CAS; (k) J. M. Edgecomb, S. N. Alektiar, N. G. W. Cowper, J. A. Sowin and Z. K. Wickens, J. Am. Chem. Soc., 2023, 145, 20169–20175 CrossRef CAS; (l) T. Wang, F. He, W. Jiang and J. Liu, Angew. Chem., Int. Ed., 2024, 63, e202316140 CrossRef CAS; (m) M. Wang, C. Zhang, C. Ci, H. Jiang, P. H. Dixneuf and M. Zhang, J. Am. Chem. Soc., 2023, 145, 10967–10973 CrossRef CAS PubMed; (n) Y.-Z. Wang, Z.-H. Wang, I. L. Eshel, B. Sun, D. Liu, Y.-C. Gu, A. Milo and T.-S. Mei, Nat. Commun., 2023, 14, 2322 CrossRef CAS PubMed; (o) F. Lian, K. Xu and C. Zeng, CCS Chem., 2023, 5, 1973–1981 CrossRef CAS.
- (a) H. Wang, X. Gao, Z. Lv, T. Abdelilah and A. Lei, Chem. Rev., 2019, 119, 6769–6787 CrossRef CAS PubMed; (b) Z. Ye and F. Zhang, Chin. J. Chem., 2019, 37, 513–528 CrossRef CAS.
- (a) X. Hu, L. Nie, G. Zhang and A. Lei, Angew. Chem., Int. Ed., 2020, 59, 15238–15243 CrossRef CAS; (b) C. Tian, L. Massignan, T. H. Meyer and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 2383–2387 CrossRef CAS; (c) J. Li, W. Huang, J. Chen, L. He, X. Cheng and G. Li, Angew. Chem., Int. Ed., 2018, 57, 5695–5698 CrossRef CAS; (d) C.-Y. Cai and H.-C. Xu, Nat. Commun., 2018, 9, 3551 CrossRef.
- (a) H. Huang and T. H. Lambert, J. Am. Chem. Soc., 2022, 144, 18803–18809 CrossRef CAS PubMed; (b) X.-A. Liang, L. Niu, S. Wang and A. Lei, Chem. Catal., 2021, 1, 1055–1064 CrossRef CAS; (c) J. D. Haupt, M. Berger and S. R. Waldvogel, Org. Lett., 2019, 21, 242–245 CrossRef CAS PubMed; (d) H. Wang, J. Zhang, J. Tan, L. Xin, Y. Li, S. Zhang and K. Xu, Org. Lett., 2018, 20, 2505–2508 CrossRef CAS PubMed.
- (a) Y. Yuan, Y. Zheng, B. Xu, J. Liao, F. Bu, S. Wang, J.-G. Hu and A. Lei, ACS Catal., 2020, 10, 6676–6681 CrossRef CAS; (b) Y. Yuan, Y. Chen, S. Tang, Z. Huang and A. Lei, Sci. Adv., 2018, 4, eaat5312 CrossRef CAS PubMed; (c) Y. Yuan, H. Jiang, Y.-N. Zhang, Y. Tao, X. Liu and C. Huo, Green Chem., 2024, 26, 10811–10817 RSC; (d) Y. Yuan, J. Qi, D. Wang, Z. Chen, H. Wan, J. Zhu, H. Yi, A. D. Chowdhury and A. Lei, CCS Chem., 2021, 3, 3027–3038 Search PubMed.
- CCDC 2375205 (2b).†.
- X.-H. Luo, X.-Z. Wang, H.-L. Jiang, J.-L. Yang, P. Crews, F. A. Valeriote and Q.-X. Wu, Fitoterapia, 2012, 83, 754–758 CrossRef CAS PubMed.
- S. Zhang, L. Li, M. Xue, R. Zhang, K. Xu and C. Zeng, Org. Lett., 2018, 20, 3443–3446 CrossRef CAS PubMed.
- (a) P. Xiong, H. Long, J. Song, Y. Wang, J.-F. Li and H.-C. Xu, J. Am. Chem. Soc., 2018, 140, 16387–16391 CrossRef CAS PubMed; (b) Y. Yuan, Z. Zhou, L. Zhang, L.-S. Li and A. Lei, Org. Lett., 2021, 23, 5932–5936 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2375205. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4gc05119j |
| This journal is © The Royal Society of Chemistry 2025 |