
Neoteric solvents for exploratory catalysis: hydrophosphination catalysis with CHEM21 solvents†
Emma J.
Finfer
 and
Rory
Waterman
*
and
Rory
Waterman
*
Department of Chemistry, University of Vermont, Burlington, VT 05405, USA. E-mail: rory.waterman@uvm.edu
First published on 22nd November 2024
Abstract
Exploratory catalytic hydrophosphination studies continue to be in toxic or environmentally harmful solvents, missing an opportunity for improved sustainability and safety. A comparative analysis of hydrophosphination catalysis using the three major categories of substrates, styrene, Michael acceptors, and unactivated alkenes, has been undertaken to assess a transition to green solvents. The compound selected, Cu(acac)2, has been identified as a highly active and most general precatalyst for hydrophosphination with known mechanistic divergence based on substrate. Additionally, three group 1 alkoxides (LiOEt, NaOEt, KOEt) have been shown to be competent hydrophosphination catalysts for these categories of alkenes; under these conditions substantially lower loadings were realized compared to prior studies with group 1 metals. Eight solvents were investigated from categories outlined in the CHEM21 guide, and seven were highly effective for most reactions, regardless of catalysts or mechanism. These results demonstrate a straightforward path to improving the sustainability of future studies in this and related catalytic reactions through bioavailable solvents, heretofore unknown in hydrophosphination catalysis. Other key findings include the improved utilization of more sustainable and low toxicity group 1 catalysts in this reaction with greater conversion (i.e., reduced waste) as well as highlighting potential pitfalls of reactions involving phosphine substrates in bioavailable solvents.
Introduction
With society's demand for phosphorus quickly increasing, novel and facile routes to synthesize organophosphines are of high importance.1,2 This circumstance arises from organophosphines being at a nexus of biologically active molecules, materials, and ligands for catalysis.3 High demand and supply strain make rectifying challenges associated with selective phosphorus–carbon (P–C) bond formation urgent. Catalytic hydrophosphination is not only atom-economical, but also an entry point to tailored steric and electronic properties in the resultant phosphine products through the selection of the substrates. Such an aim requires a broad array of catalysts,4 though access to tailor-made phosphine products by this route has not yet been realized.5Nevertheless, hydrophosphination has been rapidly developing over the last decade,4 and the substrate scope has been expanding, despite on-going challenges.5,6 Copper acetylacetonate, Cu(acac)2, is a fast and efficient precatalyst for the hydrophosphination of both primary and secondary phosphines under low intensity UV-A irradiation.7,8 Copper(II) salts are desirable for catalysis due to their recyclability, relative abundance, low toxicity, and air-stability,9,10 and these features align with our desire to develop green catalysts.11 The activity of Cu(acac)2 arises from photolysis, which amplifies activity identified in key initial discoveries of hydrophosphination with copper precatalysts.12,13 Photocatalytic conditions rid the reaction of prolonged heating, reducing the energetic burden of the catalysis. In particular, this enhanced reactivity has allowed for the experimentation to determine the limits to photoexcited copper-catalyzed hydrophosphination and truly probe the utility of this reaction.
In previous work by our group, copper-catalyzed hydrophosphination has been shown to undergo divergent mechanisms depending on the electronic structure of the alkene substrate.7 In both cases, Cu(acac)2 is reduced to generate a Cu(I)–phosphido compound. In the presence of electron rich alkenes, an alkyl copper intermediate is formed via a 2,1-insertion of the alkene into the copper–phosphorus bond. With electron deficient alkenes, the intermediate copper(I)–phosphido compound attacks the alkene substrate akin to a conjugate addition.7 These established differences represent a significant portion of the current spectrum of metal-catalyzed hydrophosphination reactions,4 and observing both mechanisms with a common catalyst is an article of convenience in this study.
While copper is a relatively benign transition metal, light group 1 salts are of greater abundance and yet lower toxicity. There are a limited number of reports on these metals as hydrophosphination catalysts, with leading efforts in systematic study led by Mulvey and coworkers.14–17 As these elements continue to be explored and understood, it is important to note that current reports leverage high loadings (10–27 mol%), and improvement in this area would be of great interest to make these viable, green catalysts.
While the substrate scope of hydrophosphination continues to progress, with copper accessing some of the substrates reputed to be the greatest challenges,5 investigation of solvent effects has lagged in comparison.4–6 It is therefore unknown if a transition to green solvents would negatively impact catalytic hydrophosphination. A significant exception to the absence of solvent effect studies in hydrophosphination is an investigation by Webster and coworkers that demonstrated how solvent can change the regioselectivity of the reaction,18 which emphasizes the value of solvent selection in this reaction. In that work, hydrophosphination performed in dichloromethane solution afforded the anti-Markovnikov product, while a similar reaction conducted in benzene solution selected for the Markovnikov product.18 These are intriguing results but utilize highly hazardous solvents by any measure.
Hydrophosphination is rife to expand, and exploring solvent effects creates an excellent opportunity to probe green solvents as alternatives to toxic, albeit more widely used, solvents in this reaction. Using NMR spectroscopy for screening is particularly convenient because many products of hydrophosphination have been well characterized and are readily and unequivocally identified by 31P NMR spectroscopy,19–23 obviating the need for deuterated solvents in this screening study.
A simple analysis of literature reports reveals that the anti-Markovnikov addition product of diphenylphosphine to styrene, the apparent benchmark reaction for catalyst screening,4,6 has been synthesized in at least 79 reports (Fig. 1). Of those 79 studies, 53 use a solvent categorized as highly hazardous according to the CHEM21 solvent guidelines either as the reaction medium or to obtain an NMR spectrum of the reaction mixture or product.24 Eight studies use solvents categorized as hazardous. A movement from these norms is critical for this reaction to be genuinely green.
 | ||
| Fig. 1 Distribution of solvent used for the reaction of diphenylphosphine and styrene. | ||
Green solvent use has gained urgency as pressure mounts to phase out traditional, toxic solvents as evidenced by the recent ban of methylene chloride by the US Environmental Protection Agency.25 The ability to conduct reactions in more benign and renewable solvents has clear potential sustainability and green chemistry advantages, but this ability may come with the potential challenge of undesired reactivity at functional groups seen on many sustainable solvents. Because photocatalytic copper hydrophosphination is not only effective, but also mechanistically divergent,7 it represents an opportunity to vary several factors in a system with understood mechanistic parameters. Furthermore, copper-catalyzed hydrophosphination catalysis has exhibited good functional group tolerance.12,13 Styrene, ethyl acrylate, and 1-hexene were used as model substrates; eight different solvents, selected in accordance with classifications from the CHEM21 solvent selection guide,24 were used for these reactions. The choice for CHEM21 as opposed to other solvent guides was based on the potential bioavailability of solvents in CHEM21,26,27 which may enhance the sustainability of exploratory catalysis. The six categories include alcohols, ketones, esters, ethers, hydrocarbons, and aprotic solvents. Each category has been represented in the solvent selection of this study. A major category that is missing from this study is a halogenated solvent. According to the CHEM21 solvent guide, no halogenated solvent is a sustainable choice for reaction screening. Furthermore, the original study of Cu(acac)2 was conducted in a halogenated solvent and noted here for comparison,7,8 but that solvent does not align with the aim to use greener and less toxic solvents. Overall, this study confirms that green solvents are viable for hydrophosphination. This observation is consistent with observed functional group tolerance of copper catalysts for hydrophosphination.7,8 Most importantly, despite changes in both mechanism and catalyst, these findings demonstrate a pathway to more sustainable exploratory hydrophosphination and related catalysis through both solvent and catalyst choice.
Results and discussion
The reaction of styrene and diphenylphosphine was selected as a benchmark transformation for solvent screening due to its widespread use in hydrophosphination catalysis.4 Styrene was treated with 1 equiv. of diphenylphosphine and 5 mol% of Cu(acac)2 in solvents from six categories of the CHEM21 guide. The reaction mixtures were then irradiated at 360 nm for 5 h (Table 1). When compared with CDCl3, the solvent from the original discovery of photocatalytic copper hydrophosphination,8 most solvents afforded increased conversion to the same product, reducing the waste via reduced unreacted starting material, therefore increasing the reaction efficiency (Table 1, entries 1–5, 7, and 8).|
|
||
|---|---|---|
| Entry | Solvent | Conversion (%) |
| a Reaction conditions: styrene (0.38 mmol), diphenylphosphine (0.38 mmol), Cu(acac)2 (0.019 mmol), solvent (400 μL). b From ref. 8. | ||
| 1 | EtOH | 96 (3.5) |
| 2 | EtOAc | 84 (1.5) |
| 3 | DMSO | 90 (1.0) |
| 4 | Heptane | 91 (2.0) |
| 5 | 2-MeTHF | 86 (2.0) |
| 6 | Cyrene | 28 (1.5) |
| 7 | CPME | 89 (1.5) |
| 8 | MEK | 93 (1.0) |
| 9 | CDCl3 | 85b |
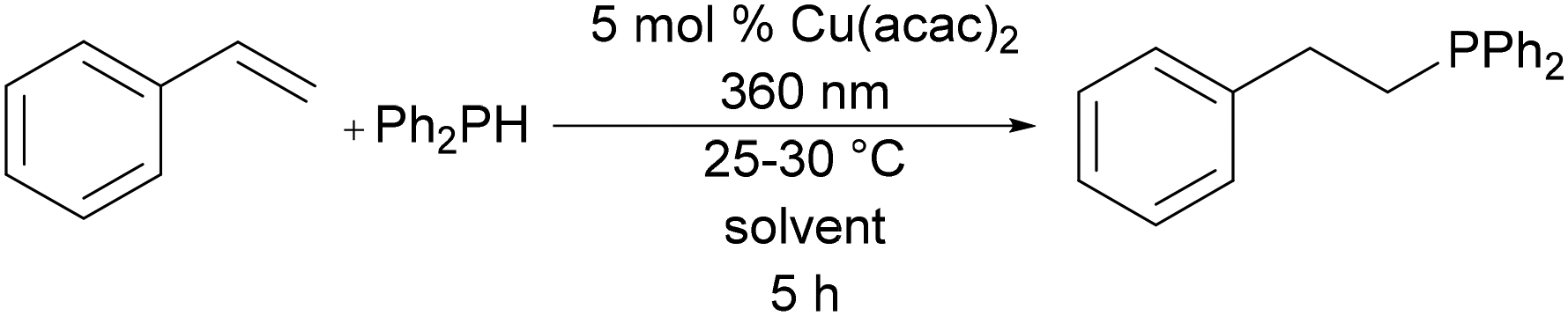
The exception to this trend was Cyrene (dihydrolevoglucosenone), which gave very low conversion (Table 1, entry 6). This is a somewhat surprising result given the prior success of Cyrene as an alternative solvent in other metal-catalyzed reactions.28 Ethyl acetate and 2-MeTHF were statistically lower in conversion than the remaining selected solvents but adequately active for use. The reaction shows no correlation of the dielectric constant and product conversion (Table S3†). In an insertion-based mechanism, it is anticipated that some solvent polarity would enhance rate. For these reactions, the improvement over chloroform indicates that such a trend is accurate but perhaps there is a maximum contribution to this effect that is reached prior to when strongly polar solvents (e.g., EtOH) are employed. Nevertheless, the green solvents are also inert in all cases aside from Cyrene and do not hinder conversion.
A preliminary effort to better understand the limited reactivity with Cyrene provided little insight. In preparing the catalytic reactions, addition of Cyrene to the mixture of reagents resulted in an apparent reaction with diphenylphosphine based on a new resonance in 31P NMR spectra, and no further conversion was observed after irradiation. Control reactions with each reagent and Cyrene revealed that diphenylphosphine was the only reagent to react with the solvent. Attempts to isolate this product were unsuccessful, and only starting materials were returned. Monitoring stoichiometric reactions by NMR spectroscopy showed only partial conversion to this new product based on Ph2PH consumption, and like the catalytic reaction, heating, cooling, or irradiating did not change the distribution of products. Subjecting these mixtures to ESI-MS provided only starting materials, which suggests that the product is unstable under ESI conditions.† Further investigation is underway.
An effort to reduce the catalyst loading of the reaction was also made to test the limits of this reactivity. Under loadings as low as 2.5 mol% and 1 mol% of Cu(acac)2, conversions of 83% and 81%, respectively, were measured (Fig. S33 and S32†). Lower catalyst loadings are viable for copper, which may be a function of functional group tolerance and in situ reduction.7
In determining optimized reaction conditions, controls were run in the absence of any catalyst. Control reactions in prior reports have already established the necessity of copper for reactions in chloroform.7,8 Styrene was treated with 1 equiv. of diphenylphosphine in the absence of Cu(acac)2 in each solvent from the selected set (Table S2†). The reaction mixtures were then irradiated at 360 nm for 5 h. Under these conditions, EtOH and DMSO both achieved close to 60% conversion to products (Table S2, entries 1 and 3†). The rest of the solvents did not reach conversion greater than 40% (Table S2†), demonstrating the catalytic activity of Cu(acac)2. While further exploration of these photo-initiated reactions is on-going, what stood out was 26% conversion to product observed when 2-MeTHF was used as a solvent (Fig. S2, entry 5†). Conversion with 2-MeTHF was anomalously low in comparison to a report in which this solvent has been reported to catalyze this transformation.29 A similar reaction to the literature report was therefore attempted. Styrene was treated with 1 equiv. of diphenylphosphine in the presence of 4 equiv. of 2-MeTHF under an N2 atmosphere on an NMR scale and heated to 90 °C for 2 h. Only 17% conversion was observed under these conditions (Fig. S51†). An effort to replicate the conditions exactly on the scale reported was also made. Styrene was added to 1 equiv. of diphenylphosphine (0.5 g) in 4 equiv. of 2-MeTHF under an atmosphere of Ar followed by stirring at 90 °C for 2 h. Under these conditions, only 20% conversion was measured (Fig. S52†). Some recent reports of catalyst-free reactions have been attributed to metal-containing residue on equipment.30 We have no data to support this, but our inability to replicate 2-MeTHF as a catalyst indicates that additional investigation is warranted.
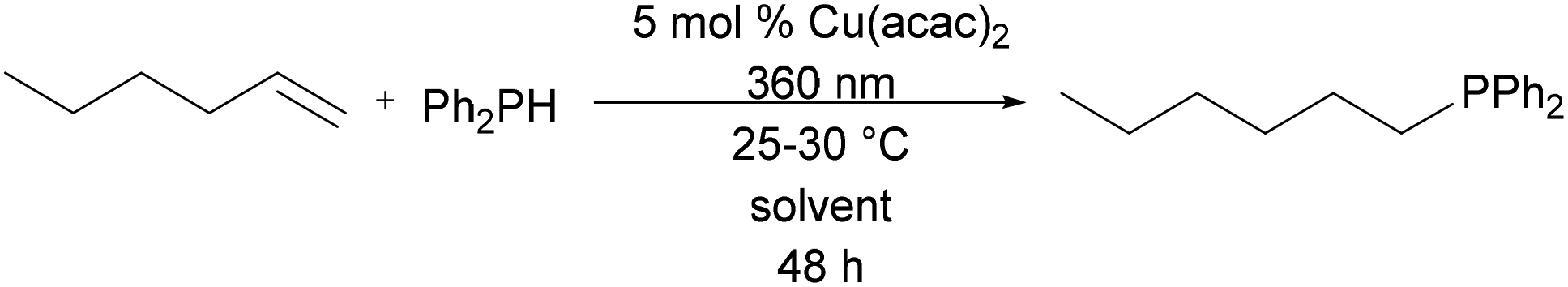
While styrene has become the benchmark substrate for hydrophosphination catalysis, the hydrophosphination of unactivated alkenes remains limited to two catalysts.4–6 To further test the robustness of the green solvents, hydrophosphination of 1-hexene was also screened in the same CHEM21 solvent set. Treatment of 1-hexene with diphenylphosphine in the presence of 5 mol% of Cu(acac)2 and solvent was followed by irradiation at 360 nm for 48 h. While only modest conversion was achieved, EtOAc showed the highest conversion to product (Table 2, entry 2). When compared with identical reaction conditions in CDCl3, reactions in most of the green solvents, EtOH, EtOAc, methyl ethyl ketone (MEK), cyclopentyl methyl ether (CPME), 2-MeTHF, and heptane, exhibited increased reactivity (Table 2, entries 1, 2, 4, 5, 7, and 8). Again, Cyrene provided the lowest reactivity and was subject to the same reaction of phosphine substrate as was observed in the hydrophosphination of styrene (vide supra).
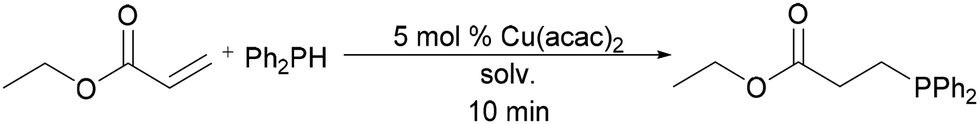
To continue testing the versatility of these findings, a Michael acceptor was selected to compare another class of alkenes commonly used in hydrophosphination catalysis. Copper, like other catalysts, can engage in a conjugate addition reaction with α,β-unsaturated or other adequately withdrawing substrates.7 Therefore, ethyl acrylate was treated with 1 equiv. of diphenylphosphine and 5 mol% of Cu(acac)2 in the same solvents. The reaction mixtures were allowed to react for 10 min at ambient temperature (Table 3). After only 10 min, greater than 95% conversion to product was achieved in all solvents except for MEK and Cyrene (Table 3, entries 1–5, and 7). Reactions in Cyrene and MEK achieved 88% and 89% conversion respectively, slower than the rest of the selected solvents (Table 3, entries 6 and 8). As demonstrated, green solvents are a viable choice for this reaction and are suitable replacements of CDCl3.
|
|
||
|---|---|---|
| Entry | Solvent | Conversion (%) |
| a Reaction conditions: ethyl acrylate (0.38 mmol), diphenylphosphine (0.38 mmol), Cu(acac)2 (0.019 mmol), solvent (400 μL). | ||
| 1 | EtOH | 95 |
| 2 | EtOAc | 99 |
| 3 | DMSO | >99 |
| 4 | Heptane | 95 |
| 5 | 2-MeTHF | 96 |
| 6 | Cyrene | 88 |
| 7 | CPME | >99 |
| 8 | MEK | 89 |
The conversion in Cyrene may appear to be an outlier as compared to the prior substrates, but this reactivity is consistent with the mechanism change between relatively unactivated alkenes like styrene and 1-hexene versus Michael acceptors, as documented for copper(I) precatalysts and outlined in our previous report in 2023.7 The latter reactions are proposed to proceed via attack of unsaturated substrate by a more nucleophilic copper(I)–phosphido (LnCuPPh2) intermediate,7 consistent with original proposals of this type by Glueck.23,31,32 In Cyrene, this nucleophilic behavior must exhibit a greater relative rate than the competitive reaction between diphenylphosphine and solvent to avoid the catalysis halting reaction seen with styrene and 1-hexene. It is remarkable that relatively reactive solvents, such as
those with ketone functionalities and relatively basic protons, are similarly robust to inert solvents (e.g., heptane) under these conditions. This observation may be a function of relative rates of the hydrophosphination reactivity as compared to competitive nucleophilic attack or acid–base chemistry (vide infra). The success of this family of solvents under these conditions is a strong indicator for their wider use in more exploratory reaction chemistry.
The success of ethanol in this catalysis raises an additional consideration. Simple group 1 salts have shown activity in other bond-forming reactions,33 including hydrophosphination.4,14 With limited but intriguing literature reports of potassium compounds achieving unique reactivity in hydrophosphination catalysis,16,34,35 it has been suggested that group 1 activity has been underexplored in hydrophosphination.4 Due to the high solubility of ethoxide salts in ethanol solutions, this study afforded an opportunity to further explore some of these observations and potentially avoid some solubility issues known to plague group 1 alkoxides as reagents through ethanol or similarly solubilizing green solvents.16 Testing these catalysts is additionally attractive for the potential risk that copper-catalyzed hydrophosphination alone may not be a good indicator for solvent effect for this transformation.36
Alkene was treated with 1 equiv. of diphenylphosphine in the presence of 5 mol% of Cu(acac)2 in either EtOH or EtOAc. The reaction mixtures were then irradiated at 360 nm for 5 h or 48 h for styrene or 1-hexene, respectively (Table 4). Greater conversions were measured for both unsaturated substrates in EtOH with either LiOEt and NaOEt as precatalysts in comparison to EtOAc (Table 4, entries 1 and 3). The opposite trend was observed with KOEt where increased conversion was measured in EtOAc solution (Table 4, entry 6). Good conversions were achieved in all cases as compared to Cu(acac)2. While the conversions were higher for Cu(acac)2, these observations suggest that a more economical if not sustainable alternative may be possible with group 1 salts after further study.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | Conversion (%) | |
| (R = Ph)b | (R = C4 H9)c | |||
| a Reaction conditions: alkene (0.38 mmol), diphenyl phosphine (0.38 mmol), ROEt (0.019 mmol), solvent (400 μL). b Conversion after 5 h. c Conversion after 48 h. | ||||
| 1 | LiOEt | EtOH | 64 | 30 |
| 2 | LiOEt | EtOAc | 56 | 16 |
| 3 | NaOEt | EtOH | 73 | 31 |
| 4 | NaOEt | EtOAc | 68 | 23 |
| 5 | KOEt | EtOH | 63 | 20 |
| 6 | KOEt | EtOAc | 80 | 31 |

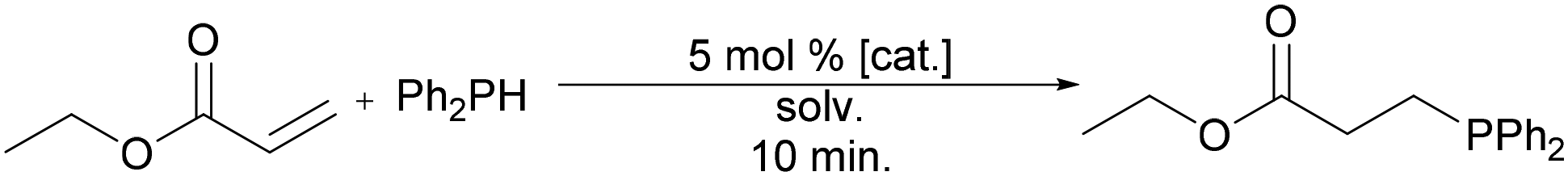
Michael acceptors are anticipated to undergo a nucleophilic addition, and these reactions are presented separately. Ethyl acrylate was treated with 1 equiv. of diphenylphosphine in the presence of 5 mol% of ethoxide catalyst in either EtOH or EtOAc solution (Table 5). As above, conversions were measured in 10 min. Unlike with styrene and 1-hexene, conversion was greater in EtOH for all three precatalysts (Table 5, entries 1, 3, and 5). This observation is perhaps unsurprising due to the higher polarity of ethanol as compared to ethyl acetate as well as the ability of ethanol to facilitate proton transfer that would complete a Michael addition. As with reactions in Table 4, KOEt afforded the greatest conversion to products in both solvents when compared with NaOEt and LiOEt (Table 5, entries 5 and 6). While conversion to products is best with KOEt in each solvent, Cu(acac)2 still demonstrates the greatest conversions under such a limited reaction time for Michael acceptors, to the best of our knowledge. Here again, a synthetic strategy involving KOEt as a precatalyst in ethanol appears to be the most green and efficient route to hydrophosphination products with Michael acceptors, though a full exploration of substrate scope and optimization is warranted.
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Solvent | Conversion (%) |
| a Reaction conditions: ethyl acrylate (0.38 mmol), diphenylphosphine (0.38 mmol), ROEt (0.019 mmol), solvent (400 μL). | |||
| 1 | LiOEt | EtOH | 88 |
| 2 | LiOEt | EtOAc | 67 |
| 3 | NaOEt | EtOH | 86 |
| 4 | NaOEt | EtOAc | 59 |
| 5 | KOEt | EtOH | 93 |
| 6 | KOEt | EtOAc | 86 |
Conclusions
The simple precursor Cu(acac)2 has been used as a precatalyst for the hydrophosphination of three different unsaturated substrates in a range of green solvents that represent broad categories under the CHEM21 solvent selection guide. To further demonstrate the utility of these solvents and expand sustainable options, group 1 ethoxide precatalysts were explored and demonstrate good conversions at lower loadings than prior reports. The CHEM21 guide was chosen for the potential bioavailability of solvents, which may enhance sustainability in exploratory catalysis. Overall, all catalysts provide as good or better conversions than reported conversions in halogenated solvents, an unequivocal statement that more toxic solvents are unnecessary for this reaction. Though only modest conversion was demonstrated with 1-hexene in all cases, conversion in these green solvents still increased when compared to conversions in CDCl3 solution. Finally, results with group 1 salts, particularly potassium, offer strategies for efficient, high-conversion preparations of these products with abundant catalysts, modest reaction conditions, and renewable solvents. Overall, the success of these solvents in meeting or exceeding the reactivity of reported halogenated or aromatic solvents in prior reports with copper or other active catalysts indicates that green solvents like these are viable candidates for exploratory reaction chemistry and augments arguments to transition to these more widely in metal-based catalysis.Experimental
General methods
Air- and moisture-sensitive reactions were carried out under an N2 atmosphere using an M. Braun glovebox or standard Schlenk techniques. Diphenylphosphine was synthesized according to a modified literature procedure.37 All other reagents were acquired from commercial sources and dried by conventional means, as necessary. Solvents were dried over calcium hydride and the distilled and stored over 3 and 4 Å molecular sieves.General procedure for catalytic experiments
In an N2 filled glovebox, 0.38 mmol of Ph2PH and 0.38 mmol of unsaturated substrate were added to a shell vial containing 0.019 mmol of catalyst and internal standard where applicable. Solvent (400 μL) was then added, and the contents were mixed via pipette. The resulting solution was transferred to an NMR tube with an external standard where applicable and covered with a disposable NMR tube cap that was subsequently wrapped with parafilm and wiped with bleach. Initial 31P{1H} NMR, and 1H NMR where applicable, spectra were obtained before placing the tube in a chamber containing a Rexim G23 UV-A (9 W) lamp. 31P{1H} spectra were collected periodically to determine reaction progress. Conversions were determined by integration of 31P{1H} NMR spectra to those of staring materials. An external standard (sealed capillary) of PPh3 was used. In reactions with DMSO-d6, 1,3,5-trimethoxybenzene was used as an internal standard.Author contributions
Study design was by E. J. F and R. W. Experimentation, data acquisition, and data analysis were conducted by E. J. F., while conceptualization and writing were conducted by both E. J. F. and R. W. Funding was secured by R. W. Both authors have read and agreed to the published version of the manuscript.Data availability
Original data for this article will be posted on-line at https://www.uvm.edu/~waterman when published.As part of the ESI,† processed spectra and experimental details have been included.
Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
This work was supported by the U.S. National Science Foundation through CHE-2348198 and CHE-2101766 (to R. W.).References
- C. E. Nedelciu, K. V. Ragnarsdottir, P. Schlyter and I. Stjernquist, Glob. Food Secur.-Agr, 2020, 26, 100426 CrossRef CAS.
- C. Alewell, B. Ringeval, C. Ballabio, D. A. Robinson, P. Panagos and P. Borrelli, Nat. Commun., 2020, 11, 4546–4558 CrossRef CAS.
- M. Kamitani, M. Itazaki, C. Tamiya and H. Nakazawa, J. Am. Chem. Soc., 2012, 134, 11932–11935 CrossRef CAS PubMed.
- B. T. Novas and R. Waterman, ChemCatChem, 2022, 14, e202200988 CrossRef CAS.
- S. Lau, T. M. Hood and R. L. Webster, ACS Catal., 2022, 12, 10939–10949 CrossRef CAS.
- C. A. Bange and R. Waterman, Chem. – Eur. J., 2016, 22, 12598–12605 CrossRef CAS PubMed.
- S. G. Dannenberg, D. M. Seth Jr., E. J. Finfer and R. Waterman, ACS Catal., 2023, 13, 550–562 CrossRef CAS.
- S. G. Dannenberg and R. Waterman, Chem. Commun., 2020, 56, 14219–14222 RSC.
- C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3464–3484 RSC.
- S. D. McCann and S. S. Stahl, Acc. Chem. Res., 2015, 48, 1756–1766 CrossRef CAS PubMed.
- M. B. Reuter, D. M. Seth, D. R. Javier-Jiménez, E. J. Finfer, E. A. Beretta and R. Waterman, Chem. Commun., 2023, 59, 1258–1273 RSC.
- A. Leyva-Pérez, J. A. Vidal-Moya, J. R. Cabrero-Antonino, S. S. Al-Deyab, S. I. Al-Resayes and A. Corma, J. Organomet. Chem., 2011, 696, 362–367 CrossRef.
- N. A. Isley, R. T. H. Linstadt, E. D. Slack and B. H. Lipshutz, Dalton Trans., 2014, 43, 13196–13200 RSC.
- M. T. Whitelaw, S. Banerjee, A. R. Kennedy, A. van Teijlingen, T. Tuttle and R. E. Mulvey, Cell Rep. Phys. Sci., 2022, 3, 100942 CrossRef CAS.
- S. Greenberg, G. L. Gibson and D. W. Stephan, Chem. Commun., 2009, 304–306, 10.1039/B817960C.
- N. T. Coles, M. F. Mahon and R. L. Webster, Chem. Commun., 2018, 54, 10443–10446 RSC.
- G. Märkl and R. Potthast, Angew. Chem., Int. Ed. Engl., 1967, 6, 86–86 CrossRef.
- A. K. King, K. J. Gallagher, M. F. Mahon and R. L. Webster, Chem. – Eur. J., 2017, 23, 9039–9043 CrossRef CAS PubMed.
- M. R. Douglass and T. J. Marks, J. Am. Chem. Soc., 2000, 122, 1824–1825 CrossRef CAS.
- P.-F. Fu, L. Brard, Y. Li and T. J. Marks, J. Am. Chem. Soc., 1995, 117, 7157–7168 CrossRef CAS.
- P. A. T. Hoye, P. G. Pringle, M. B. Smith and K. Worboys, J. Chem. Soc., Dalton Trans., 1993, 269–274 RSC.
- P. G. Pringle and M. B. Smith, J. Chem. Soc., Chem. Commun., 1990, 1701–1702 RSC.
- D. K. Wicht, I. V. Kourkine, B. M. Lew, J. M. Nthenge and D. S. Glueck, J. Am. Chem. Soc., 1997, 119, 5039–5040 CrossRef CAS.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- K. Vasquez, Chem. Eng. News, 2024, 101, 10 Search PubMed.
- N. Winterton, Clean Technol. Environ. Policy, 2021, 23, 2499–2522 CrossRef.
- F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. R. McElroy and J. Sherwood, Sustainable Chem. Processes, 2016, 4, 7 CrossRef.
- K. L. Wilson, A. R. Kennedy, J. Murray, B. Greatrex, C. Jamieson and A. J. B. Watson, Beilstein J. Org. Chem., 2016, 12, 2005–2011 CrossRef CAS PubMed.
- D. Bissessar, J. Egly, T. Achard, P. Steffanut and S. Bellemin-Laponnaz, RSC Adv., 2019, 9, 27250–27256 RSC.
- L. K. Boerner, Chem. Eng. News, 2022, 100, 20–32 CrossRef.
- L. Rosenberg, ACS Catal., 2013, 3, 2845–2855 CrossRef CAS.
- C. Scriban, D. S. Glueck, L. N. Zakharov, W. S. Kassel, A. G. DiPasquale, J. A. Golen and A. L. Rheingold, Organometallics, 2006, 25, 5757–5767 CrossRef CAS.
- M. B. Reuter, D. R. Javier-Jiménez, C. E. Bushey and R. Waterman, Chem. – Eur. J., 2023, 29, e202302618 CrossRef CAS.
- A. L. Rodriguez, T. Bunlaksananusorn and P. Knochel, Org. Lett., 2000, 2, 3285–3287 CrossRef CAS PubMed.
- J. L. Bookham and D. M. Smithies, J. Organomet. Chem., 1999, 577, 305–315 CrossRef CAS.
- J. Sherwood, Beilstein J. Org. Chem., 2020, 16, 1001–1005 CrossRef CAS.
- A. J. Roering, S. E. Leshinski, S. M. Chan, T. Shalumova, S. N. MacMillan, J. M. Tanski and R. Waterman, Organometallics, 2010, 29, 2557–2565 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc05160b |
| This journal is © The Royal Society of Chemistry 2025 |