
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Upcycling hazardous waste into high-performance Ni/η-Al2O3 catalysts for CO2 methanation†
Qaisar Maqboola,
Hamilton Uchenna Aharanwaa,
Michael Stöger-Pollachb and
Günther Rupprechter *a
*a
aInstitute of Materials Chemistry, TU Wien, Getreidemarkt 9/BC, 1060 Vienna, Austria. E-mail: guenther.rupprechter@tuwien.ac.at
bUniversity Service Center for Transmission Electron Microscopy, TU Wien, Stadionallee 2/057-02, 1020 Vienna, Austria
First published on 7th February 2025
Abstract
Transforming hazardous and difficult-to-process waste materials, like spent Ni-MH batteries and aluminium foil, into nanocatalysts (NCts) provides a sustainable solution for resource management and reducing environmental impact. This study demonstrates a novel approach by extracting nickel sulfate (NiSO4·xH2O) from battery waste and subsequently converting it into Ni(OH)2 hydrogel precursors using L-glutamic acid. Waste aluminium foil was processed into alumina (Al2O3), and combined with Ni(OH)2 to synthesize Ni/η-Al2O3 NCts with 4% and 8% Ni loading. Characterization through XRD/SAED, STEM/EFTEM, and EELS revealed a disordered cubic structure of η-Al2O3, with well-dispersed Ni particles, making it effective for CO2 hydrogenation. The 8-Ni/η-Al2O3 exhibited the best catalytic performance, with CH4 selectivity of 99.8% and space time yield (STY) of 80.3 mmolCH4 gcat−1 h−1 at 400 °C. The CO2 methanation mechanism over Ni/η-Al2O3 NCts was further explored using operando DRIFTS aligned with GC + MS. The operando investigation suggested a preferential associative CO2 methanation pathway, involving sequential adsorption and hydrogenation of CO2 to hydrogen carbonates on Ni/η-Al2O3, and their transformation into formate and methoxy intermediates leading to methane. Finally, to complete the upcycling/recycling loop, the spent Ni/η-Al2O3 NCts were recycled into Ni and Al precursors. These findings underscore the potential of upcycling waste materials for synthesizing sustainable, high-performance NCts, and offer insights into the CO2 methanation mechanism.
Green foundation1. Hazardous waste (Ni-MH batteries and aluminum foil) is transformed to nanocatalysts (NCts) for CO2 hydrogenation. To close the loop, the spent catalyst was recycled into catalyst precursors, toward a circular economy and improving resource efficiency.2. The waste-upcycled Ni/η-Al2O3 NCts achieve 99.8% CH4 selectivity and space time yield (STY) of 80.3 mmolCH4 gcat−1 h−1 at 400 °C, enabling efficient CO2 conversion into sustainable synthetic fuel. 3. Future research may focus on optimizing catalyst recycling, reducing the catalyst operation temperature to enhance energy efficiency, using H2 from renewable sources, and exploring upscaling. |
Introduction
Power-to-Methane (PtM) technology, which uses the Sabatier reaction to convert CO2 and H2 into methane (CH4), was first commercialized in the early 2010s, providing means to store excess renewable energy in the existing natural gas infrastructure.1–3 Nowadays, this process utilizes H2 produced from water electrolysis, typically using solid oxide electrolysis cells (SOECs), and CO2 captured from flue gas, biomass, or other carbon-containing resources. PtM technology, when paired with carbon capture, presents a promising solution to mitigate climate change by reducing greenhouse gas emissions.4Nevertheless, the economic feasibility of PtM plants remains a challenge, as the cost of synthesizing CH4 is currently several times higher than conventional natural gas.5 Some of the promising examples, including ‘MeGa-StoRE 2 – Optimising and Upscaling’ project in Denmark and the first PtM plant in Switzerland, which was built and operated at the ‘Institute for Energy Technology of the Hochschule für Technik Rapperswil (HSR-IET)’, both utilize Ni-based commercial catalysts for methanation.6 Still, most existing pilot plants have operated only for short periods, highlighting the need for improved efficiency to enhance economic competitiveness.3 Achieving this requires significant technical breakthroughs, particularly in the development of catalysts with high activity, selectivity, and durability, which are crucial for scaling up energy storage facilities.
Over the years, significant advancements have been made in the preparation of various materials with diverse structures and morphologies, leading to their successful application in catalysis, particularly for CO2 methanation.7,8 Among these, layered double hydroxides (LDHs) stand out as 2D materials with sheet-like structures that can be converted into mixed metal oxides. These oxides have proven to be highly effective catalysts for thermally driven CO2 methanation.9,10 Lima et al. prepared Ni–Al mixed metal oxides from LDHs, reporting the materials’ high CO2 conversion rate and CH4 selectivity, attributed to the high density of basic sites within the catalyst.11 In another study, 2D nickel@siloxene nanosheets presented a CO2 methanation rate of 100 mmol gNi−1 h−1 with over 90% selectivity when nickel dwells specifically between the sheets of siloxane.12 Moreover, Gao et al. encapsulated Ni nanoparticles (NPs) within few-layer hexagonal boron nitride (h-BN) shells for syngas methanation. The confinement effect of the h-BN shells enhances the catalytic activity and stability of the Ni catalyst.13 Similarly, Zr,14 Cu/Zr/CdS,15 Ni/CeO2,16 and Co3O4/1D TiO2 nanowires17 based metal–organic frameworks (MOFs) with complex 2D/3D network structures have been explored for their catalytic efficiency, with notable success in enhancing surface area and electron transport, crucial for facilitating the CO2 to CH4 conversion process. Regardless of these advancements, research on 2D/3D materials for CO2 methanation remains limited.
Despite ongoing promising developments,18–20 a substantial gap remains in research concerning the sustainability of these catalytic processes. Most existing catalysts, particularly those based on Ni, Rh, Pd and Ru supported on materials such as Al2O3,21–24 TiO2,22,25 SiO2/SiC,26,27 ZrO2,28 and CeO2,29,30 rely heavily on synthetic reagents for their preparation. While these catalysts exhibit remarkable selectivity and stability for CH4 production, their reliance on non-renewable resources poses a challenge to the sustainability of the process. To address this issue, it is imperative to explore alternative, sustainable routes for catalyst preparation,31 ensuring that the entire process, from catalyst synthesis to CO2 methanation, aligns with the principles of environmental sustainability, in particular, UN Sustainable Development Goals (SDGs).32 This aspect, however, has been largely overlooked in current research, underscoring the need for a more comprehensive approach to sustainable catalyst production.
The increasing demand for Ni in PtM technologies for CO2 methanation highlights the potential benefits of extracting and recovering Ni from metal waste, such as spent nickel-metal hydride (Ni-MH) batteries.33 This approach not only addresses the challenge of upcycling difficult-to-process battery waste but also provides a valuable source of Ni metal salts, essential for Ni-based catalyst preparation in PtM applications. In Ni-MH batteries, nickel oxyhydroxide (NiOOH) is utilized in both electrodes. The reversible chemical reaction at the cathode mirrors that of nickel-cadmium (NiCd) cells (Ni(OH)2 + OH− ↔ NiO(OH) + H2O + e−). However, the anode employs a hydrogen-absorbing alloy instead of cadmium (H2O + Me + e− ↔ OH− + MeH), where “Me” denotes the intermetallic compounds in the anode of an Ni-MH cell. Through processes such as acid leaching and selective precipitation, it is possible to recover high-purity Ni as NiSO4 from the cathode powder of spent Ni-MH batteries,34,35 making this method a promising avenue for sustainable resource recovery.
Still, Ni alone is insufficient for effective CO2 methanation, as it typically requires a compatible support material, such as metal oxides like Al2O3.36 In this context, aluminium (Al) can be extracted from waste aluminium foil, a common household item. In the current research, Ni was first recovered as NiSO4·xH2O from spent Ni-MH batteries and then transformed into Ni(OH)2 through L-glutamic acid and NaOH reduction. Concurrently, Al was extracted from waste aluminium foil and converted into Al2O3 via NaOH treatment of AlCl3 to Al(OH)3. The resulting Ni(OH)2 was homogenized with Al2O3 to produce Ni/η-Al2O3 nanocatalysts (NCts). The physicochemical properties of NCts were thoroughly analyzed using advanced techniques such as high-resolution transmission electron microscopy (HR-TEM), energy-filtered transmission electron microscopy/scanning transmission electron microscopy coupled with electron energy loss spectrometry (EFTEM/STEM-EELS), selected area electron diffraction (SAED), temperature-programmed reduction (TPR), X-ray diffraction (XRD), and Brunauer–Emmett–Teller (BET) analysis. CO2 methanation was investigated at 1 bar in a continuous flow fixed-bed reactor, with kinetic measurements conducted at temperatures ranging from 250 °C to 550 °C. Additionally, mechanistic studies through operando DRIFTS aligned with GC + MS analyses provided insights into the surface activity of the Ni/η-Al2O3 NCts during CO2 methanation. Lastly, to close the upcycling loop by recycling, the spent Ni/η-Al2O3 NCts were successfully transformed back into Ni and Al precursors.
Results and discussion
Battery/Al-foil waste upcycling into nanocatalysts
Upcycling hazardous and difficult-to-process waste into valuable catalysts is a promising approach for sustainable resource management and reducing the environmental impact of waste disposal.37 This process also highlights the potential for recovering valuable metals, such as Ni and Al, from end-of-life products. In this context, the upcycling of spent battery and aluminium foil waste into nanocatalysts (NCts) is illustrated in Fig. 1a. Nickel sulfate hydrate (NiSO4·xH2O) was successfully extracted from spent Ni-MH batteries, primarily from the cathode powder, through a chemical process. The extraction and crystallization process from the cathode produced NiSO4 with high phase purity of ≈100%, as determined by XRF spectroscopy (Fig. S1†). This result aligns with the phase purity achieved in previously reported Ni-extraction methods.34,35 While NiSO4 can also be recovered from a mixture of cathode and anode materials, the resulting phase purity is lower (≈84%) due to contamination mainly from Co, Fe, La and Zn (Fig. S1†). The batteries were first disassembled, and the inner components were washed with diH2O to remove the KOH electrolyte, then dissolved in dilH2SO4. The nickel hydroxide Ni(OH)2 and nickel oxyhydroxide (NiOOH) (black powder) from cathode reacts with diH2SO4 to produce nickel sulfate-hydrate (NiSO4·xH2O), which was then filtered, heat-dried (saturated), and crystalized to obtain NiSO4·6H2O (used in this study). Additionally, the nickel-based alloy (LaNi5) from anode powder reacts with diH2SO4 to produce nickel sulfate-hydrate (NiSO4·xH2O), lanthanum sulfate (La2(SO4)3), and H2 gas. The La2(SO4)3 precipitates as a slag, while the blue-green coloured NiSO4·7H2O remains in solution, which was then filtered, heat-dried (saturated), and crystalized to obtain NiSO4·7H2O (not used in this study). The recovered La2(SO4)3 precipitates were also stored for future use.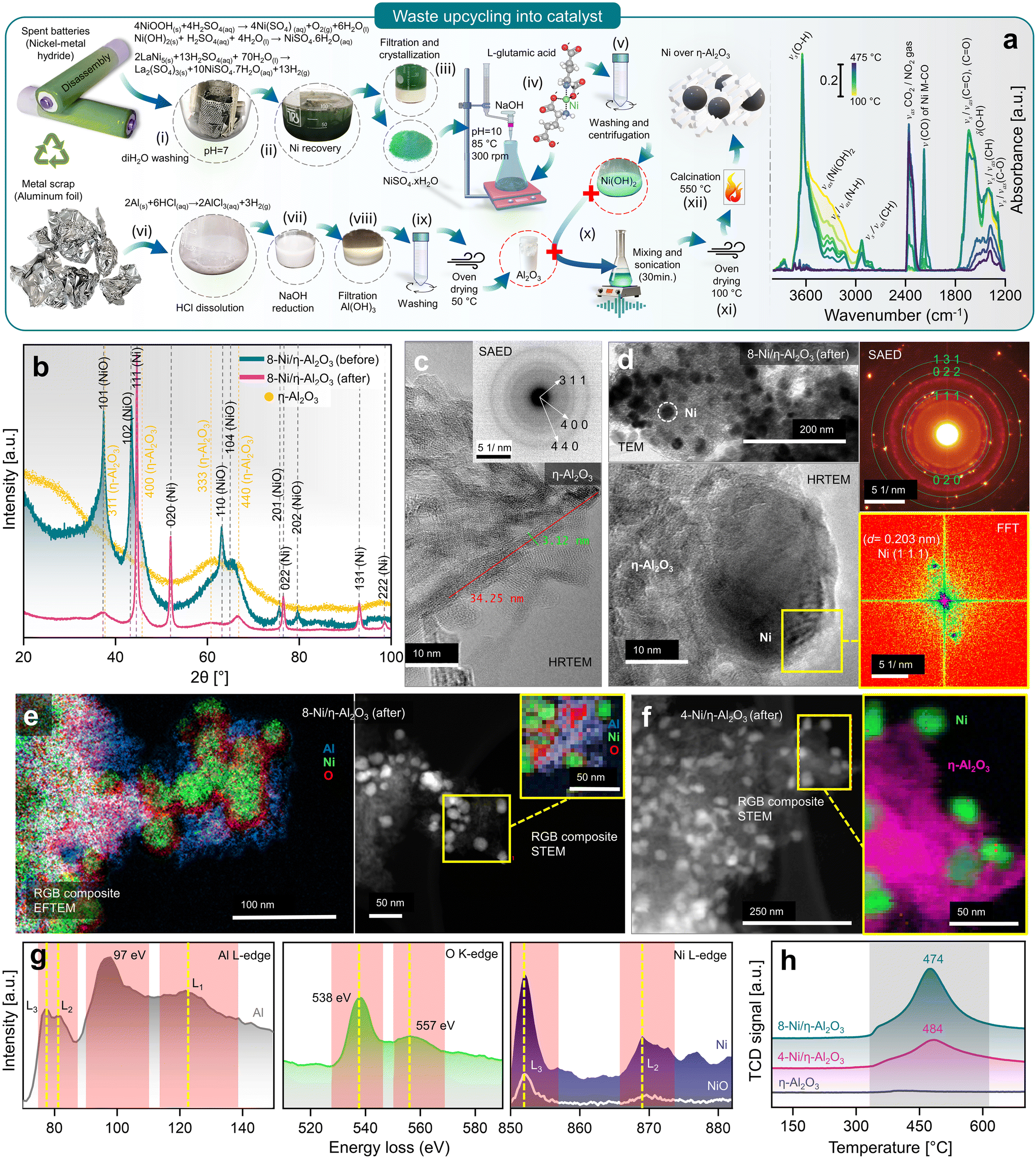 | ||
| Fig. 1 (a) Battery/aluminium waste upcycling and characterization of prepared nanocatalyst, with schematics elaborating the recovery of Ni as NiSO4·xH2O from spent Ni-MH batteries, Al2O3 from waste aluminium-foil, and recombination of Ni(OH)2 hydrogel complex and Al2O3 to obtain 4- (wt%) and 8-Ni/η-Al2O3 NCts (left), and in situ DRIFTS spectra during calcination showing thermal breakdown of Ni(OH)2 hydrogel complex at elevated temperature (100–475 °C) (right). (b) Crystal structure of η-Al2O3 and 8-Ni/η-Al2O3 NCts before and after CO2 methanation reaction (for 4-Ni/η-Al2O3 NCts, see Fig. S2†). (c) HRTEM of η-Al2O3 support. Inset: corresponding SAED pattern confirming the hkl (311), (400), and (440) of η-Al2O3. (d) TEM of 8-Ni/η-Al2O3 NCts after CO2 methanation showing homogenous Ni distribution over the η-Al2O3 support (top left), corresponding SAED pattern confirming the face-centred cubic crystal structure of Ni (top left), HRTEM of Ni lattice planes (bottom left) with FFT of the selected Ni hkl (111) (bottom right). (e) Color-coded elemental (Al, Ni, and O) mapping by EFTEM (left), and by STEM (right) of 8-Ni/η-Al2O3 NCts after CO2 methanation. (f) Color-coded elemental (pink = η-Al2O3, and green = Ni) mapping by STEM of 4-Ni/η-Al2O3 NCts after CO2 methanation. (g) EELS spectra corresponding to Al L3,2-edges, O K-edge, and Ni L3,2-edges. (h) H2-TPR spectra of 4, 8-Ni/η-Al2O3 NCts, and η-Al2O3 support. | ||
Next, a 0.5 M solution of NiSO4·7H2O obtained from cathode upcycling was dissolved in diH2O, heated to 85 °C, and 0.25 M of L-glutamic acid was added. L-Glutamic acid has two carboxyl groups (–COOH) and one amino group (–NH2) with an isoelectric point of 3.2 (pKa = 2.1, 4.3, and 9.7), which can act as a metal ion chelating agent to bind Ni ions.38 Furthermore, when 5 M NaOH is added, the pH of the mixture rises to 10, causing L-glutamic acid to exist as a doubly-negative anion, −OOC–CH(NH2)–(CH2)2–COO−.39 In this form, the carboxyl groups can donate electron pairs to Ni2+ ions, forming stable chelate complexes.40 This process results in the formation of Ni(OH)2, a hydrogel. The obtained L-glutamic acid-derived Ni(OH)2 hydrogel complex can easily attach to the substrate (e.g., Al2O3 support) and can be used as a precursor for the preparation of various Ni-based NCts.
The in situ calcination of oven-dried (100 °C) Ni(OH)2 hydrogel complex was analyzed using DRIFT spectroscopy (Fig. 1a, right). The IR spectra revealed a dominant band at 3644 cm−1, with a broad shoulder between 3370–3550 cm−1, corresponding primarily to non-hydrogen bound and hydrogen bound OH groups of Ni(OH)2.41 Bands associated with N–H and C–H stretching vibrations of the doubly-negative anion were observed in the range of 2820–3346 cm−1.42 A sharp band at 2177 cm−1 was attributed to Ni bound to carbon and oxygen (metal carbonyls, M-CO).43–45 The region between 1200–1800 cm−1 was mainly indicative of C–O, CH, O–H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, and CO groups within the Ni(OH)2 hydrogel complex.42 During the temperature increase, decomposition between 100–300 °C proceeded slowly. However, temperatures above 300 °C significantly accelerated the release of CO2 (2360 cm−1) and H2O, primarily due to the breakdown of hydroxyl and carbon species. Although nitrogen from the Ni(OH)2 hydrogel complex might have been released as NO2, it was not detected in the CO2 background. The in situ calcination confirms that the transformation of L-glutamic acid-derived Ni(OH)2 hydrogel complex to NiO2 mainly occurs at T > 300 °C.
C, and CO groups within the Ni(OH)2 hydrogel complex.42 During the temperature increase, decomposition between 100–300 °C proceeded slowly. However, temperatures above 300 °C significantly accelerated the release of CO2 (2360 cm−1) and H2O, primarily due to the breakdown of hydroxyl and carbon species. Although nitrogen from the Ni(OH)2 hydrogel complex might have been released as NO2, it was not detected in the CO2 background. The in situ calcination confirms that the transformation of L-glutamic acid-derived Ni(OH)2 hydrogel complex to NiO2 mainly occurs at T > 300 °C.
Furthermore, Al2O3 was obtained from waste aluminium foil, which is typically recycled to produce materials of the same class. The utilization of low-cost metal waste to produce high-value, functionalized materials (e.g., NCts) has not yet been fully recognized. Through a two-step process, the aluminium foil was first dissolved in HCl, and the AlCl3 solution was then reduced with 5 M NaOH to obtain Al(OH)3 pellets. The Al(OH)3 were dried at 50 °C to obtain Al2O3. NCts containing 4 and 8 (wt%) Ni were synthesized by combining Ni(OH)2 and Al2O3. The two components were sonicated, then oven-dried to remove excess water. The dried pellets were calcined at 550 °C to obtain the NCts (for more details see the Methods).
Characterization of NCts
The XRD analysis of the as-synthesized NCts, before and after CO2 hydrogenation reaction (Fig. 1b and Fig. S2a†) provides valuable insights into their structural properties. The as-synthesized Al2O3 (Fig. 1b) revealed broad diffraction peaks primarily corresponding to hkl of (311), (400), (333), and (440). These lattice planes are characteristic of the disordered and cubic spinel crystal structure of η-Al2O3 with the space group Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m.46 The broadness of these reflections suggests a small crystallite size of η-Al2O3. When NiO was introduced into the η-Al2O3 framework to form the 4 (wt%) and 8-Ni/η-Al2O3 NCts, the XRD patterns (Fig. 1b and Fig. S2†) showed broad hkl at (101), (102), (110), (104), (201), and (202), which are indicative of the hexagonal crystal structure of NiO with the space group Rm.47,48 However, after CO2 hydrogenation reaction, the NiO underwent a structural transformation, as evidenced by the appearance of sharp reflections at (111), (020), (022), (131), and (222). These Bragg peaks correspond to the face-centred cubic crystal structure of Ni with the space group Fmm.49,50 Notably, the crystal structure of η-Al2O3 remained unaffected both before and after thermal (≈550 °C) CO2 hydrogenation reaction, suggesting η-Al2O3 resistance to phase transformation.51 Moreover, by analysing the two most intense Ni peaks at 2θ = 44.65° and 52.1° (Fig. S2b†) and applying the Debye–Scherrer equation,52 we calculated the average crystallite (grain) sizes. The results show an average crystallite size of 27.4 nm for 4-Ni/η-Al2O3 and 28.9 nm for 8-Ni/η-Al2O3. These values indicate that there is no significant difference in Ni crystallite size between the two NCts.
m.46 The broadness of these reflections suggests a small crystallite size of η-Al2O3. When NiO was introduced into the η-Al2O3 framework to form the 4 (wt%) and 8-Ni/η-Al2O3 NCts, the XRD patterns (Fig. 1b and Fig. S2†) showed broad hkl at (101), (102), (110), (104), (201), and (202), which are indicative of the hexagonal crystal structure of NiO with the space group Rm.47,48 However, after CO2 hydrogenation reaction, the NiO underwent a structural transformation, as evidenced by the appearance of sharp reflections at (111), (020), (022), (131), and (222). These Bragg peaks correspond to the face-centred cubic crystal structure of Ni with the space group Fmm.49,50 Notably, the crystal structure of η-Al2O3 remained unaffected both before and after thermal (≈550 °C) CO2 hydrogenation reaction, suggesting η-Al2O3 resistance to phase transformation.51 Moreover, by analysing the two most intense Ni peaks at 2θ = 44.65° and 52.1° (Fig. S2b†) and applying the Debye–Scherrer equation,52 we calculated the average crystallite (grain) sizes. The results show an average crystallite size of 27.4 nm for 4-Ni/η-Al2O3 and 28.9 nm for 8-Ni/η-Al2O3. These values indicate that there is no significant difference in Ni crystallite size between the two NCts.
The morphology and crystal structure were further confirmed by HRTEM and selected area electron diffraction (SAED).53 The η-Al2O3 appears (Fig. 1c and Fig. S3†) as needle-like structures of l = ≈34 nm and ∅ = ≈3 nm. The SAED (inset of Fig. 1c) showed three distinct diffraction rings. The measured d-spacings (0.251, 0.196, and 0.140 nm) and the rotational average plot of the SAED (Fig. S4†) confirm the (311), (400), and (440) planes of the disordered η-Al2O3 cubic spinel crystal structure. The cubic spinel structure, commonly found in phases of Al2O3, belongs to the space group Fd3m. In a typical spinel composition, denoted as AB2O4, ‘A’ and ‘B’ represent different atomic species, such as Mg and Al in the spinel mineral (MgAl2O4).54 In a normal spinel structure, oxygen (O) atoms form a face-centred cubic (f.c.c.) sublattice, while the ‘A’ and ‘B’ atoms occupy specific interstices within this sublattice. Specifically, the O atoms occupy the (e) sites of the Fd3m space group, with ‘A’ and ‘B’ atoms residing in the tetrahedrally coordinated (a) sites and the octahedrally coordinated (d) sites, respectively.55,56 In one unit cell of spinel, there are 8 formula units of AB2O4, containing 8 A atoms, 16 B atoms, and 32 O atoms. In the case of cubic Al2O3, which has the spinel structure, there are 21 Al atoms per unit cell to fill the 8(a) and 16(d) sites. This leaves 2 vacancies, which can be distributed across the 8(a) and 16(d) interstices in various ways, introducing a degree of disorder into the structure. In γ-Al2O3, all 16 octahedrally coordinated (d) interstices of the O sublattice are fully occupied, similar to a normal spinel structure, with the remaining 5 Al ions distributed among the 8 tetrahedrally coordinated (a) interstices.57 In disordered η-Al2O3, Al atoms not only occupy the (a), (d), (f), and (c) sites typical of the normal spinel structure but also a small fraction of the 48 tetrahedrally coordinated (g) sites and octahedrally coordinated (c) sites within the Fd3m space group.58 However, the precise arrangement of Al atoms within the disordered η-Al2O3 unit cell is still an ongoing discussion.59
These metastable Al2O3 phases share several common characteristics, such as (1) they are cation-deficient spinel analogues, characterized by the distribution of Al atoms in octahedral and tetrahedral sites, with phase transitions between these forms being pseudomorphic despite marked lattice distortions in θ and κ phases,60 (2) the cubic cell, or its equivalent, has a consistent volume of approximately 7.93 Å3 across different varieties, (3) disorder occurs at various scales: in the γ, η, and θ forms, the Al sites are partially occupied without observable ordering; in the δ form, the crystal cell is fully ordered but owns a complex intergrowth from two main crystallographic variants,61,62 and (4) the crystallite size does not exceed a few tens of nanometers, which is a critical feature for applications of metastable aluminas, such as catalytic supports and absorbents, due to their high specific surface area.63 Therefore, we expect η-Al2O3 to be a perfect support for Ni in CO2 methanation, which will be discussed below.
Fig. 1d (top left) and Fig. S5, S6† demonstrate the uniform distribution of pristine NiO and Ni within the η-Al2O3 matrix, both before and after thermal CO2 hydrogenation. The Ni particles (4 and 8-Ni/η-Al2O3), as shown in Fig. S6,† were observed to be 18 to 39 nm in size and were evenly dispersed throughout the η-Al2O3 matrix, with no evidence of agglomeration. The SAED pattern (Fig. 1d, top right) revealed diffraction spots corresponding to the cubic crystal structure of Ni. Additionally, the Fast Fourier Transform (FFT) analysis (Fig. 1d, bottom right) of the HRTEM image (Fig. 1d, bottom left) confirmed the lattice spacing (d = 0.203 nm) of Ni (111). Post-thermal CO2 hydrogenation, the 4 and 8-Ni/η-Al2O3 NCts were further analyzed using EFTEM, STEM, and EELS. Elemental mapping through EFTEM (Fig. 1e, left), combined with EELS, confirmed the distribution of metallic Ni, the presence of O traces at the Ni/η-Al2O3 interfaces, and O enrichment within the η-Al2O3 lattice. Notably, the RGB composite STEM image (Fig. 1e, right) also highlighted O traces at the Ni/η-Al2O3 interfaces, suggesting potential anchoring of Ni onto the η-Al2O3 lattice. Similarly, the RGB composite STEM image of the 4-Ni/η-Al2O3 NCts (Fig. 1f) post-thermal CO2 hydrogenation also indicated the distribution of metallic Ni across the η-Al2O3 surface.
The disordered cubic crystal structure of η-Al2O3 has a lower symmetry than other Al phases (e.g. α-Al2O3)64 which can lead to different electronic band structures and, therefore, electron energy loss spectroscopy (EELS) analysis was necessary to perform, as shown in Fig. 1g. EELS of Al reflect the complex electronic structure of η-Al2O3, influenced by the mixed coordination of Al atoms and the degree of disorder in the material. The peaks at 77 and 81 eV, with 4.7 eV and 5.8 eV full width at half maximum (FWHM), respectively, correspond to the transitions from the 2p3/2 (Al–L3) and 2p1/2 (Al–L2) core levels to the unoccupied states above the Fermi level.65 The less intense and broad nature of these peaks indicates that the transitions are not sharply defined, indicating a degree of disorder in the local environment of the Al atoms which can be attributed to the mixed coordination states (tetrahedral and octahedral) of Al in η-Al2O3 lattice.66 The most intense peak at 97 eV is indicative of a strong transition to a specific unoccupied state while the broad and least intense peak at 122 eV corresponds to the Al–L1 edge. This feature represents higher energy transitions, possibly into more delocalized states further up in the conduction band. The broadness of this peak indicates that the final states available for these transitions are more spread out in energy (eV), reflecting the complex and somewhat disordered structure of η-Al2O3. This broad peak might also be influenced by multiple scattering events or by transitions involving more complex electronic states that are less localized.65 Additionally, the ratio of the most intense peak at 97 eV to the other peaks is quite different from that observed in other polymorphs of Al2O3,67–71 highlighting the unique electronic environment and structural characteristics of η-Al2O3.
O–K edge of η-Al2O3 (Fig. 1g) shows a more intense peak at 538 eV, corresponding to the transition of electrons from the O 1s core level to the unoccupied states in the conduction band, primarily the O 2p states that are hybridized with the Al 3s and 3p states.72–74 The relatively sharp and intense nature of this peak indicates a well-defined electronic transition, suggesting a strong hybridization between the O and Al atoms. The second peak, which is broader and less intense, centred at ≈557 eV, corresponds to transitions into more delocalized or higher energy states in the conduction band which possibly involve a combination of hybridized orbitals, including higher energy Al orbitals and possibly more delocalized states within the O network.75,76 The lower intensity compared to the first peak implies that there are fewer available states with O 2p character at this higher energy level.
The Ni–L edge in EELS (Fig. 1g) for metallic Ni exhibits three distinct peaks: a sharp and highly intense peak at 852 eV corresponding to the Ni–L3 edge, a broader and less intense peak at 869 eV corresponding to the Ni–L2 edge, and a third broad and least intense peak at 876 eV.77 These features arise from electronic transitions of electrons from the Ni 2p core levels (2p3/2 and 2p1/2) to unoccupied d-states. The Ni–L3 edge is more intense and sharper due to the higher density of available unoccupied d-states and stronger transition probabilities associated with the 2p3/2 to d transitions.78,79 The Ni–L2 edge, being broader and less intense, results from 2p1/2 to d-transitions,80 which have different selection rules and lower transition probabilities. The third peak at 876 eV may be attributed to higher energy transitions or multiple scattering effects that broaden and diminish its intensity. In contrast, NiO displays a less distinct separation between the first two peaks and overall lower intensities across all three peaks. This difference is due to the distinct electronic structure of NiO, where Ni exists in a +2-oxidation state, leading to filled lower Hubbard bands and empty upper Hubbard bands influenced by strong electron correlations and charge transfer from O.81,82 The increased hybridization between Ni and O in NiO results in broader and less pronounced peaks, as the transitions involve more complex interactions and a redistribution of spectral weight. Additionally, the presence of charge-transfer satellites in NiO can further obscure the distinction between the Ni–L3 and Ni–L2 edges,83,84 reducing the peak intensity and clarity compared to metallic Ni. Therefore, the EELS features reflect the fundamental differences in the electronic environments and bonding characteristics of metallic nickel and NiO. However, EELS data for 4-Ni/η-Al2O3 and 8-Ni/η-Al2O3 after CO2 methanation, including Ni L-edge, differ only in intensity due to Ni loading (Fig. S7†), confirming that no significant structural or electronic differences beyond Ni content.
The Temperature Programmed Reduction (TPR) profiles of 8 wt% and 4 wt% Ni over η-Al2O3 reveal distinct peaks (Fig. 1h), with the 8% Ni sample exhibiting a broad and intense peak at 474 °C, while the 4% Ni sample shows a similar, though less intense, peak slightly shifted to 484 °C. The TPR peaks represents the thermal energy required to reduce Ni2+ in the NiO lattice to Ni0, with the hexagonal NiO phase (space group Rm) transitioning to the more stable cubic Ni phase (space group Fmm) upon reduction.85,86 The differences in peak intensity and position reflect variations in the NiO particle size, distribution, and interaction with the η-Al2O3 support, all of which influence the reduction kinetics and the overall reduction process.87 The slight temperature shift can be attributed to the concentration of NiO and its interaction with the η-Al2O3 support. Higher NiO loading (8%) results in a more extensive and interconnected NiO phase, leading to a more pronounced reduction peak due to the larger quantity of NiO being reduced. The slightly lower reduction temperature (474 °C) for 8% NiO suggests that the NiO having higher coverage of the support surface may facilitate easier reduction, likely due to the stronger interactions between NiO particles, which promote faster electron transfer and reduce the energy barrier for the reduction process.88 The shift to a slightly higher reduction temperature (484 °C) in the 4% Ni sample suggests that the more dispersed NiO particles on the η-Al2O3 surface require slightly more energy to reduce. This may be due to the higher surface energy and the stronger metal-support interaction (MSI),89 making NiO particles more resistant to reduction.
The BET analysis, as shown in Fig. S8,† provides the specific surface area of the NCts, which is crucial for understanding their catalytic properties and overall performance. The pure η-Al2O3 support exhibits a surface area (SBET) of 53.41 m2 g−1, which increases significantly upon NiO loading, reaching 159.02 m2 g−1 for 4-Ni/η-Al2O3 and 226.82 m2 g−1 for 8-Ni/η-Al2O3, indicating that higher NiO loadings increase SBET. BJH analysis reveals that pore sizes decrease from 9.68 nm in η-Al2O3 to 6.85 nm and 6.42 nm for 4-Ni/η-Al2O3 and 8-Ni/η-Al2O3, respectively, suggesting partial pore blocking by NiO. However, the overall increase in SBET highlights that the introduction of NiO compensates for this reduction. The significant increase in SBET upon Ni loading (from 159.02 m2 g−1 for 4 wt% Ni to 226.82 m2 g−1 for 8 wt% Ni) cannot be explained solely by the NiO particles. As calculated (ESI Note 1†), the specific surface area contribution of NiO particles is not significant, even for the smallest particle sizes observed (2.5 nm, Fig. S5†). Instead, the enhancement in SBET is likely due to modifications of the η-Al2O3 support induced by the Ni loading process. These modifications may involve structural rearrangements, increased surface roughness, or interfacial effects, which enhance the overall surface area of the NCts. Further detailed investigations are required to confirm this. The type H3 isotherm hysteresis recorded during the BET analysis (Fig. S8†) further elucidates the physisorption behaviour of both the Ni/η-Al2O3 NCts and the η-Al2O3 support. The hysteresis loop, which comprises of an adsorption isotherm (type II) and a desorption isotherm,90 indicates the interaction of N2 with the NCts surfaces. Initially, the adsorption isotherm shows N2 forming a monolayer on the sample, leading to a strong increase in adsorption due to direct interaction with the porous surface. As the relative pressure (p/p0) rises, the isotherm bends, indicating a slower rate of adsorption attributed to the formation of a multilayer, which interacts less strongly with the NCts surface.91 At p/p0 ≈ 1, the multilayer reaches its critical film thickness, causing N2 molecules to interact and fill the NCts pores, a phenomenon known as capillary condensation.92 Upon reducing the relative pressure, N2 desorbs from the NCts pores through evaporation, a process distinct from capillary condensation which involves the condensation of vapor in small pores due to capillary forces, as evidenced by the hysteresis loop in the isotherm (Fig. S8†), further suggesting the mesoporous nature of the NCts.
CO2 methanation performance of nanocatalysts
Fig. 2a illustrates the setup used to measure the catalytic performance of the Ni/η-Al2O3 NCts at a gas hourly space velocity (GHSV) of 2984.16 h−1 and a CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2 ratio of 1:4. All NCts displayed a consistent increase in CO2 conversion with rising temperatures from 250 °C to 550 °C, achieving an overall CO2 conversion of 77% for both 4 and 8-Ni/η-Al2O3 (Fig. 2b). Notably, the NCts with higher Ni loading (8-Ni/η-Al2O3) significantly enhanced CH4 selectivity, reaching a maximum of 99.8% (Fig. 2b) and achieving CH4 yields of 54% and 59% at 350 °C and 400 °C, respectively (Fig. 2c). However, CH4 selectivity dropped to 49% at 450 °C and further declined to 8% at 550 °C (Fig. 2b), likely due to the thermodynamic favourability of the Reverse Water Gas Shift (RWGS) reaction (CO2 + H2 → CO + H2O, ΔH = +9 kcal mol−1),93 which produced nearly 70% CO as the side product at these higher temperatures (Fig. 2c). Interestingly, at a lower temperature of 300 °C, the CH4 yield of 4-Ni/η-Al2O3 was 2.5%, whereas it was significantly higher at 30% for 8-Ni/η-Al2O3 (Fig. 2c), underscoring the critical role of available active Ni sites over the η-Al2O3 in promoting CO2 hydrogenation toward the desired methanation pathway. Catalysts under CO2 methanation can deactivate over time or upon strong temperature changes,94 possibly due to coke formation,95 or sintering.96 Therefore, the stability of the best-performing NCts, 8-Ni/η-Al2O3, was rigorously tested, as shown in Fig. 2d. The NCts was subjected to significant temperature fluctuations (250 °C → 400 °C → 250 °C → 350 °C → 250 °C → 350 °C), held at 350 °C for 300 minutes, and then maintained at 400 °C for 600 minutes. Despite these challenging conditions, 8-Ni/η-Al2O3 demonstrated remarkable stability (Fig. 2d), with no signs of deactivation or decline in CH4 productivity, confirming its resilience under both abrupt and prolonged thermal treatments. Additionally, calculating the Space–Time Yield (STY) in CO2 methanation is crucial for assessing the efficiency and productivity of the NCts, which is vital for process optimization and potential scale-up. As expected, the 8-Ni/η-Al2O3 exhibited the highest STY, recording 79.3 mmolCH4 gcat−1 h−1 at 350 °C and 80.3 mmolCH4 gcat−1 h−1 at 400 °C, as shown in Fig. 2e. In contrast, the STY for 4-Ni/η-Al2O3 was significantly lower, with values of 11.3 mmolCH4 gcat−1 h−1 at 350 °C and 21.8 mmolCH4 gcat−1 h−1 at 400 °C. Even at the lower temperature of 250 °C, 8-Ni/η-Al2O3 achieved a STY of 19.4 mmolCH4 gcat−1 h−1. The CO2 methanation performance of 8-Ni/η-Al2O3 was also compared with literature data97–104 (Fig. 2f and Table S1†), where it was found to be highly competitive. The promising CO2 methanation performance of Ni/η-Al2O3 can be attributed to several key factors. The disordered η-Al2O3 support plays a crucial role by enhancing the activity of Ni for CO2 methanation. This disordered structure likely facilitates better adsorption of CO2, creating more favorable sites where CO2 molecules/intermediates can interact stronger with the active Ni sites. This can also facilitate electron transfer processes, which are crucial for the activation of CO2 molecules during the methanation reaction, stabilizing the Ni particles, preventing them from agglomerating and thus maintaining a high dispersion of active sites (Fig. 1d–f). Additionally, the well-dispersed and homogenous size of Ni particles, coupled with their high surface area (Fig. S5†), ensures a sufficient number of active sites are available for the H2 binding. Another important factor is the low reducibility of Ni (<500 °C) over the η-Al2O3 support, which aids in the activation of Ni at relatively lower temperatures. To support these rationalized reasons for the superior CO2 hydrogenation performance of Ni/η-Al2O3, we have conducted operando DRIFTS studies,105 as detailed below.
:H2 ratio of 1:4. All NCts displayed a consistent increase in CO2 conversion with rising temperatures from 250 °C to 550 °C, achieving an overall CO2 conversion of 77% for both 4 and 8-Ni/η-Al2O3 (Fig. 2b). Notably, the NCts with higher Ni loading (8-Ni/η-Al2O3) significantly enhanced CH4 selectivity, reaching a maximum of 99.8% (Fig. 2b) and achieving CH4 yields of 54% and 59% at 350 °C and 400 °C, respectively (Fig. 2c). However, CH4 selectivity dropped to 49% at 450 °C and further declined to 8% at 550 °C (Fig. 2b), likely due to the thermodynamic favourability of the Reverse Water Gas Shift (RWGS) reaction (CO2 + H2 → CO + H2O, ΔH = +9 kcal mol−1),93 which produced nearly 70% CO as the side product at these higher temperatures (Fig. 2c). Interestingly, at a lower temperature of 300 °C, the CH4 yield of 4-Ni/η-Al2O3 was 2.5%, whereas it was significantly higher at 30% for 8-Ni/η-Al2O3 (Fig. 2c), underscoring the critical role of available active Ni sites over the η-Al2O3 in promoting CO2 hydrogenation toward the desired methanation pathway. Catalysts under CO2 methanation can deactivate over time or upon strong temperature changes,94 possibly due to coke formation,95 or sintering.96 Therefore, the stability of the best-performing NCts, 8-Ni/η-Al2O3, was rigorously tested, as shown in Fig. 2d. The NCts was subjected to significant temperature fluctuations (250 °C → 400 °C → 250 °C → 350 °C → 250 °C → 350 °C), held at 350 °C for 300 minutes, and then maintained at 400 °C for 600 minutes. Despite these challenging conditions, 8-Ni/η-Al2O3 demonstrated remarkable stability (Fig. 2d), with no signs of deactivation or decline in CH4 productivity, confirming its resilience under both abrupt and prolonged thermal treatments. Additionally, calculating the Space–Time Yield (STY) in CO2 methanation is crucial for assessing the efficiency and productivity of the NCts, which is vital for process optimization and potential scale-up. As expected, the 8-Ni/η-Al2O3 exhibited the highest STY, recording 79.3 mmolCH4 gcat−1 h−1 at 350 °C and 80.3 mmolCH4 gcat−1 h−1 at 400 °C, as shown in Fig. 2e. In contrast, the STY for 4-Ni/η-Al2O3 was significantly lower, with values of 11.3 mmolCH4 gcat−1 h−1 at 350 °C and 21.8 mmolCH4 gcat−1 h−1 at 400 °C. Even at the lower temperature of 250 °C, 8-Ni/η-Al2O3 achieved a STY of 19.4 mmolCH4 gcat−1 h−1. The CO2 methanation performance of 8-Ni/η-Al2O3 was also compared with literature data97–104 (Fig. 2f and Table S1†), where it was found to be highly competitive. The promising CO2 methanation performance of Ni/η-Al2O3 can be attributed to several key factors. The disordered η-Al2O3 support plays a crucial role by enhancing the activity of Ni for CO2 methanation. This disordered structure likely facilitates better adsorption of CO2, creating more favorable sites where CO2 molecules/intermediates can interact stronger with the active Ni sites. This can also facilitate electron transfer processes, which are crucial for the activation of CO2 molecules during the methanation reaction, stabilizing the Ni particles, preventing them from agglomerating and thus maintaining a high dispersion of active sites (Fig. 1d–f). Additionally, the well-dispersed and homogenous size of Ni particles, coupled with their high surface area (Fig. S5†), ensures a sufficient number of active sites are available for the H2 binding. Another important factor is the low reducibility of Ni (<500 °C) over the η-Al2O3 support, which aids in the activation of Ni at relatively lower temperatures. To support these rationalized reasons for the superior CO2 hydrogenation performance of Ni/η-Al2O3, we have conducted operando DRIFTS studies,105 as detailed below.
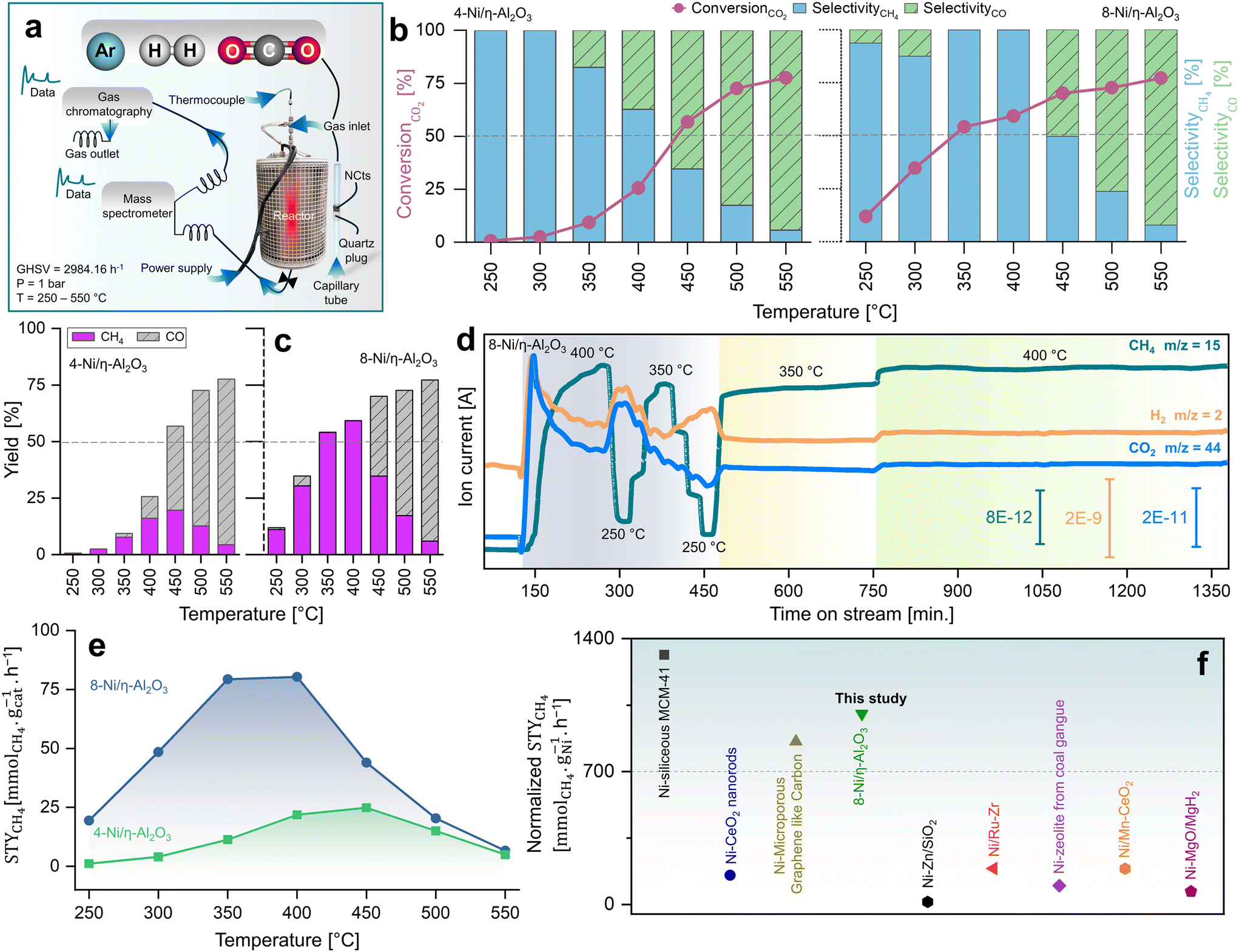 | ||
| Fig. 2 Kinetic measurements of CO2 methanation. (a) Reactor design for CO2 hydrogenation reaction at p = 1 bar and T = 250–550 °C operated at GHSV of 2984.16 h−1 and a CO2:H2 ratio of 1:4. (b) Catalytic performance of 4 and 8-Ni/η-Al2O3 NCts in terms of CO2 conversion (%), CO and CH4 selectivity (%) and (c) CO/CH4 yield (%). (d) MS spectra showing stable CH4 signal (fragment as CH3 ion, m/z = 15), for both abrupt and prolonged (400 and 600 min.) thermal (350–400 °C) treatments. (e) STY mmolCH4 gcat−1 h−1 at 250–350 °C of 4 and 8-Ni/η-Al2O3 NCts. (f) comparison of STY (mmolCH4 gNi−1 h−1) of best performing NCts (8-Ni/η-Al2O3) with Ni based NCts from literature.97–104 The literature STYCH4 values were normalized to the same Ni content (%), pressure (1.01325 bar), and temperature (400 °C) for consistency. See Table S1† for more details. | ||
Operando insights into CO2 methanation through DRIFTS/GC + MS
A mechanistic study of CO2 methanation over Ni/η-Al2O3 NCts was conducted using DRIFTS alongside catalytic performance measurements via GC + MS, in an operando setup106 (Fig. 3a). The calcined NCts underwent a pre-treatment process in a 5% H2 in Ar atmosphere at 550 °C, which effectively removed surface contaminants such as carbonates and H2O (Fig. 3b), thereby activating the NCts. This pre-treatment was crucial for ensuring the NCts’ surface was clean and ready for the subsequent CO2 methanation reaction.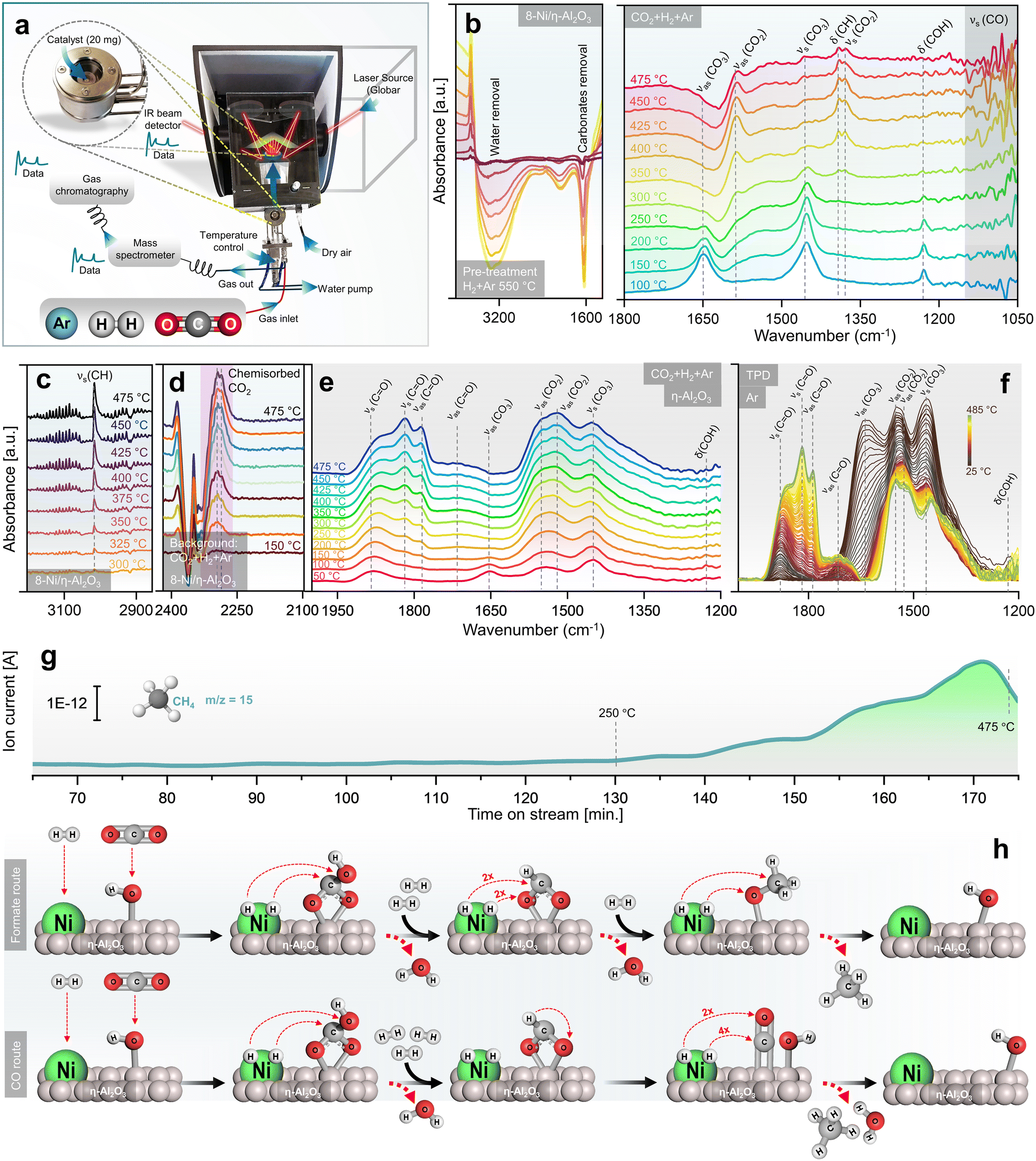 | ||
| Fig. 3 Rationalized mechanistic study of CO2 methanation by operando DRIFTS/GC + MS. (a) Experimental setup showing DRIFTS along catalytic performance measurements via GC + MS, under operando conditions. (b) DRIFTS spectra collected during the pre-treatment of NCts in a 5% H2 in Ar atmosphere at 550 °C, showing IR peaks originating from removal of carbonaceous species and surface adsorbed H2O (left), and DRIFTS spectra of 8-Ni/η-Al2O3 NCts exposed to a gas mixture of 5% CO2, 20% H2, and 75% Ar in a temperature range of 100–475 °C, showing formation of mainly hydrogen carbonates, formate and methoxy species (right). (c) DRIFTS spectra of CH4 formation in gas phase over the active 8-Ni/η-Al2O3 NCts. (d) DRIFTS spectra of chemisorbed CO2 over 8-Ni/η-Al2O3 NCts, observed taking CO2/H2/Ar as background during heat treatment (150–475 °C). (e) DRIFTS spectra of η-Al2O3 support exposed to a gas mixture of 5% CO2, 20% H2, and 75% Ar in a temperature range of 50–475 °C, showing formation of mainly hydrogen carbonates species. (f) Temperature programmed desorption (TPD) measured between 25–485 °C of (e). (g) MS spectra acquired simultaneously to the DRIFTS measurements, showing a linear increase in CH4 (fragment as CH3 ion, m/z = 15) formation with temperature. (h) CO2 methanation pathways over Ni/η-Al2O3 NCts, hypothesized based on observations in (b)–(g). | ||
Following the H2 pretreatment, the 8-Ni/η-Al2O3 NCts were exposed to a gas mixture of 5% CO2, 20% H2, and 75% Ar across a temperature range of 100–475 °C. Strong infrared bands were observed at 1648 cm−1 (νas(CO3)), 1452 cm−1 (νs(CO3)), and 1228 cm−1 (δ(COH)), which can be attributed to hydrogen carbonate species107–109 adsorbed on η-Al2O3 (Fig. 3b). As the temperature increased to 250 °C, peaks at 1585 cm−1 (νas(CO2)), 1390 cm−1 (δ(CH)), and 1377 cm−1 (νs(CO2)) began to grow, suggesting the formation of formate species,110 which are often considered as intermediates in CH4 formation. At higher temperatures, the formation of methoxy species (νs(CO)) was evident at 1050–1100 cm−1. The IR signals in this region were sharp and transient, indicating rapid formation and transformation of methoxy species to CH4.111 This suggests that methoxy species are quickly formed and rapidly converted to CH4, highlighting the dynamic nature of the reaction at elevated temperatures. Consistent with previous studies, bicarbonate species are progressively reduced by H+ spillover, transforming into formate and methoxy species, which ultimately form CH4.112 At temperatures exceeding 300 °C, the formation of gas-phase CH4 was confirmed by the growth of the νs(CH) peak at 3015 cm−1 (Fig. 3c). Additionally, bands at 2293 cm−1 and 2284 cm−1 were observed, which are related to chemisorbed CO2 on the Lewis acid sites113 of η-Al2O3 (Fig. 2d). The presence of chemisorbed CO2 on these Lewis acid sites may play an essential role in facilitating CO2 dissociation and subsequent hydrogenation.114 The formation of hydrogen carbonate species between 1400–1600 cm−1 was also noted (Fig. 3e). Interconversion of carbonate to carbonyl species (vCO2 ⇌ vCO) specific to the disordered η-Al2O3 phase was observed.115 These species permanently accumulated on the η-Al2O3 surface as the temperature increased, persisting even after the CO2/H2 cut-off and temperature-programmed desorption (TPD) under Ar up to 485 °C (Fig. 3f). Clearly, their transformation required active sites from Ni for further hydrogenation and C–O/CO bond breaking.110 The CH4 formation MS signal, parallel to the DRIFTS measurements, showed a linear increase in CH4 formation with temperature, particularly from 350–475 °C (Fig. 3g). While the study provides support for the evolution of intermediates and products, further confirmation of the role of observed surface species as active intermediates (rather than spectators) would require isotopic labelling studies via steady-state isotopic transient kinetic analysis (SSITKA)-DRIFTS-MS116,117 or, alternatively, modulation excitation spectroscopy (MES).105,118–123
Finally, the operando DRIFTS analysis (Fig. 3a–g) revealed that the CO2 hydrogenation pathway likely follows an associative CO2 methanation mechanism, involving the sequential adsorption and hydrogenation of CO2 on the NCts surface to produce CH4. The proposed formate route (Fig. 3h) begins with Ni and η-alumina adsorbing H2 and CO2 molecules, respectively. The CO2 interacts with OH ions previously adsorbed by η-Al2O3, forming a bidentate hydrogen carbonate species, which is further hydrogenated to form a bidentate formate. The amphoteric nature of η-Al2O3 plays a critical role in this process. Its surface Lewis acid sites facilitate the activation of CO2 molecules,124 while its basic hydroxyl groups enhance adsorption and stabilize intermediate species, such as hydrogen carbonates and formates.125 This dual functionality may enable η-Al2O3 to serve as an effective support, promoting CO2 activation and ensuring efficient progression along the methanation pathway. This formate is hypothesized to selectively hydrogenate into a methoxy species before CH4 is released from the NCts.126 At lower Ni loading (4-Ni/η-Al2O3), the active sites appear to favour CO formation through initial CO2 activation, leading to CH3O species or carbonyl formation and higher CO yields. However, with increased Ni loading (8-Ni/η-Al2O3), the availability of active sites may facilitate further hydrogenation of CO, promoting the conversion of methoxy species into CH4. The alternative CO route (Fig. 3d) follows similar steps for bidentate formate formation but involves formate decomposition to CO,127 which can either be desorbed or further hydrogenated to produce CH4 and H2O. The transition from bicarbonate to formate species is suggested to involve C–O bond cleavage via hydrogenation,128 indicating an associative CO2 methanation process.36 However, as described earlier, higher temperatures (≥500 °C) thermodynamically favour the formation of CO rather than CH4. Additionally, η-Al2O3 alone can also form hydrogen carbonates species as starting intermediates (Fig. 3e), but Ni as the active phase provides hydrogen atoms necessary for each hydrogenation step.
Ensuring sustainability through recycling spent NCts
Deactivation of NCts due to coke (carbon) formation or structural collapse from high-temperature sintering is often inevitable. Although no deactivation was observed in the 4 and 8 Ni/η-Al2O3 NCts during the study, it is important to consider reactivation for sustained CO2 methanation performance. Reactivation methods, such as coke removal through heating (800 °C) or chemical treatment, often lead to complete or partial structural collapse of the NCts. Thus, recycling spent Ni/η-Al2O3 NCts into Ni and Al precursors presents a more viable and sustainable option.Fig. 4a illustrates the recovery process of Al and Ni through acid leaching and selective NaOH precipitation (pH 0–14).129 The spent Ni/η-Al2O3 NCts, appearing as a black powder, were treated with H2SO4 to initiate the leaching of Ni and Al ions. This process was accelerated by heating the mixture to 90 °C for 2 hours, resulting in the formation of Al2(SO4)3 and NiSO4. Al2(SO4)3 was then recovered as Al(OH)3 by increasing the pH to 6 through NaOH treatment and centrifugation. The supernatant was stored for later Ni recovery. As shown in Fig. 4b, XRF spectroscopy confirmed that washing the recovered pellets three times with diH2O was necessary to remove Na2SO4 and fully recover Al as Al2O3, with predictable Al peaks (Kα and SKα3,5,6) visible in the XRF spectrum.
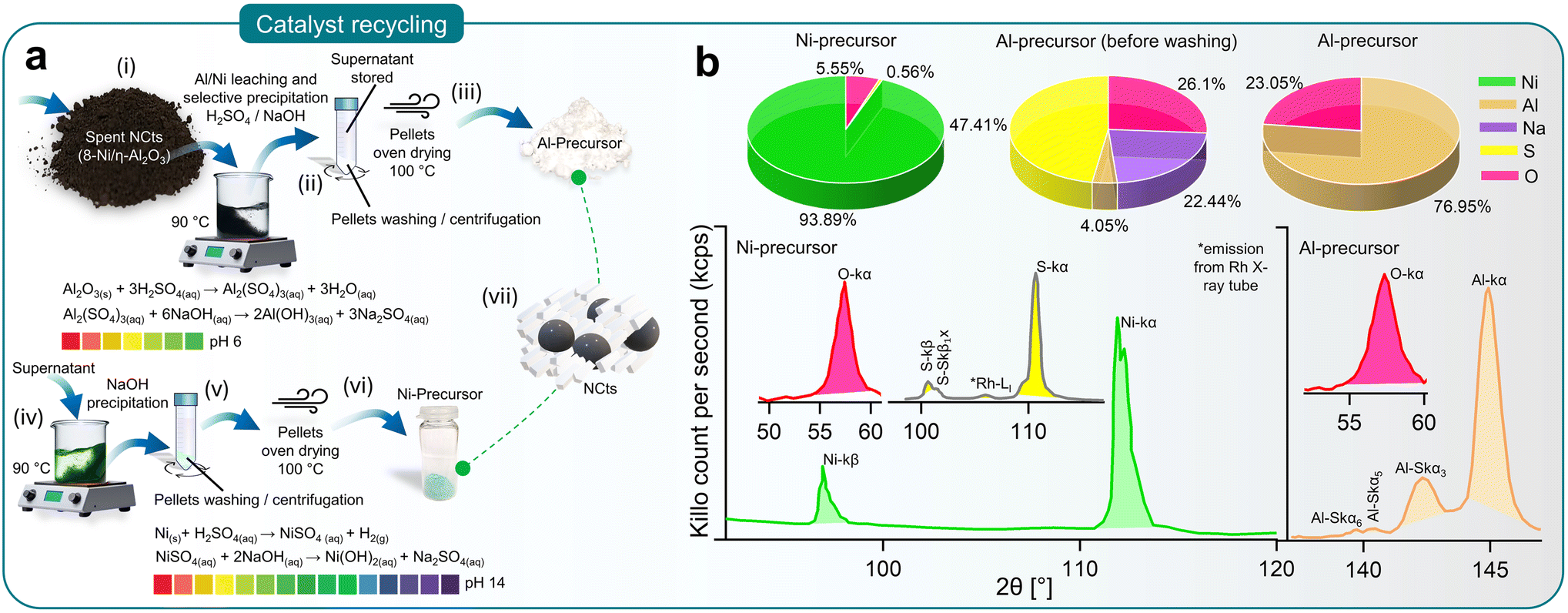 | ||
| Fig. 4 Recycling of spent NCts. (a) Schematics (steps i–vii) elaborating the recycling method of Al and Ni from spent Ni/η-Al2O3 NCts through acid leaching and selective NaOH precipitation (pH 0–14). (b) XRF spectroscopy showing elemental concentration (%) in Al and Ni precursors recovered from spent Ni/η-Al2O3 NCts (top) and characteristic XRF peaks related to Ni, Al, S and O (bottom). | ||
Further treatment of the supernatant at 90 °C with NaOH until pH 14 resulted in the precipitation of Ni(OH)2. The Ni(OH)2 pellets were washed and oven-dried to achieve a Ni/O purity of nearly 99%, with characteristics XRF peaks for Ni (Kα, and Kβ) and O (Kα), as shown in Fig. 4b. Although XRF spectroscopy is not considered reliable for lighter elements such as O, it effectively traced the Ni and Al concentrations130 (Fig. 4b). This method ensured the effective recovery of NiSO4, demonstrating the feasibility of recycling spent NCts into valuable precursors.
Conclusions
In summary, our study demonstrates the successful upcycling of hazardous waste materials, specifically spent Ni-MH batteries and aluminium foil, into high-performance NCts for CO2 methanation. Nickel sulfate was extracted from battery waste and converted into Ni(OH)2 hydrogel complex, while waste aluminium foil was processed into alumina (Al2O3). The combination of Ni(OH)2 hydrogel complex with alumina resulted in the synthesis of Ni/η-Al2O3 NCts with 4 wt% and 8 wt% Ni loading. Thorough characterization by XRD, STEM, EFTEM, HRTEM, SAED, and EELS confirmed a disordered cubic structure of η-Al2O3 and its stability during CO2 hydrogenation. The 8% Ni variant demonstrated excellent catalytic performance, achieving 99.8% selectivity, 59% yield of CH4 at 400 °C and GHSV of 2984.16 h−1, although higher temperatures (>450 °C) led to increased CO production due to the RWGS reaction.Further investigation using operando DRIFTS provided insights into the possible CO2 methanation mechanism over Ni/η-Al2O3 NCts. DRIFTS coupled with GC + MS revealed formation of key intermediates, such as hydrogen carbonates, formates, and methoxy species, illustrating the dynamic conversion of CO2 to CH4. Methane formation was observed above 300 °C, with higher Ni loading (8 wt%) enhancing CH4 production due to a combination of factors, including a larger number of Ni active sites per gram of catalyst and the influence of smaller (18–39 nm) Ni particle size. HRTEM analysis revealed that Ni nanoparticles in the 18–39 nm range were well-dispersed on the η-Al2O3 surface, ensuring a higher proportion of active surface atoms. This, possibly coupled with improved interaction between Ni and reactants, contributed to the enhanced CH4 yields at higher loadings. Moreover, the study also proposes an associative CO2 methanation pathway involving sequential adsorption and hydrogenation of CO2, with formate and methoxy intermediates leading to methane. At lower Ni loadings (4 wt%) or higher temperatures (450–550 °C), CO formation due to RWGS becomes more prevalent. To further validate the proposed mechanistic pathway, future studies could employ SSITKA-DRIFTS-MS, enabling isotopic labelling experiments to correlate the dynamics of surface and gas-phase species. Alternatively, MES could be applied. These would provide definitive evidence for the nature and role of reaction intermediates. Overall, these findings not only highlight the potential of waste-derived NCts for efficient CO2 methanation but also provide valuable insights into the reaction mechanisms involved, particularly the role of Ni over disordered η-Al2O3.
Finally, the successful recovery of Ni and Al precursors from Ni/η-Al2O3 NCts through acid leaching and selective NaOH precipitation highlights the sustainability and economic viability of this approach.
Methods
Synthesis procedures
The recovery of Ni from Ni-MH batteries, aluminium from aluminium foil, and the synthesis of NCts was accomplished through the following 12 steps, as illustrated in Fig. 1a and detailed below.i. Disassembly and washing of Ni-MH batteries: the cylindrical spent Ni-MH batteries were disassembled to extract the cathode and anode components. The disassembled materials were then thoroughly washed with diH2O to remove the KOH electrolyte, continuing until the pH of the supernatant reached neutral (pH 7).
ii. Leaching of nickel ions: the cathode material (Ni(OH)2/NiOOH), appearing as a black coiled mat, was placed in a reaction beaker. Dilute H2SO4 was added to initiate the leaching of nickel ions. This process was accelerated by heating the mixture to 80 °C for 15 minutes. The reaction is represented by the following equation (eqn (1)):
 | (1) |
iii. Separation and crystallization of NiSO4: after cooling the suspension to room temperature, the precipitated La2(SO4)3 traces settled at the bottom, while the blue-green NiSO4·xH2O solution remained at the top. The NiSO4·xH2O solution was filtered using Whatman filter paper, heated to 100 °C to concentrate the mixture, and then cooled to collect crystallized NiSO4·7H2O.
iv. Preparation of NiSO4 solution and reduction to Ni(OH)2: a 0.5 M solution of NiSO4·7H2O was prepared in diH2O, heated to 85 °C at 300 rpm, and 0.25 M L-glutamic acid was added. The mixture was then reduced to Ni(OH)2 using a 5 M NaOH solution, continuing until the pH reached 10.
v. Washing and storage of Ni(OH)2: the Ni(OH)2, forming a hydrogel, was washed with diH2O through centrifugation to remove uncoordinated reducing agents (L-glutamic acid/NaOH) and stored for further use.
vi. Dissolution of waste Al-foil: waste aluminium foil was dissolved according to the following equation (eqn (2)):
| 2Al(s) + 6HCl(aq) → 2AlCl3(aq) + 3H2(g) | (2) |
vii. Reduction to Al(OH)3 pellets: the obtained AlCl3 was slowly reduced with 5 M NaOH to form Al(OH)3 pellets. This reaction was performed at 85 °C.
viii. Recovery of Al(OH)3 pellets: after cooling the reaction mixture, the suspension was filtered using Whatman filter paper to recover the precipitated Al(OH)3 pellets.
ix. Drying of Al(OH)3 pellets: the Al(OH)3 pellets were washed with diH2O through centrifugation and dried in an oven for 24 hours to obtain Al2O3 powder.
x. Synthesis of NCts: Ni(OH)2 and Al2O3 powder were mixed with Ni at weight percentages of 4% and 8%. The mixture was first stirred and then sonicated for 30 minutes.
xi. Drying of the suspension: the resultant suspension was oven-dried at 100 °C overnight.
xii. Calcination of the dried powder: the oven-dried powder was calcined at 550 °C (4 hours) to obtain 4% or 8% (wt%) Ni over η-Al2O3, denoted as 4-Ni/η-Al2O3 and 8-Ni/η-Al2O3, respectively.
Most importantly, the NCts preparation uses minimal H2SO4 and HCl to recover nickel and aluminium from waste, employing a closed system and effluent treatment ensuring environmental safety, aligning with green chemistry principles.131
Characterization
The chemical composition and purity of the extracted Ni from spent Ni-MH batteries and spent NCts were determined using X-ray fluorescence spectrometry (XRF) on a PANalytical AxiosmAX WD-XRF™ system, equipped with a rhodium tube as the radiation source. XRF measurements were performed on pressed pellets containing approximately 10 wt% wax. The samples were irradiated with X-rays, causing the elements within the sample to emit secondary fluorescent X-rays. These emitted X-rays were detected and analyzed to identify the specific elements present and their relative abundances. Quantitative analysis was conducted using the fundamental parameter (FP) method, which corrects for matrix effects and provides accurate concentration values for each element. The data was processed to determine the weight percentages of the elements, enabling a detailed comparison of the elemental composition of the recovered Ni.X-ray diffraction (XRD) measurements were conducted to elucidate the atomic structure of various crystalline phases, including metals, and oxides. Diffractograms were obtained using a PANanalytical X'Pert Pro™ Bragg–Brentano™ powder diffractometer at the X-ray Center of TU Wien, with Cu K-α radiation (wavelength of 1.54 Å) as the source. Small amounts of each catalyst, including calcined catalysts and those subjected to three different reduction temperatures, were applied to a silicon wafer Si (111) layer fixed to a sample holder. The positions (2θ angles) of the measured reflexes were compared with diffractograms from the ICDD International Centre for Diffraction Data™ database to identify the crystalline phases.
The morphology and crystal structure of the catalysts were analyzed using a FEI TECNAI G2 F20™ microscope at the University Service Center for Transmission Electron Microscopy (USTEM) at TU Wien. This microscope, equipped with a field emission gun (X-FEG) operating at 200 kV, was used to examine NCts samples loaded onto a carbon-coated Cu grid and inserted into the TEM's inlet system with a single tilt holder. Various TEM images, including high-angle annular dark field (HAADF), high-resolution (HR) TEM, energy-filtered (EF) TEM, and scanning (S) TEM, were recorded for each NCts both before and after the reaction. Structural alterations during the reaction were identified through image comparison, with the high resolution of HRTEM images allowing precise measurement of lattice planes to identify different phases. Additionally, selected area electron diffraction (SAED) was recorded for Ni and η-Al2O3 crystal structure analysis. Electron energy loss spectroscopy (EELS) measurements were also conducted to investigate elemental distribution. Finally, micrographs were analyzed using Digital Micrograph software (Gatan™).
Temperature programmed reduction (TPR) was employed to investigate the reducibility of NCts. The H2 TPR analysis was performed in a continuous fixed-bed quartz tube reactor. Approximately 50 mg of NCts was loaded into the reactor tube, which was then placed in a heating furnace. Gas flows of argon and hydrogen (the reducing gas) were precisely controlled using calibrated mass flow controllers. A total flow of 100 mL min−1 with 10 vol% H2 in Ar was passed through the sample. During the experiment, the furnace was heated from room temperature to 500 °C at a rate of 10 °C min−1. The quartz tube reactor was connected to a quadrupole mass spectrometer (Balzers Prisma™), which recorded the mass signals of H2 (m/z = 2) and H2O (m/z = 18) over time as a function of temperature. These experiments were conducted for both synthesized NCts and the pure η-Al2O3 support.
Brunauer–Emmett–Teller (BET) analysis of the as-prepared NCts was conducted using a Micromeritics surface area and porosity analyzer. To determine the specific surface area (SSA), N2 adsorption at −196 °C was performed on an ASAP 2020 Micromeritics™ apparatus with a 0.5 g sample, preheated under vacuum (<0.013 mbar) at 150 °C for 3 hours. The SSA was evaluated based on the linear portion of the BET analysis. Pore size distributions were obtained by applying the Barrett–Joyner–Halenda (BJH) equation to the desorption branch of the isotherm, and the total pore volume was estimated from the N2 uptake at a P/P0 of 0.99.
CO2 methanation
For the kinetic measurements of the nanocatalysts (NCts), a pre-treatment process was conducted to ensure the removal of surface contaminants and activation of the NCts. Specifically, 20 mg of NCts was placed between quartz plugs inside the capillary tube, which was set up in the reactor and subjected to a 5% H2 in Ar atmosphere at 550 °C for 30 minutes, with a heating rate of 10 °C per minute (see the reactor setup in Fig. 2a). Following this pre-treatment, the temperature of the NCts bed was reduced to 250 °C, controlled precisely by a thermocouple. This step was crucial to prepare the catalyst for subsequent catalytic reactions by ensuring optimal surface conditions.For the catalytic reaction, a gas mixture comprising 5% CO2, 20% H2, and 75% Ar was introduced at 1 bar pressure, with a total flow rate of 50 ml min−1. The catalytic activity was tested across a temperature range of 250 °C to 550 °C. Effluent gases were continuously analyzed using a gas chromatography/mass spectrometry (GC + MS) system, equipped with a capillary column designed for separating light hydrocarbons and permanent gases. The quadrupole mass spectrometer (QMS, Prisma Plus QMG 220, Pfeiffer Vacuum) operated in electron ionization (EI) mode to detect and quantify reaction products online, including methane (CH4) and carbon monoxide (CO). Additionally, a gas chromatograph (GC) from Agilent Technologies, equipped with a thermal conductivity detector (TCD) and a flame ionization detector (FID), was used for product analysis. Data acquisition was performed at regular intervals using Agilent Chemstation software (B.04.03), enabling real-time monitoring of catalytic performance under steady-state conditions. Retention times and mass spectral data were utilized to accurately identify the compounds formed during the reaction.
Before the experiments, the TCD and FID detectors was calibrated using standard gas mixtures to ensure precise quantification of the detected species. Calibration curves were generated by plotting the peak areas against the known concentrations of the standards (CO2, CO, CH4, and H2). Linear regression was employed to establish the relationship between peak area and concentration. During the experiments, the peak areas corresponding to various reactants and products were recorded. These peak areas were then used to determine the concentrations of the molecules present. The calibrated peak areas from GC chromatograms were utilized for calculation of the, e.g., ConversionCO2 (%) in eqn (3),
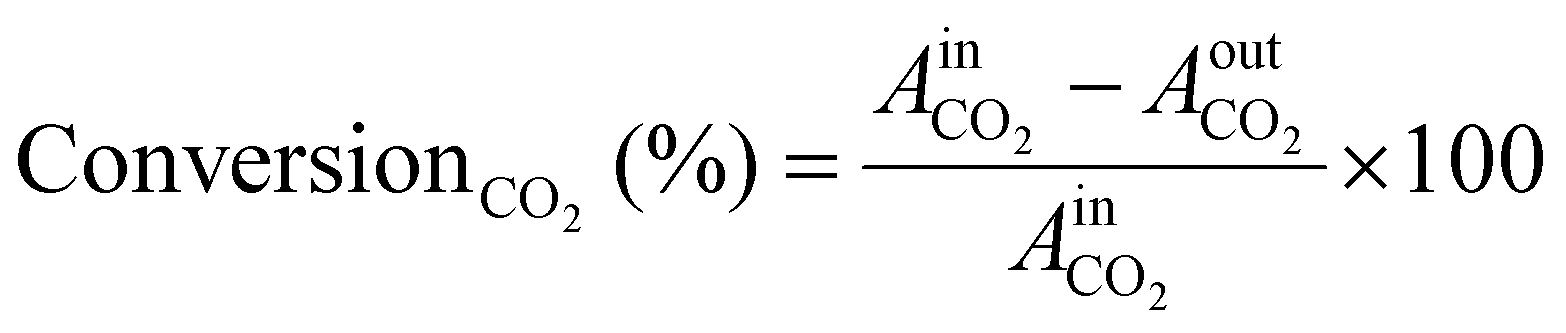 | (3) |
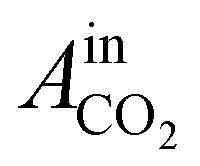 = peak area of CO2 entering the reactor;
= peak area of CO2 entering the reactor; 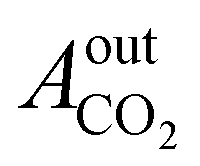 = peak area of CO2 exiting the reactor.
= peak area of CO2 exiting the reactor.
The selectivity of the catalysts for producing CH4 SelectivityCH4 (%) was determined using eqn (4),
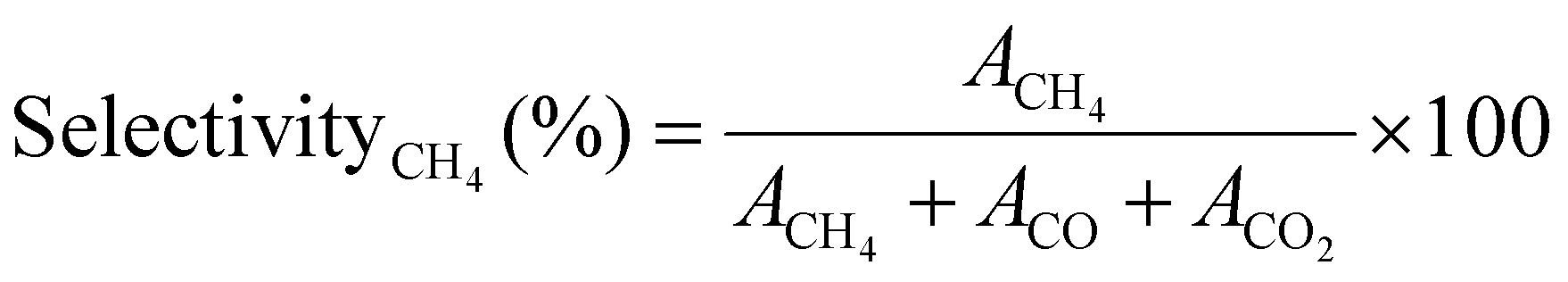 | (4) |
Similarly, the CO SelectivityCO (%) was determined using eqn (5)
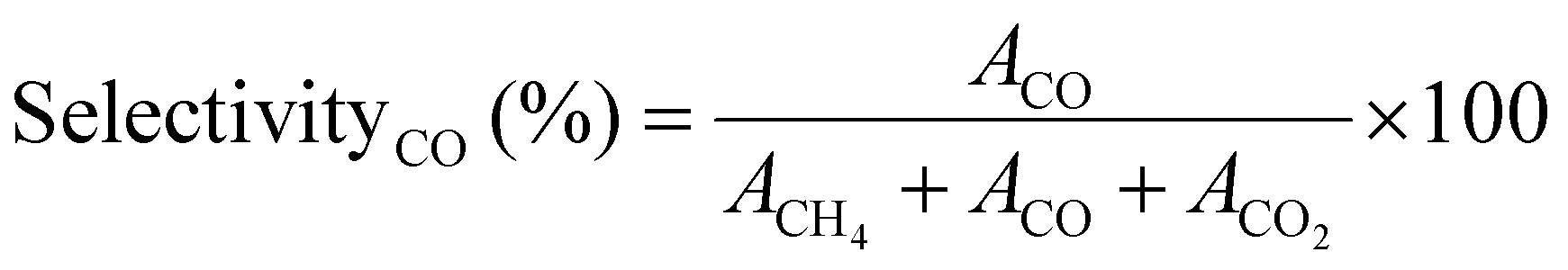 | (5) |
 | (6) |
 | (7) |
Furthermore, the GHSV can be calculated using eqn (8),
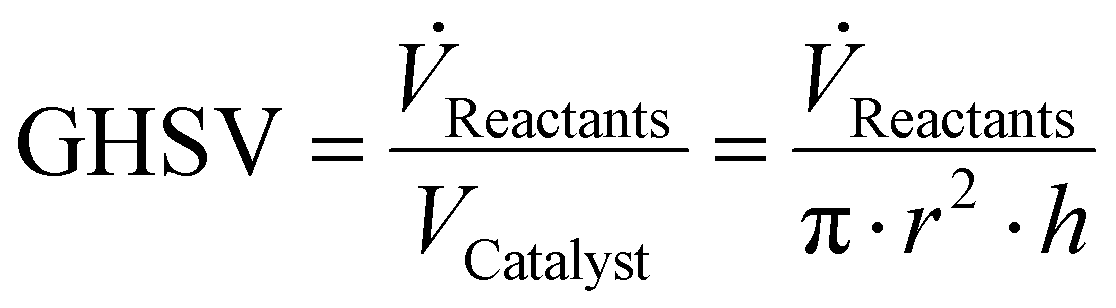 | (8) |
![[V with combining dot above]](https://www.rsc.org/images/entities/i_char_0056_0307.gif) Reactants = volumetric flow rate of reactants; VCatalyst = volume of catalytic bed; VCatalyst = volume of catalytic bed; r = internal radius of reactor; h = height of catalytic bed.
Reactants = volumetric flow rate of reactants; VCatalyst = volume of catalytic bed; VCatalyst = volume of catalytic bed; r = internal radius of reactor; h = height of catalytic bed.
Moreover, the reciprocal value of the GHSV is the residence time (τ) (eqn (9)), so that
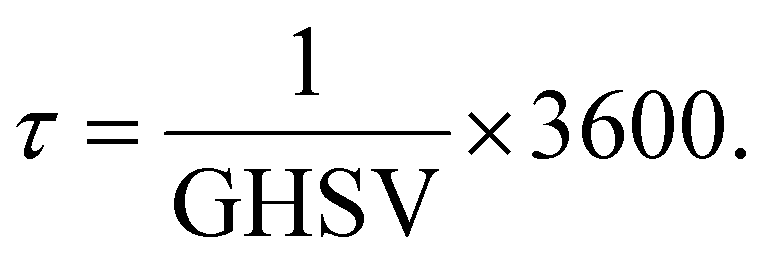 | (9) |
Finally, to determine the STY, first the volume of the reagent gas is calculated according to (eqn (10))
|
VCO2 = CO2 × τ
| (10) |
CO2 = total gas flow of CO2; τ = residence time; VCO2 = volume of CO2.
Then, using the residence time (τ) and the gas flow rate, the molar amount of the product, assuming that it equals that of the reagent gas, is determined by rearranging the universal gas equation (eqn (11)),
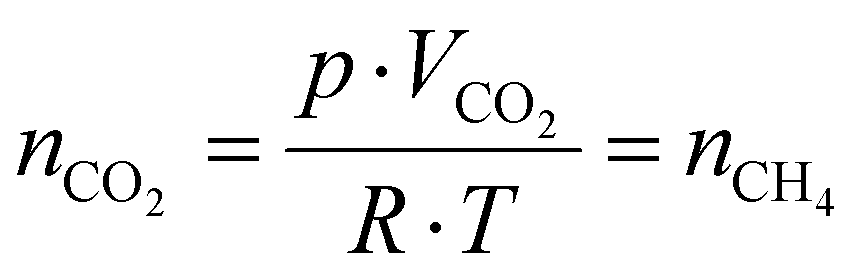 | (11) |
The STY values are finally obtained by inserting the measured CH4 in (eqn (12)),
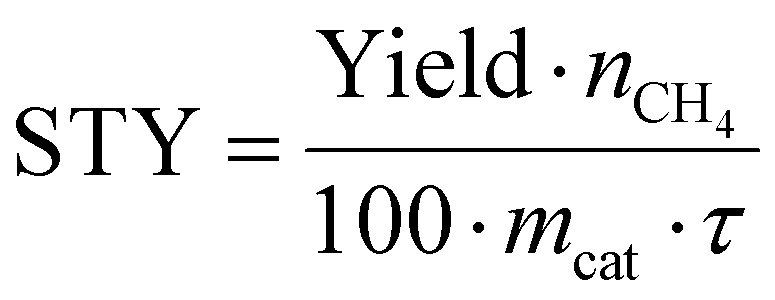 | (12) |
Detailed calculations on GHSV, residence time (τ) and STY for 4 and 8-Ni/η-Al2O3 NCts can be found in Note 2 of the ESI.†
At last, the STYCH4 of the best-performing NCts (e.g., 8-Ni/η-Al2O3 at 400 °C) was compared with the STYCH4 values of other Ni-based catalysts reported in the literature. To account for the varying Ni loading and reactions conditions, the literature STYCH4 values were normalized (eqn (13)) to the same Ni content (%), pressure (1.01325 bar), and temperature (400 °C) for consistency.
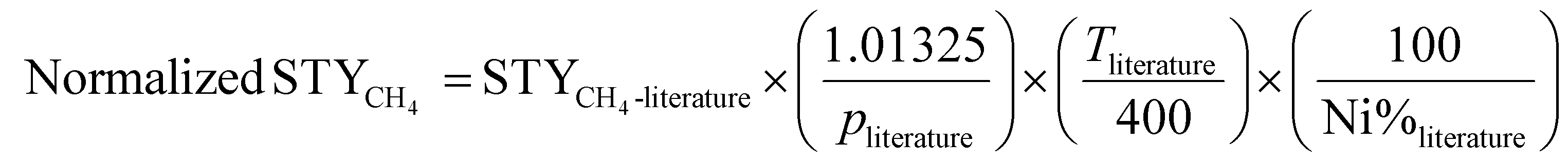 | (13) |
Operando DRIFTS/GC + MS measurements
The CO2 methanation mechanism was explored using operando Diffuse Reflectance Infrared Fourier Transform Spectroscopy (DRIFTS), conducted with a Bruker Vertex 70 spectrometer. The reaction chamber, equipped with CaF2 windows, enabled the passage of infrared light through the sample while maintaining controlled gas flow and temperature conditions. Initially, the NCts samples were loaded into the chamber and subjected to pre-treatment under a 5% H2/Ar flow at 550 °C for 30 minutes, with a heating rate of 10 °C min−1. Following this pre-treatment, the samples were cooled to the desired reaction temperature, and the gas flow was switched to a CO2 and H2 mixture (1:4 ratio) at a total flow rate of 50 mL min−1, simulating CO2 hydrogenation conditions. Additionally, for a temperature-programmed desorption study of the surface adsorbed molecules, the sample (η-Al2O3) was heated in Ar to 480 °C (10 °C min−1) in the DRIFTS cell while recording IR spectra. During the DRIFTS experiments, spectra were collected using OPUS 6.5 software at a resolution of 2 cm−1, with 128 scans recorded per spectrum. A background spectrum was recorded under pure Ar flow at the reaction temperature, and all spectra were normalized against this background to isolate signals from adsorbed species on the catalyst surface. The spectral region of interest (4000–1000 cm−1) was analyzed to monitor the formation and evolution of surface intermediates throughout the reaction. Additionally, the reactor effluent was continuously analyzed by GC + MS to ensure that the DRIFTS observations were consistent with catalytic performance. This approach allowed for direct correlation of the operando DRIFTS data with catalytic activity and selectivity under real reaction conditions.
Recycling of spent NCts
In steps i–iii (Fig. 4a), the spent Ni/η-Al2O3, appearing as a black powder, were placed in a reaction beaker where H2SO4 was added to initiate the leaching of Ni ions. This process was accelerated by heating the mixture to 90 °C for 2 hours. The reactions are represented by the following equations:| Al2O3(s) + 3H2SO4(aq) → Al2(SO4)3(aq) + 3H2O(aq) | (14) |
| Ni(s) + H2SO4(aq) → NiSO4(aq) + H2(g). | (15) |
Subsequently, Al2(SO4)3 was recovered as Al(OH)3 by increasing the pH to 6 through NaOH treatment, followed by washing with diH2O and centrifugation. The supernatant was stored for later Ni recovery, and the washed Al pellets were oven-dried at 100 °C to obtain the Al-precursor. This reaction is represented by the equation:
| Al2(SO4)3(aq) + 6NaOH(aq) → 2Al(OH)3(aq) + 3Na2SO4(aq). | (16) |
In steps iv–vii (Fig. 4a), the supernatant containing NiSO4 was further treated at 90 °C with NaOH until the pH reached 14, resulting in the precipitation of Ni(OH)2. This process is represented by the equation:
| NiSO4(aq) + 2NaOH(aq) → Ni(OH)2(aq) + Na2SO4(aq). | (17) |
The Ni precipitation was followed by washing with diH2O through centrifugation. The washed Ni pellets were then oven-dried at 100 °C to obtain the Ni-precursor. Both the Al and Ni precursors can subsequently be used to synthesize Ni/η-Al2O3 NCts.
Author contributions
Qaisar Maqbool: conceptualization, methodology, validation, software, formal analysis, investigation, data curation, writing – original draft, writing – review & editing. Hamilton Uchenna Aharanwa: methodology, validation, software, formal analysis, investigation, data curation, writing – review & editing. Michael Stöger-Pollach: methodology, validation, formal analysis, investigation, writing – review & editing. Günther Rupprechter: conceptualization, validation, resources, writing – review & editing, supervision.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was funded in part by the Austrian Science Fund (FWF) [10.55776/F81 and 10.55776/COE5] (SFB TACO and Cluster of Excellence Materials for Energy Conversion and Storage, MECS). For open-access purposes, the author has applied a CC BY public copyright license to any author-accepted article version arising from this submission.References
- M. Bailera, P. Lisbona, L. M. Romeo and S. Espatolero, Renewable Sustainable Energy Rev., 2017, 69, 292–312 CrossRef.
- A. Barbaresi, M. Morini and A. Gambarotta, Energies, 2022, 15, 5942 CrossRef.
- G. Gahleitner, Int. J. Hydrogen Energy, 2013, 38, 2039–2061 CrossRef.
- K. Ghaib and F. Z. Ben-Fares, Renewable Sustainable Energy Rev., 2018, 81, 433–446 CrossRef.
- V. Eveloy and T. Gebreegziabher, Energies, 2018, 11, 1824 CrossRef.
- E. Bargiacchi, in Power to Fuel: How to Speed Up a Hydrogen Economy, Academic Press, 2021, 211–237 Search PubMed.
- M. C. Bacariza, M. Maleval, I. Graça, J. M. Lopes and C. Henriques, Microporous Mesoporous Mater., 2019, 274, 102–112 CrossRef.
- A. Sápi, T. Rajkumar, J. Kiss, Á. Kukovecz, Z. Kónya and G. A. Somorjai, Catal. Lett., 2021, 151, 2153–2175 CrossRef.
- A. Sherryna, M. Tahir and W. Nabgan, Int. J. Hydrogen Energy, 2022, 47, 862–901 CrossRef.
- Y. R. Dias and O. W. Perez-Lopez, J. CO2 Util., 2023, 68, 102381 CrossRef.
- D. dos S. Lima, Y. R. Dias and O. W. Perez-Lopez, Sustainable Energy Fuels, 2020, 4, 5747–5756 RSC.
- X. Yan, W. Sun, L. Fan, P. N. Duchesne, W. Wang, C. Kübel, D. Wang, S. G. H. Kumar, Y. F. Li, A. Tavasoli, T. E. Wood, D. L. H. Hung, L. Wan, L. Wang, R. Song, J. Guo, I. Gourevich, F. M. Ali, J. Lu, R. Li, B. D. Hatton and G. A. Ozin, Nat. Commun., 2019, 10, 1–11 CrossRef PubMed.
- L. Gao, Q. Fu, M. Wei, Y. Zhu, Q. Liu, E. Crumlin, Z. Liu and X. Bao, ACS Catal., 2016, 6, 6814–6822 CrossRef CAS.
- H. Chen, J. B. Brubach, N. H. Tran, A. L. Robinson, F. Ben Romdhane, M. Frégnaux, F. Penas-Hidalgo, A. Solé-Daura, P. Mialane, M. Fontecave, A. Dolbecq and C. Mellot-Draznieks, ACS Appl. Mater. Interfaces, 2024, 16, 12509–12520 CrossRef CAS.
- M. E. Farshchi, K. Asgharizadeh, H. Jalili, S. Nejatbakhsh, B. Azimi, H. Aghdasinia and M. Mohammadpourfard, J. Environ. Chem. Eng., 2024, 12, 113909 CrossRef CAS.
- X. Feng, K. Wang, M. Zhou, F. Li, J. Liu, M. Zhao, L. Zhao, X. Song, P. Zhang and L. Gao, Ceram. Int., 2021, 47, 12366–12374 CrossRef CAS.
- W. K. Fan, M. Tahir and H. Alias, ACS Appl. Mater. Interfaces, 2023, 15, 54353–54372 CrossRef CAS.
- C. Shen, M. Liu, S. He, H. Zhao and C.-J. Liu, Chin. J. Catal., 2024, 63, 1–15 CrossRef CAS.
- A. Parastaev, V. Muravev, E. H. Osta, A. J. F. van Hoof, T. F. Kimpel, N. Kosinov and E. J. M. Hensen, Nat. Catal., 2020, 3, 526–533 CrossRef.
- Y. J. O. Asencios, N. Yigit, T. Wicht, M. Stöger-Pollach, A. F. Lucrédio, F. C. F. Marcos, E. M. Assaf and G. Rupprechter, Top. Catal., 2023, 66, 1539–1552 CrossRef PubMed.
- P. Shafiee, S. M. Alavi and M. Rezaei, Int. J. Hydrogen Energy, 2021, 46, 3933–3944 CrossRef.
- K. Tamimi, S. M. Alavi, M. Rezaei and E. Akbari, J. Energy Inst., 2021, 99, 48–58 CrossRef.
- S. Sharifian and N. Asasian-Kolur, Inorg. Chem. Commun., 2020, 118, 108021 CrossRef.
- L. A. Sani, H. Bai, Z. Xu, L. Fu, Y. Sun, X. Huang, H. Gao, X. Liu, D. Bai, Z. Zhang, F. Su, J. Liu and G. Xu, J. CO2 Util., 2024, 80, 102678 CrossRef.
- A. Kim, D. P. Debecker, F. Devred, V. Dubois, C. Sanchez and C. Sassoye, Appl. Catal., B, 2018, 220, 615–625 CrossRef.
- E. M. Petersen, R. G. Rao, B. C. Vance and J. P. Tessonnier, Appl. Catal., B, 2021, 286, 119904 CrossRef.
- S. Scirè, C. Crisafulli, R. Maggiore, S. Minicò and S. Galvagno, Catal. Lett., 1998, 51, 41–45 CrossRef.
- X. Jia, X. Zhang, N. Rui, X. Hu and C.-J. Liu, Appl. Catal., B, 2019, 244, 159–169 CrossRef.
- N. M. Martin, P. Velin, M. Skoglundh, M. Bauer and P. A. Carlsson, Catal. Sci. Technol., 2017, 7, 1086–1094 RSC.
- S. Lin, Z. Li and M. Li, Fuel, 2023, 333, 126369 CrossRef.
- Q. Maqbool, G. Barucca, S. Sabbatini, M. Parlapiano, M. L. Ruello and F. Tittarelli, J. Hazard. Mater., 2022, 423, 126958 CrossRef.
- THE 17 GOALS | Sustainable Development, https://sdgs.un.org/goals, (accessed 21 September 2022).
- J. A. S. Tenório and D. C. R. Espinosa, J. Power Sources, 2002, 108, 70–73 CrossRef.
- S. L. Lin, K. L. Huang, I. C. Wang, I. C. Chou, Y. M. Kuo, C. H. Hung and C. Lin, J. Air Waste Manage. Assoc., 2016, 66, 296–306 CrossRef.
- V. Innocenzi and F. Vegliò, J. Power Sources, 2012, 211, 184–191 CrossRef.
- B. Miao, S. S. K. Ma, X. Wang, H. Su and S. H. Chan, Catal. Sci. Technol., 2016, 6, 4048–4058 RSC.
- Q. Maqbool, O. Favoni, T. Wicht, N. Lasemi, S. Sabbatini, M. Stöger-Pollach, M. L. Ruello, F. Tittarelli and G. Rupprechter, ACS Catal., 2024, 14, 4820–4834 CrossRef PubMed.
- M. Stevanović, I. Savanović, V. Uskoković, S. D. Škapin, I. Bračko, U. Jovanović and D. Uskoković, Colloid Polym. Sci., 2012, 290, 221–231 CrossRef.
- B. Dutta, A. Nema, N. G. Shetake, J. Gupta, K. C. Barick, M. A. Lawande, B. N. Pandey, I. K. Priyadarsini and P. A. Hassan, Mater. Sci. Eng., C, 2020, 112, 110915 CrossRef PubMed.
- C. Zhao, S. Han, Y. Ding, Y. Yang, R. Jiang and C. Zhao, Appl. Surf. Sci., 2021, 558, 149853 CrossRef.
- M. Aghazadeh, A. N. Golikand and M. Ghaemi, Int. J. Hydrogen Energy, 2011, 36, 8674–8679 CrossRef.
- J. Coates, in Encyclopedia of Analytical Chemistry, John Wiley & Sons, Ltd, 2000 Search PubMed.
- G. Bor, J. Organomet. Chem., 1967, 10, 343–359 CrossRef.
- F. C. Meunier, Catal. Sci. Technol., 2022, 12, 7433–7438 RSC.
- J. Cui, G. Wang, X. Zhou, C. Chi, Z. H. Li, Z. Liu and M. Zhou, Phys. Chem. Chem. Phys., 2013, 15, 10224–10232 RSC.
- A. I. Osman, J. K. Abu-Dahrieh, A. Abdelkader, N. M. Hassan, F. Laffir, M. McLaren and D. Rooney, J. Phys. Chem. C, 2017, 121, 25018–25032 CrossRef CAS.
- E. Rani, P. Talebi, T. Pulkkinen, V. Pankratov and H. Singh, Nanoscale Adv., 2023, 5, 6935–6943 RSC.
- S. Huo, J. Lu and X. Wang, Energy Sci. Eng., 2021, 9, 1042–1047 CrossRef.
- N. Sengokmen-Ozsoz, R. Boston and F. Claeyssens, ACS Appl. Mater. Interfaces, 2023, 15, 30769–30779 CrossRef.
- F. Cheng and X. Li, Processes, 2022, 10, 1542 CrossRef.
- R.-S. Zhou and R. L. Snyder, Acta Crystallogr., Sect. B:Struct. Sci., 1991, 47, 617–630 CrossRef.
- Q. Maqbool, N. Yigit, M. Stöger-Pollach, M. L. Ruello, F. Tittarelli and G. Rupprechter, Catal. Sci. Technol., 2023, 13, 624–636 RSC.
- B. Kaulich, P. Thibault, A. Gianoncelli and M. Kiskinova, J. Phys.: Condens. Matter, 2011, 23, 083002 CrossRef.
- J. Sehested, A. Carlsson, T. V. W. Janssens, P. L. Hansen and A. K. Datyey, J. Catal., 2001, 197, 200–209 CrossRef.
- A. Bardaoui, I. Dhifallah, M. Daoudi, S. Aouini, M. Amlouk and R. Chtourou, J. Solid State Chem., 2024, 335, 124732 CrossRef.
- A. S. Bolokang, R. Modiba, D. E. Motaung and P. E. Ngoepe, Adv. Powder Technol., 2020, 31, 2742–2748 CrossRef.
- C. Wolverton and K. C. Hass, Phys. Rev. B:Condens. Matter Mater. Phys., 2001, 63, 024102 CrossRef.
- F. Ernst, P. Pirouz and A. H. Heuer, Philos. Mag. A, 1991, 63, 259–277 CrossRef CAS.
- M. F. Peintinger, M. J. Kratz and T. Bredow, J. Mater. Chem. A, 2014, 2, 13143–13158 RSC.
- Y. Huang, X. Peng and X. Q. Chen, J. Alloys Compd., 2021, 863, 158666 CrossRef CAS.
- L. Kovarik, M. Bowden, A. Genc, J. Szanyi, C. H. F. Peden and J. H. Kwak, J. Phys. Chem. C, 2014, 118, 18051–18058 CrossRef.
- Y. Yourdshahyan, C. Ruberto, M. Halvarsson, L. Bengtsson, V. Langer, B. I. Lundqvist, S. Ruppi and U. Rolander, J. Am. Ceram. Soc., 1999, 82, 1365–1380 CrossRef.
- G. Wallez, J. Solid State Chem., 2022, 312, 123303 CrossRef.
- Z. Li, P. R. Wray, M. P. Su, Q. Tu, H. P. Andaraarachchi, Y. J. Jeong, H. A. Atwater and U. R. Kortshagen, ACS Omega, 2020, 5, 24754–24761 CrossRef PubMed.
- R. F. Egerton, Electron Energy-Loss Spectroscopy in the Electron Microscope, Springer US, 1996 Search PubMed.
- V. Y. Gertsman and Q. S. M. Kwok, in Microscopy and Microanalysis, Cambridge University Press, 2005, vol. 11, pp. 410–420 Search PubMed.
- C. C. Wu, J. Wen, S. D. Walck, R. A. Pesce-Rodriguez and I. Arslan, J. Appl. Phys., 2021, 129, 63302 CrossRef.
- K. Elibol, M. Burghard and P. A. van Aken, Microsc. Microanal., 2024, 30, 1124–1125 Search PubMed.
- G. Noircler, F. Lebreton, E. Drahi, P. de Coux and B. Warot-Fonrose, Micron, 2021, 145, 103032 CrossRef PubMed.
- H. O. Ayoola, C. S. Bonifacio, Q. Zhu, C. H. Li, S. D. House, J. J. Kas, J. Jinschek, J. J. Rehr, W. A. Saidi and J. C. Yang, J. Phys. Chem. C, 2020, 124, 9876–9885 CrossRef.
- C. Weigel, G. Calas, L. Cormier, L. Galoisy and G. S. Henderson, J. Phys.: Condens. Matter, 2008, 20, 135219 CrossRef.
- Z. Wang, C. Li, L. Liu and T. K. Sham, J. Chem. Phys., 2013, 138, 84706 CrossRef.
- H. O. Ayoola, C. H. Li, S. D. House, C. S. Bonifacio, K. Kisslinger, J. Jinschek, W. A. Saidi and J. C. Yang, Ultramicroscopy, 2020, 219, 113127 CrossRef PubMed.
- H. O. Ayoola, C. S. Bonifacio, M. T. Curnan, S. D. House, M. Li, J. Kas, J. J. Rehr, E. A. Stach, W. A. Saidi and J. C. Yang, Microsc. Microanal., 2019, 25, 2036–2037 CrossRef.
- S. Fritz, A. Seiler, L. Radtke, R. Schneider, M. Weides, G. Weiß and D. Gerthsen, Sci. Rep., 2018, 8, 1–11 Search PubMed.
- Y. Ikuhara, P. Pirouz, A. H. Heuer, S. Yadavalli and C. P. Flynn, Proc. - Annu. Meet., Electron Microsc. Soc. Am., 1992, 50, 146–147 CrossRef.
- M. Caporali, M. Serrano-Ruiz, F. Telesio, S. Heun, G. Nicotra, C. Spinella and M. Peruzzini, Chem. Commun., 2017, 53, 10946–10949 RSC.
- N. Kitakatsu, V. Maurice, C. Hinnen and P. Marcus, Surf. Sci., 1998, 407, 36–58 CrossRef.
- G. Evmenenko, T. T. Fister, F. C. Castro, X. Chen, B. Lee, D. B. Buchholz, V. P. Dravid, P. Fenter and M. J. Bedzyk, Phys. Chem. Chem. Phys., 2019, 21, 8897–8905 RSC.
- D. P. Abraham, R. D. Twesten, M. Balasubramanian, J. Kropf, D. Fischer, J. McBreen, I. Petrov and K. Amine, J. Electrochem. Soc., 2003, 150, A1450–A1456 CrossRef.
- G. Timmer and G. Borstel, Phys. Rev. B:Condens. Matter Mater. Phys., 1991, 43, 5098–5108 CrossRef PubMed.
- D. P. Arovas, E. Berg, S. A. Kivelson and S. Raghu, Annu. Rev. Condens. Matter Phys., 2022, 13, 239–274 CrossRef.
- J. G. Chen, M. D. Weisel and R. B. Hall, Surf. Sci., 1991, 250, 159–168 CrossRef.
- R. A. Vilá, W. Huang and Y. Cui, Cell Rep. Phys. Sci., 2020, 1, 100188 CrossRef.
- H. Pourdelan, S. M. Alavi, M. Rezaei and E. Akbari, Catal. Lett., 2023, 153, 3159–3173 CrossRef.
- H. Drobná, V. Meinhardová, L. Dubnová, K. Kozumplíková, M. Reli, K. Kočí and L. Čapek, Catalysts, 2023, 13, 293 CrossRef.
- A. R. Nobakht, M. Rezaei, S. M. Alavi, E. Akbari, M. Varbar and J. Hafezi-Bakhtiari, Int. J. Hydrogen Energy, 2023, 48, 38664–38675 CrossRef.
- R. Liu, X. Zhang, T. Liu, X. Yao, Z. Zhao, C. Pei and J. Gong, Appl. Catal., B, 2023, 328, 122478 CrossRef CAS.
- G. De Piano, J. J. Andrade Gamboa, A. M. Condó and F. C. Gennari, Int. J. Hydrogen Energy, 2024, 56, 1007–1019 CrossRef CAS.
- M. A. Al-Ghouti and D. A. Da'ana, J. Hazard. Mater., 2020, 393, 122383 CrossRef CAS.
- O. P. Murphy, M. Vashishtha, P. Palanisamy and K. V. Kumar, ACS Omega, 2023, 8, 17407–17430 CrossRef CAS PubMed.
- M. Mozaffari Majd, V. Kordzadeh-Kermani, V. Ghalandari, A. Askari and M. Sillanpää, Sci. Total Environ., 2022, 812, 151334 CrossRef CAS.
- M. Langer and H. Freund, Ind. Eng. Chem. Res., 2024, 63, 10981–10996 CrossRef CAS.
- S. Ewald, M. Kolbeck, T. Kratky, M. Wolf and O. Hinrichsen, Appl. Catal., A, 2019, 570, 376–386 CrossRef.
- A. S. A. Al-Fatish, A. A. Ibrahim, A. H. Fakeeha, M. A. Soliman, M. R. H. Siddiqui and A. E. Abasaeed, Appl. Catal., A, 2009, 364, 150–155 CrossRef.
- J. Gao, Q. Liu, F. Gu, B. Liu, Z. Zhong and F. Su, RSC Adv., 2015, 5, 22759–22776 RSC.
- G. Du, S. Lim, Y. Yang, C. Wang, L. Pfefferle and G. L. Haller, J. Catal., 2007, 249, 370–379 CrossRef.
- G. Varvoutis, M. Lykaki, S. Stefa, V. Binas, G. E. Marnellos and M. Konsolakis, Appl. Catal., B, 2021, 297, 120401 CrossRef.
- J. Wu, Z. Jin, B. Wang, Y. Han, Y. Xu, Z. Liang and Z. Wang, Ind. Eng. Chem. Res., 2019, 58, 20536–20542 CrossRef.
- X. Chen, S. Ullah, R. Ye, C. Jin, H. Hu, F. Hu, Y. Peng, Z. H. Lu, G. Feng, L. Zhou and R. Zhang, Energy Fuels, 2023, 37, 3865–3874 CrossRef CAS.
- F. Lange, U. Armbruster and A. Martin, Energy Technol., 2015, 3, 55–62 CrossRef CAS.
- S. Bahraminia and M. Anbia, Int. J. Hydrogen Energy, 2024, 83, 842–855 CrossRef CAS.
- Q. Fan, S. Li, L. Zhang, P. Wang and S. Wang, J. Catal., 2022, 414, 53–63 CrossRef CAS.
- H. Chen, P. Liu, J. Liu, X. Feng and S. Zhou, J. Catal., 2021, 394, 397–405 CrossRef CAS.
- G. Rupprechter, Small, 2021, 17, 2004289 CrossRef CAS PubMed.
- M. A. Bañares, M. O. Guerrero-Pérez, J. L. G. Fierro and G. G. Cortez, J. Mater. Chem., 2002, 12, 3337–3342 RSC.
- N. A. A. Fatah, A. A. Jalil, N. F. M. Salleh, M. Y. S. Hamid, Z. H. Hassan and M. G. M. Nawawi, Int. J. Hydrogen Energy, 2020, 45, 18562–18573 CrossRef CAS.
- C. Weilach, C. Spiel, K. Föttinger and G. Rupprechter, Surf. Sci., 2011, 605, 1503–1509 CrossRef CAS.
- K. Anic, A. Wolfbeisser, H. Li, C. Rameshan, K. Föttinger, J. Bernardi and G. Rupprechter, Top. Catal., 2016, 59, 1614–1627 CrossRef CAS PubMed.
- N. Schreiter, J. Kirchner and S. Kureti, Catal. Commun., 2020, 140, 105988 CrossRef.
- A. Solis-Garcia, J. F. Louvier-Hernandez, A. Almendarez-Camarillo and J. C. Fierro-Gonzalez, Appl. Catal., B, 2017, 218, 611–620 CrossRef.
- V. Sanchez-Escribano, M. A. Larrubia Vargas, E. Finocchio and G. Busca, Appl. Catal., A, 2007, 316, 68–74 CrossRef.
- Y. Shi, S. Wang, Y. Li, F. Yang, H. Yu, Y. Chu, T. Li and H. Yin, Materials, 2022, 15, 3044 CrossRef PubMed.
- S. Qiu, Y. Wu, Y. Tan, Q. Dai, X. Gao and S. Kawi, ChemCatChem, 2023, 15, e202300420 CrossRef.
- C. Morterra, A. Zecchina, S. Coluccia and A. Chiorino, J. Chem. Soc., Faraday Trans. 1, 1977, 73, 1544–1560 RSC.
- W. Chen, R. Pestman, B. Zijlstra, I. A. W. Filot and E. J. M. Hensen, ACS Catal., 2017, 7, 8050–8060 CrossRef PubMed.
- R. Gui, C. Zhang, Y. Gao, Q. Wang and A. M. Efstathiou, Appl. Catal., B, 2025, 361, 124611 CrossRef.
- A. Urakawa, T. Bürgi and A. Baiker, Chem. Eng. Sci., 2008, 63, 4902–4909 CrossRef.
- D. Ferri, M. A. Newton and M. Nachtegaal, in Topics in Catalysis, Springer, 2011, vol. 54, pp. 1070–1078 Search PubMed.
- A. Gaur, T. M. Hartmann Dabros, M. Høj, A. Boubnov, T. Prüssmann, J. Jelic, F. Studt, A. D. Jensen and J. D. Grunwaldt, ACS Catal., 2019, 9, 2568–2579 CrossRef.
- A. Haghofer, D. Ferri, K. Föttinger and G. Rupprechter, ACS Catal., 2012, 2, 2305–2315 CrossRef.
- M. Roger, L. Artiglia, A. Boucly, F. Buttignol, M. Agote-Arán, J. A. van Bokhoven, O. Kröcher and D. Ferri, Chem. Sci., 2023, 14, 7482–7491 RSC.
- M. A. Bañares and M. Daturi, Catal. Today, 2023, 423, 114255 CrossRef.
- S. Chen, B. Wang, J. Zhu, L. Wang, H. Ou, Z. Zhang, X. Liang, L. Zheng, L. Zhou, Y. Q. Su, D. Wang and Y. Li, Nano Lett., 2021, 21, 7325–7331 CrossRef PubMed.
- O. Netskina, S. Mucha, J. Veselovskaya, V. Bolotov, O. Komova, A. Ishchenko, O. Bulavchenko, I. Prosvirin, A. Pochtar and V. Rogov, Materials, 2021, 14, 6789 CrossRef.
- A. Quindimil, J. A. Onrubia-Calvo, A. Davó-Quiñonero, A. Bermejo-López, E. Bailón-García, B. Pereda-Ayo, D. Lozano-Castelló, J. A. González-Marcos, A. Bueno-López and J. R. González-Velasco, J. CO2 Util., 2022, 57, 101888 CrossRef.
- M. Kock, E. Kowalewski, D. Iltsiou, J. Mielby and S. Kegnæs, ChemCatChem, 2024, 16, e202301447 CrossRef.
- P. Strucks, L. Failing and S. Kaluza, Chem.-Ing.-Tech., 2021, 93, 1526–1536 CrossRef.
- M. Razavian, S. Fatemi and A. T. Najafabadi, J. Environ. Chem. Eng., 2020, 8, 103660 CrossRef.
- B. Beckhoff, B. Kanngießer, N. Langhoff, R. Wedell and H. Wolff, Handbook of Practical X-Ray Fluorescence Analysis, Springer Berlin Heidelberg, 2006 Search PubMed.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2015, 18, 288–296 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary Notes 1, 2; Fig. S1–S8 and Supplementary Table S1. See DOI: https://doi.org/10.1039/d4gc05217j |
| This journal is © The Royal Society of Chemistry 2025 |