
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing the reactivity of a P450 decarboxylase with ionic liquids†
Jake H.
Nicholson
 a,
Mayara
Chagas de Avila
b,
Ricardo
Rodrigues de Melo
b,
Leticia Maria
Zanphorlin
b and
Alex P. S.
Brogan
*a
a,
Mayara
Chagas de Avila
b,
Ricardo
Rodrigues de Melo
b,
Leticia Maria
Zanphorlin
b and
Alex P. S.
Brogan
*a
aDepartment of Chemistry, King's College London, 7 Trinity Street, London, SE1 1DB, UK. E-mail: alex.brogan@kcl.ac.uk
bBrazilian Biorenewables National Laboratory, Brazilian Center for Research in Energy and Materials, Campinas, SP, Brazil
First published on 29th November 2024
Abstract
The cytochrome P450 family of enzymes have been shown to be powerful biocatalysts for a wide range of selective transformations. However, the industrial uptake of P450 enzymes has been low due to issues with enzyme stability and the requirement for exogenous cofactors to drive the reaction. Herein we describe a facile and scalable method for the stabilisation and solubilisation of a P450 decarboxylase enzyme in ionic liquids. The utilisation of ionic liquids allowed for solubilisation of the relatively water-insoluble fatty acid substrate of the enzyme and the modified enzyme was found to be significantly more thermally stable in ionic liquids relative to the enzyme in aqueous media. The shift to non-aqueous solvent allowed for the enzyme to operate in the Vmax region, which when coupled with the improved thermal stability, ultimately resulted in a 1000-fold increase in the process intensity of fatty acid decarboxylation. A novel photochemical method for driving the reaction was also discovered which removed the requirement for exogenous H2O2 to be added to the reaction. These results highlight the potential of this strategy as it facilitates a holistic process of biocatalysis engineering where by solvent consideration and increased thermal stability significantly broadens the capability of the enzyme, crucial for the wider realization of industrial biocatalysis.
Green foundation1. This work demonstrates a methodology involving chemical modification and ionic liquids for drastically enhancing biocatalysis involving P450 enzymes.2. In particular, we show that through holistic consideration of solvent and enzyme stabilization, we can increase reaction rate, dramatically increase substrate loading, and provide an alternative pathway for enzymatic action without requiring addition of peroxide. This provides a blueprint for enhancing the reactivity and usability of any P450 enzyme, which could open up biocatalysis to a broader range of reactions. 3. We have used ionic liquids, which are recognized as having key attributes of green solvents. However, future work would do well to investigate ionic liquids with improved biodegradability. Furthermore, one limitation of this study is the light based deactivation of our enzyme. This should be investigated further in order to prolong the lifetime of the enzymes such that they can be used for longer. |
Introduction
Cytochrome P450s are a family of heme-dependent enzymes which have been used for a wide range of reactions such as hydroxylation of C–H bonds, epoxidation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds, decarboxylation and carbene tranfers.1–4 P450s are of particular interest as they can perform biocatalytic transformations – that are otherwise synthetically challenging through conventional chemical means – with high enantio- and regio-selectivities, without the requirement of protecting groups.5,6 As a result of this, P450s have found applications in the synthesis of pharmaceuticals,7,8 fragrances,9 and biofuels.10 P450s have also been shown to be highly engineerable through both rational design and directed evolution, which have been used to improve substrate scope, activity, and selectivity.7,11–13
C bonds, decarboxylation and carbene tranfers.1–4 P450s are of particular interest as they can perform biocatalytic transformations – that are otherwise synthetically challenging through conventional chemical means – with high enantio- and regio-selectivities, without the requirement of protecting groups.5,6 As a result of this, P450s have found applications in the synthesis of pharmaceuticals,7,8 fragrances,9 and biofuels.10 P450s have also been shown to be highly engineerable through both rational design and directed evolution, which have been used to improve substrate scope, activity, and selectivity.7,11–13
Generally, utilization of P450s as biocatalysts requires the addition of other enzymes that act as an NAD(P)H dependent redox partner system that facilitates the reduction of the heme iron, which in-turn initiates the catalytic cycle of the enzyme.14 Some P450s are also able to utilise hydrogen peroxide as a cofactor which allows for the formation of the reactive species, compound I, from the ferric resting state of the enzyme via the shunt mechanism (Fig. S1†).14 This removes the need for additional proteins and expensive nicotinamide cofactors to the reaction. However, the addition of peroxide may result in the enzyme deactivation15 or off target oxidation of the substrate16 if used at high concentrations which may limit the synthetic applicability of this pathway. Many P450s also have poor organic solvent tolerance,5,17 which in combination with the low water solubility of many of the desired P450 substrates,18,19 means that the broader application of P450s as biocatalysts is limited.
Ionic liquids, particularly those that are organic salts with room temperature liquid phases, are a versatile class of solvents which have found application in a range of industrial applications.20,21 The properties of ionic liquids can be tuned by selection of the constituent ion allowing for a range of solvent polarities and hydrophilicities.22 This tuneability can be used to improve the solubility of substrates, allowing for process intensification.23,24 Like many non-aqueous solvents, enzymes typically have poor tolerance towards ionic liquids and limited solubility. As such, despite their advantageous properties, biocatalytic reactions are not often performed in ionic liquids. To counter this, we have developed a method to chemically modify the surface of enzymes such that they are soluble and stable in ionic liquids.25,26 Recently, we have demonstrated that modified hydrolytic enzymes solubilized in ionic liquids were able to catalyse the degradation of water insoluble polymers such as cellulose27 and poly(lactic acid).28 This was due to the partial solubilisation of the polymeric substrates by the ionic liquid and the thermal stabilisation of the enzyme allowing for the reaction to be performed above the glass transition state of the polymers. The increase in substrate availability and higher operating temperatures resulted in significant improvements (up to 30-fold27) in the activity of the enzymes relative to the activity in aqueous media.
Recently, the CYP152 peroxygenase/decarboxylase OleTPRN from Rothia nasimurium, which is highly active on unsaturated fatty acids, was discovered, representing an important step toward the biocatalytic synthesis of hydrocarbons.10,29 Additionally, it has been demonstrated that productive substrate binding involves a novel motif named the ‘hydrophobic cradle’, which modulates enzyme specificity and facilitates product release.30 The wide substrate scope and tolerance allows for the formation of hydrocarbons from a diverse range of renewable fatty acid sources such as vegetable oil and waste cooking oil.30 This biocatalytic pathway presents an attractive alternative to chemical decarboxylation of fatty acids as the biocatalytic pathway does not require the use of catalysts containing noble metals such as platinum31 or palladium,32 or the use of high temperatures and pressures33 which are typically required for chemical decarboxylation. However, traditional biocatalysis reactions rely on water as a solvent, which severely limits the fatty acid substrate scope and process intensity due to limited substrate solubility. Consequently, there remains a strong impetus to expand the scope and reactivity of this CYP152 enzyme, particularly with the aim of moving toward industrially relevant conditions, as this in turn will allow for a shift the feedstock for hydrocarbons away from petrochemicals and towards biorenewables and otherwise underutilized waste streams.
Here, using OleTPRN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 30 as a model enzymatic system, we demonstrate the formation of an enzyme-surfactant nanoconjugate containing a P450 enzyme. The resulting nanoconjugate was shown to have increased thermal stability, comparable aqueous activity to the unmodified enzyme, and was soluble and stable in a range of ionic liquids. The use of ionic liquids allowed for an over 1000-fold increase in the solubility of the fatty acid substrate relative to the solubility in aqueous media, which in turn led to significant process intensification as the enzyme could operate optimally in the Vmax region. Surprisingly, the nanoconjugates also showed photocatalytic activity when solubilized in [emim][OAc]. This allowed for conversion of the fatty acid substrate without the addition of exogenous hydrogen peroxide which is typically used to drive the decarboxylation reaction.34 The increase in process intensity, improvement in enzyme stability, and the novel photochemical activity show the importance of a holistic process of biocatalysis engineering – simultaneous consideration of catalyst, solvent, and operation conditions35 – for the creation of industrially relevant biocatalytic systems with improved scope and reactivity.
30 as a model enzymatic system, we demonstrate the formation of an enzyme-surfactant nanoconjugate containing a P450 enzyme. The resulting nanoconjugate was shown to have increased thermal stability, comparable aqueous activity to the unmodified enzyme, and was soluble and stable in a range of ionic liquids. The use of ionic liquids allowed for an over 1000-fold increase in the solubility of the fatty acid substrate relative to the solubility in aqueous media, which in turn led to significant process intensification as the enzyme could operate optimally in the Vmax region. Surprisingly, the nanoconjugates also showed photocatalytic activity when solubilized in [emim][OAc]. This allowed for conversion of the fatty acid substrate without the addition of exogenous hydrogen peroxide which is typically used to drive the decarboxylation reaction.34 The increase in process intensity, improvement in enzyme stability, and the novel photochemical activity show the importance of a holistic process of biocatalysis engineering – simultaneous consideration of catalyst, solvent, and operation conditions35 – for the creation of industrially relevant biocatalytic systems with improved scope and reactivity.
Results and discussion
Following expression and purification, the P450 fatty acid decarboxylase from Rothia nasimurium (OleTPRN)30 was chemically modified by cationisation of the surface following a modified version of the methodology established by Brogan et al.26 Optimisation of the cationisation process involved shortening the reaction time from 16 hours to 5 hours, which improved the yield of soluble cationic protein. Additionally, increasing the reaction pH from 6 to 7.4 was found to prevent the loss of the heme cofactor which resulted in the cationised enzyme having comparable activity to the unmodified enzyme (2.01 ± 0.20 μmol min−1 mg−1 and 2.11 ± 0.10 μmol min−1 mg−1 for [C-OleTPRN] and OleTPRN respectively). The cationisation of the enzyme was confirmed by measuring the ζ-potential of the protein, which showed an increase from −24.5 ± 1.0 mV to +35.8 ± 1.4 mV.Previous work has made use of surfactants with a carboxylic acid head group.27 However, when these surfactants were added to C-OleTPRN the Soret band in the UV/Vis spectra of the enzyme disappeared indicating that the heme cofactor was being lost (Fig. S2†). This sensitivity was believed to be due to the similarity of the surfactant to the fatty acid substrates of the enzyme which resulted in the surfactant binding to the enzymes active site. Therefore, a sulfated surfactant was synthesised, which was then conjugated to the surface of the cationised enzyme resulting in an enzyme-surfactant nanoconjugate ([C-OleTPRN][S]). Dynamic light scattering (DLS) experiments showed an increase in the hydrodynamic diameter of the protein in water after modification from 3.20 ± 0.59 nm for the unmodified protein to 5.56 ± 1.07 nm for the nanoconjugate (Fig. 1a). Small-angle neutron scattering (SANS) confirmed the DLS data, giving a spherical diameter of 5.64 ± 0.02 nm (Fig. S3†). This indicated that the surfactant had non-covalently bound to the surface and that the protein had not aggregated during the modification. The magnitude of the increase was consistent with previous findings which showed that the surfactant does not extend away from the surface of the protein when the nanoconjugate was dissolved in polar solvents.36 The ζ-potential of the nanoconjugate was also measured, and it had decreased to −5.5 ± 0.5 mV suggesting that a slight excess of the negatively charged surfactant had conjugated to the surface of the enzyme. The nanoconjugate was then lyophilised and thermally annealed resulting in a dark red solvent-free biofluid. The resulting biofluid was soluble in aqueous solution and a range of hydrophilic and hydrophobic ionic liquids. UV/Vis analysis of the biofluid indicated that the typical ratio of enzyme to surfactant was around 150–200 molecules of surfactant per enzyme (Fig. S4†).
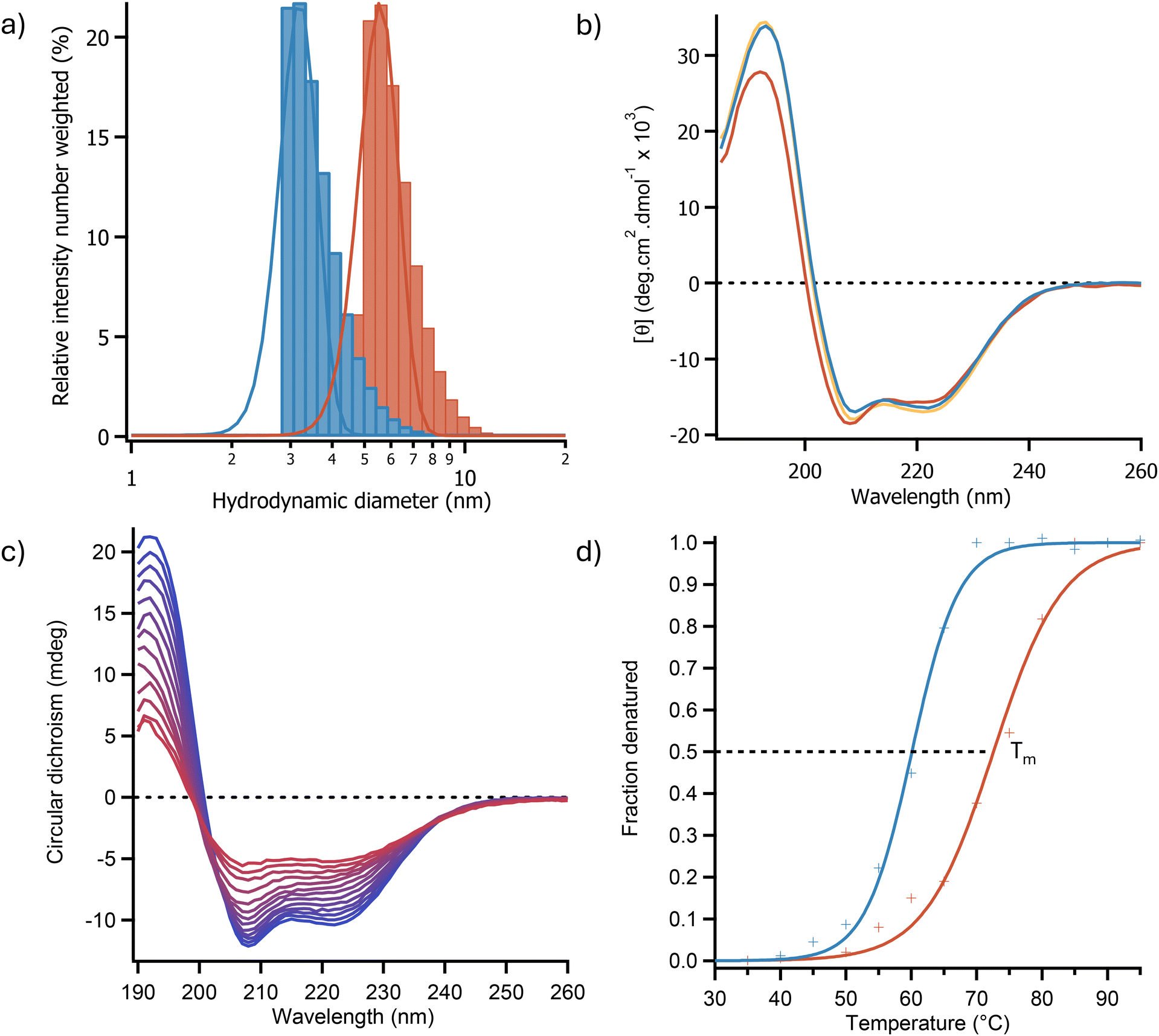 | ||
| Fig. 1 (a) Dynamic light scattering spectra showing an increase in hydrodynamic diameter between OleTPRN(blue) and [C-OleTPRN][S] (red) indicating that surfactant is bound to the surface of the enzyme. (b) Far-UV CD spectra of OleTPRN (blue), [C-OleTPRN] (orange) and [C-OleTPRN][S] (red) in aqueous solution. (c) Temperature dependent CD spectra for [C-OleTPRN][S] as it was heated from 25 °C (blue) to 95 °C (red) indicating a loss of secondary structure as temperature increased. (d) Plots of fraction denatured for OleTPRN (blue) and [C-OleTPRN][S] (red) showing an increase in thermal stability in aqueous media, data are fitted with sigmoid functions (solid lines) in accordance with a two-state model of denaturation. | ||
Circular dichroism (CD) spectroscopy was used to determine the effect of modification on the secondary structure of the protein. The CD spectra showed that the enzyme after modification and resuspension in water exhibited a near identical CD spectrum to the unmodified protein with peaks at 192 nm, 209, and 222 nm with similar intensities (Fig. 1b). This indicated that the protein had retained a predominately α-helical secondary structure (Fig. 1b). Deconvolution of the CD spectra using the BeStSel webservice37 revealed that OleTPRN and [C-OleTPRN][S] had α-helical contents of 43.8% and 44.9% respectively indicating that the secondary structure of the nanoconjugate remained largely unchanged from the native protein. Thermal denaturation CD experiments (Fig. 1c) were also used to determine if the modification of the protein had increased the thermodynamic stability of the proteins structure. Using the two-state denaturation model of denaturation, the half denaturation temperature (Tm) of the enzyme was found to increase from 60.1 ± 0.3 °C for the native enzyme to 72.5 ± 0.7 °C for [C-OleTPRN][S] (Fig. 1d). Thermodynamic analysis of the CD data revealed that the increase in thermal stability was predominately due to an increase in the enthalpy of unfolding (from 178 ± 8 kJ mol−1 for OleTPRN to 189 ± 5 kJ mol−1 for [C-OleTPRN][S]) whereas the entropy of unfolding for [C-OleTPRN][S] remained similar to OleTPRN (540 ± 18.1 J mol−1 and 532 ± 2.3 J mol−1 respectively). The activity of the P450 was determined using an ABTS assay to look at the rate of H2O2 consumption after each stage of the modification. The cationisation reaction was optimised to retain enzymatic activity which resulted in [C-OleTPRN] having a comparable activity to OleTPRN with activities of 2.01 ± 0.20 μmol min−1 mg−1 and 2.11 ± 0.10 μmol min−1 mg−1 respectively. However, conjugation of surfactant to the surface of [C-OleTPRN] had a small effect on the activity of the enzyme with the activity decreasing to 1.65 ± 0.05 μmol min−1 mg−1 for [C-OleTPRN][S]. This was believed to be due the steric bulk of the surfactant hindering access to the active site of the enzyme. Despite the small change in activity, the enzyme still retained activity towards its native substrate and based on our previous work,36 we hypothesized that the change to a non-aqueous solvent would reduce the steric hindrance of the surfactant corona, returning the activity of the native enzyme.
Prior work has demonstrated that OleTPRN has a high affinity for binding myristic acid,30 however the solubility of myristic acid is very low in water (∼20 mg L−1 at 20 °C (ref. 38)) hindering the maximum possible activity of this enzyme. We sought to overcome the issue of substrate solubility by performing the reaction in pure ionic liquids as they are typically capable of solubilizing a wide variety of molecules. To find the best ionic liquid for this reaction, we tested the solubility of myristic acid in a selection of [emim+] salts of varying polarity. Specifically, we chose the anions [OAc], [EtSO4], [OTf], and [NTf2] to cover a broad range of Kamlet–Taft polarizabilities (π*), with values of 1.05, 1.01, 0.89, and 0.85 respectively.39–41 The solubility of myristic acid was found to be highly congruent with the polarity of the anions, being most soluble in [emim][OAc] (>100 mg mL−1) and almost insoluble in [emim][NTf2] (<1 mg mL−1).
As the more polarizable ionic liquids were found to be significantly better solvents for the fatty acid substrate, the structure and stability of [C-OleTPRN][S] in pure ionic liquids containing acetate and methylsulfate anions was investigated using synchrotron radiation circular dichroism (SRCD) spectroscopy. The SRCD spectra of [C-OleTPRN][S] in [bmpyrr][OAc] and [bmpyrr][MeSO4] (UV clear surrogates of [emim] containing ionic liquids) showed negative peaks at 222 nm and 208 nm with similar intensity to the aqueous spectra (Fig. 2a). This indicated that the nanoconjugate had retained an α-helical structure in the ionic liquids that was essentially unchanged to the structure of the protein in aqueous solution (Fig. 2a).
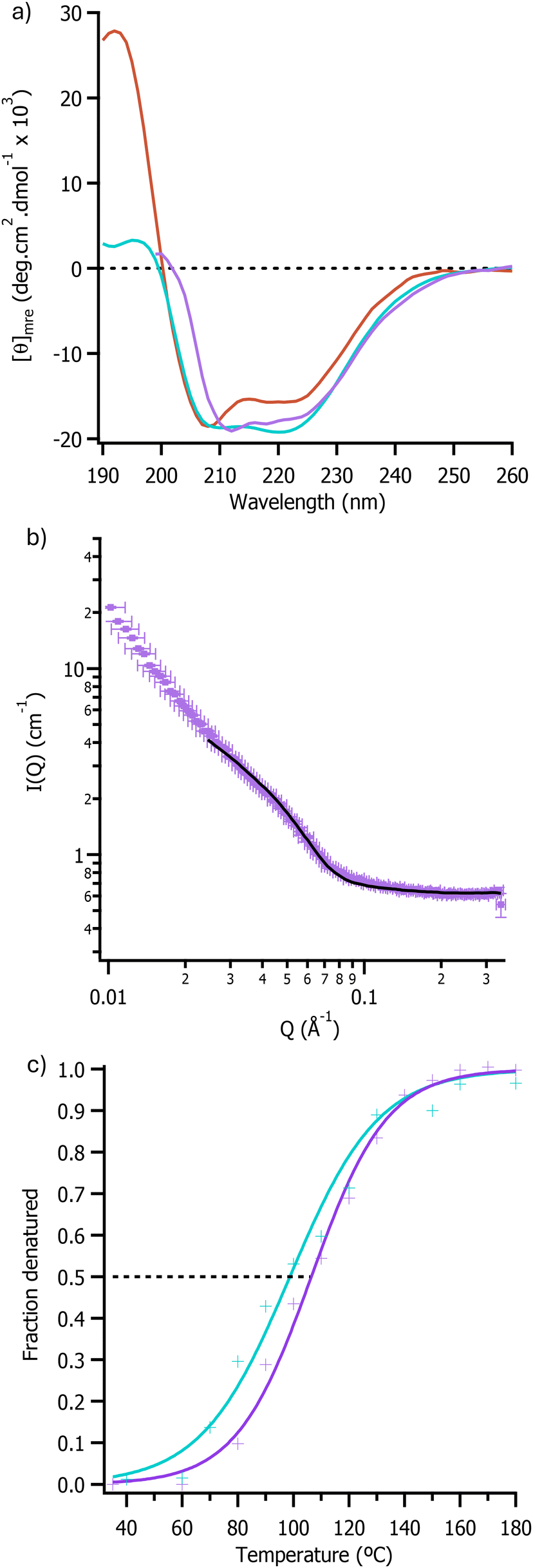 | ||
| Fig. 2 (a) Far-UV SRCD spectra of [C-OleTPRN][S] showing secondary structure in aqueous media (red), [bmpyrr][OAc] (purple), and [bmpyrr][MeSO4] (blue). (b) Neutron scattering intensity against scattering vectors for [C-OleTPRN][S] in [emim][OAc] plot was fitted using the hard sphere model in SasView and the fit is shown as a solid black line. (c) Plots of fraction denatured for [C-OleTPRN][S] in [bmpyrr][OAc] (purple) and [bmpyrr][MeSO4] (blue). | ||
SANS was then used to determine the impact of the non-aqueous environment on the tertiary structure of the enzyme. The radius of the nanoconjugate in [emim][OAc] was determined by modelling the data using the sphere model in SasView and this showed that the dimeter of [C-OleTPRN][S] was 7.20 ± 0.22 nm in [emim][OAc] (Fig. 2b). Although the value measured in the ionic liquid was larger than the radius in D2O (5.64 ± 0.02 nm), this was in agreement with previous work that showed that the surfactant corona extended away from the protein in less polar solvents.36 Taken together with the SRCD data, this demonstrated that the protein had retained both its secondary and tertiary structure, with the lack of scattering at low q in the SANS suggesting a lack of higher order aggregates.
Temperature dependent SRCD spectroscopy (Fig. S5 and S6†) was subsequently used to determine the thermal stability of the nanoconjugate in ionic liquids. These experiments indicated that the half denaturation temperature (Tm) of [C-OleTPRN][S] increased from 77.4 ± 0.7 °C in water to 106.4 ± 1.2 °C and 98.7 ± 2.0 °C in [bmpyrr][OAc] and [bmpyrr][MeSO4] respectively (Fig. 2c). The increase in thermal stability indicated that it should be possible to perform reactions at higher temperatures than in water, which in turn would result in an increase in the rate of reaction and for a higher concentration of substrate to be used.
The thermal stability of the modified enzyme was found to be the highest in acetate ionic liquids and myristic acid had the highest solubility in [emim][OAc]. Therefore, [emim][OAc] was selected as the solvent for the H2O2 dependent decarboxylation reaction as it was hoped that it would allow for the reaction to proceed as it does in water but with significantly higher loadings of substrate. However, when the reaction was performed in this ionic liquid, the conversion of fatty acid was low (<1%). Investigation of the stability of H2O2 indicated that the peroxide rapidly degraded in pure [emim][OAc] (Fig. S7†). Additionally, the enzyme was found to be sensitive to peroxide concentration, with loss of heme occurring at peroxide concentrations above 2 mM (Fig. S8†). Therefore, alternative methods for driving the reaction were explored.
It has previously been shown that the basicity of the acetate anion can deprotonate the imidazolium cation at the C2 position resulting in the formation of carbene.42,43 In addition, heme-dependent enzymes have been shown to react with carbenes44 and the heme porphyrin has been shown to act as a photoscavenger.45 Therefore, we hypothesized that it may be possible to combine these phenomena to drive the reaction photochemically. To investigate this possibility, the protein was initially exposed to UV light in [emim][OAc] and the response of the Soret band of the protein was monitored using UV/Vis spectroscopy (Fig. 3a). Analysis of the Soret band peak position revealed spectral shifts consistent with changes in the oxidation state of the heme cofactor of [C-OleTPRN][S]. Specifically, prior to illumination with UV light the Soret band peak was at 410 nm, immediately after illumination with UV light (365 nm light for 2 minutes) the peak shifted to 434 nm which indicating that the heme cofactor was being photoreduced from Fe(III) to Fe(II).46 The enzyme solution was then exposed to oxygen and the peak shifted to 422 nm suggesting that the enzyme had reacted with oxygen to form the Fe(III) state with a bound oxygen ligand (Fig. 3a).30 The Soret band of the photoreduced enzyme had the same peak position as when the enzyme was treated with H2O2 in aqueous media which further supported the idea that it should be catalytically active (Fig. S9†). When repeated in [bmpyrr][OAc] and [emim][EtSO4], the photochemical reaction did not occur suggesting that both that the imidazolium cation and acetate anion were required together. This supported the hypothesis that the carbene is the likely driver for the photochemical reaction, and that carbene formation is in turn driven by a basic anion.
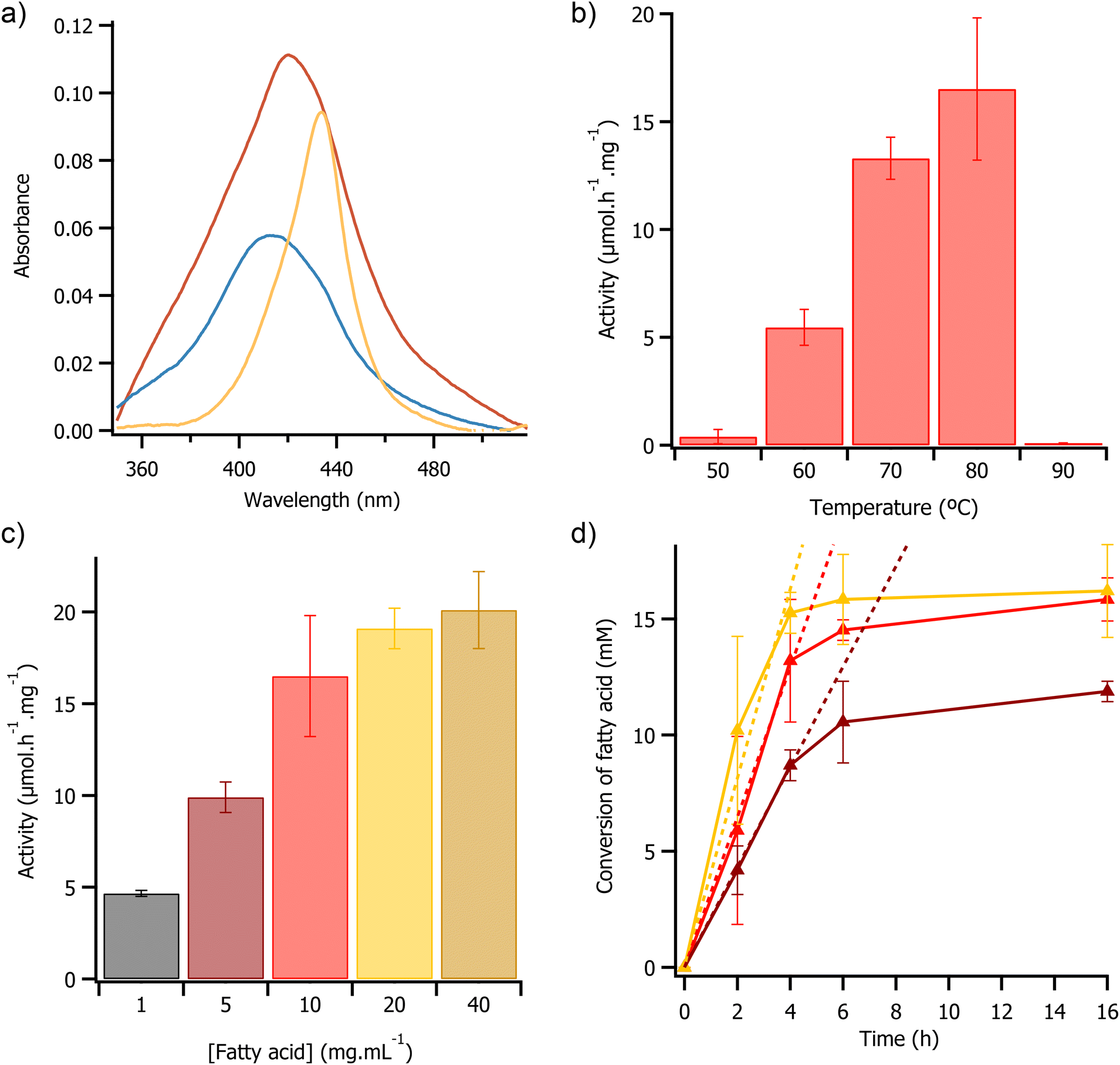 | ||
| Fig. 3 (a) UV/Vis spectra of the Soret band of [C-OleTPRN][S] in [emim][OAc] before UV exposure (blue), after 2 minutes of exposure to UV light (orange), and 10 minutes after exposure to UV light (red). (b) Plot of activity against temperature for 4 hours UV driven reactions. Reactions consisted of 1 mL [emim][OAc] containing 10 mg mL−1 myristic acid and [C-OleTPRN][S] (equivalent to 0.2 mg of C-OleTPRN). (c) Plot of activity against concentration of myristic acid for 4 hours UV driven reactions at 80 °C. Reactions consisted of 1 mL [emim][OAc] containing a range of different myristic acid concentrations and [C-OleTPRN][S] (equivalent to 0.2 mg of C-OleTPRN). (d) Plot of fatty acid conversion against time for UV driven reactions at 80 °C. Reactions consisted of 1 mL [emim][OAc] containing 5 mg mL−1 (brown), 10 mg mL−1 (red), and 20 mg mL−1 (yellow) of myristic acid and [C-OleTPRN][S] (equivalent to 0.2 mg of C-OleTPRN). Linear fits of initial activity are shown as dashed lines in matching colour. | ||
Having established that UV light could be used to photo-reduce the heme and in the presence of O2 present a potentially active form of heme, we hypothesized that this could be used for catalysis in the absence of H2O2. To test if the enzyme had photochemical activity, the nanoconjugate was dissolved in [emim][OAc] containing myristic acid and then illuminated under UV light. Due to analytical ease, the consumption of substrate was monitored by GCMS. Initial reactions showed that both the enzyme and UV light were required to consume the substrate as the control reactions in the absence of either light or enzyme showed no decrease in substrate concentration, which confirmed that the enzyme required illumination with UV light to perform the photochemical reaction (Fig. S10†). The mass spectrum of the photochemical reaction containing the enzyme suggested that the reaction was forming the intended alkene, therefore indicating that the decarboxylation reaction was proceeding in the ionic liquid and only in the presence of the enzyme (m/z 182 – Fig. S11†).
Given that we could observe the decarboxylation reaction in the ionic liquid, we repeated between the reaction at temperatures ranging from 50 °C to 90 °C, in a bid to determine the optimal temperature for the photochemical reaction. These temperatures were selected as below 50 °C the modified enzyme had limited solubility in ionic liquid, and the thermal CD analysis indicated that the protein would have begun to denature above 90 °C. As anticipated, at temperatures less than 50 °C the enzyme had low activity (<0.25 μmol h−1 mg−1), attributed to low solubility of the [C-OleTPRN][S] in [emim][OAc] at those temperatures. At 50 °C the enzyme activity was 0.40 ± 0.33 μmol h−1 mg−1, and activity then increased progressively with temperature, reaching a maximum of 16.52 ± 3.3 μmol h−1 mg−1 at 80 °C (Fig. 3b). This significant increase in activity was likely due to both higher thermal energy in the system, and a decrease in the viscosity of [emim][OAc],47 which improved the kinetics of the reaction. Again, as anticipated, at temperatures of 90 °C and above there was a no detectable activity, this coincided with both the loss of secondary structure (Fig. 2c) and heme cofactor (Fig. S12†) as the protein began to denature.49
We then sought to investigate the effect of substrate concentration on enzyme activity in a bid to determine any Michaelis Menten-like behaviour. As the concentration of substrate was increased from 1 mg mL−1 to 40 mg mL−1 there was a non-linear increase in enzyme activity from 4.67 ± 0.16 μmol h−1 mg−1 to 20.5 ± 2.1 μmol h−1 mg−1 (Fig. 3c). The observed change was likely due to saturation of the enzyme active site with substrate, consistent with Michaelis Menten type enzyme kinetics.48 The Michaelis–Menten model was fitted to the data which indicated that the Km of the reaction was 5.1 ± 1.2 mg mL−1 and a Vmax of 23.3 μmol h−1 mg−1 (Fig. S13†). During the first 4 hours of the reaction, the rate of substrate consumption was constant with initial rates of 2.2 ± 1 μmol h−1, 3.2 ± 0.1 μmol h−1, and 4.1 ± 0.4 μmol h−1 when the substrate loading was 5 mg mL−1, 10 mg mL−1, and 20 mg mL−1 respectively. However, after 4 hours there was a significant decrease in activity with all of the loadings trialled, with the reaction effectively stopping after 16 h (Fig. 3d). The turnover numbers after 4 hours were 1740, 2640, and 3060 at 5, 10, and 20 mg mL−1 of substrate respectively. The turnover numbers achieved here were greater than the reported literature values for the H2O2 driven reaction in water using OleTPRN30 and for the widely studied fatty acid decarboxylase from Jeotgalicoccus sp., OleTJE49,50 (Turnover numbers of 250 and 1099 respectively). The difference in turnover numbers with different loadings of substrate suggested that the loss of activity could not be related to active site locking (or substrate availability), but instead related to some other deactivation of the enzyme. UV/Vis spectroscopy revealed that this decrease in activity coincided with a decrease in the intensity of the Soret band (Fig. S14†), suggesting that the loss in activity may have been due to photobleaching of the heme cofactor.51 This could be mitigated by with finer control of the flux of UV light or by using pulses of UV instead of continuous illumination, however this was beyond the scope of this study.
The potential for reusing the solvent and enzyme was also investigated. At the end point of the reaction the ionic liquid solution was washed with hexane, this resulted in removal of product and unreacted substrate from the ionc liquid (Fig. S16†). After removal of the hexane layer the ionic liquid was dried and subsequently weighed, which indicated that >98% of the ionic liquid had been recovered. However, due to likely aforementioned photobleaching of the heme cofactor, it was not possible to re-use the enzyme in this current set-up.
Interestingly, during these reactions there was also a peak present in the GCMS chromatographs of enzymatic reactions which had mass spectra which matched that of an epoxide formed from the expected alkene product (m/z 199 – Fig. S17†). To investigate this further, the alkene product standard was mixed with [C-OleTPRN][S] in [emim][OAc] and illuminated with UV light. This reaction led to formation of the epoxide peak in the GCMS chromatogram that was not present in the control reaction (Fig. S14†). This indicated that, through finer control of reaction conditions (e.g. oxygen concentration), it may be possible to further convert the fatty acid to the corresponding epoxide from the alkene under the same reaction conditions. This is a fascinating prospect that, whilst beyond what we set out to achieve here, warrants further study.
Conclusions
In conclusion, we have demonstrated a chemical modification strategy for the solubilization and stabilization of a CYP152 decarboxylase, OleTPRN, in a range of pure ionic liquids. This resulted in a significant thermal stabilisation of the enzyme with an increase in Tm from 60.1 ± 0.3 °C for the native enzyme in water to up to 106.4 ± 1.2 °C for the modified enzyme in ionic liquids. This broadened operational window of the enzyme, in conjunction with considerably higher substrate concentrations, allowed for significant process intensification for the conversion of fatty acids to alkenes. This was due to enhanced enzyme activity as a result of drastically improved thermal stability coupled with substrate loadings that were far greater than the Km. The improvements in process intensity should allow for a dramatic improvement in the complete E-factor (cEF)52 of the reaction. This is due to the low solubility of the substrate (<1 mg mL−1)53 in water resulting in a cEF of over 1000 for the aqueous process, whereas in pure ionic liquids the cEF could be as low as 30. Furthermore, we also discovered a new photocatalytic reaction pathway for this heme containing enzyme that circumvents the requirement traditional requirement for exogenous H2O2. By increasing enzyme stability, improving in substrate solubility, and opening new reaction pathways, we have demonstrated the potential of this methodology for a holistic process of biocatalysis engineering, where the versatility of ionic liquids provides a unique design framework to enhance the biocatalytic process of a multitude of enzymatic reactions. In particular, application of this stabilisation technique to a range of other heme containing enzymes may allow for utilisation of this novel photochemical reactivity for a much-improved deployment of these enzymes for industrial transformations.Experimental
Materials and methods
All of the 1-ethyl-3-methylimidazolium (emim+) ionic liquids were acquired from iolitec. Anionic surfactants and 1-butyl-1-methylpyrrolidinium (bmpyrr+) ionic liquids were synthesised following literature procedures.27,54 All other chemicals were acquired from Sigma-Aldrich, and all chemicals were used without further purification. Full methodological details can be found in the ESI Materials and methods,† but key aspects are summarized below.Heterologous expression and purification of OleTPRN
Recombinant-pET-28a vector was co-transformed into E. coli BL21(DE3) with pG-TF2 plasmid (Takara Bio, Kusatsu, JPN), which encodes GroEL, GroES, and Tig chaperones. The expression and purification were performed as described by Rade et al. Briefly, the transformed strain was grown in 500 mL of a selective (chloramphenicol and kanamycin) Terrific Broth medium (TB) supplemented with 125 mg L−1 thiamine hydrochloride and 500 μL trace metals solution (containing 50 mM FeCl3, 20 mM CaCl2, 10 mM MnCl2, 10 mM ZnCl2, 2 mM CoCl2, 2 mM CuCl2, 2 mM NiSO4) at 37 °C. The growth temperature was reduced to 20 °C when the optical density (OD600 nm) of the culture reached 0.6. Then 200 μM isopropyl-β-D-thiogalactopyranoside (IPTG) (Invitrogen), 5 μM of FeCl3, 100 μM of δ-aminolevulinic acid (ALA), and 10 μg mL−1 tetracycline were added, and the culture was kept at 20 °C overnight. Cells were harvested by centrifugation at 7000g (10 min at 4 °C) and subsequently the cell pellets were resuspended in buffer A [100 mM potassium phosphate (pH 7.5), 300 mM NaCl, 10 mM imidazole, and 5% (v/v) glycerol] and disrupted by sonication in a Vibracell VCX 500 device (Sonics and Materials, Newtown, USA), by performing 2 min of sonication (40% amplitude and no pause), followed by 5 minutes of stirring at 4 °C. This procedure was repeated 5 times. The cell lysate was centrifugated at 12000g for 25 min and the clear supernatant was loaded onto a 5 mL His-Trap chelating HP column (GE Healthcare Biosciences), pre-equilibrated with buffer A. The target protein was eluted with a gradient of imidazole (5 to 300 mM) in buffer A. The eluted sample was dialyzed overnight against 100 mM phosphate buffer (pH 7.5) containing 50 mM NaCl and 5% (v/v) glycerol. Dialyzed protein was loaded onto a fast flow Q-sepharose column (Sigma-Aldrich Co., St Louis, USA) and eluted by a NaCl gradient (50 mM to 1 M). The heterologous expression and enzyme purification steps were evaluated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE, 12%) and spectrophotometric analysis of typical Soret bands.
Protein modification
OleTPRN was added to an aqueous solution of N,N,N‘-trimethyl-1,3-propanediamine (250 mM), N-hydroxysuccinimide (NHS, 50 mM) and phosphate (10 mM, pH 7). The reaction was initiated by addition of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC, 150 mM) and the pH was maintained between 7 and 7.5 by addition of dilute HCl for 5 hours. After the cationisation reaction the solution was centrifuged at 3220g for 10 minutes and subsequently syringe filtered (0.2 μm) to remove any aggregates that has formed. The protein solution was then buffer exchanged by repeated centrifugal concentration (Corning® Spin-X®, 10000 MWCO) and resuspension in phosphate buffer (10 mM pH 7) yielding a solution of cationised OleTPRN (C-OleTPRN).
C-OleTPRN was subsequently added dropwise to a stirred solution of neutralised surfactant (pH 6.5–7) and then stirred overnight to allow for the conjugation to occur. After the conjugation, the protein-surfactant nanoconjugate was syringe filtered (0.2 μm) and then buffer exchanged by repeated centrifugal concentration (Corning® Spin-X®, 10000 MWCO) and resuspension in phosphate buffer (10 mM pH 7). The purified nanoconjugate was then lyophilised and the dry nanoconjugate was then thermally annealed at 60 °C for 1 hour to yield a viscous liquid ([C-OleTPRN][S]).
Surfactant synthesis
Brij C10 sulfate ester was synthesised by reacting Brij C10 with sulfamic acid.54 An equimolar mixture of Brij C10 (10.0 g, 14.7 mmol) and sulfamic acid (1.43 g, 14.7 mmol) was heated neat at 120 °C for 18 h under nitrogen producing a brown viscous liquid. The resulting liquid was cooled to room temperature and diluted with 200 mL of dichloromethane and stirred for 1 h. The solution was subsequently filtered using filter paper, and the solvent was removed from the filtrate using a rotatory evaporator yielding a viscous oil. The oil was then washed with 3 × 100 mL of hexane to remove non-polar impurities. The hexane was then decanted, and the oil was lyophilised to give ammonium Brij C10 sulfate ester as an off-white waxy solid (8.62 g, 11.08 mmol) at a 75% yield.Synthesis of 1-butyl-1-methylpyrrolidinium acetate
1-Butyl-1-methylpyrrolidinium acetate ([bmpyrr][OAc]) was synthesized following a literature procedure.27 Briefly, silver acetate (12.81 g, 0.085 mol, 1.0 eq.) in deionized water (50 mL) was added to a flask covered in aluminium foil containing [bmpyrr][Cl] (15.00 g, 0.084 mol, 1.1 equivalents). The resulting solution was stirred at room temperature for 3 days and then filtered through filter paper. The solution was then mixed with activated charcoal for 24 h then filtered and tested for silver and chloride ions with aqueous hydrochloric acid and silver nitrate solutions respectively, no precipitate formed which indicated that no silver or chloride ions were present in the solution. The solution was then passed through a neutral alumina column (Thermo Scientific, HyperSep™ C18 cartridge). The resulting product was then lyophilised to give [bmpyrr][OAc] (9.3 g, 0.047 mol, 56.0%) as a colourless liquid. [bmpyrr][OAc]: 1H NMR (D2O, 400 MHz) δ 0.87 (t, 3H, CH3), 1.30 (h, 2H, CH2), 1.69 (m, 2H, CH2,), 1.82 (s, 3H, CH3, OAc), 2.11 (m, 4H, CH2), 2.94 (s, 3H, CH3), 3.23 (m, 2H, CH2), 3.41 (m, 4H, CH2).SRCD
Synchrotron radiation circular dichroism (SRCD) spectra of [C-OleTPRN][S] in ionic liquids were collected on beamline B23 at the Diamond Light source. Temperature dependent SCRD experiments were performed using a Linkam thermal stage. Measurements were made with a wavelength range of 180–260 nm, integration time of 2 s and 1 nm wavelength interval. For thermal denaturation experiments, spectra were collected in 10 °C intervals between 30–200 °C, samples were incubated at each temperature for 300 seconds prior to each spectrum being collected. Ionic liquid samples were cast as thin films between two quartz plates.H2O2 consumption assay
Briefly, 10 μg of decarboxylase enzyme was added to 1 mL of phosphate buffer (100 mM pH 7.2) containing 0.1 mM H2O2 and 0.5 mM myristic acid and was shaken for 5 minutes. The reaction was then quenched by addition of 0.2 mM 2,2′-Azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) and 3 U of horse radish peroxidase (HRP), which reacted with the remaining H2O2 present in the reaction. The reaction was centrifuged for 5 minutes to clarify the solution and to ensure the HRP had time to consume the residual H2O2. The concentration of oxidised ABTS was then quantified by measuring the absorbance at 420 nm (ref. 55) (ε420 = 36 mM−1 cm−1) using a nanophotometer (Implen, N60). The rate of consumption of H2O2 was then calculated by comparing the absorbance of the reaction compared to a control reaction containing no OleTPRN (see ESI Materials and methods†).UV reactions
[C-OleTPRN][S] was weighed in to 4 mL glass vials equipped with a stirrer bar, heated to 60 °C, and stirred at 200 rpm for one hour. Subsequently, 500 μL of pure ionic liquid (typically <0.5% w/w water) containing the fatty acid substrate (1–40 mg mL−1) was added to the vial containing [C-OleTPRN][S]. The solution was stirred for one hour such that the nanoconjugate was fully dissolved. Then the reaction was initiated by illuminating the solution with UV light (365 nm, 8 watts). The reaction was allowed to proceed for a predetermined amount of time, after which the UV light was removed, and the reaction was cooled to room temperature and quenched by addition of 1 mL of 3 M HCl in methanol. The quenched reactions were then heated to 60 °C and stirred at 800 rpm for one hour to form a fatty acid methyl ester from any unreacted free fatty acid remaining in the reaction. The resulting solution was then extracted with 1 mL of hexane and the hexane layer was analysed by GCMS to determine conversions. Conversions were determined by comparing the area of the peak to a calibration curve and a control reaction containing no enzyme. Both reactions and controls were performed in triplicate.GC-MS derivatisation method
After the reaction in ionic liquids the reactions were cooled to room temperature and quenched by addition of 3 M HCl in dry methanol. The samples were then stirred at 60 °C for 1 h to form a fatty acid methyl ester. The samples were then cooled to room temperature and extracted with hexane and the hexane layer was analysed by GC-MS.Michaelis–menten kinetics fitting
The Michaelis–Menten equation was fitted to the data using IGOR Prowhere v is the velocity of the reaction, Vmax is the maximum velocity of the reaction, [S] is the concentration of substrate and Km is the Michaelis constant.
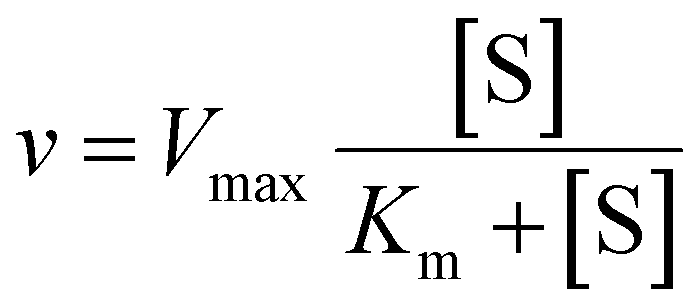
Author contributions
J. H. N. performed experiments, analysed data, and wrote the manuscript. M. C. dA. and R. R. dM. synthesized and characterized the decarboxylase. A. P. S. B. and L. M. Z. conceived the concept and developed the research. A. P. S. B. edited the manuscript. All authors provided feedback and inputted to the final manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
Leticia Maria Zanphorlin has patent pending to Brazilian National Institute of Industrial Property – INPI. Otherwise, the authors declare no other conflicts of interest.Acknowledgements
J. H. N. and A. P. S. B. thank King's College London for financial support, and L. M. Z. and M. C. A. thank LNBR – CNPEM (Campinas, Brazil) and grants from the São Paulo Research Foundation (FAPESP) #2019/08855-1; #2019/12599-0 and #2022/13956-4 for their support. The authors also thank Diamond Light Source for access to the B23 beamline for SCRD experiments, Institut Laue-Langevin for access to the D33 beamline for SANS experiments, Adrian Sanchez-Fernandez for assistance with analysing the SANS data, and Oscar Aryton for assistance with the GCMS experiments.References
- P. R. Ortiz De Montellano, Chem. Rev., 2010, 110, 932–948 CrossRef CAS PubMed.
- A. D. N. Vaz, D. F. McGinnity and M. J. Coon, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 3555–3560 CrossRef CAS PubMed.
- J. L. Grant, C. H. Hsieh and T. M. Makris, J. Am. Chem. Soc., 2015, 137, 4940–4943 CrossRef CAS PubMed.
- P. S. Coelho, E. M. Brustad, A. Kannan and F. H. Arnold, Science, 2013, 339, 307–310 CrossRef CAS PubMed.
- E. O'Reilly, V. Köhler and S. L. Flitsch, Chem. Commun., 2011, 47, 2490–2501 RSC.
- J. C. Lewis, P. S. Coelho and F. H. Arnold, Chem. Soc. Rev., 2011, 40, 2003–2021 RSC.
- Z. Li, Y. Jiang, F. P. Guengerich, L. Ma, S. Li and W. Zhang, J. Biol. Chem., 2020, 295, 833–849 CrossRef PubMed.
- L. A. Harwood, Z. Xiong, K. E. Christensen, R. Wang, L. L. Wong and J. Robertson, J. Am. Chem. Soc., 2023, 145, 27767–27773 CrossRef CAS PubMed.
- R. J. Sowden, S. Yasmin, N. H. Rees, S. G. Bell and L. L. Wong, Org. Biomol. Chem., 2005, 3, 57–64 RSC.
- S. Matthews, J. D. Belcher, K. L. Tee, H. M. Girvan, K. J. McLean, S. E. J. Rigby, C. W. Levy, D. Leys, D. A. Parker, R. T. Blankley and A. W. Munro, J. Biol. Chem., 2017, 292, 5128–5143 CrossRef CAS PubMed.
- F. H. Arnold, Angew. Chem., Int. Ed., 2018, 57, 4143–4148 CrossRef CAS PubMed.
- S. T. Jung, R. Lauchli and F. H. Arnold, Curr. Opin. Biotechnol., 2011, 22, 809–817 CrossRef CAS.
- L.-H. Xu and Y.-L. Du, Synth. Syst. Biotechnol., 2018, 3, 283–290 CrossRef.
- A. W. Munro, H. M. Girvan, A. E. Mason, A. J. Dunford and K. J. McLean, Trends Biochem. Sci., 2013, 38, 140–150 CrossRef CAS.
- H. L. Wapshott-Stehli and A. M. Grunden, Enzyme Microb. Technol., 2021, 145, 109744 CrossRef CAS PubMed.
- E. V. Pronina, Y. A. Vorotnikov, T. N. Pozmogova, A. O. Solovieva, S. M. Miroshnichenko, P. E. Plyusnin, D. P. Pishchur, I. V. Eltsov, M. V. Edeleva, M. A. Shestopalov and O. A. Efremova, ACS Sustainable Chem. Eng., 2020, 8, 5371–5379 CrossRef CAS.
- R. Tolando, A. Zanovello, R. Ferrara, J. N. Iley and M. Manno, Toxicol. Lett., 2001, 124, 101–111 CrossRef CAS PubMed.
- H. J. Ruijssenaars, E. M. G. M. Sperling, P. H. G. Wiegerinck, F. T. L. Brands, J. Wery and J. A. M. de Bont, J. Biotechnol., 2007, 131, 205–208 CrossRef CAS PubMed.
- R. Bernhardt and V. B. Urlacher, Appl. Microbiol. Biotechnol., 2014, 98, 6185–6203 CrossRef CAS PubMed.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS.
- C. J. Clarke, W. C. Tu, O. Levers, A. Bröhl and J. P. Hallett, Chem. Rev., 2018, 118, 747–800 CrossRef CAS PubMed.
- D. Singh, G. Sharma and R. L. Gardas, ChemistrySelect, 2017, 2, 3943–3947 CrossRef CAS.
- R. A. Sheldon, R. M. Lau, M. J. Sorgedrager, F. van Rantwijk and K. R. Seddon, Green Chem., 2002, 4, 147–151 RSC.
- F. Xu, J. Sun, N. V. S. N. M. Konda, J. Shi, T. Dutta, C. D. Scown, B. A. Simmons and S. Singh, Energy Environ. Sci., 2016, 9, 1042–1049 RSC.
- A. P. S. Brogan, New J. Chem., 2021, 45, 6577–6585 RSC.
- A. P. S. Brogan and J. P. Hallett, J. Am. Chem. Soc., 2016, 138, 4494–4501 CrossRef CAS PubMed.
- A. P. S. Brogan, L. Bui-Le and J. P. Hallett, Nat. Chem., 2018, 10, 859–865 CrossRef CAS PubMed.
- S. M. Meza Huaman, J. H. Nicholson and A. P. S. Brogan, Cell Rep. Phys. Sci., 2024, 5, 101783 CrossRef CAS.
- C. H. Hsieh, X. Huang, J. A. Amaya, C. D. Rutland, C. L. Keys, J. T. Groves, R. N. Austin and T. M. Makris, Biochemistry, 2017, 56, 3347 CrossRef CAS.
- L. L. Rade, W. C. Generoso, S. Das, A. S. Souza, R. L. Silveira, M. C. Avila, P. S. Vieira, R. Y. Miyamoto, A. B. B. Lima, J. A. Aricetti, R. R. de Melo, N. Milan, G. F. Persinoti, A. M. F. L. J. Bonomi, M. T. Murakami, T. M. Makris and L. M. Zanphorlin, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2221483120 CrossRef CAS.
- J. Fu, X. Lu and P. E. Savage, ChemSusChem, 2011, 4, 481–486 CrossRef CAS PubMed.
- M. Arroyo, L. Briones, H. Hernando, J. M. Escola and D. P. Serrano, Energy Fuels, 2021, 35, 17167–17181 CrossRef CAS.
- B.-S. Chen, Y.-Y. Zeng, L. Liu, L. Chen, P. Duan, R. Luque, R. Ge and W. Zhang, Renewable Sustainable Energy Rev., 2022, 158, 112178 CrossRef CAS.
- A. Dennig, M. Kuhn, S. Tassoti, A. Thiessenhusen, S. Gilch, T. Bülter, T. Haas, M. Hall and K. Faber, Angew. Chem., Int. Ed., 2015, 54, 8819–8822 CrossRef CAS PubMed.
- R. A. Sheldon and P. C. Pereira, Chem. Soc. Rev., 2017, 46, 2678–2691 RSC.
- A. P. S. Brogan, R. B. Sessions, A. W. Perriman and S. Mann, J. Am. Chem. Soc., 2014, 136, 16824–16831 CrossRef CAS PubMed.
- A. Micsonai, F. Wien, L. Kernya, Y.-H. Lee, Y. Goto, M. Réfrégiers and J. Kardos, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E3095–E3103 CrossRef CAS PubMed.
- O. Albrecht, H. Matsuda, K. Eguchi and T. Nakagiri, Thin Solid Films, 1999, 338, 252–264 CrossRef CAS.
- S. Spange, R. Lungwitz and A. Schade, J. Mol. Liq., 2014, 192, 137–143 CrossRef CAS.
- Y. Hiraga, A. Duereh and R. L. Smith, J. Supercrit. Fluids, 2018, 134, 12–20 CrossRef CAS.
- N. Weiß, C. H. Schmidt, G. Thielemann, E. Heid, C. Schröder and S. Spange, Phys. Chem. Chem. Phys., 2021, 23, 1616–1626 RSC.
- N. M. A. N. Daud, E. Bakis, J. P. Hallett, C. C. Weber and T. Welton, Chem. Commun., 2017, 53, 11154–11156 RSC.
- N. D. Clement, K. J. Cavell, N. D. Clement and K. J. Cavell, Angew. Chem., Int. Ed., 2004, 43, 3845–3847 CrossRef CAS PubMed.
- R. D. Lewis, M. Garcia-Borràs, M. J. Chalkley, A. R. Buller, K. N. Houk, S. B. J. Kan and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 7308–7313 CrossRef CAS PubMed.
- J. Chen and W. R. Browne, Coord. Chem. Rev., 2018, 374, 15–35 CrossRef CAS.
- A. Das, S. Gao, R. G. Lal, M. H. Hicks, P. H. Oyala and F. H. Arnold, J. Am. Chem. Soc., 2024, 146, 20556–20562 CrossRef CAS PubMed.
- M. Bücherl, F. Hardock, E. Kaiser, A. Netsch and K. Schaber, J. Chem. Eng. Data, 2017, 62, 720–728 CrossRef.
- K. A. Johnson and R. S. Goody, Biochemistry, 2011, 50, 8264–8269 CrossRef CAS PubMed.
- Y. Jiang, Z. Li, C. Wang, Y. J. Zhou, H. Xu and S. Li, Biotechnol. Biofuels, 2019, 12, 79 CrossRef PubMed.
- D. Bauer, I. Zachos and V. Sieber, ChemBioChem, 2020, 21, 3273–3281 CrossRef CAS PubMed.
- G. Tapia, A. Galetovic, E. Lemp, E. Pino and E. Lissi, Photochem. Photobiol., 1999, 70, 499–504 CAS.
- F. Roschangar, R. A. Sheldon and C. H. Senanayake, Green Chem., 2015, 17, 752–768 RSC.
- P. Khuwijitjaru, S. AdachiI and R. Marsuno, Biosci. Biotechnol. Biochem., 2002, 66, 1723–1726 CrossRef CAS PubMed.
- G. A. Benson and W. J. Spillane, Chem. Rev., 1980, 80, 151–186 CrossRef CAS.
- T. Kenzom, P. Srivastava and S. Mishra, Appl. Environ. Microbiol., 2014, 80, 7484–7495 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc05292g |
| This journal is © The Royal Society of Chemistry 2025 |