
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel sustainable synthesis of a formaldehyde-free thermosetting phenolic resin through solvent-free reactive extrusion†
Alex
Maokhamphiou
ab,
Matthieu
Zinet
a,
William
Guerin
b,
Arnaud
Soisson
b,
Morgane
Petit
b,
Guillaume
Jobard
a,
Fernande
da Cruz-Boisson
a,
Karim
Delage
a,
Romain
Tavernier
 a and
Véronique
Bounor-Legaré
*a
a and
Véronique
Bounor-Legaré
*a
aUniversite Claude Bernard Lyon 1, INSA Lyon, Université Jean Monnet, CNRS UMR 5223, Ingénierie des Matériaux Polymères, Villeurbanne Cédex, France. E-mail: bounor@univ-lyon1.fr
bSaint-Gobain Research Paris, département Produits Composites et Revêtements de Surface, groupe Liants Polymères, 39 Quai Lucien Lefranc, 93300 Aubervilliers, France
First published on 27th February 2025
Abstract
This study proposes a new and elegant way to synthesize thermosetting phenolic resins through a green, solvent-free and versatile reactive extrusion route. In that frame, terephthalaldehyde (TPA), a non-toxic aromatic dialdehyde, has been selected to replace formaldehyde while resorcinol has been chosen as a replacement of phenol. The syntheses were performed without solvent at temperatures between 150 and 170 °C with a reaction time of around 3 minutes. The resins were synthesized at different TPA-to-resorcinol molar ratios (0.6 and 1.6). This study investigates the mechanism and chemical reactions occurring during the reactive extrusion by characterizing the resin composition through NMR and mass spectrometry (<1500 g mol−1). In addition, differential scanning calorimetry (DSC) analyses were carried out to study the kinetics of the reactions and to estimate the activation energies (32–54 kJ mol−1) through various calculation methods (Flynn–Wall–Ozawa, Friedman, and Vyazovkin methods). It was demonstrated that multiple and consecutive reactions (electrophilic aromatic substitution and condensation) occur during the reactive extrusion process. Additionally, the resins synthesized by reactive extrusion exhibited an exothermic post-reactivity signature in DSC, enabling the estimation of the conversion degrees of 0.63 and 0.59, respectively, for ratios of 0.6 and 1.6. Finally, the resins obtained through reactive extrusion demonstrate great thermal stability even prior to post-heating.
Green foundation1. Phenolic resins are usually synthesized from non-renewable and toxic monomers phenol and formaldehyde, using solvents in batch processes. In this work, reactive extrusion is shown to be an efficient solventless process, allowing the synthesis of prepolymers in a short residence time, with potentially bio-based monomers.2. Curing kinetics allow assessing the reactivity of monomers. Then, formaldehyde-free resins were synthesized in approximately 3 minutes in a continuous process in a range of temperatures between 150 and 170 °C. The further crosslinked polymers displayed high thermal stability and a high Tg value (197 °C). 3. Kinetic modelling allows for the screening of reactivity before setting the extrusion conditions. This method opens the way for applying reactive extrusion using a large set of bio-based and non-toxic monomers. Further evaluation of the energy efficiency and the LCA of these new resins is yet to be performed. |
Introduction
For over a century, phenolic resins have been studied and developed as an important thermosetting resin material. The origin of these phenolic resins, obtained by reaction between phenol and formaldehyde, can be traced back to 1872 when they were initially discovered by Adolph von Bayer.1 Since then, phenol formaldehyde (PF) has gained worldwide attention as it is widely used in a broad range of applications (electronics, transportations, and buildings).2–4 Phenolic thermosets exhibit great mechanical properties with excellent thermal and chemical resistance5–7 as they present a highly aromatic and crosslinked network.8,9However, these resins also display a number of drawbacks. Phenolic resins are mainly synthesized from phenol and formaldehyde which are both toxic and classified as CMR (carcinogenic, mutagenic and reprotoxic) chemicals.10,11 Additionally, these monomers are fossil-based10 chemical building blocks leading to cost and availability dependence on petroleum prices, not to mention the growing environmental concerns about using limited and non-renewable resources. In turn, a lot of efforts have been made to substitute these compounds in phenolic resins. The main challenge is to find a replacement of phenol and formaldehyde by low molar mass monomers with great reactivities. This will lead to a high crosslinking and aromatic density in the final material that can be used in high-performance applications.
Previous studies have been carried out to replace formaldehyde with diverse aldehydes such as furfural11,12 or glyoxal.13,14 Nonetheless, even if both of these monomers are less toxic than formaldehyde, they are still expected to be harmful and classified as CMR.15,16 Foyer et al.17 evaluated the reactivity of phenol towards various bio-based and non-toxic aromatic dialdehydes. In this study, the phenol and the aldehyde precursors were poured into a round-bottom flask and heated at 130 °C. The resin synthesis reaction was initiated with the addition of 0.3 eq. of sodium hydroxide dissolved in 50 wt% aqueous solution. 4-Hydroxybenzaldehyde, vanillin, syringaldehyde and terephthalaldehyde (TPA) were tested as alternative aldehyde monomers. The latter showed the best reactivity owing to the second aldehyde group at the para position relative to the first one, acting as an electron withdrawing group and thus enhancing the reactivity of the opposite aldehyde. Moreover, terephthalaldehyde can react with up to four phenolic compounds, leading to a highly aromatic and crosslinked structure. Besides, the aromatic structure of TPA is favorable to obtaining a material with a high glass transition temperature and great thermal and mechanical properties. Indeed, the cured materials produced from the phenol–TPA resin exhibit a temperature of 500 °C at 10 wt% degradation, a char yield of 69% at 900 °C under a nitrogen atmosphere17 and a glass transition temperature of 160 °C.18 Thus, TPA is a promising alternative to replace formaldehyde due to its non-toxicity feature and its potential to be a bio-based compound. Indeed, TPA can be produced from para-xylene, which is involved in the mass production of bio-based polyethylene terephthalate (Fig. S1 in the ESI†).19 Therefore, there is great interest in producing this chemical component via sustainable pathways using biomass. These pathways include cycloaddition of biomass-derived furans, synthesis from isobutanol or direct conversion of lignocellulose via catalytic fast pyrolysis.20
On the other hand, phenol also has to be replaced in the synthesis of phenolic resins. Lignin represents a considerable source of renewable and bio-based phenolic compounds in nature.21,22 In 2004, the pulp and paper industry alone has generated 50 million tons of extracted lignin.23,24 Many efforts have been made towards the valorization of lignin as a raw material to produce bio-based materials, and the lignin phenolic structure can now be found in the production of some phenolic resins.25–28 However, lignin is composed of three different monolignol monomers, with varying ratios depending on the plant species, introducing possible variability in their chemical structures.29 Furthermore, the use of crude lignin as a substitute for phenol has been challenging as its reactive aromatic sites are less reactive than those of phenol. Therefore, crude lignin must be chemically modified to increase the content of phenolic hydroxyl groups to enhance the reactivity.30 Phenolation,31 methylolation32 and demethylation33 are among the various methods employed to enhance the reactivity of lignin. However, these methods are still challenging to implement, requiring additional steps. Yet, resorcinol, a smaller phenolic monomer, appears to be a better candidate to replace phenol in the synthesis of phenolic resins as it does not require any preliminary modification. Indeed, resorcinol presents an enhanced reactivity due to two hydroxyl functions on the benzene ring (located at the meta position) activating the same aromatic positions and thus increasing their nucleophilicity. Therefore, the three reactive positions are activated twice by the two hydroxyl groups. In addition to great reactivity, resorcinol is a promising monomer as it can be produced from biomass following different pathways (Fig. S2 and S3 in the ESI†).34 In fact, resorcinol can be synthesized from catechin35 or glucose36 by fermentation. Resorcinol exhibits lower acute toxicity compared to phenol, which can be fatal at higher doses, and other substituents such as catechol or pyrogallol (all of them being CMR). For all the reasons previously mentioned, resorcinol has been chosen for our proof of concept. Granado et al.18 studied the curing of different phenolic thermosets including resorcinol–TPA prepolymers prepared in a round bottomed flask using ethanol as the solvent and 0.04 eq. of NaOH (50 wt% aqueous solution) as the catalyst. The prepolymer synthesis was performed at 30 °C for 1.75 hours. The samples were cured using a two-step curing process involving pre-curing over 3 days at 100 °C (to eliminate volatiles), followed by a post-curing phase at 200 °C for 8 hours. The resulting cured materials showed not only interesting mechanical properties, with a glass transition temperature of 161 °C, but also high thermostability with a 10 wt% loss at a temperature of 394 °C and a char yield of 61% after a one-hour isotherm at 900 °C under pyrolysis conditions. These results were very promising for obtaining bio-based materials that could compete with current commercial formulations of phenol-formaldehyde resins.
Currently, most of the phenolic resins are synthesized via batch processes which present disadvantages such as long reaction times or limited range in the viscosity, resulting in the use of solvents to dilute the medium. Continuous processes involving multiple reactors connected in series have also been previously described.8 However, this approach remains uncommon for the production of PF resins due to the limited flexibility in producing a diverse range of products. As an alternative, the reactive extrusion process seems to be ideal to overcome all these drawbacks. Reactive extrusion has been used for various polymer synthesis processes,37–40 such as polycondensation, polymer modification, blend compatibilization or nanocomposite preparation. This process has the benefits of being able to work at high temperatures, without any solvent41,42 and with a wide range of raw materials displaying diverse physico-chemical properties. Moreover, the synthesis duration is highly decreased compared to the batch process.43 For all these features, reactive extrusion could be an approach to develop new generations of resins meeting the current problematics and specifications in terms of performance, health safety and environmental concerns.
In this study, we have dealt with the feasibility of synthesizing a phenolic thermosetting resin from non-toxic and potentially bio-based monomers via reactive extrusion without the use of any solvent and catalyst. The structures of the molecules obtained by reactive extrusion have been assessed by different structural characterization techniques such as Nuclear Magnetic Resonance (NMR) and Mass Spectrometry (MS). The curing kinetics have been investigated by model-free methods and the results provided deep insight into the curing mechanism and notably the activation energy dependence on the extent of conversion. The post-reactivity of the synthesized resins was also compared to that of the novolac (with 12 wt% hexamethylenetetramine) reference. Finally, the thermal stability of the resins is evaluated for post-application. From a broader perspective, this study aims to highlight the potential of reactive extrusion as an eco-friendly route for synthesizing thermosetting resins.
Results and discussion
Resin characterization and mechanistic investigation
The resins, synthesized by reactive extrusion with a TPA-to-resorcinol molar ratio of 1.6 at 150 °C with a flow rate of 1 kg h−1, a screw speed of 100 rpm, and a residence time of 3 minutes and 10 seconds (RT1.6_T150[3′10]), were first analysed by 1H and 13C-NMR spectroscopy to determine the main functional groups present in the synthesized resins (see Fig. 1). The spectra depicted a wide range of signals which can be classified as follows:- The regions between 9.6 and 10.2 ppm on the 1H-NMR spectrum and 191.8 and 193.5 ppm on the 13C-NMR spectrum correspond to the signals of the aldehyde functional groups. Notably, the 1H-NMR signal of aldehyde function from residual TPA is still observed at 10.2 ppm, indicating that this monomer has not fully reacted. The other signals appearing in the same range between 9.7 and 9.9 ppm, correspond to the aldehyde moieties present in the resin structures (Fig. 1A). Similar observations were made on the 13C-NMR spectrum with an intense aldehyde signal at 193.1 ppm from non-reacted TPA, close to several more shielded signals from the resin-bound aldehyde carbons between 192.2 and 192.7 ppm (Fig. 1B).
- In the region between 8.8 and 9.2 ppm on the 1H-NMR spectrum, the signals observed were attributed to the hydroxyl protons of the resorcinol moieties. Indeed, the 2D HSQC spectrum showed no correlation dots (Fig. S4 of the ESI†), confirming that the corresponding hydrogen atoms are not directly attached to carbons but are more likely to oxygen atoms.
- In the region between 6.0 to 8.2 ppm on the 1H-NMR spectrum, the signals observed were attributed to the protons of the aromatic rings as they correlate with signals between 102.7 and 158.5 ppm on the 13C-NMR spectrum, which are usually expected for carbons of substituted benzene rings. In this region, the signals of residual monomers can also be observed. Actually, on the 1H-NMR spectrum, the signal appearing at 8.1 ppm is attributed to the aromatic protons of the TPA. On the 13C-NMR spectrum, the aromatic carbons of the residual TPA are respectively observed at 130.0 and 139.7 ppm. In addition, the signals of the aromatic carbons of the resorcinol are also noticeable at 102.4, 106.2, 130.0 and 158.4 ppm. The precise attributions of these signals are presented in Table 1.
- Finally, the signals observed below 6.0 ppm on the 1H-NMR spectrum and at around 41.9 ppm on the 13C-NMR spectrum are attributed to methine bridges between aromatic rings of TPA and resorcinol moieties, resulting from the condensation of resorcinol with a methylenol function.
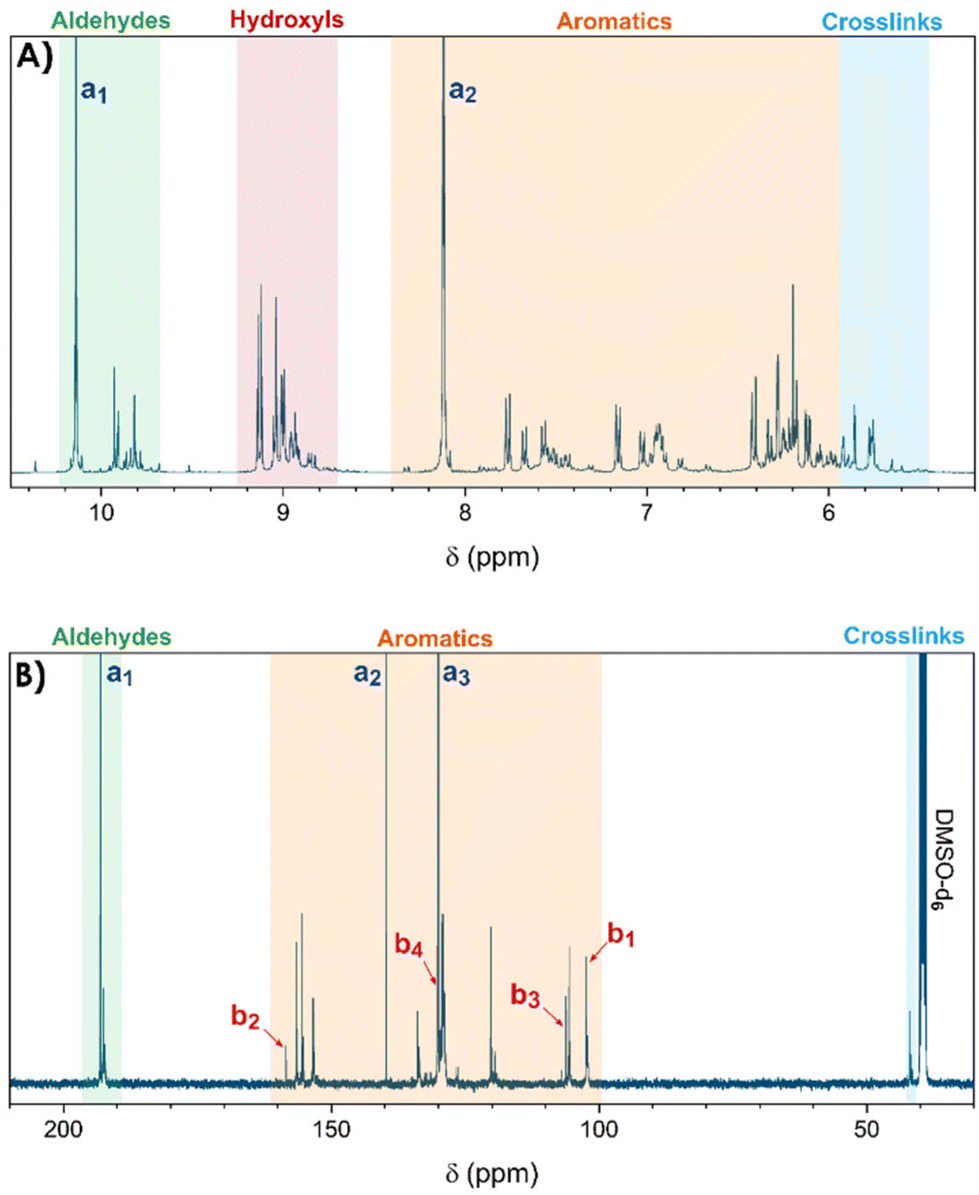 | ||
| Fig. 1 NMR spectra of the resin synthesized by reactive extrusion with a TPA-to-resorcinol molar ratio of 1.6 (RT1.6_T150[3′10]). The extrusion temperature was set to 150 °C with a flow rate of 1 kg h−1 and a screw speed of 100 rpm. (A) 1H-NMR; (B) 13C-NMR (DMSO-d6/298 K). | ||
| Compound | Structure | Labelling | Chemical shift (ppm) | |
|---|---|---|---|---|
| 1H | 13C | |||
| TPA |
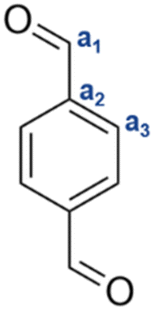
|
a1 | 10.2 | 193.1 |
| a2 | — | 139.7 | ||
| a3 | 8.1 | 130.0 | ||
| Resorcinol |
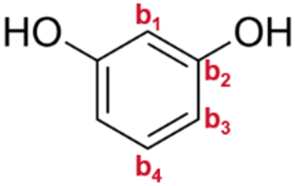
|
b1 | — | 102.4 |
| b2 | — | 158.4 | ||
| b3 | — | 103.6 | ||
| b4 | — | 130.0 | ||
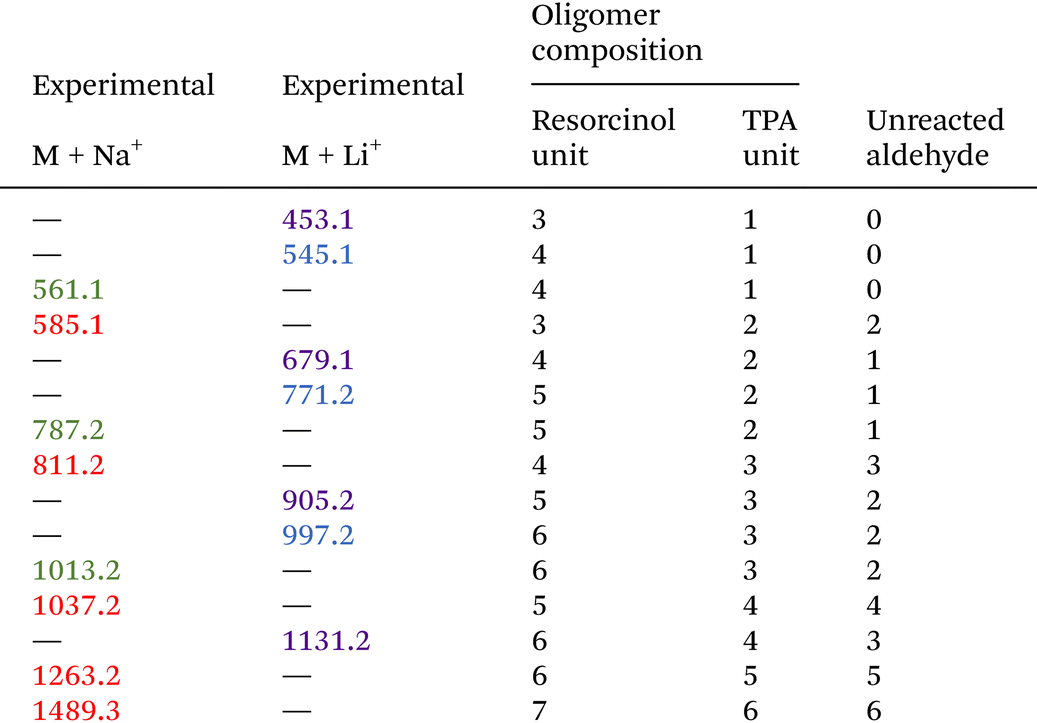
As those signals may come from a complex mixture of reaction products, 2D-DOSY NMR was performed for a more in-depth structural characterization. This technique enables the correlation of 1H-NMR chemical shifts with the self-diffusion coefficients of the corresponding molecules in solution,44,45 facilitating the differentiation of compounds based primarily on their size, which is mainly governed by their molar mass. A stimulated echo sequence with bipolar gradient pulses (ledbpgp2s Bruker pulse program) was used for data acquisition with a diffusion delay set to 0.15 s and a gradient pulse length of 2600 μs. The resulting 2D DOSY map is shown in Fig. 2. On this map, separation was evidenced by three distinct diffusion coefficients. The residual TPA appears with a diffusion coefficient of DTPA = 5.6 × 10−10 m2 s−1 while the resorcinol is not detected, in accordance with the excess of TPA used for the synthesis. The first detectable molecule (trimer 1) presents a diffusion coefficient of D1 = 2.8 × 10−10 m2 s−1. The other molecules are not well separated and have an average diffusion coefficient of Dother = 2.1 × 10−10 m2 s−1. These values are higher than the ones classically obtained with polymers of high molar masses, which are more likely one order of magnitude smaller than 2.0 × 10−11 m2 s−1.46 This observation is thus qualitative evidence that the species created in these resins have low molar masses. Additionally, with the 2D DOSY technique, a first structure has been identified by isolating the signals on the 1H-NMR spectrum which correlate with the diffusion coefficient D1. Additional NMR experiments (13C-DEPT, 2D HSQC and HMBC – see Fig. S4–S6 in the ESI†) combined with those DOSY results allowed us to solve the precise structure of trimer 1 (see Table 2). The reaction scheme leading to the creation of the first species is due to the first reaction of TPA and resorcinol through electrophilic aromatic substitution (addition) yielding a secondary alcohol (methylenol). Then, the generated methylenol function reacts with a second resorcinol monomer through a condensation reaction leading to trimer 1. Besides, the b1 position (Table 1) of resorcinol located in between the two hydroxyl groups presents a higher electron density meaning that this position is supposed to be more reactive. However, this structure involving a reaction on this position is not observed on the NMR spectra. Actually, this position is sterically hindered by the adjacent hydroxyl groups and then becomes less reactive compared to the other two para-position reactive sites47,48 as observed with the identified structure. The 2D DOSY data confirmed the presence of at least two distinct populations characterized by different average molar masses. However, the 2D DOSY analysis could not completely separate all the molecules synthesized during the extrusion as the structures corresponding to the diffusion coefficient Dothers should have higher and quite similar molar masses. For this reason, MALDI-TOF analyses were carried out on the synthesized resins in order to gain deeper insights into the mechanistic aspects of the reactions (Fig. 3). Overall, the oligomers have mainly molar masses ranging from 450 to 1500 g mol−1 (Table 3). From this analysis, different populations can be determined. The mass difference between the oligomers can provide valuable information about the polymerization processes. Notably, the MALDI TOF spectrum clearly shows a repetitive pattern of peaks that enables the identification of specific adduct series present in the resins. For each oligomer series, there is an increase of +226 m/z corresponding to the condensation of a single resorcinol–TPA dimer onto the oligomer with the release of water (as illustrated in Scheme 1). Those successive condensation reactions prevail over electrophilic aromatic substitution (addition) reactions. In contrast, Granado et al.18 obtained different structures by the MALDI-TOF analysis of the resorcinol–TPA resin synthesized in a batch reactor and in the presence of a base catalyst. Even though they pointed out some condensation reactions, they also observed addition involving TPA or addition of the phenol to the dangling aldehyde moiety of the oligomer. Herein, the condensation reactions are more dominant, which can be explained by the selected temperature of extrusion. Indeed, the high temperature set at 150 °C supports polycondensation whereas lower temperatures favor methylolation49 (addition reaction). Yet, it is still possible to observe some addition reactions of resorcinol onto the available second aldehyde function of TPA, resulting in the generation of a methylenol group.
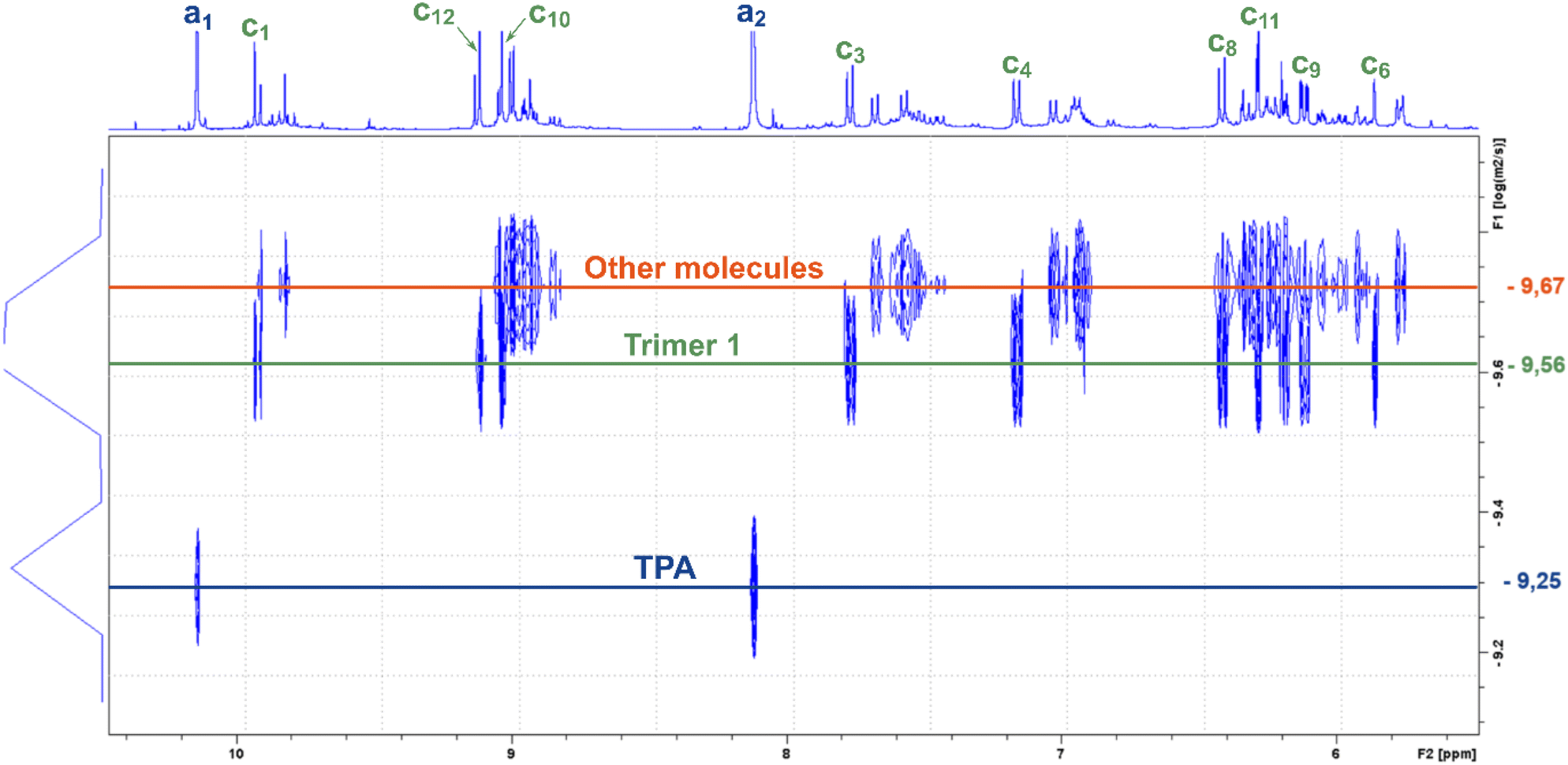 | ||
| Fig. 2 2D DOSY map of the resin synthesized by reactive extrusion with a TPA-to-resorcinol molar ratio of 1.6. The extrusion temperature was set to 150 °C with a flow rate of 1 kg h−1 and a screw speed of 100 rpm (RT1.6_T150[3′10]). Diffusion time Δ = 0.15 s and gradient pulse length δ = 2600 μs. Solvent: DMSO-d6 at 298K. | ||
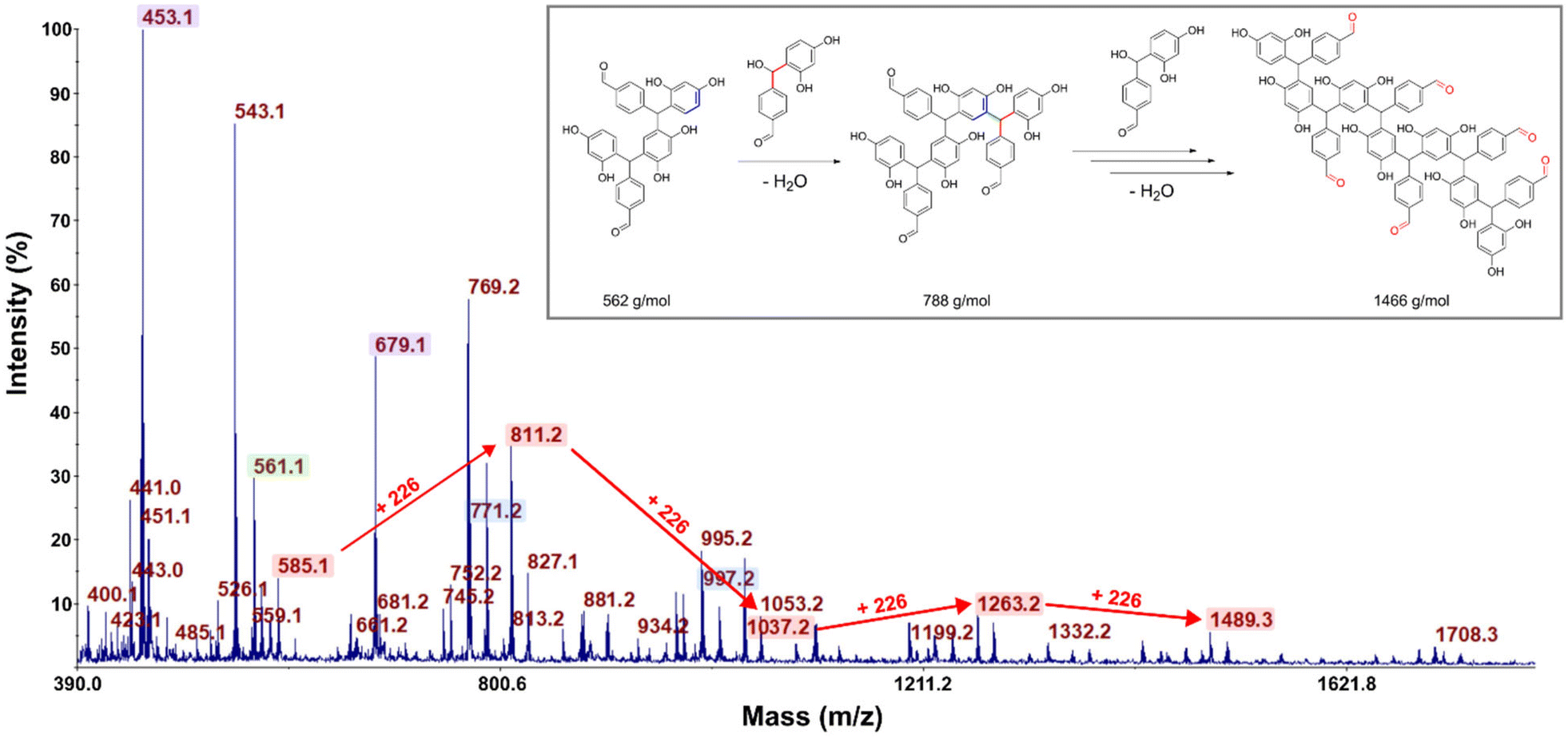 | ||
| Fig. 3 MALDI-TOF spectra of the resin synthesized by reactive extrusion for a TPA-to-resorcinol molar ratio of 1.6 (RT1.6_T150[3′10]) in the positive mode. An illustration of identified structures involving condensation reactions with a resorcinol TPA dimer with the release of water leading to a molar mass difference of 226 g mol−1 is displayed. The bond created by condensation is colored in green. The unreacted pending aldehydes are highlighted in red for the structure with a higher molar mass. | ||
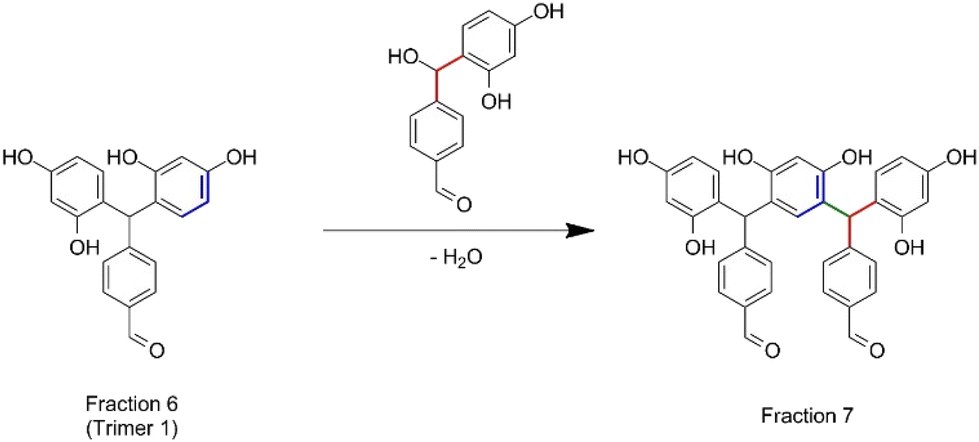 | ||
| Scheme 1 Condensation reaction between trimer 1 with a resorcinol–TPA dimer with the release of a water molecule. | ||
| Compound | Structure | Labelling | Chemical shift (ppm) | |
|---|---|---|---|---|
| 1H | 13C | |||
| Trimer 1 |
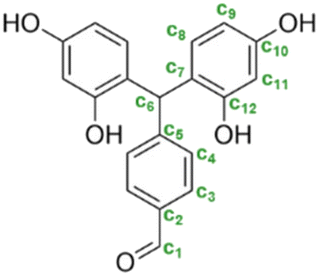
|
c1 | 9.9 | 192.5 |
| c2 | — | 134.0 | ||
| c3 | 7.7 | 129.0 | ||
| c4 | 7.1 | 129.0 | ||
| c5 | — | 153.0 | ||
| c6 | 5.9 | 41.9 | ||
| c7 | — | 120.3 | ||
| c8 | 6.4 | 129.0 | ||
| c9 | 6.1 | 105.6 | ||
| c10 | 9.0 | 156.5 | ||
| c11 | 6.3 | 102.4 | ||
| c12 | 9.1 | 155.4 | ||
|
Based on the structures determined by MALDI-TOF, the presence of unreacted aldehyde functions is confirmed among the structures of the oligomers synthesized, which is in accordance with the NMR analyses and the literature.18 These unreacted aldehyde groups might be important for the post-curing application as they can be involved in additional electrophilic aromatic substitution reactions to form the final crosslinked network. From this MALDI-TOF analysis, it is clearly evidenced that different oligomer structures are synthesized throughout the extrusion process.
To complete this characterization and give some quantitative information, flash chromatography has been performed on the resin to recover 10 distinct fractions (see Fig. S7 in the ESI†) that have been weighted and analyzed by 1H-NMR spectroscopy (Fig. 4). From the NMR spectra, it is clear that the first 3 fractions are attributed to the unreacted TPA and resorcinol (Fig. 4A). Nonetheless, fractions 1 and 2 are not completely pure, as additional signals appear in the spectra. These signals are attributed to acetals and hemiacetals produced by the reaction of methanol (employed as the polar eluent for the flash chromatography analysis) with the aldehydes. It should be noted that the unreacted TPA represents around 40 wt% of the total recovered fractions. Regarding the initial ratio, it was expected to reach 35 wt% of residual TPA. Yet, fraction 3 corresponds to 1.3 wt% of non-reacted resorcinol. In contrast to the 3 first fractions, it was found to be more difficult to assign accurate structures to fractions 4 and 5 (Fig. S8 in the ESI†). However, these two fractions account for less than 2 wt% of the purified resin obtained through flash chromatography.
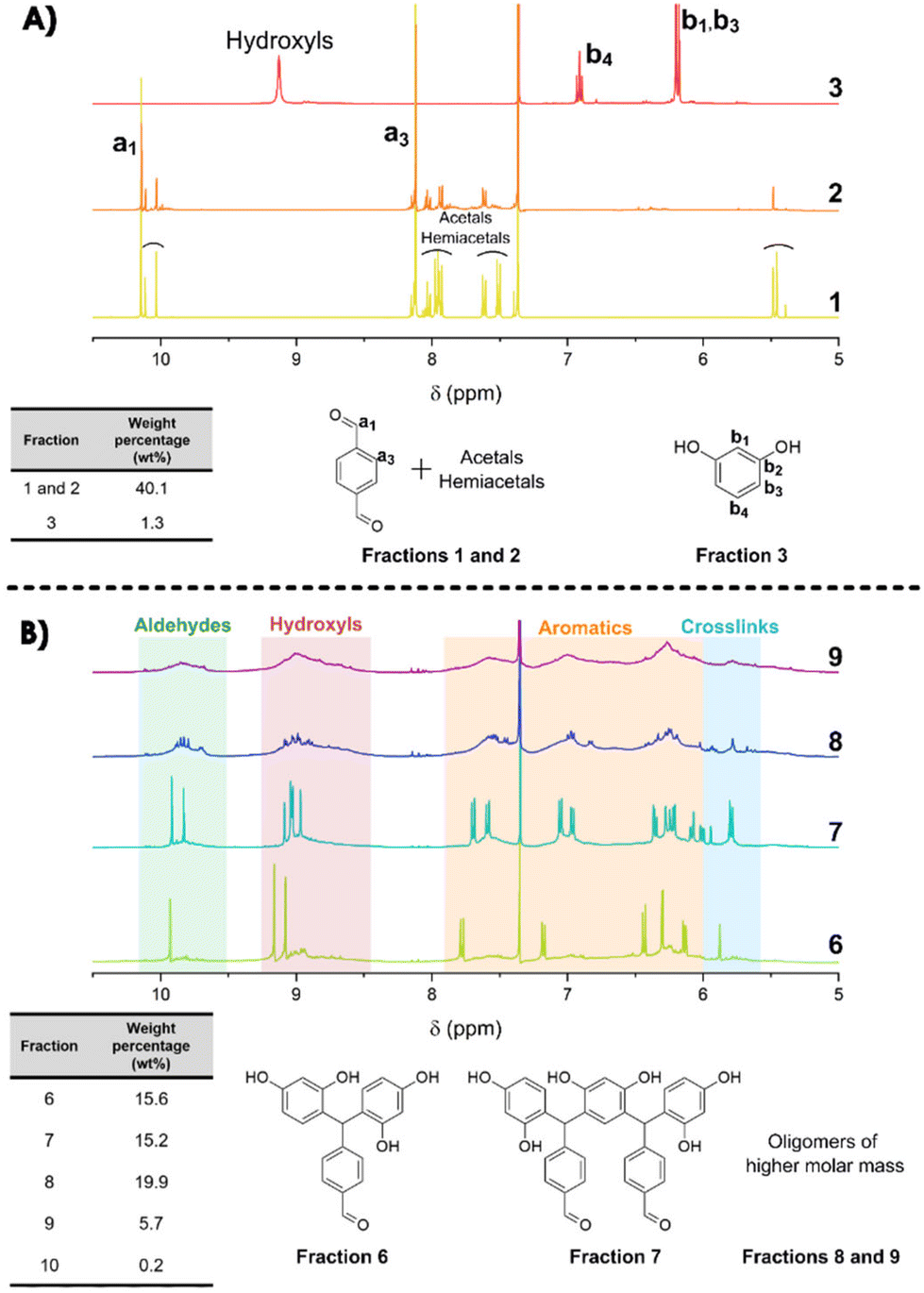 | ||
| Fig. 4 1H-NMR spectra (DMSO-d6) and weight distribution of the main isolated fractions recovered by flash chromatography of the resin synthesized by extrusion (RT1.6_T150[3′10]). For more clarity, fractions 4, 5 and 10 (2.8% of the total weight) are not presented in this figure. (A) Fractions 1 to 3 of the residual monomers; (B) fractions 6 to 9 of the oligomers synthesized. | ||
The synthesized oligomers constituting fractions 6, 7, 8 and 9 represent 56.4 wt% of the overall resin (Fig. 4B). The identification of the chemical structures of oligomers from fractions 6 and 7 was managed by combining the NMR and mass spectrometry measurements (see in Fig. S9 in the ESI†). The oligomers of fraction 6 exhibit a main molar mass of 336 g mol−1. Regarding the corresponding signals in the NMR spectrum, fraction 6 contains trimer 1 previously identified with the 2D DOSY technique. The oligomer structure from fraction 7 exhibits a main molar mass of 562 g mol−1. This structure is obtained by the condensation reaction of trimer 1 identified in fraction 6 with a resorcinol–TPA adduct (Scheme 1), confirming the mechanism of oligomerization highlighted previously.
In general, the hydroxyl group (located on the ternary carbon) is considered to be a poor leaving group. Nevertheless, theoretical calculations based on quantum chemistry theory have revealed that the short-lived quinonemethide intermediate formation is energetically favorable.50,51 The structural attribute of this intermediate species can produce resonance forms that, in turn, enhance the stability of the carbocation resulting from the departure of the hydroxyl group. In addition, the electrophilic aromatic substitution on the second aldehyde is less favored. Indeed, the analysis of the NMR spectra has indicated the presence of pending aldehyde moieties within the oligomer structure. Indeed, after the reaction of the first aldehyde group, the second one is no longer activated in the para-position by electron withdrawing effects.17 It can also be noted that the oligomer structures of fractions 6 and 7 have been previously observed with MALDI-TOF analyses as well as the condensation reaction pattern (Table 3).
Finally, fractions 8 and 9 mainly exhibit broader signals, which indicate more complex structures from oligomers of higher molar mass. Yet, the observed larger signals appear in the same range of chemical shifts as aldehyde, hydroxyl, aromatic and crosslinking regions. It should be noted that they were already detectable as minor contributions in fractions 4 to 7.
Regarding the various structural analyses performed on the synthesized resin by extrusion, it can be concluded that the main reaction mechanism occurring within the extruder is the condensation reactions yielding oligomers with pending aldehydes. However, we did not observe the first methylenol dimer resorcinol-TPA obtained by electrophilic aromatic substitution, which may be due to an already higher conversion (cf. DSC analyses of the resin) obtained at the end of the first extrusion process. In addition, at the high extrusion temperature chosen, the methylenol dimer resorcinol–TPA might react instantly and cannot be directly observed using NMR or MALDI-TOF techniques. Additionally, MALDI-TOF analysis revealed that the highest molar mass depicted was 1466 g mol−1 for a TPA-to-resorcinol molar ratio of 1.6 and an extrusion temperature of 150 °C. In contrast, according to the literature, the higher molar mass novolac has been used in order to improve the thermal and mechanical properties of phenolic resins, with number-average molar masses between 600 and 4000 g mol−1, and weight-average molar masses between 2000 and 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1.49,52,53 Thus, it would be worth exploring the influence of different parameters such as the molar ratio of monomers, the temperature of extrusion, the screw speed or the flow rate in order to increase the oligomer size. To evaluate the average molar masses, the resins synthesized through reactive extrusion were characterized by size-exclusion chromatography and compared to the novolac reference (Fig. 5, Table 4).
000 g mol−1.49,52,53 Thus, it would be worth exploring the influence of different parameters such as the molar ratio of monomers, the temperature of extrusion, the screw speed or the flow rate in order to increase the oligomer size. To evaluate the average molar masses, the resins synthesized through reactive extrusion were characterized by size-exclusion chromatography and compared to the novolac reference (Fig. 5, Table 4).
 | ||
| Fig. 5 Chromatograms of novolac and the resorcinol–TPA resins synthesized by reactive extrusion under different extrusion parameters. (A) Influence of the molar ratio of TPA-to-resorcinol; (B) influence of the extrusion temperature; and (C) influence of the screw speed and the flow rate. | ||
| Sample | Molar ratio | Extrusion temperature (°C) | Screw speed (rpm) | Flow rate (kg h−1) | Residence time | M n (g mol−1) | M w (g mol−1) |
|---|---|---|---|---|---|---|---|
| Novolac + 12 wt% HMTA (reference) | — | — | — | — | — | 4716 | 9137 |
| RT0.6_T150[3′10] | 0.6 | 150 | 100 | 1 | 3′10 | 699 | 848 |
| RT1.6_T150[3′10] | 1.6 | 150 | 100 | 1 | 3′10 | 761 | 993 |
| RT1.6_T160[3′10] | 1.6 | 160 | 100 | 1 | 3′10 | 712 | 887 |
| RT1.6_T170[3′10] | 1.6 | 170 | 100 | 1 | 3′10 | 735 | 1029 |
| RT1.6_T150[2′50]_200rpm | 1.6 | 150 | 200 | 1 | 2′50 | 753 | 969 |
| RT1.6_T150[2′]_2kgh | 1.6 | 150 | 100 | 2 | 2′ | 771 | 981 |
The novolac chromatogram reveals two main populations. The first one appearing at a retention time of 27 minutes corresponds to large oligomer structures, with an estimated number average molar mass of around 4700 g mol−1. The second population centered at a retention time of 37 minutes represents smaller chemical structures, with an estimated number average molar mass of around 490 g mol−1. It has been observed in the literature that free phenol, dimer and trimer are generally separated in SEC from the population of higher molar mass which presents higher polydispersity due to the isomers formed.8,49,53,54 Therefore, the population appearing at a retention time of 37 minutes is more likely composed of a mixture of dimer, trimer, and tetramer species that have not been fully separated. In contrast, the other population corresponds to bigger oligomer structures with higher branching, characteristic of a random novolac and defining the resin's performance such as its mechanical strength and thermal stability.
Regarding the resins synthesized by reactive extrusion, distinct peaks can be observed for the oligomers at 36.5 minutes, the resorcinol at 38.5 minutes and TPA at 40 minutes of retention time. For a TPA-to-resorcinol molar ratio of 1.6 (RT1.6_T150[3′10]), the chromatogram shows a clear peak for residual TPA, with no residual resorcinol, which is in accordance with previous flash chromatography results (Fig. 5A). In contrast, for a TPA-to-resorcinol molar ratio of 0.6 (RT0.6_T150[3′10]), where resorcinol is in excess, no residual TPA is observed and a small amount of resorcinol remains. In both cases, the oligomers formed are more monodisperse and have lower molar masses compared to the novolac reference, which is in accordance with the small and linear structures identified previously by NMR and MALDI-TOF analysis. A slight increase in molar mass is observed when the synthesis is performed with an excess of TPA (ratio 1.6), with a molar mass of 761 g mol−1 for a ratio of 1.6 compared to 699 g mol−1 for a ratio of 0.6. However, this difference is likely not significant.
Increasing the extrusion temperature from 150 °C to 170 °C was expected to produce bigger oligomers. Yet, the chromatogram shows minimal changes, with an average molar mass in number of around 750 g mol−1 (Fig. 5B). At 170 °C (RT1.6_T170[3′10]), TPA seems to be slightly more converted, resulting in a smaller peak. A slight increase in the peak at 33 minutes retention time is also noted even though it is minimal. Thus, this suggests that temperature does not appear to be the primary factor in enhancing the system's reactivity.
Reactive extrusion offers flexibility to adjust various parameters, such as screw speed and flow rate, to optimize the synthesis process. Using a ratio of 1.6 and an extrusion temperature of 150 °C, resorcinol–TPA resins were produced by varying the screw speed between 100 and 200 rpm and the flow rate between 1 and 2 kg h−1 (Fig. 5C). The chromatograms indicate that even with a screw speed of up to 200 rpm (RT1.6_T150[2′50]_200 rpm) or a flow rate of 2 kg h−1 (RT1.6_T150[2′]_2kgh), the oligomer size remains unchanged. At 2 kg h−1, TPA appears to be slightly more converted, leading to an increase in oligomer formation. In fact, a higher flow rate increases the extruder's filling degree, which benefits oligomer synthesis. Additionally, it shows that increasing the productivity to 2 kg h−1 did not impact the resin synthesis which means that production can be increased without altering the resin's structure.
Despite the varying extrusion conditions, the oligomers obtained are linear with a number average molar mass lower than 1000 g mol−1 in contrast to the novolac reference which presents oligomers of higher molar masses and more branching. The short residence time inside the extruder may limit the formation of bigger oligomers. While the increased screw speed did not affect the oligomer size, it seems that increasing the flow rate leads to higher conversion of TPA with a slight increase in oligomer formation. Although there is a slight increase in molar mass with increasing temperature, raising the temperature does not seem to be the optimal parameter to adjust. An alternative approach to achieve higher molar mass could involve adding a catalyst to enhance system reactivity and can also allow for a decrease in extrusion temperature.
Reactivity and kinetics studies
Phenolic resins fall within the category of thermosetting polymers that are thermally curable. Consequently, DSC analyses proved to be the most suitable and simple approach for investigating the curing kinetics of the synthesized resins. In fact, these analyses provide quantitative data, facilitating a deeper understanding of the underlying mechanisms. On the one hand, the curing kinetics investigation facilitates the evaluation of the reactivity between resorcinol and TPA, along with the determination of the activation energies during the synthesis process. The activation energy readily provides valuable and predictive insights into both kinetics and mechanisms. On the other hand, the DSC analysis is also a useful instrument for assessing both the conversion degree and the post-reactivity characteristic of the resin synthesized by reactive extrusion. Furthermore, the curing kinetics study gives precious information for the design of further curing cycles. A good understanding of the curing mechanism coupled with a control of the kinetics is essential for achieving a precise targeted conversion.The exothermic peaks observed in the thermograms are indicative of the heat release associated with the various reactions that occur within thermosetting polymers. Hence, it is interesting to scrutinize the exothermic peaks as it can bring important information on the mechanism underlying the distinct chemical reaction steps. DSC analyses at different heating rates were first performed on the pristine system resorcinol–TPA (TPA-to-resorcinol molar ratio of 1.6) prior to the extrusion (Fig. 6).
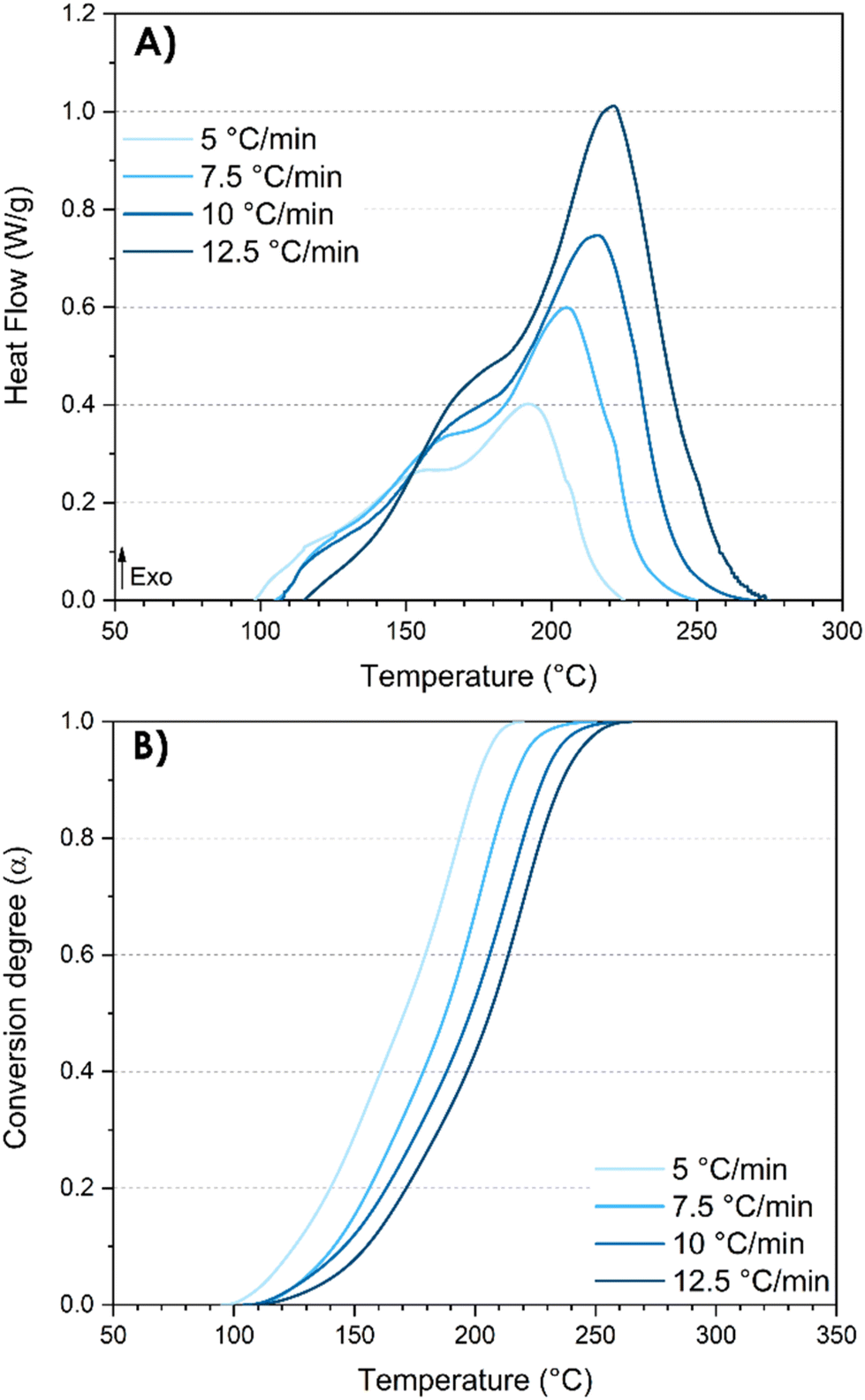 | ||
| Fig. 6 DSC analyses of the pristine system resorcinol–TPA (TPA-to-resorcinol molar ratio of 1.6) before extrusion. (A) DSC thermograms after baseline corrections; (B) nonisothermal kinetic profiles. | ||
It was observed that increasing the heating rate from 5 to 12.5 °C min−1 resulted in a shift of both the initial (Ti) and maximum (Te) temperatures of the exotherm to higher values (Table 5). As expected, the intensity of the exothermic peak is higher with higher heating rates, given the greater energy released from the reaction per unit of time. Remarkably, the total enthalpy of the reaction is not dependent on the heating rates (309 ± 5 J g−1) suggesting that the temperature program does not influence the reaction mechanism. The thermograms revealed three overlapping exothermic peaks (Fig. 6A). Actually, multiple successive reactions of electrophilic aromatic substitutions and condensation reactions can be considered regarding the study of the oligomer structures obtained during the extrusion. By integrating the exothermal peaks, the degree of curing α can be plotted as a function of the temperature giving a series of sigmoidal curves (Fig. 6B). The curves shift to elevated temperatures as the heating rates increase.
| β (°C min−1) | T i (°C) | T e (°C) | ΔH (J g−1) |
|---|---|---|---|
| 5 | 98 | 191 | 315.3 |
| 7.5 | 105 | 205 | 308.7 |
| 10 | 107 | 215 | 300.3 |
| 12.5 | 115 | 220 | 311.2 |
The three model-free methods enable the calculation of the apparent activation energies throughout the conversion degree (displayed in Fig. 7). The calculated activation energy is not only characteristic of the chemical reaction but may also be influenced by diffusion effects. This is the reason why we refer to it as “apparent activation energy”. The apparent activation energies calculated at a conversion degree below 0.1 and above 0.9 are not represented due to the increased approximations in the integration of the exotherm at the initial and final stages of the reactions. These activation energies are spanning over a broad range of values from 32 to 54 kJ mol−1.
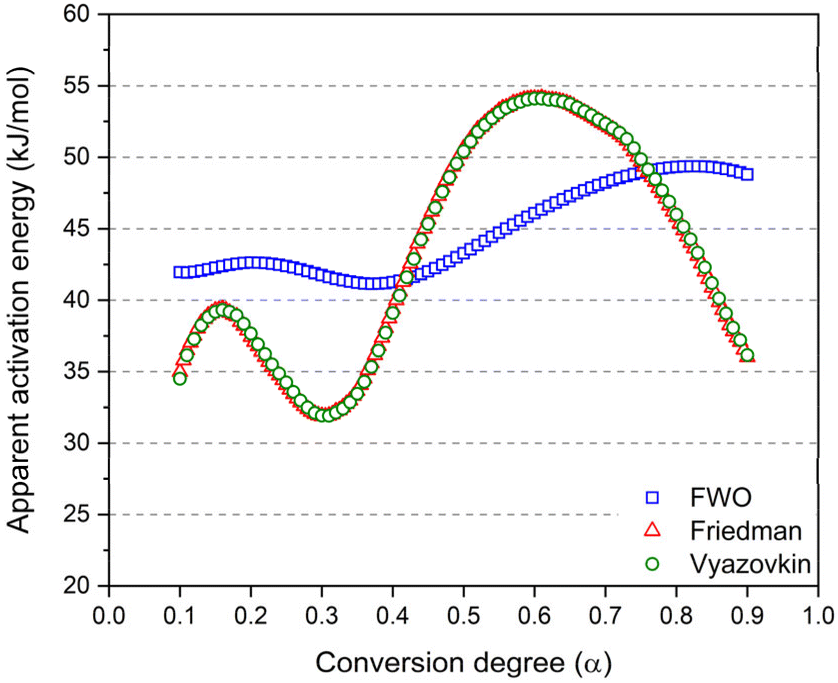 | ||
| Fig. 7 Apparent activation energy as a function of the conversion degree calculated using 3 different model free methods. The Friedman (red) and the Vyazovkin (green) methods are superimposed. | ||
The Friedman and Vyazovkin methods consistently yield the same activation energy depending on the conversion degree. This remarkable concordance underlines the accuracy of the obtained result. In fact, both Friedman and Vyazovkin methods respectively solve the differential and the integral kinetic equations (eqn (7)) without approximations.55 In contrast, while the Flynn–Wall–Ozawa (FWO) method is widely employed due to its ease of use, it remains that the method has a low accuracy in evaluating Eα.56 Indeed, as the integral in eqn (7) does not have an analytical solution, the FWO method uses Doyle's linear approximation of the temperature integral introducing a source of error in the activation energy calculated from the isoconversional method.57 Therefore, the important integral approximation results in imprecise values of Eα. For that reason, in order to reach a better understanding of the chemical mechanism occurring during the reaction, the preference leans toward employing the Vyazovkin and Friedman methods over the FWO method.56,58
Regarding the Vyazovkin and Friedman methods, the activation energy spans over a wider range and the curves display more complex trends. This can be attributed to the transition from one reaction mechanism to another. From a conversion degree of 60%, an unexpected decrease in activation energy is observed, as we would expect an increase for higher conversion degrees. Typically, increasing activation energy can be assigned to a rising energy barrier due to viscosity increase during the synthesis and the change in Eα indicates a switch from a chemically controlled reaction to diffusion control.56 Normally, in thermosetting systems, the diffusion contribution should become particularly significant towards the latter stage of the curing process, during which the viscosity increases considerably as a result of the growing polymer network.59 In addition, the progressive increase of the glass transition during the reaction should also affect the kinetics, leading to higher activation energy in the latter stages of the crosslinking. It is known that phenolic thermosets can achieve glass transition temperatures as high as 175–185 °C.60–62 However, this phenomenon is not observed in the present study and this decrease in activation energies has been previously reported in the literature.63 For instance, Granado et al.64 observed a similar trend in the synthesis of PF resole using 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as a base catalyst.
They attributed the decrease in Eα to the absence of the diffusion contribution to the activation energy and suggested that the reactive sites are located on short still mobile monomers and that the polymer network does not hinder the curing kinetics with additional diffusion barriers. Nevertheless, further investigation is needed to explain this decrease in activation energy.
Compared to the literature, the activation energies we observed are lower than those reported for phenol-formaldehyde and phenol–TPA resins, which were previously noted as 78 kJ mol−1.18 This reduction can be attributed to the +M electron-donating effect of the additional hydroxyl group in resorcinol. Additionally, in the same work, they determined the activation energy for the resorcinol–TPA system to be between 56 and 65 kJ mol−1. The difference between their higher values and our findings might be explained by their approach, which involved calculating the activation energy based on a prepolymer already formed under solvent conditions (ethanol) with the use of a base catalyst (NaOH).
Isothermal predictions using isoconversional analysis
Furthermore, the kinetics study offers valuable information for the post-curing process and the optimization of the thermal cycle required to complete the 3D network formation. Indeed, the Vyazovkin method enables kinetic predictions of the conversion degree under isothermal conditions. The predicted time tα*,T0 necessary to reach a specific conversion degree α* by isothermal curing at temperature T0 is calculated by summing the times necessary to obtain the successive elementary conversion increments (Δα = 0.005) according to the following equation:65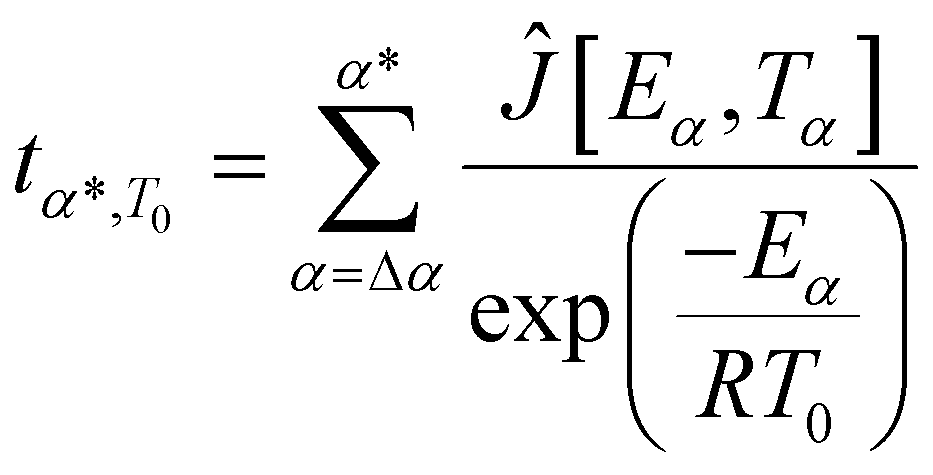 | (1) |
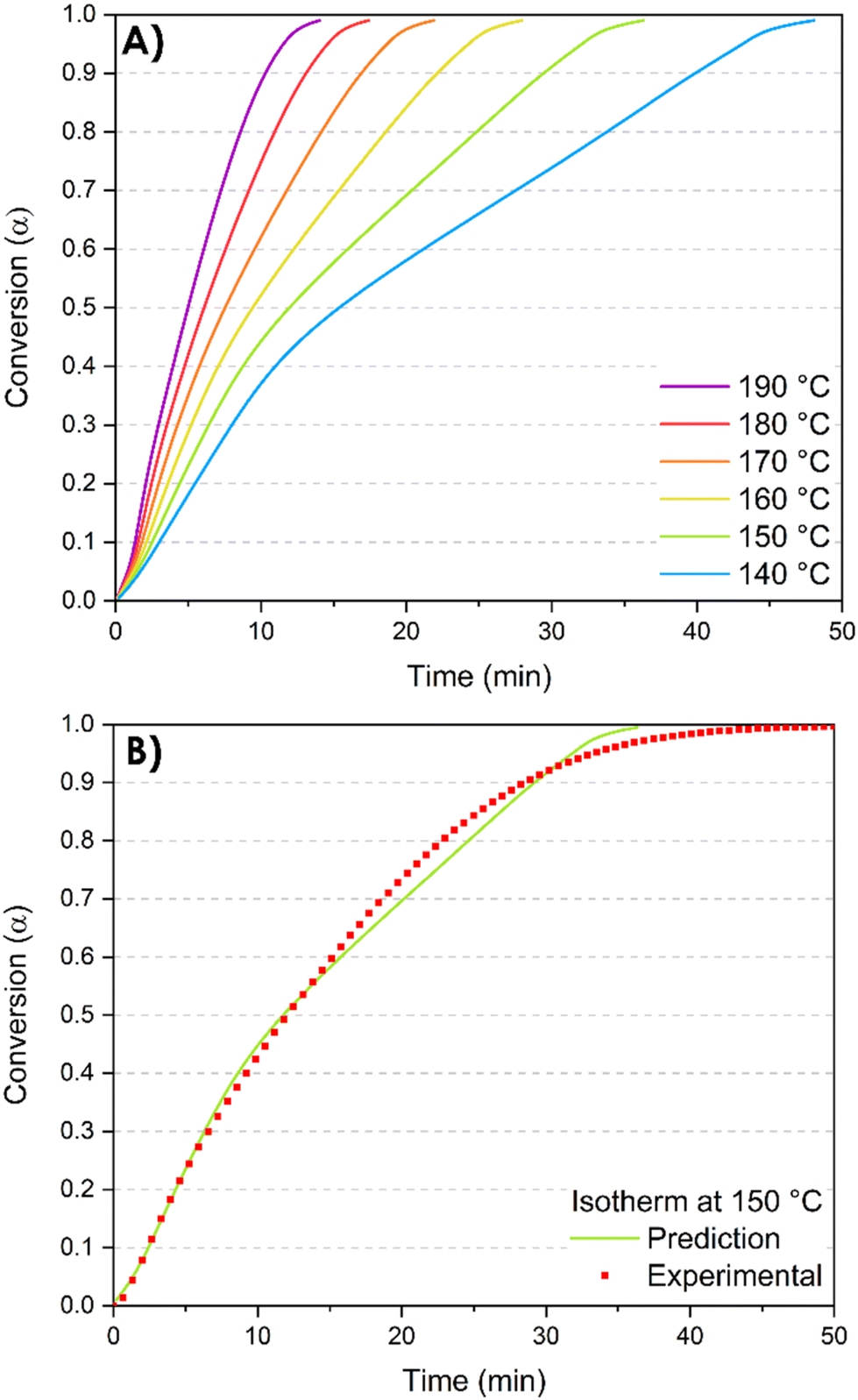 | ||
| Fig. 8 Isothermal prediction curves. (A) Isothermal predictions at different temperatures; (B) comparison of predicted and experimentally measured isothermal kinetic profiles at 150 °C. | ||
Regarding the isothermal predictions at different temperatures (Fig. 8A), at the chosen isothermal temperature of 140 °C, the complete conversion is reached in 48 minutes while at 190 °C, the total conversion is reached in 14 minutes. These predictions are crucial in the context of the reactive extrusion process. Indeed, they provide valuable insights for the optimal selection of temperature and residence time within the extruder. These parameters play a critical role in preventing excessively high conversion degrees that may lead to the complete blockage of the extruder due to the formation of not only insoluble but also infusible crosslinked materials. On the other hand, low temperature of extrusion results in extended reaction time to reach a targeted conversion degree, which is incompatible with the limited residence time constraints imposed by the extruder (less than 4 minutes).
To evaluate the prediction accuracy, one of the simulated curves is compared to experimental results. An isothermal temperature of T0 = 150 °C has been chosen and the exotherm of the reaction has been monitored for 50 minutes. The accordance between experimental and predicted curves demonstrates that the Vyazovkin method accurately describes the curing kinetics (Fig. 8B).
Nevertheless, it is essential to consider that these predictions are generated from DSC analyses where the reactions occur without any shearing or mixing throughout the process. In contrast, reactive extrusion implies screw rotation, and the kneading elements induce high shear rates that facilitate effective mixing of the monomers and enhance interactions among them. Consequently, the prediction data can be taken into account to determine a specific extrusion temperature. However, the conversion degree of the resin at the end of the extruder might deviate from the predicted value. This hypothesis was verified by performing additional DSC analyses of the synthesized resin.
Conversion and post-reactivity study
As previously mentioned, DSC analyses is useful not only as a valuable tool to elucidate the mechanisms of the various reaction steps but also to provide insights into the post-reactivity of the resin. Actually, DSC analyses enable the evaluation of the extent of conversion of the resin after its synthesis by reactive extrusion (Fig. 9).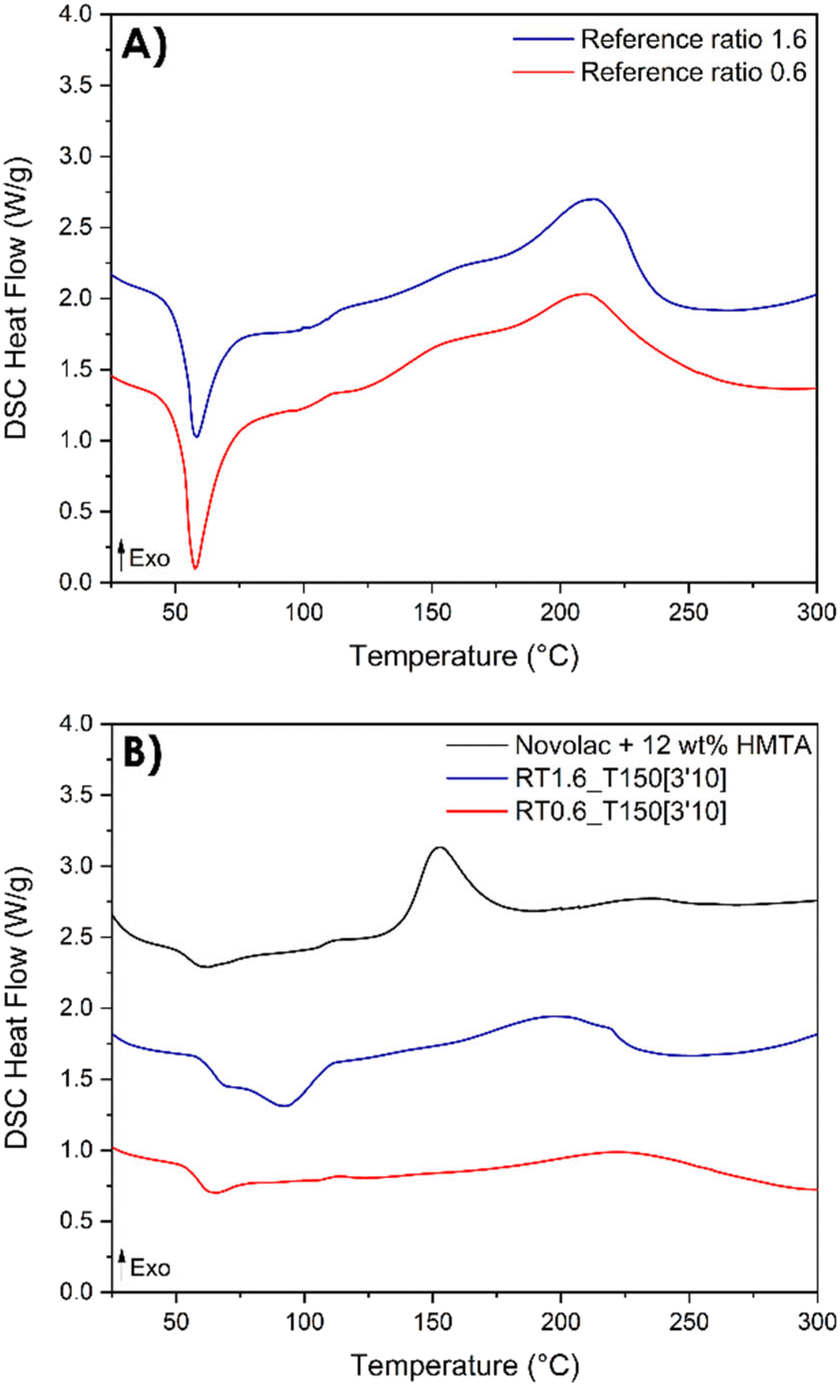 | ||
| Fig. 9 DSC thermograms of resorcinol–TPA formulations with different TPA-to-resorcinol molar ratios. (A) DSC thermograms of the resorcinol–TPA formulations prior to the extrusion; (B) DSC thermograms of the synthesized resins by extrusion at 150 °C, 100 rpm and a flow rate of 1 kg h−1. | ||
It is noteworthy to point out that the resins synthesized by reactive extrusion exhibit a residual enthalpy of reaction (Fig. 9B), indicating that the obtained resin can be further post-cured. In contrast, the Novolac does not have any functional group within its oligomer structure and therefore requires the use of a crosslinker. In this case, hexamethylenetetramine (HMTA) is employed for this purpose. The exothermic peak of the Novolac is shifted toward lower temperatures (150 °C) compared to the resorcinol–TPA system.
The conversion degree α of the resin obtained by reactive extrusion can be determined using the simple following equation:
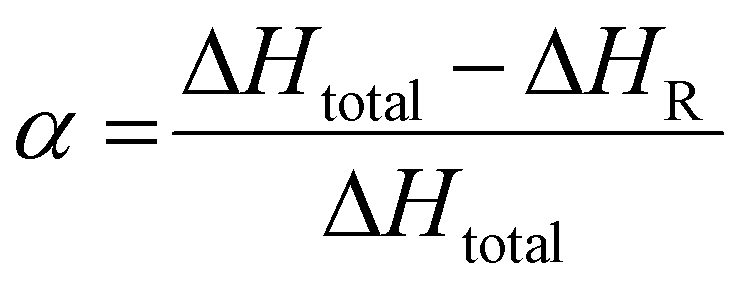 | (2) |
The thermograms show that the pristine system displayed a total enthalpy of 260.3 and 301.3 J g−1 for TPA-to-resorcinol molar ratios of 1.6 and 0.6, respectively. The heat released for the synthesized resins was measured at 104.3 J g−1 for the 1.6 ratio and 110.0 J g−1 for the 0.6 ratio. Thus, the conversion degrees of the formulations, obtained through the reactive extrusion process, is determined to be 0.59 and 0.63 respectively for the ratios 1.6 and 0.6. These measured values are noteworthy when compared to previous predictions based on DSC data analyses for the ratio 1.6 (Fig. 8). Under isothermal conditions at 150 °C, the predicted time required to reach a conversion degree of 0.59 was 15 minutes and 20 seconds. Yet, in reactive extrusion, this degree of conversion is reached with an estimated residence time of 3.2 minutes. This evidences a significant influence of the shear brought by the extrusion process leading to faster reaction rates for the synthesis of phenolic resin, in a continuous process.
Considering the impact of the extrusion process, it is interesting to look at the potential effects of various extrusion parameters on the conversion degree, including the extrusion temperature, the screw speed and the flow of materials. The corresponding conversion degrees for the ratio 1.6 under different extrusion parameters are presented in Table 6.
| Sample | Temperature (°C) | Screw speed (rpm) | Flow rate (kg h−1) | Residual enthalpy ΔHR (J g−1) | Conversion α (%) |
|---|---|---|---|---|---|
| RT1.6_T150[3′10] | 150 | 100 | 1 | 104.3 | 59 |
| RT1.6_T170[3′10] | 170 | 100 | 1 | 94.7 | 64 |
| RT1.6_T150[2′50]_200rpm | 150 | 200 | 1 | 104.0 | 59 |
| RT1.6_T150[2′]_2kgh | 150 | 100 | 2 | 99.0 | 62 |
Increasing the temperature from 150 to 170 °C resulted in an increase in conversion degree from 59 to 64%, as we would expect from the kinetics. Additionally, the screw speed and flow rate influence the shear rate and the residence time within the extruder.66 In fact, increasing the screw rotation speed leads to an increase in the shear rate. However, despite an increase in the screw speed from 100 to 200 rpm, the conversion degree remains constant at 59%. On the other hand, a higher flow rate usually leads to a higher degree of filling, which is in favor of the oligomer synthesis. Indeed, an increased degree of filling leads to higher shear rates because the material is forced through a confined space under greater pressure. However, increasing the flow rate in reactive extrusion shortens the residence time of the resin within the extruder due to the higher volume of monomers introduced, which tends to accelerate the transport of the material through the extruder. Herein, the data indicate that an increase in flow rate from 1 to 2 kg h−1 raises the conversion degree from 59 to 62%. It appears that the enhanced shear rate from higher filling has a more significant effect than the reduced residence time. The changes in temperature, screw speed, and flow rate led to only slight improvements in conversion. These observations correlate with the previous SEC analyses showing slight increases in molar mass (Table 5). Herein, the extrusion conditions tested were not sufficient enough to push the conversion to significantly higher levels. Therefore, further investigation into increasing the screw rotation speed and the flow rate could be interesting to observe a more significant impact on the conversion degree. Additionally, modifying the screw profile to increase the shear rate could also be beneficial in increasing the resin conversion. If these extrusion parameters turn out to effectively govern the conversion degree, varying these parameters might be more relevant than increasing the temperature of extrusion, enabling lower energy consumption during synthesis.
Thermal stability of the resin synthesized by reactive extrusion
The thermal stability of the synthesized resins was assessed through TGA (Fig. 10). At 400 °C, the resin with a TPA-to-resorcinol molar ratio of 1.6 presents 20 wt% of weight loss, while the resin of ratio 0.6 exhibits a loss of 15 wt%. The 20 wt% weight loss observed for the ratio 1.6 can be attributed to different phenomena. Firstly, as indicated previously in Fig. 4, the resin contains residual monomers (mostly TPA) that have not reacted during the synthesis in the extruder. In addition, it has been measured on a Kofler bench that the TPA sublimates around 116 °C. Therefore, volatile evaporation of these unreacted monomers could contribute to the weight loss at <200 °C. Moreover, as the resin is still reactive when subjected to temperature (cf. DSC analyses), there is a possibility that condensation reactions still occur during the analysis, resulting in the release of water. The higher conversion degree of 0.63 for RT0.6_T150[3′10] might explain the lower weight loss around 200 °C compared to RT1.6_T150[3′10], which has a conversion degree of 0.59. Both of these hypotheses have been clarified using TGA analysis coupled with the infrared spectroscopy (TGA-IR) technique (Fig. S10–S13 in the ESI†). The evolution of the IR absorbance peaks corresponding to water (3745 cm−1) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration (1716 cm−1) characteristics of the aldehyde function of the TPA moiety have been monitored through the heating ramp. It has been noted that the first weight loss at 107 °C correlates with the sublimation of TPA. Water release has also been observed at the same temperature, which is indicative of the water adsorbed by the resin. Then, a second weight loss at 200 °C also shows the release of TPA and water. The evaporation of TPA at this stage corresponds to the monomer volatilization (as observed in Fig. S10 in the ESI†). On the other hand, the signal of the water corresponds to its release during the condensation reactions.
O stretching vibration (1716 cm−1) characteristics of the aldehyde function of the TPA moiety have been monitored through the heating ramp. It has been noted that the first weight loss at 107 °C correlates with the sublimation of TPA. Water release has also been observed at the same temperature, which is indicative of the water adsorbed by the resin. Then, a second weight loss at 200 °C also shows the release of TPA and water. The evaporation of TPA at this stage corresponds to the monomer volatilization (as observed in Fig. S10 in the ESI†). On the other hand, the signal of the water corresponds to its release during the condensation reactions.
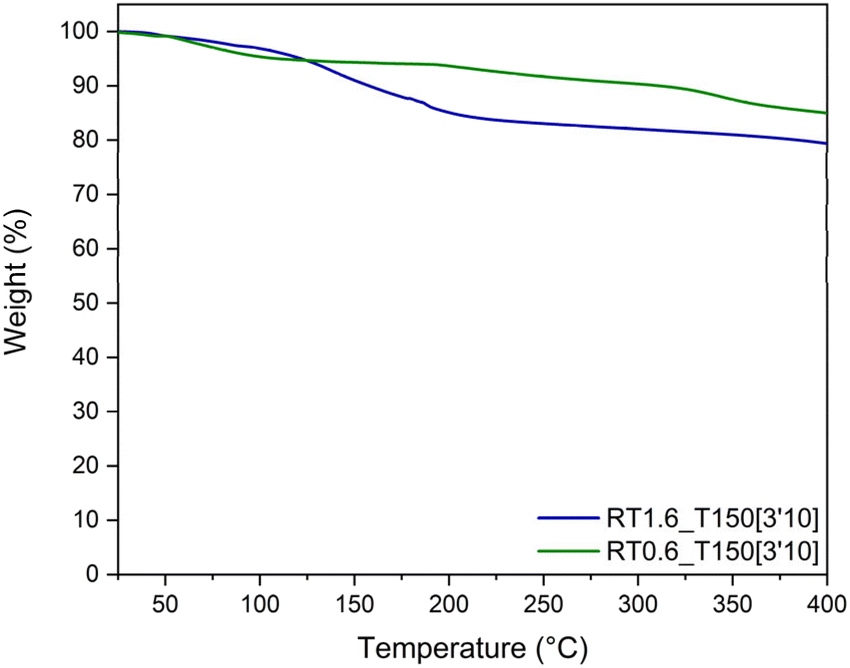 | ||
| Fig. 10 Thermogravimetric analyses of the resins obtained by extrusion with different TPA-to-resorcinol molar ratios. The extrusion temperature was set to 150 °C with a flow rate of 1 kg h−1 and a screw speed of 100 rpm. The thermograms were recorded at 10 °C min−1 under an inert atmosphere. | ||
At temperatures higher than 230 °C, the resins are both stable up to 400 °C. The resin obtained through reactive extrusion demonstrates great thermal stability even prior to post-heating. In fact, Granado et al.18 already demonstrated the great thermal stability of their resorcinol–TPA phenolic resin synthesized in ethanol showing high degradation temperature and high char yield. The remarkable thermal stability of this resorcinol–TPA phenolic resin is mostly due to the high aromatic content brought by both monomers and the high crosslink density.
Mechanical properties of the cured resin
Given the structure with an elevated aromatic content of the phenolic networks, examining the final properties of the cured material was particularly relevant. The mechanical properties of a cured sample were analyzed using DMA on a bar-shaped sample cured at 110 °C for 24 hours. The results, shown in Fig. 11, reveal a glassy plateau at ca. 0.6 GPa and a Tα value of around 197 °C. In comparison, Granado et al. reported a higher glassy plateau of 1.7 GPa for their resorcinol–TPA resin but a lower Tα value of 161 °C.18 It is important to note that the DMA in this study was conducted in three-point bending mode, whereas Granado et al. performed their analysis in tensile mode, complicating direct comparisons. Additionally, they obtained defect-free samples by performing a slow curing program (3 days at 100 °C followed by 8 hours at 200 °C in an autoclave), which may also account for the observed differences. Additionally, they also evaluated the properties of the phenol-formaldehyde resole resin, which exhibited a higher storage value at the glassy plateau of 2.8 GPa but a comparable Tα value of 196 °C to our cured resin. Although the storage modulus of our cured resin is lower compared to the PF resin, the obtained values are still high enough for high-performance applications, regarding the high glass transition temperature. However, with a curing at 110 °C, the material might not be completely crosslinked. The resin likely vitrified when Tg exceeded the curing temperature. The insoluble fraction of this cured resin is approximately 86%. To fully complete the curing process and potentially enhance the properties of the cured material, an additional curing step between 180 and 200 °C (regarding the DSC thermograms in Fig. 9) could be beneficial.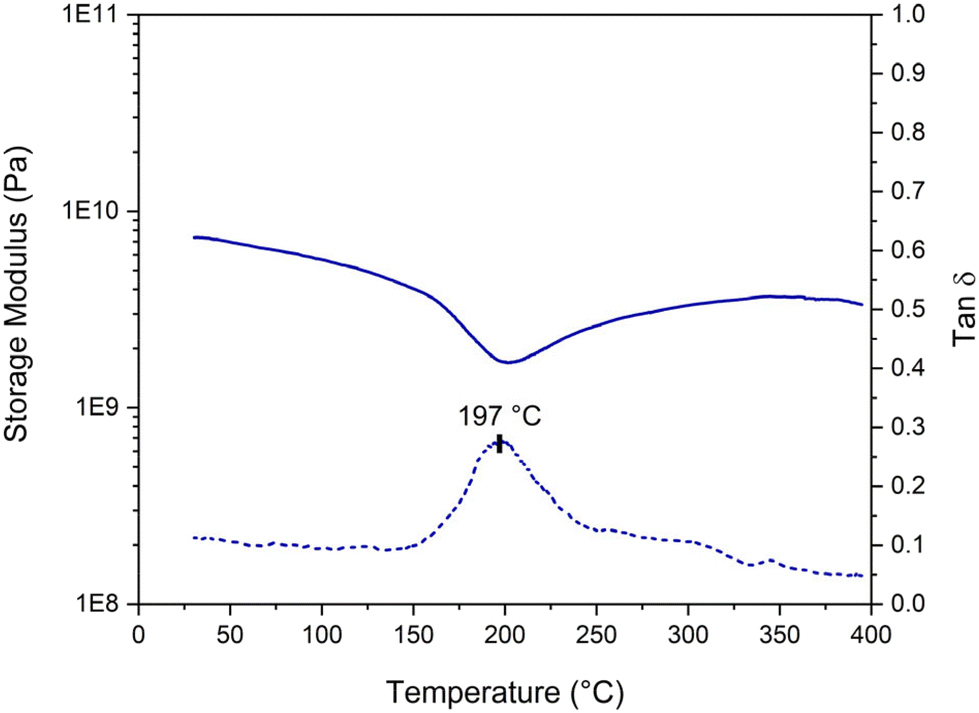 | ||
| Fig. 11 DMA analysis of the cured resorcinol–TPA resin at a ratio of 1.6 (RT1.6_T150[3′10]), cured at 110 °C for 24 hours. | ||
Conclusions
In this work, we have developed a method for the synthesis of thermosetting resins by reactive extrusion, which is a significant advance over traditional batch synthesis.Furthermore, phenolic resins were obtained using monomers that are potentially bio-based. Oligomers of different molar masses have been synthesized in a short residence time (around 3 minutes). The mechanistic investigations revealed that the main reactions occurring within the extruder are condensation reactions accompanied by water release. This insight is valuable for optimizing the design of the extrusion process, involving the presence of vents or vacuum pumps to facilitate water elimination.
The kinetics study and the methods implemented for calculating the apparent activation energies through the degree of conversion provided key information on the reactivity of the formulations. In fact, given the very short reaction time imposed by a continuous process of extrusion, it is necessary to use monomer systems with high reactivity to achieve advanced conversion. Terephthalaldehyde and resorcinol displayed great reactivity towards each other without additional catalysts. It will be interesting to look at the reactivity of other bio-based phenolic and aldehyde monomers in the future. Besides, the resins exhibited post-curing abilities without the addition of any crosslinker, in contrast to novolac resins.
Deeper investigations could be conducted to determine the influence of the extrusion parameters on the conversion degree of phenolic resins. Additionally, the screw profile could also be tailored to increase the shearing rate or extend the residence time during the extrusion process. Finally, the use of catalysts could be explored to address the challenge of achieving a high conversion degree in a short time residency.
The outcomes of this study are very promising for the future of bio-based phenolic resins obtained via reactive extrusion. The synthesized phenolic resins are expected to be suitable for traditional applications, such as adhesives in the wood industry, as well as coatings and organic binders for various composite materials. Their physical form, whether liquid or solid, determines their applicability in different contexts requiring phenolic prepolymers. Liquid resins are ideal for use in prepregs for structural composites, while solid resins are better suited for applications involving novolacs. Notably, solid resins find utility in the production of porous carbons, ion-exchange resins, and carbon nanoparticles.
Moreover, the advantages of this process could make these formaldehyde-free formulations competitive and ultimately replace phenol and formaldehyde ones in the current syntheses, thanks to the potential increase in productivity achieved due to this inherent continuous process. Besides, the solvent-free approach qualitatively suggests a more environmentally friendly method, as the E-factor (environmental factor), defined as the ratio of total waste (kg) to total product (kg), should be close to 0, indicating that no waste is generated relative to the total product weight. Further life cycle analysis of resin synthesis via reactive extrusion could provide a complete assessment, considering energy efficiency and enabling a comparison with conventional PF resin synthesis.
Experimental part
Materials
The chemical structures of the monomers and their main physico-chemical properties are presented in Fig. 12. | ||
| Fig. 12 Chemical structures and physico-chemical properties of resorcinol and terephthalaldehyde. | ||
Terephthalaldehyde (99%) was supplied by Synthon Chemicals GmbH. Resorcinol (>99%) and deuterated dimethyl sulfoxide (DMSO-d6) were supplied by Sigma-Aldrich. Methanol and dichloromethane were supplied by Carlo Erba. The novolac (with 12 wt% hexamethylenetetramine) reference resin was supplied by Prefere. All chemical products were used as received without any further purification.
Resin synthesis
The resin is synthesized by reactive extrusion using a corotative twin-screw extruder (Leistritz ZSE 18 HPe model, a screw diameter of 18 mm, L/D = 68). The twin-screw profile is presented in Fig. 13. The monomers are introduced through the hopper in the extruder separately via two gravimetric feeders. Four zones of high shear rates are implemented with kneading elements. Three open vents are placed in the last part of the extruder for efficient devolatilization of potential residual monomers and reaction sub-products such as water released by the condensation reactions occurring during the synthesis. The mean residence time of the resin is estimated at approximately 3 minutes 10 seconds for a screw rotation speed of 100 rpm and a flow rate of 1 kg h−1 (simulation on Ludovic 6.1 software). For a screw rotation speed of 200 rpm and a flow rate of 1 kg h−1, the residence time is decreased to 2 minutes 50 seconds. For a screw rotation speed of 100 rpm and a flow rate of 2 kg h−1, the residence time is decreased to 2 minutes. The resin samples are named according to the following format: RTX_TY[residency time], where “R” stands for resorcinol, “T” represents terephthalaldehyde (TPA), X refers to the TPA-to-resorcinol molar ratio, and TY refers to the extrusion temperature. The mean residency time is indicated in brackets, formatted as [minutes'seconds].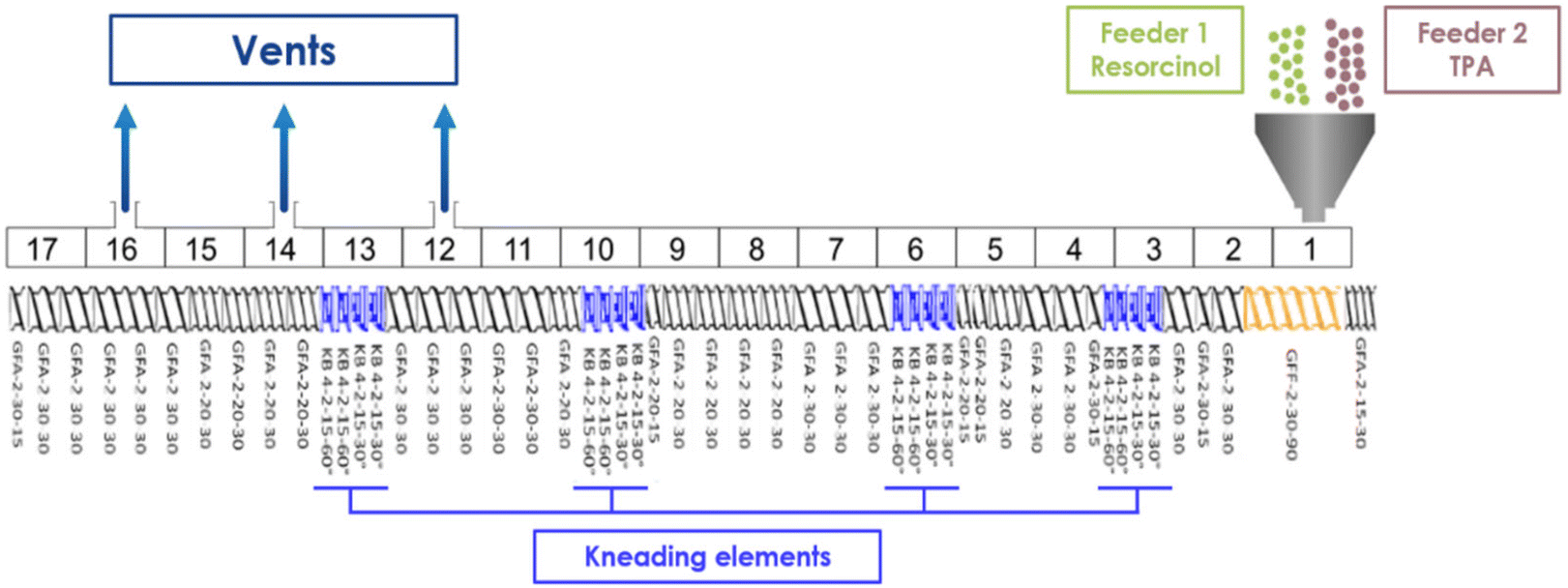 | ||
| Fig. 13 Twin-screw profile. | ||
All the syntheses were realized without any solvent and catalyst. The syntheses were carried out under various conditions. First, resins were synthesized by varying the TPA-to-resorcinol molar ratio from 0.6 (excess of resorcinol building blocks) to 1.6 (excess of TPA building blocks). These ratios were selected to observe potential variations in reactivity and final thermal properties. The temperature of extrusion is modulated from 150 to 170 °C. Different global flow rates have been tested from 1 to 2 kg h−1 as well as different screw speeds from 100 to 200 rpm. The complete resin characterization and the mechanistic investigation have been carried out on the resin synthesized by extrusion at 150 °C with a flow rate of 1 kg h−1, a screw speed of 100 rpm and a TPA-to-resorcinol molar ratio of 1.6 (RT1.6_T150[3′10]).
Curing kinetics theory and computations
The degree of curing (or conversion) α is assumed to be proportional to the heat release measured by differential scanning calorimetry (DSC) analysis under non-isothermal conditions. It can be determined using the following equation: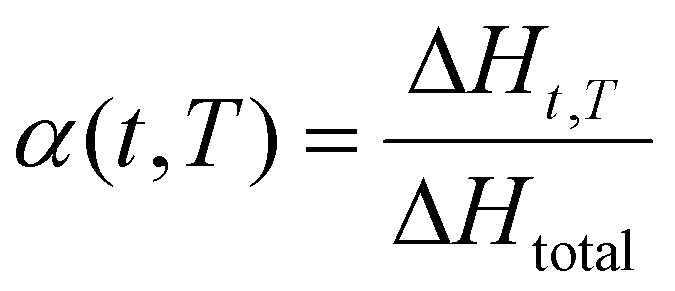 | (3) |
The general equation in kinetic methods considers the reaction rate to be a function of only two variables, the temperature T and the extent of conversion α:
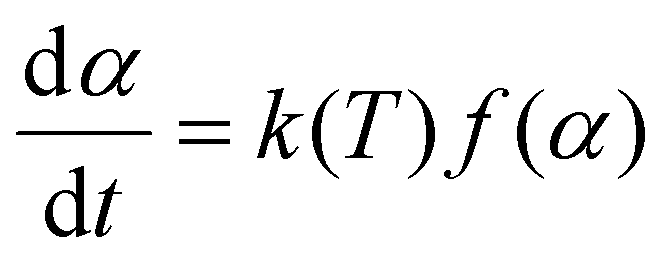 | (4) |
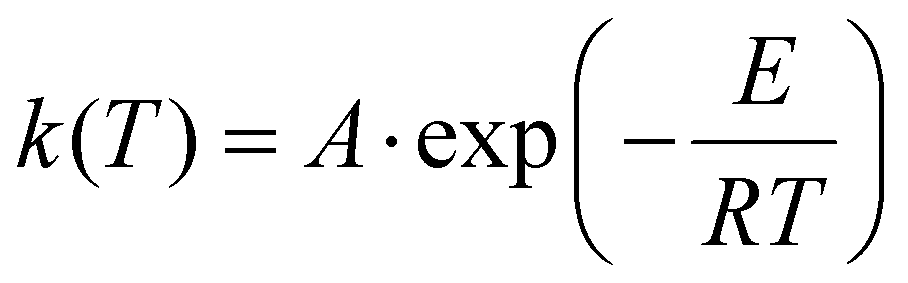 | (5) |
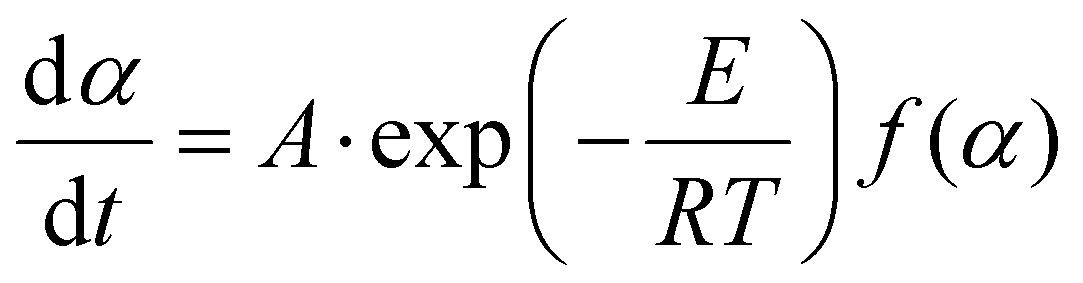 | (6) |
Integration of eqn (6) leads to:
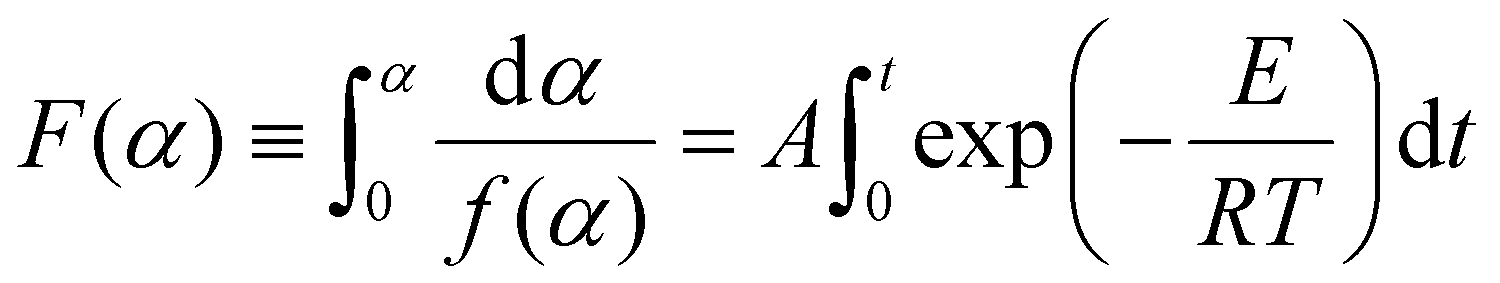 | (7) |
where F(α) is the integral form of the reaction model.
Under non-isothermal conditions, for a constant heating rate β = dT/dt, the integral in eqn (7) can be rewritten with respect to temperature:
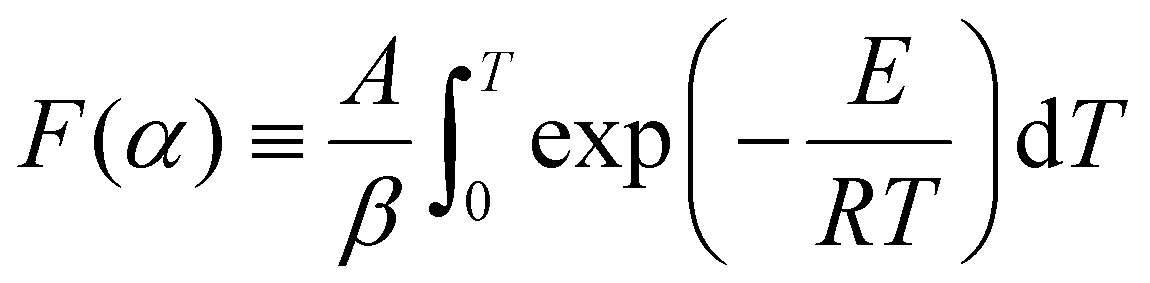 | (8) |
To avoid the necessity of identifying a reaction model, the isoconversional principle is frequently employed. Indeed, isoconversional methods stand out as the most reliable methods to study the kinetics of thermally activated processes. These methods do not require identifying the reaction model (model-free kinetic approach). The fundamental assumption of the isoconversional method is that the reaction model is not dependent on the temperature or the heating rate.55,56 This assumption is verified from the different linear heating rates data revealing a constant reaction enthalpy and parallel kinetic profiles. Consequently, three different isoconversional methods using non-isothermal DSC experimental data were tested to evaluate the activation energy of the system throughout the synthesis and the cross-linking process: Flynn–Wall–Ozawa (FWO), Friedman and Vyazovkin methods.
The first isoconversional method tested relies on a number of approximate solutions because the integral in eqn (7) does not have an analytical solution.56,57 The Doyle linear approximation of the temperature integral gives rise to the Flynn–Wall–Ozawa (FWO) equation:
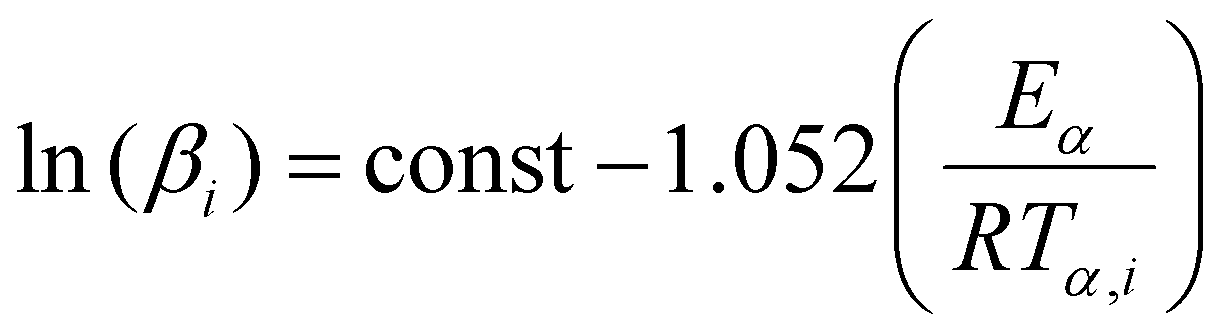 | (9) |
(βi)) data points obtained from several (a minimum of three is recommended) distinct temperature programs.
The Friedman method is another common differential isoconversional method.65,67 The Friedman's equation is derived from eqn (6) without any approximation and is expressed as follows:
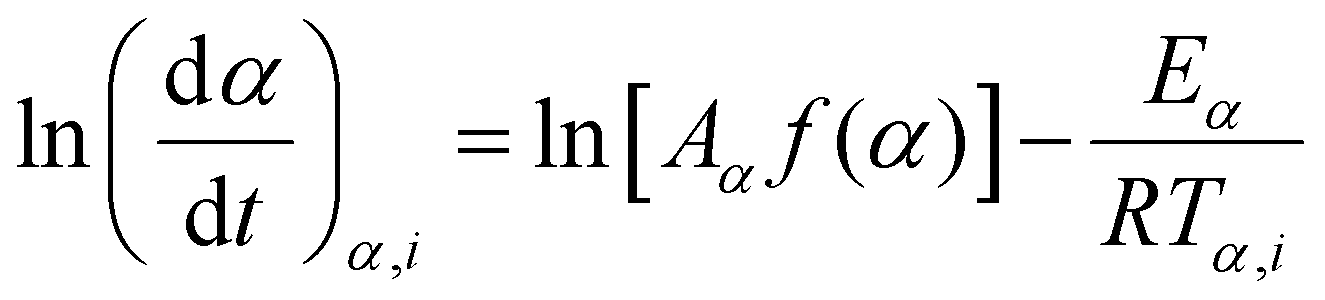 | (10) |
Hence, similar to the FWO method, performing linear regression on the (ln(dα/dt)α,i; ln(βi)) data points acquired from multiple temperature programs enables the determination of the activation energy Eα at a specific conversion.
The Vyazovkin method67,68 is a numerical integral method also based on the isoconversional assumption and displays numerous advantages: it takes into account the self-heating/cooling that can occur during DSC analyses and it can be applied to any program (including cooling and multi-step programs).56 Additionally, the integral method avoids inaccuracies coming from noisy data. To obtain the best accuracy in the determination of the activation energy, small conversion degree increments Δα are selected for numerical integration. For a series of n temperature programs, the activation energy Eα at a specific extent of conversion α is obtained as the value for which the following function Φ is minimized:
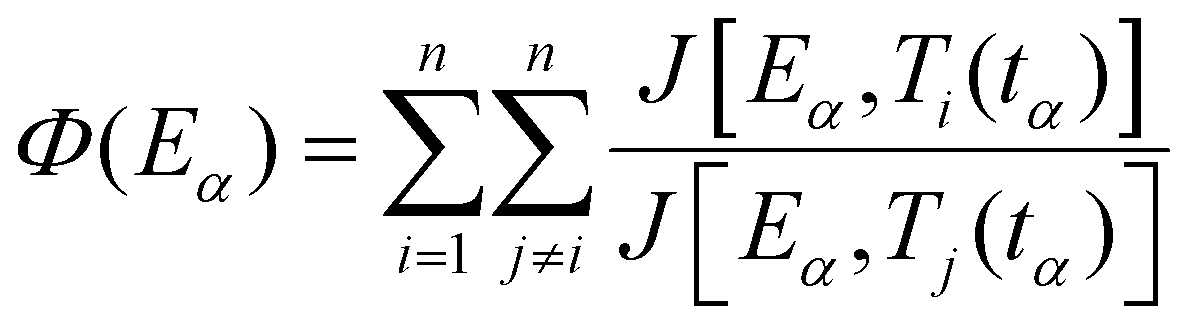 | (11) |
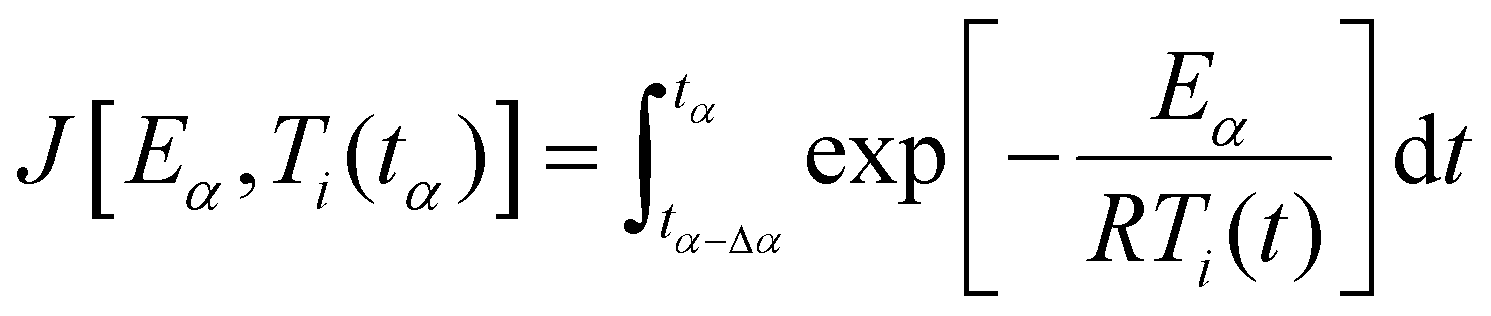 | (12) |
The Vyazovkin method code was implemented using MATLAB software. Integral functions and activation energy values were calculated with an increment of Δα = 0.005. The J integrals were evaluated using the built-in quad function based on a recursive adaptive Simpson quadrature method. The minimization of the ϕ functions was performed using the built-in fminsearch function implementing the Nelder–Mead Simplex direct search method.
Characterization techniques
The separation of the different species from the resin synthesized by extrusion was realized by a flash chromatography technique using a Reveleris X2 (Büchi Labortechnik AG) instrument. The separation was realized through solid deposition prepared with a silica gel (particle size 63–200 μm) and 10 g of the sample. The species were separated with a gradient program of solvent A (dichloromethane) and solvent B (methanol). The separation was monitored with two types of detectors: a UV detector and an evaporative light scattering detector (ELSD). The fractions obtained were recovered and dried by evaporating the solvents under reduced pressure.1D-1H, 2D-1H/13C HSQC and HMBC and 1H-DOSY Nuclear Magnetic Resonance (NMR) spectra were recorded using a Bruker Avance III spectrometer equipped with a 5 mm BBFO+ robe operating at 400.1 MHz for 1H observation. 13C-NMR spectra were recorded using a Bruker Avance II equipped with a 10 mm 13C-selective probe operating at 100.6 MHz for 13C observation. All samples were solubilized in deuterated dimethyl sulfoxide (DMSO-d6) and analyzed at 298 K. The chemical shifts (δ) were given in ppm and were referenced to the tetramethylsilane (TMS) signal at 0.00 ppm.
Mass spectrometry was performed using a high-resolution hybrid quadrupole-time of flight mass spectrometer (Impact II, Bruker, Bremen, Germany) equipped with an atmospheric pressure chemical ionization (APCI) ion source. Samples were diluted 10 times in methanol (>99.9% Optima LC/MS grade, Fisher Chemical) and injected at 10 μL min−1 using a syringe pump. The APCI ion source was operated in the negative ion mode and the parameters were set as follows: corona discharge, 4500 nA; dry heat, 200 °C; and nitrogen flow of the nebulizer gas, 4.0 L min−1. The calibration of the mass spectrometer was performed with an APCI/APPI Tuning mix from Agilent Technologies (reference G2432A). The data were processed using DataAnalysis 5.0 software.
The matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry measurements were carried out using a Voyager-DE Pro (AB Sciex, Framingham, MA) equipped with a nitrogen laser emitting at 337 nm. Ions were accelerated to a final potential of 20 kV. Mass spectra were the sum of 300 shots, and an external mass calibration was used (a mixture of peptides from Sequazyme™ standards kit, AB Sciex). Analyses were recorded in the positive and reflector mode. The matrix used was α-cyano-4-hydroxy-cinnamic acid (CHCA) dissolved in a mixture of water:acetonitrile (50:50 v:v). The cationizing agent used was Li2SO4 at 10 g L−1 in water. The samples were dissolved in acetone at a concentration of 10 g L−1. Then, a mixture of 45 μL of the matrix solution, 5 μL of the sample solution, and 5 μL of the Li2SO4 solution was realized. Finally, 1 μL of this final mixture was deposited on the MALDI sample plate, and then dried in ambient air.
Molar masses of the different resins were determined by SEC using an Agilent 1200 Series device with Agilent Polargel-M 300 × 7.5 mm columns. Differential index refraction was measured with a Wyatt Optilab rEX (λ = 658 nm) refractometer. Polystyrene samples of different molar masses were used as standards for calibration. Solutions with a concentration of 5 mg mL−1 were prepared in DMF with LiNTF2 salt. The solutions were filtered with 0.45 μm polytetrafluoroethylene (PTFE) filters and eluted through columns with DMF at a flow of 0.5 mL min−1.
Differential scanning calorimetry (DSC) was used to study the reactivity of the monomers and the curing kinetics. DSC runs were performed with a DSC Q200 from TA instruments equipped with a cooler module. The atmosphere was dry nitrogen at a flow rate of 50 mL min−1. High-pressure gold plated pans and lids (15 MPa, 25 μL) were used to prevent parasite signals from volatile evaporation. The samples were weighed to be around 5 mg. In this work, four heating rates were selected in order to carry out the kinetic study: β = 5, 7.5, 10, and 12.5 °C min−1. To evaluate the conversion degree of the resin synthesized by extrusion, we selected a heating ramp of β = 10 °C min−1.
Thermogravimetric analyses (TGAs) were carried out on an SDT Q600 from TA instruments. The heating ramp was set from room temperature to 400 °C at a heating rate of 10 °C min−1. The atmosphere was dry nitrogen at a flow rate of 50 mL min−1. The samples were weighed to be around 10 mg in open alumina pans.
Dynamic mechanical analyses (DMAs) were carried out using a DMA Q800 from TA Instruments using the three-point bending mode. The clamp with a 50 mm span was used. Dynamic sample deformation was performed at an amplitude of 20 μm at a frequency of 5 Hz. The tests were conducted with a preload of 0.1 N. The analyses were performed from RT to 400 °C at 5 °C min−1. The sample dimensions were 60.0 × 13.0 × 4 mm.
Author contributions
All of the experiments were conducted by A. M. and G. J., and the processes were supervised by K. D. All the authors contributed to the preparation of this manuscript. Conceptualization: R. T. and V. B.-L.; and writing – original draft, review, and editing: A. M., M. Z., F. B., R. T., and V. B.-L.Data availability
The data that support the findings of this study are available from the corresponding author on reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the ANRT (Association Nationale de la Recherche et de la Technologie) for financial support. They thank the Axel'One Campus for providing facilities to perform reactive extrusion and NMR analyses. Acknowledgment is given to “La Plateforme Lyonnaise de Caractérisation des Polymères” for the SEC analyses. Special thanks to Catherine LADAVIÈRE for her experimental support in MALDI-TOF mass spectrometry and her involvement in the interpretation of our results. The authors would also like to thank the CCSM (Centre Commun de Spectrométrie de Masse) and especially Elodie FROMENTIN for her help and the interesting discussions in mass spectrometry. Finally, we would like to extend our warmest thanks to Professor Philippe Cassagnau for our enriching exchanges and discussions on this subject.References
- L. Pilato, React. Funct. Polym., 2013, 73, 270–277 CrossRef CAS.
- K. Hirano and M. Asami, React. Funct. Polym., 2013, 73, 256–269 CrossRef CAS.
- S. Rimdusit and H. Ishida, Polymer, 2000, 41, 7941–7949 CrossRef CAS.
- E. Frollini, C. G. Silva and E. C. Ramires, Advanced Fibre-Reinforced Polymer (FRP) Composites for Structural Applications, Elsevier, 2013, pp. 7–43 Search PubMed.
- D. Liu, H. Wang, H. Jiang and D. Zhou, J. Appl. Polym. Sci., 2016, 133, app.42734 CrossRef.
- F.-Y. Wang, C.-C. M. Ma and W.-J. Wu, J. Appl. Polym. Sci., 1999, 73, 881–887 CrossRef CAS.
- C. Mougel, T. Garnier, P. Cassagnau and N. Sintes-Zydowicz, Polymer, 2019, 164, 86–117 CrossRef CAS.
- A. Gardziella, L. A. Pilato and A. Knop, Phenolic Resins: Chemistry, Applications, Standardization, Safety and Ecology, Springer Berlin Heidelberg, Berlin, Heidelberg, 2000 Search PubMed.
- Y. Wang, S. Wang, C. Bian, Y. Zhong and X. Jing, Polym. Degrad. Stab., 2015, 111, 239–246 CrossRef CAS.
- A. Knop and L. A. Pilato, Phenolic Resins: Chemistry, Applications and Performance, Springer Berlin Heidelberg, Berlin, Heidelberg, 1985 Search PubMed.
- J. Liu, D. Xuan, J. Chai, D. Guo, Y. Huang, S. Liu, Y. T. Chew, S. Li and Z. Zheng, ACS Omega, 2020, 5, 10011–10020 CrossRef CAS PubMed.
- F. B. Oliveira, C. Gardrat, C. Enjalbal, E. Frollini and A. Castellan, J. Appl. Polym. Sci., 2008, 109, 2291–2303 CrossRef CAS.
- I. Van Nieuwenhove, T. Renders, J. Lauwaert, T. De Roo, J. De Clercq and A. Verberckmoes, ACS Sustainable Chem. Eng., 2020, 8, 18789–18809 CrossRef CAS.
- S. Amirou, A. Pizzi and X. Xi, Eur. J. Wood Wood Prod., 2019, 77, 453–463 CrossRef CAS.
- European Chemical Agency , Substance Infocard - Furfural, https://echa.europa.eu/nl/substance-information/-/substanceinfo/100.002.389, (accessed May 2, 2024).
- European Chemicals Agency , Substance Infocard - Glyoxal, https://echa.europa.eu/nl/substance-information/-/substanceinfo/100.003.160, (accessed May 2, 2024).
- G. Foyer, B.-H. Chanfi, D. Virieux, G. David and S. Caillol, Eur. Polym. J., 2016, 77, 65–74 CrossRef CAS.
- L. Granado, R. Tavernier, S. Henry, R. O. Auke, G. Foyer, G. David and S. Caillol, ACS Sustainable Chem. Eng., 2019, 7, 7209–7217 CrossRef CAS.
- V. Siracusa and I. Blanco, Polymers, 2020, 12, 1641 CrossRef CAS PubMed.
- A. Maneffa, P. Priecel and J. A. Lopez-Sanchez, ChemSusChem, 2016, 9, 2736–2748 CrossRef CAS PubMed.
- T. V. Lourençon, S. Alakurtti, T. Virtanen, A.-S. Jääskeläinen, T. Liitiä, M. Hughes, W. L. E. Magalhães, G. I. B. Muniz and T. Tamminen, Holzforschung, 2020, 74, 175–183 Search PubMed.
- B. Lochab, S. Shukla and I. K. Varma, RSC Adv., 2014, 4, 21712–21752 RSC.
- H. Younesi-Kordkheili and A. Pizzi, J. Adhes., 2020, 96, 1385–1395 CrossRef CAS.
- S. Gillet, M. Aguedo, L. Petitjean, A. R. C. Morais, A. M. da Costa Lopes, R. M. Lukasik and P. T. Anastas, Green Chem., 2017, 19, 4200–4233 RSC.
- W. Zhang, Y. Ma, C. Wang, S. Li, M. Zhang and F. Chu, Ind. Crops Prod., 2013, 43, 326–333 CrossRef CAS.
- A. Bansode, M. Barde, O. Asafu-Adjaye, V. Patil, J. Hinkle, B. K. Via, S. Adhikari, A. J. Adamczyk, R. Farag, T. Elder, N. Labbé and M. L. Auad, ACS Sustainable Chem. Eng., 2021, 9, 10990–11002 CrossRef CAS.
- Y. Zhang, Z. Yuan, N. Mahmood, S. Huang and C. (Charles) Xu, Ind. Crops Prod., 2016, 79, 84–90 CrossRef CAS.
- W. Li, H. Sun, G. Wang, W. Sui, L. Dai and C. Si, Green Chem., 2023, 25, 2241–2261 RSC.
- B. M. Upton and A. M. Kasko, Chem. Rev., 2016, 116, 2275–2306 CrossRef CAS PubMed.
- Z. Gao, X. Lang, S. Chen and C. Zhao, Energy Fuels, 2021, 35, 18385–18395 CrossRef CAS.
- M. V. Alonso, M. Oliet, F. Rodríguez, J. García, M. A. Gilarranz and J. J. Rodríguez, Bioresour. Technol., 2005, 96, 1013–1018 CrossRef CAS PubMed.
- H. Paananen, L. Alvila and T. T. Pakkanen, Sustainable Chem. Pharm., 2021, 20, 100376 CrossRef CAS.
- J. Li, W. Wang, S. Zhang, Q. Gao, W. Zhang and J. Li, RSC Adv., 2016, 6, 67435–67443 RSC.
- C. Gioia, M. B. Banella, M. Vannini, A. Celli, M. Colonna and D. Caretti, Eur. Polym. J., 2015, 73, 38–49 CrossRef CAS.
- N. Vasudevan and A. Mahadevan, Curr. Sci. Assoc., 1990, 59, 1323–1325 CAS.
- C. A. Hansen and J. W. Frost, J. Am. Chem. Soc., 2002, 124, 5926–5927 CrossRef CAS PubMed.
- P. Cassagnau, V. Bounor-Legaré and F. Fenouillot, Int. Polym. Process., 2007, 22, 218–258 CrossRef CAS.
- P. Cassagnau, V. Bounor-Legaré and B. Vergnes, Mech. Ind., 2019, 20, 803 CrossRef.
- B. Guerdener, V. Ayzac, S. Norsic, P. Besognet, V. Bounor-Legaré, V. Monteil, V. Dufaud, J. Raynaud and Y. Chalamet, Green Chem., 2023, 25, 6355–6364 RSC.
- A. Breuillac, F. Caffy, T. Vialon and R. Nicolaÿ, Polym. Chem., 2020, 11, 6479–6491 RSC.
- J. Raquez, R. Narayan and P. Dubois, Macromol. Mater. Eng., 2008, 293, 447–470 CrossRef CAS.
- P.-I. Dassie, R. Haddad, M. Lenez, A. Chaumonnot, M. Boualleg, P. Legriel, A. Styskalik, B. Haye, M. Selmane, D. P. Debecker, C. Sanchez, C. Chaneac and C. Boissiere, Green Chem., 2023, 25, 2800–2814 RSC.
- G. Moad, Prog. Polym. Sci., 1999, 24, 81–142 CrossRef CAS.
- H. Walderhaug, O. Söderman and D. Topgaard, Prog. Nucl. Magn. Reson. Spectrosc., 2010, 56, 406–425 CrossRef CAS PubMed.
- C. S. Johnson, Prog. Nucl. Magn. Reson. Spectrosc., 1999, 34, 203–256 CrossRef CAS.
- A. Lucas, A. Tauleigne, F. Da Cruz-Boisson, A. Crépet, A. Bergeron-Vanhille, G. Martin, N. Garois, P. Cassagnau and V. Bounor-Legaré, Ind. Eng. Chem. Res., 2020, 59, 16579–16590 CrossRef CAS.
- S. Pratihar and S. Roy, J. Org. Chem., 2010, 75, 4957–4963 CrossRef CAS PubMed.
- R. B. Durairaj, Resorcinol: chemistry, technology, and applications, Springer, Berlin, New York, 2005 Search PubMed.
- Phenolic Resins: A Century of Progress, ed. L. Pilato, Springer Berlin Heidelberg, Berlin, Heidelberg, 2010 Search PubMed.
- T. Li, M. Cao, J. Liang, X. Xie and G. Du, Polymers, 2017, 9, 45–55 CrossRef PubMed.
- T. Li, M. Cao, J. Liang, X. Xie and G. Du, Polymers, 2017, 9, 426–439 CrossRef PubMed.
- Polymer science: a comprehensive reference, ed. K. Matyjaszewski and M. Möller, Elsevier, Amsterdam, 2012 Search PubMed.
- T.-A. Yamagishi, M. Nomoto, S. Ito, S. Ishida and Y. Nakamoto, Polym. Bull., 1994, 32, 501–507 CrossRef CAS.
- S. Podzimek and L. Hroch, J. Appl. Polym. Sci, 1993, 47, 2005–2012 CrossRef CAS.
- L. Granado, R. Tavernier, G. Foyer, G. David and S. Caillol, Thermochim. Acta, 2018, 667, 42–49 CrossRef CAS.
- S. Vyazovkin, A. K. Burnham, J. M. Criado, L. A. Pérez-Maqueda, C. Popescu and N. Sbirrazzuoli, Thermochim. Acta, 2011, 520, 1–19 CrossRef CAS.
- J. H. Flynn, J. Therm. Anal., 1983, 27, 95–102 CrossRef CAS.
- J. Wang, M.-P. G. Laborie and M. P. Wolcott, Thermochim. Acta, 2005, 439, 68–73 CrossRef CAS.
- L. Granado, S. Kempa, L. J. Gregoriades, F. Brüning, A.-C. Genix, N. Fréty and E. Anglaret, Thermochim. Acta, 2018, 667, 185–192 CrossRef CAS.
- P. W. Kopf, Phenolic resins, in Kirk-Othmer Encyclopedia of Chemical Technology, Wiley, 2003 Search PubMed.
- E. Bellinetto, N. Fumagalli, M. Astorri, S. Turri and G. Griffini, ACS Appl. Polym. Mater., 2024, 6, 1191–1203 CrossRef CAS PubMed.
- S. B. Shen and H. Ishida, J. Appl. Polym. Sci., 1996, 61, 1595–1605 CrossRef CAS.
- N. Gabilondo, M. López, J. A. Ramos, J. M. Echeverría and I. Mondragon, J. Therm. Anal. Calorim., 2007, 90, 229–236 CrossRef CAS.
- L. Granado, R. Tavernier, G. Foyer, G. David and S. Caillol, Chem. Eng. J., 2020, 379, 122237 CrossRef CAS.
- S. Vyazovkin and N. Sbirrazzuoli, Macromol. Rapid Commun., 2006, 27, 1515–1532 CrossRef CAS.
- E. Fel, V. Massardier, F. Mélis, B. Vergnes and P. Cassagnau, Int. Polym. Process., 2014, 29, 71–80 CrossRef CAS.
- N. Sbirrazzuoli, Molecules, 2019, 24, 1683 CrossRef CAS PubMed.
- S. Vyazovkin and D. Dollimore, J. Chem. Inf. Comput. Sci., 1996, 36, 42–45 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc05352d |
| This journal is © The Royal Society of Chemistry 2025 |