
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organonitrogen platform chemicals and pharmaceutical precursors: a perspective on sustainable chitin utilization
Ting
Wang
 a,
Junnan
Wei
*b and
Peter J.
Deuss
*a
a,
Junnan
Wei
*b and
Peter J.
Deuss
*a
aDepartment of Chemical Engineering (ENTEG), University of Groningen, Nijenborgh 3, 9747 AG Groningen, The Netherlands. E-mail: p.j.deuss@rug.nl
bCollege of Food Science and Pharmaceutical Engineering, Nanjing Normal University, Nanjing, 210009, China. E-mail: wei_junnan@njnu.edu.cn
First published on 30th January 2025
Abstract
Chitin is an abundant biopolymer sourced from, among others, crustacean shell waste and contains biologically-fixed nitrogen. Its conversion into valuable nitrogen-containing organic compounds has gained growing interest. These compounds can serve as platform chemicals and offer a sustainable alternative to fossil-based chemical production. In this review, we discuss the general pathways for chitin valorisation, including chitin pyrolysis, deacetylation to chitosan, depolymerisation into N-acetyl chitin oligosaccharides (COSs) and N-acetylglucosamine (NAG) and subsequent conversion to furans, heterocyclic compounds, polyols, amines and amino acids. Special emphasis is placed on the recent progress in expanding the chemical space attainable from chitin-derived furan platforms, particularly focusing on the synthesis of benzenoid aromatic compounds via Diels–Alder and subsequent dehydration reactions. By highlighting these transformation pathways and the resulting high-value products, this review provides a comprehensive overview of recent developments and future opportunities for the sustainable production of nitrogen-containing compounds from chitin.
Green foundation1. Organonitrogen platform chemicals are vital components of numerous chemical products, including polymers and pharmaceuticals. However, their current industrial synthesis is energy-intensive, relying on the Haber–Bosch process, which generates significant carbon emissions.2. Chitin, a natural polysaccharide derived from crustacean shell waste, offers a renewable alternative for producing organonitrogen chemicals without the Haber–Bosch process. Hence, the review advances green chemistry by utilizing chitin, as a sustainable and renewable resource for producing nitrogen-containing (2x) chemicals, potentially reducing the environmental footprint of nitrogen-rich chemical production. 3. By showcasing the potential of chitin as a feedstock for organonitrogen chemicals, the insights from this review highlight opportunities to meet high market demand for nitrogenous pharmaceuticals and agrochemicals through scalable processes, catalytic innovations, integrated biorefineries, and expanded product portfolios. |
1. Introduction
Resource shortages and environmental degradation are increasingly driving industry to prioritize sustainable and green technologies. Traditional reliance on fossil resources for chemical production has not only strained non-renewable resources but also contributed to significant carbon dioxide emissions, further exacerbating climate change.1,2 This situation underscores the urgent need for alternative feedstocks, such as biomass that can address both environmental and resource-related challenges.The formation of bonds between carbon skeletons and nitrogen atoms is crucial for the synthesis of nitrogen-containing compounds and is considered one of the top five most important reactions for the production of value-added materials.3 Nitrogen-containing compounds are integral to the structural framework of numerous pharmaceuticals and play a pivotal role across diverse fields, particularly aromatic compounds.4 The current synthesis of these compounds is predominantly reliant on the Haber–Bosch process which after a long history still remains the cornerstone of nitrogen fixation. This process, however, is highly energy-intensive, requiring significant fossil fuel consumption, which results in significant environmental and economic costs.5
Additionally, the global demand for aromatic compounds is projected to grow significantly, with the market size increasing from $25.67 billion in 2024 to an estimated $38.67 billion by 2032.6 This includes many important nitrogen-containing aromatic building blocks such as anilines. However, this rising demand is predominantly met through fossil carbon sources, such as petroleum and coal, which serve as the primary feedstocks for aromatic compound synthesis. In light of these challenges, there is a pressing need to develop sustainable and eco-friendly alternatives for producing both nitrogen-containing and aromatic compounds.
Biomass-derived feedstocks have emerged as promising candidates for sustainable chemical production, offering potential routes to reduce CO2 emissions and alleviate the depletion of non-renewable resources. Among these, chitin stands out due to its abundance and naturally fixed nitrogen content.7 Chitin is derived from a diverse array of natural sources, including the exoskeletons of crustaceans (such as crabs, shrimp, and lobsters), the cell walls of fungi, and the cuticles of insects.8 These sources contribute to its abundance as a renewable biopolymer.9 Global fisheries alone generate an estimated 6–8 million tons of crustacean shell waste annually, providing a substantial source of chitin and thus of fixed organo-nitrogen for utilization.10
Chitin is a linear polysaccharide composed of β-1,4-linked 2-(acetylamino)-2-deoxy-D-glucose units, structurally similar to cellulose but with an acetamido group at the C2 position (Scheme 1).11 This unique structure forms a network of strong intra- and intermolecular hydrogen bonds, imparting high crystallinity and robustness to chitin. Chitin exists in three different structural forms, α-chitin, β-chitin, and γ-chitin.12 α-Chitin is predominantly sourced from the exoskeletons of crustaceans, particularly shrimps and crabs. β-Chitin is typically extracted from squid pens, while γ-chitin is obtained from sources like fungi and yeast.13 These forms differ in their crystalline structures and how the molecular chains are arranged (Fig. 1). α-Chitin has an anti-parallel chain arrangement that forms a tightly packed structure with strong hydrogen bonds. In β-chitin, the polymer chains are arranged in a parallel configuration, and this arrangement results in a less ordered and more loosely packed structure. Besides, γ-chitin is a less common form of chitin, with a structure involving a mix of both α and β forms. Of these different chitin forms, α-chitin is the most abundant, crystalline, and compact form. To disrupt this robust structure, various pretreatment methods have been developed to increase its accessibility and enhance its reactivity for its further conversion to nitrogen-containing compounds.
 | ||
| Scheme 1 Chitin biomass valorisation for high-value nitrogen-containing chemicals. | ||
 | ||
| Fig. 1 Different structural form of chitin (a) α-chitin, (b) β-chitin, and (c) γ-chitin.12 | ||
Beyond these pretreatment strategies, this review provides a comprehensive overview of the recent advances in the transformation of chitin into high-value nitrogen-containing compounds. We begin by briefly discussing the primary processes used to modify the chitin polymer structure, including its deacetylation to produce chitosan, a versatile derivative with numerous applications in biomedicine, agriculture, and water treatment.14 Additionally, we examine the depolymerisation of chitin into its monomer N-acetylglucosamine (NAG) and also N-acetyl chitin oligosaccharides (N-acetyl COSs), which have gained attention for their bioactive properties and potential use in pharmaceuticals and functional materials.15–18 The pyrolysis of chitin, which offers a pathway to generate a diverse array of nitrogen-rich compounds through thermochemical conversion, is also reviewed, highlighting the product scope and the possible pyrolysis pathways.
The core focus of this review is on the synthesis of high-value nitrogen-containing products derived from chitin. We explore the synthesis of various nitrogenous compounds, such as furans, heterocyclic compounds, polyols, amines, and amino acids (Scheme 1). Furthermore, a particular focus is given to the latest advancements in expanding the chemical space attainable from these nitrogen-containing furans. Finally, the generation of benzenoid aromatic compounds is highlighted as for example their formation from furans via Diels–Alder reactions and subsequent aromatization represents a promising emerging area for the utilization of chitin. By highlighting these transformation pathways and the resulting high-value products, this review aims to provide insights into the potential of chitin as a renewable feedstock for the production of valuable nitrogen-containing chemicals, thereby contributing to the development of sustainable and environmentally friendly processes in the chemical industry.
2. Depolymerisation and deacetylation
Chitin pretreatment, including deacetylation and depolymerisation, can yield various oligomers and chitosan with diverse degrees of deacetylation (DD), respectively, which have a significant impact on the subsequent production of low-molecular-weight chemicals. This section provides an overview of the synthesis of N-acetyl chitin oligosaccharides (COSs) and N-acetylglucosamine (NAG) from chitin biomass, highlighting the distinctions among enzymatic, chemical, and mechanochemical approaches. Furthermore, the deacetylation of chitin is discussed, including the heterogenous and homogenous chemical, and enzymatic approaches. The various methods used to determine DD, are briefly summarized as well.2.1. Chitin depolymerisation
By breaking down chitin into smaller components such as N-acetyl COSs and monomer NAG, researchers can tap into its significant potential for producing bioactive compounds. These compounds are highly valuable in fields ranging from medicine to agriculture due to their unique biological properties.15–18 A variety of depolymerisation techniques, including enzymatic, chemical, and mechanochemical methods, have been explored to achieve selective and efficient conversion of chitin into its desired products. Given the high crystallinity of chitin, these approaches are often combined with pretreatment methods to reduce chitin crystallinity, thereby improving solubility, swelling, enzyme or reagent accessibility, and thereby enhancing the efficiency of further chemical conversions.19 To obtain nitrogen-containing compounds, retention of the N-acetyl group is often desired as this masks the reactivity of the amine group. This means that one of the challenges is to optimize depolymerisation methods to minimize deacetylation and other undesired side reactions.Chitinases and other specific enzymes can selectively degrade chitin into N-acetyl COSs.16 Chitinases specifically catalyse the cleavage of β-1,4 linkages in chitin, typically producing monomers or dimers.20 For instance, chitinase PbCHi70 was reported to afford a total monomer and dimer yield of 89.6% from colloidal chitin after treatment at 50 °C for 24 h.21 In contrast, certain chitinases, such as SmChiC-D139A, have been shown to produce N-acetyl COSs with higher DP with 38.1% yield of DP6-9 and 5.9% yield of DP10-12 (37 °C, 18 h).22 Furthermore, β-N-acetylhexosaminidases exhibit strong transglycosylation activity, enabling the production of N-acetyl COSs with DP lower than 3 at around 35 °C within 4.5–6.5 h.16 Additionally, several non-specific enzymes, such as cellulase, lysozyme, lipase, papain, and pepsin, have been reported to produce N-acetyl COSs, albeit with relatively low yield. Enzymatic depolymerisation strategies enable the production of specific N-acetyl COSs with minimal side reactions under mild conditions. However, the high cost of enzymes and their sensitivity to reaction parameters, such as temperature and pH, often pose challenges to large scale and wide-spread application. Notably, a more comprehensive discussion of enzymatic methods for producing N-acetyl COSs and NAG from chitin can be found in the review by Wang and colleagues.16 This review thoroughly summarizes the specific reaction conditions and the results over various enzymes.
Depolymerisation using chemical approaches generally involves acid hydrolysis, where strong acids like hydrochloric acid are used to cleave the glycosidic bonds in chitin.15 Acid hydrolysis with HCl remains the most common and effective method. However, it often results in low yields of N-acetyl COSs and predominantly favours the production of monomeric NAG and D-glucosamine (GlcN). Comprehensive overviews and discussions are available in the studies by Niranjan et al.23 and Schmitz et al.24
Recently, mechanochemical methods have emerged as effective tools for producing N-acetyl COSs and NAG from chitin (Fig. 2). Mechanical-force-assisted depolymerisation of H2SO4-treated chitin produces soluble N-acetyl COSs, which can be further transformed into NAG (53% yield) in water at 443 K and 1-O-methyl-N-acetylglucosamine (MeGlcNAc, 70% yield) in methanol at 463 K.25 Additionally, a total yield of 66% of N-acetyl COSs with DP ranging from 2 to 7, along with 8.3% of NAG, was achieved by using activated carbon materials as catalysts during the milling process.26 In the mechanocatalytic hydrolysis process, the ball-milling first disrupts the crystallinity of chitin, which enhances the accessibility of catalysts to the chitin structure. Additionally, the mechanical force works along the polymer chain of a chitin molecule, facilitating the breaking of polymer linkages (Fig. 2b). DFT calculations also suggest that tensile forces as well as compressible forces decrease the activation energy required for the hydrolysis of glycosidic bonds.27–29
 | ||
| Fig. 2 Mechanochemical hydrolysis of chitin to N-acetyl COSs and NAG (a) and proposed depolymerization scheme (b). | ||
Molten salts hydrates (MSH) emerge as effective media for chitin hydrolysis due to their ability to disrupt the robust inter- and intra-hydrogen bonding networks in chitin. Acidic LiBr MSH (AMSH) containing 40 mM HCl was shown to predominantly produce NAG with a yield of 71.5%, along with 3% of dimers at 120 °C in 30 min.30 The Li+ ions in AMSH break the hydrogen bonding network in chitin, facilitating dissolution and promoting the acid-mediated hydrolysis process. Furthermore, Yan et al. highlighted the potential of zeolites as solid acid catalysts for chitin hydrolysis in LiBr MSH, achieving a 63% yield of NAG using zeolite SAPO-34.31 The low density of Lewis acid sites in SAPO-34 minimized undesired side reactions, while appropriate Brønsted acid sites enhanced the hydrolysis reaction. Similarly, Chen et al. employed the more abundant and inexpensive CaCl2-based MSH to selectively convert chitin into a 51% yield of NAG at 393 K in 1 h.32 With ZnBr2 as the co-salt, the yield was further increased to 67%.
Overall, these recent studies on chitin depolymerisation highlight the potential of tuning the chitin-derived products. Acid hydrolysis methods are cost-efficient, but less sustainable. Enzymatic methods can achieve tuneable DP in chitin-derived oligomers by selecting specific enzymes. On the other hand, mechanochemical and MSH systems significantly improve the efficiency of chitin hydrolysis, especially for the production of the NAG monomer. While enzymatic methods excel in specificity, conventional acid hydrolysis, mechanochemical and MSH approaches are advantageous for scalability and cost-effectiveness in large-scale applications to some extent.
2.2. Deacetylation
Chitosan is a natural biopolymer derived from chitin through the removal of acetyl groups via chemical or enzymatic processes, resulting in a polymer composed of β-1,4 linked GlcN and (<50%) NAG units (Scheme 1).33Both acidic and alkaline conditions can facilitate deacetylation. However, alkaline deacetylation is more commonly employed because glycosidic bonds in chitin are highly susceptible to degradation in acidic environments.34 The use of alkalis is more suitable to preserve the polymer backbone, making it the preferred method for producing chitosan.
The alkali deacetylation is typically achieved in either heterogeneous or homogeneous methods through alkaline treatment using concentrated NaOH or KOH aqueous solution.14 In the heterogeneous method, high aqueous concentrations of NaOH (often 40–50%) are mixed with solid chitin using elevated temperatures (80–120 °C) for several hours resulting in insoluble chitosan with the degree of deacetylation (DD) ranging from 80% to 90%. The heterogeneous conditions proceed preferentially in the amorphous region leading to an uneven distribution of deacetylated regions and insoluble chitosan with residual crystalline part.8,34,35 Besides, high temperatures may cause some degradation of the polymer chains, reducing the molecular weight.
The homogeneous method involves two steps: a well-dispersed chitin mixture in a highly concentrated alkali solution (>50%) at room temperature, followed by dissolution through a freezing (−27 to 35 °C) and thawing (18–22 °C) process.36 The alkali solution swells the chitin particles at low temperatures and affords highly soluble chitosan with 40–50% DD with amorphous structures,35 leading to a more uniform deacetylation throughout the polymer matrix. Each of these methods has its own advantages and limitations depending on the desired properties of the final chitosan product and the specific application requirements.
The DD is influenced by several other factors as well, including the structure of the chitin substrate, reaction time, temperature, and base concentration. For instance, under the tested conditions (80–110 °C and 30–70% w/v base concentrations), higher temperatures and base concentrations resulted in higher DD values, ranging from 20% to 80%.37 Additionally, β-chitin shows better deacetylation activity than α-chitin due to its weaker intermolecular hydrogen bonding, which is attributed to the parallel arrangement of the main chains.38 However, increasing the severity of the conditions, chitin may undergo more extensive hydrolysis, decreasing the molecular weight (Mw) of chitosan.39
Chitin deacetylases provide a promising enzymatic alternative for the selective removal of acetyl groups from NAG units, resulting in the formation of chitosan without significant chain scission. These enzymes operate effectively at mild conditions with the optimum temperature at around 50–60 °C, making it environmental-friendly.12 However, their use has so far been restricted to low molecular weight and amorphous chitin on a laboratory scale.40 Additional information can be found in studies focusing on chitin deacetylases.41,42
The DD of chitosan is a critical parameter that significantly affects its physical, chemical, and biological properties.43,44 Accurate quantification of DD is essential to determine whether it can be used in a particular application. Various techniques are employed to quantify the DD of chitosan, each with its own advantages and limitations. Titration methods, including acid–base and conductometric titrations, are effective for determining the DD of chitosan, though these can be time-consuming.45 In acid–base titration, DD is quantified by titrating the free amine groups (–NH2) in chitosan which are generated during the deacetylation process. Conductometric titration assesses DD by measuring the changes in the conductivity during the titration of chitosan with a strong acid or base, corresponding to the protonation or deprotonation of the amine groups. UV spectrometry is an appropriate technique for quantitative analysis of soluble samples, particularly for highly deacetylated chitosan (DD: 0–50%).33 Fourier transform infrared spectroscopy (FT-IR) is another method used to measure DD, by analysing absorbance peaks at specific wavenumbers, such as 1655 cm−1 (amide I band) and 3450 cm−1 (amine group). FT-IR allows rapid measurements and consistent baseline correction is crucial for reproducible quantitative results.46 Nuclear magnetic resonance (NMR) spectroscopy is considered one of the most accurate methods for determining the DD of chitosan.46 In particular, quantitative 1H NMR measures the DD of chitosan by analysing the chemical shifts of the acetyl group protons (CH3) and the glucosamine protons in the chitosan backbone. However, 1H NMR is limited to a DD range of 0–60% due to the low solubility of chitosan with lower DD in deuterated solvents. In contrast, solid-state 13C NMR and 15N NMR spectroscopy can measure DD across the entire range (0–100%). Solid-state 13C NMR gives DD values by measuring the integral of the carbonyl or methyl group divided by the integral of all the carbon atoms in the backbone.47 In solid-state 15N NMR spectra, the amide and amine groups in chitin and chitosan display distinct peaks at approximately 110 ppm and 10 ppm, respectively, enabling accurate DD determination.47 Similarly, elemental analysis is another method for determining DD, particularly effective for samples with lower DD. Elemental analysis quantifies the nitrogen content in chitosan, by calculating the N/C ratio and comparing it to the theoretical values for fully acetylated chitin and fully deacetylated chitosan. However, the presence of nitrogen from non-chitosan sources can affect the accuracy of the results.45 In conclusion, selecting an appropriate method for determining DD is crucial, as each technique offers different advantages and limitations depending on the chitosan's specific characteristics.
3. Pyrolysis of chitin and its derivatives
Pyrolysis, a thermochemical decomposition process, has emerged as a key method for converting chitin into bio-oils, biochar, and various gases with applications in biofuels, chemical synthesis, and environmental remediation.48,49 Notably, the chitin pyrolysis oils are reported to contain valuable nitrogen-containing compounds. Herein, we introduce the thermal behaviour of chitin, pyrolysis product distributions, and possible pyrolysis pathways to highlight the potential of the use of pyrolysis for chitin valorisation.Thermogravimetric analysis shows that the thermal decomposition of chitin can be divided into three distinct degradation stages.50 The first stage, occurring below 150 °C, involves the loss of moisture, typically resulting in a 5–10% weight loss. This stage corresponds to the evaporation of absorbed water as the temperature increases. The main thermal degradation of chitin begins at around 250 °C and peaks between 300 °C and 400 °C. This stage is characterized by the breakdown of chitin's polymeric structure, including the decomposition of N-acetyl groups, glycosidic bonds, and side chains. The weight loss during this stage is significant, as volatile components such as acetamide, acetic acid, and ammonia are released. After the major weight loss, the TG curve stabilizes, with some minor mass loss continuing at higher temperatures. At the final stage, residual char, primarily carbonaceous material, is formed. The overall mass loss by the end of pyrolysis is typically 70–80 wt%, leaving around 20–30 wt% of char residue.
In the early years, studies primarily focused on exploring the thermal behaviours of chitin and identifying the key pyrolysis products. Franich, Goodin and Wilkins explored the pyrolysis of the chitin monomer, NAG at 400 °C, achieving isolated yields of 5%, 2%, and 3% for 3-acetamidofuran (3AF), 3-acetamido-5-acetylfuran (3A5AF) and acetamido acetaldehyde, respectively in addition to the confirmed formation of acetamide (Fig. 3, Group 1, No. 1–4).51 It should be noted that acetamido acetaldehyde was isolated as its dimethyl acetal due to the applied isolation process. Fabbri et al. proposed the possible production of 3-acetamido-5-methylfuran (3A5MF), acetamido-substituted 2- and 4-pyrones and hydroxydihydropyran-4-one from chitin pyrolysis based on mass spectra (Fig. 3, Group 2, No. 5–8).52 Simoneit et al. found that burning/charring of shrimp exoskeletons produced dominantly 1,6-anhydro-2-acetamido-2-deoxyglucose (Fig. 3, Group 3, No. 9),53 showing it can be a key marker for identifying emissions from cooking and burning crustaceans. Borchers et al. studied the pyrolysis of chitin in high-boiling solvents, tetraethyleneglycol dimethyl ether, as well as in supercritical acetone.54 Tetraethyleneglycol dimethyl ether allowed the production of soluble oligomers and inhibited secondary reactions. Besides, the degradation of chitin in supercritical acetone yielded about 85% decomposition, forming a 5.6% yield of 1,6-anhydro-2-acetamido-2-deoxyglucose (Fig. 3, Group 3 No. 9) as well as minor amounts of numerous nitrogen-containing heterocycles (e.g., N-acetylpyrrole, N-acetylpyrrolidine, 2-formylpyrrole, and 2-formyl-5-methyl-pyrrole, Fig. 3, Group 3, No. 10–13). Additionally, Knorr et al. specifically demonstrated the potential formation of pyrazines from chitin pyrolysis at temperatures ranging from 300 °C to 500 °C.55 The key pyrazines identified in this study included 2-methylpyrazine, 2,3-dimethylpyrazine, 2,5-dimethylpyrazine, and 2,3,5-trimethylpyrazine (Fig. 3, Group 4, No. 14–17). The concentration of these pyrazines increased with higher pyrolysis temperatures and longer durations.
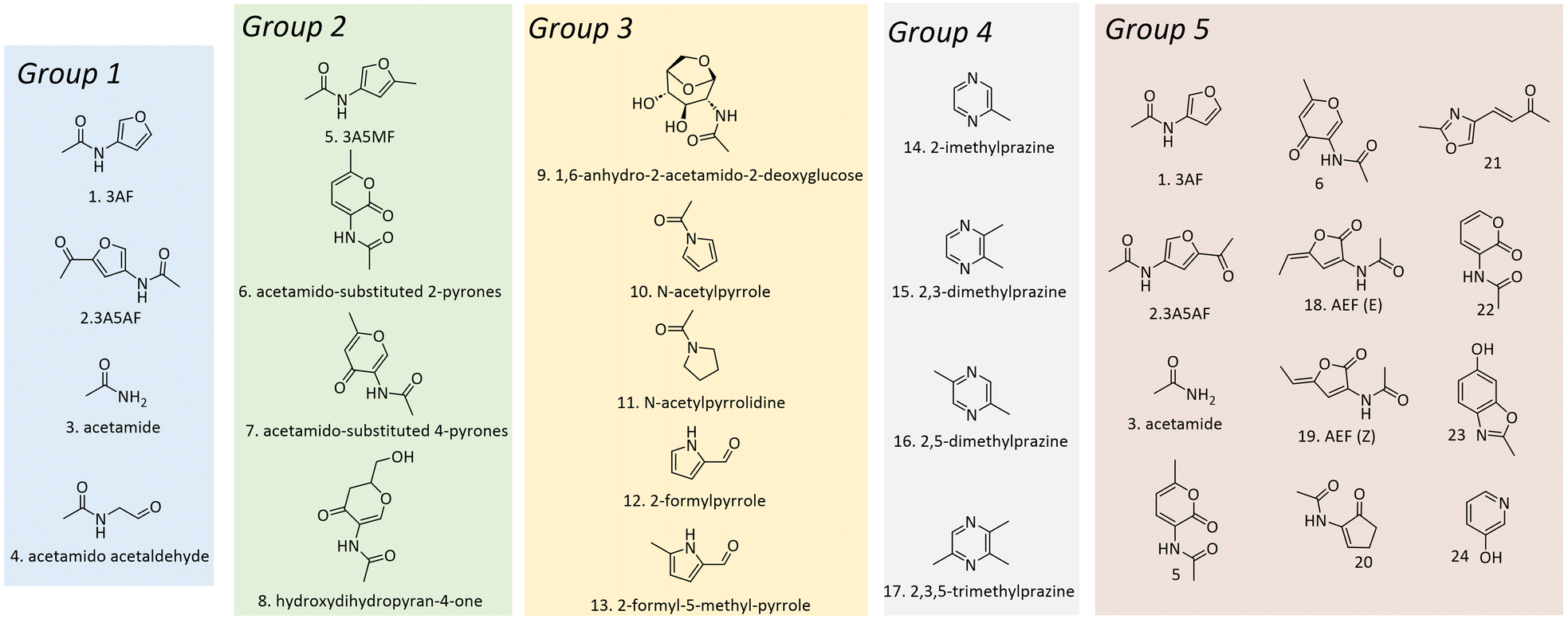 | ||
| Fig. 3 Representative products from pyrolysis of chitin or its derivatives. Products in Group 1–Group 5 were reported by Franich et al.,51 Fabbri et al.,52 Simoneit et al.,53 Knorr et al.,55 and Banwell et al., respectively.56 | ||
In recent years, the research on chitin pyrolysis has increasingly shifted its focus towards co-pyrolysis and catalytic pyrolysis. Liu et al. explored the co-pyrolysis of chitin with various nitrogen carriers, including urea, ammonium acetate, glutamic acid, and polyurethane, aiming to improve the production of nitrogen-rich bio-oil and biochar.50 Co-pyrolysis with urea increased the selectivity for nitrogen-containing compounds, primarily amines in the bio-oil, whereas the addition of polyurethane negatively affected the production of these nitrogenous compounds. Additionally, co-pyrolysis with ammonium acetate promoted the cleavage of C–N and then afforded a high concentration of acetic acid in bio-oil, possibly leading to less stable bio-oil. Notably, the nitrogen in urea was more easily converted to volatile nitrogen leading to the production of nitrogen-containing compounds through the Maillard reaction, whereas the nitrogen from other carriers tended to be retained in the biochar. Kwon and colleagues found the presence of CO2 shifted more carbon to the gas phase and reduced the nitrogen-containing pyrolytic oil in chitin pyrolysis.57
Catalytic chitin pyrolysis effectively reduced the activation energy (Ea) for the pyrolysis process, enhancing the efficiency of the pyrolysis process. Shen et al. found that the calcinated dolomite enhanced the conversion of large-molecular compounds into small-molecular products, likely due to the numerous basic sites of CaO and MgO in calcinated dolomite allowing the cleavage of glucosidic bond, C–O, and C–C bond.58 Specifically, the introduction of calcinated dolomite greatly increased the relative content of N-acetamido and N-heterocyclic compounds. Furthermore, Shen's group explored the catalytic pyrolysis of chitin using waste cathode material (BC) recovered from spent lithium-ion batteries as a catalyst.59 Wet-impregnation by acidic pickle liquor enhanced the production of small-molecule compounds such as acetamides, acetonitriles, and hydrocarbons while reducing the formation of heavy components, N-heterocycles (e.g., pyridines, pyrroles, and their derivatives). Cabrera-Barjas et al. showed that phosphorylation of chitin inhibited the formation of nitrogenous compounds leading to enhanced selectivity to long-chain hydrocarbons (C12–C17) during pyrolysis.60
Advanced techniques, such as single-crystal X-ray analysis and two-dimensional gas chromatography coupled with time-of-flight mass spectrometry (GC-GC-TOFMS) enable the identification of complex compounds that were previously difficult to detect. Umamaheswaran and Dutta et al. studied the GC-GC-TOFMS technique for chitin pyrolysis products, which allows enhanced separation and identification of complex N-containing compounds in low abundances.61 Notably, Banwell et al. explored the pyrolysis of raw chitin, pretreated chitin, and its derivatives. The product distribution varied greatly depending on the pretreatment of chitin (Fig. 2, Group 5).56 For instance, pyrolysis of raw chitin mainly gave acetamides (Fig. 3, Group 5, No. 3 & 18–20), while phosphoric acid-treated chitin afforded products 3, 5, and 18. Glyoxal solution pretreatment resulted in a completely different product distribution, mainly producing levoglucosenone and acetamide. Additionally, pyrolysis of untreated NAG only provided acetamide and product 23, while NaH3PO4 treated one afforded higher amounts of 3AF (Fig. 3, No.1). It is worth mentioning that this study provided a thorough structural characterization of the pyrolysis products (Fig. 2, Group 5) following their isolation using techniques such as single-crystal X-ray diffraction, NMR, and IR spectroscopy, allowing for detailed insight into the molecular structures of the compounds formed.
Recent studies have delved deeper into the pyrolysis pathways of chitin, offering a more detailed understanding of the thermal decomposition process.50,58,59,61 Chitin pyrolysis typically yields a wide range of products, including acids (e.g., acetic acid), alcohols (e.g., glycidol), aldehydes, ketones (e.g., acetone), hydrocarbons, carbohydrates (e.g., levoglucosenone), and especially N-containing compounds, such as nitriles, acetamides (e.g., acetamido acetaldehyde), and N-heterocycles (e.g., pyridines, pyrroles). The possible formation pathways of volatile products from chitin pyrolysis are illustrated in Scheme 2. The formation of acetamides is primarily attributed to the breakdown of glycosidic bonds, as well as the cleavage of C–C and C–O–C linkages (Scheme 2, Path 1). These compounds can also be produced from precursors such as acetonitrile and acetone through deoxygenation and hydrogenation reactions (Scheme 2, Path 1). In addition to the mentioned compounds, acetic acid is another significant product in chitin pyrolysis, primarily formed via the removal of side chain acetyl groups (Scheme 2, Path 1). The cleavage of the glycosidic bond and the reconstruction of 1,4-oxygen-bridged bonds play a crucial role in the formation of the oxygenates with five-membered ring structures (Scheme 2, Path 3). Levoglucosenone can be formed from the D-glucose moiety of the chitin monomer during the pyrolysis process (Scheme 2, Path 5). This compound is particularly important due to its potential applications as a bio-based chemical platform molecule. Its formation pathway involves the dehydration and rearrangement of glucose units, making it a key intermediate in the pyrolysis process. Moreover, the N-heterocycles can be generated by the intramolecular nucleophilic addition reaction, especially at relatively high temperatures (Scheme 2, Path 4 & 6). Furthermore, other light components such as hydrocarbons, acids, aldehydes, and ketones, can be formed through multiple reactions such as dehydration, ring-opening fragmentation, glycosidic bond breakage, and ring-opening reconstructions.
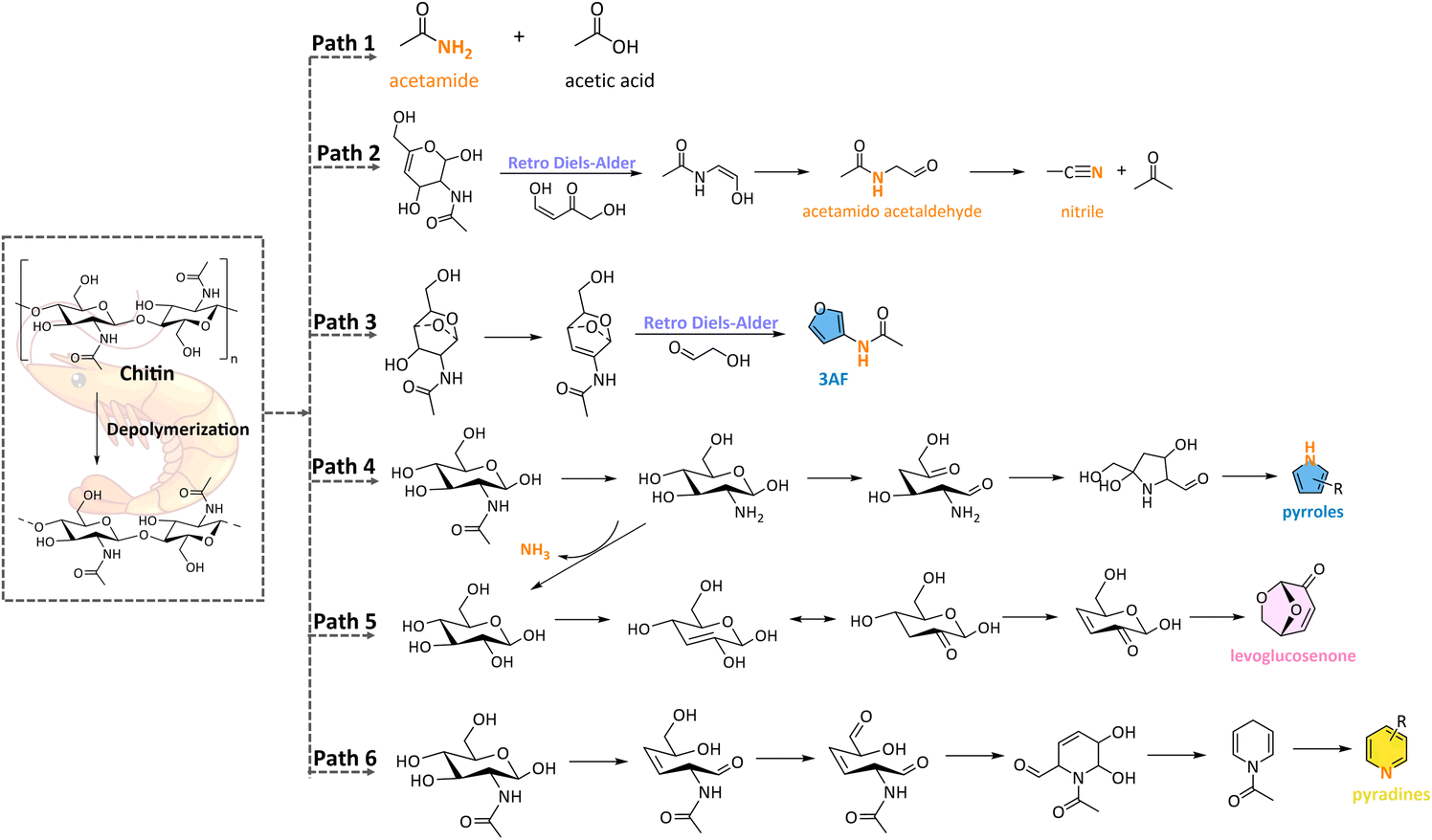 | ||
| Scheme 2 Proposed reaction pathways of forming representative products from chitin pyrolysis. | ||
The pyrolysis of chitin is a complex process that generates a variety of valuable products. Considerable progress has been made in understanding its thermal behaviour and identifying key pyrolysis products. However, there are still many challenges in optimizing product yield and selectivity. Much of the current research focuses on relative selectivity and product distributions, often using relative area measurements from GC(-MS) spectra. However, the absence of standardized calibration methods hinders accurate quantification and comparison of yields. Therefore, the few studies that provide isolated yields are of extreme value. To overcome these challenges, future research should prioritize the confirmation of complex product structures and the development of reliable calibration techniques that allow for precise quantification of pyrolysis products. This will enable more consistent and meaningful comparisons between studies and enhance the efficiency of chitin pyrolysis for industrial applications.
4. Chitin-derived monomers
Chitin pyrolysis typically requires high temperatures and often produces a wide range of products with low selectivity. In contrast, wet chemistry offers more controlled conditions, enabling the selective production of specific compounds. Chitin-derived monomers exhibit unique chemical functionality, making them versatile building blocks for the synthesis of various value-added products. Nitrogen-free monomers, such as 5-hydroxymethylfurfural (HMF) and other platform chemicals commonly derived from lignocellulosic biomass, can also be produced from chitin. These compounds have been extensively reviewed in the literature.62,63 However, converting chitin into such monomers results in the loss of its naturally-fixed nitrogen. Hence, in this section, we will explore the different catalytic methods used to derive nitrogen-containing monomers from chitin or chitin derivatives, highlighting recent advancements in their synthesis and plausible reaction pathways.4.1. N-containing furans
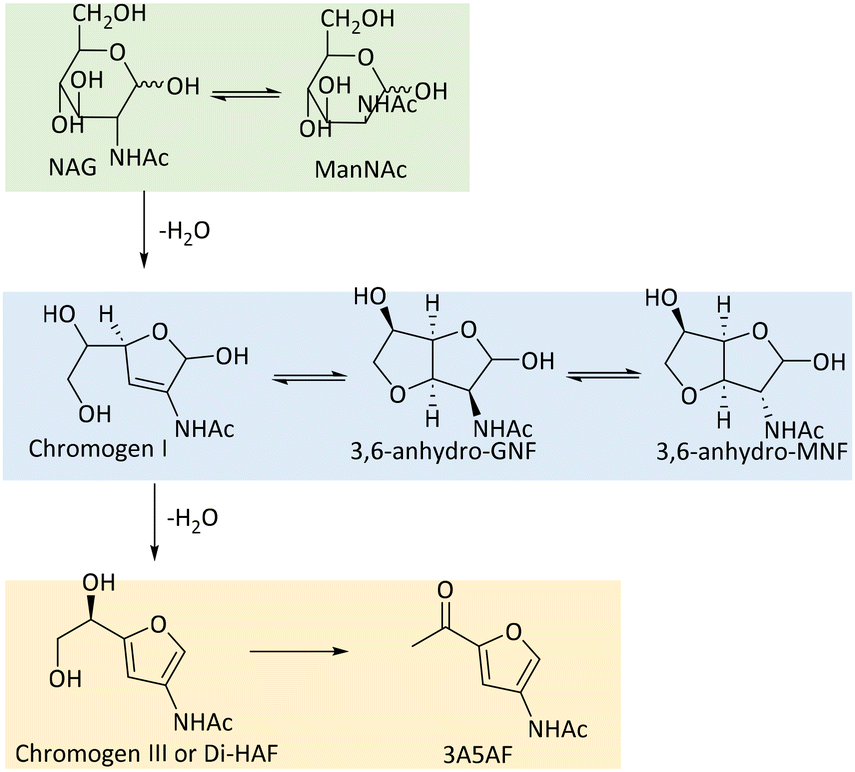 | ||
| Scheme 3 Derivatives from dehydration of NAG or ManNAc. | ||
In early studies, Chromogen I was synthesized through alkali treatment of NAG or N-acetylmannosamine (ManNAc) based on the Morgan–Elson reactions.65–67 Notably, Derevitskaya et al. demonstrated that sodium carbonate treatment at 70 °C for 2.5 h converted NAG to approximately 10% of isolated yield of 3,6-anhydro-MNF after recrystallization.67 However, selective formation of Chromogen I was not achieved in this alkaline dehydration system due to the co-occurrent generation of 3,6-anhydro-MNF and 3,6-anhydro-GNF. Use of the manno-isomer led to the production of a similar molar ratio of products compared to NAG showing that substrate stereo conformation is not of significance. Furthermore, a borate solution was also found to selectively dehydrate NAG to Chromogen I, yielding 56% of Chromogen I, 12% of 3,6-anhydro-MNF and 12% of 3,6-anhydro-MNF at 100 °C for 2 h.68 Also here, NAG and ManNAc gave a similar product distribution.
Zheng and coworkers developed an effective method for producing and purifying Chromogen I from NAG by utilizing metal-ion coordination strategy.69 They synthesized two nonanuclear lanthanide complexes featuring doubly deprotonated chiral Chromogen I as the organic ligand. A block-shaped pale-yellow crystal of Chromogen I-Ln cluster complex was gained with a yield of 40% after easy filtration followed by evaporation under ambient conditions. The decomposition of cluster complexes using HCl in methanol followed by trituration with NaHCO3 resulted in the precipitation of lanthanide carbonate. This process afforded Chromogen I with a 40% yield after solvent evaporation.
Since 2013, Osada and coworkers have focused on the synthesis of Chromogen I and Chromogen III from NAG/chitin in high-temperature water systems.64,70–72 The highest yields of 37% and 35% of Chromogen I and Chromogen III from NAG, respectively, were achieved at temperatures ranging from 180 to 280 °C with residence times of 5–34 s.71 The H+ and OH− ions generated from water dissociation promote two key reactions: the acid-promoted conversion of Chromogen I to Chromogen III and the base-promoted dehydration of NAG to Chromogen I. However, the concentrations of H+ or OH− generated by high-temperature water were inefficient for further dehydration of Chromogen III, resulting in almost no 3A5AF (<1%). Besides, high-temperature water treatment of chitin produced only 3% of Chromogen I and less than 0.5% of Chromogen III, even at 360 °C.73 Most of the chitin was converted to unidentified water-soluble compounds via decomposition reactions. Kinetic modelling demonstrated the activation energy for converting NAG to Chromogen I (85.7 kJ mol−1) was lower than that required for converting Chromogen I to Chromogen III (105 kJ mol−1), indicating that harsher conditions are necessary for the latter process.64 Notably, NAG decomposition occurs more readily than glucose decomposition, which requires 121 kJ mol−1,74 possibly due to the N-acetyl group in NAG lowering its decomposition energy to 95.6 kJ mol−1.
The deep eutectic solvent (DES), choline chloride (ChCl)-glycerol can also achieve the dehydration of NAG, leading to the formation of Chromogen III with a maximum yield of 31% at 140 °C within 140 min.75 In contrast, the ternary DES (proline-glycerol-lactic acid) significantly enhanced the dehydration process, achieving a maximum yield of 61% at 120 °C in 30 min.76
Recently, Minnaard et al. presented the first multi-gram synthesis of enantiopure Di-HAF from NAG (50 g) at significantly improved yields77 compared to an earlier report by Kuhn and Kruger.78 A good yield of 73% and >99% ee were achieved by dehydrating NAG in pyridine with phenylboronic acid and triflic acid (Scheme 4). Notably, the use phenylboronic acid enhanced chemoselectivity, while triflic acid significantly accelerated the reaction rate. The scalable synthesis opens up new possibilities for Di-HAF as a building block for the synthesis of other fine chemicals.
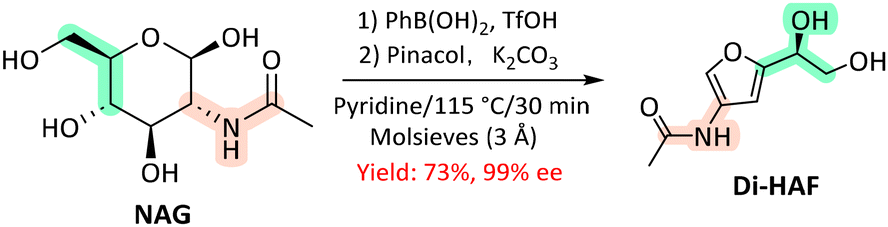 | ||
| Scheme 4 Synthesis of enantiopure Di-HAF by dehydration of NAG.77 | ||
The pyrolysis of chitin was initially employed to obtain 3A5AF, albeit with yields typically ≤5%, accompanied by various side-products such as 3-acetamidofuran, 2-acetylfuran and pyrazine (section 3).51,81 However, the extremely high reaction temperatures required for pyrolysis often lead to low selectivity and high tar content, necessitating further processing. These drawbacks have restricted its development in 3A5AF synthesis.
In pursuit of more efficient methods, researchers have explored various additives and catalysts. For instance, the combination of boric acid (B(OH)3) or B2O3 with (Lewis acidic) chloride salts were initially identified as the optimal formulation for synthesizing 3A5AF from NAG.19,82 The effectiveness of boric acid and borates is particularly noteworthy, as their action is believed to involve the formation of stable chelate adducts with substrate hydroxyl groups.83 These borate-hexose complexes facilitate the isomerization of aldoses to ketoses, likely by stabilizing intermediates or transition states.84
The synthesis of 3A5AF was initially constrained to high-boiling dipolar aprotic solvents such as N,N-dimethylacetamide (DMA), N,N-dimethylformamide (DMF), and N-methyl-2 pyrrolidone (NMP).79,85,86 In the presence of 1 eq. of B(OH)3 and 4 eq. of NaCl, a maximum yield of 3A5AF (62%) was gained from NAG (0.24 g) at 220 °C for 15 min using DMA as the solvent (Table 1 entry 1).79 A similar result (43% yield of 3A5AF) was obtained in a scaled-up experiment using 5.0 g of NAG at 220 °C for 30 min. Increasing the NaCl content was found to significantly enhance the yield of 3A5AF. For example, the yield increased by nearly 25% when the NaCl was increased from 0.5 to 4 eq. relative to NAG. Notably, NAG from different suppliers contains varying amounts of boron and chloride impurities, and higher levels of these elements in NAG led to a higher yield of 3A5AF. The combination of SrCl2 and B(OH)3 produced the highest yield of 3A5AF (52%) from NAG at 180 °C within 60 min (Table 1, entry 2).87 The in situ generated HCl gas and strontium borate likely promoted the selective dehydration process. Additionally, a combined additive system of B2O3 and MgCl2 can also produce a maximum yield of 42% 3A5AF from NAG at 180 °C within 60 min (Table 1, entry 3).82
| Entry | Solvent | Substrate | Catalyst | Additive | T (°C) | t (min) | Yield (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Conditions: conventional heating. b Microwave. c Isolated yield. d Proline-glycerol-lactic acid DESs. | ||||||||
| 1b | DMA | NAG | B(OH)3 | NaCl | 220 | 15 | 62 | 79 |
| 2 | NMP | NAG | B(OH)3 | SrCl2 | 180 | 60 | 52 | 87 |
| 3 | NMP | NAG | B2O3 | MgCl2 | 180 | 60 | 42 | 82 |
| 4b | [Bmim]Cl | NAG | B(OH)3 | — | 180 | 3 | 60, 57c | 88 |
| 5 | DMA | NAG | B(OH)3 + [TEA]Cl | — | 170 | 60 | 62 | 89 |
| 6 | DMA | NAG | [GlyCl] | CaCl2 | 200 | 10 | 53 | 86 |
| 7 | NMP | NAG | B2O3+[CMPy]Cl | CaCl2 | 180 | 20 | 67 | 90 |
| 8 | DMA | NAG | B(OH)3 + [Pyz]Cl | CaCl2 | 190 | 60 | 70 | 91 |
| 9 | NMP | NAG | ChCl-citric acid | CaCl2·2H2O | 210 | 20 | 47 | 92 |
| 10 | — | NAG | ChCl-PEG200-B(OH)3 | — | 180 | 15 | 18 | 93 |
| 11 | Ternary DESsd | NAG | Ternary DESs | — | 120 | 150 | 39 | 76 |
| 12 | DMF | NAG | AlCl3·6H2O | — | 120 | 30 | 30, 18c | 94 |
| 13 | NMP | NAG | [BuPy]2WCl8 + B(OH)3 | 120 | 120 | 60 | 95 | |
| 14 | DMF | NAG | NH4Cl | LiCl | 160 | 5 | 43, 45c | 96 |
| 15 | GVL | NAG | NH4SCN | HCl | 140 | 120 | 75, 41c | 97 |
| 16 | Pyridine | Di-HAF | Pyridine hydrochloride | — | 116 | 180 | 64c | 77 |
| 17 | DMF | NAG | Al-exchanged montmorillonite | NaCl | 135 | 120 | 14 | 98 |
| 18 | NMP | Chitin | B(OH)3 | NaCl + HCl | 215 | 120 | 8 | 85 |
| 19 | [Bmim]Cl | Chitin | B(OH)3 | NaCl + HCl | 180 | 60 | 6 | 99 |
| 20 | NMP | Chitin | [Gly]Cl | — | 180 | 60 | <1 | 86 |
| 21 | NMP | Chitin | [CMPy]Cl | — | 180 | 60 | 1 | 90 |
| 22 | NMP | Chitin | [Pyz]Cl | — | 180 | 60 | 2 | 91 |
| 23 | NMP | Chitin | [PDCMPi]Cl | — | 180 | 60 | <1 | 100 |
| 24 | NMP | Chitin | ChCl-citric acid | CaCl2·2H2O | 210 | 120 | 5 | 92 |
| 25 | [Bmim]Cl | BM-Chitin | B(OH)3 | HCl | 180 | 10 | 29, 20c | 19 |
Ionic liquids (ILs) have emerged due to their unique properties, such as low vapor pressure, high thermal stability, better capability for chitin dissolution, and particularly Brønsted and Lewis acidity as a solvent in chitin biomass conversion.101 Kerton et al. were the first to explore the dehydration of NAG in ILs, selectively achieving 60% and 38% yields of 3A5AF at 180 °C within 3 min in the presence of B(OH)3 or NaCl, respectively, using [Bmim]Cl as the solvent (Table 1 entry 4).88 Extraction by utilizing a two-layer solvent system with ILs ([Bmim]Cl-1-hexanol) containing the feedstock at the bottom and an extracting organic solvent on top effectively achieved a yield of 3A5AF of 40% starting from NAG. The system facilitated the separation of the desired product from the reaction mixture preventing secondary reactions.99 Zang et al. utilized ethanolamine-based ILs for the efficient conversion of NAG into 3A5AF (Table 1, entry 5).89 They achieved a high yield of over 62% under optimized conditions (4 weight equivalents IL, 1 weight equivalent of boric acid, 170 °C, 20 min, in DMA) using triethanolamine hydrochloride ([TEA]Cl), which was successfully reused for 5 times with a drop in yield from 60% to 40%. It was found that by replenishing with fresh boric acid activity could be recovered. Jiao Wang et al. showcased the efficient synthesis of 3A5AF using amino acid ILs.86 Specifically, the use of 200 wt% of glycine chloride [GlyCl] ILs resulted in a 43% yield of 3A5AF at 200 °C within 10 min using DMA as the solvent (Table 1, entry 6). The yield was further enhanced to 53% upon the addition of CaCl2. In contrast, lysine chloride [Lys]Cl2 yielded only 2% 3A5AF, likely due to its alkalinity. Moreover, [GlyCl] exhibited excellent recyclability, maintaining high yields over eight cycles. Additionally, Zang et al. conducted a series of studies to optimize the synthesis of 3A5AF from NAG using various catalytic systems and conditions with boron and chloride-containing additives. They first reported a maximum yield of 42% 3A5AF from NAG at 180 °C within 60 min using a combined additive system of B2O3 and MgCl2.82 Based on these results, they further explored acidic ILs for NAG dehydration. Without additional additives, the highest yield of 3A5AF reached 38% and 37% using 100 wt% of 1-carboxymethyl pyridinium chloride ([CMPy]Cl)90 and 200 wt% of pyrazine hydrochloride ([Pyz]Cl),91 respectively with NMP and DMA as solvents. Furthermore, the addition of the composite additives, such as B2O3-CaCl2 or B(OH)3-CaCl2 nearly doubled the 3A5AF yields, achieving 67% at 180 °C within 20 min (ref. 90) and 70% at 190 °C within 60 min,91 respectively (Table 1, entries 7 and 8). Compared with ILs containing chloride ions, IL catalysts with the HSO4− anion achieved significantly lower yields of less than 7%. These findings further underscore the critical roles of boron and chlorine elements in enhancing 3A5AF formation.
Deep eutectic solvents (DES), as analogues of ILs, are typically formed by mixing a hydrogen-bond donor and a hydrogen-bond acceptor.102 Due to their hydrogen-bonding capabilities, DES can effectively disrupt the inter-/intramolecular hydrogen bonding networks in polysaccharides, making them wildly used in upgrading lignocellulosic biomass.103 However, the application of DES in the conversion of chitin biomass to 3A5AF have been seldom reported. One notable example involves the use of a DES composed of ChCl and citric acid, which yielded 47% of 3A5AF from NAG at 210 °C in 20 min with NMP as the solvent and CaCl2·2H2O as the additive (Table 1, entry 9).92 Furthermore, acidic DESs, such as those composed of ChCl-citric acid, ChCl-lactic acid, and malic acid, have demonstrated superior performance in NAG dehydration compared to DESs containing polyols and urea. A DES consisting of ChCl, polyethylene glycol-200 (PEG200), and boric acid produced an 18% yield of 3A5AF at 180 °C within 15 min (Table 1, entry 10), even in the absence of additional solvents and additives.93 Binary and ternary DESs can been obtained using natural components such as proline, arginine, glycerol, and lactic acid.76 Among these, ternary DESs (e.g., Pro-Gly-Lactic Acid) was recently demonstrated to achieve a 3A5AF yield of 39% at 120 °C within 150 min (Table 1, entry 11). Interestingly also the Di-HAF yield was monitored over time reaching a maximum of 43% after 80 min in the same reaction. Lactic acid was claimed to play pivotal role in promoting the reaction by strengthening hydrogen-bonding networks, providing mild acidity to catalyse dehydration, and interacting with specific GlcNAc sites, thereby facilitating its transition to the β-pyranose tautomer. Despite these relatively promising results, the potential of DES in chitin biomass conversion remains to be fully explored, offering a fertile area for further research and development.
The synthesis of 3A5AF has traditionally required high temperatures exceeding 180 °C. However, Fukuoka et al. proposed an alternative approach using AlCl3·6H2O in DMF as the solvent, which allows for lower temperature conditions.94 For instance, this approach achieved a 30% yield of 3A5AF from a 2.5 wt% concentration of NAG at 120 °C in 30 min (Table 1, entry 12). Even at a higher NAG concentration of 10 wt%, the yield remained at 20%, demonstrating the method's potential for scalability. Remarkably, AlCl3·6H2O was able to produce a 7% yield of 3A5AF even at a significantly low temperature of 80 °C. In this reaction system, two main by-products were identified, 3-acetamido-5-ethylidene furanone (AEF) (Fig. 3, No. 18 & 19) and HMF. Notably, in contrast to HMF, AEF was more favoured at higher NAG concentrations, which is possibly enhanced by the stronger intermolecular interactions of NAG at higher concentrations. Compared to the 2%–4% yield of AEF over AlCl3·6H2O, higher AEF yield can be obtained in the presence of Al(OTf)3, which could be a good starting point for AEF synthesis. In another study, Zang and colleagues employed a combination of WCl6 and B(OH)3 as additives for NAG dehydration, resulting in a 34% yield of 3A5AF at 120 °C within 1 h using NMP as the solvent.95 The introduction of 1-butylpyridinium chloride [BuPy]Cl further improved 3A5AF yield to 45%. Building on these findings, this group synthesized Brønsted-Lewis acidic ILs, by mixing WCl6 with different ILs at certain temperatures.95 Under optimal conditions, which included a premixing time of 3 h under a nitrogen atmosphere, they achieved a 60% yield of 3A5AF using [BuPy]2WCl8 as the catalyst with B(OH)3 as an additive (Table 1, entry 12). The study also highlighted that ILs synthesis conditions, such as the nitrogen atmosphere and premixing time, significantly influence the performance of [BuPy]2WCl8. For example, reducing the premixing time of WCl6 and [BuPy]Cl to one hour decreased the 3A5AF yield to 53%, and the yield further declined to 44% in an air atmosphere.
Ammonium salts have also demonstrated promising results in the dehydration of NAG to 3A5AF. Zhang and Chen reported that using NH4Cl as the catalyst yielded 43% yield of 3A5AF in DMF, with LiCl as an additive at 160 °C within 5 min (Table 1, entry 13).96 Notably, the reaction system exhibited good scalability, maintaining a consistent yield of 42% for 3A5AF even at a scaled-up volume of 200 mL. Chen et al. identified NH4SCN as the most effective catalyst for enhancing 3A5AF production from NAG using γ-valerolactone (GVL) as the solvent.97 GVL, a bio-based and non-toxic solvent, serves as an efficient and sustainable alternative to hazardous solvents such as DMF and DMA. The dehydration of NAG to 3A5AF was optimized at 140 °C for 2 h, achieving a high yield of 75% (Table 1, entry 15). This reaction was facilitated by a dissolution-dehydration effect, where NH4SCN significantly enhanced NAG's solubility in GVL, thereby promoting rapid conversion and dehydration. Specifically, the NAG solubility in GVL increased from 14% to 99% with the addition of 200 mol% of NH4SCN. The proposed reaction pathway involved the formation of intermediates Chromogen I and Chromogen III before final dehydration to 3A5AF, as evidenced by UPLC-TOF-MS and NMR analyses. Minnaard and colleagues proposed an alternative strategy for the dehydration of NAG to 3A5AF by going via Di-HAF. Isolated Di-HAF was converted to 3A5AF in a 64% yield using pyridine hydrochloride in pyridine at 116 °C for 3 h (Table 1, entry 16).77 These works collectively represent significant advances in the synthesis of 3A5AF, showcasing the potential for lower temperature processes and more sustainable, scalable methods. Most studies on 3A5AF synthesis have focused on homogeneous catalysts offering relatively high efficiency and yield, but these typically involve complex separation and recovery processes. In contrast, heterogeneous catalysts offer the advantage of easy recovery and reuse across multiple reaction cycles, leading to reduced costs and waste. Recently, Yamaguchi et al. investigated the catalytic efficiency of various solid catalysts for NAG dehydration in the presence of NaCl and DMA as a solvent. The Al-exchanged montmorillonite catalyst outperformed others tested catalysts (H-ZSM-5, TiO2, and γ-Al2O3). The Al-exchanged montmorillonite catalyst is believed to perform better due to its Brønsted and Lewis acid sites that facilitate the dehydration reaction.98 The highest yield of 3A5AF (14%) was achieved at 135 °C within 2 h, meanwhile forming around 16% of HMF (Table 1, entry 17). Despite the relatively low 3A5AF yield, this study highlights the potential of using solid catalysts for NAG dehydration.
Compared to using NAG as a substrate, the yield of 3A5AF significantly decreases when chitin is directly converted. In Yan et al.'s study, the combination of B(OH)3 and NaCl was also shown to be the most effective additive system for the direct conversion of chitin (50%) into 3A5AF (8% yield) at 215 °C within 2 h in NMP (Table 1, entry 18).85 The addition of HCl further facilitated chitin hydrolysis. Furthermore, the introduction of ethylene glycol (EG) and 1,3-propanediol as poison test significantly inhibited 3A5AF formation in contrast to the stable yield observed with ethanol. This suggests the involvement of boron-containing five- or six-membered ring intermediates during the dehydration process. The disappearance of two -OH peaks in 1H NMR and the arisen new peaks in 13C and 11B NMR further confirmed the formation of chelate complexes between the hydroxyl groups in NAG subunits and B(OH)3. Notably, a minor product, 4-(acetylamino)-1,3-benzenediol, was also detected, indicating the potential to derive organonitrogen benzenediols from chitin (Scheme 5) in analogy to HMF-derived 1,2,4-benzenetriol.104 Later, they screened 10 ILs with different anions/cations and 25 additives for the direct conversion of chitin into 3A5AF.99 Only chloride-containing ILs formed 3A5AF from chitin. Under milder conditions (180 °C, 1 h), a comparable yield of 3A5AF (6%) was achieved in [Bmim]Cl (Table 1, entry 19) similar to the yield obtained using NMP at higher temperatures (215 °C, 2 h) (Table 1, entry 18). While the distillation and extraction methods proved effective for NAG conversion, they were unsuccessful in the direct conversion of chitin, likely due to the higher viscosity of the reaction mixture. Similarly, ILs such as [Gly]Cl, [CMPy]Cl, [Pyz]Cl, and [PDCMPi]Cl both demonstrated good performance in NAG dehydration,86,90,91,100 but yielded almost no 3A5AF (Table 1, entries 20–23). Similarly, acidic DES, ChCl-citric acid, gave a maximum 3A5AF yield of 5% from chitin (Table 1 entry 24).92 Subsequently, various methods, such as ball milling, steam explosion, alkaline/acid treatment, and ILs dissolution and regeneration, have been explored to disrupt the structural rigidity of chitin and enhance its reactivity and its ability to be solubilized.19 Among these, ball-milling made a great difference. Under milder conditions (180 °C, 10 min, [Bmim]Cl), a significantly increased 3A5AF yield (29 wt%) was obtained from ball-milled chitin (Table 1, entry 25). This improvement is likely due to the substantially reduced crystallinity of chitin (crystallinity index: 28%) by the disruption of internal hydrogen bonding interactions. Consistent with findings from reference,26 ball milling appears to facilitate the hydrolysis of chitin into oligosaccharides or even the monomers, thereby enhancing the efficiency of the subsequent dehydration process due to improved access and dissolutions. These results highlight the challenge of directly converting chitin into 3A5AF and chitin hydrolysis is the limiting step, underscoring the need for alternative strategies or effective hydrolysis methods to address the limitations posed by chitin's structural integrity.
 | ||
| Scheme 5 Proposed pathway of the formation of 4-(acetylamino)-1,3-benzenediol from NAG.85 | ||
 | ||
| Scheme 6 Potential chemical transformation of NAG or 3A5AF to other N-rich furans. | ||
3A5F has been recognized as a versatile building block for N-containing heterocycles and aromatics as well as a bio-conjugation agent.105 Its synthesis is notably efficient, with periodate oxidation of Di-HAF readily producing 3A5F at room temperature. Minnaard et al. demonstrated an 83% isolated yield of 3A5F from Di-HAF on a 10 g scale (Table 2, entry 1).77 Similarly, Gomes and co-workers achieved a 76% yield of 3A5F through a one-pot transformation of NAG on a 2 g scale (Table 2, entry 2).105
| Entry | Substrate | Product | Catalyst | T (°C) | t (h) | Yield (%) | Ref. |
|---|---|---|---|---|---|---|---|
| a Isolated yield. | |||||||
| 1 | Di-HAF | 3A5F | NaIO4 | RT | 1 | 83a | 77 |
| 2 | NAG | 3A5F | B(OH)3, NaIO4 | RT | 1.5 | 76a | 105 |
| 3 | NAG | 3AF | Ba(OH)2, B(OH)3 | 180 | 0.3 | 74/86a | 106 |
| 4 | NAG | 3AF | La2O3 | 180 | 3 | 50/50a | 107 |
| 5 | 3A5AF | 3A5HEF | Iridium complex | RT | 2.5 | 80a | 108 |
| 6 | 3A5AF | (S)-3A5HEF | Noyori catalyst | 50 | 7 days | 91a | 109 |
| 7 | 3A5AF | (S)-3A5HEF | YueD | 30 | 9 | 90 | 110 |
| 8 | 3A5AF | (R)-3A5HEF | ScCR | 30 | 9 | >99 | 110 |
| 9 | NAG | (R)-3A5HEF | M4 from ScCR | 30 | 12 | 53 | 111 |
| 10 | 3A5AF | 2A4AF | NaOH | 80 | 1 | 57 | 108 |
| 11 | 3A5AF | 2A4AF | MmH | 37 | 12 | 79 | 112 |
| 12 | 3A5AF | 3A5AEF | ATA117 | 45 | 6 | 84 | 112 |
3AF can be obtained through chitin pyrolysis, as discussed in detail in Section 3, but also has been formed more selectively using wet chemistry. Cao and Gan et al. developed an efficient catalytic system to selectively convert NAG to 3AF.106 By employing a combination of barium hydroxide (Ba(OH)2), B(OH)3, and NaCl, they achieved a 74% yield of 3AF in NMP at 180 °C in 20 min (Table 2, entry 3). Notably, the recrystallization process in a 2-methyltetrahydrofuran (MeTHF)/water solution resulted in an isolated yield of 86%. A key finding of their study was that the formation of 3AF stemmed from the retro-aldol breakdown of NAG, catalysed by bases, such as Ba(OH)2 (Scheme 7), rather than through the degradation of intermediates like 3A5AF or Chromogen III. The intermediate glycolaldehyde was successfully trapped in situ with acetylacetone to obtain the corresponding furan 2-methyl-3-acetylfuran (MAF), confirming the formation of glycolaldehyde as an intermediate (Scheme 7). The mass spectroscopy studies also detected the N-acetylerythrosamine (NAE) cyclization intermediate (Scheme 7, dash line), gaining more insights into the reaction pathway (Scheme 7). In contrast, using 3A5AF as a substrate under a NaOH catalytic system produced the amino-substituted furan, 2A4AF, with a yield of 57% at 80 °C in 1 h (Table 2, entry 10).108 These results further confirm that 3A5AF is not an intermediate in the dehydration of NAG to 3AF and that basic conditions favour the conversion of 3A5AF to 2A4AF. Additionally, La2O3, a catalyst with notable Brønsted basicity and nanopores, was also able to facilitate this conversion, achieving a 50% yield of 3AF at 180 °C after 3 h (Table 2, entry 4).107
 | ||
| Scheme 7 Proposed formation pathway of 3AF from NAG.106 | ||
The N-containing chiral furfuryl alcohol 3A5HEF serves as a crucial precursor for the synthesis of the rare 2-amino sugar L-rednose, as well as various nitrogen-based scaffolds important in medicinal chemistry.113 Kerton et al. first reported the synthesis of 3A5HEF using sodium borohydride (NaBH4) or via the transfer hydrogenation with chiral Ir-based catalysts from 3A5AF (Scheme 8a).108 They reported a high yield of 80% at room temperature within 2.5 h (Table 2, entry 5), although no enantioselectivity was reported. Later, the asymmetric reduction of 3A5AF to (S)-3A5HEF was achieved using a Ru-based Noyori catalyst under high-pressure (65 psi) hydrogen conditions (Scheme 8b), yielding 91% of 3A5HEF with 91% ee over a 7-day period (Table 2, entry 6).109 Recently, Li and Wang et al. described a two-step process for converting NAG into 3A5HEF, integrating chemical catalysis with enzyme catalysis to achieve excellent yields and enantioselectivities for 3A5HEF.110 The first step involves the dehydration of NAG to 3A5AF using [Bmim]Cl-B(OH)3 catalytic system, yielding around 31% of 3A5AF. Subsequently, carbonyl reductases, Streptomyces coelicolor (ScCR) (Scheme 8c) and CR from Bacillus sp. ECU0013 (YueD) (Scheme 8d), respectively catalysed the asymmetric reduction of 3A5AF into (R)- and (S)-3A5HEF, leading to yields of >99% with excellent ee values of >99% (Table 2, entries 7 and 8). Furthermore, Chen and co-workers developed a one-pot chemobiocatalytic method to convert NAG to (R)-3A5HEF, combining chemical dehydration of NAG to 3A5AF with tyrosine hydrochloride (TyrHCl) and CaCl2, followed by the asymmetric enzymatic reduction of 3A5AF to (R)-3A5HEF (yield: 53%, ee: >99%) using an engineered carbonyl reductase (M4) from ScCR (Scheme 8e, Table 2, entry 9).111
 | ||
| Scheme 8 Chemical and chemoenzymatic routes toward 3A5HEF from NAG.108–111 | ||
The biocatalytic approach was also employed for the synthesis of 2-acetyl-4-aminofuran (2A4AF) and 3-acetylamino-5-(α-aminoethyl)-furan (3A5AEF) from 3A5AF.112 The cited study employs two enzymes, an amidase (MmH) and aminotransferase (ATA117), to transform 3A5AF into 2A4AF and 3A5AEF, respectively catalysing the deacetylation of 3A5AF to yield 2A4AF (79.3%) (Table 2, entry 11) and facilitating the reductive amination to produce (R)-3A5AEF (84.0%) with high enantioselectivity (over 99% ee) (Table 2, entry 12). In terms of these results, the integration of chemical and enzymatic routes holds great potential for synthesizing chiral alcohols and amines.
4.2. Fructosazine and deoxyfructosazine
Pyrazine derivatives, such as deoxyfructosazine (DOF) and fructosazine (FZ) are typically synthesized through the self-condensation of glucosamine (Scheme 9a). These two compounds have garnered significant attention due to their potential applications in the food flavour industry.114 Another advantage of their use in food applications is the recently discovered antimicrobial activity of DOF and FZ against both Gram-positive and Gram-negative bacteria.115 The self-condensation reactions typically occur under basic conditions, with dihydrofructosazine serving as a key intermediate. The reaction pathways leading to DOF and FZ are influenced by various factors, such as basic conditions, hydrogen bonding interactions, boron-transfer or other coordination, promoting the formation of specific intermediates or final products (Scheme 9b). | ||
| Scheme 9 Overview of the self-condensation pathway (a); proposed mechanism for the formation of DOF and FZ from GlcNH2via dihydrofructosazine as the intermediate (b). | ||
Fujii et al. initially reported the formation of FZ from GlcNH2 and D-mannosamine in a methanolic alkaline solution.116 The yield of FZ from D-mannosamine was 39% at 70 °C in 4 h, while that from GlcNH2 was 25% (Table 3, entries 1 and 2). The results indicated that epimerization played a critical role in the reaction mechanism and may account for the yield differences observed between the two substrates.
| Entry | Substrate | Product | Catalyst | Solvent | T (°C) | t (h) | Yield (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | GlcNH2 | FZ | CH3ONa | Methanol | 70 | 4 | 25 | 116 |
| 2 | D-Mannosamine | FZ | CH3ONa | Methanol | 70 | 4 | 39 | 116 |
| 3 | GlcNH2-HCl | DOF | 3-Aminophenyl-boronic acid | Water | 60 | 6 | 86 | 117 |
| 4 | GlcNH2-HCl | DOF | 4-Pyridylboronic acid | Water | 60 | 6 | 89 | 117 |
| 5 | GlcNH2 | FZ + DOF | [Bmim]OH | DMSO | 120 | 3 | 49 | 118 |
| 6 | GlcNH2 | FZ + DOF | [C2C1Im][OAc] | DMSO | 100 | 1.5 | 37 | 119 |
| 7 | GlcNH2 | DOF | [C2C1Im][OAc] + B(OH)3 | DMSO | 90 | 3 | 40 | 120 |
| 8 | GlcNH2 | DOF + FZ | [C2C1Im][OAc] + 0.3 mL H2O2 | DMSO | 90 | 3 | 6, 25 | 120 |
| 9 | GlcNH2 | FZ | [C2C1Im][OAc] + 0.5 mL H2O2 | DMSO | 90 | 3 | ∼16 | 120 |
| 10 | GlcNH2 | DOF | ChCl-urea | — | 100 | 2.5 | 14 | 121 |
| 11 | GlcNH2 | DOF | ChCl-urea + arginine | — | 100 | 1.5 | 30 | 121 |
| 12 | GlcNH2 | DOF + FZ | [Ch][Pro] | DMSO | 100 | 60 | 34, 15 | 122 |
Additionally, arylboronic acids, such as 3-aminophenylboronic acid and 4-pyridylboronic acid, demonstrated high selectivity for DOF formation, achieving yields of 86% and 89%, respectively, at 60 °C in 6 h (Table 3, entries 3 and 4).117 The enhanced condensation rate was attributed to hydrophobic aggregation facilitated by boronic acids and hydrogen bonding interactions induced by the amino or pyridine groups. Furthermore, the pH-dependent boron transfer, either through direct transfer or dissociation, was deemed crucial in the catalytic cycle.
Basic ILs, have demonstrated excellent performance as both solvents and catalysts in the synthesis of FZ and DOF from GlcNH2. For instance, 1-butyl-3-methylimidazolium hydroxide ([Bmim]OH) achieved a total yield of 49% for FZ and DOF with DMSO as the co-solvent at 120 °C in 180 min (Table 3, entry 5).118 Comparable results were obtained using [C2C1Im][OAc] as catalyst, which yielded 37% of FZ and DOF at 100 °C within 90 min (Table 3, entry 6).119 The basic [C2C1Im][OAc] strongly coordinated with the hydroxyl and amino groups of GlcNH2, thereby promoting the formation of DOF and FZ. Furthermore, in the presence of basic ILs [C2C1Im][OAc], the extra addition of B(OH)3 and H2O2 can enhance selectivity for the transformation of GlcNH2 into DOF and FZ, respectively.120 B(OH)3 significantly enhanced the yield of DOF (40.2%) by facilitating the dehydration reaction (Table 3, entry 7). The B(OH)3-GlcNH2 coordination is responsible for the DOF selectivity as evidenced by the NMR results, especially the 1H diffusion-ordered NMR spectroscopy (DOSY). DFT calculations further confirmed the impact of boron coordination, significantly lowering the energy barrier for the dehydration progress. Conversely, proper amounts of H2O2 act as oxidizing agent, increasing the yield of FZ to 25% (Table 3, entry 8). Hou et al. combined in situ NMR, mass spectroscopy, and DFT calculations to understand the reaction pathways and mechanisms involved in the [C2C1Im][OAc] catalysed system.123 They utilized site-selectively labelled 13C-GlcNH2 to monitor the formation of reactive intermediates identifying two main pathways for DOF and FZ synthesis from a common intermediate, dihydrofructosazine (Scheme 8). The DFT results highlighted the dual roles of ILs, where the acidic cation centre reduced the energy barriers of the dehydration reaction, while the basic centre promoted the dehydrogenation process.
Wang et al. explored ChCl-based DES combined with various hydrogen bond donors such as urea, dimethylurea, thiourea, glycerol, and levulinic acid for DOF synthesis.121 The ChCl-urea DES was found to be the most effective, achieving a 14% yield of DOF at 100 °C in 150 min without additional catalysts (Table 3, entry 10). The addition of basic amino acids (e.g., arginine) doubled the DOF yield (30%) at 100 °C in 90 min (Table 3, entry 11). Later, this group synthesized [Ch][Pro] (cholinium as the cation and L-proline as the anion) as a catalyst, yielding 34% and 15% of DOF and FZ, respectively, at 100 °C in 60 min (Table 3, entry 12).122 NMR analysis revealed that the [Pro]− anion had the main interaction with the substrate, as evidenced by the same diffusion of [Pro]− and GlcNH2 in 1H DOSY spectra. In situ1H and 13C NMR were conducted to monitor the reaction progress, suggesting the formation of the intermediate, dihydrofructosazine, which subsequently converted to DOF and FZ.
4.3. Other N-containing heterocyclic compounds
Beyond the N-heterocycles discussed above, other N-heterocyclic compounds and amines have been reported, which are important chemical intermediates and products.124Lin and co-workers realized the formation of various amines from the deoxygenation of NAG catalyzed by Ru/C in the presence of H2 and H3PO4 (Scheme 10).125 The protonated glucosamine underwent further reactions, including decarbonylation, dehydration, and cyclization, leading to the formation of a mixture of various cyclic and linear amines. Besides, the optimal reaction conditions, specifically at 250 °C, 33 bar H2 pressure with a molar ratio of H3PO4 to NAG of 4.45, allowed the production of a 50% yield of various amines. Notably, the addition of H3PO4 played a crucial role in stabilizing the amino group, preventing deamination, and enhancing the selectivity toward desired amine products.
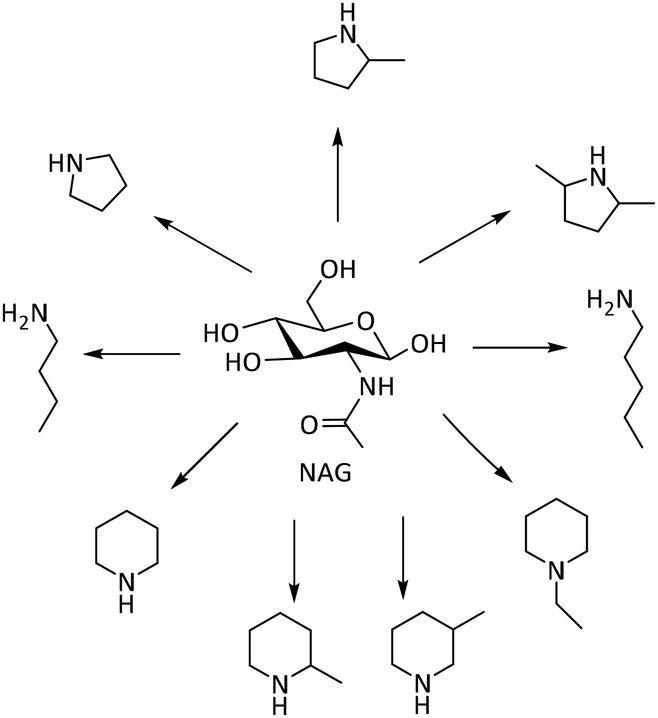 | ||
| Scheme 10 Various amines from deoxygenation reactions of NAG.126 | ||
Liu et al. aimed to develop a novel method for utilizing the N-acetyl groups in chitin directly in transamidation reactions with amines to produce amides, avoiding the need for pre-removal of the N-acetyl groups and generating amide products directly.126 The study used various amine derivatives, including aliphatic amines, cyclic amines, and functionalized aromatic amines, which were selectively converted into their corresponding amides with yields exceeding 75% for most reactions. For instance, aniline was converted to an 87% yield of acetanilid in the presence of chitin using Cu(OAc)2 as a catalyst.
Gao and co-workers successfully realised a one-step synthesis of pyrrole from chitin.127 A metal oxide-based catalytic system (CuO) that operates in alkaline water, allowed the direct transformation of chitin into a 2% yield of pyrrole at 573 K for 5 min. The pyrrole yield was increased to 6% after 1 min at 598 K when ammonia was introduced as an external nitrogen source.
Pyrrole is regarded as a key precursor or intermediate for the synthesis of pyrrole-2-carboxylic acid (PCA), a platform intermediate and building block for the amino acid proline, bioactive marine natural products, and various synthetic bioactive compounds.128 The condensation reaction between GlcNH2 and pyruvic acid (PA) was originally reported in 1957 by Gottschalk.129 Using Ba(OH)2 as the base, the PCA yield was 20%, as determined by UV/Vis spectroscopy. More recently, Yan and Lapkin et al. utilized an automated synthesis planning tool to identify new routes for the production of PCA from biomass feedstocks, which were subsequently tested experimentally.128 The condensation of GlcNH2 with PA was identified as the most promising route. Upon performing the experiments, the highest PCA yield (51%) was gained under highly concentrated base conditions (12 eq. LiOH) at 100 °C for 240 min, where the LiOH solution was dropwise added to preheated water over 150 min. A mechanistic investigation, utilizing temperature-variable NMR studies, revealed that the formation of PCA was driven by aldol condensation between GlcNH2 and PA. Additionally, the synthesis scope was extended to include various 3-substituted PCA derivatives using biomass-derived α-keto acids, as well as a range of ester and amide derivatives. This work offers access to bio-based alcohols, amines, and drug-like compounds, including pyrrolizine, pyrroloimidazole, pyrroloazepine, and benzopyrrolooxazine derivatives, all of which have significant relevance in pharmaceutical applications (Scheme 11).
 | ||
| Scheme 11 Conversion of GlcNH2 into valuable N-containing compounds through PCA. CDI: 1,1′-carbonyldiimidazole. | ||
4.4. N-containing polyols and amino acids
In general, the hydrolysis of chitin typically yields NAG, which can then be hydrogenated to produce ADS, a sugar alcohol. ADS can further undergo dehydration condensation reactions to form ADI (Scheme 12a). A two-step protocol has been developed to convert chitin into ADS with yields reaching 52%, using H2SO4 and Ru/TiO2 as catalysts (Table 4, entry 1).132 The optimization of the reaction conditions revealed that the hydrolysis and hydrogenation steps require significantly different parameters. In the hydrolysis step, higher temperatures (175 °C) and lower pH levels (2) were favoured, whereas in the hydrogenation step, a temperature of 120 °C and pH values between 3 and 4 were optimal. Lower pH values (2) during hydrogenation led to multiple side reactions involving the amide and hemiacetal groups, while higher pH levels (6.8) induced breakdown via a retro-aldol reaction, yielding erythritol (16%) and N-acetylethanolamine (AMEA, 31%). These results highlight the critical role that pH plays in these hydrolysis and hydrogenation processes.
 | ||
| Scheme 12 Reaction pathways for the conversion of chitin to ADI (a); proposed reaction mechanisms for the dehydration of ADS to ADI by Brønsted acids (b) and H3PO3 (c).131,133 | ||
Kobayashi and Fukuoka et al. explored the catalytic dehydration of ADS to ADI using strong acids, particularly trifluoromethanesulfonic acid (HOTf), under vacuum conditions.131 Unlike the simpler dehydration of sorbitol to isosorbide, ADS requires harsher conditions for dehydration (Scheme 12a and b). For instance, sorbitol achieved nearly complete conversion with a 72% yield of isosorbide in the presence of H2SO4 (substrate to catalyst ratio (S/C) = 150 mol mol−1) at 120 °C for 3 h, while ADS showed only 20% conversion, with no detectable ADI under the same conditions. The dehydration of ADS required the use of HOTf as super acid, resulting in a 33% yield of ADI under harsher conditions (150 °C, 3 h, S/C = 2) (Table 4, entry 2). DFT calculations revealed that the acetamide group in ADS traps acid protons, raising the activation energy required for dehydration.
Subsequently, Kobayashi and Fukuoka et al. explored various strategies to overcome the proton-neutralization effect and further enhance the efficiency and selectivity of ADS dehydration process. They first explored the combined effects of Brønsted and Lewis acids to enhance the dehydration process's efficiency and selectivity.134 Brønsted acids alone, such as HOTf, tended to produce unwanted side products, particularly 1,5-anhydro-ADS (17%), leading to a modest 22% yield of ADI. The addition of Lewis acids, particularly ytterbium triflate (Yb(OTf)3), greatly improved ADI yield up to 40% at 150 °C in 1 h (Table 4, entry 3). DFT studies were performed to investigate the reason for this improvement. Specifically, the interactions between Yb(OTf)3 and the amide group of substrate were identified to lower the activation energy and alter the regioselectivity of the reaction, favouring 3,6-dehydration over 1,5-dehydration. Later, they further investigated the use of H3PO3 as a catalyst for ADS dehydration, providing a more efficient and less corrosive alternative to the previously used HOTf.133 For instance, while HOTf yielded 20% ADI at 150 °C for 3 h with S/C = 2.0, H3PO3 achieved a higher yield of 25% under milder conditions (130 °C, 3 h, S/C = 4.0) (Table 4, entry 4). DFT calculations suggested that the formation of phosphite esters as intermediates significantly reduced the activation energy (82 kJ mol−1) compared to traditional acid catalysed dehydration (118 kJ mol−1), leading to a more efficient reaction (Scheme 12b and c). Although yields remain moderate, the development of efficient catalytic systems for ADS dehydration to ADI has made significant progress. Studies underscore the role of acids in overcoming challenges such as proton-trapping effects and steering selectivity.
In addition, Yan and co-workers described the liquefaction of chitin to two polyols in EG at 165 °C with sulfuric acid as a catalyst.135 Up to 75% of chitin was liquefied after 90 min and more than 30% yield of hydroxyethyl-2-amino-2-deoxyhexopyranoside (HADP, around 24%, Scheme 1) and hydroxyethyl-2-acetamido-2-deoxyhexopyranoside (HAADP, around 7%, Scheme 1) was achieved in 1 h. Kinetic and mechanistic studies showed that EG promoted the depolymerisation of chitin to produce EG-derived NAG units (HAADP), followed by the sequential hydrolysis of the acetyl amide group to produce the major product, HADP. Using ball-milled chitin as a substrate resulted in comparable results with 28% for HADP and 4% for HAADP under milder conditions (150 °C, 20 min), while native chitin yielded only 8% in total yield under the same conditions.19
In addition to the enzymatic synthesis of GlcNA from oxidative fermentation of GlcNH2,139,140 the production of GlcNA or its derivatives is currently limited to noble metal-supported catalytic systems. Xiu and Shui reported the oxidation of GlcNH2-HCl to GlcNA in water by molecular oxygen over a Pd–Bi supported on cylindrical active charcoal (Pd–Bi/C) catalyst at 30 °C allowing a highly isolated yield of GlcNA (70%) (Table 5, entry 1).141 However, base, such as NaOH and KHCO3 was required in this reaction system. Au nanoparticles with a 2–6 nm diameter exhibited high catalytic activity for oxidation reactions. The electrocatalytic oxidation of GlcN to GlcNA was catalysed by gold nanoparticles with a 2 nm core size, modified on carbon felt electrodes.142 In NaOH aqueous solutions, at a potential of −0.2 V, GlcNA was formed with a current efficiency of close to 100%.
| Entry | Substrate | Product | Catalyst | T (°C) | t (h) | Yield (%) | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | GlcNH2-HCl | GlcNA | Pd–Bi/C | 30 | 6 | 70 | 141 |
| 2 | GlcNH2 | GlcNA | Au/carbon electrode NaOH solution | — | — | 100 | 142 |
| 3 | GlcNH2 | GlcNA | Au/carbon electrode Phosphate buffer | — | — | 70 | 142 |
| 4 | GlcNH2-HCl | GlcNA | Au/HT | 40 | 3 | 89 | 143 |
| 5 | GlcNH2-HCl | GlcNA | Au/MgO | 20 | 2 | 93 | 143 |
| 6 | GlcNH2-HCl | GalNA | Au/MgO | 20 | 2 | 99 | 143 |
| 7 | NAG | GlcNAcA | Au/HT | 25 | 5 | 95 | 143 |
| 8 | GalNAc | GalNAcA | Au/MgO | 20 | 2 | 99 | 143 |
| 9 | ManNAc | ManNAcA | Au/HT | 25 | 5 | 87 | 143 |
| 10 | Chitosan-H2SO4 | GlcNA | (1) Amberlyst 15 | (1) 160 | (1) 2 | 36 | 144 |
| (2) Au/MgO | (2) 40 | (2) 3 | |||||
| 11 | NAG | AMEA | Ru/C, NaHCO3 | 120 | 1 | 15 | 145 |
| 12 | AMEA | AcGly | Ru/C, NaHCO3 | 120 | 1 | 18 | 145 |
Electrolysis in neutral solutions (phosphate buffer) at 0.4 V resulted in a current efficiency of around 70% (Table 5, entries 2 and 3). Ebitani et al. reported an aerobic base-free oxidation of glucosamine derivatives into their corresponding amino acids GlcNA using Au nanoparticles supported on basic support catalysts.143 The Au/HT and Au/MgO catalysts showed high catalytic activity for the selective oxidation of GlcNH2-HCl to GlcNA achieving yields of up to 89% at 40 °C in 3 h and 93%, respectively, at 20 °C in 2 h (Table 5, entries 4 and 5). However, Au/HT showed reduced activity after reusing it twice due to the growth of the size of the Au nanoparticles. In contrast, the more basic surface of MgO possibly prevents the growth of Au species, leading to its more stable properties. The method was extended to synthesize other α-amino acids, such as galactosaminic acid (GalNA), N-acetyl-glucosaminic acid (GlcNAcA), and N-acetyl-galactosaminic acid (GalNAcA) from their corresponding glucosamine derivatives with the yield of 99%, 95%, 99% and 87%, respectively (Table 5, entries 6–9).
Later, Yan et al. focused on the direct conversion of chitosan into GlcNA through a two-step catalytic process involving hydrolysis and oxidation.144 Amberlyst-15 and Au/MgO were identified as optimal catalysts for hydrolysis and oxidation, respectively. Notably, a detoxification step using activated carbon to remove humin-like byproducts significantly improves the efficiency of the catalytic oxidation step, increasing the overall yield of GalNA from 10% to 36% (Table 5, entry 10).
Glycine is a versatile compound extensively utilized in the production of food products, agricultural chemicals, chelating agents, and cosmetics.146 One of its derivatives, N-acetylglycine (AcGly), has significant potential for these applications. Fukuoka and Kobayashi et al. proposed a two-step synthesis strategy for converting NAG to AcGly, which involved the transformation of NAG into AMEA under hydrogenation conditions followed by the oxidation of AMEA to AcGly (Table 5, entries 11 and 12).145 Under optimal conditions (120 °C, 1 h, p(H2) = 4 MPa), 15% of AMEA was achieved catalysed by Ru/C in aqueous NaHCO3. It is noteworthy that the reactor was not pressurized with H2 until the temperature reached 120 °C, effectively minimizing the formation of ADS. In the subsequent step, AMEA is oxidized to AcGly under oxygen pressure using the same catalytic system, resulting in an 18% yield of AcGly at 120 °C in 1 h. The Ru species were found to be present as small RuO2 clusters with hydroxo, aqua, and oxo ligands, likely serving as the catalyst for the oxidation of AMEA.
5. Conversion of chitin-derived furans to value-added chemical compounds
Organonitrogen aromatics are paramount in the pharmaceutical industry, with over 80% of the best-selling pharmaceutical products containing nitrogen in their chemical formula.147 Chitin-derived compounds, composed of carbon, hydrogen, oxygen, and nitrogen, offer an efficient and renewable alternative for producing organonitrogen chemicals. As discussed earlier, furans represent the most prominent class of compounds that can be obtained from chitin and its derivatives. To emphasize their significance as platform chemicals, we provide an overview of the broader chemical landscape of nitrogen-containing aromatics that can be synthesized from key chitin-derived furans. Notably, special attention is given to recent advances in the conversion of these compounds into benzenoid aromatic structures.5.1. Further chemical space attainable from chitin-derived furans
Chitin-derived platforms, particularly 3A5AF, 3A5F, and Di-HAF have been explored to access bio-based nitrogen-containing building blocks. Notably, Jonathan Sperry and colleagues successfully synthesized the anticancer alkaloid proximicin A (Scheme 13a) from 3A5AF,148 exhibiting cytotoxic effects against several cancer cell lines. Its ability to inhibit cancer cell proliferation indicates its potential to develop new anticancer drugs, especially for cancer types resistant to existing therapies.149 Later, the same group investigated the chemical reactivity of 3A5AF with various aliphatic ketones, leading to the formation of dihydrodifuropyridine, which represents a significant advancement in sustainable chemical space (Scheme 13b).150 Additionally, the group also functionalized 3A5AF into series of 3-amidofurans at different positions, such as C5-ketone, C2 of furan ring, and C3-acetamide (Scheme 13c).5,151 This research establishes 3A5AF as a prominent platform for producing diverse relevant nitrogen-containing compounds. | ||
| Scheme 13 Representative chemical space attainable from chitin-derived furans.5,105,109,113,148,150–157 | ||
Expanding their work, the group explored previously untapped 2-aminosugars, such as N-acetyl-L-rednose (RedNAc), synthesized from 3A5AF via a Noyori reduction and Achmatowicz rearrangement (Scheme 13d).109 RedNAc serves as a versatile platform for further derivatisation, enabling the synthesis of glycosyl donors and various stereochemically pure derivatives. Moreover, the acetamide hydrolysis of RedNAc yields L-rednose, a rare 2-aminosugar found in certain antibiotics, including anthracyclines and angucyclines.152,153 However, the presence of a methyl group at the C5 position in RedNAc limits its functionalization. To address this, the team leveraged the Achmatowicz rearrangement of Di-HAF, a chiral synthon, as an alternative route to produce 2-aminosugars (Scheme 13e).154 This approach eliminates the need for the Noyori reduction step and introduces a versatile hydroxymethyl group at the C5 position, expanding its potential for further functionalization and its utility in chemical synthesis. In addition, they further illustrated the utility of Di-HAF in the asymmetric synthesis of the marine alkaloid epi-leptosphaerin, highlighting its notable structural similarity to ascorbic acid (vitamin C) (Scheme 13f).155
The reduction of 3A5AF to the corresponding alcohol allows its use as building blocks with increased reactivity and decreased steric hindrance.105 Sperry's group introduced a novel variation of the Piancatelli rearrangement, converting 3A5AF-derived furfuryl alcohol (3A5HEF) into a versatile 3-aminocyclopentenone (Scheme 13g).113 This compound serves as a key intermediate for synthesizing other 3 or 4-aminocyclopentenones and 4-aminocyclopentene-1,3-diones. The study further broadens chemical diversity by transforming these intermediates into medicinally relevant scaffolds, highlighting their significant potential for drug discovery applications. The oxidative cleavage of one hydroxyethyl group in Di-HAF produces 3A5F, a highly reactive aldehyde platform suitable for diverse synthetic applications and rapid functionalization.105,158 Jonathan Sperry advanced the utility of 3A5F through an innovative oxidative esterification process catalysed by N-heterocyclic carbene (NHC) under molecular oxygen, producing 4-acetamidofuran-2-carboxylates with a wide range of alcohols (Scheme 13h).156 Subsequent reduction of 3A5F, followed by photooxygenation and further reduction, yielded 3-acetamidobutenolide (Scheme 13h), a scaffold present in natural products like fritenolides.159 Another transformation produced 5-vinylfuran-3-acetamide, a key intermediate with applications in polymerisation, cross-metathesis, and cycloaddition studies (Scheme 13h).160–162 Gomes and colleagues further demonstrated the versatility of 3A5F by showcasing its ability to undergo numerous chemical transformations, including reduction, reductive amination, aldol condensation, olefination, cyclopentenone formation, and thiazolidine synthesis (Scheme 13i).105 These transformations highlight 3A5F's potential for diverse applications in fields such as biofuels, polymer development, and chemical biology. In addition to these synthetic capabilities, the study emphasized 3A5F's utility as a bioconjugation handle, specifically targeting N-terminal cysteine residues in peptides and proteins. The researchers demonstrated its high reactivity and remarkable site-selectivity, achieving stable and precise modifications without affecting internal cysteine residues or other reactive sites. This makes 3A5F one of the few sustainable and bio-derived reagents capable of such specific bioconjugation.
Additionally, Jonathan Sperry also described the conversion of 3A5AF derived furfural and (methyl 4-acetamidofuran-2-carboxylate, M4A2C) respectively into furo[3,2-d]pyrimidin-4-one and furo[3,2-d]pyrimidin-4-amine (Scheme 13j and k).154 While these transformations introduced additional functionality, only one nitrogen atom originated from chitin. Lately, Gan et al. presented the acid-catalysed reaction of 3AF with various aldehydes to synthesize difuropyridines via bis(furan-2,3-diyl)diacetamides as intermediates (Scheme 13l).157 These difuropyridines demonstrate practical utility in fluorescence-based probes and photocatalytic applications, exemplifying the potential of bio-derived nitrogen-rich compounds in sustainable chemical synthesis.
These advancements highlight the transformative potential of chitin-derived furan platforms in sustainable chemical synthesis, positioning them as pivotal resources for developing nitrogen-rich compounds with broad applications. Further expansion of the library of nitrogen-rich scaffolds, coupled with the exploration of their applications in advanced materials, catalysis, and biotechnological processes, can significantly enhance their versatility.
5.2. Benzenoid aromatics
The Diels–Alder reaction is a pivotal tool for the sustainable upgrading of bio-based furan derivatives with high atom efficiency.163 The subsequent dehydration process can transform the resulting polycyclic compounds into aromatic compounds featuring a benzene ring, thereby expanding their utility and applicability. The DA/aromatisation cascade reaction is emerging as an interesting strategy for leveraging chitin-derived furans in the synthesis of nitrogen-containing benzenoid aromatic compounds, allowing direct access to nitrogen-substituted benzene rings. Recently, Gomes and co-workers conducted a study on the DA reaction involving 3A5AF and 3A5HEF as dienes, employing various dienophiles in buffered systems and deuterated dimethyl sulfoxide as the solvent.164 Their investigation aimed to enhance yields, delineate the reaction scope, and elucidate the formation and structure determination of hydration products. Additionally, they reported exo-selectivity for 3A5AF including a subsequent hydration product of this exo-isomer, while no endo-adduct or derived product was found. They also explored the aromatization of 3A5AF adducts under acidic or basic conditions but reported no success.Later, Deuss et al. synthesized five different amido-furans from the chitin monomer subjected to a subsequent DA reaction with a range of N-substituted maleimides under different conditions.165 Detailed insights into the formation profiles of both exo and endo isomers were obtained. This reaction is initially kinetically endo-selective for all the tested maleimides but shifts to exo-selectivity at thermodynamic equilibrium. Besides, carbonyl and alkyl hydroxyl substituents on the furans changed the DA rate significantly and shifted stereoselectivity from the exo- to the endo-product. DFT calculations revealed that the presence of a hydroxyl group in N-[5-(1-hydroxymethyl)-3-furanyl] acetamide (HMFA) leads to a more stable endo-isomer, evidenced by a decreased ΔGendo compared to 3A5AF containing a carbonyl group. Stronger hydrogen bonding interactions and van der Waals interaction in HMFA-involved TS are responsible for its lower ΔG‡ values as evidenced by noncovalent interaction analysis, probably promoting the cycloaddition rate in the HMFA case. This work demonstrated the successful formation of both exo- and endo-adducts from a series of chitin derivatives by adjusting the substituents on the 2-position of furan. The stereoselective control to either the exo or the endo isomer opens new opportunities for synthesizing N-containing derivatives with specific configurations.
Pastre et al. were the first to address the challenges of aromatization from chitin-derived furans.166 A reduced analogue of 3A5AF, 3-acetamido-5-ethylfuran (3A5EF) was used to synthesize several substituted anilides via an acetic anhydride-mediated DA/aromatisation cascade reaction under acidic conditions. For instance, an 80% yield of product 25 was formed at 50 °C in 10 h under acidic conditions using acetic anhydride as an additive, while the absence of acetic anhydride drove the reaction into the ketone derivative (Scheme 14a). The results highlight the important role of acetic anhydride in the aromatisation process by stabilizing the reactive intermediates and benefiting the elimination of the hydroxyl group. In contrast, no aromatisation was achieved for 3A5AF. The ketone as the electron-withdrawing group in 3A5AF hinders the dehydration reaction as evidenced by its higher ΔG‡ (36.8 kcal mol−1) in comparison to that of 3A5EF (26.2 kcal mol−1). Furthermore, they investigated the fluorescence properties of the product phthalimides, laying the groundwork for their potential applications in molecular fluorescence.
 | ||
| Scheme 14 Representative pathways to synthesize benzenoid aromatic compounds from chitin derivatives.158,166–168 | ||
Minnaard and co-workers reported a DA/aromatisation cascade reaction of chitin-derived 3A5F, leading to the formation of di- and tri-substituted anilides through formyl derivatisation with hydrazones.158 The study emphasized the dual role of acetic anhydride, serving as a stabilizer to trap the intermediate, stopping the back-ward reaction, and thereby preventing the retro-DA reactions. Furthermore, converting the carbonyl group into its pyrrolidine hydrazone enabled the utilization of mono-activated dienophiles, such as ethyl acrylate (Scheme 14b), as the nitrogen electron delocalisation in the hydrazone significantly enhances the orbital coefficient at the C-5 position of furan. The product scope was further expanded by converting the hydrazone into an aldehyde or cyano function. Aromatising DA products of 3A5AF remains challenging due to steric hindrance from the methyl group of the ketone. Reverting back to dimethyl hydrazone, aromatization could still be achieved using a reactive dienophile.
Later, Pastre et al. also employed the two-step protocol to realize the 3A5F aromatisation with the assistance of mechanochemistry (Scheme 14c).167 The hydrazone approach successfully promoted the DA/aromatisation reaction of 3A5F under the ball-milling conditions. However, the retro-cycloaddition of 3A5AF adducts prioritise ring-opening in both solution phases or under ball-milling conditions, limiting the realization of its aromatisation. The developed methodology enables chitin-derived furans to access nitrogen-substituted benzenoid aromatic compounds, thereby broadening their application prospects.
While the Diels–Alder reaction provides a robust tool for creating cyclic frameworks, bioprocesses introduce a sustainable route to benzenoid aromatics, offering a complementary strategy for aromatic compound synthesis. Despite the potential of bioprocesses, only limited work has been reported in this domain. Yan and Zhou developed an integrated biorefinery process utilizing mild pretreatment and enzymatic/fermentative techniques to convert shrimp shell waste into tyrosine (Tyr) and L-3,4-dihydroxyphenylalanine (L-DOPA) (Scheme 14d).168 The process involved efficient demineralisation and depolymerisation of shrimp shell waste producing water-soluble chitin hydrolysates containing N-acetyl COSs and protein hydrolysates. Metabolic engineering of Escherichia coli strains facilitated the direct conversion of 22.5 g L−1 unpurified chitin hydrolysates into 0.91 g L−1 Tyr and 0.41 g L−1L-DOPA. A scale-up process utilizing 40 g L−1 of unpurified chitin hydrolysates further validated the robustness of the method, achieving a yield of 1.3 g L−1 Tyr. Notably, Tyr, an essential aromatic amino acid, serves as a precursor for neurotransmitters, hormones, flavonoids, and alkaloids,169–171 while L-DOPA is a key drug for Parkinson's disease treatment and a precursor for various alkaloid natural products.169 Hence, this work highlights the potential of chitin-rich biomass as a sustainable resource for synthesizing nitrogen-containing benzenoid aromatics, with important applications in pharmaceuticals and medical treatments. Moreover, it lays a solid foundation for future advancements in bioprocess optimization and scale-up production strategies.
6. Conclusion and future outlook
In conclusion, the conversion of chitin and its derivatives into high-value nitrogen-containing compounds offers a compelling pathway for sustainable chemical production. This approach leverages an abundant and renewable resource while aligning with the principles of green chemistry by minimizing dependence on fossil-based feedstocks and eliminating reliance on the energy-intensive Haber–Bosch process. By unlocking the potential of chitin-derived platforms, researchers can contribute to the development of advanced materials, pharmaceuticals, and bio-based chemicals, paving the way for innovative applications across multiple industries.Despite these promising advances, significant challenges remain. Due to its crystalline structure and strong intra- and intermolecular hydrogen bonds, the conversion of raw chitin often yields significantly lower amounts of products compared to reactions using its monomer, NAG. To address this issue, it is essential to develop innovative strategies that improve the accessibility and reactivity of chitin. One promising direction is the optimisation of advanced pretreatment methods to break down its rigid structure. Additionally, the development of bifunctional catalysts that can simultaneously depolymerise chitin and promote its further conversion into desired products is an option.
Chitin pyrolysis is a promising approach to generate nitrogen-rich compounds. However, pyrolysis of chitin often yields a complex mixture of products, complicating the identification and quantification of individual compounds. These issues hinder the development of standardised and scalable processes for chitin utilisation. The development of advanced computational tools for predicting reaction pathways and product distribution can complement experimental efforts, enabling a deeper understanding of the pyrolysis process. Besides, the use of selective catalysts can direct the chitin pyrolysis towards specific target products, thereby simplifying the product mixture and enhancing yield selectivity.
The demonstrated production of 3A5F and Di-HAF on gram scales by Minnaard et al. (10 g for 3A5F from Di-HAF and 50 g for Di-HAF from NAG) and Gomes et al. (2 g for 3A5F from NAG), and Kerton et al. (5 g for 3A5AF from NAG) underscore the first steps to scaling up chitin-derived platforms for practical applications. These achievements highlight significant progress in the gram-scale synthesis of nitrogen-rich intermediates, bridging the gap between laboratory-scale research and industrial relevance. Further advancements in process optimization by changing the types and amounts of reagents and solvents used in scalability will be crucial for realizing the full potential of this sustainable approach, focusing on efficient, cost-effective methods like continuous flow systems to boost yield, lower energy use, and minimize waste.
For example, the production of Di-HAF process relies on pyridine, a toxic solvent. The aromatisation or other modification processes often involve harmful reagents and solvents, raising sustainability and safety concerns for large-scale applications. There is a pressing need to develop green and sustainable methodologies that minimize or eliminate the use of harmful substances. One promising approach is to explore enzymatic or fermentative bioprocesses as alternatives to traditional chemical methods. These biological systems can operate under milder conditions and use water as a solvent, significantly reducing the environmental impact and improving the safety profile of the process. However, the implementation of bioprocesses can be costly and time-consuming. Therefore, by integrating bioprocesses and other innovative green chemical process, we can pave the way for the development of more sustainable and environmentally friendly methods for the production of valuable nitrogen-containing chemicals.
Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are grateful for funding supported by China Scholarship Council (Grant Numbers 202006310031 for Ting Wang) and funding supported by National Natural Science Foundation of China (22008166). The Topconsortium voor Kennis en Innovatie Chemie (TKI Chemie, PGT.2018.003 and PGT.2020.014) is also acknowledged for funding.References
- M. Höök and X. Tang, Energy Policy, 2013, 52, 797–809 CrossRef.
- D. Welsby, J. Price, S. Pye and P. Ekins, Nature, 2021, 597, 230–234 CrossRef CAS PubMed.
- A. Ruffoni, F. Juliá, J. J. Douglas, D. Leonori, T. D. Svejstrup and A. J. McMillan, Nat. Chem., 2019, 11, 426–433 CrossRef CAS PubMed.
- X. Chen, S. Song, H. Li, G. Gozaydin and N. Yan, Acc. Chem. Res., 2021, 54, 1711–1722 CrossRef CAS PubMed.
- T. T. Pham, A. C. Lindsay, X. Chen, G. Gözaydin, N. Yan and J. Sperry, Sustainable Chem. Pharm., 2019, 13, 100143 CrossRef.
- C. Jaiswal, Aromatic Market Research Report Information By Type (P-Xylene, O-Xylene, Toluene, Benzene, and Others), By Application (Additive and Solvent), By Industry (Paint & Coatings, Adhesives, Pharmaceuticals, Chemicals, and Others), and By Region (North America, Europe, Asia-Pacific, and Rest Of The World) – Market Forecast Till 2032, 2024 Search PubMed.
- X. Chen, H. Yang and N. Yan, Chemistry, 2016, 22, 13402–13421 CrossRef CAS PubMed.
- S. M. Joseph, S. Krishnamoorthy, R. Paranthaman, J. A. Moses and C. Anandharamakrishnan, Carbohydr. Polym. Technol. Appl., 2021, 2, 10036–10050 Search PubMed.
- I. Hamed, F. Özogul and J. M. Regenstein, Trends Food Sci. Technol., 2016, 48, 40–50 CrossRef CAS.
- M. Feng, X. Lu, J. Zhang, Y. Li, C. Shi, L. Lu and S. Zhang, Green Chem., 2019, 21, 87–98 RSC.
- J. L. Shamshina, Green Chem., 2019, 21, 3974–3993 RSC.
- V. Y. Novikov, S. R. Derkach, I. N. Konovalova, N. V. Dolgopyatova and Y. A. Kuchina, Polymers, 2023, 15, 1729 CrossRef CAS PubMed.
- G. A. F. Roberts, in Chitin Chemistry, MacMillan, 1992, ch. 1–53 Search PubMed.
- M. Pakizeh, A. Moradi and T. Ghassemi, Eur. Polym. J., 2021, 159, 110709 CrossRef CAS.
- L. Zhao, Oligosaccharides of chitin and chitosan, 2019 Search PubMed.
- Y. Liu, Z. Qin, C. Wang and Z. Jiang, Carbohydr. Polym., 2023, 315, 121019 CrossRef CAS PubMed.
- H. Yin, Y. Du and Z. Dong, Front. Plant Sci., 2016, 7, 1–4 Search PubMed.
- R. Chambon, G. Despras, A. Brossay, B. Vauzeilles, D. Urban, J. M. Beau, S. Armand, S. Cottaz and S. Fort, Green Chem., 2015, 17, 3923–3930 RSC.
- X. Chen, Y. Gao, L. Wang, H. Chen and N. Yan, ChemPlusChem, 2015, 80, 1565–1572 CrossRef CAS PubMed.
- Y. Yang, R. Xing, S. Liu, Y. Qin, K. Li, H. Yu and P. Li, Mar. Drugs, 2019, 17, 452 CrossRef PubMed.
- S. Yang, X. Fu, Q. Yan, Y. Guo, Z. Liu and Z. Jiang, Food Chem., 2016, 192, 1041–1048 CrossRef CAS PubMed.
- C. Alsina, M. Faijes and A. Planas, Carbohydr. Res., 2019, 478, 1–9 CrossRef CAS PubMed.
- M. Z. Abidin, M. P. Junqueira-Gonçalves, V. V. Khutoryanskiy and K. Niranjan, J. Chem. Technol. Biotechnol., 2017, 92, 2787–2798 CrossRef CAS.
- C. Schmitz, L. G. Auza, D. Koberidze, S. Rasche, R. Fischer and L. Bortesi, Mar. Drugs, 2019, 17, 1–22 CrossRef PubMed.
- M. Yabushita, H. Kobayashi, K. Kuroki, S. Ito and A. Fukuoka, ChemSusChem, 2015, 8, 3760–3763 CrossRef CAS PubMed.
- H. Kobayashi, Y. Suzuki, T. Sagawa, M. Saito and A. Fukuoka, Angew. Chem., Int. Ed., 2023, 62, e202214229 CrossRef CAS PubMed.
- H. Kobayashi, Y. Suzuki, T. Sagawa, K. Kuroki, J. Y. Hasegawa and A. Fukuoka, Phys. Chem. Chem. Phys., 2021, 23, 15908 RSC.
- S. Amirjalayer, H. Fuchs and D. Marx, Angew. Chem., Int. Ed., 2019, 58, 5232–5235 CrossRef CAS PubMed.
- D. De Chavez, H. Kobayashi, A. Fukuoka and J. Y. Hasegawa, J. Phys. Chem. A, 2021, 125, 187–197 CrossRef CAS PubMed.
- G. Gözaydın, S. Song and N. Yan, Green Chem., 2020, 22, 5096–5104 RSC.
- G. Gözaydın, Q. Sun, M. Oh, S. Lee, M. Choi, Y. Liu and N. Yan, ACS Sustainable Chem. Eng., 2023, 11, 2511–2519 CrossRef.
- Y. Wang, J. Kou, X. Wang and X. Chen, Green Chem., 2023, 25, 2596–2607 RSC.
- M. R. Kasaai, J. Agric. Food Chem., 2009, 57, 1667–1676 CrossRef CAS PubMed.
- B. T. Iber, N. A. Kasan, D. Torsabo and J. W. Omuwa, J. Renewable Mater., 2022, 10, 1097–1123 CAS.
- K. Kurita, T. Sannan and Y. Iwakura, Makromol. Chem., 1977, 178, 3197–3202 CrossRef CAS.
- S. V. Nemtsev, A. I. Gamzazade, S. V. Rogozhin and V. M. Bykova, Appl. Biochem. Microbiol., 2002, 38, 521–526 CrossRef CAS.
- G. Galeda, E. Diazb, F. M. Goycooleab and A. Herasa, Nat. Prod. Commun., 2008, 3, 543–550 Search PubMed.
- A. Tolaimate, J. Desbriéres, M. Rhazi, A. Alagui, M. Vincendon and P. Vottero, Polymer, 2000, 41, 2463–2469 CrossRef CAS.
- G. Lamarque, M. Cretenet, C. Viton and A. Domard, Biomacromolecules, 2005, 6, 1380–1388 CrossRef CAS PubMed.
- I. Tsigos, A. Martinou and D. K. V. Bouriotis, Trends Biotechnol., 2000, 18, 305–312 CrossRef CAS PubMed.
- L. Grifoll-Romero, S. Pascual, H. Aragunde, X. Biarnes and A. Planas, Polymers, 2018, 10, 352 CrossRef PubMed.
- Y. Zhao, R. D. Park and R. A. Muzzarelli, Mar. Drugs, 2010, 8, 24–46 CrossRef CAS PubMed.
- C. Carrera, C. Bengoechea, F. Carrillo and N. Calero, Food Hydrocolloids, 2023, 137, 108383 CrossRef CAS.
- K. Gupta and F. Jabrail, Carbohydr. Polym., 2006, 66, 43–54 CrossRef CAS.
- J. Weisspflog, D. Vehlow, M. Muller, B. Kohn, U. Scheler, S. Boye and S. Schwarz, Int. J. Biol. Macromol., 2021, 171, 242–261 CrossRef CAS PubMed.
- R. Czechowska-Biskup, D. Jarosińska, B. Rokita, P. Ulański and J. M. Rosiak, Determination of degree of deacetylation of chitosan - comparison of methods, 2012 Search PubMed.
- L. Heux, J. Brugnerotto, J. Desbriéres, M.-F. Versali and M. Rinaudo, Biomacromolecules, 2000, 1, 746–751 CrossRef CAS PubMed.
- S. Y. Foong, R. K. Liew, P. N. Y. Yek, Y. H. Chan and S. S. Lam, Curr. Opin. Green Sustainable Chem., 2024, 47, 100920 CrossRef CAS.
- N. Bossons and R. F. A. Gomes, Curr. Opin. Green Sustainable Chem., 2024, 100961, DOI:10.1016/j.cogsc.2024.100961.
- J. Liu, S. Liu, A. Zhao, D. Bi, D. Yao and R. Kong, J. Anal. Appl. Pyrolysis, 2023, 169, 105863 CrossRef CAS.
- R. A. Franich, S. J. Goodin and A.L. Wilkins, J. Anal. Appl. Pyrolysis, 1984, 7, 91–100 CrossRef CAS.
- D. Fabbri, S. Prati, I. Vassura and G. Chiavari, J. Anal. Appl. Pyrolysis, 2003, 68–69, 163–171 CrossRef CAS.
- B. R. T. Simoneit, D. R. Oros and V. O. Elias, Chemosphere, 2000, 2, 101–105 CAS.
- P. Köll, G. Borchers and J. O. Metzger, J. Anal. Appl. Pyrolysis, 1991, 19, 119–129 CrossRef.
- D. Knorr, T. P. Wampler and R. A. Teutonico, J. Food Sci., 2006, 50, 1762–1763 CrossRef.
- M. Nikahd, J. Mikusek, L. J. Yu, M. L. Coote, M. G. Banwell, C. Ma and M. G. Gardiner, J. Org. Chem., 2020, 85, 4583–4593 CrossRef CAS PubMed.
- G. Kwon, D. C. W. Tsang, J.-I. Oh, E. E. Kwon and H. Song, Energy, 2019, 177, 136–143 CrossRef CAS.
- P. Wang and Y. Shen, Fuel, 2022, 312, 122875 CrossRef CAS.
- L. Chen and Y. Shen, J. Environ. Manage., 2022, 315, 115133 CrossRef CAS PubMed.
- G. Cabrera-Barjas, R. Jimenez, R. Romero, O. Valdes, A. Nesic, R. Hernandez-Garcia, A. Neira, S. Alejandro-Martin and A. F. de la Torre, Int. J. Biol. Macromol., 2023, 238, 124130 CrossRef CAS PubMed.
- R. Umamaheswaran, S. Dutta, H. Singh and S. Kumar, J. Anal. Appl. Pyrolysis, 2022, 161, 105362 CrossRef CAS.
- H. Huang, G. Zhou, S. Luo and S. Xie, J. Ind. Eng. Chem., 2023, 128, 127–142 CrossRef CAS.
- D. Zhou, D. Shen, W. Lu, T. Song, M. Wang, H. Feng, J. Shentu and Y. Long, Molecules, 2020, 25, 541–556 CrossRef CAS PubMed.
- M. Osada, K. Kikuta, K. Yoshida, K. Totani, M. Ogata and T. Usui, Green Chem., 2013, 15, 2960–2966 RSC.
- W. T. J. Morgan and L. A. Elson, Biochem. J., 1934, 28, 988–995 CrossRef CAS PubMed.
- J. M. Beau, P. Rollin and P. Sinaÿ, Carbohydr. Res., 1977, 53, 187–195 CrossRef CAS PubMed.
- V. A. Derevitskaya, L. M. Likhosherstov, V. A. Schennikov and N. K. Kochetkov, Carbohydr. Res., 1971, 20, 285–291 CrossRef CAS PubMed.
- M. Ogata, T. Hattori, R. Takeuchi and T. Usui, Carbohydr. Res., 2010, 345, 230–234 CrossRef CAS PubMed.
- X. Y. Zheng, J. B. Peng, M. M. Livera, Y. Luo, Y. Y. Wang, X. J. Kong, L. S. Long, Z. Zheng and L. S. Zheng, Inorg. Chem., 2017, 56, 110–113 CrossRef CAS PubMed.
- M. Osada, K. Kikuta, K. Yoshida, K. Totani, M. Ogata and T. Usui, RSC Adv., 2014, 4, 33651 RSC.
- M. Osada, S. Shoji, S. Suenaga and M. Ogata, Fuel Process. Technol., 2019, 195, 106154 CrossRef CAS.
- M. Osada, H. Kobayashi, T. Miyazawa, S. Suenaga and M. Ogata, Int. J. Biol. Macromol., 2019, 136, 994–999 CrossRef CAS PubMed.
- L. Zhang, T. Gao, Q. Song, Y. Qiu, C. Lin and H. Shi, Synth. Chem., 2012, 20, 366–368 Search PubMed.
- Y. Matsumura, S. Yanachi and T. Yoshida, Ind. Eng. Chem. Res., 2006, 45, 1875–1879 CrossRef CAS.
- J. Zhao, C. M. Pedersen, H. Chang, X. Hou, Y. Wang and Y. Qiao, iScience, 2023, 26, 106980 CrossRef CAS PubMed.
- J. Zhao, Z. Guo, C. M. Pedersen, L. Jia, S. Jia, X. Hou, Y. Wang and Y. Qiao, J. Mol. Liq., 2024, 413, 126006 CrossRef CAS.
- C. H. M. van der Loo, M. L. G. Borst, K. Pouwer and A. J. Minnaard, Org. Biomol. Chem., 2021, 19, 10105 RSC.
- R. Kuhn and G. Kruger, Chem. Ber., 1957, 90, 264–277 CrossRef CAS.
- K. W. Omari, L. Dodot and F. M. Kerton, ChemSusChem, 2012, 5, 1767–1772 CrossRef CAS PubMed.
- X. Ji, Y. Lu and X. Chen, Chem. Commun., 2025, 6, 1303 RSC.
- J. Chen, M. Wang and C.-T. Ho, J. Agric. Food Chem., 1998, 46, 3207–3209 CrossRef CAS.
- H. Zang, Y. Feng, M. Zhang, K. Wang, Y. Du, Y. Lv, Z. Qin and Y. Xiao, Carbohydr. Res., 2022, 522, 108679 CrossRef CAS PubMed.
- R. v. d. Berg, J. A. Peters and H. v. Bekkum, Carbohydr. Polym., 1994, 253, 1–12 CrossRef PubMed.
- T. Stahlberg, S. Rodriguez-Rodriguez, P. Fristrup and A. Riisager, Chemistry, 2011, 17, 1456–1464 CrossRef CAS PubMed.
- X. Chen, S. L. Chew, F. M. Kerton and N. Yan, Green Chem., 2014, 16, 2204–2212 RSC.
- J. Wang, H. Zang, S. Jiao, K. Wang, Z. Shang, H. Li and J. Lou, Sci. Total Environ., 2020, 710, 136293 CrossRef CAS PubMed.
- H. Zang, Z. Liu, C. Wu, Y. Chang, X. Zhu, X. Zhu and M. Yan, Sustainable Chem. Pharm., 2024, 37, 101388 CrossRef CAS.
- M. W. Drover, K. W. Omari, J. N. Murphy and F. M. Kerton, RSC Adv., 2012, 2, 4642 RSC.
- H. Zang, H. Li, S. Jiao, J. Lou, Y. Du and N. Huang, ChemistrySelect, 2021, 6, 3848–3857 CrossRef CAS.
- H. Zang, J. Lou, S. Jiao, H. Li, Y. Du and J. Wang, J. Mol. Liq., 2021, 330, 115667 CrossRef CAS.
- Y. Du, H. Zang, Y. Feng, K. Wang, Y. Lv and Z. Liu, J. Mol. Liq., 2022, 347, 117970 CrossRef CAS.
- C. Wu, C. Wang, A. Zhang, K. Chen, F. Cao and P. Ouyang, React. Chem. Eng., 2022, 7, 1742 RSC.
- K. Wang, Y. Xiao, C. Wu, Y. Feng, Z. Liu, X. Zhu and H. Zang, Carbohydr. Res., 2023, 524, 108742 CrossRef CAS PubMed.
- D. Padovan, H. Kobayashi and A. Fukuoka, ChemSusChem, 2020, 13, 3594–3598 CrossRef CAS PubMed.
- C. Wu, X. Zhu, H. Zang, Z. Liu, X. Zhu and Y. Chang, Mol. Catal., 2024, 566, 114404 CrossRef CAS.
- C. Wang, C. Wu, A. Zhang, K. Chen, F. Cao and P. Ouyang, ChemistrySelect, 2022, 7, e202104574 CrossRef CAS.
- X. Ji, J. Kou, G. Gözaydın and X. Chen, Appl. Catal., B, 2024, 342, 123379 CrossRef CAS.
- K. Yamazaki, N. Hiyoshi and A. Yamaguchi, ChemistryOpen, 2023, 12, e202300148 CrossRef CAS PubMed.
- X. Chen, Y. Liu, F. M. Kerton and N. Yan, RSC Adv., 2015, 5, 20073 RSC.
- H. Zang, Y. Feng, J. Lou, K. Wang, C. Wu, Z. Liu and X. Zhu, J. Mol. Liq., 2022, 366, 120281–120289 CrossRef CAS.
- L. Korampattu, N. Ghosh and P. L. Dhepe, Green Chem., 2024, 26, 5601–5634 RSC.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060 CrossRef CAS PubMed.
- Y. Chen and T. Mu, Green Energy Environ., 2019, 4, 95–115 CrossRef.
- A. J. Kumalaputri, C. Randolph, E. Otten, H. J. Heeres and P. J. Deuss, ACS Sustainable Chem. Eng., 2018, 6, 3419–3425 CrossRef CAS PubMed.
- R. F. A. Gomes, B. M. F. Goncalves, K. H. S. Andrade, B. B. Sousa, N. Maulide, G. J. L. Bernardes and C. A. M. Afonso, Angew. Chem., Int. Ed., 2023, 62, e202304449 CrossRef CAS PubMed.
- C. Lin, H. Yang, X. Gao, Y. Zhuang, C. Feng, H. Wu, H. Gan, F. Cao, P. Wei and P. Ouyang, ChemSusChem, 2023, 16, e202300133 CrossRef CAS PubMed.
- S. S. Shaikh, C. R. Patil, N. Lucas, V. V. Bokade and C. V. Rode, Waste Biomass Valorization, 2023, 14, 4201–4214 CrossRef CAS.
- Y. Liu, C. Stähler, J. N. Murphy, B. J. Furlong and F. M. Kerton, ACS Sustainable Chem. Eng., 2017, 5, 4916–4922 CrossRef CAS.
- T. T. Pham, G. Gözaydın, T. Söhnel, N. Yan and J. Sperry, Eur. J. Org. Chem., 2018, 1355–1360 Search PubMed.
- Y. C. Hao, M. H. Zong, Z. L. Wang and N. Li, Bioresour. Bioprocess, 2021, 8, 80–89 CrossRef PubMed.
- Y.-C. Hao, M.-H. Zong, Q. Chen and N. Li, Green Chem., 2023, 25, 5051–5058 RSC.
- C. Wu, X. Zhang, W. Liu, C. Wang, Q. Jiang, F. Chen, Q. Liu, F. Cao, G. Zheng, A. Zhang and K. Chen, ACS Sustainable Chem. Eng., 2024, 12, 11145–11154 CrossRef CAS.
- T. T. Pham, X. Chen, T. Söhnel, N. Yan and J. Sperry, Green Chem., 2020, 22, 1978–1984 RSC.
- M. L. Scotter, Colour Additives for Foods and Beverages, Amsterdam: Elsevier, 2015, pp. 61–74 Search PubMed.
- M. A. Croxen, R. J. Law, R. Scholz, K. M. Keeney, M. Wlodarska and B. B. Finlay, Clin. Microbiol. Rev., 2013, 26, 822–880 CrossRef CAS PubMed.
- S. Fujii, R. Kikuchi and H. Kushida, J. Org. Chem., 1966, 31, 2239–2241 CrossRef CAS.
- M. Wang, G. Zhu, Y. Li and L. Gu, Curr. Res. Green Sustainable Chem., 2022, 5, 100308 CrossRef CAS.
- L. Jia, Y. Wang, Y. Qiao, Y. Qi and X. Hou, RSC Adv., 2014, 4, 44253 RSC.
- L. Jia, C. M. Pedersen, Y. Qiao, T. Deng, P. Zuo, W. Ge, Z. Qin, X. Hou and Y. Wang, Phys. Chem. Chem. Phys., 2015, 17, 23173 RSC.
- L. Jia, Z. Zhang, Y. Qiao, C. M. Pedersen, H. Ge, Z. Wei, T. Deng, J. Ren, X. Liu, Y. Wang and X. Hou, Ind. Eng. Chem. Res., 2017, 56, 2925–2934 CrossRef CAS.
- M. Wu, H. Ma, Z. Ma, Y. Jin, C. Chen, X. Guo, Y. Qiao, C. M. Pedersen, X. Hou and Y. Wang, ACS Sustainable Chem. Eng., 2018, 6, 9434–9441 CrossRef CAS.
- P. Liu, J. Zhang, Y. Qiao, X. Hou, Y. Liu and Y. Wang, J. Agric. Food Chem., 2021, 69, 2403–2411 CrossRef CAS PubMed.
- L. Jia, X. Liu, Y. Qiao, C. M. Pedersen, Z. Zhang, H. Ge, Z. Wei, Y. Chen, X. Wen, X. Hou and Y. Wang, Appl. Catal., B, 2017, 202, 420–429 CrossRef CAS.
- V. Froidevaux, C. Negrell, S. Caillol, J. P. Pascault and B. Boutevin, Chem. Rev., 2016, 116, 14181 CrossRef CAS PubMed.
- S. Xie, C. Jia, S. S. G. Ong, Z. Wang, M. J. Zhu, Q. Wang, Y. Yang and H. Lin, iScience, 2020, 23, 101096 CrossRef CAS PubMed.
- Y. Xin, X. Shen, H. Liu and B. Han, Front. Chem. Eng., 2021, 2, 634983–634991 CrossRef.
- X. Gao, X. Chen, J. Zhang, W. Guo, F. Jin and N. Yan, ACS Sustainable Chem. Eng., 2016, 4, 3912–3920 CrossRef CAS.
- T. T. Pham, Z. Guo, B. Li, A. A. Lapkin and N. Yan, ChemSusChem, 2024, 17, e202300538 CrossRef CAS PubMed.
- A. Gottshalk, Arch. Biochem. Biophys., 1957, 59, 37–44 CrossRef PubMed.
- H. Kobayashi, H. Yokoyama, B. Feng and A. Fukuoka, Green Chem., 2015, 17, 2732–2735 RSC.
- T. Sagawa, H. Kobayashi, C. Murata, Y. Shichibu, K. Konishi and A. Fukuoka, ACS Sustainable Chem. Eng., 2019, 7, 14883–14888 CrossRef CAS.
- H. Kobayashi, K. Techikawara and A. Fukuoka, Green Chem., 2017, 19, 3350–3356 RSC.
- C. Yang, T. Sagawa, A. Fukuoka and H. Kobayashi, Green Chem., 2021, 23, 7228–7234 RSC.
- T. Sagawa, H. Kobayashi and A. Fukuoka, Mol. Catal., 2020, 498, 111282 CrossRef CAS.
- Y. Pierson, X. Chen, F. D. Bobbink, J. Zhang and N. Yan, ACS Sustainable Chem. Eng., 2014, 2, 2081–2089 CrossRef CAS.
- O. Varela, A. P. Nin and R. M. d. Lederkremer, Tetrahedron Lett., 1994, 35, 9359–9362 CrossRef CAS.
- M. Gerspacher and H. Rapoport, J. Org. Chem., 1991, 56, 3700–3706 CrossRef CAS.
- Y. Kanekiyo, S. Aizawa, N. Koshino and S. Funahashi, Inorg. Chim. Acta, 2000, 298, 154–164 CrossRef CAS.
- F. Pezzotti, H. Therisod and M. Therisod, Carbohydr. Res., 2005, 340, 139–141 CrossRef CAS PubMed.
- B. Wu, Z. Bai, X. Meng and B. He, Biotechnol. Prog., 2011, 27, 32–37 CrossRef CAS PubMed.
- G. Wen-xiu and X. Wen-shui, J. Carbohydr. Chem., 2006, 25, 297–301 CrossRef.
- M. Tominaga, M. Nagashima and I. Taniguchi, Electrochem. Commun., 2007, 9, 911–914 CrossRef CAS.
- Y. Ohmi, S. Nishimura and K. Ebitani, ChemSusChem, 2013, 6, 2259–2262 CrossRef CAS PubMed.
- J. Dai, G. Gözaydın, C. Hu and N. Yan, ACS Sustainable Chem. Eng., 2019, 7, 12399–12407 CAS.
- K. Techikawara, H. Kobayashi and A. Fukuoka, ACS Sustainable Chem. Eng., 2018, 6, 12411–12418 CrossRef CAS.
- Global Glycine Market Report 2018.
- A. Ricci, Amino Group Chemistry: From Synthesis to the Life Sciences, Wiley, 2008 Search PubMed.
- A. D. Sadiq, X. Chen, N. Yan and J. Sperry, ChemSusChem, 2018, 11, 532–535 CrossRef CAS PubMed.
- K. Schneider, S. Keller, F. E. Wolter, L. Roglin, W. Beil, O. Seitz, G. Nicholson, C. Bruntner, J. Riedlinger, H. P. Fiedler and R. D. Sussmuth, Angew. Chem., Int. Ed., 2008, 47, 3258–3261 CrossRef CAS PubMed.
- T. T. Pham, X. Chen, N. Yan and J. Sperry, Monatsh. Chem. – Chem. Mon., 2017, 149, 857–861 CrossRef.
- T. T. Pham, A. C. Lindsay, S. W. Kim, L. Persello, X. Chen, N. Yan and J. Sperry, ChemistrySelect, 2019, 4, 10097–10099 CrossRef CAS.
- O. Johdo, T. Yoshioka, H. Naganawa, T. Takeuchi and A. Yoshimoto, J. Antibiot., 1996, 49, 669–675 CrossRef CAS PubMed.
- H. S. Kim, Y. H. Kim, O. J. Yoo and J. J. Lee, Biosci. Biotechnol. Biochem., 1996, 60, 906–908 CrossRef CAS PubMed.
- T. A. Rossa, J. C. Neville, S. P. Jun, T. Söhnel and J. Sperry, Chemistry, 2023, 5, 1998–2008 CrossRef CAS.
- J. C. Neville, M. Y. Lau, T. Sohnel and J. Sperry, Org. Biomol. Chem., 2022, 20, 6562 RSC.
- J. C. Neville, C. N. Y. Kieu and J. Sperry, Sustainable Chem. Pharm., 2024, 42, 101838 CrossRef CAS.
- H. Gan, P. Ma, T. Yang, F. Cao and J. Zhu, Adv. Synth. Catal., 2024, 366, 1–8 CrossRef.
- C. H. M. van der Loo, J. P. Kaniraj, T. Wang, J. O. P. Broekman, M. L. G. Borst, K. Pouwer, A. Heeres, P. J. Deuss and A. J. Minnaard, Org. Biomol. Chem., 2023, 21, 8372 RSC.
- J. Liu, C. Peng, C.-J. He, J.-L. Liu, Y.-C. He, L. Guo, Q.-M. Zhou, H. Yang and L. Xiong, Fitoterapia, 2014, 98, 53–58 CrossRef CAS PubMed.
- M. G. B. Drew, A. Jahans, L. M. Harwood and S. A. B. H. Apoux, Eur. J. Org. Chem., 2002, 3589–3594 CrossRef CAS.
- T. Kawai, M. Komaki and T. Iyoda, Presented in part at the 15th Int. Symp. Olefin Metathesis Relat. Chem., 2002.
- N. Yoshida, N. Kasuya, N. Haga and K. Fukuda, Polym. J., 2008, 40, 1164–1169 CrossRef CAS.
- V. Froidevaux, M. Borne, E. Laborbe, R. Auvergne, A. Gandini and B. Boutevin, RSC Adv., 2015, 5, 37742 RSC.
- J. G. Pereira, J. M. J. M. Ravasco, J. R. Vale, F. Queda and R. F. A. Gomes, Green Chem., 2022, 24, 7131–7136 RSC.
- T. Wang, C. H. M. van der Loo, Z. Zhang, J. O. P. Broekman, Q. Yuan, J. G. M. Winkelman, A. Heeres, R. W. A. Havenith, A. J. Minnaard and P. J. Deuss, ACS Sustainable Chem. Eng., 2024, 12, 11195–11205 CrossRef CAS.
- C. S. Santos, R. R. Mattioli, J. S. Baptista, V. H. Menezes da Silva, D. L. Browne and J. C. Pastre, Green Chem., 2023, 25, 5059–5067 RSC.
- R. Mattioli, C. Santos, B. De Souza, P. Branco, R. Bolt, S. Raby-Buck, T. Cabral, C. Tormena, D. Browne and J. Pastre, ChemRxiv, 2023. DOI:10.26434/chemrxiv-2023-h2cb8.
- X. Ma, G. Gozaydin, H. Yang, W. Ning, X. Han, N. Y. Poon, H. Liang, N. Yan and K. Zhou, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 7719–7728 CrossRef CAS PubMed.
- A. Nakagawa, H. Minami, J.-S. Kim, T. Koyanagi, T. Katayama, F. Sato and H. Kumagai, Nat. Commun., 2011, 2, 326 CrossRef PubMed.
- D. Juminaga, E. E. K. Baidoo, A. M. Redding-Johanson, T. S. Batth, H. Burd, A. Mukhopadhyay, C. J. Petzold and J. D. Keasling, Appl. Environ. Microbiol., 2012, 78, 89–98 CrossRef CAS PubMed.
- T. Lütke-Eversloh and G. Stephanopoulos, Appl. Microbiol. Biotechnol., 2007, 75, 103–110 CrossRef CAS PubMed; Derevitskaya, L. M. Limosherstov, V. A. Schennikov and N. K. Kochetkov, Carbohydr. Res., 1971, 20, 285–291 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2025 |