
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photo-induced enhancement of hydrogenation activity for ruthenium nanoparticles immobilized on carbon dots†
Carlotta Campalani a,
Manisha Duraiab,
Walter Leitnerab and
Alexis Bordet*a
a,
Manisha Duraiab,
Walter Leitnerab and
Alexis Bordet*a
aMax Planck Institute for Chemical Energy Conversion, Stiftstraße 34–36, 45470, Mülheim an der Ruhr, Germany. E-mail: alexis.bordet@cec.mpg.de
bInstitut für Technische und Makromolekulare Chemie, RWTH Aachen University, Worringerweg 2, 52074 Aachen, Germany
First published on 10th January 2025
Abstract
Ruthenium nanoparticles immobilized on environmentally benign luminescent carbon dots act as hydrogenation catalysts, whereby light irradiation results in greatly enhanced activity. Nitrogen-doped carbon dots were prepared by hydrothermal treatment, and served as supports for the organometallic synthesis of 2.1 nm Ru NPs. UV-irradiated Ru@CDs proved 1.5–13 times more active for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hydrogenation of α–β unsaturated ketones under mild conditions (3.5 bar H2, 60 °C, in water/butan-1-ol) than when used in the dark. Reference experiments and mechanistic investigations rationalize this activity enhancement by a transfer of electron density from the irradiated CDs to the Ru NPs. The strong synergistic effects arising from the combination of CDs and Ru NPs under UV are potentially more broadly accessible with other CDs structures and metal NPs compositions.
C hydrogenation of α–β unsaturated ketones under mild conditions (3.5 bar H2, 60 °C, in water/butan-1-ol) than when used in the dark. Reference experiments and mechanistic investigations rationalize this activity enhancement by a transfer of electron density from the irradiated CDs to the Ru NPs. The strong synergistic effects arising from the combination of CDs and Ru NPs under UV are potentially more broadly accessible with other CDs structures and metal NPs compositions.
Green foundation1. We investigate the synthesis, characterization, and application in catalysis of photo-responsive multifunctional catalytic systems composed of metal nanoparticles immobilized on carbon dots.2. We demonstrate a photo-induced enhancement of hydrogenation activity (up to ×13) for ruthenium nanoparticles immobilized on biodegradable, non-toxic, and luminescent carbon dots. Mechanistic investigations involving references experiments and CO-IR measurements revealed a transfer of electron density from CDs to supported Ru nanoparticles resulting in a boost of the metal nanoparticles’ hydrogenation activity 3. The recyclability of NPs@CDs catalysts should be improved, and the demonstrated capability of CDs to act as electron reservoirs for Ru nanoparticles should be explored for other CDs and nanoparticles compositions, in particular non-noble metals. |
Catalytic hydrogenation with molecular hydrogen (H2) is a crucial atom economic transformation for the chemical industry that is applied in the production of most chemicals such as fuels, fine chemicals, agrochemicals, and pharmaceuticals.1 The development of sustainable catalysts with enhanced and tailor-made reactivity is thus a long-lasting goal in the field.2 In heterogeneous catalysis, the modulation of catalytic activity can be achieved through the alteration of the structural and electronic properties of catalytic surfaces.3 For example, traditional strategies involve the control of particle size, structure and dispersion,4 the preparation of doped/multimetallic particles,5,6 the use of promoters,7 and metal support interactions.8,9 Photo-irradiation is also attractive in this context, as it offers sustainable means for activating or boosting performances of appropriately designed catalytic systems under mild conditions.10 In particular, photocatalysis is considered as an inherently sustainable approach, as it leverages light as a renewable energy source to drive chemical transformations, thereby reducing reliance on energy-intensive chemicals and processes.11 Promising advances have been made in the use of irradiation in photocatalysis for the valorization of biomass and plastics to renewable chemicals in so-called “photorefineries”.12 TiO2-based catalysts are typical and extensively studied examples of photocatalysts, for which ultraviolet (UV) irradiations generate surface reactive electron–hole pairs that facilitate redox reactions.13
In recent years, carbon-based nanomaterials emerged as potentially more versatile and environmentally benign alternatives to TiO2 and classical semi-conductors for photocatalytic applications.14,15 In particular, carbon dots (CDs) possessing excellent photo-responsive properties are attracting increasing interest among the scientific community for applications in catalysis, and also in optoelectronic, bioimaging, drug delivery, and energy storage.16–21 CDs are biodegradable, non-toxic and luminescent carbon nanoparticles (NPs) with a spherical shape comprising a carbogenic core and a surface rich in organic functionalities such as hydroxyls, carbonyls and carboxylic groups.22–24 They can be prepared from various abundant and renewable carbon feedstock such as fish scales, waste paper, algae, etc.25 Their ability to absorb/emit light at wavelengths ranging from UV to visible (Vis) and infrared (IR) allows their use as both electron donor and acceptor photocatalysts.26 Their photoredox properties can be easily tailored through doping with various heteroatoms (e.g. nitrogen, sulphur, boron).27,28 The combination of CDs with metal NPs (NPs@CDs) can generate complementary interaction resulting in materials with enhanced thermo/photo-catalytic properties.29–32 For example, CDs have been used as supports for rhodium NPs, improving their stability in thermocatalytic alkene hydrogenation.33 CDs can also act as e− reservoirs,34 and the potential of photo-induced e− density transfer from CDs to metal NPs has been hypothesized as a way to boost the catalytic performance of metal NPs.29,32 Experimental demonstrations and fundamental understanding of such effects are stills lacking, however.
Herein, we present a novel photo-responsive hydrogenation catalyst composed of ruthenium (Ru) NPs supported on nitrogen-doped CDs (Ru@CDs). Ru@CDs are prepared, characterized, and applied to the hydrogenation of various α–β unsaturated ketones (Fig. 1). The focus is placed on the impact of light irradiation on catalytic performance, and on the understanding of underlying mechanisms.
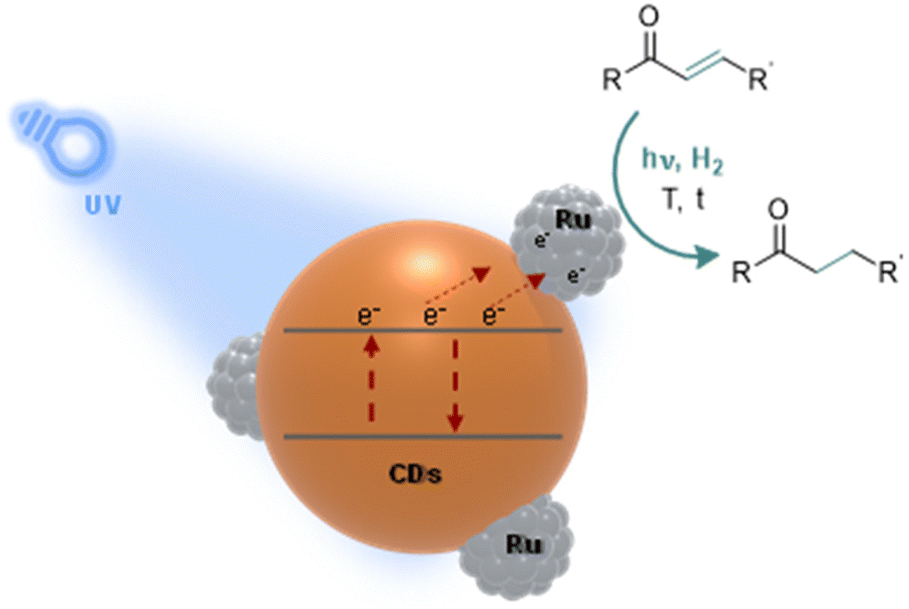 | ||
| Fig. 1 Illustration of our approach: study of photo-responsive Ru@CDs applied to the model hydrogenation of α–β unsaturated ketones. | ||
Nitrogen-doped CDs were, indeed, prepared following a previously reported simple bottom-up hydrothermal treatment, using citric acid and diethylenetriamine as widely available precursors and microwave as a green heating method (see ESI,† for experimental procedures).35–37
Scanning Transmission Electron Microscopy (STEM) showed fairly spherical CDs with a diameter of 86.7 ± 21.5 nm (Fig. S1†), consistent with literature data.38–40 CDs’ surface properties were probed by Fourier Transform infrared spectroscopy (FT-IR) in ATR mode, revealing the presence of various organic functionalities including hydroxyls, carbonyls, carboxyls, but also aminic and amidic groups, consistent with the desired nitrogen doping (Fig. S2†). Successful N-doping of the CDs was confirmed by elemental analysis that gave the following weight composition: 48% C, 27% O, 18% N, and 7% H. CDs’ optical characteristics were studied by Ultraviolet–Visible (UV-Vis) and Photoluminescence–Photoluminescence Emission (PL–PLE) spectroscopies, evidencing two main absorption peaks (Fig. S3†). The first at 250 nm was attributed to the π–π* transition of the aromatic carbogenic core, while the second at 350 nm was assigned to the n–π* transition of the functional groups on the surface. PLE spectra showed a maximum emission in the blue region of the visible spectrum at 440 nm (Fig. S4†). The absence of blue or red shifts as a function of the excitation wavelength indicates that the emission originates from well-defined electronic states (i.e. functional groups or fluorophores) and not from surface defects.
The obtained N-doped CDs were used as photo-responsive supports for the preparation of Ru NPs by adapting an organometallic approach using [Ru(2-methylallyl)2(cyclooctadiene)] as precursor (see ESI† for experimental details).41,42 The resulting Ru@CDs material contained 5 wt% of Ru as determined by Inductively-Coupled Plasma Mass Spectrometry (ICP-MS) analyses, and retained the chemical and optical properties of the CDs (Fig. S5† for FT-IR spectrum and Fig. S6† for UV-Vis spectrum). STEM analysis showed the presence of spherical Ru NPs with an average diameter of 2.1 ± 0.5 nm and narrow size distribution on the CDs (Fig. 2a and b). The blue emission at 440 nm observed for the pristine CDs was conserved also in Ru@CDs (Fig. 2c), confirming the retention of CDs’ luminescent properties upon deposition of Ru NPs at their surface.
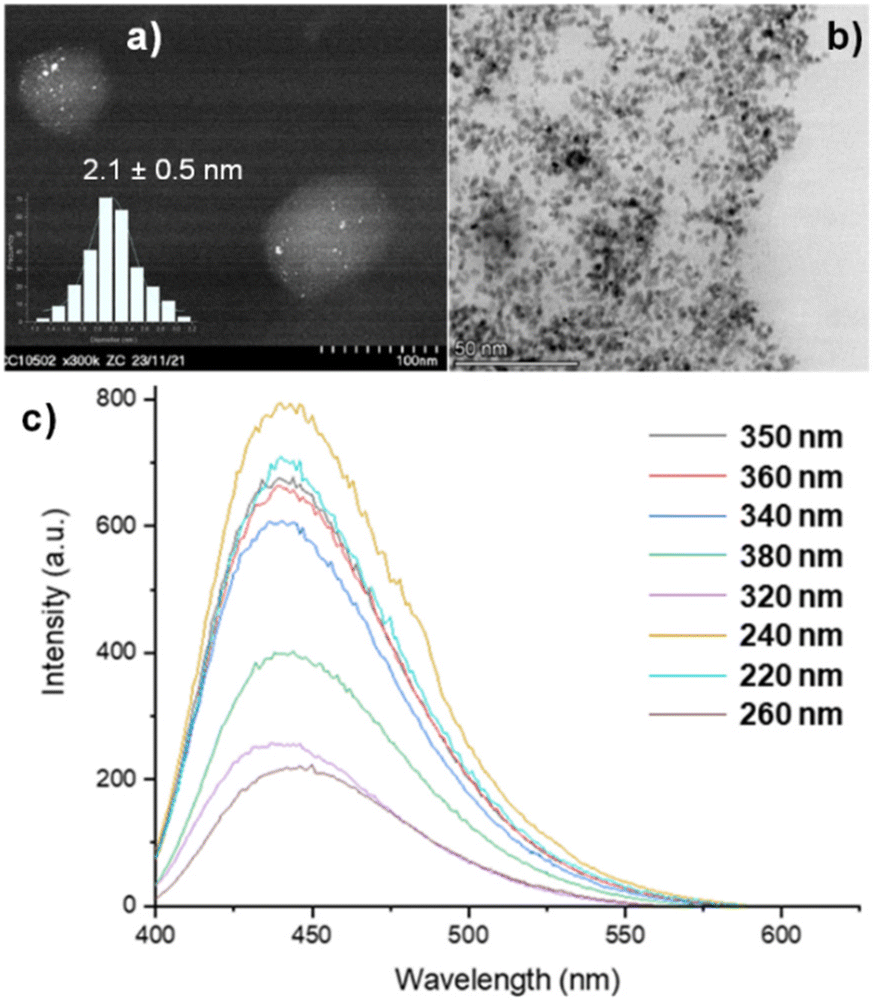 | ||
| Fig. 2 Characterization of Ru@CDs by (a) STEM-HAADF, (b) STEM-BF, (c) PLE at different excitation wavelengths. | ||
Potential effects of light irradiation on the catalytic properties of Ru@CDs were investigated using the hydrogenation of biomass-derived furfuralacetone (FFA, 1) as a model reaction (Scheme 1) under mild conditions.
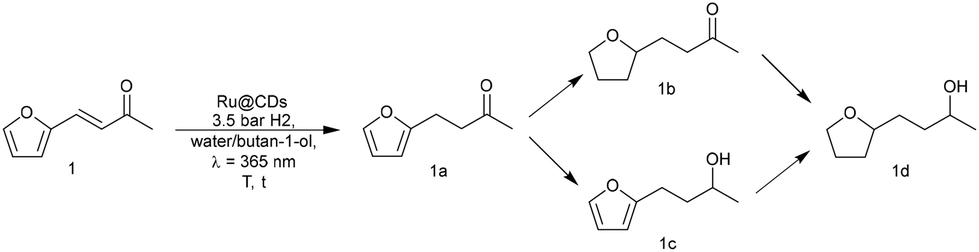 | ||
| Scheme 1 Reaction sequence for the hydrogenation of furfuralacetone (1) using Ru@CDs in the presence of hydrogen and light irradiation. | ||
FFA was selected as the model substrate because it contains several reducible moieties that allow probing easily changes in catalytic activity and selectivity. Standard reaction conditions were set at 3.5 bar H2, 60 °C, 4 h, 700 rpm, water/butan-1-ol as a green solvent mixture, without or with light irradiation at λirradiation = 365 nm (see Fig. S7–S10 in the ESI† for optimization steps).
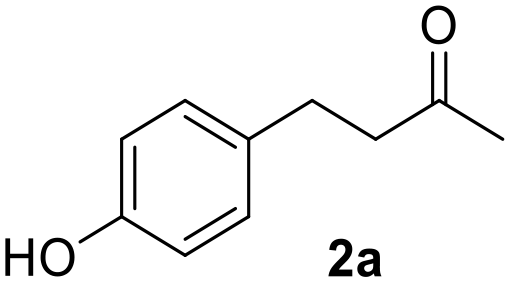
Without irradiation, Ru@CDs was found poorly active under these conditions, giving only 28% conversion of 1 toward product 1a (Table 1, entry 1). Light irradiation resulted in strikingly higher activity, with a conversion reaching 82% and the following distribution of products: 72% of 1a, 7% of 1b, 3% of 1c (Table 1, entry 2). Time profiles revealed a five-fold increase in the initial CC hydrogenation rate upon light exposure (r0(CC) = 0.06 s−1 under irradiation vs. 0.012 s−1 in the dark Fig. S11 and S12†).
| Entry | Catalyst | hν (nm) | Time (h) | Conv.a (%) | Product yieldsa (%) | |||
|---|---|---|---|---|---|---|---|---|
| 1a | 1b | 1c | 1d | |||||
| a Determined by GC-FID using tetradecane as internal standard. | ||||||||
| 1 | Ru@CDs | — | 4 | 28 | 28 | 0 | 0 | 0 |
| 2 | Ru@CDs | 365 | 4 | 82 | 72 | 7 | 3 | 0 |
| 3 | CDs | 365 | 4 | 0 | 0 | 0 | 0 | 0 |
| 4 | None | 365 | 4 | 0 | 0 | 0 | 0 | 0 |
| 5 | Ru@CDs | 450 | 4 | 30 | 30 | 0 | 0 | 0 |
| 6 | None | 450 | 4 | 0 | 0 | 0 | 0 | 0 |
| 7 | Ru@CDs | 400–700 | 4 | 63 | 63 | 0 | 0 | 0 |
| 8 | Ru/C | — | 2 | 86 | 18 | 0 | 68 | 0 |
| 9 | Ru/C | 365 | 2 | 87 | 22 | 0 | 65 | 0 |
Potential photothermal contributions to this substantial enhancement were investigated by performing blank experiments at a fixed temperature (30 °C) and monitoring any variation in the measured values of the reaction media (Table S1†). In all experiments, temperature variations remained within measurement error, suggesting poor photothermal properties of CDs at the used excitation wavelength.43,44 These reference experiments indicate that the contribution of thermal effects to catalytic performance is negligible.
Reactions performed with irradiation and only CDs or without any catalyst gave negligible conversion (Table 1, entries 3 and 4), confirming the need for Ru NPs and the absence of spontaneous photo-induced hydrogenation of the substrate. Changing the irradiation wavelength to 450 nm led to lower conversion with Ru@CDs, and still no activity in the absence of catalyst (Table 1, entries 5 and 6). Interestingly, using white light from a simple torchlight gave 63% yield of 1a (Table 1, entry 7), significantly higher than for a reaction in the dark. However, the enhancement is lower than for an irradiation at 365 nm due to the limited ability of our CDs to absorb the visible and infrared portions of the white light. Most importantly, no significant effect of light irradiation was observed when using a reference non-luminescent Ru/C catalyst (Table 1, entries 8 and 9), indicating that photothermal effects from the carbon support or plasmonic effects coming from the Ru NPs are negligible. Taken together, these results reveal a strong synergistic effect arising from the combination of Ru NPs and luminescent CDs. Satisfyingly, the reaction (conditions of Table 1, entry 2) could be scaled up by a factor of 40 (1.1 g (8 mmol) of substrate 1, 800 mg of catalyst) while maintaining the same level of performance (84% conversion, 82% yield of 1a, Scheme S1†). Recyclability was found challenging (Fig. S13†), although no substantial Ru NPs size increase (2.3 nm, Fig. S14†) nor Ru leaching (XRF) were detected. Thus, the observed deactivation is presumably due to the aggregation of the catalyst during washing steps, consistent with the known challenge associated with CDs separation and aggregation.45,46
To gather additional insight, the systematic activity enhancement of Ru@CDs under light irradiation was investigated by FT-IR using carbon monoxide (CO) as a molecular probe (Fig. 3, see ESI† for experimental procedure). FT-IR spectra show CO absorption bands at 2289 cm−1 for Ru@CDs in the dark (black line), and at 2216 cm−1 for the irradiated sample (red line). Such a shift toward lower wavenumbers is characteristic of electron density being transferred from a metal center to the CO bond, weakening it and lowering the vibration frequency.47,48 This data evidences that irradiated CDs act as electron donors increasing the electron density on the surface of Ru NPs, which can then be transferred to CO or other molecules, thereby facilitating their reaction. Interestingly, the CO band shift was found fully reversible upon turning on or off light irradiation. Most importantly, such behavior was not observed when using reference Ru/C or metal-free CDs (Fig. S15 and S16†), demonstrating that intimate contact between CDs and metal NPs is required to observed synergistic effects in hydrogenation. Thus, the enhancement of hydrogenation activity observed for Ru@CDs under light irradiation is attributed to a transfer of electron density from CDs to the Ru NPs, resulting in facilitated activation and/or transfer of hydrogen to the CC bond of FFA.
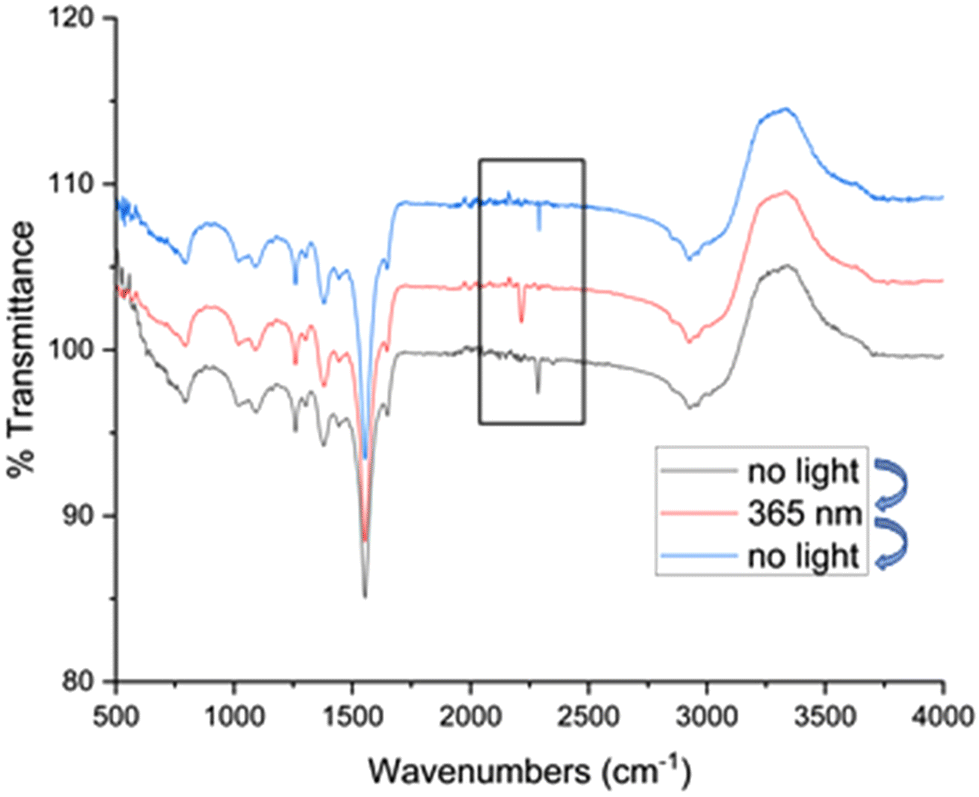 | ||
| Fig. 3 Comparison between the FT-IR spectra of CO-adsorbed Ru@CDs in water under irradiation (365 nm, red spectrum) and in the dark (blue and black spectra). The black rectangle highlights the region with CO absorption bands. | ||
The optical properties of the catalyst, assessed via UV-Vis spectroscopy, were retained after reaction (Fig. S17†). On–off experiments were conducted to highlight the dependence of the reaction on the presence of UV irradiation and the adaptivity of the catalyst to intermittent electricity supply. As can be seen in Fig. 4, when the light source was switched off the reaction became slow and then started again with the restart of the irradiation.
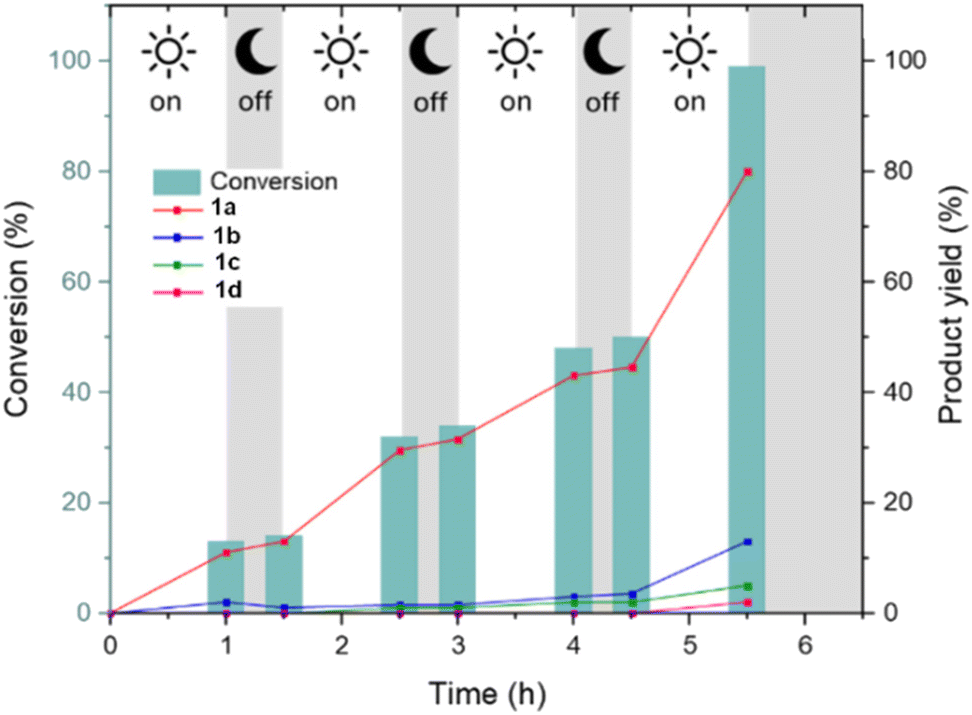 | ||
| Fig. 4 Conversion and product yields versus time for on–off experiments for the hydrogenation of FFA with Ru@CDs. White areas represent the time frame conducted with irradiation at 365 nm, grey areas are in the dark. | ||
The versatility of Ru@CDs as photo-enhanced hydrogenation catalysts was tested for other α–β unsaturated ketones (Table 2). Reactions were conducted under conditions adapted for each substrate, at incomplete conversion to facilitate the observation of irradiation effect (for more conditions, see Tables S2–S5†). With benzylideneacetone (3), conversion reached 70% under irradiation, and only 46% in the absence of light (Table 2, entries 3 and 4). The conversion levels of the cyclic α–β unsaturated ketone methylcyclopentenone (4) remained moderate, but still substantially higher under irradiation (31%) than in the dark (9%) (Table 2, entries 5 and 6). Similar photo-induced enhancements were observed for chalcone (5) and its methoxy (6) and fluorinated (7) derivatives (Table 2, entries 7–12). Conversion reached 54–66% under light irradiation, and remained under 10% in the dark.
| Entry | Substrate | Product Xa | t (h) | T (°C) | λ | Conversiona (%) | YieldXa![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (%) a (%) |
|---|---|---|---|---|---|---|---|
| Conditions: 3.5 bar of H2, 20 mg catalyst (0.01 mmol Ru), 0.2 mmol substrate, water/butan-1-ol, 700 rpm, 365 nm.a Calculated via GC-FID using tetradecane as internal standard.b Water/toluene used as solvent. | |||||||
| 1 | 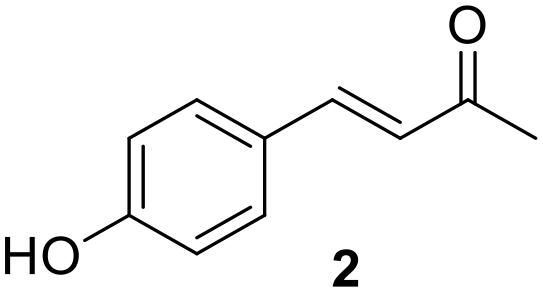 |
 |
4 | 60 | Yes | 100 | 100 |
| 2 | 4 | 60 | No | 13 | 13 | ||
| 3 | 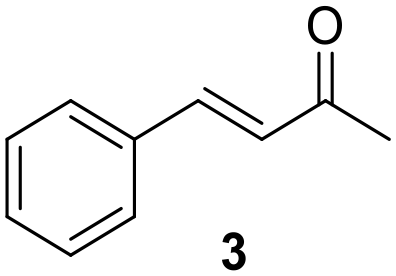 |
 |
4 | 80 | Yes | 70 | 67 |
| 4 | 4 | 80 | No | 46 | 43 | ||
| 5 |  |
 |
6 | 80 | Yes | 31 | 27 |
| 6 | 6 | 80 | No | 9 | 7 | ||
| 7 |  |
 |
6 | 80 | Yes | 63 | 63 |
| 8 | 6 | 80 | No | 8 | 8 | ||
| 9 |  |
 |
6 | 80 | Yes | 66 | 66 |
| 10 | 6 | 80 | No | 5 | 5 | ||
| 11 |  |
 |
6 | 80 | Yes | 54 | 54 |
| 12 | 6 | 80 | No | 6 | 6 | ||
| 13 |  |
 |
6 | 80 | Yes | 21 | 21 |
| 14 | 6 | 80 | No | 2 | 2 | ||
| 15b |  |
 |
6 | 80 | Yes | 11 | 11 |
| 16b | 6 | 80 | No | 0 | 0 | ||
| 17 |  |
 |
4 | 80 | Yes | 88 | 50 |
| 18 | 4 | 80 | No | 27 | 11 | ||
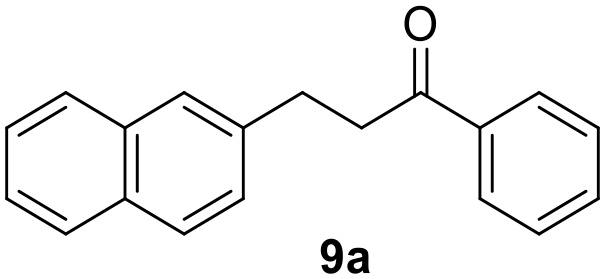
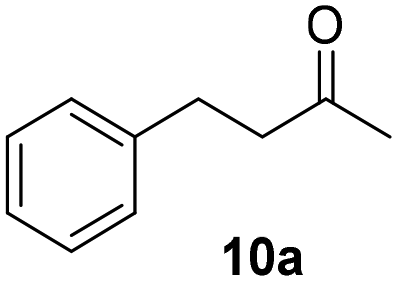
Trifluoromethyl- (8) and naphthyl-substituted (9) chalcones were less reactive under these reaction conditions (Table 2, entries 13–16), showing respectively 21% and 11% conversion. However, these substrates were not converted in the dark. Finally, the ability of Ru@CDs to reduce triple C–C bonds was assessed using phenylbutynone (10) as a substrate reaching 88% conversion under irradiation (with 50% yield of 10a, 22% of the corresponding alkene and 16% of the equivalent alcohol, Table 2, entry 17 and Table S6† entry 3) and 27% conversion without light (11% yield of 10a, Table 2, entry 18). No arene hydrogenation was observed for these substrates. Light-induced activity enhancement was observed irrespective of the substrates’ absorption at 365 nm (Fig. S18–S27†), indicating the absence of correlation between catalytic performance and substrates’ response to UV irradiations. No coupling product resulting from potential radical pathways was observed.
In conclusion, a multifunctional catalytic system composed of Ru NPs immobilized on luminescent and environmentally benign CDs has been prepared and characterized. The performances of the Ru@CDs catalyst in the hydrogenation of α–β unsaturated ketones under mild conditions were found substantially enhanced by UV irradiation. Mechanistic investigations involving references experiments and CO-IR measurements revealed a transfer of electron density from CDs to supported Ru NPs resulting in a boost of the metal NPs’ hydrogenation activity. The present demonstration of the capability of irradiated CDs to act as electron reservoirs for Ru NPs is potentially applicable to other CDs compositions and metal NPs, and may pave the way toward the development of a wide variety of photo-responsive NPs@CDs with enhanced catalytic performance.
Author contributions
A. B. and C. C. conceptualized and developed the project. C. C. and M. D. performed experiments, data analysis. C. C., W. L. and A. B. wrote and edited the manuscript. All authors contributed and agreed on the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors acknowledge financial support by the Max Planck Society and by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – Exzellenzcluster 2186 “The Fuel Science Center” ID: 390919832. We thank Alina Jakubowski, Annika Gurowski and Justus Werkmeister for GC, GC-MS and elemental analysis. The authors would like to thank Alin Benice Schöne and Norbert Pfänder for TEM and STEM/EDX measurements. Open Access funding provided by the Max Planck Society.References
- H. U. Blaser, C. Malan, B. Pugin, F. Spindler, H. Steiner and M. Studer, Adv. Synth. Catal., 2003, 345, 103–151 CrossRef CAS.
- K. Liu, R. Qin and N. Zheng, J. Am. Chem. Soc., 2021, 143, 4483–4499 CrossRef CAS PubMed.
- A. T. Bell, Science, 2003, 299(5613), 1688–1691 CrossRef CAS PubMed.
- G. A. Somorjai and J. Y. Park, Angew. Chem., Int. Ed., 2008, 47, 9212–9228 CrossRef CAS PubMed.
- P. Buchwalter, J. Rose and P. Braunstein, Chem. Rev., 2015, 115, 28–126 CrossRef CAS PubMed.
- L. He, F. Weniger, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 12582–12594 CrossRef CAS PubMed.
- G. J. Hutchings, Catal. Lett., 2001, 75, 1–12 CrossRef CAS.
- P. Liu, R. Qin, G. Fu and N. Zheng, J. Am. Chem. Soc., 2017, 139, 2122–2131 CrossRef CAS PubMed.
- W. Karim, C. Spreafico, A. Kleibert, J. Gobrecht, J. VandeVondele, Y. Ekinci and J. A. van Bokhoven, Nature, 2017, 541, 68–71 CrossRef CAS PubMed.
- M. Tarr and P. Zito, in Photochemistry of Nanomaterials, ed. American Chemical Society, ACS In Focus, 2022, vol. 15 Search PubMed.
- D. Ravelli, D. Dondi, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2009, 38, 1999–2011 RSC.
- S. Feng, P. T. T. Nguyen, X. Ma and N. Yan, Angew. Chem., Int. Ed., 2024, 63, e202408504 CrossRef CAS PubMed.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- J. Ackermann, J. T. Metternich, S. Herbertz and S. Kruss, Angew. Chem., Int. Ed., 2022, 61, e202112372 CrossRef CAS PubMed.
- Y. Liang, Y. Li, H. Wang and H. Dai, J. Am. Chem. Soc., 2013, 135, 2013–2036 CrossRef CAS PubMed.
- S. N. Baker and G. A. Baker, Angew. Chem., Int. Ed., 2010, 49, 6726–6744 CrossRef CAS PubMed.
- G. A. M. Hutton, B. C. M. Martindale and E. Reisner, Chem. Soc. Rev., 2017, 46, 6111–6123 RSC.
- S. Cailotto, E. Amadio, M. Facchin, M. Selva, E. Pontoglio, F. Rizzolio, P. Riello, G. Toffoli, A. Benedetti and A. Perosa, ACS Med. Chem. Lett., 2018, 9, 832–837 CrossRef CAS PubMed.
- H. Yu, R. Shi, Y. Zhao, G. I. Waterhouse, L. Z. Wu, C. H. Tung and T. Zhang, Adv. Mater., 2016, 28, 9454–9477 CrossRef CAS PubMed.
- C. Xia, S. Zhu, T. Feng, M. Yang and B. Yang, Adv. Sci., 2019, 6, 1901316 CrossRef CAS PubMed.
- J. Liu, R. Li and B. Yang, ACS Cent. Sci., 2020, 6, 2179–2195 CrossRef CAS PubMed.
- T. C. Wareing, P. Gentile and A. N. Phan, ACS Nano, 2021, 15, 15471–15501 CrossRef CAS PubMed.
- V. Mishra, A. Patil, S. Thakur and P. Kesharwani, Drug Discovery Today, 2018, 23, 1219–1232 CrossRef CAS PubMed.
- C. Campalani, E. Cattaruzza, S. Zorzi, A. Vomiero, S. You, L. Matthews, M. Capron, C. Mondelli, M. Selva and A. Perosa, Nanomaterials, 2021, 11, 524 CrossRef CAS PubMed.
- Z. L. Wu, Z. X. Liu and Y. H. Yuan, J. Mater. Chem. B, 2017, 5, 3794–3809 RSC.
- (a) X. Wang, L. Cao, F. Lu, M. J. Meziani, H. Li, G. Qi, B. Zhou, B. A. Harruf, F. Kermarrec and Y.-P. Sun, Chem. Commun., 2009, 3774–3776 RSC; (b) C. Campalani, G. Petit, J. C. M. Monbaliu, M. Selva and A. Perosa, ChemPhotoChem, 2022, e202200234 Search PubMed.
- S. Cailotto, R. Mazzaro, F. Enrichi, A. Vomiero, M. Selva, E. Cattaruzza, D. Cristofori, E. Amadio and A. Perosa, ACS Appl. Mater. Interfaces, 2018, 10, 40560–40567 CrossRef CAS PubMed.
- W. K. Li, J. T. Feng and Z. Q. Ma, Carbon, 2020, 161, 685–693 CrossRef CAS.
- C. Lu, Q. Zu, X. Zhang, H. Ji, Y. Zhou, H. Wang, Q. Liu, J. Nie, W. Han and X. Li, ACS Sustainable Chem. Eng., 2019, 7, 8542–8553 CrossRef CAS.
- S. Sadjadi, M. M. Heravi, L. Mohammadi and M. Malmir, ChemistrySelect, 2019, 4, 7300–7307 CrossRef CAS.
- J. Shen, W. Chen, G. Lv, Z. Yang, J. Yan, X. Liu and Z. Dai, Int. J. Hydrogen Energy, 2021, 46, 796–805 CrossRef CAS.
- Y. Ren, C. Hao, Q. Chang, N. Li, J. Yang and S. Hu, Green Chem., 2021, 23, 2938–2943 RSC.
- J. Zhang, Y. Chen, J. Tan, H. Sang, L. Zhang and D. Yue, Appl. Surf. Sci., 2017, 396, 1138–1145 CrossRef CAS.
- S. Deshmukh, A. Deore and S. Mondal, ACS Appl. Nano Mater., 2021, 4, 7587–7606 CrossRef CAS.
- K. Jiang, Y. Wang, X. Gao, C. Cai and H. Lin, Angew. Chem., Int. Ed., 2018, 57, 6216–6220 CrossRef CAS PubMed.
- T. V. de Medeiros, J. Manjoudakis, F. Noun, J.-R. Macairan, F. Victoria and R. Naccache, J. Mater. Chem. C, 2019, 7, 7175–7195 RSC.
- L. Vallan and H. Himahori, ACS Appl. Electron. Mater., 2022, 4, 4231–4257 CrossRef CAS.
- Y. Liu, S. Roy, S. Sarkar, J. Xu, Y. Zhao and J. Zhang, Carbon Energy, 2021, 23, 795–826 CrossRef.
- S.-C. Wei, Y.-W. Lin and H.-T. Chang, J. Food Drug Anal., 2020, 28, 558–574 CAS.
- J. C. G. Esteves da Silva and H. M. R. Gonçalves, TrAC, Trends Anal. Chem., 2011, 30(8), 1327–1336 CrossRef CAS.
- A. Bordet and W. Leitner, Acc. Chem. Res., 2021, 54, 2144–2157 CrossRef CAS PubMed.
- A. Bordet, G. Moos, C. Welsh, P. Licence, K. L. Luska and W. Leitner, ACS Catal., 2020, 10, 13904–13912 CrossRef CAS PubMed.
- S. Balou, P. Shandilya and A. Priye, Front. Chem., 2022, 10, 1023602 CrossRef CAS PubMed.
- T. Zhang, J. Wu, Z. Tang and S. Qu, Mater. Chem. Front., 2023, 7, 2359–2372 RSC.
- J. Gao, M. Zhu, H. Huang, Y. Liu and Z. Kang, Inorg. Chem. Front., 2017, 4, 1963 RSC.
- L. L. Mokoloko, R. P. Forbes and N. J. Coville, Catalysts, 2023, 13, 1201 CrossRef CAS.
- J. A. Anderson, C. H. Rochester and Z. Wang, J. Mol. Catal. A: Chem., 1999, 139, 285–303 CrossRef CAS.
- S. Xu, S. Chansai, S. Xu, C. E. Stere, Y. Jiao, S. Yang, C. Hardacre and X. Fan, ACS Catal., 2020, 10, 12828–12840 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc05468g |
| This journal is © The Royal Society of Chemistry 2025 |