
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMonolithic, hybrid and particulate lignin-based hydrogels for sustainable CO2 capture†
Adrian
Moreno
 *,
Javier
Delgado-Lijarcio
,
Juan C.
Ronda
,
Marina
Galià
and
Gerard
Lligadas
*
*,
Javier
Delgado-Lijarcio
,
Juan C.
Ronda
,
Marina
Galià
and
Gerard
Lligadas
*
Universitat Rovira i Virgili, Departament de Química Analítica i Química Orgànica, Laboratory of Sustainable Polymers, Tarragona 43007, Spain. E-mail: adrian.moreno@urv.cat; gerard.lligadas@urv.cat
First published on 16th January 2025
Abstract
Amine-infused hydrogels (AIHs) represent a promising platform for developing solid absorbents with improved CO2 absorption capacity. However, most of them rely on petroleum-based and toxic monomers. Lignin nanoparticles (LNPs) are becoming prominent players at the interface between sustainable nanomaterials technology and chemical science due to their high surface-area-to-mass ratio, which allows them to interact with multiple active compounds. Capitalizing on this spherical morphology and high surface area, the present work presents a strategy to prepare hybrid and particulate lignin-based hydrogels that can act as amine carriers for CO2 capture. The entire process is based on the internal stabilization of LNPs via intraparticle cross-linking process and subsequent base-catalyzed ring-opening reaction between LNPs and poly(ethylene glycol) diglycidyl ether in aqueous media. Upon swelling the hydrogel with an amine solution, hybrid and particulate lignin-based AIHs rapidly capture CO2 with a higher overall uptake compared to commonly used aqueous amine solutions under similar experimental conditions, while also stand and in some cases surpass the performance of other AIHs reported in the literature. Additionally, these new materials can be easily regenerated multiple times with minimal decrease in CO2 absorption capacity, demonstrating their potential application in decarbonization capture technologies.
Green foundation1. In this work, lignin nanoparticles (LNPs) are used as covalent cross-linking agents to prepare lignin-based particulate hydrogels with enhanced mechanical properties. These new hydrogels are capable of swelling in amine aqueous solutions and shown a rapidly and quantitatively absorption of CO2. Typically, hydrogels used for CO2 capture are composed of monomers derived from fossil resources and toxic cross-linking agents. Thus, our approach represents a promising alternative by utilizing lignin, a natural and abundant biopolymer with no food competition, and typically discarded by the pulp and paper industry.2. We have developed a particulate hydrogel with up to 60% biobased content and demonstrated that stand or, in some cases, surpasses the performance of petroleum-derived hydrogels in CO2 capture experiments. 3. Future experiments should address issues such as the effect of particle size and uniformity on the mechanical properties and performance of the material. Additionally, exploring the possibility of increasing the biobased content should also be considered to minimize or eliminate the use of petroleum-sourced resources completely. |
Introduction
The unstoppable development of modern industrial civilization and unrestrained consumption of fossil oil sources have triggered a severe energy crisis and global environmental concerns. In particular, the greenhouse effect resulting from a continuous increase in carbon dioxide (CO2) emissions in the atmosphere imposes a heavy burden on the Earth.1,2 In this context, efforts to capture CO2 from anthropogenic sources are being intensified to combat the greenhouse effect and climate change.3,4 Among the various CO2 capture techniques, amine scrubbing is the most widely used due to its high absorption capacity at very low CO2 concentrations, its ability to be recycled multiple times, and its relatively benign nature in contrast to other alternatives such as alkali solutions.5–7 However, like all ventures, amine scrubbing also faces certain challenges.8,9 These include lower overall capture efficiency compared to other capture agents, potential degradation into volatile byproducts through undesired reactions at high temperatures, and the loss of solvents –most commonly water– needed for amine activation.10To address this challenge, the use of hydrogels as an amine carrier for CO2 absorption has garnered interest in recent years. Pioneering works reported by Wood et al. demonstrated that hydrogels swelled in aqueous amine solutions, can act as macro-recipients where CO2 and amines can interact easily.11–14 This fact is due to the effective absorption of amines within the cross-linked polymeric matrix –owing to the swelling ability of hydrogels–, and the increase in surface area that facilitates CO2 diffusion and its reaction with the amine solution in situ. These relatively new CO2 absorbents were coined amine-infused hydrogels (AIHs), given the fact that their preparation simply involves the swelling “infusion” of the hydrogel with the corresponding amine solution. These systems have demonstrated quantitative and short absorption times (<1 hour),11 and the versatility to carrier greener materials for CO2 capture such as amino acid salts in contrast to conventional amines such as monoethanolamine (MEA).14 However, when it comes to the hydrogel part, the systems reported so far are based in cross-linked polymers derived from fossils resources such as poly(acrylamide)/poly(sodium acrylate) that are synthetized from toxic monomers (acrylamide) and employing also toxic cross-linking agents such as N,N′-methylenebis(acrylamide) (MBA).12,13 Here, it is worth mentioning that the use of inorganic capsules or organic hydrogel beads is also emerging as a notably approach.15–17 These materials tend to have a more remarkable porous structure and large surface area, which, upon swelling with the corresponding amine aqueous solution, shown high efficiency towards CO2 capture.
Therefore, it can be deduced that the next logical steps to upgrade this strategy should involve the utilization of hydrogels derived from biomass (i.e., bio-based hydrogels) and combine them with nanoparticles to maximize the efficiency of CO2 capture. In this sense, particulate hydrogels, where polymeric nanoparticles act as the cross-linking sites, have demonstrated improved mechanical properties and a large surface area in contrast to monolithic hydrogels.18 Thus, one can envision that particulate hydrogels could have great potential when applied to CO2 capture after swelling with amine aqueous or organic solutions, owing to the inherent porosity of hydrogels and the high surface area of nanoparticles that would facilitate the interaction with the amine and promote the reaction with CO2.
Among possible raw materials, lignin is one of the most promising components for the development of bio-based hydrogels, as it is often discarded in pulp and paper processes and has an inherently partially cross-linked structure rich in aliphatic, phenolic, and carboxylic acid groups, making it an excellent cross-linking agent.19–21 The formation of lignin-based hydrogels has been explored previously, showing their potential to develop functional materials for diverse applications, such as filtration membranes,22–24 drug delivery systems,25–27 or thermoelectric materials,28–30 among others.31–33 However, so far, lignin nanoparticles (LNPs) have not been applied in the preparation of particulate hydrogels, despite having a well-defined spherical shape, a large surface area-to-mass ratio and remarkable colloidal stability.34–37 In addition, LNPs can circumvent issues such as heterogeneous structure and broad molecular weight distribution, which are known hurdles in developing lignin-based materials.
In this work, we present a robust and straightforward preparation method to develop hybrid and particulate lignin-based hydrogels composed primarily of internally stabilized LNPs. In these new particulate hydrogels, LNPs act as physical cross-linking points, increasing the surface area of the hydrogels. This represents a promising strategy to enhance the interaction of active molecules within the hydrogel matrix (Fig. 1). We show that these bio-based particulate hydrogels can swell in the presence of aqueous or organic amine solutions and perform an efficient CO2 absorption, surpassing in some cases the performance of commercially available hydrogels. We also demonstrate that the incorporation of LNPs is a key factor not only in increasing the kinetics of the CO2 absorption process but also in improving the mechanical properties of the final material, which is critical for further applications.
 | ||
Fig. 1 Schematic illustration of the preparation of particulate lignin-based hydrogels (c-hy-LNPs@gel) and their application as amine carrier for CO2 absorption: (1) codissolution of SKL and TECy in acetone-water (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, w/w). (2) Gradually coprecipitation of SKL and TECy against water to form hybrid LNPs (hy-LNPs). (3) Hydrothermal curing of hy-LNPs to obtain stabilized hy-LNPs (c-hy-LNPs). (4) Base-catalyzed ring opening reaction between c-hy-LNPs and PEGDGE to form c-hy-LNPs@gel. (5) Hydration of c-hy-LNPs@gel with aqueous/organic amine solution and application as CO2 absorbent. :1, w/w). (2) Gradually coprecipitation of SKL and TECy against water to form hybrid LNPs (hy-LNPs). (3) Hydrothermal curing of hy-LNPs to obtain stabilized hy-LNPs (c-hy-LNPs). (4) Base-catalyzed ring opening reaction between c-hy-LNPs and PEGDGE to form c-hy-LNPs@gel. (5) Hydration of c-hy-LNPs@gel with aqueous/organic amine solution and application as CO2 absorbent. | ||
Results and discussion
The overarching objective of this work was to develop particulate lignin-based hydrogels using LNPs as building blocks and apply them in CO2 capture, as shown in Fig. 1. To achieve this, our work begins with the preparation of internally stabilized LNPs (i.e., cross-linked LNPs) since the preparation of the particulate hydrogels requires markedly basic conditions and LNPs tend to dissolve in such an environment.34 A commercially available and previously characterized Softwood Kraft lignin (SKL) (BioPiva 100), was used as the starting raw material for the preparation of cross-linked hybrid LNPs (c-hy-LNPs).38 Several approaches have been reported recently to render LNPs stable in basic conditions, thus increasing their application window.39–43 Among these approaches, the use of discrete molecules as cross-linkers (e.g., epoxides) during the self-assembly of LNPs emerges as one of the most attractive due to its operational simplicity and potential scalability.39,43–45 Here, a triepoxide derived from the bio-based and green solvent dihydrolevoglucosenone, commercialized under the trade name of Cyrene™, has been synthesized and selected as the cross-linker agent (TECy in Fig. 1). Details for TECy preparation and full characterization are found in the Experimental section (Fig. S1–S15†). The rational choice of TECy as a cross-linker was made to minimize the amount of cross-linker needed to ensure an efficient intraparticle cross-linking process to make LNPs resistant to strong alkaline pH. Thus, TECy was loaded into LNPs via a co-precipitation methodology, which involves the co-dissolution of SKL and TECy in a binary solvent mixture of acetone and water at a mass ratio of 3:1 to ensure complete dissolution of the starting materials. Hybrid particles (hy-LNPs) were formed by rapidly pouring water into the initial binary solvent mixture containing SKL and TECy (Fig. 1, steps 1–2). Following that, stable colloidal hy-LNPs dispersions with a TECy content of 5 wt% were obtained, and their characterization was performed via dynamic light scattering (DLS) and transmission electron microscopy (TEM). DLS results revealed a narrow particle size distribution (Fig. 2a), while zeta potential measurements demonstrated the strongly negative charged nature of the prepared hy-LNPs due to the accumulation of low molecular weight charged lignin residues on their surface (Fig. S16†). TEM imaging of hy-LNP dispersions revealed a core–shell structure attributed to the presence of TECy in liquid form inside the hy-LNPs, thus confirming the successful loading of the cross-linker (Fig. 2c, red arrows).
 | ||
| Fig. 2 Preparation and internal stabilization of hy-LNPs: (a) hydrodynamic diameter of hy-LNPs and c-hy-LNPs prepared in this work. (b) FTIR spectra of hy-LNPs before (black plot) and after (red plot) the hydrothermal curing process. Transmission electron microscopy (TEM) images of (c) hy-LNPs before curing and (d) c-hy-LNPs after being exposed to basic conditions (pH = 13) for 72 hours. Red arrows in c point the presence of TECy in liquid state. | ||
With the colloidal stable hy-LNPs available, our next step was to explore the thermal-induced intraparticle cross-linking process and investigate their stability in alkaline pH. To achieve this, hy-LNPs were subjected to hydrothermal curing at 100 °C for 48 hours to produce cross-linked hy-LNPs (c-hy-LNPs) (Fig. 1, step 3). The intraparticle cross-linking process takes place between the phenolic OH of SKL and the oxirane groups of TECy, as reported previously when epoxides are used as cross-linkers.39 The success of the cross-linking process was evaluated via FTIR analysis before and after the curing process from isolated freeze-dried hy-LNPs and c-hy-LNPs. Among the FTIR bands, the most characteristic is the band corresponding to the oxirane group around 910 cm−1. As can be seen from Fig. 2b, after the curing process, this band vanishes completely, thus confirming the effective ring-opening of TECy and the cross-linking reaction. In addition, DSC thermogram recorded from hy-LNPs also revealed the presence of an exothermic peak associated with the ring-opening processes between 60–90 °C confirming the curing process in this range of temperature (Fig. S17†). DLS analysis of the colloidal c-hy-LNPs dispersions revealed negligible variation in particle size, thus confirming that the curing step was limited to an intraparticle, and not interparticle, cross-linking processes (Fig. 2a). The structural stability of c-Hy-LNPs after being exposed to a strong alkaline environment (pH = 13) for 24 hours was initially evaluated by DLS. DLS analysis indicates that c-hy-LNPs remain colloidal, as evidenced by only a slight increase in particle size that can be attributed to the swelling of the particles (Fig. 2a). In addition, TEM imaging of c-hy-LNPs before and after exposure to alkaline conditions did not reveal any significant change in the shape and size of the particles, thus confirming their colloidal stability and resistance against marked basic pH (Fig. 2d and Fig. S18 and S19†). It is worth mentioning that colloidal stability against basic pH was achieved by employing only 5 wt% of cross-linker, thus leading to the obtention of stabilized LNPs with a lignin content as high as 95%. Previous reports required a minimum of 20 wt% of cross-linker to achieve internal stabilization of LNPs.39,43 This difference can be attributed to the increased reactivity of triepoxides compared to diepoxides, which could lead to a reduction in the production cost of stabilized LNPs while maintaining a higher biobased content.
With the internally stabilized c-hy-LNPs in our hands, we directed our efforts to the preparation of lignin-based particulate hydrogels (c-hy-LNPs@gel) via a base-catalyzed ring-opening reaction between c-hy-LNPs and poly(ethylene glycol) diglycidyl ether (PEGDGE) (Fig. 1, step 4). Accordingly, c-hy-LNPs@gel was prepared by simple homogenization of previously basified c-hy-LNPs dispersions with PEGDGE at room temperature, followed by hydrothermal curing at 40 °C for 16 hours to trigger the base-catalyzed epoxy ring-opening reaction. After gelation, the resulting mechanically stable gels were peeled off from the containers in the form of discs (d = 1 cm, h = 0.5 cm), soaked, and rinsed in pure water prior to further testing (Fig. 3b). Here it is important to mention that in order to obtain mechanically robust and flexible hydrogels, it is essential to add the optimal amount of cross-linker (PEGDGE). Considering that the total amount of phenolic hydroxyl groups in pristine SKL is 4.04 mmol g−1 lignin, as quantified by 31P NMR spectroscopy (Table S1 and Fig. S20†), and assuming that some of them have been consumed during the internal stabilization of hy-LNPs and not all are exposed on the surface of the particles, the optimal amount of PEGDGE to obtain self-standing hydrogels was found to be 2 mmol g−1 lignin. The cross-linking process was confirmed by FTIR, which, as discussed above for the stabilization of hy-LNPs, shows a complete disappearance of the oxirane bands at 910 cm−1 and 758 cm−1 after the hydrothermal curing, thus confirming the success of the cross-linking process (Fig. 3a). To study the morphology of c-hy-LNPs@gel scanning electron microscopy (SEM) and atomic force microscopy (AFM) analysis was employed. The SEM images of the surface and cross sections of c-hy-LNPs@gel revealed microscopic pore structures were c-hy-LNPs are homogeneously dispersed within the hydrogel matrix without any micrometer sized agglomerates (Fig. 3c and Fig. S21†). Similarly, AFM micrograph also prove the particulate nature of the synthetized hydrogel (Fig. 3d). One possible explanation for this well-defined particle dispersion could be attributed to an increase viscosity of the system during the curing process due to the evaporation of water, which likely restricted particle diffusion and, consequently, reduced their tendency to agglomerate due to the density difference compared to the hydrogel matrix. For comparative purposes, monolithic lignin-based hydrogels without LNPs (SKL@gel) and lignin-based hybrid hydrogels (hybrid@gel) using a mixture of c-hy-LNPs and SKL (1:1 wt%) were also prepared. SEM imaging of both control hydrogels revealed a microscopic porous structure, with an obvious reduction of c-hy-LNPs content in the case of the hybrid hydrogel and a complete absence in the case of the monolithic hydrogel (Fig. S22†). In addition, regardless of the hydrogel composition (monolithic, hybrid, or particulate), a gel content exceeding 95% was determined through extraction with an organic solvent (tetrahydrofuran) (Fig. S23†), indicating extensive cross-linking network formation during the synthetic step.
 | ||
| Fig. 3 Preparation and characterization of particulate lignin-based hydrogels (c-hy-LNPs@gel): (a) FTIR spectra of softwood kraft lignin (SKL), poly(ethylene glycol) diglycidyl ether (PEGDGE) and c-hy-LNPs@gel. (b) Digital image showing the free-standing c-hy-LNPs@gel. (c) Scanning electron microscopy (SEM) micrograph of top surface of c-hy-LNPs@gel. (d) AFM height image of top surface of c-hy-LNPs@gel. (e) Tensile stress–strain curves of monolithic (SKL@gel), hybrid (hybrid@gel) and particulate (c-hy-LNPs@gel) lignin-based hydrogels. (f) Water absorbency of lignin-based hydrogels prepared in this work after immersion in water for 2 hours. | ||
Mechanical properties are crucial and largely define the range of applications for hydrogels. In this sense, and intrigued by the impact that c-hy-LNPs could have in this aspect, uniaxial tensile testing was carried out to investigate the mechanical properties of the synthetized hydrogels. The c-hy-LNPs@gel exhibited a marked improvement in both tensile strength and elongation at break compared to the monolithic hydrogel without LNPs and the hybrid hydrogel (Fig. 3e). Notably, direct comparison with the monolithic (SKL@gel) and hybrid (hybrid@gel) hydrogels evidenced an increase in elongation at break by a factor of 4 and 2, respectively. This was accompanied by a more moderate increase in tensile strength (50% for the monolithic and 25% for the hybrid), demonstrating that particulate hydrogels are tougher and more resistant materials.
These significant differences in mechanical properties can be attributed to the well-defined spherical shape and high surface area-to-mass ratio of c-hy-LNPs, which would act as physical cross-linking points and establish effective interactions within the hydrogel matrix via non-covalent bond formation. These non-covalent interactions, mainly hydrogen bonding between the hydroxyl groups of both c-hy-LNPs and PEGDGE, would play a crucial role as sacrificial bonds that form new interactions during the deformation process, ultimately explaining the positive reinforcing effect in the hybrid and particulate hydrogels in contrast to monolithic hydrogel. While many previous works have reported the positive effects of incorporating LNPs into polymeric matrixes,46–48 none have explored their use in the preparation of covalent cross-linked particulate hydrogels. In this context, it is important to highlight that our approach represents a simple methodology to access lignin-based particulate hydrogels with improved mechanical properties, thus expanding their potential application to fields where marked mechanical properties are needed, such as membrane filtration preparation.24 Aligned with this, the water absorbency capacity (i.e., swelling) of the hydrogels was also evaluated. The monolithic (SKL@gel) hydrogel swelled and absorbed water quickly, reaching a water content of over 230% after 2 hours. When immersing the hybrid (hybrid@gel) and the particulate (c-hy-LNPs@gel) hydrogels, a systematic decrease in the water absorbency capacity was observed as the wt% of c-hy-LNPs in the system increased (200% for the hybrid and 170% for the particulate). This decrease could be attributed to a denser packing and higher cross-linked density in the hydrogels containing c-hy-LNPs, as the particles act as physical cross-linking points, thereby limiting the pore size around them.
As discussed in the introduction, amine-infused hydrogels (AIHs) allow fast and efficient CO2 uptake due to their enlarged surface area, which promotes effective diffusion of CO2.11 Intrigued by the effect that the incorporation of LNPs with a favorable and large surface-area-to-mass ratio could have on these systems, we decided to investigate lignin-based particulate hydrogels as AIHs for CO2 uptake. Regardless of whether the hydrogel employed was monolithic, hybrid, or particulate, AIHs were prepared by simply grounding and swelling of the hydrogel with an amine aqueous solution for 1 hour, highlighting the operational simplicity of preparing these CO2 capture absorbents (Fig. 4a). Diethanolamine (DEA) was selected as the amine due to its water solubility and high boiling point (271 °C). It is important to note that a minimum amount of hydrogel (15 wt% based on the liquid amine solution) is essential to ensure a high adsorption of the amine (91% based on titration experiments) within the hydrogel matrix in order to prevent the undesired squeezing of the amine solution.
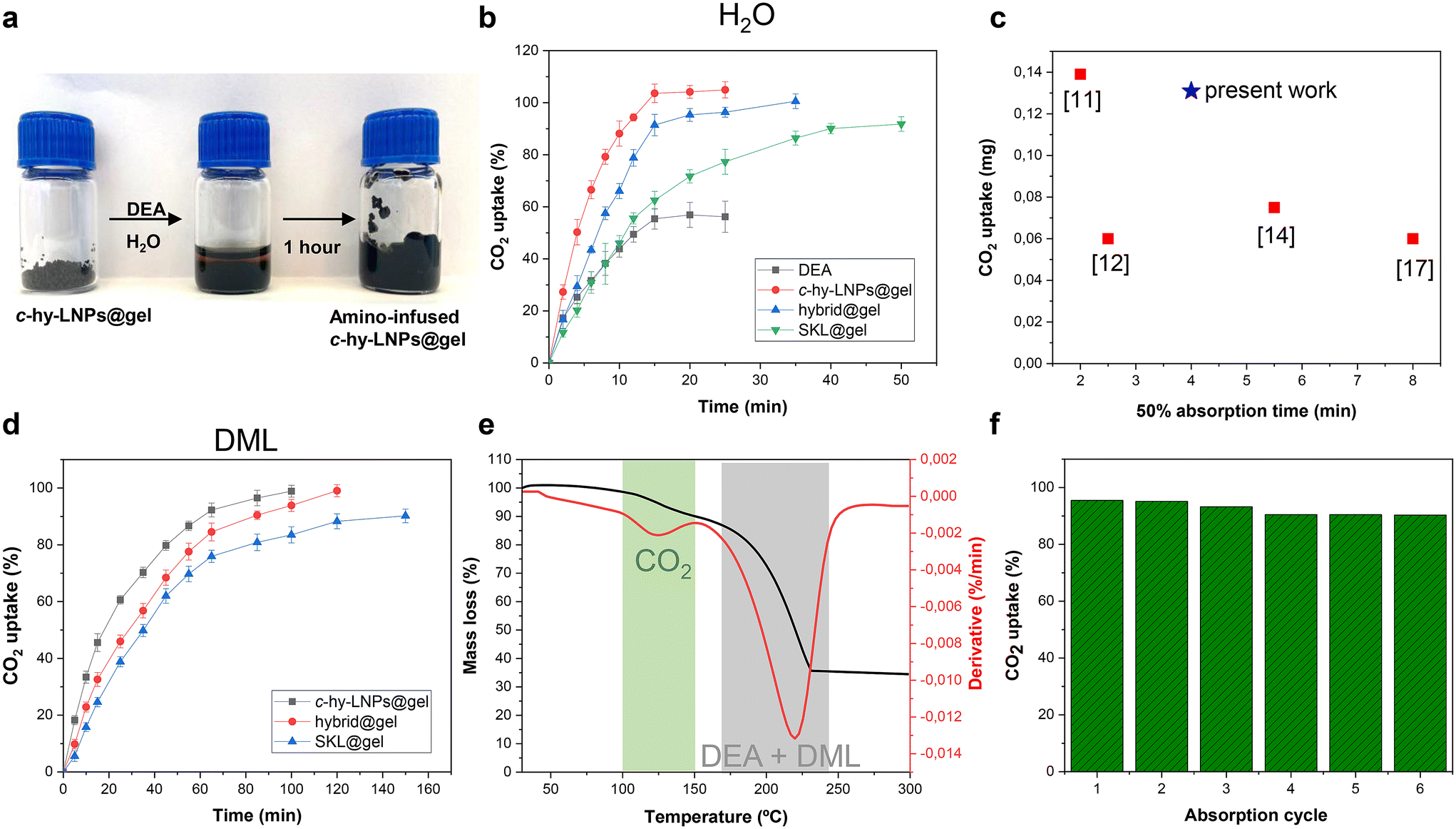 | ||
| Fig. 4 Preparation of lignin-based AIHs and their application in CO2 absorption: (a) digital images of the procedure to prepare lignin-based AIHs. (b) Gravimetric CO2 uptake of diethanolamine (DEA) aqueous solution (black squares) and DEA-lignin-based AIHs, where the only difference was the hydrogel employed during the amine-swelling step (monolithic, hybrid and particulate). (c) CO2 uptake vs. absorption time for previous reported AIHs and c-hy-LNPs@gel. (d) Gravimetric CO2 uptake of DEA-lignin-based AIHs, when non-aqueous solvent (N,N-dimethylactamide, DML) is employed. (e) Thermogravimetric analysis (TGA) showing the weight ratio loss and derivative weight loss of DEA-lignin-based AIHs. (f) Regeneration of DEA-lignin-based AIHs at 120 °C. In e and f, c-hy-LNPs@gel and DML were used as hydrogel and solvent respectively. The error bars in b and d represent ± standard deviation (SD) from the mean values (n = 3). DEA concentration in CO2 uptake experiments was fixed to 30 wt%. | ||
Attempts to reduce the hydrogel content in an effort to optimize the process result in the squeezing out of the amine solution over time, which could negatively impact the CO2 absorption capacity of the system (Fig. S24†).
The initial CO2 capture experiments using AIHs were conducted using a DEA aqueous solution at a fixed concentration of 30 wt%. The experimental conditions and corresponding results are summarized in Table 1 (entries 1–4), while the absorption kinetics as a function of time are shown in Fig. 4b.
| Entry | DEA (g) | Water (g) | DML (g) | Hydrogela | Theoretical absorptionb (g) | Determined absorption (g) | 50% absorption timec (min) | CO2 loading (mol of CO2 mol−1 of DEA) |
|---|---|---|---|---|---|---|---|---|
|
a 0.3 g of hydrogel were used in each experiment.
b Assuming the reaction mole ratio of DEA to CO2 is 2:1.
c Time required to achieve the 50% of actual CO2 absorption.
|
||||||||
| 1 | 0.6 | 1.4 | — | — | 0.124 | 0.070 | 6 | 0.28 |
| 2 | 0.6 | 1.4 | — | SKL@gel | 0.124 | 0.115 | 10 | 0.46 |
| 3 | 0.6 | 1.4 | — | hybrid@gel | 0.124 | 0.120 | 7 | 0.48 |
| 4 | 0.6 | 1.4 | — | c-hy-LNPs@gel | 0.124 | 0.131 | 4 | 0.53 |
| 5 | 0.6 | — | 1.4 | SKL@gel | 0.124 | 0.101 | 35 | 0.38 |
| 6 | 0.6 | — | 1.4 | hybrid@gel | 0.124 | 0.124 | 24 | 0.50 |
| 7 | 0.6 | — | 1.4 | c-hy-LNPs@gel | 0.124 | 0.124 | 17 | 0.50 |
For comparative purposes, a DEA aqueous solution was also studied, and as can be observed, it showed the lowest absorption capacity (56% CO2 uptake) (Table 1, entry 1 and Fig. 4b). This is mainly associated with the formation of viscous carbamate salts, which hampers the mass transfer inside the DEA solution and therefore slows the interaction of CO2 with DEA, as reported previously.49,50 When it comes to using AIHs, it is clear that regardless of the lignin-based hydrogel employed, higher CO2 uptake compared to the DEA solution was observed (Table 1, entries 2–4 and Fig. 4b). For instance, the monolithic hydrogel (SKL@gel) showed a 90% CO2 uptake in around 1 hour, while in the case of the hybrid hydrogel (hybrid@gel) and particulate hydrogel (c-hy-LNPs@gel), quantitative CO2 uptake was observed in 35 minutes and 25 minutes, respectively (Fig. 4b). These significant differences are associated with the increased surface area, which enables a complete and maximum interaction of CO2 with DEA. Here it is worth mentioning that the theoretical molar ratio of secondary amines to CO2 is 2:1, as reported previously,11 thus the maximum loading for DEA was 0.5 mol CO2 per mol of DEA. As can be observed in Table 1 (entries 2–4), the CO2 uptake of lignin-based AIHs was in the range of 0.5 mol of CO2 per mol of DEA, which proves the excellent absorption efficiency of lignin-based hydrogels. In contrast, the DEA solution showed a CO2 uptake of 0.28 mol per mol of DEA, which is significantly lower than that of AIHs. The effect of the introduction of LNPs in the hydrogel formulation was also evident, as increasing the amount of c-hy-LNPs systematically showed faster CO2 uptake kinetics. For instance, the particulate hydrogel reached 50% CO2 uptake in 4 minutes and complete uptake in 25 minutes, while the hybrid hydrogel showed 50% CO2 uptake in 7 minutes and complete uptake in 35 minutes (Table 1, entries 3 and 4 and Fig. 4b). These results can be rationalized by the fact that c-hy-LNPs have a favorable large surface area enriched with hydroxyl and phenolic groups. These functional groups would allow an efficient immobilization of the amine via non-covalent interactions (e.g., hydrogen bonding) at the particle surface, improving their accessibility to interact with CO2, thus accelerating the in situ reaction between CO2 and DEA. It is important to highlight that the results (e.g., CO2 uptake values and kinetic absorption) obtained with the particulate hydrogels (c-hy-LNPs@gel) not only stand the comparison but also, in some cases, surpass those of hydrogels derived from petroleum resources, thus highlighting the potential of lignin-based particulate hydrogels for decarbonization technologies (Fig. 4c). To confirm the interaction between the CO2 gas and DEA in lignin-based AIHs, FTIR analysis before and after CO2 absorption was conducted. After CO2 absorption, the characteristic bands of carbamate formation, which are the symmetric and asymmetric COO− stretching vibrations at 1540 cm−1 and 1255 cm−1, respectively, together with the band assigned to the NR3+ deformation at 1307 cm−1, were observed, thus validating the CO2 absorption in lignin-based AIHs (Fig. S25†).51
In order to develop a competitive CO2 capture technology, the captured CO2 must be easily removed from the sorbents so they can be recycled and used again as circular materials. AIHs swollen with amine aqueous solutions suffer from water evaporation during thermal-induced CO2 desorption, which leads to a poor sorbent cycling process. To avoid that, high-boiling point non-aqueous solvents such as ethylene glycol have been reported to show notable performance in the recycling process.13 Here, we selected Agnique AMD 3L (N,N-dimethylactamide, DML), a green bio-solvent derived from the lactic acid platform and commercialized by BASF, as a high-boiling point (200 °C) solvent to explore the recyclability of lignin-based AIHs. Thus, the hydrogels were swollen under the same conditions used in the previous experiments, simply replacing the amine aqueous solution with an amine DML solution. Kinetic absorption experiments and experimental details are shown and summarized in Fig. 4d and Table 1 (entries 5–7), respectively. Kinetic experiments revealed an identical trend to that observed previously when amine aqueous solutions were used. For instance, the particulate hydrogel (c-hy-LNPs@gel) exhibited the fastest CO2 uptake with a 50% absorption time of 17 minutes, in contrast to 25 minutes and 35 minutes for hybrid (hybrid@gel) and monolithic (SKL@gel) hydrogels, respectively (Fig. 4d and Table 1, entries 5–7). Additionally, it was noted that the presence of c-hy-LNPs is crucial to reach quantitative CO2 absorption, as hybrid and particulate hydrogels achieved levels of 0.5 mol of CO2 per mol of DEA, in contrast to the monolithic hydrogel that exhibited lower values (0.38 mol of CO2 per mol of DEA), as can be observed in Table 1, entries 5–7. Here it is worth mentioning that when non-aqueous solvents are used, lignin-based AIHs clearly surpass the performance of commercial hydrogels. For instance, poly(acrylamide)-based hydrogels exhibited a 50% absorption time of 150 minutes compared to a 50% absorption time of 17 minutes for particulate lignin-based hydrogels.13 This confirms the exceptional performance of lignin-based hydrogels for CO2 capture and highlights their potential for industrial carbon neutralization processes.52
To further confirm the occurrence of CO2 absorption in lignin-based AIHs when non-aqueous solvents are employed, and also assess the temperature conditions for desorption of CO2 and evaporation of the DEA/DML mixture, thermogravimetric analysis (TGA) was performed. Thus, particulate lignin-based AIHs (c-hy-LNPs@gel) were presaturated with CO2 to assess the desorption conditions. Fig. 4e displays the mass change and derivative mass, which indicates the rate of mass loss for the CO2-loaded particulate lignin-based AIHs during the heating process. Two peaks are clearly distinguishable in the plot of derivative weight (Fig. 4e, green and gray shadows). The one at a lower temperature around 100–130 °C corresponds to the characteristic CO2 desorption as reported previously for similar systems,11,12 while the second starting around 170 °C corresponds to the evaporation process of both DEA and DML. These results confirm the absorption of CO2, but more importantly, they also show the possibility to remove CO2 from the sorbent without significant loss of amine and solvent, which is crucial for assessing the recyclability of lignin-based AIHs. The recyclability of particulate lignin-based AIHs was investigated by exposing the particulate hydrogel to CO2 for 2 hours before being placed in an oven at 120 °C for 1 hour to remove the CO2. After that, and without adding additional DEA and DML, it was used for the next absorption cycle (Fig. 4f, absorption cycle 1). A minor decrease in CO2 uptake (less than 10%) was observed over 6 cycles associated to minor evaporation process of solvent and amine, indicating a notable capability of lignin-based AIHs to be reused and demonstrating their potential application in the circular capture of CO2 (Fig. 4f).
Conclusions
We have reported the preparation of hybrid and particulate lignin-based hydrogels via a simple and robust base-catalyzed ring-opening reaction between commercially available poly(ethylene glycol) diglycidyl ether (PEGDGE) and internally stabilized lignin nanoparticles (c-hy-LNPs). These hydrogels have shown great potential as amine carriers for CO2 absorption in both aqueous and organic solvents. The large surface-area-to-mass ratio of the c-hy-LNPs promotes effective interaction between the amine and CO2, enabling fast absorption kinetics at room temperature. It is worth mentioning that the CO2 absorption capabilities of particulate lignin-based hydrogels not only compare favourably with those derived from petroleum sources but also surpass some of them. Additionally, we demonstrated that when water is replaced by sustainable organic solvents (e.g. N,N-dimethylactate amide), the particulate hydrogels can be easily regenerated and reused up to six times with only a minimal loss of CO2 absorption capacity, making them promising circular lignin-based materials. Finally, beyond the scope of our current study, we anticipate that particulate lignin-based hydrogels will open new avenues for the development of value-added lignin materials, such as anticorrosion particulate coatings or particulate membrane filtration devices. However, further work is needed to scale up hydrogel production while minimizing energy-intensive processes (e.g., lyophilization). In addition, and as discussed in the sections above, we hypothesize that the positive effect on mechanical properties and CO2 absorption capacity is related to the increased surface area provided by LNPs and their ability to interact with amines. Therefore, we believe that the size and uniformity of LNPs is critical in these aspects and can affect the performance of the final material, thus warranting further investigation.Experimental section
Preparation of hy-LNPs
hy-LNPs were prepared by replacing SKL with the corresponding weight percentage of TECy (5 wt%), but otherwise following the same procedure for the preparation of hy-LNPs described previously.53 Briefly, TECy and SKL were separately dissolved in an acetone/water mixture (3:1 mass ratio), with insoluble impurities filtered out, and the soluble fractions combined in the predetermined ratio (95% SKL and 5% TECy). hy-LNPs were then formed by rapidly adding deionized water to the TECy and SKL solution under stirring. The dispersions were concentrated using rotary evaporation and dialyzed against water for 24 hours to remove any remaining organic solvent. The final aqueous dispersion of hy-LNPs (5 g L−1) was achieved with a lignin mass yield of 87%.
Internal stabilization of hy-LNPs: preparation of c-hy-LNPs
A total of 500 mL of hy-LNPs dispersion (5 g L−1) was hydrothermally cured in a Teflon-sealed reactor at 100 °C for 16 hours to induce the intraparticle cross-linking process and form c-hy-LNPs. After the curing process, c-hy-LNPs dispersion was filtered to remove any insoluble precipitated, concentrated using rotary evaporation to a concentration of 10 g L−1 and lyophilized to be used for the preparation of hydrogel.Preparation of particulate (c-hy-LNPs@gel), hybrid (hybrid@gel) and monolithic (SKL@gel) lignin-based hydrogels
In the case of c-hy-LNPs@gel, 600 mg of lyophilized c-hy-LNPs were dispersed in 3 mL of basic solution (NaOH, 0.1 M, pH = 13.0). Next, 0.6 mL of PEGDGE (2 mmol g−1 of c-hy-LNPs) was added and the mixture was allowed to stir for 30 minutes. After that, the mixture was transferred to a Petri dish (d = 1 cm, h = 0.5 cm) and cured in an oven at 50 °C for 16 hours in order to trigger the cross-linking process and obtain free-standing c-hy-LNPs@gel. In the case of the hybrid@gel, same synthetic protocol was applied, but instead of 600 mg of c-hy-LNPs, 300 mg of SKL and 300 mg of c-hy-LNPs were used. The preparation of monolithic hydrogel (SKL@GEL) follows also the same protocol, replacing c-hy-LNPs for pristine SKL. To assess the gel content, pre-weighed lignin-based hydrogels (0.5 g) were extracted with tetrahydrofuran (150 mL) for 48 h at 70 °C. The insoluble fraction was then vacuum-dried at 50 °C until a constant weight was reached. The gel content was calculated according to the following equation| Gel content (%) = (M1 − M2)/M1 × 100% |
Preparation of lignin-based AIHs and their application in CO2 absorption
2 g of amine (DEA) solution (using water or DML as solvents) at a fixed concentration of 30 wt% was placed into a vial together with 0.3 g of the corresponding lignin-based hydrogel (SKL@gel, hybrid@gel, or c-hy-LNPs@gel), which had been previously lyophilized, ground into powder using a food blender, and sieved through a 425 μm metal sieve. Next, the vial (with a headspace of 3 cm) was sealed with a septum and mechanically shacked with a vibromatic instrument during 15 minutes. After that, the lignin-based hydrogel was allowed to swell for additional 45 minutes at room temperature. For CO2 uptake experiments, the vial was weighted (M1) and subjected to CO2 capture. CO2 was introduced into the vial using a balloon with one needle for injection and a second needle for purging. After purging for 1 minute, the second needle was removed. The samples were weighted (M2) at predetermined times as CO2 was absorbed into the polymer matrix under balloon pressure at ambient conditions. This process continued up to 50 minutes for the systems using water as solvent and 180 minutes when DML was used as solvent, with the balloon being refilled as needed when depleted. At least three measurements were performed for each sample. To determine the amine content adsorbed by the hydrogel, the swollen hydrogels were placed in a beaker, diluted with 15 mL of water, and stirred for 30 minutes. This was followed by titration with a standardized hydrochloric acid (0.1 M) solution.Regeneration of lignin-based AIHs
In a typical cycle, DEA-lignin-based AIHs underwent a CO2 uptake experiment at ambient temperature. Afterward, the CO2-loaded hydrogel was placed in an oven preheated to 120 °C for 1 hour. Subsequently, the hydrogel was allowed to cool down to room temperature before the CO2 uptake experiment was repeated. Multiple absorption cycles (six in total) were performed to fully evaluate the circularity of the lignin-based AIHs. For regeneration experiments only the use of c-hy-LNPs@gel was considered for the investigation.Author contributions
Javier Delgado-Lijarcio: methodology, validation, investigation of the preparation of cross-linker (TECy), lignin-based hydrogels and formal analysis of part of the CO2 absorption studies. Juan Carlos Ronda: review & editing. Marina Galià: funding acquisition and review & editing. Adrian Moreno: methodology, validation, investigation, formal analysis, figure preparation, conceptualisation, methodology, supervision (lead), and writing, review & editing. Gerard Lligadas: funding acquisition, conceptualisation, methodology, supervision (lead), and writing, review & editing. All authors have given approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported through project PID2020-114098RB-100 and PID2023-149489OB-100 to G. L and M. G., all funded by MICIU/AEI/10.13039/501100011033. The authors also thank the Serra Hunter Programme of the Government of Catalonia to G. L., Universitat Rovira i Virgili (2021PMF-BS-13) grant (to J. D.-L.) and the “Ramón y Cajal” contract (RYC2022-035322-I) funded by MICIU/AEI/ 10.13039/501100011033 and ESF+ to A. M. The authors also thank BASF SE, Ludwigshafen, Germany (Dr O. Gronwald) and UPM Biochemicals, Finland (Dr Leo Airaksinen) for kindly supplying Agnique® AMD 3L (DML) solvent and Softwood Kraft lignin (SKL) (BioPiva 100), respectively.References
- M. Redlin and T. Gries, Theor. Appl. Climatol., 2021, 146, 713–721 CrossRef.
- Z. Liu, Z. Deng, S. J. Davis, C. Giron and P. Ciais, Nat. Rev. Earth Eniron., 2022, 3, 217–219 CrossRef PubMed.
- J. Chen, L. Zhang, L. Wang, M. Kuang, S. Wang and J. Yang, Matter, 2023, 6, 3322–3347 CrossRef CAS.
- J. Zheng, X. Hou, H. Duan and S. Wang, Fundam. Res., 2024, 4, 1696–1709 CrossRef CAS PubMed.
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef CAS PubMed.
- J. Hack, N. Maeda and D. M. Meier, ACS Omega, 2022, 44, 39520–39530 CrossRef PubMed.
- H. M. Stowe and G. S. Hwang, Ind. Eng. Chem. Res., 2017, 56, 6887–6899 CrossRef CAS.
- V. Ramar and A. Balraj, Energy Fuels, 2022, 36, 13479–13505 CrossRef CAS.
- N. Yi, S. Li, X. Wang, F. Kong, X. Li, Y. Ren, H. Liu and S. Xu, Sep. Purif. Technol., 2025, 354, 129295 CrossRef CAS.
- A. M. Varghese and G. N. Karanikolos, Int. J. Greenhouse Gas Control, 2020, 96, 103005 CrossRef CAS.
- X. Xu, C. Heath, B. Pejcic and C. D. Wood, J. Mater. Chem. A, 2018, 6, 4829–4838 RSC.
- X. Xu, Y. Yang, L. Pompeo, A. Falcon, P. Hazewinkel and C. D. Wood, Int. J. Greenhouse Gas Control, 2019, 88, 226–232 CrossRef CAS.
- C. White, E. Adam, Y. Sabri, M. R. Myers, B. Pejcic and C. D. Wood, Ind. Eng. Chem. Res., 2021, 60, 14758–14767 CrossRef CAS.
- X. Xu, M. N. Myers, F. G. Versteeg, E. Adam, C. White, E. Crooke and C. D. Wood, J. Mater. Chem. A, 2021, 9, 1692–1704 RSC.
- X. Xu, M. B. Myers, F. G. Versteeg, B. Pejcic, C. Heath and C. D. Wood, Chem. Commun., 2020, 56, 7151–7154 RSC.
- M. Chen, M. Li, Y. Liang, W. Meng, Z. Zhang, Y. Wu, X. Li and F. Zhang, Langmuir, 2023, 40, 14451–14458 CrossRef PubMed.
- X. Xu, B. Pejcic, C. Heath and C. D. Wood, J. Mater. Chem. A, 2018, 6, 21468–21474 RSC.
- L. Andrée, P. Bertsch, R. Wang, M. Becker, J. Leijten, P. Fischer, F. Yang and S. C. G. Leeuwenburgh, Biomacromolecules, 2023, 24, 2755–2765 CrossRef PubMed.
- A. K. Mondal, M. T. Uddin, S. M. A. Sujan, Z. Tang, D. Alemu, H. A. Begum, J. Li, F. Huang and Y. Ni, Int. J. Biol. Macromol., 2023, 245, 125580 CrossRef CAS PubMed.
- D. Rico-García, L. Ruiz-Rubio, L. Pérez-Alvarez, S. L. Hernández-Olmos, G. L. Guerrero-Ramírez and J. L. Vilas-Vilela, Polymers, 2020, 12, 81 CrossRef PubMed.
- Q. Wang, J. Gao, X. Lu, X. Ma, S. Cao, X. Pan and Y. Ni, Int. J. Biol. Macromol., 2021, 181, 45–50 CrossRef CAS PubMed.
- L. Dai, W. Zhu, J. Lu, F. Kong, C. Si and Y. Ni, Green Chem., 2019, 21, 5222–5230 RSC.
- Z. Lv, J. Xu, C. Li, L. Dai, H. Li, Y. Zhong and C. Si, ACS Sustainable Chem. Eng., 2021, 41, 13972–13978 CrossRef.
- L. Dai, M. Ma, J. Xu, C. Si, X. Wang, Z. Li and Y. Ni, Chem. Mater., 2020, 32, 4324–4330 CrossRef CAS.
- A. A. Khan, J. K. Nayak, B. U. Amin, M. Muddasar, M. Culebras, V. V. Ranade and M. N. Collins, Int. J. Biol. Macromol., 2024, 281, 136292 CrossRef CAS PubMed.
- E. Larrañeta, M. Imízcoz, J. X. Toh, N. J. Irwin, A. Ripolin, A. Perminova, J. Domínguez-Robles, A. Rodríguez and R. F. Donnelly, ACS Sustainable Chem. Eng., 2018, 6, 9037–9046 CrossRef PubMed.
- A. Moreno and M. H. Sipponen, Mater. Horiz., 2020, 7, 2237–2257 RSC.
- M. Muddsar, N. Menéndez, A. Quero, M. A. Nasiri, A. Cantarero, J. García-Cañadas, C. M. Gómez, M. N. Collins and M. Culebras, Adv. Compos. Hybrid Mater., 2024, 7, 47 CrossRef.
- M. Muddsar, M. A. Nasiri, A. Cantarero, C. Gómez, M. Culebras and M. N. Collins, Adv. Funct. Mater., 2024, 34, 2306427 CrossRef.
- H. Zhou, L. Yan, D. Tang, T. Xu, L. Dai, C. Li, W. Chen and C. Si, Adv. Mater., 2024, 36, 2403876 CrossRef CAS PubMed.
- H. Zao, S. Hao, Q. Fu, X. Zhang, L. Meng, F. Xu and J. Yang, Chem. Mater., 2022, 34, 5258–5272 CrossRef.
- X. Han, Y. Su, G. Che, J. Zhou and Y. Li, ACS Sustainable Chem. Eng., 2023, 11, 8255–8270 CrossRef CAS.
- Y. Zheng, A. Moreno, Y. Zhang, M. H. Sipponen and L. Dai, Trends Chem., 2024, 6, 62–78 CrossRef CAS.
- A. Moreno and M. H. Sipponen, Acc. Chem. Res., 2024, 57, 918–1930 CrossRef PubMed.
- E. Lizundia, M. H. Sipponen, L. G. Greca, M. Balakshin, B. L. Tardy, O. J. Rojas and D. Puglia, Green Chem., 2021, 23, 6698–6760 RSC.
- B. Zhao, M. Borghei, T. Zou, L. Wang, L.-S. Johansson, J. Majoinen, M. H. Sipponen, M. Österberg, B. D. Mattos and O. J. Rojas, ACS Nano, 2021, 15, 6774–6786 CrossRef CAS PubMed.
- A. Moreno and M. H. Sipponen, Nat. Commun., 2020, 11, 5599 CrossRef CAS PubMed.
- A. Moreno, J. Delgado-Lijarcio, J. C. Ronda, V. Cádiz, M. Galià, M. H. Sipponen and G. Lligadas, Small, 2023, 19, 2205672 CrossRef CAS PubMed.
- T. Zou, M. H. Sipponen, A. Henn and M. Österberg, ACS Nano, 2021, 15, 4811–4823 CrossRef CAS PubMed.
- A. Moreno, J. Liu, R. Gueret, S. E. Hadi, L. Bergström, A. Slabon and M. H. Sipponen, Angew. Chem., Int. Ed., 2021, 60, 20897–20905 CrossRef CAS PubMed.
- M. Morsali, A. Moreno, A. Loukovitou, I. Pylypchuck and M. H. Sipponen, Biomacromolecules, 2022, 23(11), 4597–4606 CrossRef CAS PubMed.
- A. Moreno, I. Pylypchuck, Y. Okahisa and M. H. Sipponen, ACS Macro Lett., 2023, 12, 759–766 CrossRef CAS PubMed.
- Y. Fan, H. Ji, X. Ji, Z. Tian and J. Chen, Small, 2024, 20, 2405754 CrossRef CAS PubMed.
- K. A. Henn, S. Forssell, A. Pietläinen, N. Forsman, I. Smal, P. Nousiainen, R. Prasad, B. Ashok, P. Oinas and M. Österberg, Green Chem., 2022, 24, 6487–6500 RSC.
- F. Wang, M. Morsali, J. Rizikovs, I. Pylypchuck, A. P. Mathew and M. H. Sipponen, Mater. Horiz., 2024, 11, 6504–6515 RSC.
- A. Moreno, M. Morsali, J. Liu and M. H. Sipponen, Green Chem., 2021, 23, 3001–3014 RSC.
- E. Kimiaei, M. Farooq, R. Grande, K. Meinander and M. Österberg, Adv. Mater. Interfaces, 2022, 9, 2200988 CrossRef CAS.
- L. Wang, Q. Wang, E. Rosqvist, J.-H. Smått, Q. Yong, L. Lassila, L. J. Peltonen, T. Rosenau, M. Toivakka, S. Willför, P. Eklund, C. Xu and X. Wang, Small, 2023, 19, 2207085 CrossRef CAS PubMed.
- J. Yang, H. Y. Han, Q. X. Low, B. P. Binks and J. M. Chin, J. Mater. Chem. A, 2015, 3, 6440–6446 RSC.
- O. M. Al-Azzawi, C. M. Hofmann, G. A. Baker and S. N. J. Baker, J. Colloid Interface Sci., 2012, 385, 154–159 CrossRef CAS PubMed.
- D. L. Frasco, J. Chem. Phys., 1964, 41, 2134–2140 CrossRef CAS.
- D. Barker-Rothschild, J. Chen, Z. Wan, S. Renneckar, I. Burgert, Y. Ding, Y. Lu and O. J. Rojas, Chem. Soc. Rev., 2025 10.1039/D4CS00923A , Advance Article.
- A. Moreno, I. Pylypchuck, Y. Okahisa and M. H. Sipponen, ACS Macro Lett., 2023, 12, 759–766 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc05489j |
| This journal is © The Royal Society of Chemistry 2025 |