
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBatch and flow synthesis of sulfides and sulfoxides using green solvents and oxidant through visible-light photocatalysis†
Jin Park‡
b,
Su Hyeon Kim‡b,
Jun-Young Cho‡c,
Shafrizal Rasyid Atriardi a,
Jae-Young Kimb,
Hanifah Mardhiyaha,
Boyoung Y. Park*c and
Sang Kook Woo*a
a,
Jae-Young Kimb,
Hanifah Mardhiyaha,
Boyoung Y. Park*c and
Sang Kook Woo*a
aDepartment of Chemistry and Chemistry Institute for Functional Materials, Pusan National University, Busan 46241, Korea. E-mail: skwoo@pusan.ac.kr
bDepartment of Chemistry, University of Ulsan, 93 Daehak-Ro, Nam-Gu, Ulsan 44610, Korea
cDepartment of Fundamental Pharmaceutical Science, College of Pharmacy, Kyung Hee University, Seoul 02447, South Korea. E-mail: boyoungy.park@khu.ac.kr
First published on 20th February 2025
Abstract
Herein, we report an environmentally friendly, one-pot, scalable method for the synthesis of sulfides and sulfoxides via photocatalyzed C–C bond-formation and oxidation. Employing mild photoredox conditions, green solvents, and oxygen as the oxidant, the approach is sustainable and efficient. The integration of a microreactor-based flow system enabled large-scale production, addressing the typical scalability issues of photocatalytic and liquid–gas phase reactions. Mechanistic studies confirmed that C–C bond formation occurs via an α-thiomethyl radical through a single-electron transfer (SET) pathway, while oxidation involves both SET and energy transfer (EnT) mechanisms.
Green foundation1. This method is a sustainable synthetic approach that employs mild, atom-economical reaction conditions with green solvents and oxidant, facilitated by visible-light photocatalysis. Additionally, we improved the scalability and reproducibility of photocatalytic and liquid–gas phase reactions through a flow system, making them suitable for large-scale synthesis while minimizing waste and environmental impact.2. This method employed ethanol and water as reaction solvents, with oxygen from the air serving as the oxidant. The green metrics exhibited notably high values, with AE (61%), AEf (58%), and RME (64%). Under alkene, catalyst, and solvent reuse conditions, an E-factor of 9.6 and a PMI of 10.6 were achieved. 3. Through further research, we aim to enhance the sustainability of the reaction conditions by optimizing alkene, catalyst, and solvent reuse strategies in flow chemistry while also expanding the scope of this methodology to include sulfone synthesis. |
Introduction
Sulfur-containing compounds, including sulfides, sulfoxides, and sulfones, are found in a wide range of natural products, bioactive compounds, pharmaceuticals, pesticides, and functional organic materials.1 Generally, sulfides are formed through alkylation, arylation, and thiol–ene reactions of thiols2 and can be transformed into sulfoxides and sulfones via oxidation.3 Additionally, sulfoxides and sulfones can be structurally diversified through sulfoxidation4 and sulfonylation5 of their simpler forms.Over the past two decades, visible-light photocatalytic reactions have garnered significant attention as environmentally friendly synthetic approaches.6 This includes various photocatalytic reactions for the synthesis and functionalization of sulfur-containing compounds. Sulfides have been synthesized through addition reactions between alkenes and thiyl radicals7 or α-alkylthiomethyl radicals,8 which are generated during photocatalysis. The oxidative generation of sulfoxides and sulfones has been investigated similarly to conventional syntheses.9 In particular, in the case of sulfones, a three-component reaction of alkyl radical precursors, SO2 surrogates, and alkenes under photocatalytic conditions has been developed to synthesize complex sulfones from simple starting materials.10 In contrast to sulfides and sulfones, the synthesis of complex sulfoxides from simple starting materials through bond-forming reactions has rarely been reported (Scheme 1a).11 Therefore, the synthesis of sulfoxides via bond-forming reactions under mild reaction conditions is an important challenge in organic synthesis.
 | ||
| Scheme 1 Synthetic approaches for sulfur-containing compounds. | ||
We devised a strategy to synthesize complex and diverse sulfoxide derivatives through functionalization at the α-position of sulfoxides in milder and greener conditions. Previous studies have reported approaches utilizing the α-sulfinyl anion as a key intermediate, wherein the sulfoxide (pKa ≈ 33)4c is deprotonated at the α-position, and then alkylated by reacting with an electrophile such as an alkyl halide,4a,b aldehyde,12 or alkene.13 However, owing to the lower acidity of the α-proton of sulfoxides compared with that of ketones and sulfones, strong bases such as lithium diisopropylamide or n-BuLi are required. The resulting sulfinyl anions are unstable and sensitive to moisture, making them difficult to handle. In 2013, Walsh and co-workers reported an arylation at the α-position of the sulfoxide with aryl halides using a palladium catalyst and alkoxide base.4c However, this method could not introduce alkyl groups and required high-temperature conditions (Scheme 1b).
Our goal was to develop an environmentally friendly and scalable method for the synthesis of sulfoxides. We considered α-sulfinyl radicals14 as key intermediates, serving as a synthetic equivalent to α-sulfinyl anions that are less sensitive to moisture and do not require strong bases. This allows milder reaction conditions, easier handling, and greater suitability for large-scale production. Our research group has developed organosilicon compounds as radical precursors to generate various alkyl radicals via photoredox catalysis.15 The introduction of an alkyl silyl group facilitates oxidation through the β-silicon effect, and the alkyl silyl cation acts as a good leaving group, facilitating the formation of alkyl radicals. However, α-trimethylsilyl sulfoxides still have a high oxidation potential (Eox ≈ +2.0 V vs. SCE), making them unsuitable as α-sulfinyl radical precursors. To address this challenge, we plan to use α-trimethylsilyl thioethers, which have a lower oxidation potential (Eox ≈ +1.0 V vs. SCE) and are more easily oxidized. These thioethers will act as α-sulfinyl radical surrogates, enabling the desired sulfoxides to be synthesized through photocatalyzed, one-pot C–C bond formation and oxidation (Scheme 1c).
To achieve these goals, we aimed to use oxygen as an oxidant, employ green solvents,16 and establish scalable conditions for large-scale production. However, when conducting photocatalytic liquid–gas reactions in a batch system, reactivity is usually significantly reduced as the scale increases. This decrease in reactivity can be attributed to the characteristics of photocatalytic multiphasic reactions, where longer light penetration results in uneven and low photon concentrations, and the enlarged headspace between the liquid and gas phases leads to reduced mass transfer.17 To overcome these limitations, we have integrated photocatalytic liquid–gas reactions into a flow system, making large-scale production feasible. Although the application of photocatalytic liquid reactions in flow systems is relatively straightforward, reactions involving gases pose a significant challenge. Using a microreactor, we can maintain a uniform and high photon concentration, significantly reduce the headspace between the liquid and gas phases, and increase gas solubility through easy pressure control. Ultimately, this enhances reactivity and reproducibility, enabling large-scale production.
In this paper, we present an environmentally friendly and scalable method for the synthesis of sulfoxides via photocatalyzed C–C bond formation and oxidation. This method enables the synthesis of diverse sulfides and sulfoxides through a controlled one-pot process. By employing mild reaction conditions, green solvents, and oxygen as an oxidant, we established a sustainable approach. Furthermore, the integration of an efficient flow system enabled us to overcome the scalability limitations inherent in photocatalytic and liquid–gas phase reactions.
Results and discussion
In our initial study, we investigated the Giese addition of PMPSCH2TMS 1a and acrylonitrile 2a in the C–C bond-forming reaction (Table 1). PMPSCH2TMS 1a (Eox = +0.98 V vs. SCE in MeCN) was chosen as a radical precursor owing to its low oxidation potential, which allows for facile oxidation and the generation of an α-thioalkyl radical. Based on the oxidation potential of the substrate and previous research findings,15f we selected Ir(dF(CF3)ppy)2(dtbpy) (Ir(III)*/Ir(II) = +1.21 V vs. SCE) as the photocatalyst. Fortunately, under blue LED irradiation in acetonitrile, the iridium catalyst efficiently facilitated the Giese reaction, resulting in the formation of the sulfide product in 36% yield (Table 1, entry 1). To develop an environmentally friendly reaction, we explored various green solvents and found that methanol provided 50% yield (Table 1, entry 3). However, the desilylation byproduct PMPSCH3 1a-I was formed, and the yield did not further improve. We hypothesized that PMPSCH3 1a-I was formed via the protonation of the α-thioalkyl radical. Assuming that a slight increase in the pH of the reaction solution could prevent protonation, we used mildly basic NaHCO3, which increased the yield to 75% and decreased the reaction time (Table 1, entry 4). However, when ethanol was used as the solvent, the yield decreased to 66% owing to the lower solubility of NaHCO3 in ethanol (Table 1, entry 5). To address this issue, we used a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of ethanol and water as the cosolvent, which resolved the solubility problem and increased the yield to 85% (Table 1, entry 6). Next, we examined the one-pot Giese addition and oxidation reactions by opening the reaction tube to air after the Giese reaction was complete. Consequently, the argon atmosphere was replaced with air, and we fortuitously obtained sulfoxide 3a in 75% yield (Table 1, entry 7). To develop more practical reaction conditions, we replaced NaHCO3 with a buffer solution at pH 9 mixed with ethanol in a 5:1 volume ratio, which increased the yield of 3a to 95% (Table 1, entry 8). By reducing the catalyst loading from 1 to 0.5 mol% and extending the reaction time to 4 h, 3a was obtained in excellent yield (92%; Table 1, entry 9). However, further reducing the catalyst loading to 0.1 mol% necessitated a prolonged reaction time of 20 h to achieve a yield of 86% (Table 1, entry 10). Considering both efficiency and cost, we opted for a catalyst loading of 0.5 mol% as the optimal condition. As reducing the amount of acrylonitrile 2a significantly lowered the yield, we used 2.0 equiv. 2a. Under the optimized conditions for sulfide (Table 1, entry 9), followed by the introduction of an air atmosphere for an additional 6 h, an excellent yield of 95% was achieved for sulfoxide 4a (Table 1, entry 13). Finally, we confirmed that the reaction did not proceed in the absence of light or a photocatalyst, demonstrating that both were essential to the reaction (Table 1, entries 14 and 15). Through this optimization, we successfully developed a green solvent- and oxidant-based method for synthesizing sulfides and sulfoxides via one-pot C–C bond formation and oxidation.
a
:1 mixture of ethanol and water as the cosolvent, which resolved the solubility problem and increased the yield to 85% (Table 1, entry 6). Next, we examined the one-pot Giese addition and oxidation reactions by opening the reaction tube to air after the Giese reaction was complete. Consequently, the argon atmosphere was replaced with air, and we fortuitously obtained sulfoxide 3a in 75% yield (Table 1, entry 7). To develop more practical reaction conditions, we replaced NaHCO3 with a buffer solution at pH 9 mixed with ethanol in a 5:1 volume ratio, which increased the yield of 3a to 95% (Table 1, entry 8). By reducing the catalyst loading from 1 to 0.5 mol% and extending the reaction time to 4 h, 3a was obtained in excellent yield (92%; Table 1, entry 9). However, further reducing the catalyst loading to 0.1 mol% necessitated a prolonged reaction time of 20 h to achieve a yield of 86% (Table 1, entry 10). Considering both efficiency and cost, we opted for a catalyst loading of 0.5 mol% as the optimal condition. As reducing the amount of acrylonitrile 2a significantly lowered the yield, we used 2.0 equiv. 2a. Under the optimized conditions for sulfide (Table 1, entry 9), followed by the introduction of an air atmosphere for an additional 6 h, an excellent yield of 95% was achieved for sulfoxide 4a (Table 1, entry 13). Finally, we confirmed that the reaction did not proceed in the absence of light or a photocatalyst, demonstrating that both were essential to the reaction (Table 1, entries 14 and 15). Through this optimization, we successfully developed a green solvent- and oxidant-based method for synthesizing sulfides and sulfoxides via one-pot C–C bond formation and oxidation.
a
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | 2a (equiv.) | Solvent (0.1 M) | Additive (equiv.) | Oxidant | Time (h) | Yieldb (%) | By-productc (%) |
| 3a/4a | 1a-I | |||||||
| a Reaction conditions: 1a (0.2 mmol), 2a (quantity noted), catalyst (quantity noted), and solvent (0.1 M) irradiated with 10 W blue LED (452 nm) at room temperature under argon in a pressure tube.b Isolated yield determined by flash column chromatography.c NMR yield using styrene as internal standard.d The solvent ratio is expressed as volume/volume (v/v).e After 1a was consumed, the reaction tube was left open in air.f In the absence of light source. nd = not detected. | ||||||||
| 1 | 1 | 2.0 | MeCN | — | — | 14 | 36/nd | 43 |
| 2 | 1 | 2.0 | EtOAc | — | — | 14 | 30/nd | 52 |
| 3 | 1 | 2.0 | MeOH | — | — | 14 | 50/nd | 35 |
| 4 | 1 | 2.0 | MeOH | NaHCO3 (1) | — | 2 | 75/nd | nd |
| 5 | 1 | 2.0 | EtOH | NaHCO3 (1) | — | 3 | 66/nd | nd |
| 6 | 1 | 2.0 | EtOH/H2O (5:1)d |
NaHCO3 (1) | — | 3 | 85/nd | nd |
| 7 | 1 | 2.0 | EtOH/H2O (5:1)d |
NaHCO3 (1) | Aire | 3/16 | nd/75 | nd |
| 8 | 1 | 2.0 | EtOH/pH 9 buffer (5:1)d |
— | — | 3 | 95/nd | nd |
| 9 | 0.5 | 2.0 | EtOH/pH 9 buffer (5:1)d |
— | — | 4 | 92/nd | nd |
| 10 | 0.1 | 2.0 | EtOH/pH 9 buffer (5:1)d |
— | — | 20 | 86/nd | nd |
| 11 | 0.5 | 1.5 | EtOH/pH 9 buffer (5:1)d |
— | — | 8 | 58/nd | nd |
| 12 | 0.5 | 1.2 | EtOH/pH 9 buffer (5:1)d |
— | — | 20 | 30/nd | nd |
| 13 | 0.5 | 2.0 | EtOH/pH 9 buffer (5:1)d |
— | Aire | 4/6 | nd/95 | nd |
| 14 | — | 2.0 | EtOH/pH 9 buffer (5:1)d |
— | — | 4 | nd/nd | nd |
| 15f | 0.5 | 2.0 | EtOH/pH 9 buffer (5:1)d |
— | — | 4 | nd/nd | nd |
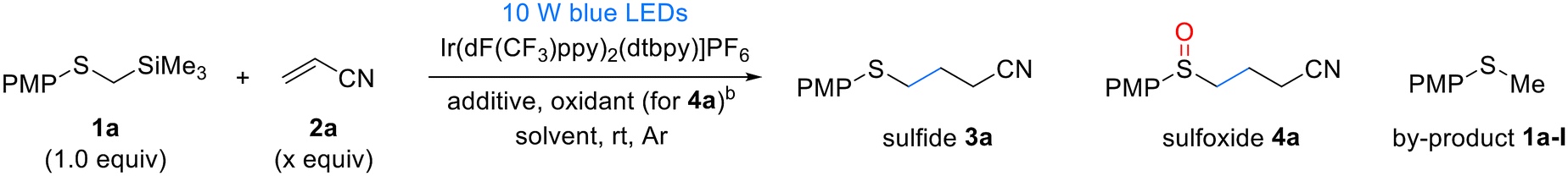
The substrate scope of α-silyl sulfides 1 was evaluated under the optimized reaction conditions (Table 2). Electron-rich methoxy-substituted aryl groups in α-silyl sulfides 1a–c exhibited good reactivity regardless of the substitution position (ortho, meta, or para), yielding sulfide products 3a–c in excellent yields. However, in the one-pot sulfoxide synthesis, the meta-methoxy aryl substrate 1b afforded lower sulfoxide yields than the ortho- and para-methoxy aryl substrates 1a and c. These results indicate that sulfides 3 formed via the Giese addition are more difficult to oxidize than the α-silyl sulfides 1, making they more sensitive to the electronic properties of the substituents. α-Silyl sulfides containing halogens in the aryl group also reacted efficiently, yielding sulfides 3d–f and sulfoxides 4d–f in good yields. However, the fluorine-substituted derivative 1d produced a slightly lower sulfoxide 4d yield than sulfide 3d yield. Phenyl- and tolyl-substituted substrates 1g,h were similarly well tolerated, affording high yields of the corresponding sulfides 3g,h and sulfoxides 4g,h. However, sterically hindered substrates such as mesityl- and 1-naphthyl-substituted α-trimethylsilyl thioethers exhibited low yields or no reaction under the same conditions (see details in the ESI†). By contrast, the alkyl-substituted substrate 1i exhibited decreased reactivity. Although it produced the corresponding sulfide 3i in moderate yields, it failed to yield the desired sulfoxide, suggesting that the oxidation step is more challenging for alkyl-substituted substrates than aryl-substituted substrates owing to the electronic properties of the former. For another alkyl-substituted substrate 1j, the corresponding sulfide 3j and sulfoxide 4j were obtained in moderate yields by extending the reaction time and increasing the catalyst loading.
a
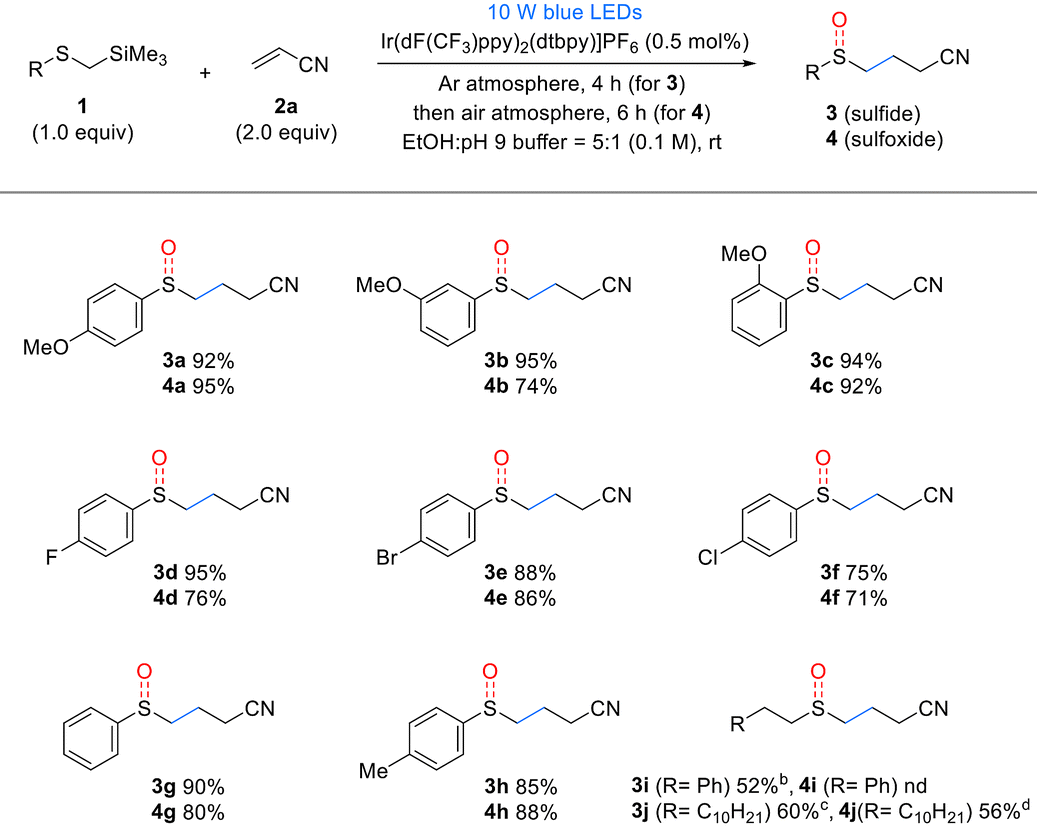
Next, the substrate scope of the Michael acceptors 2 was evaluated under the optimized reaction conditions (Table 3). Various α,β-unsaturated esters, such as methyl, ethyl, n-butyl acrylate, and hexafluoroisopropyl, demonstrated good reactivity, and both corresponding sulfide products 3k–n and sulfoxide products 4k–n were obtained in good yields. Additionally, methyl methacrylate, when substituted at the α-position of the α,β-unsaturated ester, barely affected the reactivity, yielding results similar to the unsubstituted substrate. Afterwards, we used the developed method to investigate the effect of β-substitution in Michael acceptors. Crotonitrile 2p, a β-methyl acrylonitrile, exhibited low reactivity and yields, following a similar trend to Giese reactions. However, benzalmalononitrile 2q, a highly activated Michael acceptor containing a dinitrile, produced sulfide 3q and sulfoxide 4q in excellent yields using 1 mol% catalyst and methanol as the solvent, despite the β-phenyl substituent. In this reaction, 2- and 4-vinyl pyridines acted as effective Michael acceptors, producing the corresponding sulfides 3r and 3s in high yields. However, oxidation of the resulting sulfides was inhibited by pyridine, with 2-vinyl pyridine producing only trace amounts of sulfoxide and 4-vinyl pyridine, resulting in a reduced yield. Lastly, α,β-unsaturated ketones also served as effective Michael acceptors in the developed reaction. In particular, cyclic enones such as cyclohexenone 2u and cyclopentenone 2v, despite having substituents at the β-position, did not exhibit the reactivity drop observed with crotonitrile 2p owing to their Z-alkene configuration, which allowed for the formation of both sulfides and sulfoxides in excellent yields. However, vinyl phenyl sulfone, dimethyl maleate, maleonitrile, and N-cyclohexylmaleimide failed to yield the desired products (see details in the ESI†).
a

Performing photocatalytic multiphasic reactions in a batch system, particularly during a scaled-up process, can present significant challenges. For photochemical reactions, as the size of the batch reactor increases, a longer distance between the light source and reaction solution can lead to uneven irradiation, resulting in insufficient photon concentration. In the case of multiphase reactions on a larger scale, the interfacial area per unit volume can decrease, reducing mass transfer between the liquid and gas phases. To address these challenges, we introduced a flow system. This system, which utilizes microreactors for mass production, maintains a short distance between the light source and reactants to maintain a uniform and high photon concentration. Furthermore, by using a back-pressure regulator for easy pressure control, the liquid and gas are continuously mixed at a consistent ratio while maintaining a low headspace and high solubility of the oxygen gas.
Initially, the reaction conditions for the synthesis of sulfide 3a in the flow process were optimized based on the conditions of the batch reaction. Through optimization, the concentration of the reaction solution was adjusted to 0.05 M to reduce viscosity. Finally, sulfide 3a was synthesized with a residence time of 40 min and 92% yield (Table S1, entry 6; see ESI† for details). After determining the optimal conditions for synthesizing sulfide 3a through photochemical reactions in the flow system, we determined the optimal conditions for the synthesis of sulfoxide 4a by performing additional photoinduced multiphase reactions under continuous flow using oxygen gas as the oxidant. Various oxygen gas flow rates (7.0, 5.0, and 3.0 mL min−1) were investigated (Table 4, entries 1–3) using a backpressure regulator set at 2.8 bar. As a result, 4a was obtained in 75% NMR yield at a gas flow rate of 5.0 mL min−1 (Table 4, entry 2). However, at a relatively low flow rate of 3.0 mL min−1, the reaction solution backflowed into the gas line, disrupting the reaction progress (Table 4, entry 3). To prevent such backflow while achieving more precise control of the oxygen gas flow rate, the back pressure was reduced from 2.8 bar to 1.4 bar. After conducting experiments at various oxygen flow rates (Table 4, entries 4–6), the desired sulfoxide 4a was obtained in 88% isolated yield at an oxygen flow rate of 1.7 mL min−1.
|
||||
|---|---|---|---|---|
| Entry | Set gas flow (mL min−1) | BPRb (bar) | System pressure (bar) | Yield (%) |
| 3a/4ac |
||||
| a Reaction conditions: 1a (0.2 mmol), 2a (2.0 equiv.) catalyst (0.5 mol%), solvent (0.05 M), flow rate (0.125 mL min−1), and temperature (25 °C).b Pressure setting of the backpressure regulator components.c Yield determined by 1H NMR analysis of the crude mixture using 1,3-benzodioxole as the internal standard.d Isolated yield determined using flash column chromatography. | ||||
| 1 | 7.0 | 2.8 | 3.6 | 35/44 |
| 2 | 5.0 | 2.8 | 3.3 | 14/75 |
| 3 | 3.0 | 2.8 | — | Backflow into gas line |
| 4 | 2.0 | 1.4 | 2.1 | 2/87 |
| 5 | 1.7 | 1.4 | 2.1 | −/90 (88)d |
| 6 | 1.3 | 1.4 | 1.5 | 7/83 |
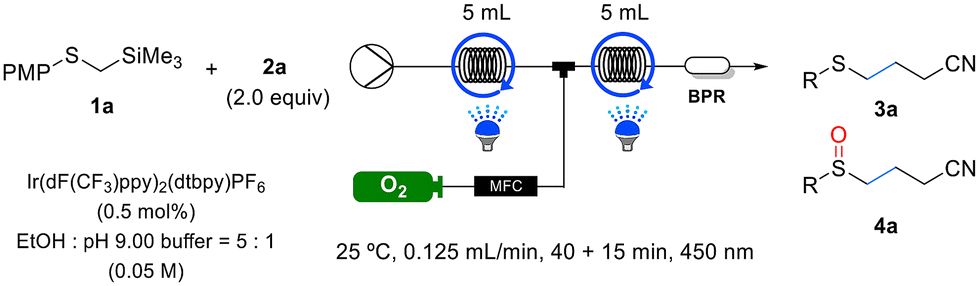
To evaluate the effectiveness of this continuous flow system, experiments were conducted using selected samples from Tables 2 and 3 for the reactions listed in Table 5. Remarkably, all the substrates exhibited shorter residence times under continuous flow than in batch reactions, while maintaining similar yields. To assess the scalability of the flow system, we performed 5 mmol reactions for the synthesis of sulfide 3a and sulfoxide 4a, as outlined in Scheme 2. The yields achieved for both 3a and 4a in the 5 mmol scale reactions were comparable to those obtained in the 0.2 mmol scale reactions. Furthermore, the productivity for each compound was measured to be 0.339 mmol−h and 0.307 mmol−h, respectively (as illustrated in Scheme 2).
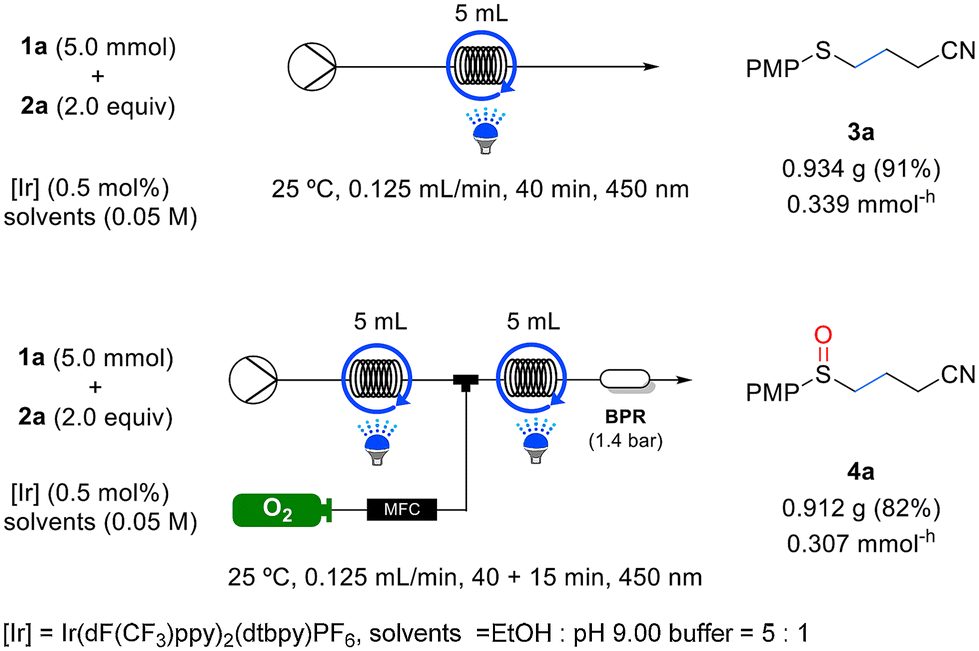 | ||
| Scheme 2 Gram-scale reaction in flow process. | ||
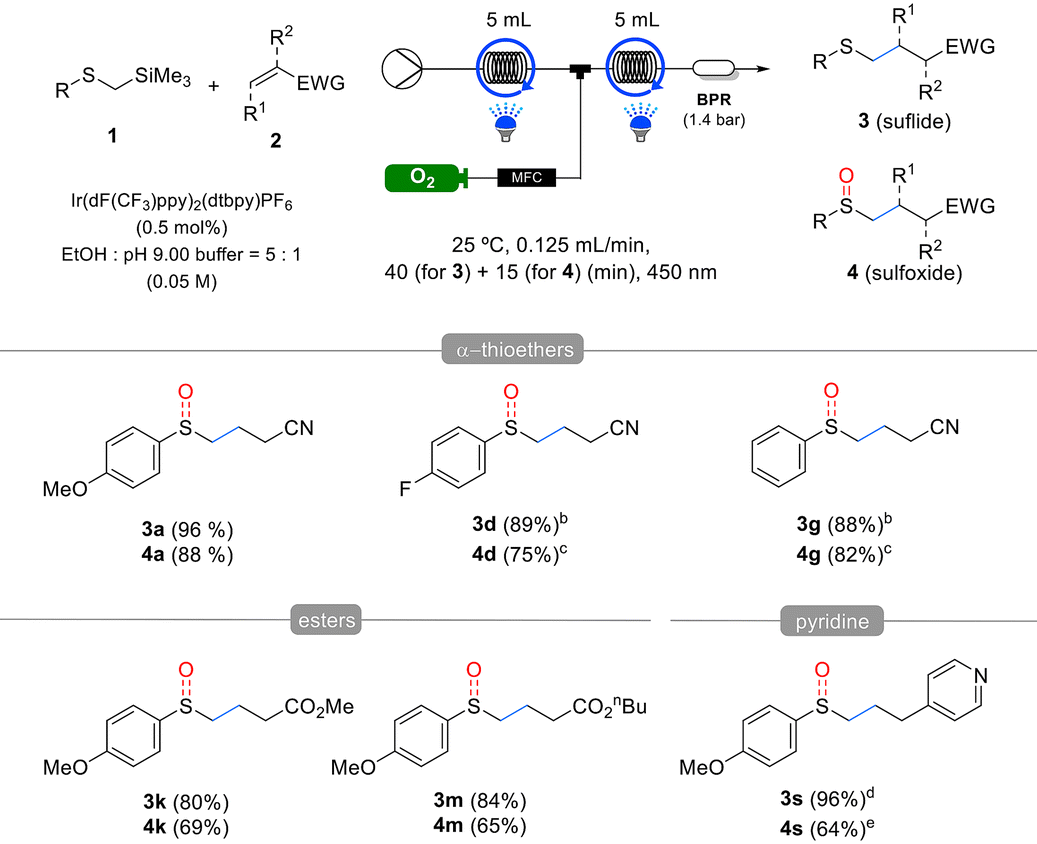
We propose a reaction mechanism based on controlled experiments and previous studies8a,b,9a,c,d,11a (Scheme 3). The excited-state iridium photocatalyst can oxidize 1a via single-electron transfer (SET) to form the cationic radical 1a˙+, which then undergoes desilylation to produce the α-thiomethyl radical I. This step was validated by luminescence quenching and TEMPO-mediated radical trapping experiments (see details in the ESI,† Scheme 3a). Furthermore, we measured the oxidation potential of 1a, which was found to be +0.98 V vs. SCE in MeCN, indicating that it can be oxidized by the excited-state Ir(dF(CF3)ppy)2(dtbpy) photocatalyst (Ir(III)*/Ir(II) = +1.21 V vs. SCE). The generated α-thiomethyl radical I adds to acrylonitrile 2a, forming the radical intermediate II, which is then reduced by the Ir(II) photocatalyst (Ir(III)/Ir(II) = −1.37 V vs. SCE) to produce the anionic intermediate III. Subsequently, this intermediate is protonated by the solvent to yield the sulfide product 3a. The protonation process was confirmed using methanol-D as the solvent; 100% of the deuterium was incorporated, indicating that the proton originated from the solvent (Scheme 3b). We additionally measured the quantum yields for sulfide synthesis step in both batch and flow reactions. In both cases, the quantum yields were greater than 1, with the batch reaction (Φ ≈ 3) and the flow reaction (Φ > 7). While the chain propagation pathway within the mechanism remains unclear, its involvement cannot be definitively excluded (see details in the ESI†). The oxidation of sulfide 3a to sulfoxide 4a can occur under photocatalytic conditions in the presence of oxygen. In this process, oxygen can facilitate oxidation either through the generation of a superoxide anion via SET, or through the formation of singlet oxygen via energy transfer (EnT). The possible oxidation pathways were validated through controlled experiments (Scheme 3d). In the SET pathway, the oxidation of sulfide 3a and the generation of superoxide are essential. This process is feasible based on the redox potentials of the photocatalyst, sulfide (3a, Eox = +1.13 V vs. SCE in MeCN), and oxygen. The use of a sulfide oxidation inhibitor, 1,4-dimethoxybenzene,9c and a superoxide quencher, benzoquinone,18 resulted in significant suppression or complete cessation of the oxidation process (Scheme 3d, entries 2 and 3). These results indicate that the SET pathway is a suitable route for oxidation. Generation of singlet oxygen is essential for the EnT pathway. Ir(dF(CF3)ppy)2(dtbpy) is a well-known, effective EnT photocatalyst for converting triplet oxygen into singlet oxygen.19 We conducted controlled experiments to quench the generated singlet oxygen using DABCO20 and NaN3,11b which resulted in significant suppression or complete cessation of the oxidation process (Scheme 3d, entries 4 and 5). Based on these results, we propose that sulfide oxidation proceeds via both SET and EnT pathways. Lastly, the potential byproduct PMPSCH3 1a-I did not form any sulfide 3a product under the standard reaction conditions (Scheme 3e). This confirms that the key intermediate, α-thiomethyl radical I, is generated from 1a rather than 1a-I in the Giese addition reaction.
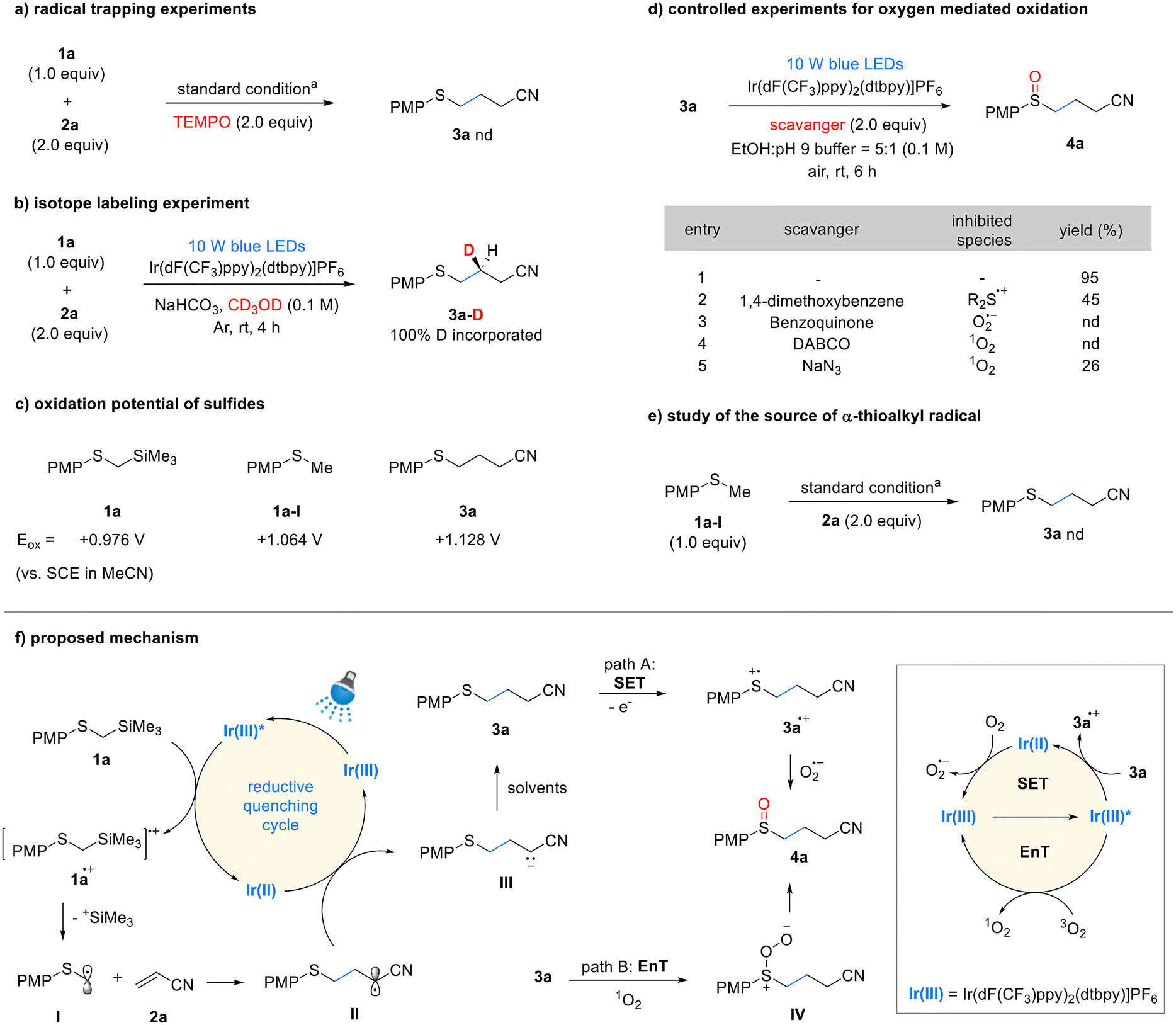 | ||
| Scheme 3 Controlled experiments and proposed mechanism. aStandard conditions: 1a (0.2 mmol), 2a (2.0 equiv.), Ir(dF(CF3)ppy)2(dtbpy)PF6 (0.5 mol%), EtOH:pH = 9 buffer (5:1, 0.1 M), Ar, room temperature, 4 h. See ESI† for details. | ||
To enhance the efficiency of the developed reaction, we conducted experiments on the reuse and recovery of reactants, solvents, and catalysts (Scheme 4). To evaluate the reusability, a reaction system was designed as shown in Scheme 4a. After one reaction cycle, 1 equivalent each of 1a and 2a was added, yielding 3a with a slightly reduced yield of 84% compared to the original reaction (n = 1). An additional 0.5 mol% of catalyst was introduced after every two reaction cycles, resulting in a yield of 79% after four cycles (n = 3) and 89% after six cycles (n = 5). Furthermore, after four cycles (n = 3), oxidation under aerobic conditions yielded sulfoxide 4a in a high yield of 82%. Next, alkene recovery experiments were conducted. Benzylidenemalononitrile (BMN, 2q) was recovered in high yields after the synthesis of sulfides and sulfoxides (Scheme 4b). These experiments established an efficient process for the reuse of reagents and solvent, thereby validating the sustainability of the developed reaction system.
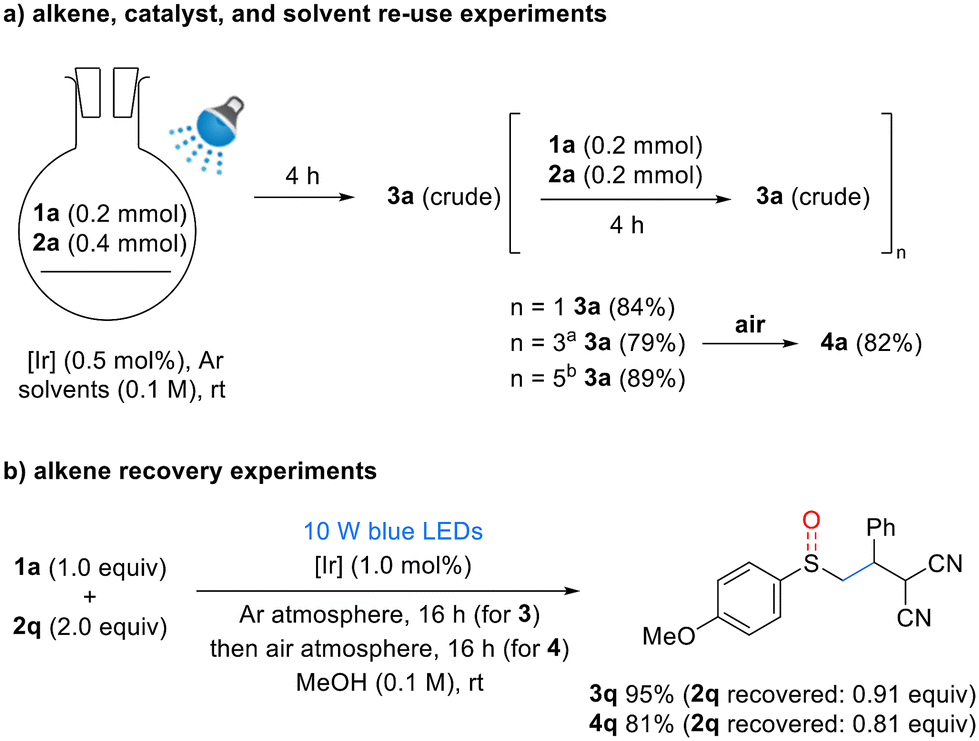 | ||
| Scheme 4 Reusability and recovery of reaction components aAfter n = 1, 0.5 mol% of [Ir] was added. bAfter n = 1 and n = 3, 0.5 mol% of [Ir] was added each time. [Ir] = Ir(dF(CF3)ppy)2(dtbpy)PF6 (0.5 mol%), solvents = EtOH:pH = 9 buffer (5:1). See ESI† for details. | ||
To evaluate the green chemistry aspects of the developed reaction, we calculated key green chemistry metrics, including Atom Economy (AE), Atom Efficiency (AEf), Reaction Mass Efficiency (RME), E-factor, and Process Mass Intensity (PMI) (Table 6).21 The metrics showed notably high values, with AE (61%), AEf (58%), and RME (64%), compared to previously reported methods.4c,11a However, due to the use of 0.1 M solvent concentration, the solvent-inclusive metrics, E-factor (31.6) and PMI (32.3), were slightly higher than those in prior studies. By employing the developed alkene, catalyst, and solvent re-use strategy, we significantly improved the metrics, achieving an E-factor of 9.6 and a PMI of 10.6 (see details in the ESI†).
Conclusions
In summary, we have developed an environmentally friendly and scalable synthetic method for both sulfides and sulfoxides through photocatalyzed, controllable one-pot C–C bond formation and oxidation. The optimized reaction conditions, utilizing mild photoredox conditions, green solvents, and oxygen as the oxidant, provided a sustainable approach to C–C bond formation and oxidation. Moreover, the integration of a microreactor-based flow system enables the scalable application of photocatalytic and liquid–gas phase reactions. Mechanistic studies revealed that C–C bond formation proceeds via an α-thiomethyl radical generated through a SET pathway, while the oxidation step involves both SET and EnT mechanisms. This methodology, with its use of green solvents and minimal catalyst loading, offers a promising solution for sustainable, large-scale sulfide and sulfoxide production, and has significant potential for broader applications in synthetic organic chemistry.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (NRF-2022R1A2C1005108, and 2021R1A4A3022415).References
- (a) P. Devendar and G.-F. Yang, Top. Curr. Chem., 2017, 375, 82 CrossRef; (b) K. A. Scott and J. T. Njardarson, Top. Curr. Chem., 2018, 376, 5 CrossRef; (c) D. Kaiser, I. Klose, R. Oost, J. Neuhaus and N. Maulide, Chem. Rev., 2019, 119, 8701–8780 CrossRef CAS PubMed; (d) S. Han, Bull. Korean Chem. Soc., 2023, 44, 172–201 CrossRef CAS; (e) Y. Kim, S. Y. Kim and S.-G. Kim, Bull. Korean Chem. Soc., 2023, 44, 619–628 CrossRef CAS; (f) S. Na and A. Lee, Bull. Korean Chem. Soc., 2023, 44, 921–925 CrossRef CAS; (g) A. S. Anderton, O. J. Knowles, J. A. Rossi-Ashton and D. J. Procter, ACS Catal., 2024, 14, 2395–2401 CrossRef CAS.
- (a) C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS; (b) A. K. Sinha and D. Equbal, Asian J. Org. Chem., 2019, 8, 32–47 CrossRef CAS; (c) F. Abedinifar, S. Bahadorikhalili, B. Larijani, M. Mahdavi and F. Verpoort, Appl. Organomet. Chem., 2022, 36, e6482 CrossRef CAS.
- (a) S. Choi, J.-D. Yang, M. Ji, H. Choi, M. Kee, K.-H. Ahn, S.-H. Byeon, W. Baik and S. Koo, J. Org. Chem., 2001, 66, 8192–8198 CrossRef CAS PubMed; (b) K. Kaczorowska, Z. Kolarska, K. Mitka and P. Kowalski, Tetrahedron, 2005, 61, 8315–8327 CrossRef CAS; (c) B. Karimi, M. Ghoreishi-Nezhad and J. H. Clark, Org. Lett., 2005, 7, 625–628 CrossRef CAS PubMed; (d) C. O. Kinen, L. I. Rossi and R. H. de Rossi, Green Chem., 2009, 11, 223–228 RSC; (e) K. A. Stingl and S. B. Tsogoeva, Tetrahedron: Asymmetry, 2010, 21, 1055–1074 CrossRef CAS.
- (a) P. Pitchen, C. J. France, I. M. McFarlane, C. G. Newton and D. M. Thompson, Tetrahedron Lett., 1994, 35, 485–488 CrossRef CAS; (b) G. Solladie, F. Colobert and F. Somny, Tetrahedron Lett., 1999, 40, 1227–1228 CrossRef CAS; (c) T. Jia, A. Bellomo, K. E. L. Baina, S. D. Dreher and P. J. Walsh, J. Am. Chem. Soc., 2013, 135, 3740–3743 CrossRef CAS PubMed; (d) G. Ludwig, T. Rüffer, A. Hoppe, T. Walther, H. Lang, S. G. Ebbinghaus and D. Steinborn, Dalton Trans., 2015, 44, 5323–5330 RSC.
- (a) J. Choi, P. Martín-Gago and G. C. Fu, J. Am. Chem. Soc., 2014, 136, 12161–12165 CrossRef CAS PubMed; (b) T. Knauber and J. Tucker, J. Org. Chem., 2016, 81, 5636–5648 CrossRef CAS.
- (a) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC; (b) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS; (c) S. Fukuzumi and K. Ohkubo, Org. Biomol. Chem., 2014, 12, 6059–6071 RSC; (d) R. A. Angnes, Z. Li, C. R. D. Correia and G. B. Hammond, Org. Biomol. Chem., 2015, 13, 9152–9167 RSC; (e) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS; (f) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed; (g) S. Crespi and M. Fagnoni, Chem. Rev., 2020, 120, 9790–9833 CrossRef CAS PubMed; (h) J. D. Bell and J. A. Murphy, Chem. Soc. Rev., 2021, 50, 9540–9685 RSC; (i) S. R. Atriardi and S. K. Woo, Aldrichimica Acta, 2023, 56, 25–33 Search PubMed; (j) A. T. Nguyen, H. Kang, T. G. Luu, S.-E. Suh and H.-K. Kim, Bull. Korean Chem. Soc., 2024, 45, 738–758 CrossRef CAS.
- (a) E. L. Tyson, M. S. Ament and T. P. Yoon, J. Org. Chem., 2013, 78, 2046–2050 CrossRef CAS PubMed; (b) M. H. Keylor, J. E. Park, C.-J. Wallentin and C. R. J. Stephenson, Tetrahedron, 2014, 70, 4264–4269 CrossRef CAS; (c) E. L. Tyson, Z. L. Niemeyer and T. P. Yoon, J. Org. Chem., 2014, 79, 1427–1436 CrossRef CAS PubMed.
- (a) Y. Li, K. Miyazawa, T. Koike and M. Akita, Org. Chem. Front., 2015, 2, 319–323 RSC; (b) E. Alfonzo and S. M. Hande, ACS Catal., 2020, 10, 12590–12595 CrossRef CAS; (c) E. Alfonzo and S. M. Hande, Org. Lett., 2021, 23, 6115–6120 CrossRef CAS PubMed; (d) O. J. Knowles, L. O. Johannissen, G. E. M. Crisenza, S. Hay, D. Leys and D. J. Procter, Angew. Chem., Int. Ed., 2022, 61, e202212158 CrossRef CAS PubMed; (e) M. J. Rourke, C. T. Wang, C. R. Schull and K. A. Scheidt, ACS Catal., 2023, 13, 7987–7994 CrossRef CAS PubMed.
- (a) C. Ye, Y. Zhang, A. Ding, Y. Hu and H. Guo, Sci. Rep., 2018, 8, 2205 CrossRef; (b) Y. Li, S. A.-e.-A. Rizvi, D. Hu, D. Sun, A. Gao, Y. Zhou, J. Li and X. Jiang, Angew. Chem., Int. Ed., 2019, 58, 13499–13506 CrossRef CAS PubMed; (c) C. Li, N. Mizuno, K. Murata, K. Ishii, T. Suenobu, K. Yamaguchi and K. Suzuki, Green Chem., 2020, 22, 3896–3905 RSC; (d) W. Zhao, C. Yang, J. Huang, X. Jin, Y. Deng, L. Wang, F. Su, H. Xie, P. K. Wong and L. Ye, Green Chem., 2020, 22, 4884–4889 RSC.
- (a) T. Liu, Y. Ding, X. Fan and J. Wu, Org. Chem. Front., 2018, 5, 3153–3157 RSC; (b) T. Liu, Y. Li, L. Lai, J. Cheng, J. Sun and J. Wu, Org. Lett., 2018, 20, 3605–3608 CrossRef CAS PubMed; (c) G. Qiu, L. Lai, J. Cheng and J. Wu, Chem. Commun., 2018, 54, 10405–10414 RSC.
- (a) Y. Li, M. Wang and X. Jiang, ACS Catal., 2017, 7, 7587–7592 CrossRef CAS; (b) D. H. Kim, J. Lee and A. Lee, Org. Lett., 2018, 20, 764–767 CrossRef CAS PubMed.
- S. Kusuda, Y. Ueno and T. Toru, Tetrahedron, 1994, 50, 1045–1062 CrossRef CAS.
- J.-L. Marco, I. Fernandez, N. Khiar, P. Fernandez and A. Romero, J. Org. Chem., 1995, 60, 6678–6679 CrossRef CAS.
- A. Nishida, M. Nishida and O. Yonemitsu, Tetrahedron Lett., 1990, 31, 7035–7038 CrossRef CAS.
- (a) N. Khatun, M. J. Kim and S. K. Woo, Org. Lett., 2018, 20, 6239–6243 CrossRef CAS PubMed; (b) A. Gontala and S. K. Woo, Adv. Synth. Catal., 2020, 362, 3223–3228 CrossRef CAS; (c) S. B. Nam, N. Khatun, Y. W. Kang, B. Y. Park and S. K. Woo, Chem. Commun., 2020, 56, 2873–2876 RSC; (d) S. R. Atriardi, J.-Y. Kim, Y. Anita and S. K. Woo, Bull. Korean Chem. Soc., 2023, 44, 50–54 CrossRef CAS; (e) A. Gontala, H. Huh and S. K. Woo, Org. Lett., 2023, 25, 21–26 CrossRef CAS PubMed; (f) Y. W. Kang, R. H. Kim, S. R. Atriardi and S. K. Woo, J. Org. Chem., 2023, 88, 3555–3566 CrossRef CAS PubMed.
- D. Prat, J. Hayler and A. Wells, Green Chem., 2014, 16, 4546–4551 RSC.
- (a) M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed; (b) C. A. Hone and C. O. Kappe, Top. Curr. Chem., 2018, 377, 2 CrossRef PubMed; (c) C. Sambiagio and T. Noël, Trends Chem., 2020, 2, 92–106 CrossRef CAS; (d) A. Sivo, V. Ruta, V. Granata, O. Savateev, M. A. Bajada and G. Vilé, ACS Sustainable Chem. Eng., 2023, 11, 5284–5292 CrossRef CAS PubMed.
- M. Hayyan, M. A. Hashim and I. M. AlNashef, Chem. Rev., 2016, 116, 3029–3085 CrossRef CAS PubMed.
- F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC.
- K.-J. Liu, J.-H. Deng, J. Yang, S.-F. Gong, Y.-W. Lin, J.-Y. He, Z. Cao and W.-M. He, Green Chem., 2020, 22, 433–438 RSC.
- N. Fantozzi, J.-N. Volle, A. Porcheddu, D. Virieux, F. García and E. Colacino, Chem. Soc. Rev., 2023, 52, 6680–6714 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc05769d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |