DOI:
10.1039/D4GC05781C
(Paper)
Green Chem., 2025, Advance Article
Process intensification of heterogeneous dehydrochlorination of β-chlorohydrin using phase transfer catalysts
Received
12th November 2024
, Accepted 29th January 2025
First published on 11th February 2025
Abstract
Terminal epoxides are important monomers and modifiers that are widely used in the polymer and coating industries. Apart from direct oxidation of alkenes to produce ethylene oxide or propylene oxide, many terminal epoxides are still synthesized through the chlorohydrin method. Due to the limited mass transfer performance and complex thermodynamics and kinetics in this heterogeneous reaction system, the dehydrochlorination reaction still suffers from low reaction efficiency, large amounts of solid precipitation waste, and highly excessive use of alkali. Herein, the dehydrochlorination process of 3-chloro-2-hydroxypropyl neodecanoate (CHPDA), selected as a typical β-chlorohydrin with extremely poor solubility in water, was intensified using phase transfer catalysts (PTCs). The structure–function relationship of PTCs was elucidated and it was demonstrated that tetrabutylammonium chloride (TBACl) is one of the most effective catalysts for heterogeneous dehydrochlorination. Due to the significant promotion of mass transfer by applying a catalytic amount of TBACl, the reaction time was shortened from several hours to just 3 min, and the usage of the reactant sodium hydroxide can be reduced from multiple equivalents to nearly stoichiometric with β-chlorohydrin, while the selectivities of the side products, neodecanoic acid (NDA) and diester, were only 0.4 and 1 mol%, respectively. Compared to existing works, this study could be very helpful for the development of continuous and green synthesis technology for epoxides.
Green foundation
1. Using a TBACl catalyst in a dehydrochlorination reaction has reduced the reaction time from hours to 3 min, decreased the excess NaOH amount from 0.8 equiv. to 0.05 equiv., and lowered the selectivity of diester.
2. A shorter reaction time reduces energy consumption, while a lower excess of NaOH decreases both costs and energy consumption for NaOH recycling. Lower selectivity of diester prevents EPDA from undergoing distillation to meet quality standards.
3. We can develop a continuous EPDA synthesis with a microreactor system, which can further increase the reaction efficiency through a higher mass transfer rate. Additionally, optimizing the overall process, such as aligning the temperatures of the ring-opening and dehydrochlorination reactions or adjusting the TBACl/CHPDA molar ratio, given that TBACl is recoverable, might further reduce energy consumption.
|
1 Introduction
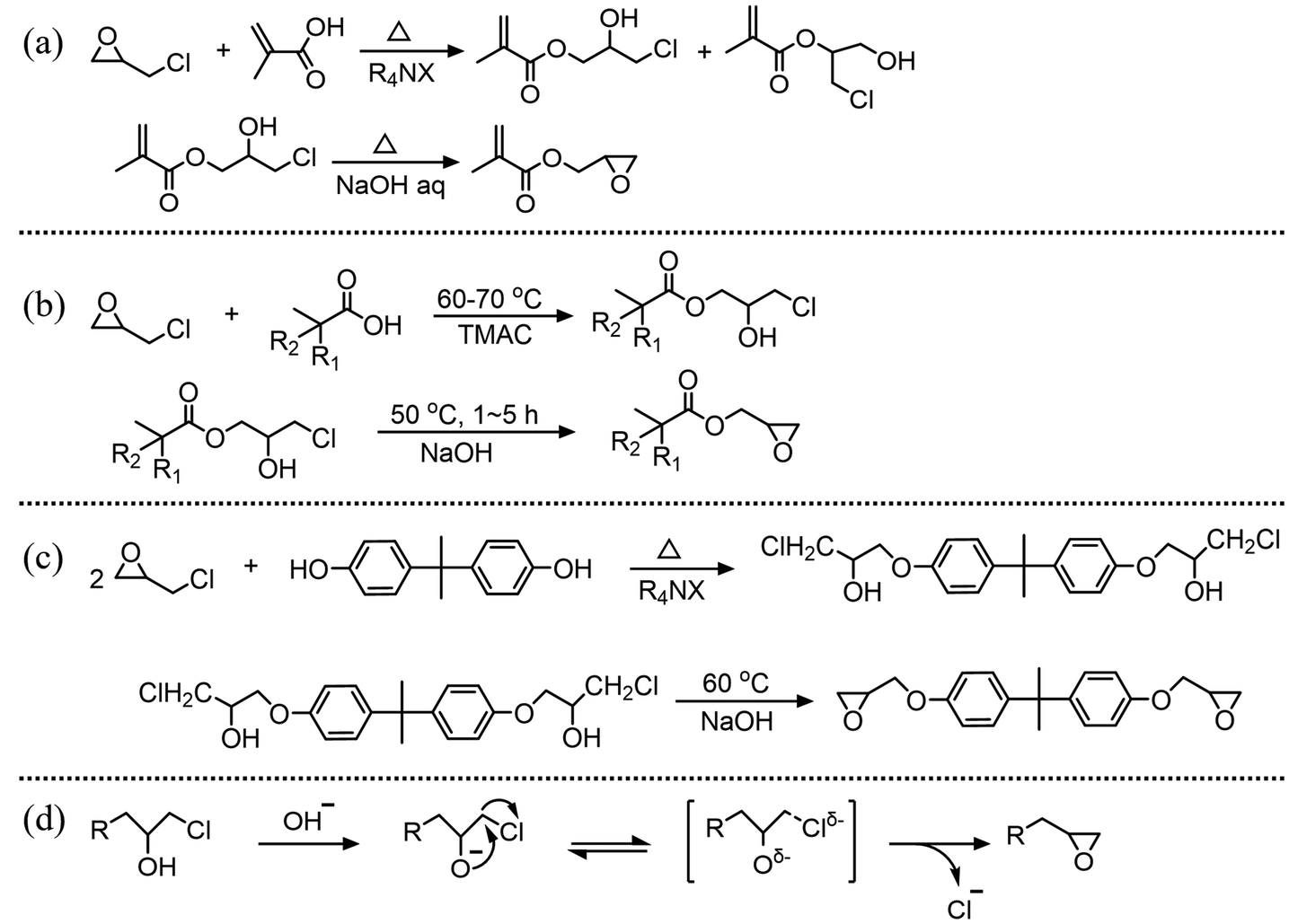
Terminal epoxides, which mainly include glycidyl esters and glycidyl ethers, are a class of important chemicals. They serve as building blocks for organic synthesis,1–3 such as monomers in the ring-opening polymerization to produce or modify epoxy resins4,5 and coatings.6,7 Additionally, they are widely used in the pharmaceutical community8–10 and environmental engineering.11,12 Terminal epoxides are usually prepared through olefin epoxidation reactions, such as epoxidation of ethylene oxide13 and propylene oxide,14 and the chlorohydrin method. The chlorohydrin method starts with the ring-opening reaction of epichlorohydrin (ECH) with the corresponding acids or phenols to produce β-chlorohydrins, followed by a dehydrochlorination process,15,16 as shown in Fig. 1(a)–(c). The common procedure for the dehydrochlorination process is that β-chlorohydrins are reacted with alkaline solutions, typically NaOH solution and lime slurry solution.17–19
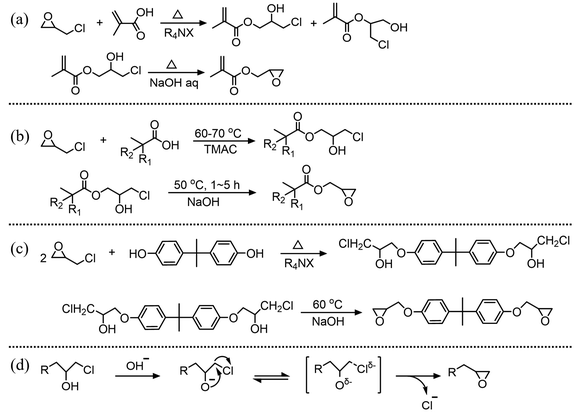 |
| | Fig. 1 Synthesis of terminal epoxides: (a) glycidyl methacrylate,20 (b) 2,3-epoxypropyl neodecanoate,21,22 and (c) diglycidyl ester of bisphenol A23 by a two step method, and (d) the dehydrochlorination mechanism of β-chlorohydrin. | |
The mechanism of the dehydrochlorination reaction is illustrated in Fig. 1(d). Initially, the hydroxyl group in β-chlorohydrin donates a proton to the base, forming an alkoxide ion (–O−). This alkoxide ion serves as an internal nucleophile, attacking the carbon with a good leaving group and subsequently converting β-chlorohydrin into a terminal epoxide.24,25 For smaller β-chlorohydrins, such as dichloropropanol24 or those with large hydrophobic side groups such as 3-chloro-2-hydroxypropyl neodecanoate (CHPDA),26 dehydrochlorination is an inherently rapid reaction, completing within 1 min in kinetic studies conducted under homogeneous conditions.
Unlike in kinetic studies,27 the dehydrochlorination of β-chlorohydrins in industry is generally conducted in a heterogeneous system. The reaction conditions of industrial dehydrochlorination reactions may be affected by the hydrolysis rate of the resulting epoxy products.28 For instance, the production of epichlorohydrin (ECH) from dichloropropanol (DCP) requires an accurate dosing of NaOH solution and is carried out in a reactive-distillation unit to limit the contact time to within one minute.29–32 In contrast, the dehydrochlorination of 3-chloro-2-hydroxypropyl methacrylate is typically conducted in a batch reactor, but requires prolonged feeding and reaction times as a highly concentrated NaOH solution (1.0–1.2 mol equivalents) is added intermittently over 3 hours to avoid localized high alkalinity.33,34 For glycidyl esters of branched monocarboxylic acids, such as CHPDA,21 the hydrolysis reaction is weak, and the interface mass transfer rate during the middle and later stages of the dehydrochlorination reaction is very limited due to the poor solubility of NaOH in the epoxy products.35 Despite using high alkali concentrations (40%), high molar ratios of NaOH (1.8 mol equivalents of NaOH), and elevated temperatures (50 °C), the reaction still requires several hours.36 The use of high alkali concentration leads to NaCl precipitation, which complicates both batch and continuous operations. It causes erosion of agitator parts in batch reactors37 and clogging in continuous tubular reactors.38 Besides, further processing is required for the precipitation, and the product might remain in the solid waste during the filtration. Excess alkali also requires additional recovery steps, such as membrane electrolysis.29 The resulting saturated alkali brine prolongs phase separation and increases alkali entrainment in the product, leading to further hydrolysis of ester products during post-treatment. Thus, it is essential to explore strategies to overcome these shortcomings.
Compared to the preceding ring-opening reaction, research on the process intensification of the dehydrochlorination reaction of β-chlorohydrins, excluding DCP, has received less attention, as it is believed that the ring-opening reaction is more critical to the yield and quality of the final product.33 However, the dehydrochlorination reaction is one of the most crucial steps in determining the overall reaction efficiency. The preliminary effort of our group in the dehydrochlorination of CHPDA has improved the reaction performance, with a focus on enhancing the mass transfer of NaOH from the aqueous phase to the organic phase. For instance, a microtube reactor system combined with a highly concentrated alkali solution and 4 molar equivalents of the alkali has been established.26 Alternatively, we used n-propanol as a co-extractant after realizing that the EPDA produced cannot extract NaOH from the water phase.35 Although the reaction efficiency has been elevated in these works, the use of excess alkali and the precipitation of NaCl are still unavoidable if we aim to complete the reaction within minutes.
Phase transfer catalysis is one of the most important methods in organic chemistry.39 It can increase the rate and selectivity of reactions, facilitate the interphase mass transfer between immiscible reactants, and prevent the usage of polar aprotic solvents.40 Moreover, it is theoretically recyclable.41 The most common types of PTCs include quaternary ammonium salts, phosphonium amphiphilic salts, and crown ethers.32 Among them, quaternary ammonium salts are always used in ring-opening reactions.36 However, to the best of our knowledge, there has been no systematic research on the effect of catalysts in the dehydrochlorination of β-chlorohydrins. In this work, based on the major factor that glycidyl esters can hardly extract alkali from the aqueous phase, we focus on the effect of PTCs on the dehydrochlorination reaction of CHPDA to produce 2,3-epoxypropyl neodecanoate (EPDA) as the specific example of the dehydrochlorination reaction of β-chlorohydrin. The reaction mechanism based on PTCs has been presented, and the effect of the structure of PTCs has been studied, subsequently determining the optimal catalyst. Additionally, the reaction conditions for the PTC-assisted dehydrochlorination reaction have been optimized. Finally, a comparison of this work with the literature has been conducted from the perspective of green chemistry.
2 Materials and methods
2.1 Chemicals
CHPDA was synthesized via the acidolysis ring-opening reaction of epichlorohydrin with neodecanoic acid, as described in our previous work,42 and the purity was between 90 and 93 wt%, characterized by gas chromatography. EPDA was purchased from Guangdong Weng Jiang Reagent, and its epoxy group content was 4195 mmol kg−1 (corresponding to 95.8 wt% purity). KH2PO4 was bought from Sinopharm Chemical Reagent Co., Ltd and classified as analytical grade. All other reagents were purchased from Aladdin Chemistry Co., Ltd and classified as analytical grade.
2.2 Experimental procedures
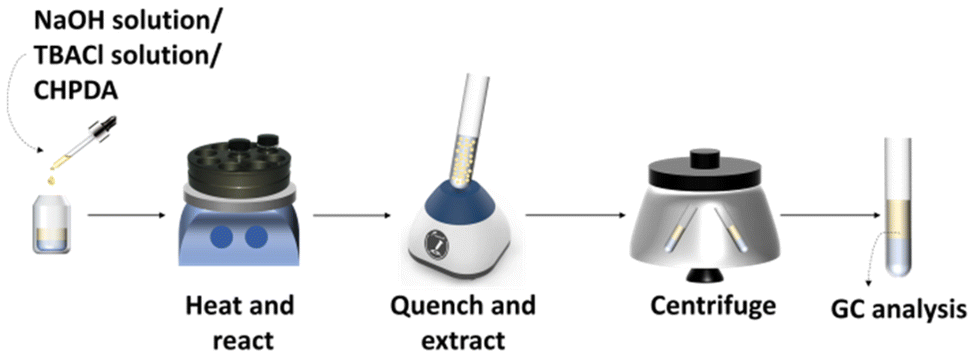
The heterogeneous dehydrochlorination reaction was performed in a 4 mL glass vial sealed with a plastic cap. A heating platform (IKA, RCT basic) and a stainless-steel module were applied to maintain the reaction temperature within an error of ±3 °C, as shown in Fig. 2. The speed of magnetic stirring could be adjusted in the range of 0–1500 rpm. Before the reaction, the NaOH solution, the TBACl solution and CHPDA were sequentially added into the glass vial, which was then placed into the preheated stainless-steel module in the magnetic heating stirrer for the reaction. The mass fraction of TBACl solution added was always identical to that of NaOH solution. An excess amount of phosphate buffer solution (KH2PO4 and K2HPO4 dissolved in water, pH = 7.0) was used to quench the reaction by neutralizing hydroxide ions, while n-octane was used to extract the dissolved and dispersed organic substances into the oil phase. Both the buffer solution and n-octanol were pre-added to the sample vessel and the reaction mixture from the glass vial was quickly poured into the sample vessel and shaken for 3 min in a vortex mixer (JOANLAB, VM-300S) to quench the reaction and extract the organic compounds. After 2 min of phase separation in a centrifuge at 5000 rpm, the oil phase was analyzed by gas chromatography to determine the concentrations of CHPDA and EPDA and the testing conditions are listed in Table 1.
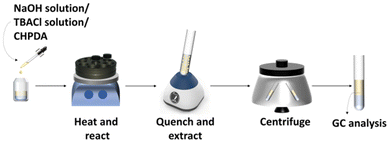 |
| | Fig. 2 Schematic of the experimental procedure and equipment. | |
Table 1 GC testing conditions
| Device |
Agilent GC-7890A |
Injection temperature |
300 °C |
| Detector |
FID |
Dejection temperature |
300 °C |
| Carrier gas |
N2, 1 mL min−1 |
Sample |
1 μL |
| Split ratio |
30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
Quantitative method |
Internal standard |
| Column |
HP-5/30 m × 0.32 mm × 0.25 μm |
Internal standard substance |
Methyl benzoate |
| Column temperature |
Start at 50 °C, maintain for 1 min; 10 °C min−1 to 160 °C; 5 °C min−1 to 200 °C; 10 °C min−1 to 300 °C, maintain for 1 min |
|
|
3 Results and discussion
3.1 Conventional dehydrochlorination of CHPDA
The conversion of CHPDA, along with the mass fractions of neodecanoic acid (NDA) and diester during the two-step alkali treatment process, is shown in Fig. 3(a) and (b). It is clear that several hours of reaction time are required under the conventional conditions. The selectivity of NDA increases from 0.17 mol% to 0.6 mol%, demonstrating the ongoing hydrolysis of CHPDA (eqn (1)) and EPDA (eqn (2)) in the highly alkaline reaction system.26,43 The side reaction accompanying the hydrolysis reaction is the acidolysis ring-opening of EPDA, with the neodecanoate generated by the hydrolysis of ester bonds (eqn (3)).42,43 In the industrial process, due to non-ideal behavior during phase separation and the saturated concentration of the aqueous phase, the entrainment of alkali brine is far more severe than that under laboratory conditions. This results in a higher incidence of side reactions during the post-treatment processes.
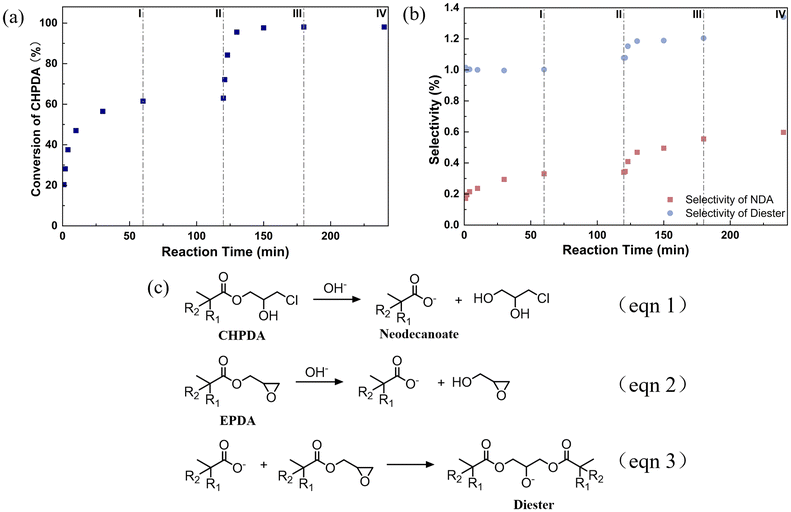 |
| | Fig. 3 (a) Conversion of CHPDA and (b) mass fraction of NDA and diester during the two-stage cross-flow dehydrochlorination reaction of CHPDA, where each stage involves a reaction, followed by a settling phase separation, corresponding to steps I, II, III, and IV. All these steps are conducted at 50 °C with an initial NaOH solution concentration of 40 wt%. The molar ratios for the two stages of the dehydrochlorination reaction are n(NaOH)/n(CHPDA) = 0.8 and 1, respectively. Note that the molar ratio for the second stage is based on the initial amount of CHPDA in the first stage. (c) Side reactions in the dehydrochlorination of CHPDA, including the hydrolysis of CHPDA (eqn (1)) and EPDA (eqn (2)), as well as the ring-opening reaction of NDA and EPDA to form diester (eqn (3)). | |
In our experiment, the selectivity of side products (neodecanoate and diester) resulting from hydrolysis is lower than 1 mol%. Thus, the side reactions can be neglected, and the conversion of CHPDA becomes the key parameter used to evaluate the reaction performance in the subsequent work. It is important to note that when the selectivity of diester exceeds 1.7 mol% (equivalent to 3 wt% in EPDA), distillation is required to separate diester from EPDA to meet quality standards. Since diester is a heavy component, this distillation process is likely to consume a great amount of energy, as it requires the distillation of the entire EPDA component.44
3.2 Reaction mechanism
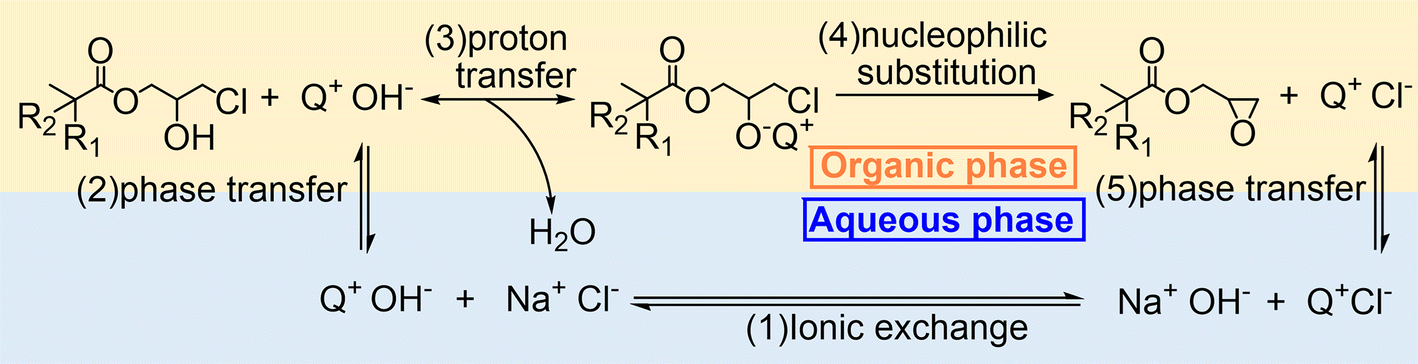
As mentioned in the Introduction, the reaction rate of the heterogeneous dehydrochlorination of β-chlorohydrin is primarily limited by the interphase mass transfer of NaOH.35 Therefore, it is natural to consider the influence of PTCs in this reaction, which can theoretically facilitate the transfer of the base from the aqueous phase to the oil phase. By determining the partition coefficient of OH− in varying doses of TBACl (Table 2) as the specific PTC, it was demonstrated that the increase in the partition coefficient is directly proportional to the amount of TBACl, indicating its ability to transport OH− into the oil phase. On the other hand, the logK value of OH− in n-octanol and EPDA presents that without a PTC, OH− can migrate easily to the oil phase at the initial stage, but it barely moves to the oil phase at the final stage. In contrast, the logK value of OH− in EPDA with TBACl is comparable to that of OH− in n-octanol without a catalyst, suggesting that the catalyst significantly assists in the transfer of OH− at the final stage of the reaction. Hence, we introduce the mechanism of the dehydrochlorination reaction of β-chlorohydrin with PTCs, as illustrated in Fig. 4.
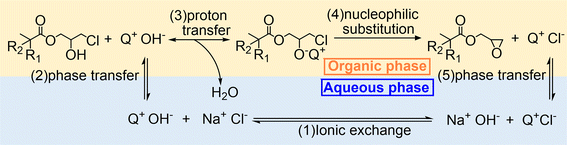 |
| | Fig. 4 Mechanism of the dehydrochlorination reaction of β-chlorohydrin using Q+Cl− (starting from (1)) or Q+OH− (starting from (2)) as a phase transfer catalyst. | |
Table 2 Variation of the partition coefficient of OH− (logK = log([OH−]org/[OH−]aq)) with different amounts of TBACl
| TBACl/NaOH |
LogK of OH− |
| LogK of OH− in the n-octanol/NaOH/TBACl/water system. LogK of OH− in the EPDA/NaOH/TBACl/water system. The logK values are measured at ambient temperature, and the mass fraction of water in the aqueous solution is 90 wt%. |
| 0a |
−2.6 |
| 0.025a |
−1.7 |
| 0.05a |
−1.5 |
| 0.1a |
−1.2 |
| 0b |
— |
| 0.05b |
−2.3 |
In the mechanism scheme, the PTC is denoted as Q+Cl−. Initially, Q+Cl− undergoes an ionic exchange with NaOH, converting to Q+OH−, which has a higher partition coefficient than NaOH and can transfer into the organic phase more readily. Once Q+OH− enters the organic phase, a proton transfer between CHPDA and Q+OH− takes place immediately, followed by an intramolecular nucleophilic substitution of the intermediate, leading to the formation of EPDA and the side product Q+Cl−. The Q+Cl then returns to the water phase and undergoes ionic exchange with NaOH, thus reactivating the catalytic cycle. If Q+OH− is used as the catalyst, the reaction cycle begins with its phase transfer from the aqueous phase to the organic phase.
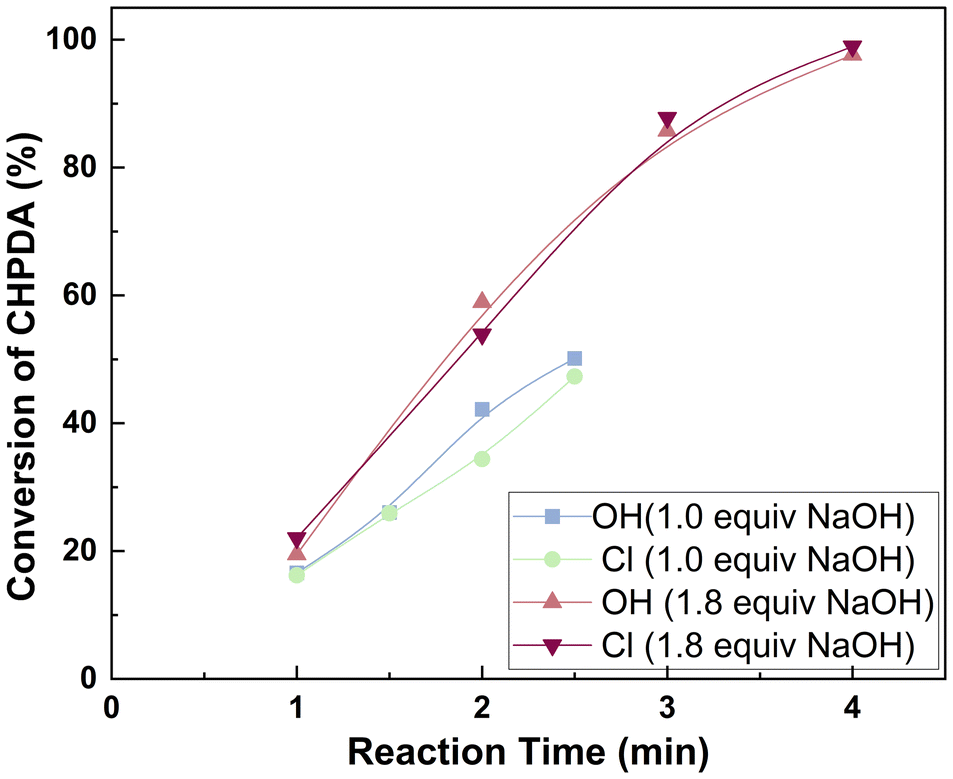
3.3 The selection of the catalyst
The effect of PTCs is related to their anion, cation, and molar ratio.45 Among these, the anion and cation of PTCs are particularly related to their activity, making their investigation essential for selecting the most suitable catalyst. In this work, we focus on quaternary ammonium chlorides and quaternary ammonium hydroxides as potential catalysts because they are among the most widely used PTCs and have been reported to act as catalysts in ring-opening reactions for the synthesis of CHPDA.21,36
3.4 Optimization of the process conditions for the PTC-based dehydrochlorination reaction of CHPDA
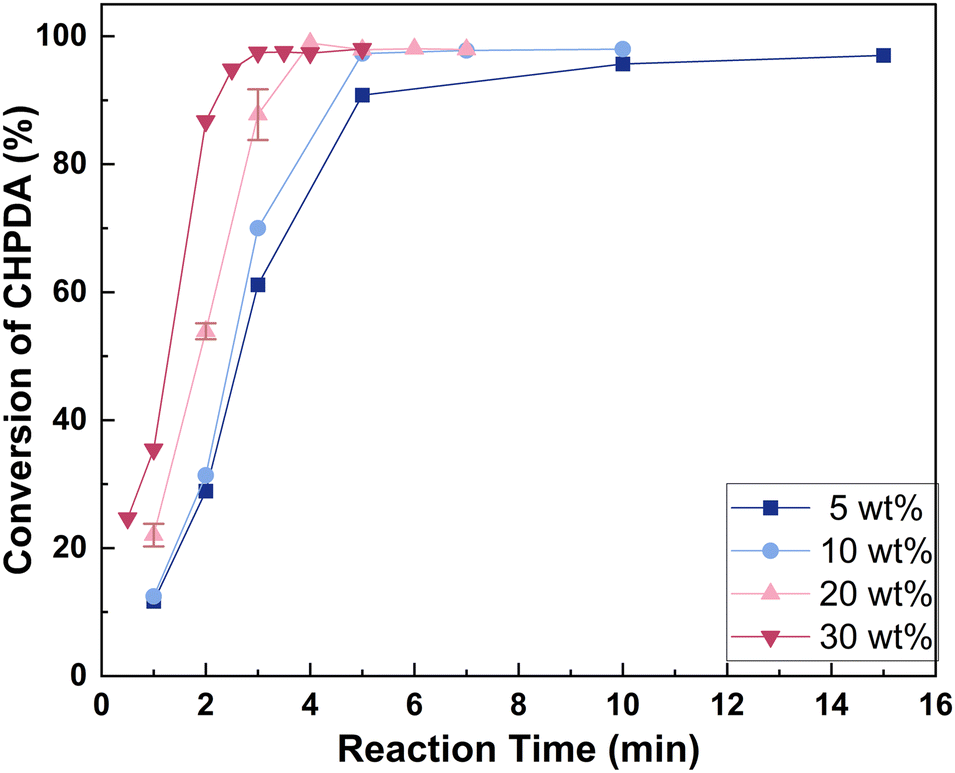
3.4.1 Effect of alkali concentration. Fig. 7 shows the effect of reactant NaOH concentration on the reaction rate within the range of 5 wt% to 30 wt%. With the assistance of TBACl, the conversion of CHPDA exceeds 95% within 15 minutes even at a base concentration as low as 5 wt%, which is significantly lower than that typically used in industrial reaction processes. Higher NaOH concentrations enhance the reaction rate by increasing the driving force for the OH− mass transfer from the water phase to the oil phase. Consequently, a high concentration of alkali is preferred for a shorter reaction time. However, a high concentration of alkali will cause NaCl precipitation, longer phase separation time, and entrainment of the alkali brine in the oil phase. When using a 30 wt% alkali concentration, the by-product NaCl may precipitate at lower temperatures and with a reduced amount of base. Therefore, an alkali concentration range of 10–20 wt% is more appropriate, and the reaction can be completed within 5 min. This concentration is reduced by 50–75% compared to the industrial conditions and the precipitation of NaCl is prevented. It is believed to be not only suitable for continuous operation and capable of avoiding the treatment of solid waste because no NaCl precipitates, but also effective in reducing the side products in the post-treatment due to a shorter settling time and less entrainment in the oil phase.
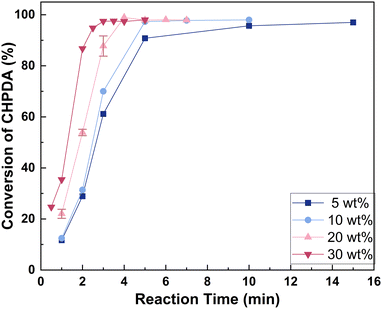 |
| | Fig. 7 Effect of alkali concentrations on the dehydrochlorination rate. Reaction conditions: n(TBACl):n(CHPDA):n(NaOH) = 0.05:1:1.8 and T = 50 °C. The concentrations of the TBACl and NaOH solutions are 20 wt%. | |
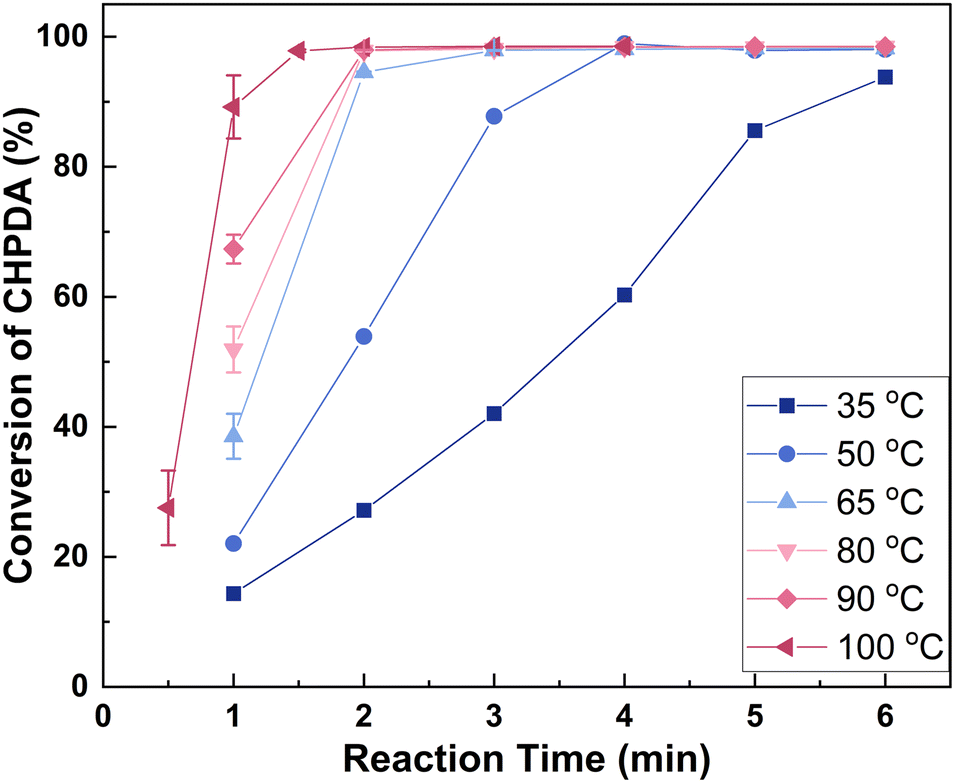
3.4.2 Effect of reaction temperature. Fig. 8 shows the effect of reaction temperature on the reaction rate within a range of 35 to 100 °C. The upper limit of 100 °C was set to prevent the alkali solution from boiling, which could affect the alkali concentration and lead to NaCl precipitation. Within this range, the reaction rate consistently increases with rising temperature. This increase is likely due not only to enhanced reaction kinetics but also to the elevated temperature increasing the mutual solubility of the two phases, reducing interfacial tension, and thereby facilitating interphase mass transfer.26 As mentioned in section 3.1, the reaction exhibits high selectivity, so a higher reaction rate is desirable. Consequently, the optimal temperature range is 50–100 °C, within which the reaction can be completed in 1.5–4 min. The temperature selected for the subsequent experiments is 100 °C. It is worth noting that the reaction temperature affects not only the reaction rate, but also the energy consumption and energy efficiency of industrial processes. Therefore, in addition to reaction time, the choice of reaction temperature should consider the temperatures of the preceding ring-opening reaction and the subsequent washing process.
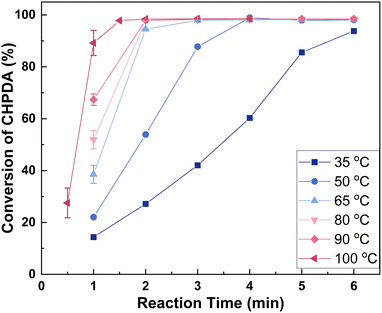 |
| | Fig. 8 Effect of reaction temperature on the reaction rate. Reaction conditions: n(TBACl):n(CHPDA):n(NaOH) = 0.05:1:1.8. The concentrations of TBACl and NaOH solutions are 20 wt%. | |
3.4.3 Effect of the molar ratio of n(NaOH)/n(CHPDA). The dehydrochlorination reaction without PTCs requires an excess of NaOH to maintain a high alkali concentration during the later stages of the reaction to reduce the overall reaction time. However, the use of excess NaOH causes entrainment in the product and necessitates subsequent treatment for recovery. Hence, the excess NaOH should be minimized, provided that the reaction rate remains acceptable, which is within 5 minutes in this work. Based on Table 4, when the n(NaOH)/n(CHPDA) molar ratio is within the range of 1.0–1.3, the reaction rate increases with a higher molar ratio. Beyond this range, the change in the reaction rate with respect to the molar ratio becomes minimal. Besides, when the molar ratio is precisely 1.0, the conversion of CHPDA cannot reach 97% because the dehydrochlorination reaction also occurs in 1,3-DCP impurities in CHPDA, leading to the formation of ECH. As a consequence, if the reaction termination condition is a conversion of 97%, a slightly excess amount of NaOH is inevitable and it is preferable for n(NaOH)/n(CHPDA) to be within 1.05–1.1 to complete the reaction within 2–3 min. On the other hand, if a 95% conversion is designated as the end point of the reaction, the optimal n(NaOH)/n(CHPDA) range is 1.0–1.1 and the reaction time is 2–5 min. Notably, the recommended NaOH dosage of 1.0–1.1 mol equivalents is much lower than the 1.8 mol equivalents used in industry. This reduction not only minimizes the cost of recovering NaOH, but also decreases the entrainment of NaOH in the oil phase.
Table 4 Effect of n(NaOH)/n(CHPDA) on the reaction. Reaction conditions: n(TBACl):n(CHPDA) = 0.05:1 and T = 100 °C. The concentrations of TBACl and NaOH solutions are 20 wt%
| n(NaOH):n(CHPDA) |
Reaction time (min) |
Conversion (%) |
Reaction time (min) |
Conversion (%) |
| 1.0 |
1 |
56.7 |
5 |
95.2 |
| 1.05 |
1 |
61.7 |
3 |
97.4 |
| 1.1 |
1 |
61.3 |
2 |
97.3 |
| 1.3 |
1 |
87.2 |
1.5 |
97.6 |
| 1.8 |
1 |
88.2 |
1.5 |
97.8 |
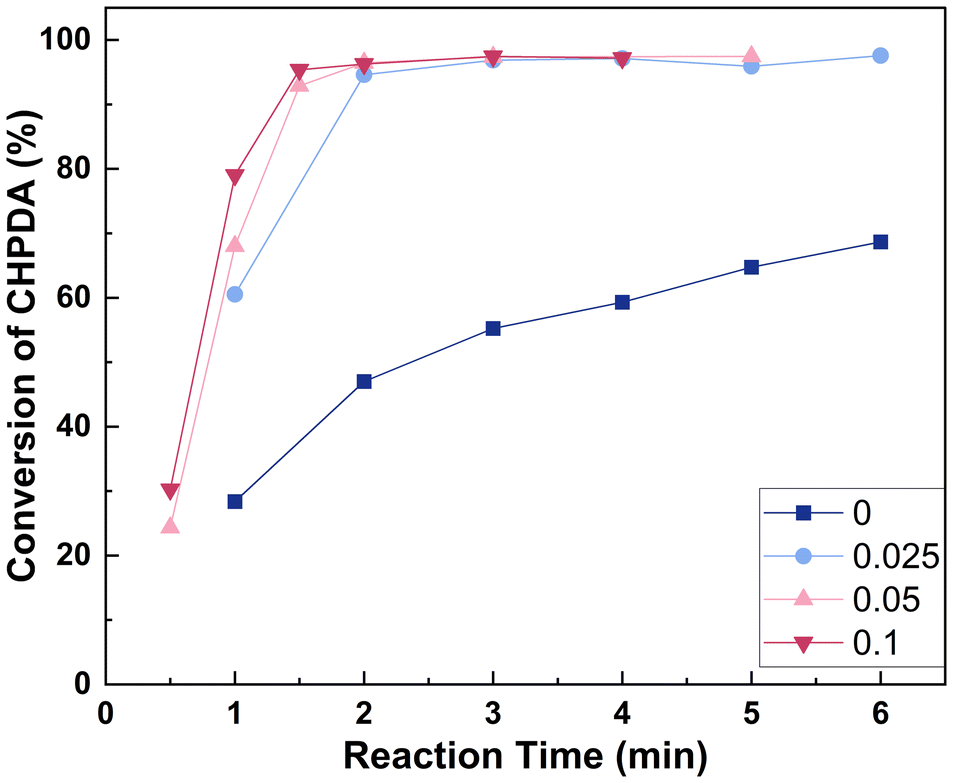
3.4.4 Effect of the molar ratio of n(TBACl)/n(CHPDA). The typical amount of PTC used ranges from 0.01 to 0.1 equivalent molar. This is because although the quaternary ammonium salt catalyst is recoverable and can be reused in the subsequent reactions,41,46,47 exceeding this range usually has a minimal effect on improving the reaction efficiency.45 The effect of n(TBACl)/n(CHPDA) within this range has been investigated, as shown in Fig. 9. It can be observed that within this range, the reaction rate significantly increases with higher catalyst dosage, because at lower NaOH concentrations, TBACl is the main method to transfer OH− to the oil phase. To ensure that the reaction is completed within 5 minutes, n(TBACl)/n(CHPDA) should be between 0.025 and 0.1. If the reaction is aimed to be completed within 3 min, n(TBACl)/n(CHPDA) should be in the range of 0.05–0.1.
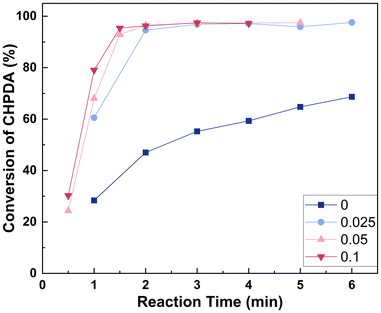 |
| | Fig. 9 Effect of n(TBACl)/n(CHPDA) on the reaction. Reaction conditions: n(NaOH):n(CHPDA) = 1.05:1 and T = 100 °C, and the concentrations of the TBACl and NaOH solutions are 20 wt%. | |
3.5 Green advances in dehydrochlorination with PTC
Table 5 lists the comparison of this work with 0.05–0.1 equiv. of TBACl to previous literature without PTC. The comparison between No. 1, 2 and 5 clearly shows the astonishing effect of PTC; as the reaction time is reduced by more than 95%, the excess amount of NaOH is reduced by over 92% and the concentration of NaOH is decreased by 50%. A shorter reaction time contributes to lower energy consumption and smaller reactor size, while a reduced excess of NaOH decreases the material consumption and energy consumption of NaOH recycling. A lower NaOH concentration prevents the filtration and post-treatment of NaCl solid waste, which may contain organic compounds such as product EPDA, further increasing the yield and making the process more feasible for continuous operation. However, compared to the reaction system without PTC, where the catalyst recovered from the system is always tetramethylammonium chloride (TMAC),46 more water may be required to wash the catalyst from the products, as the solubility of TBACl in the product is higher than that of TMAC. On the other hand, the composition of the product after a 2-hour reaction, followed by 5 days of settling, was analyzed and is listed in Table 6. It shows that much less extensive hydrolysis and subsequent ring-opening reactions would happen when the TBACl catalyst was used. This implies that the new reaction system and conditions employed in this study can result in fewer side products during post-treatment, due to the lower concentration of the alkali brine solution. As mentioned in section 3.1, lower selectivity for side products, especially diester, might eliminate the need for further distillation of EPDA to meet quality standards.
Table 5 Comparisons of this work with previous literature results without PTC
| No. |
Reactor types |
NaOH concentration (wt%) |
Molar ratio n(NaOH):n(CHPDA) |
Temperature (°C) |
Reaction time (min) |
Conversion (%) |
| 10 wt% n-propanol was added. n(TBACl):n(CHPDA) = 0.05–0.1. |
| 1 |
Batch21 |
40 |
1.65:1 |
70 |
60 |
95 |
| 2 |
Batch36 |
40 |
2:1 |
50 |
90 |
95.6 |
| 3 |
Microreactor26 |
40 |
4:1 |
50 |
2.7 |
93.8 |
| 4a |
Batch35 |
30 |
8:1 |
50 |
3 |
97.5 |
| 5b |
Batch (this work) |
20 |
1.05:1 |
50–100 |
3–7 |
>96 |
Table 6 Comparisons of side product selectivity with and without PTC after 5 days of settling
| No. |
NaOH concentration (wt%) |
Molar ratio n(NaOH):n(CHPDA) |
T (°C) |
Reaction time (min) |
Conversion (%) |
Selectivity of NDA (mol%) |
Selectivity of diester (mol%) |
| NaOH is added all at once. n(TBACl):n(CHPDA) = 0.05. |
| 1a |
40 |
1.8:1 |
50 |
120 |
99 |
0.64 |
2.3 |
| 2b |
20 |
1.05:1 |
50 |
5 |
98 |
0.59 |
1.6 |
To complete the reaction within 5 min, a large excess of NaOH is always used to make sure that the concentration of NaOH remains above 26 wt% throughout the reaction, even when using a microreactor (Table 5, No. 3), which can provide a higher specific surface area or a co-extractant (Table 5, No. 4), which can facilitate alkali transportation. However, microreactors can only enhance the mass transfer rates, but cannot violate the thermodynamic rules to transfer alkali when the driving force is extremely low. Meanwhile, a co-extractant can help in extraction of alkali, but it decreases the space-time yield of the reactor and requires an additional post-treatment process to separate alcohol from the product. Therefore, using PTCs is an effective process intensification method, as it can complete the reaction within 5 min without excessive NaOH, NaCl precipitation or the introduction of additional pre- or post-treatment procedures. This is because PTCs can greatly enhance mass transfer thermodynamically by raising the partition coefficient of OH− throughout the reaction, especially in the middle and final stages. Moreover, TBACl can catalyze both the ring-opening and dehydrochlorination processes.
4 Conclusion
In this work, we first investigated the effects of PTCs on dehydrochlorination of CHPDA as a specific reaction to produce glycidyl ethers, based on the fact that the reaction efficiency is remarkably constrained by the interphase mass transfer of alkali. By determining the partition coefficients of catalysts, we proposed the reaction mechanism based on PTCs. TBACl was identified as the most efficient catalyst that can catalyze both ring-opening and dehydrochlorination reactions, and 0.05–0.1 equiv. of TBACl can complete the heterogeneous dehydrochlorination reaction in 3 min without solid precipitation (20 wt% of NaOH solution) and with a minimal excess of the reactant NaOH (1.05 equiv. of NaOH). The reaction time decreased by 95% under the conditions where the NaOH concentration is reduced by 50% and the excess amount of NaOH decreased by 92%. The selectivity of side products after the reaction and phase separation was proven to be lower than that in the conventional reaction system. This work represents a significant breakthrough compared to the existing works, which always necessitated excessive amounts and saturated NaOH solutions to achieve high reaction efficiency. This work demonstrates great potential for continuous and green production, particularly in terms of reduced energy consumption and avoiding the post-treatment of solid waste.
Author contributions
Grace Dai Zhen Lee: conceptualization, data curation, investigation, methodology, validation, writing – original draft, and writing – review & editing; Zifei Yan: writing – review & editing and validation; Jing Song: writing – review & editing and validation; Jian Deng: writing – review & editing, resources and supervision; and Guangsheng Luo: conceptualization, methodology, writing – review & editing, supervision, and funding acquisition.
Data availability
All data included in this article are included within the figures and tables of this article and no ESI is provided.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors wish to gratefully acknowledge the financial support for this project from the National Natural Science Foundation of China (No. 21991100 and 21991104).
References
- A. Berkessel, C. Rollmann, F. Chamouleau, S. Labs, O. May and H. Gröger, Adv. Synth. Catal., 2007, 349, 2697–2704 CrossRef.
- S. E. Schaus, B. D. Brandes, J. F. Larrow, M. Tokunaga, K. B. Hansen, A. E. Gould, M. E. Furrow and E. N. Jacobsen, J. Am. Chem. Soc., 2002, 124, 1307–1315 CrossRef.
- M. Tokunaga, J. F. Larrow, F. Kakiuchi and E. N. Jacobsen, Science, 1997, 277, 936–938 CrossRef.
- J. Huang and X. Nie, Polym. Int., 2016, 65, 835–840 CrossRef.
- Z. S. Pour and M. Ghaemy, Prog. Org. Coat., 2014, 77, 1316–1324 CrossRef.
- D. Ai, R. Mo, H. Wang, Y. Lai, X. Jiang and X. Zhang, Prog. Org. Coat., 2019, 136, 105258 CrossRef.
- Y. Zhou, S. Ming, L. Tang and J. Qu, J. Coat. Technol. Res., 2021, 18, 1591–1601 CrossRef.
- E. D. Babot, C. Aranda, J. Kiebist, K. Scheibner, R. Ullrich, M. Hofrichter, A. T. Martínez and A. Gutiérrez, Antioxidants, 2022, 11, 522 CrossRef PubMed.
- M. W. Bedore, N. Zaborenko, K. F. Jensen and T. F. Jamison, Org. Process Res. Dev., 2010, 14, 432–440 CrossRef CAS.
- Z. Yan, J. Tian, K. Wang, K. D. P. Nigam and G. Luo, Chem. Eng. Sci., 2021, 229, 116071 CrossRef CAS.
- K. Kiatkittipong, M. A. A. M. Shukri, W. Kiatkittipong, J. W. Lim, P. L. Show, M. K. Lam and S. Assabumrungrat, Processes, 2020, 8, 548 CrossRef CAS.
- X. Sun, L. Yang, H. Xing, J. Zhao, X. Li, Y. Huang and H. Liu, Chem. Eng. J., 2013, 234, 338–345 CrossRef CAS.
- T. Pu, H. Tian, M. E. Ford, S. Rangarajan and I. E. Wachs, ACS Catal., 2019, 9, 10727–10750 CrossRef CAS.
- A. Blanckenberg and R. Malgas-Enus, Catal. Rev., 2019, 61, 27–83 CrossRef.
- J. Otera and S. Matsuzaki, Synthesis, 1986, 1019–1020 CrossRef.
- A. Bukowska, W. Bukowski and B. Mossety-Leszczak, J. Chem. Technol. Biotechnol., 1999, 74, 1145–1148 CrossRef.
- A. Krzyżanowska and E. Milchert, Chem. Pap., 2013, 67, 1218–1224 Search PubMed.
- E. I. Muresan, D. Lutic, M. Dobrota, A. Coroaba, F. Doroftei and M. Pinteala, Appl. Catal., A, 2018, 556, 29–40 CrossRef.
- R. Morodo, R. Gérardy, G. Petit and J.-C. M. Monbaliu, Green Chem., 2019, 21, 4422–4433 RSC.
- S. Wu, B. Wang, Y. Zhang, E. H. M. Elageed, H. Wu and G. Gao, J. Mol. Catal. A: Chem., 2016, 418–419, 1–8 Search PubMed.
- J. Gouman, S. Rens-van der Lee and R. van t Sand, United States Pat., 8802872, 2014 Search PubMed.
- Z. Yan, Z. Ma, J. Deng and G. Luo, Chem. Eng. Sci., 2021, 242, 116746 CrossRef.
- R. P. Shirtum, United States Pat., 5245057, p. 1993 Search PubMed.
- J. S. Zhang, Y. C. Lu, Q. R. Jin, K. Wang and G. S. Luo, Chem. Eng. J., 2012, 203, 142–147 CrossRef.
- K. P. C. Vollhardt and N. E. Schore, Organic Chemistry: Structure and Function, W.H. Freeman and Company, New York, 8th edn, 2018 Search PubMed.
- Z. Yan, C. Du, Y. Wang, J. Deng and G. Luo, Chem. Eng. J., 2022, 444, 136498 CrossRef.
- M. N. Makhin and M. S. Klebanov, Kinet. Catal., 2020, 61, 238–241 CrossRef.
- P. G. Gassman and W. N. Schenk, J. Org. Chem., 1977, 42, 918–920 CrossRef CAS.
- A. M. Krzyżanowska, E. Milchert and W. M. Paździoch, Ind. Eng. Chem. Res., 2013, 52, 10890–10895 CrossRef.
- M. Dargelos, M. Borredon and A. Gaset, Aust. J. Chem., 1992, 45, 1327 CrossRef.
- G. M. Lari, G. Pastore, C. Mondelli and J. Pérez-Ramírez, Green Chem., 2018, 20, 148–159 RSC.
- R. Wang, Synthesizing Epichlorohydrin from Dichloropropanol in Heterogeneous System in The Microreactor, Master thesis, Tsinghua University, 2014 Search PubMed.
- S. Du, Research on The Synthesis and Separation of Glycidyl Methacrylate, Master thesis, Tianjin University, 2019 Search PubMed.
- W. Zhao, Research on Synthesis Process of Glycidyl Methacrylate, Master thesis, Dalian University of Technology, 2020 Search PubMed.
- Z. Yan, G. D. Z. Lee, J. Deng and G. Luo, Chem. Eng. Process., 2024, 203, 109906 CrossRef.
- Z. Yan, J. Deng, Y. Chen and G. Luo, Ind. Eng. Chem. Res., 2020, 59, 19168–19176 CrossRef.
- T. Jirout and I. Fořt, Acta Polytech., 2008, 48, 16–26 Search PubMed.
- J. Claes, A. Vancleef, M. Segers, B. Brabants, M. E. Leblebici, S. Kuhn, L. Moens and L. C. J. Thomassen, Chem. Eng. Process., 2023, 183, 109252 CrossRef.
- T. Shibuguchi, Y. Fukuta, Y. Akachi, A. Sekine, T. Ohshima and M. Shibasaki, Tetrahedron Lett., 2002, 43, 9539–9543 CrossRef.
- S. P. Neofotistos, A. Tzani and A. Detsi, Catalysts, 2023, 13, 474 CrossRef.
- C. M. Starks, C. L. Liotta and M. E. Halpern, Phase-Transfer Catalysis, Springer Netherlands, 1994 Search PubMed.
- Z. Yan, C. Du, G. Luo and J. Deng, React. Chem. Eng., 2021, 6, 2159–2169 RSC.
- Z. Yan, Y. Wang, C. Du, J. Deng and G. Luo, AIChE J., 2022, 68, e17791 CrossRef.
- Z. Yan, Green Synthesis Technology of 2,3-Epoxypropyl Neodecanoate and Process Intensification, Master thesis, Tsinghua University, 2022 Search PubMed.
- D. Zhao, Phase Transfer Catalysis: Principle and Applications, Chemical Industry Press, 2007 Search PubMed.
- Z. Yan, Y. Hu, C. Du, J. Deng, K. Wang and G. Luo, J. Catal., 2020, 385, 44–51 CrossRef.
- R. A. Jones, Quaternary ammonium salts: their use in phase-transfer catalysed reactions, Academic Press, 2001 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Zifei Yan
,
Zifei Yan