
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A chemoselective electrochemical birch carboxylation of pyridines†
Soumik
Sarkar
,
Rohit
and
Michael W.
Meanwell
 *
*
Department of Chemistry, University of Alberta, 11227 Saskatchewan Dr NW, T6G 2N4, Edmonton, AB, Canada. E-mail: meanwell@ualberta.ca
First published on 19th February 2025
Abstract
Nucleophilic addition to pyridiniums, metal-catalyzed hydrogenation, and cycloadditions constitute a valuable toolbox of modern pyridine dearomatization strategies. Though, in recent years, there have been notable improvements and variations of the canonical Birch reduction to address its notorious safety hazards and poor chemoselectivity, it remains an unexplored mode of reactivity for controlled pyridine dearomatization. Here, we report a simple and safe protocol for the electrochemical Birch carboxylation of pyridines utilizing a sustainable approach and CO2 as a green C1 building block. This reaction is highly selective for pyridine reduction in the presence of several functional groups incompatible with the canonical Birch reduction and enables direct access to decorated piperidine scaffolds.
Green foundation1. Our chemistry addresses many of the safety issues associated with the canonical Birch while also using CO2, a sustainable C1 building block, as an electrophilic trap. CO2 valorization is an important modern area of research in advancing sustainable synthetic methods and our work contributes a unique example to this.2. Our reaction aligns with many fundamental principles of green chemistry including waste prevention, atom efficiency, less hazardous reaction design, and reagent renewability. When placed in contrast with the canonical Birch, our reaction is clearly a much greener alternative that also enables new reactivity. 3. Our work could be made greener by developing greener downstream chemistry for derivatizing the dihydropyridine products that we make using our electrochemical Birch carboxylation. |
Pyridine and piperidine were the two most common heterocycles found in FDA approved drugs of the past decade.1 Due to their growing popularity in drug discovery, pyridine dearomatization has become an area of intense research in synthetic chemistry for enabling direct access to piperidine cores and their related precursors.2 Classical dearomatization strategies such as the Birch reduction3 and metal-catalyzed hydrogenation4 pose obvious safety concerns (i.e., pyrophoric reagents and cryogenic liquid ammonia) and offer poor chemoselectivity, thus limiting their broader deployment in complex settings.3,4 Another classical approach, nucleophilic addition to activated pyridines, while powerful, requires the use of stoichiometric electrophilic activators that are inherently incompatible with substrates containing additional nucleophilic sites and can proceed with poor regioselectivity.2a In recent years, innovative advances in both pyridine activation/nucleophilic addition chemistry2,5 and catalytic hydrogenation6 have improved upon many of these aforementioned shortcomings. Dearomative cycloadditions of pyridines have contributed towards providing more elaborative piperidine frameworks.7
In its traditional form, the Birch reduction employs pyrophoric alkali metals (i.e. Na, K, and Li), with cryogenic liquid ammonia, to generate solvated electrons for arene reduction leading to the formation of a carbanion that is subsequently trapped with either a judiciously chosen proton source or carbon-based electrophile (see Scheme 1A).8 While the strong reduction potential of solvated electrons (∼−3.3 V vs. SCE)9a allows for dearomatization to occur, this also renders the Birch reduction poorly chemoselective in the presence of functional groups with higher reduction potentials (e.g., alkenes, esters, ketones, heteroaromatics, etc.).9f Despite its long-standing use in synthetic chemistry, only recently have new methods emerged to address the safety hazards and chemoselectivity issues associated with the Birch reduction.9 Baran and coworkers disclosed an electrochemical Birch-type reduction of heteroarenes and electron-deficient arenes where they used rapid-alternating polarity (rAP) to control chemoselectivity.9f Photochemical variants, like Baran's electrochemical approach, avoid the use of cryogenic ammonia and pyrophoric metals.9b,c Though these advances undoubtedly represent substantial improvements over the canonical Birch reduction, there still remain many unsolved challenges. For instance, recent approaches only have demonstrated compatibility with proton electrophiles as Birch alkylations remain unexplored in this modern context. Further, even among these many noted improvements, there is an absence of literature on pyridine Birch reductions. In fact, the scarce examples of classical Birch reduction of pyridines (6) show that it is difficult to control selectivity as both dimerization products (8) and N-alkylation (7) complicate the product distribution (Scheme 1C).3
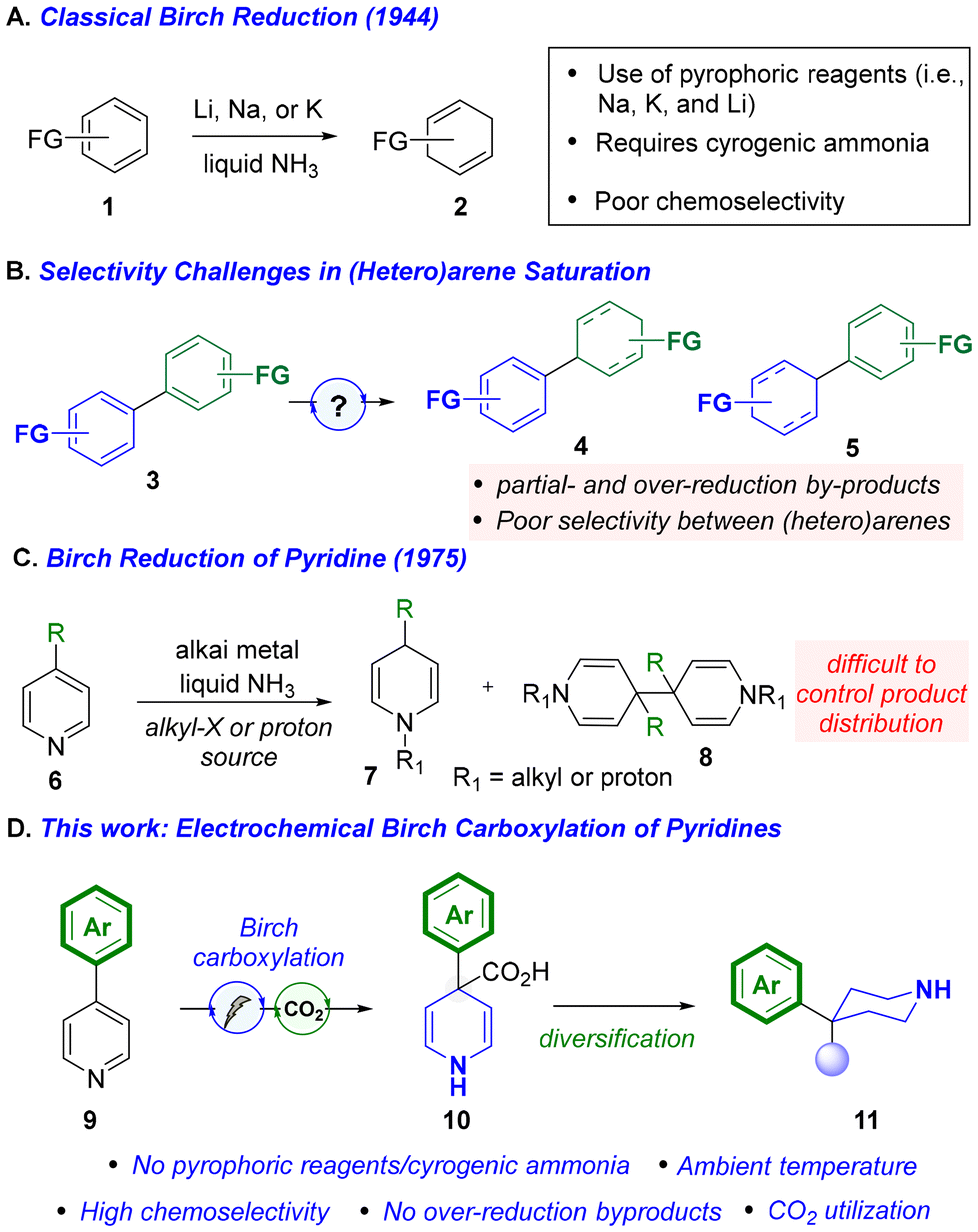 | ||
| Scheme 1 A–C) Challenges with the classical Birch reduction; (D) our approach towards pyridine dearomatization. | ||
In this article, we report a mild and selective electrochemical Birch carboxylation of pyridines. This protocol enables exclusive partial reduction of pyridine in the presence of (hetero)aromatics and other functional groups typically incompatible with the canonical Birch reduction (and other modes of dearomatization) while employing CO2, a sustainable C1 building block, as an electrophilic trap. The deployment of CO2 as an electrophilic trap in Birch reductions overall remains unexplored despite environmental and economic factors motivating its utilization. Since classical Birch alkylations of pyridines are complicated by competitive N-alkylation, CO2 represents the ideal electrophilic trap as undesired N-carboxylation is entropically disfavoured and reversible. Our approach provides a greener method for pyridine dearomatization by eliminating the need for stoichiometric activators and reductants, which lead to unnecessary waste generation, while also minimizing the safety risks commonly associated with conventional dearomatization strategies.
The selective reduction of one aromatic system in the presence of another remains a common challenge in dearomatization chemistry. As reported by Doyle, pyridine, owing to its increased aromatic character compared to other heteroaromatics, is notably difficult to reduce chemoselectively in this setting (see Scheme 1B).4 With this in mind, we choose 4-phenyl pyridine (12; −2.15 V vs. SCE)10 as our study substrate for exploring electrochemical Birch/carboxylations. While only rare examples exist of selective Birch reductions in biaryl systems of any kind,11 we posited that, under controlled electroreducing conditions, the pyridine fragment in the 4-phenyl pyridine would undergo selective reduction due to the increased stabilization of the formed radical anion intermediate (i.e., benzylic radical and nitrogen anion).12 We proceeded to examine several parameters including electrolyte, solvent, temperature, and electrode material (see ESI† for full optimization details) to effect the desired Birch carboxylation. Strikingly, in an undivided cell, employing a platinum cathode and magnesium anode in DMF at ambient temperature, (entry 1) we obtained 1,4-dihydropyridine (DHP, 13) in 63% yield. Attempts to use greener solvents in place of DMF such as MeCN were unsuccessful.13 Under these conditions (entry 1), no phenyl reduction was observed and carboxylation occurred exclusively at C4. The regioselectivity observed here is analogous to what would be expected from a canonical Birch reduction. Running the reaction at lower or higher temperatures, as well as with other electrode combinations, afforded little product. Other electrolytes (e.g., TBABr, TBAOAc, and TMABF4) performed poorly as replacements for TBABF4. However, the magnesium anode could be replaced with zinc with only a modest decrease in yield (entry 2). Though it was later found that replacing the platinum cathode with a nickel foam cathode led to a modest improvement in yield (entry 3), this condition did not translate to other substrates (<20%). As such, subsequent substrate evaluation was carried out with the conditions depicted in entry 1, which proved to be scalable up to 500 mg.
The reaction's generality is shown in Scheme 2. Bi- and tri-aryl pyridines were all reduced, with complete chemoselectivity and regioselectivity, to their corresponding 1,4-DHPs. Unlike the classical Birch reduction, which for biaryl systems can lead to unpredictable mixtures of reduced products, no (hetero)arene reduction nor dimerization products were observed in our reaction.3,11 Furthermore, the simplicity, avoidance of expensive additives (i.e., 4′,4′-di-tert-butylbiphenyl (DBB), 15-crown-5, and TPPA (tri(pyrrolidin-1-yl))),9g and safety of our protocol provides an attractive solution for pyridine Birch reduction. Unsurprisingly, electron-neutral and electron-rich arenes were all well-tolerated as these systems require lower reduction potentials (more negative). Even electron-deficient arenes and heteroarenes (30–33, 37), all of which have relatively higher reduction potentials (more positive), were not reduced under our mild conditions. Heteroarenes, in particular, are notoriously incompatible or reactive under canonical Birch reduction and other modes of pyridine dearomatization (i.e., metal-catalyzed hydrogenation). For instance, canonical Birch reduction can lead to the fragmentation of furan and thiophene rings.9f,14 Having achieved orthogonal pyridine reduction versus other (hetero)aromatics, we proceeded to assess other functional groups known to pose compatibility issues with the canonical Birch reduction. Our milder conditions enabled broad functional group tolerance to aryl trifluoromethyls (15, 24), aryl fluoride (18), ketone (35), and alkene (26). As noted by Glorius, in metal catalyzed hydrogenation, sulfur-containing motifs, which are widely prevalent in Nature and drug discovery, often lead to catalyst poisoning thus limiting the use of hydrogenation-based dearomatization strategies with sulfur-containing molecules.15 This drawback has inspired considerable effort in search of sulfur-resistant hydrogenation catalysts. However, for pyridine dearomatization our protocol provides a convenient, alternative solution to this problem as sulfur-containing motifs, including thioethers (28, 29), benzothiophene (30), and benzothiazole (31) were all tolerated in our reaction. In recent years, late-stage saturation (LSS), a concept aimed at converting aromatics into their saturated counterparts in complex molecules, has shown promise as a powerful tool for improving a drug candidate's physiochemical properties.6c,d Towards this goal, natural product-derived substrates were readily converted into their corresponding 1,4-DHP analogues (34, 35, and 36). Notably, the estrone-derived substrate highlights the vastly improved chemoselectivity of our reaction over the canonical Birch reduction as neither the arene nor ketone were reduced. Overall, this process aligns with many foundational principles of green chemistry including waste prevention, atom efficiency, less hazardous reaction design, and reagent renewability.16 In fact, standard green chemistry metrics further support this assertion; the atom economy (AE %) and carbon efficiency (CE %) of this reaction were determined to be 93–96% and 100%, respectively, for all substrates. Further, the products can be used in subsequent steps as crude materials or after simple recrystallization, reducing waste and enhancing efficiency. By avoiding chromatographic purification, which generates significant solvent and silica waste, the process achieves a lower E-factor, improving sustainability.17
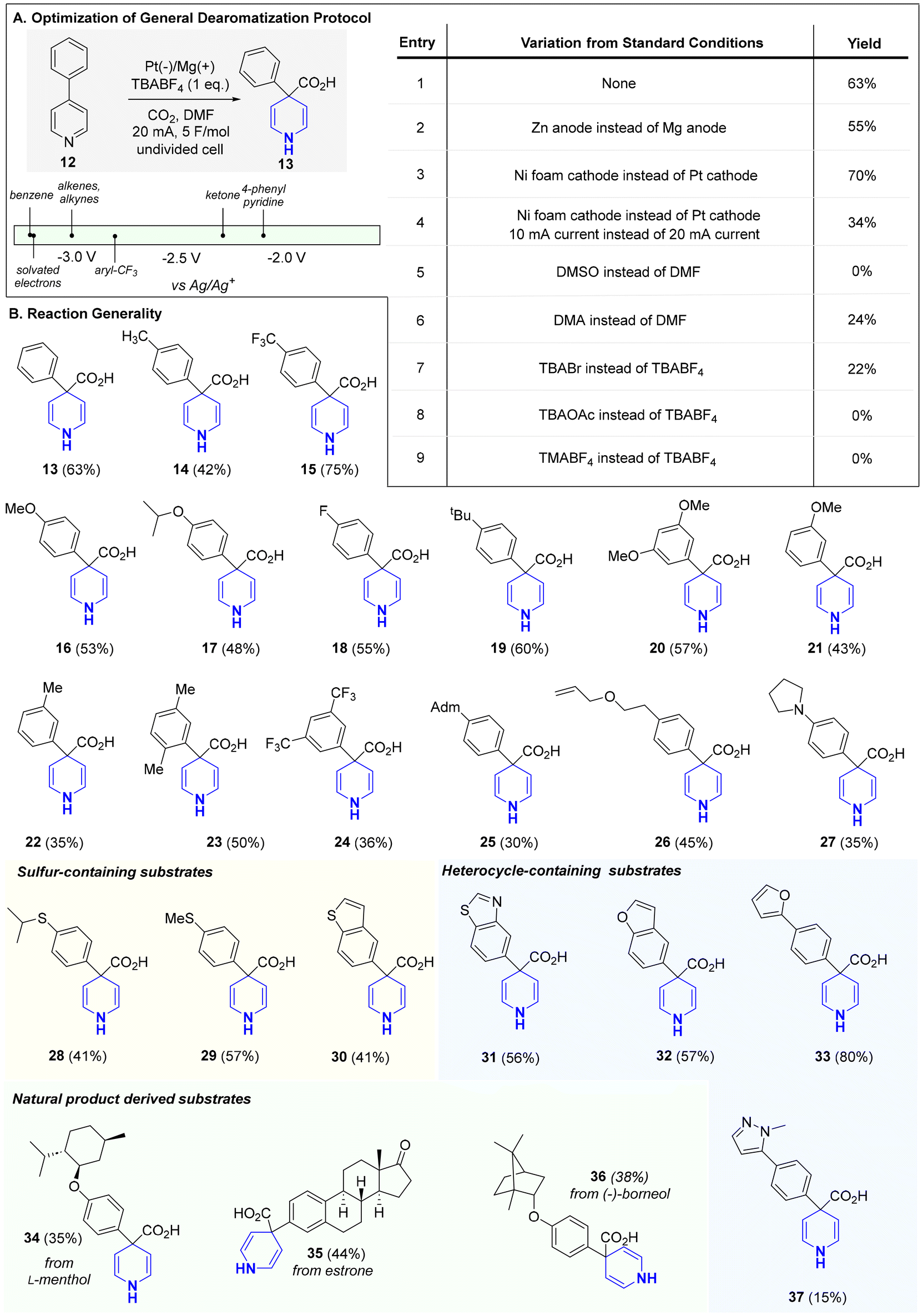 | ||
| Scheme 2 Optimization table and substrate scope. Reaction conditions: TBABF4 (1 eq.) and 4-aryl-pyridine in DMF (0.10 M) in a 5 mL ElectraSyn vial with a Pt cathode and an Mg anode. CO2 was bubbled into the reaction mixture as it was electrolyzed at 20 mA for 5 F mol−1. Adm = adamantyl. | ||
1,4-DHPs are widely utilized in catalysis and as building blocks for piperidine synthesis.18 As such, we sought to provide examples of useful downstream chemistry for making popular piperidine frameworks using the 1,4-DHPs generated from our Birch carboxylation. 4,4-disubstituted piperidines are privileged scaffolds found in a plethora of FDA-approved drugs (e.g., haloperidol, loperamide, traxoprodil, etc.) and investigational therapeutics.19 Our protocol allows for streamlined access to 4,4-disubstituted piperidines from their corresponding biaryl frameworks. Bristol Myers Squibb (BMS) previously identified 38 and analogues thereof (e.g.39 and 40) as nanomolar dual NK1/serotonin receptor antagonists for treating depression.19b,c Towards making the piperidine core, the crude reaction mixtures obtained from the electrochemical Birch carboxylation were treated with sodium cyanoborohydride and TFA, followed by moderate heating with borane-THF, to afford several analogues (41–51) of the central piperidine fragment in a convenient sequence that avoids chromatographic purification and requires only a single purification via recrystallization from diethyl ether after the final step. A selection of the products are shown in Scheme 3 (see ESI† for many more additional examples). 4-aryl-4-carbonyl piperidines (e.g.52–57) constitute an important subclass of opioid-based painkillers and antidiarrheals.19d,e After once again treating the crude reaction mixture obtained from Birch carboxylation with sodium cyanoborohydride, the resulting product (58) was then esterified and methylated to deliver merperidine (56), a clinically approved opioid used for treating severe pain.19d Overall, these examples highlight how the electrochemical Birch carboxylation can be deployed as a tool in drug discovery for directly accessing derivatives with increased three-dimensional character from their respective planar analogues. Finally, we considered opportunities to pair our Birch carboxylation with other reductive reactions in a tandem process. Pyridine N-oxides not only represent a subclass of pyridine-containing therapeutics but are also common synthetic intermediates used to alter pyridine reactivity in electrophilic and nucleophilic aromatic substitutions. We showed, without any changes to the reaction conditions, that a tandem electrochemical N-oxide reduction/Birch carboxylation enables the streamlined conversion of pyridine N-oxides (60) into their corresponding 1,4-DHPs.
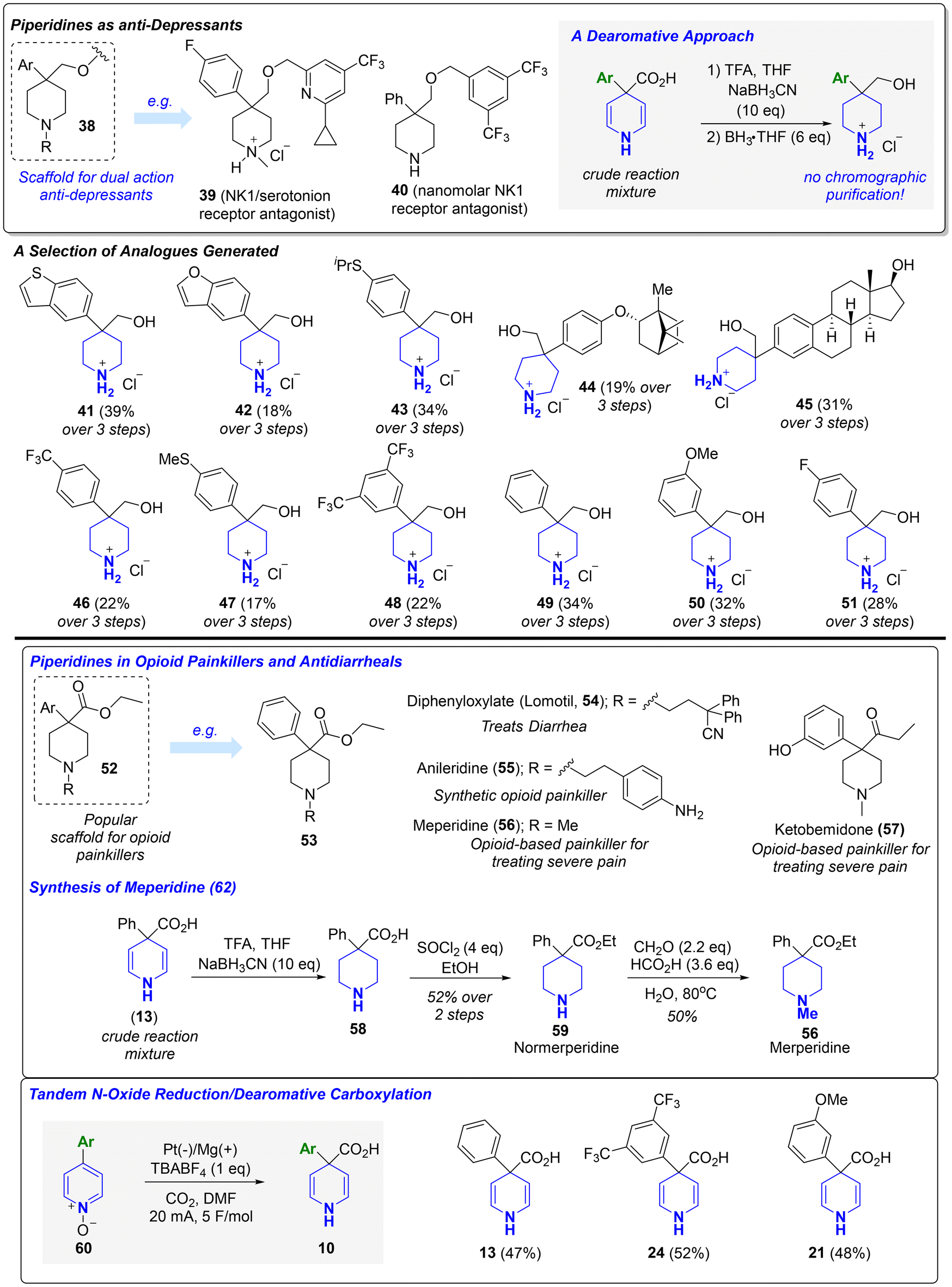 | ||
| Scheme 3 Utility of electrochemical Birch carboxylation for synthesis of popular piperidine scaffolds. | ||
Lastly, we sought to provide a mechanistic rationale for the exclusive C4 selectivity observed in the Birch carboxylation. While we presumed that this dearomative carboxylation occurred through an electrochemical Birch-type mechanism (pathway 1), we considered a Minisci-like radical addition (pathway 2) as a possible alternative. As shown in Scheme 4, pathway 1 proposes a Birch-type reduction of the pyridine (12) to the radical anion (61). The generated benzylic radical is stabilized through electron delocalization into the arene substituent, thus lowering the barrier towards dearomatization. This hypothesis is supported by the fact that 4-methyl-pyridine, which would lead to the formation of a less stabilized radical, failed to undergo dearomative carboxylation under our conditions. The negative charge of the radical anion sits predominantly on the nitrogen leading to its trapping with CO2. Subsequent cathodic reduction of the benzylic radical (62) and trapping with CO2 affords 63 where the carbamic acid moiety readily undergoes decarboxylation to deliver the product 13. The alternative Pathway 2, on the other hand, involves a Minisci-type addition of CO2 radical anion after either activation of the pyridine ring via coordination to either CO2 or Mg2+ cation (see 64 in Scheme 4). However, this pathway fails to rationalize the exclusive C4 selectivity that we observe. Furthermore, no oxalate dianion, which is associated with CO2 radical anion dimerization, was detected by 13C NMR. To confirm the presence of the benzylic anion proposed in pathway 1, we ran the reaction with D2O and observed C4′ deuteration (65, 76% deuterium incorporation). As such, mechanistic evidence strongly supports pathway 1 (Birch reduction) as being the operative mode of dearomative carboxylation.
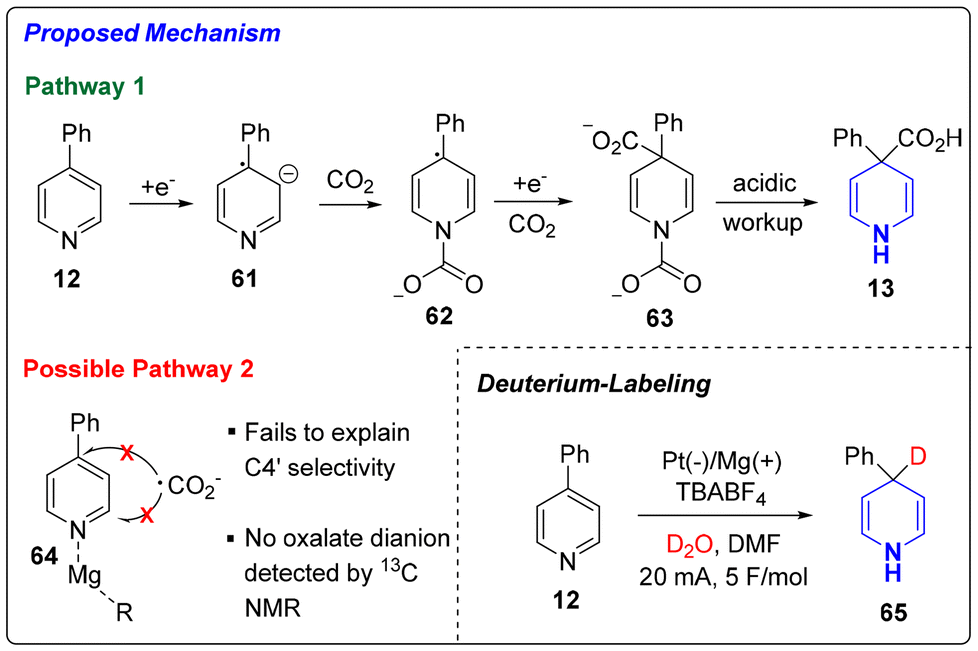 | ||
| Scheme 4 Proposed mechanism. | ||
Conclusions
In summary, we have developed an operationally simple, electrochemical Birch carboxylation of pyridines. This process is additive free, utilizes CO2 as a green C1 building block, and is compatible with a range of functional groups not tolerated in the classical Birch reduction and other dearomatization strategies (i.e., metal-catalyzed hydrogenation). Ultimately, these mild conditions address the well-known safety hazards of the canonical Birch to provide a new avenue for directly transforming pyridines into their corresponding piperidine analogues.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that there are no conflicts interests.Acknowledgements
This work was supported by the Manley and Marian Johnston Professorship in Chemistry and an NSERC Discovery Grant.References
- C. M. Marshall, J. G. Federice, C. N. Ball, P. B. Cox and J. T. Njardarson, J. Med. Chem., 2024, 67, 11622–11655 CrossRef CAS PubMed.
- (a) M. Escolano, D. Gavina, G. Alzuet-Pina, S. Díaz-Oltra, M. Sánchez-Roselló and C. del Pozo, Chem. Rev., 2024, 124, 1122–1246 CrossRef CAS PubMed; (b) L. Lückemeier, M. Pierau and F. Glorius, Chem. Soc. Rev., 2023, 52, 4996–5012 RSC.
- (a) T. J. Donohue, A. J. McRiner and P. Sheldrake, Org. Lett., 2000, 2, 3861–3863 CrossRef PubMed; (b) A. J. Birch and E. A. Karakhanov, J. Chem. Soc., Chem. Commun., 1975, 480–481 RSC.
- T. W. Lyons, I. N.-M. Leibler, C. Q. He, S. Gadamsetty, G. J. Estrada and A. G. Doyle, J. Org. Chem., 2024, 89, 1438–1445 CrossRef CAS PubMed.
- (a) A. Heusler, J. Fliege, T. Wagener and F. Glorius, Angew. Chem., Int. Ed., 2021, 60, 13793–13797 CrossRef CAS PubMed; (b) M. W. Gribble Jr., S. Guo and S. L. Buchwald, J. Am. Chem. Soc., 2018, 140, 5057–5060 CrossRef PubMed.
- (a) Z. Nairoukh, M. Wollenburg, C. Schlepphorst, K. Bergander and F. Glorius, Nat. Chem., 2019, 11, 264–270 CrossRef CAS PubMed; (b) M. P. Wiesenfeldt, Z. Nairoukh, T. Dalton and F. Glorius, Angew. Chem., Int. Ed., 2019, 58, 10460–10476 CrossRef CAS PubMed; (c) F. Zhang, H. Sasmal, D. Rana and F. Glorius, J. Am. Chem. Soc., 2024, 146, 18682–18688 CrossRef CAS PubMed; (d) D.-H. Liu, P. M. Pflüger, A. Outlaw, L. Lückemeier, F. Zhang, C. Regan, H. Nodeh, T. Cernak, J. Ma and F. Glorius, J. Am. Chem. Soc., 2024, 146, 11866–11875 CrossRef CAS PubMed; (e) N. Shida, Y. Shimizu, A. Yonezawa, J. Harada, Y. Furutani, Y. Muto, R. Kurihara, J. N. Kondo, E. Sato, K. Mitsudo, S. Suga, S. Iguchi, K. Kamiya and M. Atobe, J. Am. Chem. Soc., 2024, 146, 30212–30221 CrossRef CAS PubMed.
- (a) Z. Siddiqi, T. Bingham, T. Shimakawa, K. D. Hesp, A. Shavnya and D. Sarlah, J. Am. Chem. Soc., 2024, 146, 2358–2363 CrossRef CAS PubMed; (b) J. Petrovcic, D. Y. Boyko, A. S. Shved, G. Lenardon, C. Salome, Q. Lefebvre, T. Fessard and D. Sarlah, ChemRxiv, 2023 DOI:10.26434/chemrxiv-2023-jgr4j.
- P. W. Rabideau and Z. Marcinow, The Birch Reduction of Aromatic Compounds, in Organic Reactions, 2004, DOI:10.1002/0471264180.or042.01.
- (a) T. Yuan, L. Sun, Z. Wu, D. Wang, X. Cai, W. Lin, M. Zheng and X. Wang, Nat. Catal., 2022, 5, 1157–1168 CrossRef CAS; (b) A. Chatterjee and B. König, Angew. Chem., Int. Ed., 2019, 58, 14289–14294 CrossRef CAS PubMed; (c) J. P. Cole, D.-F. Chen, M. Kudisch, R. M. Pearson, C.-H. Lim and G. M. Miyake, J. Am. Chem. Soc., 2020, 142, 13573–13581 CrossRef CAS PubMed; (d) B. K. Peters, R. X. Rodriguez, S. H. Reisberg, S. B. Beil, D. P. Hickey, Y. Kawamata, M. Collins, J. Starr, L. Chen, S. Udyavara, K. I. Klunder, T. J. Gorey, S. L. Anderson, M. Neurock, S. D. Minteer and P. S. Baran, Science, 2019, 363, 838–845 CrossRef CAS PubMed; (e) E. Y. K. Tan, A. S. Mat Lani, W. Sow, Y. Liu, H. Li and S. Chiba, Angew. Chem., Int. Ed., 2023, 62, e202309764 CrossRef CAS PubMed; (f) K. Hayashi, J. Griffin, K. C. Harper, Y. Kawamata and P. S. Baran, J. Am. Chem. Soc., 2022, 144, 5762–5768 CrossRef CAS PubMed; (g) J. Burrows, S. Kamo and K. Koide, Science, 2021, 374, 741–746 CrossRef CAS PubMed.
- C. Combellas, F. Kanoufi and D. Mazouzi, J. Phys. Chem. B, 2004, 108, 19260–19268 CrossRef CAS.
- K. Koide, Synthesis, 2023, 707–718 CrossRef CAS.
- G.-Q. Sun, P. Yu, W. Zhang, W. Zhang, Y. Wang, L.-L. Liao, Z. Zhang, L. Li, Z. Li, D. G. Yang and S. Lin, Nature, 2023, 615, 67–72 CrossRef CAS PubMed.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- S. F. Birch and D. T. McAllan, J. Chem. Soc., 1951, 2556–2563 RSC.
- L. Lückemeier, T. De Vos, L. Schlichter, C. Gutheil, C. G. Daniliuc and F. Glorius, J. Am. Chem. Soc., 2024, 146, 5864–5871 CrossRef PubMed.
- R. A. Sheldon, ACS Sustainable Chem. Eng., 2018, 6, 32–48 CrossRef CAS.
- R. A. Sheldon, Green Chem., 2003, 25, 1704–1728 RSC.
- (a) V. K. Sharma and S. K. Singh, RSC Adv., 2017, 7, 2682–2732 RSC; (b) P.-Z. Wang, J.-R. Chen and W.-J. Xiao, Org. Biomol. Chem., 2019, 17, 6936–6951 RSC.
- (a) L. Yet, 4-(Hetero)Arylpiperidines, in Privileged Structures in Drug Discovery, ed. L. Yet, 2018, DOI:10.1002/9781118686263.ch5; (b) G. I. Stevenson, A. M. MacLeod, I. Huscroft, M. A. Cascieri, S. Sadowski and R. Baker, J. Med. Chem., 1995, 38, 1264–1266 CrossRef CAS PubMed; (c) G. I. Stevenson, I. Huscroft, A. M. MacLeod, C. J. Swain, M. A. Cascieri, G. G. Chicchi, M. I. Graham, T. Harrison, F. J. Kelleher, M. Kurtz, T. Ladduwahetty, K. J. Merchant, J. M. Metzger, D. E. MacIntyre, S. Sadowski, B. Sohal and A. P. Owens, J. Med. Chem., 1998, 41, 4623–4635 CrossRef CAS PubMed; (d) N. Elbaridi, A. D. Kaye, S. Choi and R. D. Urman, Pain Physician, 2017, 20, SE23–SE31 Search PubMed; (e) A. Karim, R. E. Ranney, K. L. Evensen and M. L. Clark, Clin. Pharmacol. Ther., 1972, 13, 407–419 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc05976j |
| This journal is © The Royal Society of Chemistry 2025 |