
The Piancatelli rearrangement of AMF (5-azidomethylfurfural) derivatives: a biobased opportunity for the synthesis of nitrogenous cyclopentenones†
Clémentine
Mayet
,
Jérôme
Bignon
and
Jean-François
Betzer
 *
*
Institut de Chimie des Substances Naturelles (ICSN), CNRS-UPR2301, Université Paris-Saclay, Gif-sur-Yvette, France. E-mail: jean-francois.betzer@cnrs.fr
First published on 19th March 2025
Abstract
In the field of renewable materials to substitute petroleum-based products, one of the main areas of research is the study and transformation of small molecules derived from biomass into high value-added compounds. In this context, we report the preparation of 4-(azidomethyl)-cyclopentenones starting from AMF (5-azidomethylfurfural) derivatives, directly obtained from the well-known biosourced renewable material CMF (5-chloromethylfurfural). The Piancatelli rearrangement of these AMF derivatives, catalyzed by Dy(OTf)3 and under microwave activation, affords substituted cyclopentenones with two contiguous stereogenic centres, one of which is quaternary, exhibiting high diastereoselectivity. We were able to exploit the azidomethyl side chain to produce cyclopentenones with other nitrogenous functionalities, such as amine or triazole moieties. Moreover, these 4-(azidomethyl)-cyclopentenones exhibited desired cytotoxic activity against HCT116 and HL60 cancer cell lines with nanomolar IC50 values.
Green foundation1. The use of bio-based materials derived from biomass (CMF = 5-chloromethylfurfural) for high value-added cyclopentenones2. Our synthetic method provides efficient access to nitrogen-based substrates through the Piancatelli rearrangement, via a catalytic process under microwave activation using CHEM21 recommended solvents, tert-butanol and water. 3. We described the first Piancatelli rearrangement on nitrogen-based substrates. Other nitrogen functionalities might be considered to extend the scope of this major advance in the development of the Piancatelli rearrangement. |
Introduction
In recent decades, increased attention from scientific and industrial communities has been focused on reducing dependence on oil-based fossil raw materials. Given their environmental impact, fossil fuels significantly contribute to carbon dioxide and other greenhouse gas emissions, and their extraction generates pollutants that degrade air and water quality. Thus, replacing fossil fuels with renewable materials is indeed a key element in developing a green and circular economy. The use of renewable biomass resources for the production of fuels, chemicals, and materials offers a valuable alternative to the fossil-based industry.1 The two main lines of research in this field focus, firstly, on the transformation of biomass into molecular building blocks and, secondly, on the conversion of these building blocks into high value-added compounds for many applications such as precursors for pharmaceutical compounds, monomers for polymer synthesis, surfactants, lubricants, paints, plasticizers, paper additives and biocarburants.In the field of renewable materials, lignocellulosic biomass is a cheaper and the most abundant material. This sustainable lignocellulose is composed of cellulose (50%), hemicellulose (30%), and polyphenolic lignins (20%).2 For the transformation of this lignocellulosic biomass, the production of furfural (FFR), furfuryl alcohol (FA), and 5-hydroxymethyl-furfural (HMF) provides versatile sustainable and valuable substitutes for petrochemical building blocks.3 HMF is regarded by the scientific community as a versatile building block platform and is considered to be one of the “Top 14” bio-based building blocks listed by the US Department of Energy.4
Despite significant interest and the synthetic potential of HMF to replace petroleum-based products, this compound suffers from significant drawbacks. First, HMF is a polar and hydrophilic molecule that is not easily extracted from aqueous media. Second, HMF is sensitive to the acidic conditions under which it is produced, leading to the formation of numerous by-products. Among these, we can mention OBMF, which results from HMF dimerization, as already observed by Ananikov5 and Afonso.6 Another major drawback is the formation of levulinic acid associated with the concomitant release of formic acid.7 The major difficulty encountered when using HMF as the starting material is the formation of oligomeric and polymeric materials that are commonly called humins. These humins are formed by multiple combinations of the HMF hydrolyzed product and HMF itself to produce dark-colored solid materials.8
In this context, other versatile sustainable and valuable substitutes of petrochemical compounds have been prospected to identify biomass molecular building blocks for chemical applications and the conversion of these building blocks into high value-added compounds. These studies have allowed establishing a more convenient and stable derivative of HMF, namely 5-(chloromethyl)furfural (CMF) 1.9 Among the benefits of CMF over HMF, CMF is produced in high yield under mild conditions directly from raw biomass, and its stability and hydrophobicity markedly facilitate isolation.10 Very recently, a comparative study of the potential between HMF and CMF was reported, clearly highlighting the advantages of CMF in terms of ease of production, stability, and reactivity.11
Different synthetic transformations have been described, supporting the versatility of CMF as a building block platform.12 From them, we have highlighted the substitution reaction of the chloromethyl group into the azidomethyl function in quantitative yield by a substitution reaction, described for the first time by Mascal,13 thus providing a nitrogenous biobased furan derivative AMF 2.
Considering the derivatization of AMF 2 and taking advantage of azidomethyl, aldehyde and furan functionalities, only a few selective synthetic transformations have been described. The aldehyde group can participate in the condensation reaction with ammonia for carbon–nitrogen bond formation (Fig. 1a), to produce diamine derivatives after reduction.14 This same aldehyde group can also undergo a change in the oxidation state, to the carboxylic acid or alcohol functionalities. For instance, Ananikov has described the transformation of AMF 2 into methyl 5-(azidomethyl)furan-2-carboxylate through the Corey–Gilman–Ganem oxidative esterification process using NaCN/MnO2/MeOH and AcOH (Fig. 1b).15 Ananikov has also benefited from the azide function to perform a copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) to produce 1,4-substituted 1,2,3-triazole derivatives (Fig. 1c).16 The use of bis-terminal alkyne allowed the formation of bis-(1,2,3-triazolyl) aldehyde derivatives (not shown).17 The furan ring itself is able to undergo [4 + 2] cycloaddition with singlet oxygen to produce a 1,2,4-trioxolane intermediate, which is known to decompose into the corresponding butenolide derivatives. In this way, starting from AMF 2, Mascal13 has described the two step synthesis of δ-aminolevulinic (Fig. 1d), an effective non-toxic biodegradable herbicide and insecticide,18 as well as a precursor for biosynthesis of tetrapyrroles, the core structure of porphyrins.13
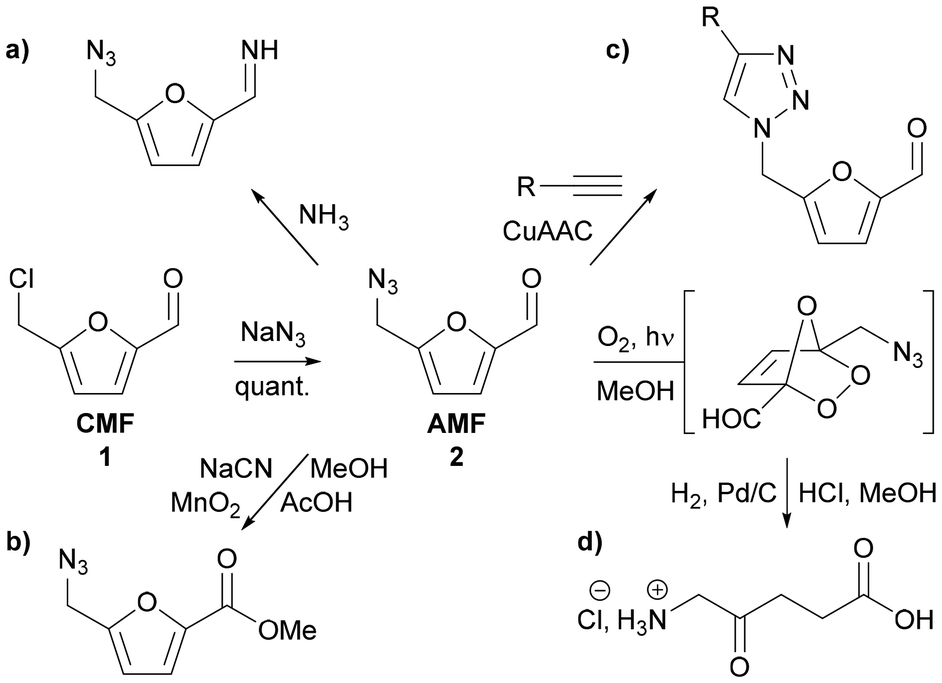 | ||
| Fig. 1 Transformation of bio-sourced CMF 1 into AMF 2, and aldehyde, azide and the furan ring reactivity of AMF 2. | ||
Taking advantage of the great synthetic potential of AMF 2, this compound, or its close furanic derivatives, could be converted into cyclopentenone with 4-(azidomethyl) and 4-hydroxy functionalities via the Piancatelli rearrangement.19 Despite the recent number of studies on the Piancatelli rearrangement and the effectiveness of this process to produce cyclopentenones from 2-furylcarbinols,20 C-5 substituted 2-furylcarbinol substrates have scarcely been explored. Except for the favoured intramolecular version with a tethered nucleophile at the C-5 position, the intermolecular Piancatelli rearrangement with C-5 substituted 2-furylcarbinol has only been described with substrates containing a methyl,21 a phenyl22 and an ethoxymethyl group.23 The challenge in using C-5 substituted 2-furylcarbinol as the starting material for the Piancatelli rearrangement is due to this C-5 substitution, thereby significantly reducing the reactivity during the elementary step of the catalytic cycle for the C-5 addition on the furanoxonium ion intermediate.20 In previous studies, we have shown that HMF derivatives, meaning those with a hydroxymethyl C-5 substitution, can deliver cleanly 5-substituted 4-hydroxymethyl-4-hydroxycyclopentenones.24 We have highlighted that the combination of microwave activation and the use of a catalytic amount of DyCl3 was necessary to provide high chemical yields and to ensure control of the relative configuration of the two stereogenic centers that are created in the rearrangement. In this study, we would like to extend this synthetic methodology for the preparation of 5-substituted-4-(azidomethyl)-4-hydroxycyclopentenones, starting from (5-(azidomethyl)furan-2-yl)-carbinol derivatives.
Results and discussion
Initial studies were conducted by examining the Piancatelli rearrangement directly on AMF 2 and on its simple reduced derivative 4 obtained in one step by selective reduction of the aldehyde function into carbinol using sodium borohydride in 95% yield. The implemented experimental conditions for this Piancatelli rearrangement are those optimized for the transformation of furfuryl alcohol (FA) into 4-hydroxy-2-cyclopentenone (HCP), specificallyusing microwave (MW) activation. The use of MW irradiation allows performing this rearrangement within a short time, thus leading to an efficient rearrangement while minimizing side reactions and the formation of humins.25 An attempt to directly transform AMF 2, or its hydrated form, which could be generated in situ in the reaction medium, into a cyclopentenone failed (Scheme 1). Under these conditions, no reaction was observed and the starting material was cleanly recovered unchanged. Only traces of a few new products could be observed, but none corresponded to a cyclopentenic core structure. In contrast, with the use of carbinol derivative 4 we have observed a complete conversion of the starting material and the formation of the desired 4-(azidomethyl)-4-hydroxycyclopentenone 5 in 37% yield. | ||
Scheme 1 Attempted Piancatelli rearrangement on AMF 1 and on its reduced derivative 4. Piancatelli rearrangement was performed on a 1 mmol scale in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 t-BuOH/H2O mixture at 0.1 M under microwave (MW) heating for 5 min. NR = no reaction. :5 t-BuOH/H2O mixture at 0.1 M under microwave (MW) heating for 5 min. NR = no reaction. | ||
Encouraged by this first result and in order to apply this methodology to the preparation of 5-substituted 4-(azidomethyl)-4-hydroxycyclopentenones, we next turned our attention to reactions involving (5-(azidomethyl)furan-2-yl)-α-substituted carbinol derivatives. The first reaction pathway to introduce a phenyl substituent on the aldehyde moiety of AMF 2 was performed with phenylmagnesium bromide. The desired carbinol adduct was obtained in a 85% yield, in the yield range of the Grignard 1,2-addition reaction on the carbonyl function for substrates also bearing an azide function.26 Actually, it is well known in the literature that organic azides may act as nitrogen sources for electrophilic amination reactions for organometallic species such as Grignard reagents, leading to secondary amines.27 For these reasons, we would like to take advantage of alternative milder conditions involving a transition metal-catalyzed 1,2-addition of arylboronic acids.
To identify the best reaction conditions of this 1,2-addition of benzeneboronic acid on AMF 2, we have first used the conditions described by Miyaura, which imply the use of rhodium(I) as a catalyst combined with the bidentate phosphine 1,1′-bis(diphenylphosphino)ferrocene as the ligand. The use of Rh(acac)(CO)2 led to no reaction (Table 1, entry 1),28 while the use of more labile ligands on rhodium, specifically Rh(acac)(coe)2,29 led to a low conversion but without any formation of the desired product 6a (entry 2).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Ligand | Base | T (°C) | Time (h) | Conv. (%) | Yield 6aa (%) |
| Acac = acetylacetonate. Dppf = 1,1′-bis(diphenylphosphino)ferrocene. Coe = cyclooctene. IPrHCl = 1,3-bis(2,6-diisopropylphenyl) imidazolium chloride. Pfb = perfluorobutyrate. P(1-Nap)3 = tri-1-naphthylphosphine.a Isolated yield.b Reaction performed in a 1:1.6 DME/H2O mixture at 0.2 M.c Reaction performed in a 3:2 DME/H2O mixture at 0.2 M.d Reaction performed in a 4:1 DME/H2O mixture at 0.2 M.e Reaction performed in a 5:1 DME/H2O mixture at 0.3 M.f Reaction performed in toluene at 0.3 M.g Reaction performed in THF at 0.2 M. |
|||||||
| 1b | Rh(acac)(CO)2 (3 mol%) | dppf (3 mol%) | — | 80 | 18 | 0 | NA |
| 2c | Rh(acac)(coe)2 (3 mol%) | dppf (3 mol%) | — | 80 | 18 | 30 | 0 |
| 3d | RhCl3·3H2O (3 mol%) | IPrHCl (3 mol%) | NaOMe (1 eq.) | 80 | 2 | 0 | NA |
| 4e | [Rh2(OAc)4] (3 mol%) | IPrHCl (3 mol%) | KOtBu (1 eq.) | 90 | 0.5 | 100 | 0 |
| 5f | Pd2(dba)3·CHCl3 (5 mol%) | PPh3 (5 mol%) | Cs2CO3 (1 eq.) | 60 | 24 | 70 | <5 |
| 6g | PdCl2 (5 mol%) | P(1-Nap)3 (5 mol%) | K2CO3 (3 eq.) | 65 | 16 | 100 | 20 |
| 7g | PdCl2 (5 mol%) | P(1-Nap)3 (5 mol%) | K2CO3 (3 eq.) | 20 | 16 | 80 | 60 |

The same reaction was then attempted with the use of rhodium(III) as the catalyst combined with N-heterocyclic carbene IPr (1,3-bis(2,6-diisopropylphenyl)-imidazol-2-ylidene) as the ligand, which is easily generated in basic media from its corresponding imidazolium salts, under reaction conditions described by Fürstner,30 but no reaction was observed (entry 3). This 1,2-addition reaction was also described by Gois with the use of dirhodium(II) complexes with axial N-heterocyclic carbene (NHC) ligands,31 but while total conversion was observed, no identified product could be isolated from the crude reaction mixture (entry 4). We next turned our attention to the use of palladium as a catalyst, first described by Ohta with the use of Pd2(dba)3·CHCl3/PPh332 and investigated by Wu and Cheng with the use of PdCl2/P(1-Naphtyl)3.33 Under the optimized conditions described by these authors, the use of Pd2(dba)3·CHCl3/PPh3 led to the desired compound 6a in low yield despite a satisfactory conversion (entry 5), while the use of PdCl2/P(1-Naphtyl)3 led to a complete conversion but the desired compound 6a was isolated in only 20% yield (entry 6). Gratifyingly, by changing the conditions described in the literature, specifically by performing the reaction at a lower temperature, a cleaner reaction was observed and the compound 6a was isolated in a satisfactory 60% yield (entry 7). Thus, this study demonstrated that it is possible to perform 1,2-addition reactions of boronic acids on AMF, opening up a wide range of possibilities considering the large number of commercially available boronic acid derivatives.
The reactivity control of substrate 6a for the Piancatelli rearrangement is a real challenge that requires overcoming three major difficulties. First is the low reactivity of furan substrates substituted at the C-5 position. Second is the control of the diastereoselectivity of the two newly formed stereogenic centers, including one quaternary center. The final challenge is preserving the stability and integrity of the alkyl azide functional group. First, we examined the reactivity of substrate 6a, under the same microwave conditions (MW-190) applied to the unsubstituted substrate 4, but despite total conversion, only the decomposition of this substrate was observed (Part II in the ESI, Table S1,† entry 1). In contrast, under milder microwave conditions (MW-100), no reaction was observed and the starting material was cleanly recovered unchanged (Table 2, entry 1).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Conv. (%) | NMR yieldb (%) | Isolated yield (%) | drc7a/8a |
| MW = microwave heating. CH = conventional heating. ND = not determined. NA = not applicable.a Reactions performed on the 0.2 mmol scale at 0.1 M.b Yield determined by 1H NMR with trimethoxybenzene as the internal standard added at the end of the reaction.c The diastereoisomeric ratio (dr) was determined by 1H NMR on the crude mixture.d (C6HF4)B(OH)2 (20 mol%) at CH-90 for 24 h.e CH-50 for 18 h.f TFA (20 mol%).g 1 mmol scale and sodium dithionite (Na2S2O4) in a 3 mass percentage was added.h 37% of the starting material 6a was recovered. | |||||
| 1 | — | 0 | — | — | — |
| 2 | DyCl3 | 40 | 10 | ND | 80:20 |
| 3 | Dy(OTf) 3 | 60 | 50 | 46 |
>95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :5 :5
|
| 4 | Yb(OTf)3 | 70 | 30 | ND | 85:15 |
| 5 | Ce(OTf)3 | 80 | 20 | ND | ND |
| 6 | Sc(OTf)3 | 75 | 35 | ND | 80:20 |
| 7 | ScCl 3 | 70 | 40 | 37 |
95:5
|
| 8 | Y(OTf)3 | 65 | 35 | ND | 90:10 |
| 9 | Fe(OTf)3 | 80 | 20 | ND | 90:10 |
| 10 | Co(OTf)3 | 70 | 30 | ND | 85:15 |
| 11 | HAuCl4·3H2O | 100 | 10 | ND | ND |
| 12 | Al(OTf)3 | 85 | 25 | ND | 85:15 |
| 13 | Zn(OTf)3 | 65 | 10 | ND | ND |
| 14 | In(OTf)3 | 80 | 20 | ND | 90:10 |
| 15 | Bi(OTf)3 | 75 | 15 | ND | ND |
| 16 | Ca(NTf)2 | 0 | NA | NA | NA |
| 17 | Ba(OTf)3 | 70 | 20 | ND | ND |
| 18 | B(C6F5)3 | 50 | 25 | ND | 90:10 |
| 19d | (C6HF4)B(OH)2 | 70 | 0 | NA | NA |
| 20e | BINOL·PA | 90 | 25 | ND | ND |
| 21f | TFA | 100 | 15 | ND | 90:10 |
| 22g | Dy(OTf)3 | 60 | 50 | 34h | >95:5 |
| 23g | ScCl3 | 65 | 25 | 18 | 95:5 |

We then applied the conditions previously developed for the Piancatelli rearrangement for the C-5 substituted 2-furylcarbinol substrates, specifically the use of a catalytic amount of DyCl3 under microwave activation in a mixture of t-BuOH/H2O,24 but a low conversion was observed associated with a poor NMR yield and modest diastereoisomeric ratio.
In order to improve this conversion reaction, Dy(OTf)3 was used as a dysprosium catalyst21b,23,34 under microwave activation24, which allowed the formation of the desired cyclopent-2-enone 7a with a satisfactory 46% yield and high diastereoselectivity, despite an incomplete conversion (entry 3). All attempts to achieve completion, which included the increase of catalyst loading, time, temperature, conventional heating (CH) and also the use of TFA as a co-catalyst (since a combination of Lewis and Brønsted acids has been well known to improve the yield of these rearrangements),21b and also the change of the counteranion failed (Part II in the ESI, Table S1,† entries 6–11). We considered other lanthanide catalysts used for the Piancatelli rearrangement like ytterbium35 or cerium,36 but no improvement was observed (entries 4 and 5), either under the described conditions or under our microwave conditions.
We next expanded our screening to include transition metals as catalysts for this Piancatelli rearrangement with the most commonly used catalyst, that is Sc(OTf)3,21b,34a but once again an incomplete reaction was observed and unfortunately associated with poor diastereoselectivity (entry 6). This lack of stereoselectivity can be attributed to the high reactivity of scandium triflate.37 In order to moderate this reactivity, scandium trichloride, a milder catalyst, was considered for the first time to promote the Piancatelli rearrangement. With the use of ScCl3 as the catalyst, we have observed an incomplete but clean reaction, and the desired cyclopent-2-enone 7a was obtained in 37% isolated yield with high diastereoselectivity (entry 7). An increase in temperature or the use of scandium triflimide did not improve the conversion of the reaction (Part II in the ESI, Table S1,† entries 17 and 18). The use of a transition metal from the row below, yttrium,38 as well as other transition metals previously used with success for the Piancatelli rearrangement in the literature, namely iron,39 cobalt,40 or even gold,41 did not improve the efficiency of the rearrangement of 6a (entries 8–11).
The use of non-noble metals also known to trigger the Piancatelli rearrangement, such as aluminum,36 zinc,42 indium36 and bismuth,43 also led to disappointing results for the formation of compound 7a (entries 12–15). The use of alkaline earth metals, such as calcium44 or barium,45 is also well known to promote the Piancatelli rearrangement, but is inadequate for our substrate 6a, whether under standard conditions or under our microwave conditions (entries 16 and 17).
We have speculated that using a non-metallic Lewis acid such as B(C6F5)3, applied recently in the literature for the Piancatelli rearrangement,45,46 might be more compatible with the azide function, but its use also led to a low reaction conversion associated with a low yield in the formation of compound 7a (entry 18). Brønsted acids, such as (C6HF4)B(OH)2,47 BINOL-derived phosphoric acid48 and TFA,34b,35 were also considered, but despite the high level of conversion of substrate 6a, compound 7a was formed in very low yields (entries 19–21).
We chose to retain the two conditions using Dy(OTf)3 as the lanthanide catalyst and ScCl3 as the transition metal catalyst under microwave activation. These two selected optimized conditions have been applied to a 1 mmol scale with sodium dithionite (at a 3 mass percentage). Sodium dithionite (Na2S2O4) is a well-known additive for HMF derivative reactions, acting as a reductive agent able to avoid oxidative pathways that are involved in the oligomerization of HMF derivatives and the resulting humin formation.6b
For this 1 mmol scale experiment performed with Dy(OTf)3 as the lanthanide catalyst, high diastereoselectivity was maintained but the yield decreased slightly to 34%. But more interestingly, we were able to recover 37% yield of the starting material 6a, thus leading to 7a in a 54% corrected yield, based on the conversion of 6a (entry 22).
For this same 1 mmol scale experiment performed with ScCl3 as the transition metal catalyst and Na2S2O4 as the additive, the yield was severely reduced to 18% (entry 23).
The trans relative stereochemistry of cyclopentenone 7a is the result of the relative stability of pentadienyl cation intermediates and the pericyclic conrotatory 4π-electrocyclization process, closely related to the Nazarov reaction (Part III in the ESI, Scheme S1†).49 This trans relative stereochemistry was assigned based on NMR NOESY 2D experiments (Part IX in the ESI†).
Thus, for the continuation of our study, we examined the scope of (5-(azidomethyl)furan-2-yl)-α-substituted carbinol derivatives 6. These substrates were prepared starting from AMF 2, using the two identified conditions in Table 1, namely the use of Grignard reagents or the palladium-catalyzed 1,2-addition of boronic acids. The details of these preparations are provided in the ESI (Part VI in the ESI).†
Under the optimized reaction conditions for the Piancatelli rearrangement (Table 2, entry 3), namely Dy(OTf)3 10 mol%, t-BuOH/H2O 5:1, microwave heating, 100 °C, and 1.5 h, the variety of (5-(azidomethyl)furan-2-yl)-α-substituted carbinols 6a–n delivered the expected C5-substituted 4-(azidomethyl)-4-hydroxycyclopentenone 7a–n in moderate to good isolated yields (Scheme 2). The rearrangement of carbinols 6a–f into cyclopentenones 7a–f does not appear to depend on the steric hindrance of the substituent R, as the same range of yields was observed, along with good diastereoselectivities. The best yield was obtained with substrate 6d containing a 9-phenanthrenyl substituent that provides cyclopentenone 7d in 62% yield.
 | ||
| Scheme 2 Substrate scope in the Dy(OTf)3-catalyzed MW-100 °C Piancatelli rearrangement of (5-(azidomethyl)furan-2-yl)-α-substituted carbinols 6a–n. | ||
The reaction proved to be quite general, since both electron-withdrawing and electron-donating groups were tolerated on the aromatic substituent of 6, affording products 7 in the same range of yields. However, with an electron-withdrawing group, a slight decrease of diastereoselectivity was observed, which could be attributed to the higher acidity of the proton in the benzylic position, specifically for compound 7j with the perfluorophenyl substitution. The high level of diastereoselectivity was maintained with electron-donating groups for compounds 7k–m.
The use of 2-thienyl as the heteroaryl group resulted in a loss of diastereoselectivity for compound 7n. We would also like to mention that when the aryl group was replaced with an alkyl substituent, such as n-butyl, t-butyl or cyclopropyl, no reaction was observed and the starting material was cleanly recovered unchanged. The same Piancatelli rearrangements were also performed on carbinols 6 under optimized reaction conditions for ScCl3 as the catalyst (Table 2, entry 7). However, as observed for compound 6a, slightly lower yields were observed for the formation of cyclopentenones 7 compared to that with the use of Dy(OTf)3 as the catalyst (Part VII in the ESI†).
In the case of substrate 6l, in addition to the formation of cyclopentenone 7l, we also observed the formation of aldehyde compound 9 in 5% yield, which is the outcome of the decomposition of the primary alkyl azide followed by alpha hydrogen deprotonation to generate an aldimine, which, upon hydrolysis, releases the aldehyde function (Scheme 3). This oxidative conversion of primary azides to aldehydes, useful for the installation of aldehydes from a non-oxygenated precursory functional group, was previously described using molybdenum xanthate50 or iron(III) chloride51 under harsh reaction conditions, and more recently under milder conditions involving the use of metalloproteins such as cytochrome P450s and myoglobin.52
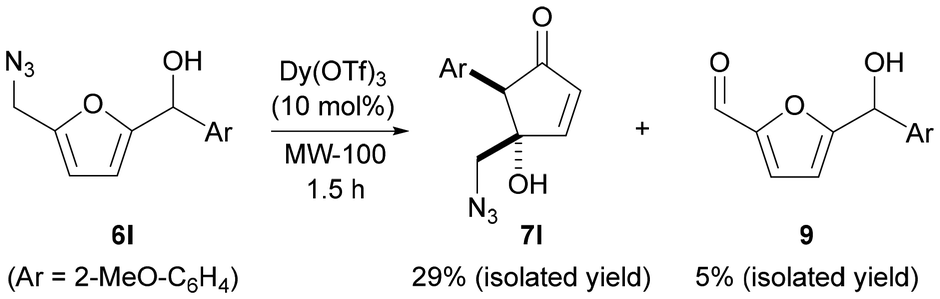 | ||
| Scheme 3 Denitrogenative reaction of 6l leading to the formation of aldehyde compound 9. | ||
To confirm the relative stability of this alkyl azide function under our reaction conditions, the azide function was introduced at an sp2-hybridized carbon center, both on the aromatic and vinylic parts of the cyclopentenone, leading to better yields in the Piancatelli rearrangement (Part IV in the ESI†). To overcome this issue of azide function stability, other nitrogen functions such as aliphatic or aromatic amines and protected the amine function in phthalimide derivatives have been considered, but all attempts for the Piancatelli rearrangement on these substrates failed (Part V in the ESI†).
To showcase the synthetic versatility of the obtained cyclopentenones 7 with the azidomethyl side chain, further functional group elaborations were performed (Scheme 4). First, the carbonyl function was selectively reduced towards the double bond and azide function under Luche53 conditions to provide the secondary alcohol 10 in 76% yield, without stereoselectivity as previously observed in the literature for similar substrates.54 Azide and double reduction were performed via the mild catalytic hydrogenation in the presence of Boc2O,55 which gave the cyclopentanone 11 with the N-Boc-protected-methyl side chain in 40% yield.
 | ||
| Scheme 4 Synthetic transformation of 4-(azidomethyl)-cyclopentenones 7. | ||
Moreover, subjecting 7a and 7e to the conditions of copper-catalyzed azide–alkyne cycloaddition (CuAAC)56 with phenylacetylene resulted in the formation of cyclopentenones with a triazole-methyl side chain 12 and 13 in 88% and 99% yields, respectively.
As some natural products with a hydroxylated cyclopentenone scaffold, such as the marine natural products trichodenone A,57 didemnenone D58 and Terrein59 from the fungi Aspergillus terreus, have exhibited significant cytotoxicity against cancer cell lines, we evaluated the cytotoxicity activity of the newly synthesized compounds 7a–n. Moreover, we also intend to take advantage of the azidomethyl side chain, as many well-known chemotherapeutic drugs contain these metabolically active azide groups.60
Biological preliminary results are highly promising as compounds 7a–n exert antiproliferative effects at nanomolar concentrations for two different tumor cell lines. Concentrations inducing 50% cell growth inhibition (IC50) are given in Tables 3 and 4. The first observation is that substitution with an aromatic moiety on the C5 position of the cyclopentenone is necessary, as the unsubstituted compound 5 does not exhibit cytotoxic activity.
| Compound | IC50a (nM) | Compound | IC50a (nM) |
|---|---|---|---|
| a Data are the mean ± standard error (SEM) of three independent experiments. | |||
| 5 | >10000 |
7h | 2462 ± 316 |
| 7a | 2607 ± 243 | 7i | 742 ± 16 |
| 7b | 1570 ± 241 | 7j | 811 ± 43 |
| 7c | 2700 ± 222 | 7k | 294 ± 129 |
| 7d | 1080 ± 144 | 7l | 3163 ± 243 |
| 7e | 491 ± 16 | 7m | 1320 ± 16 |
| 7f | 3295 ± 880 | 7n | 568 ± 29 |
| 7g | 3724 ± 465 | ||
| Compound | IC50a (nM) | Compound | IC50a (nM) |
|---|---|---|---|
| a Data are the mean ± standard error (SEM) of three independent experiments. | |||
| 5 | 10200 ± 180 |
7h | 126 ± 3.7 |
| 7a | 145 ± 4.3 | 7i | 144 ± 64 |
| 7b | 220 ± 33 | 7j | 194 ± 19 |
| 7c | 136 ± 10.6 | 7k | 535 ± 43 |
| 7d | 131 ± 8.4 | 7l | 149 ± 7.5 |
| 7e | 104 ± 1.5 | 7m | 190 ± 16 |
| 7f | 264 ± 64 | 7n | 330 ± 10 |
| 7g | 160 ± 2.8 | ||
For all the C5 substituted compounds, the antiproliferative effects are more pronounced on the HL60 non-adherent leukemia cell line. Moreover, some compounds such as 7c, 7d and 7h behave differently towards these two tumor cell lines, which may indicate some tissue specific antiproliferative activity of these molecules.
Conclusions
In conclusion, we have shown for the first time that AMF (5-azidomethylfurfural) derivatives, biosourced renewable materials directly obtained from CMF (5-chloromethylfurfural), could lead to nitrogenous cyclopentenones with an azidomethyl side chain. These cyclopentenones with two contiguous stereogenic centres, one of which is quaternary, were obtained in moderate to good yields with high diastereoselectivity via the Piancatelli rearrangement. ScCl3 as a transition metal catalyst was used for the first time to catalyse this rearrangement, and even better results were obtained using lanthanide Dy(OTf)3 as the catalyst.The introduction of this azido group is particularly innovative in these Piancatelli rearrangements. On the one hand, it allows taking advantage of the high synthetic potential of this functional group, which can be derivatized into a wide variety of other functionalities and for the synthesis of a range of nitrogen-based scaffolds of biologically active compounds and functional materials. Moreover, these 4-(azidomethyl)-cyclopentenones 7 exhibited relevant cytotoxic activity against HCT116 and HL60 cancer cell lines with nanomolar IC50 values.
This work has led to the first proof of concept that a nitrogen-containing substrate can participate in the Piancatelli rearrangement. We have no doubt that this methodology, based on the Piancatelli rearrangement on a substrate with the azido function, will subsequently be used to construct other nitrogen derivatives with multiple applications.
Author contributions
J-FB designed the experiments and supervised the project. CM performed all chemical syntheses and characterization of data of compounds. JB carried out the bioassays for cytotoxicity activities. J-FB wrote the manuscript and CM revised the manuscript. All the authors have given approval to the final version of the manuscript.Data availability
The authors confirm that the data supporting this article and the findings of this study have been included as part of the ESI.† The software used for drawing chemical structures is ChemDraw 23.0. The dose–response curves were plotted using GraphPad Prism software and the IC50 values were calculated from these polynomial curves using the same software.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support of the “Centre National de la Recherche Scientifique” (CNRS), the “Institut de Chimie des Substances Naturelles” (ICSN) and the “Agence Nationale de la Recherche” (ANR) for financial support (AntiCyp project, Grant N° ANR-21-CE07-0055).References
- (a) D. C. Elliott, Historical Developments in Hydroprocessing Bio-oils, Energy Fuels, 2007, 21, 1792–1815 CrossRef CAS; (b) R. Mori, Replacing all petroleum-based chemical products with natural biomass-based chemical products: a tutorial review, RSC Sustainability, 2023, 1, 179–212 CAS.
- C. Liu, H. Wang, A. M. Karim, J. Sun and Y. Wang, Catalytic fast pyrolysis of lignocellulosic biomass, Chem. Soc. Rev., 2014, 43, 7594–7623 CAS.
- S. Chen, R. Wojcieszak, F. Dumeignil, E. Marceau and S. Royer, How Catalysts and Experimental Conditions Determine the Selective Hydroconversion of Furfural and 5-Hydroxymethylfurfural, Chem. Rev., 2018, 118, 11023–11117 Search PubMed.
- J. J. Bozell and G. R. Petersen, Technology development for the production of biobased products from biorefinery carbohydrates-the US Department of Energy's “Top 10” revisited, Green Chem., 2010, 12, 539–554 CAS.
- K. I. Galkin, E. A. Krivodaeva, L. V. Romashov, S. S. Zalesskiy, V. V. Kachala, J. V. Burykina and V. P. Ananikov, Critical Influence of 5-Hydroxymethylfurfural Aging and Decomposition on the Utility of Biomass Conversion in Organic Synthesis, Angew. Chem., Int. Ed., 2016, 55, 8338–8342 CAS.
- (a) A. Páez, H. A. Rojas, O. Portilla, G. Sathicq, C. A. M. Afonso, G. P. Romanelli and J. J. Martínez, Preyssler Heteropolyacids in the Self-Etherification of 5-Hydroxymethylfurfural to 5,5′-[Oxybis(methylene)]bis-2-furfural Under Mild Reaction Conditions, ChemCatChem, 2017, 9, 3322–3329 Search PubMed; (b) R. F. A. Gomes, Y. N. Mitrev, S. P. Simeonov and C. A. M. Afonso, Going Beyond the Limits of the Biorenewable Platform: Sodium Dithionite-Promoted Stabilization of 5-Hydroxymethylfurfural, ChemSusChem, 2018, 11, 1612–1616 CAS.
- (a) C. Rosenfeld, J. Konnerth, W. Sailer-Kronlachner, P. Solt, T. Rosenau and H. W. G. van Herwijnen, Current Situation of the Challenging Scale-Up Development of Hydroxymethylfurfural Production, ChemSusChem, 2020, 13, 3544–3564 Search PubMed; (b) M. Sajid, U. Farooq, G. Bary, M. M. Azim and X. Zhao, Sustainable production of levulinic acid and its derivatives for fuel additives and chemicals: progress, challenges, and prospects, Green Chem., 2021, 23, 9198–9238 Search PubMed.
- S. Constant, C. S. Lancefield, W. Vogelzang, R. K. Pazhavelikkakath Purushothaman, A. E. Frissen, K. Houben, P. de Peinder, M. Baldus, B. M. Weckhuysen, D. S. van Es and P. C. A. Bruijnincx, Molecular structure and composition elucidation of an industrial humin and its fractions, Green Chem., 2024, 26, 7739–7751 Search PubMed.
- M. Mascal, 5-(Chloromethyl)furfural is the New HMF: Functionally Equivalent But More Practical in Terms of its Production From Biomass, ChemSusChem, 2015, 8, 3391–3395 Search PubMed.
- D. Soukup-Carne, F. S. Bragagnolo, C. S. Funari and J. Esteban, Production and Synthetic Possibilities of 5-Chloromethylfurfural as Alternative Biobased Furan, Catalysts, 2024, 14, 117 CAS.
- S. Karimi, S. Gharouni Fattah, Z. Li, M. Zuo, M. Nasrollahzadeh and X. Zeng, A comparative study of 5-(chloromethyl)furfural and 5-(hydroxymethyl)furfural, Green Chem., 2025, 27, 379–402 Search PubMed.
- M. Mascal, 5-(Chloromethyl)furfural (CMF): A Platform for Transforming Cellulose into Commercial Products, ACS Sustainable Chem. Eng., 2019, 7, 5588–5601 CAS.
- M. Mascal and S. Dutta, Synthesis of the natural herbicide δ-aminolevulinic acid from cellulose-derived 5-(chloromethyl)furfural, Green Chem., 2011, 13, 40–41 Search PubMed.
- M. N. Masuno, D. A. Hirsch-Weil, R. L. Smith and J. A. Bissell II, Methods of producing compounds from 5-(halomethyl)furfural, WO2015175528, 2015.
- K. S. Kozlov, L. V. Romashov and V. P. Ananikov, A tunable precious metal-free system for selective oxidative esterification of biobased 5-(hydroxymethyl)furfural, Green Chem., 2019, 21, 3464–3468 RSC.
- B. Y. Karlinskii, L. V. Romashov, K. I. Galkin, P. G. Kislitsyn and V. P. Ananikov, Synthesis of 2-Azidomethyl-5-ethynylfuran: A New Bio-Derived Self-Clickable Building Block, Synthesis, 2019, 1235–1242 CAS.
- A. B. Popov, I. Stolić, L. Krstulović, M. C. Taylor, J. M. Kelly, S. Tomić, L. Tumir, M. Bajić and S. Raić-Malić, Novel symmetric bis-benzimidazoles: Synthesis, DNA/RNA binding and antitrypanosomal activity, Eur. J. Med. Chem., 2019, 173, 63–75 CrossRef PubMed.
- D. Gupta and B. Saha, Carbon nanosphere supported Ru catalyst for the synthesis of renewable herbicide and chemicals, Catal. Commun., 2017, 100, 206–209 Search PubMed.
- G. Piancatelli, A. Scettri and S. Barbadoro, A useful preparation of 4-substituted 5-hydroxy-3-oxocyclopentene, Tetrahedron Lett., 1976, 17, 3555–3558 CrossRef.
- (a) C. Verrier, S. Moebs-Sanchez, Y. Queneau and F. Popowycz, The Piancatelli reaction and its variants: recent applications to high added-value chemicals and biomass valorization, Org. Biomol. Chem., 2018, 16, 676–687 Search PubMed; (b) S. Zhong, L. Xu and Y. Cai, Recent Advances on Piancatelli Reactions and Related Cascade Processes, Synthesis, 2022, 589–599 Search PubMed.
- (a) G. Piancatelli, A. Scettri, G. David and M. D'Auria, A new synthesis of 3-oxocyclopentenes, Tetrahedron, 1978, 34, 2775–2778 Search PubMed; (b) D. Fisher, L. I. Palmer, J. E. Cook, J. E. Davis and J. Read de Alaniz, Efficient synthesis of 4-hydroxycyclopentenones: dysprosium(III) triflate catalyzed Piancatelli rearrangement, Tetrahedron, 2014, 70, 4105–4110 Search PubMed.
- M. D'Auria, F. D'Onofrio, G. Piancatelli and A. Scettri, A convenient synthesis of 2-bromo-4-hydroxy-2-cyclopenten-1-ones and 3-bromo-4-hydroxy-2-cyclopenten-1-ones, Gazz. Chim. Ital., 1986, 116, 173–175 Search PubMed.
- A. Bredihhin, S. Luiga and L. Vares, Application of 5-Ethoxymethylfurfural (EMF) for the Production of Cyclopentenones, Synthesis, 2016, 4181–4188 Search PubMed.
- F. Cacheux, G. Le Goff, J. Ouazzani, J. Bignon, P. Retailleau, A. Marinetti, A. Voituriez and J.-F. Betzer, The Piancatelli rearrangement of non-symmetrical furan-2,5-dicarbinols for the synthesis of highly functionalized cyclopentenones, Org. Chem. Front., 2021, 8, 2449–2455 Search PubMed.
- (a) K. Ulbrich, P. Kreitmeier and O. Reiser, Microwave- or Microreactor-Assisted Conversion of Furfuryl Alcohols into 4-Hydroxy-2-cyclopentenones, Synlett, 2010, 2037–2040 Search PubMed; (b) A. Saitman and E. A. Theodorakis, Synthesis of a Highly Functionalized Core of Verrillin, Org. Lett., 2013, 15, 2410–2413 Search PubMed; (c) S. Specklin, A. Dikova, A. Blanc, J.-M. Weibel and P. Pale, Chemoenzymatic routes to cyclopentenols: the role of protecting groups on stereo- and enantioselectivity, Tetrahedron Lett., 2014, 55, 6987–6991 CrossRef.
- (a) J. Maibaum, S. Stutz, R. Göschke, P. Rigollier, Y. Yamaguchi, F. Cumin, J. Rahuel, H.-P. Baum, N.-C. Cohen, C. R. Schnell, W. Fuhrer, M. G. Gruetter, W. Schilling and J. M. Wood, Structural Modification of the P2′ Position of 2,7-Dialkyl-Substituted 5(S)-Amino-4(S)-hydroxy-8-phenyl-octanecarboxamides: The Discovery of Aliskiren, a Potent Nonpeptide Human Renin Inhibitor Active after Once Daily Dosing in Marmosets, J. Med. Chem., 2007, 50, 4832–4844 CrossRef PubMed; (b) E. K. W. Tam, Z. Li, Y. L. Goh, X. Cheng, S. Y. Wong, S. Santhanakrishnan, C. L. L. Chai and S. Q. Yao, Cell-Based Proteome Profiling Using an Affinity-Based Probe (AfBP) Derived from 3-Deazaneplanocin A (DzNep), Chem. –Asian J., 2013, 8, 1818–1828 CrossRef PubMed; (c) F. Požgan, B. Štefane, D. Kiđemet, J. Smodiš and R. Zupet, A New Synthetic Route Towards Aliskiren Intermediates, Synthesis, 2014, 3221–3228 Search PubMed.
- (a) B. M. Trost and W. H. Pearson, Sulfur activation of azides toward addition of organometallics. Amination of aliphatic carbanions, J. Am. Chem. Soc., 1983, 105, 1054–1056 Search PubMed; (b) G. W. Kabalka and G. Li, Synthesis of aromatic amines using allyl azide, Tetrahedron Lett., 1997, 38, 5777–5778 CrossRef; (c) H. M. S. Kumar, B. V. S. Reddy, S. Anjaneyulu and J. S. Yadav, A novel and efficient approach to mono-N-alkyl anilines via addition of grignard reagents to aryl azides, Tetrahedron Lett., 1999, 40, 8305–8306 CrossRef.
- M. Sakai, M. Ueda and N. Miyaura, Rhodium-Catalyzed Addition of Organoboronic Acids to Aldehydes, Angew. Chem., Int. Ed., 1998, 37, 3279–3281 CrossRef.
- M. Ueda and N. Miyaura, A Large Accelerating Effect of Tri(tert-butyl)phosphine in the Rhodium-Catalyzed Addition of Arylboronic Acids to Aldehydes, J. Org. Chem., 2000, 65, 4450–4452 CrossRef PubMed.
- A. Fürstner and H. Krause, Practical Method for the Rhodium-Catalyzed Addition of Aryl- and Alkenylboronic Acids to Aldehydes, Adv. Synth. Catal., 2001, 343, 343–350 CrossRef.
- P. M. P. Gois, A. F. Trindade, L. F. Veiros, V. André, M. T. Duarte, C. A. M. Afonso, S. Caddick and F. G. N. Cloke, Tuning the Reactivity of Dirhodium(II) Complexes with Axial N-Heterocyclic Carbene Ligands: The Arylation of Aldehydes, Angew. Chem., Int. Ed., 2007, 46, 5750–5753 Search PubMed.
- T. Yamamoto, T. Ohta and Y. Ito, Palladium-Catalyzed Addition of Arylboronic Acids to Aldehydes, Org. Lett., 2005, 7, 4153–4155 CrossRef PubMed.
- C. Qin, H. Wu, J. Cheng, X. a. Chen, M. Liu, W. Zhang, W. Su and J. Ding, The Palladium-Catalyzed Addition of Aryl- and Heteroarylboronic Acids to Aldehydes, J. Org. Chem., 2007, 72, 4102–4107 Search PubMed.
- (a) G. K. Veits, D. R. Wenz and J. Read de Alaniz, Versatile Method for the Synthesis of 4-Aminocyclopentenones: Dysprosium(III) Triflate Catalyzed Aza-Piancatelli Rearrangement, Angew. Chem., Int. Ed., 2010, 49, 9484–9487 Search PubMed; (b) D. Yu, V. T. Thai, L. I. Palmer, G. K. Veits, J. E. Cook, J. Read de Alaniz and J. E. Hein, Importance of Off-Cycle Species in the Acid-Catalyzed Aza-Piancatelli Rearrangement, J. Org. Chem., 2013, 78, 12784–12789 Search PubMed.
- S. Gouse, N. R. Reddy and S. Baskaran, A Domino Aza-Piancatelli Rearrangement/Intramolecular Diels–Alder Reaction: Stereoselective Synthesis of Octahydro-1H-cyclopenta[cd]isoindole, Org. Lett., 2019, 21, 3822–3827 CrossRef PubMed.
- B. V. S. Reddy, Y. V. Reddy, P. S. Lakshumma, G. Narasimhulu, J. S. Yadav, B. Sridhar, P. P. Reddy and A. C. Kunwar, In(OTf)3-catalyzed tandem aza-Piancatelli rearrangement/Michael reaction for the synthesis of 3,4-dihydro-2H-benzo[b][1,4]thiazine and oxazine derivatives, RSC Adv., 2012, 2, 10661–10666 RSC.
- S.-W. Li and R. A. Batey, Mild lanthanide(III) catalyzed formation of 4,5-diaminocyclopent-2-enones from 2-furaldehyde and secondary amines: a domino condensation/ring-opening/electrocyclization process, Chem. Commun., 2007, 3759–3761 RSC.
- L. Xu, Q. Yang, S. Zhong, H. Li, Y. Tang and Y. Cai, Ln(III)/Chiral Brønsted Acid Catalyzed Asymmetric Cascade Ring Opening/Aza-Piancatelli Rearrangement of D–A Cyclopropanes, Org. Lett., 2020, 22, 9016–9021 CrossRef PubMed.
- J. Liu, Q. Shen, J. Yu, M. Zhu, J. Han and L. Wang, A Concise Domino Synthesis of Benzo-1,4-heterocycle Compounds via a Piancatelli/C–N Coupling/Michael Addition Process Promoted by La(OTf)3, Eur. J. Org. Chem., 2012, 6933–6939 CrossRef.
- B. Shen, Q. He, S. Dong, X. Liu and X. Feng, A chiral cobalt(II) complex catalyzed enantioselective aza-Piancatelli rearrangement/Diels–Alder cascade reaction, Chem. Sci., 2020, 11, 3862–3867 RSC.
- M. A. Tzani and I. N. Lykakis, Gold(III) Chloride-Mediated Transformation of Furfural to the trans-N,N-4,5-Diaminocyclopent-2-enones in the Presence of Anilines, Chemistry, 2023, 5, 393–405 CrossRef.
- J. P. Henschke, Y. Liu, X. Huang, Y. Chen, D. Meng, L. Xia, X. Wei, A. Xie, D. Li, Q. Huang, T. Sun, J. Wang, X. Gu, X. Huang, L. Wang, J. Xiao and S. Qiu, The Manufacture of a Homochiral 4-Silyloxycyclopentenone Intermediate for the Synthesis of Prostaglandin Analogues, Org. Process Res. Dev., 2012, 16, 1905–1916 CrossRef.
- S. Dhiman and S. S. V. Ramasastry, Taming furfuryl cations for the synthesis of privileged structures and novel scaffolds, Org. Biomol. Chem., 2013, 11, 4299–4303 Search PubMed.
- D. Lebœuf, L. Marin, B. Michelet, A. Perez-Luna, R. Guillot, E. Schulz and V. Gandon, Harnessing the Lewis Acidity of HFIP through its Cooperation with a Calcium(II) Salt: Application to the Aza-Piancatelli Reaction, Chem. – Eur. J., 2016, 22, 16165–16171 Search PubMed.
- P. R. Solanke, P. Kumar, P. S. Mainkar, K. Nayani and S. Chandrasekhar, Synthesis of cis-fused cyclopentenone-pyrrolidine scaffolds via sequential aza-Piancatelli and Conia-ene type reactions in one pot, Chem. Commun., 2024, 60, 4234–4237 Search PubMed.
- S. Raju, P. Ghosh, K. Nayani, J. Prashanth, B. Sridhar, P. S. Mainkar and S. Chandrasekhar, Construction of Octahydro-4H-cyclopenta[b]pyridin-6-one Skeletons using Pot, Atom, and Step Economy (PASE) Synthesis, Chem. – Eur. J., 2023, 29, e202301058 Search PubMed.
- W.-B. Tang, K.-S. Cao, S.-S. Meng and W.-H. Zheng, Boronic Acid Catalysis for Aza-Piancatelli Rearrangement, Synthesis, 2017, 3670–3675 Search PubMed.
- (a) Y. Cai, Y. Tang, I. Atodiresei and M. Rueping, Catalytic Asymmetric Piancatelli Rearrangement: Brønsted Acid Catalyzed 4π Electrocyclization for the Synthesis of Multisubstituted Cyclopentenones, Angew. Chem., Int. Ed., 2016, 55, 14126–14130 Search PubMed; (b) H. Li, R. Tong and J. Sun, Catalytic Enantioselective Aza-Piancatelli Rearrangement, Angew. Chem., Int. Ed., 2016, 55, 15125–15128 CrossRef PubMed.
- O. Nieto Faza, C. Silva López, R. Álvarez and Á. R. de Lera, Theoretical Study of the Electrocyclic Ring Closure of Hydroxypentadienyl Cations, Chem. – Eur. J., 2004, 10, 4324–4333 CrossRef PubMed.
- M. Maddani and K. R. Prabhu, A chemoselective aerobic oxidation of benzylic azides catalyzed by molybdenum xanthate in an aqueous medium, Tetrahedron Lett., 2008, 49, 4526–4530 CrossRef.
- H.-P. Zhang, Y.-Z. Dai and L.-M. Tao, Oxidation of Benzylic Azides Catalysed by Iron(III) Chloride with Hydrogen Peroxide as Oxidant, J. Chem. Res., 2011, 35, 720–722 Search PubMed.
- S. Giovani, R. Singh and R. Fasan, Efficient conversion of primary azides to aldehydes catalyzed by active site variants of myoglobin, Chem. Sci., 2016, 7, 234–239 RSC.
- Z. Wang, Luche Reduction, in Comprehensive Organic Name Reactions and Reagents, 2010, pp. 1782–1786 Search PubMed.
- M. T. Barros, C. M. Alves, A. G. Santos, L. S. Godinho and C. D. Maycock, On the diastereoselectivity of the 1,2-reduction of 2-alkyl-4-hydroxycyclopentenones with sodium borohydride in the presence of cerium(III): Synthesis of prostaglandin precursors, Tetrahedron Lett., 1995, 36, 2321–2324 CrossRef.
- V. V. Semeno, V. O. Vasylchenko, I. M. Fesun, L. Y. Ruzhylo, M. O. Kipriianov, K. P. Melnykov, A. Skreminskyi, R. Iminov, P. Mykhailiuk, B. V. Vashchenko and O. O. Grygorenko, Bicyclo[m.n.k]alkane Building Blocks as Promising Benzene and Cycloalkane Isosteres: Multigram Synthesis, Physicochemical and Structural Characterization, Chem. – Eur. J., 2024, 30, e202303859 CrossRef PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 Search PubMed.
- T. Amagata, Y. Usami, K. Minoura, T. Ito and A. Numata, Cytotoxic substances produced by a fungal strain from a sponge: physico-chemical properties and structures, J. Antibiot., 1998, 51, 33–40 Search PubMed.
- K.-D. Feussner, K. Ragini, R. Kumar, K. M. Soapi, W. G. Aalbersberg, M. K. Harper, B. Carte and C. M. Ireland, Investigations of the marine flora and fauna of the Fiji Islands, Nat. Prod. Rep., 2012, 29, 1424–1462 Search PubMed.
- W.-Y. Liao, C.-N. Shen, L.-H. Lin, Y.-L. Yang, H.-Y. Han, J.-W. Chen, S.-C. Kuo, S.-H. Wu and C.-C. Liaw, Asperjinone, a Nor-Neolignan, and Terrein, a Suppressor of ABCG2-Expressing Breast Cancer Cells, from Thermophilic Aspergillus terreus, J. Nat. Prod., 2012, 75, 630–635 CrossRef PubMed.
- P. Sivaguru, Y. Ning and X. Bi, New Strategies for the Synthesis of Aliphatic Azides, Chem. Rev., 2021, 121, 4253–4307 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc06306f |
| This journal is © The Royal Society of Chemistry 2025 |