
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTailoring the electronic structure and acid–base properties of MgO by Ce doping promotes biomass-derived formic acid production at room temperature†
Yiqi
Geng
a,
Wenhua
Xue
a,
Jian
Ye
a,
Ruilong
Zhang
a,
Puranjan
Mishra
a and
Jun
Zhao
 *ab
*ab
aSino-Forest Applied Research Centre for Pearl River Delta Environment, Department of Biology, Hong Kong Baptist University, Kowloon Tong, Hong Kong SAR. E-mail: zhaojun@hkbu.edu.hk
bInstitute of Advanced Materials, Hong Kong Baptist University, Kowloon Tong, Hong Kong SAR
First published on 18th February 2025
Abstract
Biomass-based monosaccharide oxidation for formic acid production is significant due to its potential to provide a sustainable, bio-based alternative to traditional fossil fuel-derived methods of formic acid synthesis. In this study, we developed a Ce–MgO catalyst by incorporating Ce to enhance the oxidation of glucose to formic acid. Compared to unmodified MgO, the Ce–MgO catalyst exhibits an increased number of basic sites and higher charge densities at the Mg and O sites. These modifications facilitate the selective dissociation of hydrogen peroxide to form ˙OOH species and enhance the adsorption of ˙OOH at the MgO sites. The electron-rich nature of these Mg(OH)(OOH) active sites lowers the energy barrier for the C–C cleavage and oxidation reaction through more efficient electron transfer. Consequently, the reaction can be conducted at room temperature, achieving a 97.34% conversion of glucose and 93.65% yield of formic acid, which represents the highest performance among all glucose oxidation catalysts for formic acid production. Furthermore, the Ce–MgO catalyst demonstrated its efficacy in catalyzing the oxidation of a mixed sugar solution derived from corncob, achieving a formic acid yield of 49.13% at 30 °C. Additionally, the formic acid produced via this process enables in situ hydrogen production at room temperature, highlighting an effective and sustainable approach for generating green hydrogen from biomass.
Green foundation1. This work developed a Ce-doped MgO catalyst for efficient formic acid production from biomass-derived glucose at room temperature. The catalyst improves selectivity and energy efficiency, reducing reliance on traditional fossil fuel-based methods.2. The study's key achievement is achieving 97.34% glucose conversion and 93.65% formic acid yield under mild conditions, the highest among glucose oxidation catalysts for formic acid production. 3. Further exploration into the use of renewable biomass feedstocks, such as agricultural waste or other non-food sources, and integrating the process with green hydrogen production could make the process even more sustainable. |
1. Introduction
Biomass resources, recognized as green and renewable raw materials, offer a wide range of potential applications, particularly in the synthesis of chemicals such as formic acid (FA). FA serves as a liquid hydrogen carrier, demonstrating considerable promise due to its high hydrogen content, safety profile, ease of transportation, renewability, and efficient hydrogen release capabilities.1–3 The production of FA from biomass not only reduces reliance on fossil fuels but also effectively mitigates greenhouse gas emissions. Carbohydrates present in biomass provide abundant precursors for FA synthesis, which can be efficiently converted through catalytic and advanced technological processes.4–6 Therefore, the advancement of efficient and economically viable technologies for the conversion of carbohydrates to FA holds substantial theoretical significance and practical value.Recent research on the conversion of carbohydrates, particularly glucose, into FA via oxidative reactions has garnered significant attention.7–10 Glucose, a widely available monosaccharide derived from renewable biomass sources, exhibits excellent biocompatibility and biodegradability. The conversion of glucose to FA presents a novel approach for the efficient utilization of biomass resources. Moreover, the oxidation of glucose can be enhanced through the use of catalysts and the optimization of reaction conditions, thereby improving reaction efficiency and reducing both energy consumption and costs. Among various techniques, including catalytic oxidation, photocatalysis, and electrocatalysis, the base-catalyzed oxidative conversion of carbohydrates to FA stands out due to its mild reaction conditions and minimal energy consumption.11–15 Alkali catalysts typically utilize hydrogen peroxide (H2O2) as a green oxidizing agent to generate reactive species, including superoxide anions (O2˙), hydroxyl radicals (˙OH), and perhydroxyl radicals (˙OOH), thereby facilitating the cleavage of C–C bonds in glucose and promoting its oxidation.16–18 Among these alkali catalysts, lithium hydroxide (LiOH) demonstrates excellent catalytic performance, achieving an FA yield of 91.3% at 35 °C. However, the use of homogeneous catalysts poses significant challenges in the catalyst separation and recovery processes. To mitigate the issues, heterogeneous alkali catalysts have been explored, with magnesium oxide (MgO) emerging as a promising candidate for the oxidation of glucose to FA. Nevertheless, MgO demonstrates optimal catalytic performance only at temperatures exceeding 50 °C due to the limited H2O2 activation and oxidation capacity.19–22 Compared to homogeneous catalysts, there remains potential for further improvement in reaction conditions using heterogeneous catalysts. Therefore, it is expected that the MgO catalyst would be modified to enhance its catalytic oxidation ability and facilitate the reaction under milder conditions.
Herein, cerium (Ce) was introduced into the MgO catalyst to modulate the charge density around the magnesium (Mg) and oxygen (O) atoms. Experimental results and simulation calculations indicated that this modification significantly enhanced the selective activation of H2O2 to form ˙OOH species, which adsorbed on the Mg sites to form Mg(OH)(OOH) active sites. The electron-rich states of Mg and O contribute to the improved catalytic oxidation capability of Mg(OH)(OOH), effectively lowering the energy barriers associated with the cleavage and oxidation of glucose C–C bonds. In this manner, the synthesized Ce–MgO catalyst exhibits superior catalytic activity in converting various sugars into FA, surpassing the performance of previously reported catalysts.
2. Experimental section
2.1 Preparation of Ce–MgO catalysts
The Ce–MgO catalysts with different Ce/Mg molar ratios (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :200, 1:100, 1:50, and 1:25) were prepared using a hydrothermal method and named Ce0.5%–MgO, Ce1.0%–MgO, Ce2.0%–MgO and Ce4.0%–MgO, respectively. In a typical process, 0.024 mol of magnesium chloride (MgCl2) and a calculated amount of cerium nitrate hexahydrate (Ce(NO3)3·6H2O) were dissolved in 60 mL of deionized water. Then, the mixed solution was slowly added dropwise to 0.7 mol L−1 (35 mL) potassium carbonate (K2CO3) solution. After thorough stirring and precipitation, the resulting precipitate was hydrothermally heated in an oven at 180 °C for 2 h. The solid was washed several times and then calcined in a muffle furnace at 800 °C for 5 h. MgO catalysts without Ce addition were prepared by the same method for comparison.
:200, 1:100, 1:50, and 1:25) were prepared using a hydrothermal method and named Ce0.5%–MgO, Ce1.0%–MgO, Ce2.0%–MgO and Ce4.0%–MgO, respectively. In a typical process, 0.024 mol of magnesium chloride (MgCl2) and a calculated amount of cerium nitrate hexahydrate (Ce(NO3)3·6H2O) were dissolved in 60 mL of deionized water. Then, the mixed solution was slowly added dropwise to 0.7 mol L−1 (35 mL) potassium carbonate (K2CO3) solution. After thorough stirring and precipitation, the resulting precipitate was hydrothermally heated in an oven at 180 °C for 2 h. The solid was washed several times and then calcined in a muffle furnace at 800 °C for 5 h. MgO catalysts without Ce addition were prepared by the same method for comparison.
2.2 Conversion of glucose to FA over Ce–MgO catalysts
The catalytic oxidation of glucose to FA was carried out in a clear glass bottle. Typically, 120 mg of Ce–MgO catalyst, 0.5 mmol of glucose in 1.25 mL of water, 0.45 mL of H2O2 and 7 mL of deionized water were added into the reactor, followed by stirring at 30 °C. After the reaction, the solid catalysts were separated by centrifugation, washed several times and calcined in a muffle furnace at 500 °C for 4 h for the next run. The liquid product was analyzed by high performance liquid chromatography (HPLC, Agilent 1260 Infinity II) equipped with a refractive index detector (RID). The Aminex HPX-87H Organic Acid column was used for chromatographic separation in the mobile phase (5 mM H2SO4). The methods for glucose conversion, product yields, FA selectivity and kinetic calculations are detailed in the ESI.†To assess the scope of the reaction system, corncob was used as the raw material and subjected to hydrothermal treatment in a 0.1 M H2SO4 solution at 120 °C for 2 hours. The resulting hydrolyzed solutions containing glucose, xylose, and mannose were further studied for the oxidation reaction to FA over Ce–MgO catalysts under the same conditions.
2.3 Characterization
Scanning Electron Microscopy (SEM, Zeiss Sigma 500) and Transmission Electron Microscopy (TEM, FEI Tecnai G2 f20 S-Twin 200 kV) were employed to obtain the morphology and inner structure of the Ce–MgO catalysts. The chemical states were performed by X-ray Photoelectron Spectroscopy (XPS, Thermo K-Alpha+). X-ray diffraction (XRD, Thermo ESCALAB 250Xi) with Cu Kα source irradiation was used to observe the physical phases of the catalysts. CO2-temperature programmed desorption (CO2-TPD) and ammonia temperature-programmed desorption (NH3-TPD) were measured on TP-5080-B. The specific surface area and pore parameters of the Ce–MgO catalysts were calculated by the BET method and the BJH method (Micromeritics ASAP 2460 apparatus), respectively. Furthermore, the oxidation reaction over Ce–MgO catalyst was studied by in situ Fourier transform infrared spectroscopy (in situ FTIR, Invenio, Bruker, Germany). All spectra are absorption spectra, net of background.2.4 Theoretical calculations
The VASP software was used to perform density-functional theory (DFT) calculations. The PBE generalized gradient approximation (GGA) functional was used to determine the exchange–correlation energies. Stable structural configurations were obtained by structural relaxation. The energy cut-off and Monkhorst–Pack k-point grid settings were 450 eV and 4 × 4 × 1, respectively. During the geometry optimization process, the convergence criteria were established with an energy tolerance of 1.0 × 10−5 eV per atom and a maximum force threshold of 0.03 eV Å−1. To construct the surface model, a vacuum of 15 Å was implemented to mitigate interactions between periodic images.The charge density difference of MgO and Ce–MgO was calculated according to the following equation:
| Δρ = ρAB − ρA − ρB | (1) |
The adsorption energy (ΔEads) was calculated using the following equation:
| Eads = Etotal − Eslab − Eob | (2) |
The CI-NEB method was employed to investigate the transition state, enhancing the accuracy of force calculations by performing structural relaxation to 1 × 10−7. An optimization algorithm integrated within VASP was also utilized to interpolate points at four distinct positions. To validate the identified transition state, its vibrational frequencies were analyzed, confirming the presence of only one imaginary frequency.
3. Results and discussion
3.1 Preparation and characterization of Ce–MgO catalysts
The synthesis procedure of Ce–MgO catalysts is illustrated in Fig. 1a. The co-precipitated Ce- and Mg-containing solids were hydrothermally treated at 180 °C for 2 h and calcined at 800 °C for 5 h to obtain the Ce uniformly dispersed MgO catalyst.23 SEM images showed that the typical Ce–MgO catalysts possessed a pompom structure composed of irregularly stacked nanosheets, with a diameter of ∼55 μm (Fig. 1c, S1b and c†). The pristine MgO showed a similar pompom structure to that of Ce–MgO, which consisted of vertically growing interconnected nanosheets. Notably, the diameter of the MgO pompom was only ∼16 μm (Fig. S1a†), which suggested that the addition of Ce affected the growth of MgO particles. However, the incorporation of Ce into the catalyst did not have a significant effect on the BET surface area (Fig. S2†), which was 24.53 m2 g−1 for MgO, 26.47 m2 g−1 for Ce0.5%–MgO, 20.56 m2 g−1 for Ce1.0%–MgO, and 22.97 m2 g−1 for Ce4.0%–MgO, respectively. XRD patterns (Fig. 1b and S3†) indicated that the diffraction peaks of MgO exhibited a slight blue shift after the addition of the Ce element, which was attributed to the crystal lattice expansion due to the larger radius of Ce4+ (0.92 Å) than Mg2+ (0.65 Å).24 Meanwhile, with the increase in Ce doping, the intensity of diffraction peaks belonging to CeO2 increases, indicating that Ce gradually acted in the form of oxides in the Ce–MgO composites. Fig. S4† illustrates that CeO2 itself did not possess the ability to oxidize glucose to FA, with a yield of only 1.63%, which was almost equivalent to the blank yield (1.07%) observed without any catalyst. The promotion of the MgO catalyst by CeO2 loading was less than the synergistic effect of Ce doping and CeO2, with an enhanced FA yield of 46.61% compared to 72.88%%. Therefore, the doping ratio of Ce needs to be controlled. HRTEM images (Fig. 1e) of Ce–MgO revealed lattice fringes with spacings of 0.22 and 0.30 nm, corresponding to the (200) planes of MgO and (311) planes of CeO2, respectively. The distribution of particles with varying brightness shown in the TEM images (Fig. 1d and S1d–f†) and corresponding elemental maps (Fig. 1f) illustrated that the Ce element is uniformly confined in MgO.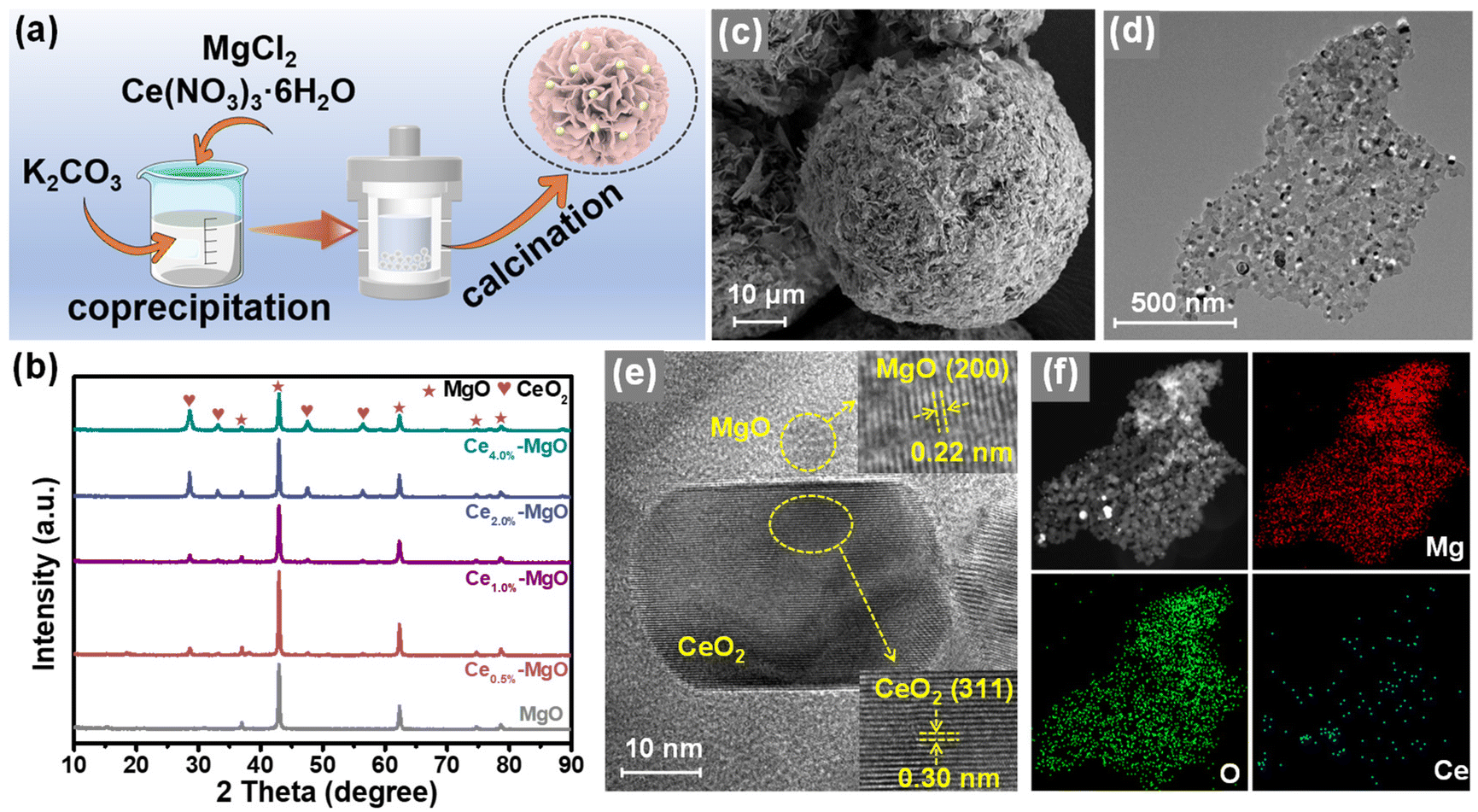 | ||
| Fig. 1 (a) Schematic illustration of the Ce–MgO catalyst synthesis process. (b) XRD patterns of the as-prepared MgO and Ce–MgO catalysts. (c) SEM, (d) TEM and (e) HRTEM images of the Ce1.0%–MgO catalyst and (f) corresponding elemental mapping. | ||
3.2 Promotion mechanism of Ce introduction
The catalytic activities of Ce–MgO catalysts with varying Ce ratios were assessed for the oxidation of glucose to FA in aqueous solution (Fig. 2a). It can be observed that the addition of Ce significantly enhanced the ability of MgO to oxidize glucose to FA at room temperature. Specifically, the FA yield increased from 31.80% (MgO) to 62.65% (Ce0.5%–MgO), 72.88% (Ce1.0%–MgO), 70.49% (Ce2.0%–MgO) and 53.18% (Ce4.0%–MgO). The conversion of glucose improved from 42.21% over MgO to 73.16%, 78.95%, 77.71% and 65.63% over Ce0.5%–MgO, Ce1.0%–MgO, Ce2.0%–MgO and Ce4.0%–MgO, respectively. Notably, the addition of 1.0% Ce exhibited the most significant enhancement in catalytic performance. It can be observed from XPS spectra that the doping of Ce resulted in a shift of the Mg 1s and lattice oxygen (Olatt) to a lower binding energy (Fig. 2d and e). In particular, for the Ce1.0%–MgO catalyst, the binding energy of Mg 1s was observed to shift from 1303.96 eV in pure MgO to 1303.35 eV in the Ce-doped sample. Similarly, the binding energy of Olatt decreased from 529.71 eV for MgO to 529.00 eV for Ce1.0%–MgO (Table S1†). This indicated that Mg and O in Ce1.0%–MgO exist in a more electron-rich state, thereby facilitating the activation of C–C bonds in glucose.25–27 This can be further corroborated by changes in the electronic structure before and after Ce incorporation, as determined through DFT simulations (Fig. 2g). The results indicated a significant increase in electron density (depicted in yellow) of the Ce1.0%–MgO catalyst, highlighting the enhanced electron aggregation associated with the modified catalyst. Correspondingly, the peak attributed to Ce4+ 3d3/2, observed at a binding energy of 915.60 eV in Ce1.0%–MgO, shifted towards a higher binding energy position (Fig. 2f and Table S1†). This suggested that Ce functions as an electron donor to enhance the electron richness of the MgO, which facilitates more efficient electron transfer between the active site and substrate.28 Additionally, the peaks observed around ∼884.05, 899.68, and 902.26 eV originate from the Ce3+ species, indicating that the introduced Ce exists in a multivalent state.29 The reversible valence shifts between Ce4+ and Ce3+ may enhance the mobility of lattice oxygen, facilitate the activation of H2O2, and improve the oxygen storage capacity of the catalyst.29,30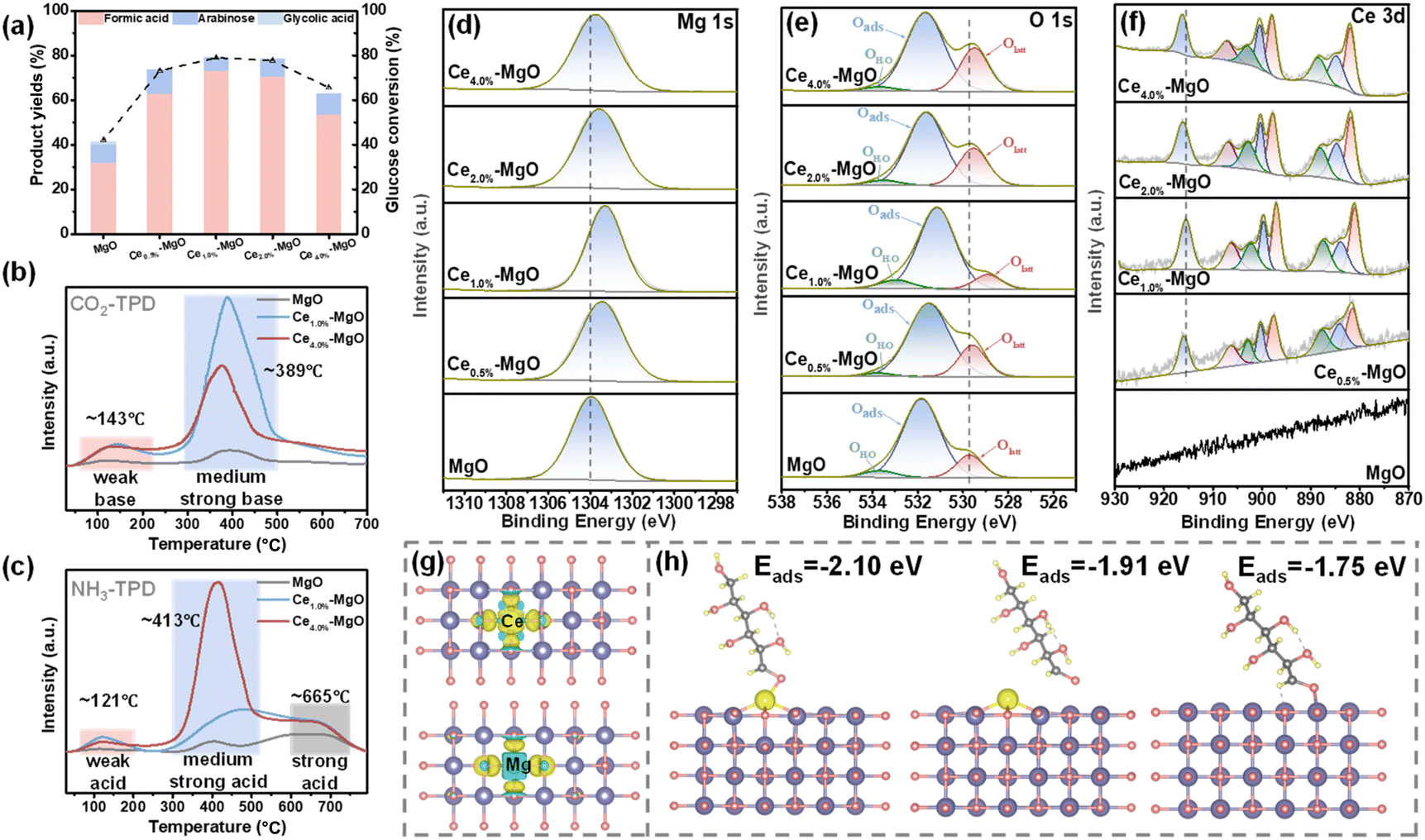 | ||
| Fig. 2 (a) Product yields and glucose conversion of different Ce–MgO catalysts (reaction conditions: 10 g L−1 glucose, 120 mg catalysts, 100% H2O2, 30 °C, 4 h). (b) CO2-TPD and (c) NH3-TPD of the Ce1.0%–MgO catalyst. XPS spectra of (d) Mg 1s, (e) O 1s and (f) Ce 3d of as-prepared MgO and Ce–MgO catalysts. (g) Differential charge density of Ce1.0%–MgO (above) and MgO (below). Yellow regions, electron accumulation and blue regions, electron depletion. (h) Adsorption energies of glucose on Ce site and Mg site of Ce1.0%–MgO, and Mg site in MgO. | ||
Fig. 2b and c show that the Ce–MgO catalysts exhibited distinctly different acid–base properties from MgO. The addition of Ce resulted in a significant enhancement in the number of both basic and acidic sites on the catalyst surface, particularly with regard to medium-strength acid and base sites. For CO2-TPD, the total alkalinity increases from 0.183 mmol g−1 (MgO) to 1.393 mmol g−1 (Ce1.0%–MgO) and then decreases to 1.069 mmol g−1 (Ce4.0%–MgO) with the further addition of Ce (Table S2†). The observed enhancement in surface alkalinity can be primarily attributed to the defects introduced by the incorporation of Ce, along with the increased electron density at Mg and O sites. These unsaturated Mg and O ions exhibit a greater capacity for proton adsorption, leading to the formation of more robust alkaline sites.31–33 The increased alkalinity can enhance the dissociation of H2O2 into highly oxidizable species ˙OOH, thereby promoting the glucose oxidation reaction.17,18,34,35 Consequently, Ce1.0%–MgO, which possesses the highest alkalinity, demonstrates the greatest yield of FA. The absence of a continued increase in basic sites with further additions of Ce can be attributed to the limitation imposed by the MgO content. In contrast, the total acidity exhibits a progressive increase with the addition of Ce, rising from 0.780 mmol g−1 for MgO to 1.956 mmol g−1 for Ce1.0%–MgO and subsequently to 3.667 mmol g−1 for Ce4.0%–MgO (Table S2†). The acid sites on the surface of the initial MgO primarily arise from hydroxyl groups (–OH) generated by the adsorption of water molecules in humid environments. The introduction of a small amount of Ce (1%) resulted in a substantial increase in acid sites on the catalyst surface, indicating that the incorporation of Ce likely enhances the formation of –OH groups on the MgO surface. Furthermore, a more significant increase persisted with the larger quantity of Ce (4%) introduced. This phenomenon may be attributed to the propensity of CeO2 to create abundant oxygen vacancies, which can interact with protons or water molecules, thereby facilitating the formation of –OH groups or other acidic species.36–38 Acidic sites can interact with the oxygen atoms of H2O2 by protonation, thereby enhancing their stability and inhibiting the spontaneous dissociation of H2O2 into H2O and O2.39–43 With the cooperation of basic sites, the selectivity for the formation of ˙OOH active species was significantly improved. However, excessive acidic sites can be detrimental to the dissociation of H2O2 into ˙OOH. Consequently, the Ce1.0%–MgO catalyst exhibits the most favorable surface acid–base properties for the glucose oxidation reaction. Furthermore, it was found that Ce doping significantly enhanced the adsorption of glucose onto the material surface (Fig. 2h). The adsorption energy of glucose at Ce sites was calculated to be −2.10 eV, which is considerably greater than that on pure MgO (−1.75 eV). Additionally, the adsorption energy at the Mg sites in Ce–MgO was also higher than that at the Mg sites in pure MgO. The strong adsorption of the substrate is a factor contributing to the efficient execution of the oxidation reaction at ultra-low temperatures.
3.3 Enhanced catalytic oxidation of glucose to FA over Ce1.0%–MgO
Ce1.0%–MgO, which exhibited optimal surface acidity and alkalinity, along with the highest electron density of Mg and O atoms, demonstrated superior performance as a catalyst for the oxidation of glucose to FA. Based on these findings, the effects of temperature, catalyst amount, glucose concentration, and reaction time on the yield of FA over the Ce1.0%–MgO catalyst were investigated. Ce1.0%–MgO demonstrated exceptional catalytic performance at extremely low temperatures, achieving high FA yields of 63.66% and 72.88% at 25 °C and 30 °C, respectively, with the highest yield of 80.64% recorded at 40 °C. However, as the temperature further increased to 50 °C, glucose conversion increased from 88.52% to 90.95%. Conversely, FA conversion unexpectedly declined to 75.51%, which can be attributed to the excessive decomposition of FA occurring over the active catalyst in the oxidative environment at elevated temperatures (Fig. 3a). The reaction achieved the highest FA yield with a catalyst dosage of 120 mg (Fig. 3b). FA accumulated rapidly during the initial 4 hours of the reaction, achieving a yield of 72.88%. Then, the rate of increase in yield diminished, ultimately reaching 76.00% after 8 h (Fig. 3d). The Ce1.0%–MgO catalyst exhibited superior conversion efficiency at low glucose concentrations. However, as the substrate concentrations increased to 15 g L−1 and 20 g L−1, the yield of FA decreased to 58.73% and 49.20%, respectively (Fig. 3c). At lower glucose concentrations, particularly at 5 g L−1, the catalyst demonstrated a remarkable ability to convert glucose to FA with high selectivity (96.21%) at 30 °C, achieving a conversion of 97.34% and a FA yield of 93.65%. The carbon balance (Fig. S5†) was determined to consist of unconverted glucose (2.66%), formic acid (93.65%), and a minor by-product, arabinose (0.44%). Other by-products, such as glycolic acid, were not monitored. This indicates that the Ce–MgO catalyst exhibits high selectivity for the cleavage of the C1–C2 bond in glucose at low concentrations, thereby favoring the production of FA. Notably, this represents the lowest reaction temperature required and the highest FA yield achieved among all reported catalysts (Fig. 3e).9,11,17,19,21,35,44–48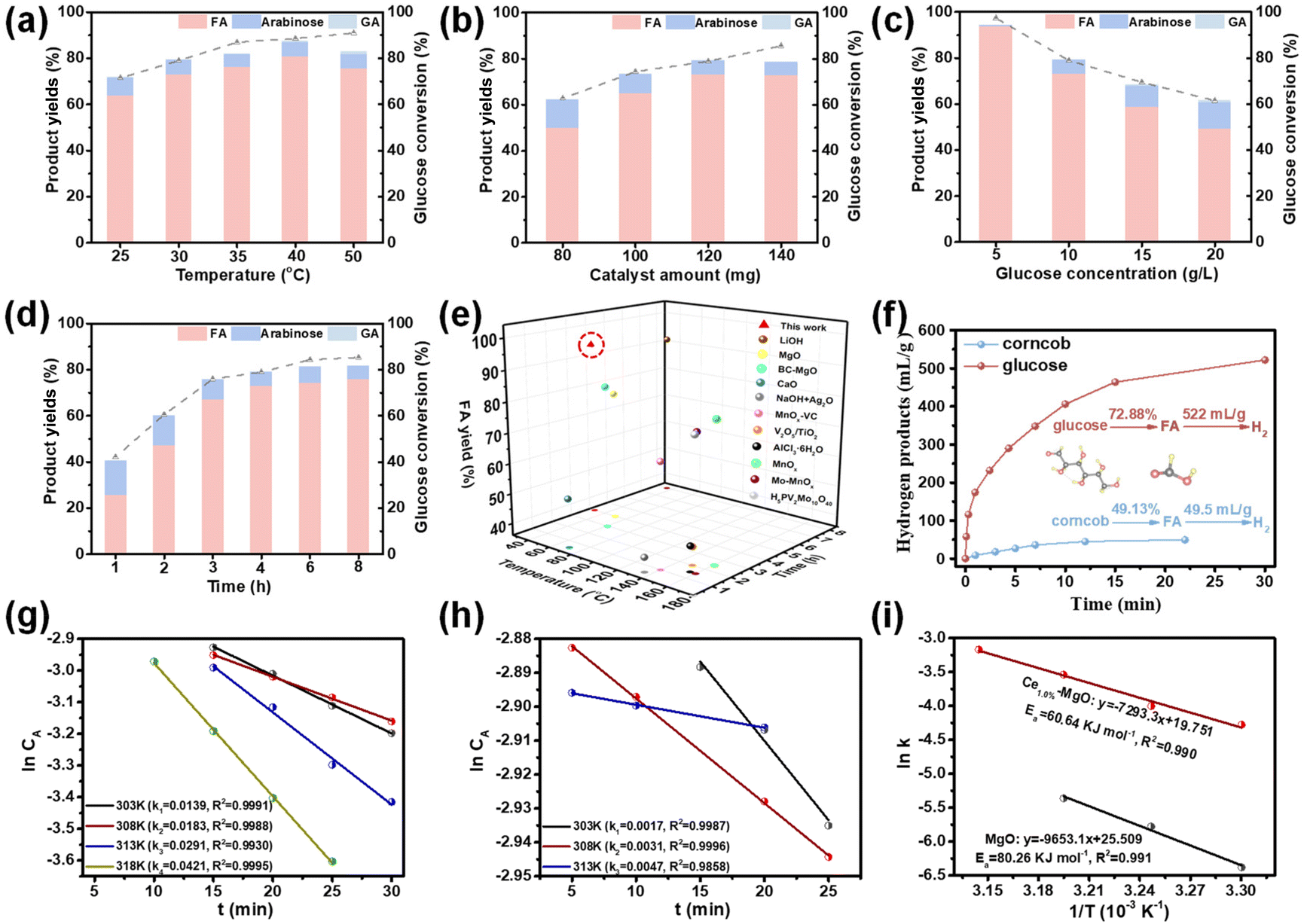 | ||
| Fig. 3 Effects of (a) temperature (reaction conditions: 10 g L−1 glucose, 120 mg catalysts, 100% H2O2, 4 h), (b) catalyst amount (reaction conditions: 10 g L−1 glucose, 120 mg catalysts, 30 °C, 4 h), (c) glucose conversion (reaction conditions: 120 mg catalysts, 100% H2O2, 30 °C, 4 h) and (d) time (reaction conditions: 10 g L−1 glucose, 120 mg catalysts, 100% H2O2, 30 °C) on product yields and glucose conversion over the Ce1.0%–MgO catalyst. (e) Oxidation of glucose to FA over Ce1.0%–MgO and other reported catalysts at optimal conditions. (f) Dehydrogenation of FA prepared from glucose and corncob-derived carbohydrates. Rate equation fitting (lnCAversus t) of glucose oxidation reaction over (g) the Ce1.0%–MgO catalyst and (h) MgO catalyst. (i) Arrhenius plot (lnk versus 1/T) and corresponding activation energy over the Ce1.0%–MgO catalyst (red line) and MgO (blue line). | ||
To elucidate the catalytic enhancement afforded by Ce introduction in the oxidation reaction, kinetic analyses were conducted on both Ce1.0%–MgO and MgO catalysts (Fig. S6†). The results indicated that the rate constant for the reaction over Ce1.0%–MgO increased by more than eightfold at 25 °C, while there was an approximate six-fold increase at 30 °C and 35 °C (Fig. 3g and h). Additionally, the activation energies for the two catalysts were determined using the Arrhenius equation, yielding values of 60.64 kJ mol−1 for Ce1.0%–MgO and 80.26 kJ mol−1 for MgO (Fig. 3i). These findings clearly demonstrated that the Ce-doped catalysts are significantly more effective in lowering the activation energy required for the glucose oxidation reaction, thereby facilitating a more rapid reaction at room temperature.
The Ce1.0%–MgO catalyst also demonstrated significant efficacy in the catalytic oxidation of biomass-derived carbohydrates (Fig. 3f). Corncob was selected for hydrothermal treatment in the presence of H2SO4, resulting in hydrolyzed xylose (0.021 mol L−1), mannose (0.003 mol L−1), and glucose (0.003 mol L−1) in the liquid phase (Fig. S7a and Table S3†). Following the adjustment of the pH of the hydrothermal solution to 6.15, an oxidation reaction was conducted under the same conditions (30 °C) using the Ce1.0%–MgO catalyst, yielding a FA production of 49.13% (Fig. S7b†). As illustrated in Fig. S8,† the initial orange-yellow sugar-containing solution was transformed into a clear and transparent liquid by the conclusion of the reaction. This observation further supports the potential capability of the Ce1.0%–MgO catalyst to oxidize biomass to FA while demonstrating a comparable conversion efficiency for other carbohydrates. Interestingly, it was found that the FA solutions derived from the oxidation of corncob and glucose can be dehydrated to produce green hydrogen over a palladium-based catalyst at 30 °C.49 It was estimated that this conversion route could yield 522 mL and 49.5 mL of H2 from 1 g of glucose and corncob, respectively. GC-TCD analyses (Fig. S9†) and HPLC spectra (Fig. S7c†) confirmed that FA was entirely converted into H2 and CO2, with no gas by-products detected. This conversion pathway for hydrogen production from biomass-derived carbohydrates at room temperature holds significant promise.
3.4 Mechanism of glucose oxidation to FA over the Ce–MgO catalyst
The reaction mechanism of glucose oxidation to FA over the Ce1.0%–MgO catalyst was further investigated by in situ FTIR and radical trapping experiments. In previous studies on the base-catalyzed oxidation of glucose, it was commonly assumed that the base facilitated the dissociation of H2O2 as a weak acid, as represented by the reaction: H2O2 + OH− → HOO− + H2O. The highly reactive HOO− species generated in this process subsequently attacked the carbon atoms in the aldehyde or ketone functional groups of glucose, ultimately converting all carbon atoms into FA.16,18 However, the reaction in this study exhibited minimal inhibition following the removal of free radicals using 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as a trapping agent (Fig. 4d). This observation indicated that the catalytic mechanism of Ce–MgO catalysts differs fundamentally from the free radical reaction mechanism associated with homogeneous bases. Wu et al. proposed that the active site of the MgO catalyst is Mg(OH)(OOH), based on the observation of Mg(OH)+ intermediates and the redshift of the Mg–O bond monitored by in situ FTIR.21 Building upon this insight, we recorded the changes in the FTIR absorption spectra of our Ce1.0%–MgO catalyst during the catalytic conversion of glucose to FA at 30 °C (Fig. 4a). The results revealed a band at approximately 3751 cm−1, corresponding to the surface hydroxyl groups associated with Mg (Mg–OH).50–53 During the initial 10 minutes, the intensity of the Mg–OH bands increased, suggesting that –OH species were predominantly formed on the surface of the Ce1.0%–MgO catalyst. Subsequently, this hydroxyl peak diminished, suggesting that –OH species were consumed during the oxidation reaction (Fig. 4b). Additionally, the bands observed around 1062 cm−1 (attributed to –OOH) and around 1419 cm−1 (associated with CH2 bonds in glucose) exhibited a consistent downward trend, which indicated that the depletion of –OH, –OOH and glucose species on the surface of the Ce1.0%–MgO catalyst.54–57 Notably, a blue shift in Mg–OH (3751 to 3743 cm−1) and a red shift in CH2 of glucose (1406 to 1419 cm−1) were observed, indicating the coordination of glucose and the Mg(OH)(OOH) active site (Fig. 4a and c). Based on these findings, the conversion pathway of glucose to FA at the Ce–MgO active site is hypothesized in Fig. S10.† The Ce-promoted Mg(OH)(OOH) active site interacts with the aldehyde terminus of glucose, facilitating the cleavage of the C1–C2 bond and resulting in the formation of one molecule of FA and one molecule of arabinose. Subsequently, arabinose undergoes successive cleavages, further producing FA. It is noteworthy that the bands corresponding to the CH2 groups in glucose (1419 cm−1) become discernible only after 10 minutes, coinciding with the depletion of Mg–OH species. This observation suggested that the Mg(OH)+ species may play a significant role in the adsorption of the substrate. Furthermore, upward bands corresponding to C–H bonds (∼2944 and 2838 cm−1) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds of carboxylic acid (∼1647 cm−1) were observed (Fig. 4a), providing clear evidence for the formation of FA over the Ce1.0%–MgO catalyst.58,59
O bonds of carboxylic acid (∼1647 cm−1) were observed (Fig. 4a), providing clear evidence for the formation of FA over the Ce1.0%–MgO catalyst.58,59
 | ||
| Fig. 4 (a) In situ FTIR spectra of glucose oxidation to FA over the Ce1.0%–MgO catalyst with H2O2 (reaction conditions: 10 g L−1 glucose, 120 mg catalysts, 100% H2O2, 30 °C). (b) and (c) Amplified in situ spectra. (d) Effect of DMPO radical scavengers on glucose oxidation to FA in the Ce1.0%–MgO catalyst and H2O2 system. (e) Product yields and glucose conversion over Ce1.0%–MgO before use, after use and after calcination (reaction conditions: 10 g L−1 glucose, 120 mg catalysts, 100% H2O2, 30 °C, 4 h). (f) XRD patterns of catalysts before use, after use and after calcination. | ||
The free radical burst experiments (Fig. 4d) demonstrated that while the DMPO trap agent did not significantly affect the oxidation reaction, the yields of FA slightly decreased to 64.62%, 59.23%, and 59.09% with increasing additions of DMPO. This observation provided compelling evidence that –OH and –OOH species are catalytically active, but not in the form of free radicals. The minor reduction in FA yield can be attributed to the partial deactivation of –OH and –OOH groups adsorbed on the catalyst surface due to their binding with DMPO. These findings suggested that MgO exhibited a strong affinity for ˙OOH free radicals, thereby effectively enhancing its stability and promoting the oxidation reaction.60 To eliminate the influence of the Mg–OH bond in Mg(OH)2, the catalytic performance of the reacted Ce1.0%–MgO catalyst, which primarily contained Mg(OH)2 (Fig. 4f), was assessed in the subsequent reaction. The results revealed that the used catalyst exhibited poor performance, achieving only 13.32% glucose conversion and 8.29% FA yield (Fig. 4e). In contrast, following the calcination treatment of the catalyst, Mg(OH)2 in Ce1.0%–MgO was converted back to MgO (Fig. 4f), resulting in a nearly complete restoration of catalytic performance (conversion of 77.72% and a FA yield of 69.90%). This finding not only offers new evidence supporting the confirmation of the active site but also demonstrates the excellent reusability of Ce1.0%–MgO catalysts.
3.5 Enhanced Mg(OH)(OOH) formation and C–C bond activation
In the previous discussion, the modulation of Ce doping was confirmed to influence the electron density, surface acidity, and alkalinity of Ce–MgO catalysts. Herein, DFT calculations were supplemented, revealing that these enhancements directly promote the formation of Mg(OH)(OOH) active sites and facilitate the activation of C–C bonds in glucose. As illustrated in Fig. 5a and c, the dissociation energy for the decomposition of H2O2 to ˙OOH decreased significantly from 98.3 kcal mol−1 for MgO to 90.1 kcal mol−1 for Ce–MgO. The facilitated dissociation of H2O2 can be attributed to the enhancement of MgO surface alkalinity through the doping of Ce, as well as the direct reaction of Ce(IV) with H2O2. The increase in basic sites on the surface of the catalyst can enhance the binding capacity for H+ in the aqueous solution. This interaction promotes the ionization of H2O, resulting in an increased concentration of OH+ within the system. Consequently, this effect facilitates the formation of Mg(OH)+ intermediates and the equilibrium reaction H2O2 + OH− ⇌ HOO− + H2O.61,62 Besides, H2O2 also undergoes a reaction with Ce(IV) as follows: Ce4+ + H2O2 ⇌ [CeIV(HOO)]3+ + H+ and [CeIV(HOO)]3+ → Ce3+ + HOO˙.63–66 These two promotional effects contributed to the reduced dissociation energy of H2O2 over Ce–MgO catalysts. Notably, the adsorption energy of ˙OOH on the surface of Ce–MgO (−0.18 eV) is lower than that on MgO (−0.16 eV), indicating that Ce–MgO exhibits a stronger interaction with ˙OOH, which is advantageous for the generation of Mg(OH)(OOH).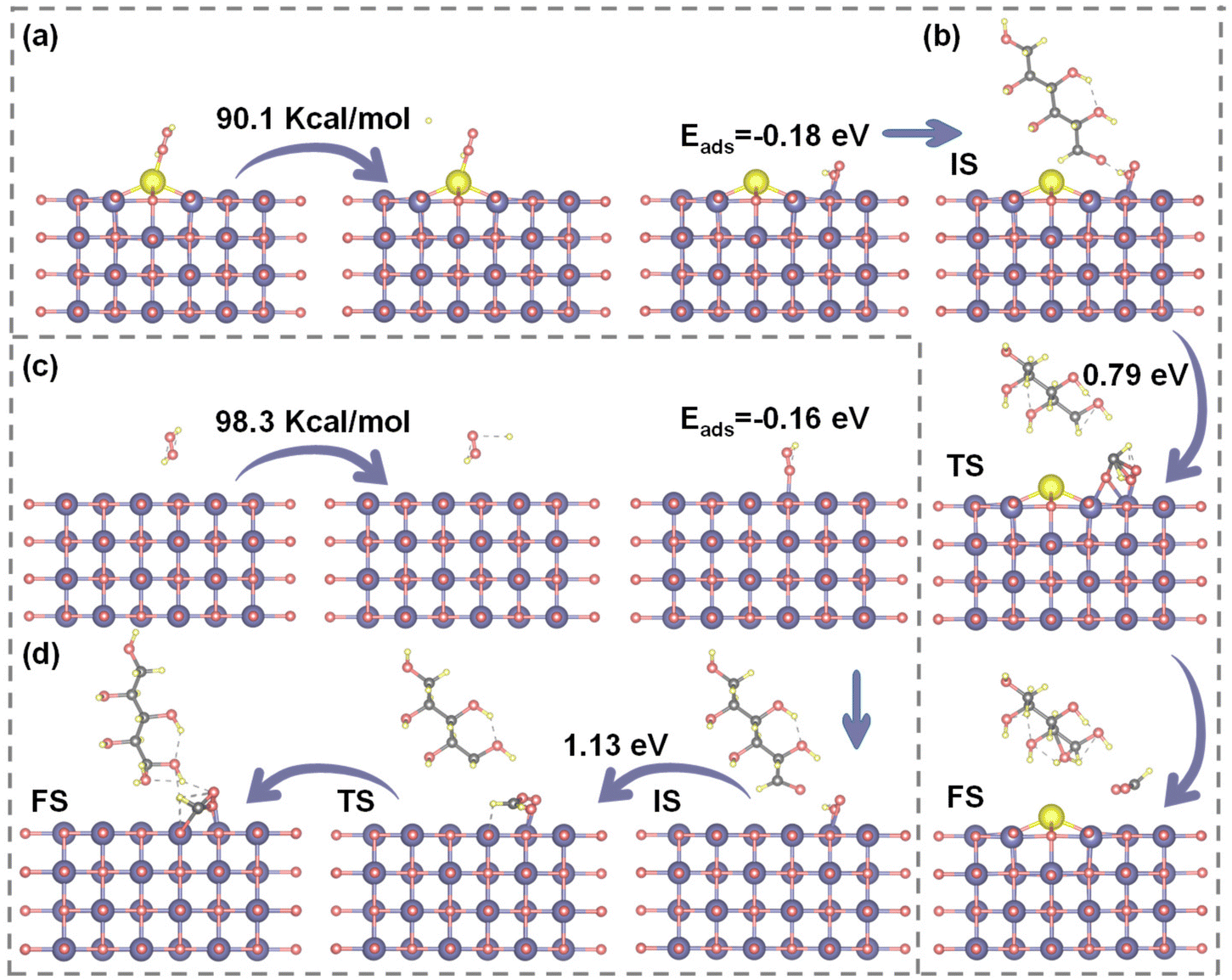 | ||
| Fig. 5 (a) Dissociation energy of H2O2 over the Ce1.0%–MgO catalyst and the adsorption energy of OOH. (b) Reaction energy barriers of glucose C1–C2 bond cleavage over the Ce1.0%–MgO catalyst. (c) Dissociation energy of H2O2 over the MgO catalyst and the adsorption energy of OOH. (d) Reaction energy barriers of glucose C1–C2 bond cleavage over the MgO catalyst. | ||
After the activation of H2O2 over the Ce–MgO catalyst, ˙OOH was generated and subsequently absorbed onto the Mg sites, leading to the formation of active sites identified as Mg(OH)(OOH). These active species then interacts with glucose adsorbed on the catalyst surface, facilitating the cleavage of C–C bonds and promoting oxidation reactions (Fig. 5b). The oxidation of glucose at Mg(OH)(OOH) active sites typically initiates at the aldehyde terminus. The electron-rich Mg and O, facilitated by Ce promotion, promoted the cleavage of the C–C bond at the aldehyde terminus through effective electron transfer. The resulting aldehyde fragment subsequently bound to the –OOH species, forming a transition state. Following the interaction, an oxygen atom was inserted into the single-carbon aldehyde group, leading to its oxidation to format. Meanwhile, the remaining –OH group continued to interact with the five-carbon sugar moiety, converting the terminal carbon into an aldehyde group. This process was iterated, resulting in the sequential cleavage of C–C bonds in glucose and culminating in its complete oxidation. DFT simulations (Fig. 5b and d) demonstrated that the energy barrier for C–C bond cleavage and subsequent oxidation to FA on the Ce–MgO catalyst (0.79 eV) is lower than that observed on MgO (1.13 eV). These findings provided strong evidence that the Mg(OH)(OOH) active site was enhanced by the Ce-promoted effect, consistent with previous analyses. Ce acts as the electron donor, increasing the electron densities of both Mg and O, which in turn facilitated electron transfer between the Mg(OH)(OOH) site and the substrate. This enhancement significantly improved the C–C bond-breaking and oxidizing capabilities of active sites. Consequently, the intensified Mg(OH)(OOH) active site was a critical factor enabling the Ce–MgO catalyst to efficiently oxidize glucose to FA at room temperature.
4. Conclusion
A Ce-modified MgO heterogeneous catalyst was developed in this study to facilitate the oxidation of biomass-derived sugars into formic acid. The incorporation of Ce significantly enhanced the charge densities of the Mg and O atoms on the catalyst's surface, thereby improving its acidic and basic properties. These optimized acid–base sites effectively inhibited the spontaneous dissociation of hydrogen peroxide while promoting the selective generation of the active species ˙OOH. The resulting Mg(OH)(OOH) active sites demonstrated higher charge densities and enhanced electron transfer capabilities, significantly facilitating the cleavage of the glucose C–C bond and lowering the energy barrier for the oxidation reaction. Leveraging the enhanced Mg(OH)(OOH) active sites, the Ce–MgO catalyst exhibited remarkable efficiency in converting various carbohydrates at room temperature, achieving an impressive 97.34% conversion for glucose and a formic acid yield of 93.65%, surpassing all reported catalysts. Furthermore, the formic acid generated by the Ce–MgO catalyst was capable of in situ hydrogen generation at room temperature, laying a crucial foundation for the conversion of biomass into green hydrogen.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the Hong Kong Innovation and Technology Fund (Ref. ITS-065-22MX) and the Environment and Conservation Fund (Ref. 2021-127).References
- C. Cheng, S. Zhang, J. Zhang, L. Guan, M. E. El-Khouly and S. Jin, Mixed Crystalline Covalent Heptazine Frameworks with Built-in Heterojunction Structures towards Efficient Photocatalytic Formic Acid Dehydrogenation, Angew. Chem., Int. Ed., 2024, 63(43), e202411359 CrossRef CAS PubMed.
- P. Kumar, K. Kannimuthu, A. S. Zeraati, S. Roy, X. Wang, X. Wang, S. Samanta, K. A. Miller, M. Molina, D. Trivedi, J. Abed, M. A. Campos Mata, H. Al-Mahayni, J. Baltrusaitis, G. Shimizu, Y. A. Wu, A. Seifitokaldani, E. H. Sargent, P. M. Ajayan, J. Hu and M. G. Kibria, High-Density Cobalt Single-Atom Catalysts for Enhanced Oxygen Evolution Reaction, J. Am. Chem. Soc., 2023, 145(14), 8052–8063 CrossRef CAS PubMed.
- H. Zhang, X. Wang, C. Chen, X. Yang, C. Dong, Y. Huang, X. Zhao and D. Yang, Selective CO2-to-formic acid electrochemical conversion by modulating electronic environment of copper phthalocyanine with defective graphene, Chin. J. Struct. Chem., 2023, 42(10), 100089 CrossRef.
- Y. Chen, Y. Yang, X. Liu, X. Shi, C. Wang, H. Zhong and F. Jin, Sustainable production of formic acid and acetic acid from biomass, Mol. Catal., 2023, 545, 113199 CrossRef CAS.
- W. Tang, L. Zhang, T. Qiu, H. Tan, Y. Wang, W. Liu and Y. Li, Efficient Conversion of Biomass to Formic Acid Coupled with Low Energy Consumption Hydrogen Production from Water Electrolysis, Angew. Chem., Int. Ed., 2023, 62(30), e202305843 CrossRef CAS PubMed.
- R. Zhang, J. Zhang, H. Liu, Z. Jiang, X. Liu, W. Wang, L. Peng and C. Hu, Toward Value-Added Chemicals from Carbohydrates via C–C Bond Cleavage and Coupling Transformations, ACS Catal., 2024, 14(7), 5167–5197 CrossRef CAS.
- Z. He, Y. Hou, H. Li, J. Wei, S. Ren and W. Wu, Novel chemical looping oxidation of biomass-derived carbohydrates to super-high-yield formic acid using heteropolyacids as oxygen carrier, Renewable Energy, 2023, 207, 461–470 CrossRef CAS.
- Z. He, Y. Hou, H. Li, J. Wei, S. Ren and W. Wu, Catalytic aerobic oxidation of carbohydrates to formic acid over H5PV2Mo10O40: Rate relationships among catalyst reduction, catalyst re-oxidation and acid-catalyzed reactions and evidence for the Mars-van Krevelen mechanism, Chem. Eng. Sci., 2023, 280, 119005 CrossRef.
- J. Reichert, B. Brunner, A. Jess, P. Wasserscheid and J. Albert, Biomass oxidation to formic acid in aqueous media using polyoxometalate catalysts – boosting FA selectivity by in situ extraction, Energy Environ. Sci., 2015, 8(10), 2985–2990 RSC.
- X. Wei, Q. Wang, X. Zhang, Y. Chen, N. Jin and Y. Zhao, Highly selective oxidation of glucose to formic acid and exploring reaction pathways using continuous flow microreactors, Fuel, 2024, 372, 132198 CrossRef CAS.
- D. Álvarez-Hernández, S. Ivanova, A. Penkova and M.Á Centeno, Influence of vanadium species on the catalytic oxidation of glucose for formic acid production, Catal. Today, 2024, 441, 114906 CrossRef.
- X. Liu, Y. Wang, Z. Dai, D. Gao and X. Zhao, Electrochemical reduction of carbon dioxide to produce formic acid coupled with oxidative conversion of biomass, J. Energy Chem., 2024, 92, 705–729 CrossRef CAS.
- C. Shi, Y. An, G. Gao, J. Xue, H. Algadi, Z. Huang and Z. Guo, Insights into Selective Glucose Photoreforming for Coproduction of Hydrogen and Organic Acid over Biochar-Based Heterojunction Photocatalyst Cadmium Sulfide/Titania/Biochar, ACS Sustainable Chem. Eng., 2024, 12(7), 2538–2549 CrossRef CAS.
- K. Wang, Z. Guo, M. Zhou, Y. Yang, L. Li, H. Li, R. Luque and S. Saravanamurugan, Biomass valorization via electrocatalytic carbon–carbon bond cleavage, J. Energy Chem., 2024, 91, 542–578 CrossRef CAS.
- W.-M. Zhang, K.-W. Feng, R.-G. Hu, Y.-J. Guo and Y. Li, Visible-light-induced iron redox-catalyzed selective transformation of biomass into formic acid, Chem, 2023, 9(2), 430–442 CAS.
- P. Pullanikat, S. J. Jung, K. S. Yoo and K. W. Jung, Oxidative degradation of reducing carbohydrates to ammonium formate with H(2)O(2) and NH(4)OH, Tetrahedron Lett., 2010, 51(47), 6192–6194 CrossRef CAS PubMed.
- A. Takagaki, W. Obata and T. Ishihara, Oxidative Conversion of Glucose to Formic Acid as a Renewable Hydrogen Source Using an Abundant Solid Base Catalyst, ChemistryOpen, 2021, 10(10), 954–959 CrossRef CAS PubMed.
- J. Yun, G. Yao, F. Jin, H. Zhong, A. Kishita, K. Tohji, H. Enomoto and L. Wang, Low-temperature and highly efficient conversion of saccharides into formic acid under hydrothermal conditions, AIChE J., 2016, 62(10), 3657–3663 CrossRef CAS.
- G. Guo, H. Zhong, X. Shi, L. Tai, Y. Yang and F. Jin, Synthesis of biochar-doped MgO catalyst for highly efficient conversion of glucose into formic acid at low temperatures, J. Cleaner Prod., 2024, 471, 143393 CrossRef CAS.
- H. Lan, F. Wang, M. Lan, X. An, H. Liu and J. Qu, Hydrogen-Bond-Mediated Self-Assembly of Carbon-Nitride-Based Photo-Fenton-like Membranes for Wastewater Treatment, Environ. Sci. Technol., 2019, 53(12), 6981–6988 CrossRef CAS PubMed.
- L. Wu, Y. Yang, J. Cheng, X. Shi, H. Zhong and F. Jin, Highly Efficient Conversion of Carbohydrates into Formic Acid with a Heterogeneous MgO Catalyst at Near-Ambient Temperatures, ACS Sustainable Chem. Eng., 2022, 10(47), 15423–15436 CrossRef CAS.
- R. Zhang, J. Zhao, J. Ye, X. Tian, L. Wang, J. Pan and J. Dai, Role of tea polyphenols in enhancing the performance, sustainability, and catalytic cleaning capability of membrane separation for water-soluble pollutant removal, J. Hazard. Mater., 2024, 468, 133793 CrossRef CAS PubMed.
- J. Zhao, J. Liu, H. Li, M. Zeng, X. Yang, X. Liu and Z. Yang, Porous MgO pompons as a binder for the molten electrolyte applied in thermal batteries, Ionics, 2021, 27(3), 1271–1278 CrossRef CAS.
- J. Ye, W. Xue, P. Wang, Y. Geng, J. Dai, J. Pan, X. Duan and J. Zhao, Intensify Mass Transfer and Molecular Oxygen Activation by Defect–Bridged Asymmetric Catalytic Sites Toward Efficient Membrane–Based Nanoconfined Catalysis, Adv. Funct. Mater., 2024, 34(37), 2403964 CrossRef CAS.
- J. Jiang, D. Shi, S. Niu, S. Liu, Y. Liu, B. Zhao, Y. Zhang, H. Liu, Z. Zhao, M. Li, M. Huo, D. Zhou and S. Dong, Modulating electron density enable efficient cascade conversion from peroxymonosulfate to superoxide radical driven by electron-rich/poor dual sites, J. Hazard. Mater., 2024, 468, 133749 CrossRef CAS PubMed.
- Y. F. Liang, M. Bilal, L. Y. Tang, T. Z. Wang, Y. Q. Guan, Z. Cheng, M. Zhu, J. Wei and N. Jiao, Carbon-Carbon Bond Cleavage for Late-Stage Functionalization, Chem. Rev., 2023, 123(22), 12313–12370 CrossRef CAS PubMed.
- J. Xu, J. Qiu, L. Zhang and J. Yao, Research progress on carbon nitride-based photocatalysts for lignin C–C bond cleavage: Material design and cleavage mechanism, Appl. Catal. B: Environ. Energy, 2025, 361, 124639 CrossRef CAS.
- J. Ye, J. Yang, Y. Liu, W. Xue, J. W. C. Wong, J. Dai, J. Zhao and J. Crittenden, Synergy of pore confinement and co-catalytic effects in Peroxymonosulfate activation for persistent and selective removal of contaminants, Chem. Eng. J., 2024, 496, 154034 CrossRef CAS.
- X. Ding, Y. Yang, Z. Li, P. Huang, X. Liu, Y. Guo and Y. Wang, Engineering a Nickel–Oxygen Vacancy Interface for Enhanced Dry Reforming of Methane: A Promoted Effect of CeO2 Introduction into Ni/MgO, ACS Catal., 2023, 13(23), 15535–15545 CrossRef CAS.
- C. Shuai, K. Wang, S. Peng, J. Zan, J. Xiao, S. Hu and Q. Zhong, Accelerating Ce3+/Ce4+ Conversion in CeO2 via Mn doping to Endow Scaffolds with Chemodynamic Therapy Properties, Surf. Interfaces, 2024, 45, 103846 CrossRef CAS.
- V. J. Ferreira, P. Tavares, J. L. Figueiredo and J. L. Faria, Effect of Mg, Ca, and Sr on CeO2 Based Catalysts for the Oxidative Coupling of Methane: Investigation on the Oxygen Species Responsible for Catalytic Performance, Ind. Eng. Chem. Res., 2012, 51(32), 10535–10541 CrossRef CAS.
- A. S. Ivanova, B. L. Moroz, E. M. Moroz, Y. V. Larichev, E. A. Paukshtis and V. I. Bukhtiyarov, New binary systems Mg–M–O (M=Y, La, Ce): Synthesis and physico-chemical characterization, J. Solid State Chem., 2005, 178(11), 3265–3274 CrossRef CAS.
- G. I. Siakavelas, N. D. Charisiou, S. AlKhoori, A. A. AlKhoori, V. Sebastian, S. J. Hinder, M. A. Baker, I. V. Yentekakis, K. Polychronopoulou and M. A. Goula, Highly selective and stable nickel catalysts supported on ceria promoted with Sm2O3, Pr2O3 and MgO for the CO2 methanation reaction, Appl. Catal., B, 2021, 282, 119562 CrossRef CAS.
- F. Jin, J. Yun, G. Li, A. Kishita, K. Tohji and H. Enomoto, Hydrothermal conversion of carbohydrate biomass into formic acid at mild temperatures, Green Chem., 2008, 10(6), 612–615 RSC.
- C. Wang, X. Chen, M. Qi, J. Wu, G. Gözaydın, N. Yan, H. Zhong and F. Jin, Room temperature, near-quantitative conversion of glucose into formic acid, Green Chem., 2019, 21(22), 6089–6096 RSC.
- A. Davó-Quiñonero, M. Navlani-García, D. Lozano-Castelló, A. Bueno-López and J. A. Anderson, Role of Hydroxyl Groups in the Preferential Oxidation of CO over Copper Oxide–Cerium Oxide Catalysts, ACS Catal., 2016, 6(3), 1723–1731 CrossRef.
- Y. Zhang, J. Lu, L. Zhang, T. Fu, J. Zhang, X. Zhu, X. Gao, D. He, Y. Luo, D. D. Dionysiou and W. Zhu, Investigation into the catalytic roles of oxygen vacancies during gaseous styrene degradation process via CeO2 catalysts with four different morphologies, Appl. Catal., B, 2022, 309, 121249 CrossRef CAS.
- M. Ziemba, C. Schilling, M. V. Ganduglia-Pirovano and C. Hess, Toward an Atomic-Level Understanding of Ceria-Based Catalysts: When Experiment and Theory Go Hand in Hand, Acc. Chem. Res., 2021, 54(13), 2884–2893 CrossRef CAS PubMed.
- Y. Ding, J. Wang, S. Xu, K.-Y. A. Lin and S. Tong, Oxygen vacancy of CeO2 improved efficiency of H2O2/O3 for the degradation of acetic acid in acidic solutions, Sep. Purif. Technol., 2018, 207, 92–98 CrossRef CAS.
- Y. Liu, W. Xue, A. Chowdhury, A. P. Rangappa and J. Zhao, S-vacancy regulation over ultra-thin ZnIn2S4 for enhanced photocatalytic valorization of biomass-derived 5-hydroxymethylfurfural to 2,5-diformylfuran, Chem. Eng. J., 2024, 497, 154613 CrossRef CAS.
- R. S. Ribeiro, A. M. T. Silva, J. L. Figueiredo, J. L. Faria and H. T. Gomes, The influence of structure and surface chemistry of carbon materials on the decomposition of hydrogen peroxide, Carbon, 2013, 62, 97–108 CrossRef CAS.
- T. Shen, W. Su, Q. Yang, J. Ni and S. Tong, Synergetic mechanism for basic and acid sites of MgMxOy (M = Fe, Mn) double oxides in catalytic ozonation of p-hydroxybenzoic acid and acetic acid, Appl. Catal., B, 2020, 279, 119346 CrossRef CAS.
- Y. Wang, X. Shen and F. Chen, Improving the catalytic activity of CeO2/H2O2 system by sulfation pretreatment of CeO2, J. Mol. Catal. A: Chem., 2014, 381, 38–45 CrossRef CAS.
- A. Bayu, G. Guan, S. Karnjanakom, X. Hao, K. Kusakabe and A. Abudula, Catalytic synthesis of levulinic acid and formic acid from glucose in choline chloride aqueous solution, ChemistrySelect, 2016, 1(2), 180–188 CrossRef CAS.
- Y. Geng, W. Xue, J. Ye, R. Zhang, P. Mishra and J. Zhao, Enhanced Oxidation of Glucose to Formic Acid under Mild Conditions Using an Oxygen-Deficient MnOx-Based Catalyst and a Novel Catalyst Regeneration Strategy, ACS Sustainable Chem. Eng., 2024, 12(41), 15182–15192 CrossRef CAS.
- H. Guo, J. Li, S. Xu, J. Yang, G.-H. Chong and F. Shen, Mo-modified MnOx for the efficient oxidation of high-concentration glucose to formic acid in water, Fuel Process. Technol., 2023, 242, 107662 CrossRef CAS.
- R. He, T. Ma, J. Cheng, B. Jin and J. Xu, Formation of Formic Acid from Glucose with Simultaneous Conversion of Ag(2)O to Ag under Mild Hydrothermal Conditions, ACS Omega, 2021, 6(17), 11260–11265 CrossRef CAS PubMed.
- J. Li, R. L. Smith, S. Xu, D. Li, J. Yang, K. Zhang and F. Shen, Manganese oxide as an alternative to vanadium-based catalysts for effective conversion of glucose to formic acid in water, Green Chem., 2022, 24(1), 315–324 RSC.
- Y. Geng, D. Johnravindar, W. Xue, J. Ye and J. Zhao, Efficient Dehydrogenation of Formic Acid at Room Temperature Using a Pd/Chitosan-Derived Nitrogen-Doped Carbon Catalyst: Synthesis, Characterization, and Kinetic Study, Ind. Eng. Chem. Res., 2023, 62(47), 20213–20222 CrossRef CAS.
- D. L. Carvalho, L. E. P. Borges, L. G. Appel, P. Ramírez de la Piscina and N. Homs, In situ infrared spectroscopic study of the reaction pathway of the direct synthesis of n-butanol from ethanol over MgAl mixed-oxide catalysts, Catal. Today, 2013, 213, 115–121 CrossRef CAS.
- A. Kondo, R. Kurosawa, J. Ryu, M. Matsuoka and M. Takeuchi, Investigation on the Mechanisms of Mg(OH)2 Dehydration and MgO Hydration by Near-Infrared Spectroscopy, J. Phys. Chem. C, 2021, 125(20), 10937–10947 CrossRef CAS.
- R. Kurosawa, M. Takeuchi and J. Ryu, Fourier-transform infrared and X-ray diffraction analyses of the hydration reaction of pure magnesium oxide and chemically modified magnesium oxide, RSC Adv., 2021, 11(39), 24292–24311 RSC.
- J. Lichtenberger, S. Hargroveleak and M. Amiridis, In situ FTIR study of the adsorption and reaction of 2′-hydroxyacetophenone and benzaldehyde on MgO, J. Catal., 2006, 238(1), 165–176 CrossRef CAS.
- T. Faverge, B. Gilles, A. Bonnefont, F. Maillard, C. Coutanceau and M. Chatenet, In Situ Investigation of d-Glucose Oxidation into Value-Added Products on Au, Pt, and Pd under Alkaline Conditions: A Comparative Study, ACS Catal., 2023, 13(4), 2657–2669 CrossRef CAS.
- C. Feng, Z. Zhang, D. Wang, Y. Kong, J. Wei, R. Wang, P. Ma, H. Li, Z. Geng, M. Zuo, J. Bao, S. Zhou and J. Zeng, Tuning the Electronic and Steric Interaction at the Atomic Interface for Enhanced Oxygen Evolution, J. Am. Chem. Soc., 2022, 144(21), 9271–9279 CrossRef CAS PubMed.
- Y. Lin, L. Yu, L. Tang, F. Song, R. Schlögl and S. Heumann, In Situ Identification and Time-Resolved Observation of the Interfacial State and Reactive Intermediates on a Cobalt Oxide Nanocatalyst for the Oxygen Evolution Reaction, ACS Catal., 2022, 12(9), 5345–5355 CrossRef CAS.
- W. Xue, J. Ye, Z. Zhu, R. Kumar and J. Zhao, Harnessing trace water for enhanced photocatalytic oxidation of biomass-derived alcohols to aldehyde, Energy Environ. Sci., 2025, 18, 214–226 RSC.
- R. Zhang, H. Wang, S. Tang, C. Liu, F. Dong, H. Yue and B. Liang, Photocatalytic Oxidative Dehydrogenation of Ethane Using CO2 as a Soft Oxidant over Pd/TiO2 Catalysts to C2H4 and Syngas, ACS Catal., 2018, 8(10), 9280–9286 CrossRef CAS.
- G. Zhou, J. Yang, X. Zhu, Q. Li, Q. Yu, W. El-alami, C. Wang, Y. She, J. Qian, H. Xu and H. Li, Cryo-induced closely bonded heterostructure for effective CO2 conversion: The case of ultrathin BP nanosheets/g-C3N4, J. Energy Chem., 2020, 49, 89–95 CrossRef.
- Y. Pan, H. Xu, M. Chen, K. Wu, Y. Zhang and D. Long, Unveiling the Nature of Room-Temperature O2 Activation and O2˙− Enrichment on MgO-Loaded Porous Carbons with Efficient H2S Oxidation, ACS Catal., 2021, 11(10), 5974–5983 CrossRef CAS.
- R. J. Buszek, M. Torrent-Sucarrat, J. M. Anglada and J. S. Francisco, Effects of a single water molecule on the OH + H2O2 reaction, J. Phys. Chem. A, 2012, 116(24), 5821–5829 CrossRef CAS PubMed.
- S. Xu, T. He, J. Li, Z. Huang and C. Hu, Enantioselective synthesis of D-lactic acid via chemocatalysis using MgO: Experimental and molecular-based rationalization of the triose's reactivity and preliminary insights with raw biomass, Appl. Catal., B, 2021, 292, 120145 CrossRef CAS.
- W. Chen, J. Xie, X. Li and L. Li, Oxygen vacancies and Lewis sites activating O(3)/H(2)O(2) at wide pH range via surface electron transfer over CeO(x)@SiO(2) for nitrobenzene mineralization, J. Hazard. Mater., 2021, 406, 124766 CrossRef CAS PubMed.
- S. Li, J. Huang, Z. Ye, Y. Wang, X. Li, J. Wang and L. Li, The mechanism of Metal-H2O2 complex immobilized on MCM-48 and enhanced electron transfer for effective peroxone ozonation of sulfamethazine, Appl. Catal., B, 2021, 280, 119453 CrossRef CAS.
- M. Xu, J. Wei, X. Cui, J. Li, G. Pan, Y. Li, Z. Jiang, X. Niu, N. Cui and J. Li, High-efficiency electro-Fenton process based on in situ grown CoFeCe-LDH@CFs free-standing cathodes: Correlation of cerium and oxygen vacancies with H2O2, Chem. Eng. J., 2023, 455, 140922 CrossRef CAS.
- Z. Zhu, X. Xing, Q. Qi, W. Shen, H. Wu, D. Li, B. Li, J. Liang, X. Tang, J. Zhao, H. Li and P. Huo, Fabrication of graphene modified CeO2/g-C3N4 heterostructures for photocatalytic degradation of organic pollutants, Chin. J. Struct. Chem., 2023, 42(12), 100194 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Calculation methods; SEM images; nitrogen adsorption/desorption isotherms; partially enlarged XRD patterns; performance comparison experiments; kinetic experiments; schematic diagrams; GC spectra; binding energies; and quantitative analysis. See DOI: https://doi.org/10.1039/d5gc00008d |
| This journal is © The Royal Society of Chemistry 2025 |