
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProduction of high-carbon-number hydrocarbon bio-aviation fuels via catalytic hydrogenation of vanillin and non-catalytic condensation: a mechanistic study with DFT and experimental insights†
Jina
Eun‡
 ab,
Jeonghun
Kim‡
ac,
Han Byeol
Kim
d,
Do Heui
Kim
e,
Jae-Wook
Choi
a,
Kwang Ho
Kim
f,
Chun-Jae
Yoo
acgh,
Seongmin
Jin
a,
Kyeongsu
Kim
ac,
Hyunjoo
Lee
ac,
Chang Soo
Kim
ac,
Kwan-Young
Lee
bi,
Jong Suk
Yoo
*jk,
Seo-Jung
Han
*dl,
Keunhong
Jeong
*m and
Jeong-Myeong
Ha
*ac
ab,
Jeonghun
Kim‡
ac,
Han Byeol
Kim
d,
Do Heui
Kim
e,
Jae-Wook
Choi
a,
Kwang Ho
Kim
f,
Chun-Jae
Yoo
acgh,
Seongmin
Jin
a,
Kyeongsu
Kim
ac,
Hyunjoo
Lee
ac,
Chang Soo
Kim
ac,
Kwan-Young
Lee
bi,
Jong Suk
Yoo
*jk,
Seo-Jung
Han
*dl,
Keunhong
Jeong
*m and
Jeong-Myeong
Ha
*ac
aClean Energy Research Center, Korea Institute of Science and Technology, Seoul 02792, Republic of Korea. E-mail: jmha@kist.re.kr
bDepartment of Chemical and Biological Engineering, Korea University, Seoul 02841, Republic of Korea
cDivision of Energy and Environment Technology, KIST School, University of Science and Technology, Seoul 02792, Republic of Korea
dChemical and Biological Integrative Research Center, Korea Institute of Science and Technology, Seoul 02792, Republic of Korea. E-mail: sjhan@sogang.ac.kr
eSchool of Chemical and Biological Engineering, Institute of Chemical Process, Seoul National University, Seoul 08826, Republic of Korea
fDepartment of Wood Science, University of British Columbia, 2424 Main Mall, V6T1Z4 Vancouver, Canada
gSchool of Chemical Engineering, Sungkyunkwan University, Suwon 16419, Republic of Korea
hKIST-SKKU Carbon-Neutral Research Center, Sungkyunkwan University, Suwon 16419, Republic of Korea
iCenter for Hydrogen and Fuel Cells, Korea Institute of Science and Technology, Seoul 02792, Republic of Korea
jDepartment of Chemical Engineering, University of Seoul, Seoul 02504, Republic of Korea. E-mail: jsyoo84@uos.ac.kr
kCenter for Innovative Chemical Processes, Institute of Engineering, University of Seoul, Seoul, 02504 Republic of Korea
lDivision of Bio-Medical Science & Technology, KIST School, University of Science and Technology, Seoul 02792, Republic of Korea
mDepartment of Chemistry, Korea Military Academy, Seoul 01805, Republic of Korea. E-mail: doas1mind@gmail.com
First published on 7th May 2025
Abstract
Lignocellulose or lignin present significant potential as sustainable feedstocks to replace petroleum-derived resources through catalytic upgrading. Hydrodeoxygenation of phenolic molecules derived from lignocellulose or lignin can produce cycloalkanes, but often forms low-carbon-number hydrocarbons, which are more suitable for gasoline rather than high-carbon-number diesel or aviation fuels. This study investigates the production of high-carbon-number hydrocarbons in the aviation fuel range from lignin-derived compounds, using vanillin as a model. A two-step process was performed to achieve this: selective hydrogenation of vanillin to vanillyl alcohol and creosol using 1 wt% ruthenium on carbon, followed by non-catalytic condensation and subsequent hydrodeoxygenation of the condensates to cycloalkanes using 3 wt% ruthenium on HZSM-5. This process yielded C14 aviation fuel precursor (19%) and C14 deoxygenated hydrocarbon (5%) whereas the one-step process without the condensation step did not yield any C14 compounds. The reaction pathway was elucidated through density functional theory calculations and control experiments with intermediates, providing insights into the mechanisms of upgrading lignin-derived compounds for sustainable aviation fuel production.
Green foundation1. This study proposes a sustainable approach to replacing petroleum-derived aviation fuels by producing bio-aviation fuels from lignocellulose-derived vanillin. The deoxygenated C14 hydrocarbons synthesized in this work have the potential to substitute for naphthenes in current aviation fuel formulations.2. While most current bio-aviation fuels primarily consist of paraffins, this study demonstrates the potential to produce high-carbon-number naphthenes, enabling biofuels that closely resemble petroleum-based fuels. 3. Further research leveraging lignin- or lignocellulose-derived feedstocks to produce petroleum-like renewable bio-aviation fuels will help sustain the existing petroleum-based industry infrastructure. |
1. Introduction
In response to growing concerns about achieving net-zero emissions, particularly in the aviation sector, significant research efforts have focused on developing bio-aviation fuels from biomass.1 While lipds are used for producing commercial paraffin-based sustainable aviation fuels meeting international regulatory standards,2 lignocellulosic biomass has emerged as a promising alternative for carbon-neutral liquid fuels because of its abundance and non-food nature. Composed of cellulose, hemicellulose, and lignin, lignocellulosic biomass has been studied for aviation fuel synthesis via platform compounds derived from cellulose and hemicellulose.3,4 However, lignin, the second most abundant component in agricultural and forest residues, remains underutilized, highlighting the need for its valorization.5 Although many studies have explored the hydrodeoxygenation and hydrogenation of lignin-derived model compounds to produce gasoline-range alkanes,6,7 these processes predominantly yield low-carbon-number hydrocarbons, unsuitable for diesel and aviation fuel applications.8 Recent research has shifted toward producing higher-carbon-number hydrocarbons, suitable for jet and diesel fuels, through strategies such as alkylation9 and aldol-condensation.10 The preparation of high-carbon-number phenolic compounds from lignin is expected to improve the production of high-carbon-number hydrocarbons.11 For example, the high-carbon-number phenolic compounds were obtained via reductive catalytic fractionation of lignin, which were further converted to high-carbon-number naphthenes (cycloalkanes) using Ru/C, suppressing significant cracking.12 The condensation of lignin-derived cycloalkenes using H form of zeolite Y was also suggested to obtain C12 or larger hydrocarbons.13 In addition to the catalysts, deep eutectic solvent was also used for the aldol condensation of phenolic compounds to prepare the aviation fuels.14Vanillin (VAN), a lignin-derived chemical, features various oxygenated functional groups (aldehyde, methoxy, and hydroxyl), making it a versatile precursor for forming higher-carbon-number hydrocarbons. Recent studies have demonstrated the potential for fractionating VAN from lignin with a yield of 21.1 wt%,15 offering a foundation for large-scale VAN production despite its relatively low yield from lignin. Most research on VAN has focused on monomeric products such as vanillyl alcohol (VANOL),16 creosol (CRSOL),17 methylcyclohexanol,18 and methylcyclohexane.19 Additionally, VAN condensation has been proposed for producing higher-carbon-number hydrocarbons,20,21 suitable for jet and diesel fuels, though the detailed reaction mechanisms remain unclear. This underscores the importance of further research to optimize the production of such compounds.
Catalytic hydrotreating has been widely developed for producing hydrocarbons from biomass-derived compounds. Noble metals such as ruthenium (Ru),22 palladium (Pd),23 and rhenium (Re),24 along with non-noble transition metals such as nickel (Ni),25 copper (Cu),26 and cobalt (Co),27 have been used in these processes. Among these, Ru has exhibited exceptional efficiency in hydrogenating oxygenated model compounds, including guaiacol (GUA),22 levoglucosan,28 furfuryl alcohol,29 eugenol,30 and VAN,31 as well as in converting actual biomass-derived oils.9 Ru catalysts selectively hydrogenate C–O bonds while minimizing undesired cleavage of C–C and C–(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) bonds. The choice of support material significantly influences catalytic performance by adjusting metal dispersion, promoting metal–support interactions, and tuning activity toward specific substrates.32 Activated carbon, a widely used catalyst support, offers advantages such as high surface area, low cost, and stability at elevated temperatures in oxygen-free environments. Moreover, its economic benefits include ease of noble metal recovery through combustion, making it preferable to traditional supports such as alumina and silica.33
O) bonds. The choice of support material significantly influences catalytic performance by adjusting metal dispersion, promoting metal–support interactions, and tuning activity toward specific substrates.32 Activated carbon, a widely used catalyst support, offers advantages such as high surface area, low cost, and stability at elevated temperatures in oxygen-free environments. Moreover, its economic benefits include ease of noble metal recovery through combustion, making it preferable to traditional supports such as alumina and silica.33
The efficiency of hydrogenation, hydrodeoxygenation, and alkylation reactions can be modulated by solvent choice, which stabilizes reactants and intermediates, dissolves substrates, disperses catalysts, and sometimes acts as a reactant or promoter.34 Biphasic reaction systems, particularly those involving water and organic solvents, are advantageous for biomass-derived oils, as they enable product extraction from the monophasic solvent, enhancing product yield and protection.35 Because of the high water content (15–30%) in biomass pyrolysis oil,36 biphasic systems provide a realistic simulation of actual biomass-derived feedstocks. Water, because of its environmental compatibility and ability to dissolve polar reactants and intermediates, is commonly used in hydrodeoxygenation of VAN, often in combination with an organic phase for continuous hydrophobic product extraction.19,37,38 This biphasic approach improves separation efficiency and product stability, making it a practical solution for catalytic processes.
This study investigates the synthesis of high-carbon-number hydrocarbons from VAN based on catalytic upgrading of VAN and other biomass-derived reactants. In addition to the production of VANOL,39 CRSOL,40,41 cycloalkanes,19 and cyclic alcohols42 from VAN, the production of high-carbon-number hydrocarbons is achieved via the condensation of VANOL and CRSOL, both obtained through the selective hydrogenation/hydrodeoxygenation of VAN (Scheme 1). The resulting C14 cycloalkane is suitable for use in aviation and diesel fuels. This study elucidates the detailed reaction mechanism through experimental results and density functional theory (DFT) calculations, providing critical insights for the selective production of high-carbon-number aviation fuels from lignin- or lignocellulose-derived feedstocks.
 | ||
| Scheme 1 Aviation biofuels from lignocellulose-derived VAN. | ||
The condensation of lignin-derivatives to higher-carbon-number compounds has been suggested in the literature (Table 1). The condensation of VAN to its dimers was reported. While the alkylation using homogeneous catalysts was reported,43 the heterogeneous catalysts produced the dimers via oxygen-functionality-involved alkylation.20,21 Notably, the dimer reported from the homogenous catalysis differed from that observed in this study.43 The condensation of saturated compounds of cyclohexanone and cyclohexane was also suggested, which requires the saturation of phenyl rings prior to the condensation. In addition to these single compound reactants, the condensation of phenolic compounds in the lignocellulose fast pyrolysis oil was also suggested.9 While several studies have been reported for the condensation of lignin-derivatives, the reaction mechanism has not been well understood which can be important to improve the condensation. Based on these observations, the goals of this study are (i) to understand the reaction mechanism of VAN condensation and (ii) to suggest how to improve the production of dimers from phenolic monomers, VAN in this study.
| Reactant | Catalyst | Reaction conditions | Yield and product |
|---|---|---|---|
| a GC-MS-measured area% assuming 100% yields of deoxygenated monomers and dimers. | |||
| VAN (2 mmol) in 20 mL water and 10 mL n-octane21 | Ru/Hβ | Batch, 50 bar H2, 200 °C, 1 h | 7% oxygen-free dimers |
| VAN (3.3 mmol) in 15 mL water with a microwave43 | K2SO4, FeSO4·7H2O | Batch, 100 °C, 5 min | 95% oxygenated dimer |
| VAN (1.8 mmol) in 30 mL water20 | Al2O3-cogelled Ru | Batch, 40 bar H2, 270 °C, 2 h | 48 area% deoxygenated dimersa |
| Cyclohexanone44 | HRF5015 resin (2 wt%) | Batch, 100 °C, 10 h | 50% dimers |
| Cyclohexene45 | Re2O7-B2O3/Al2O3 | Batch, 75–80 °C | 95% dimers |
| Lignocellulose fast pyrolysis oil9 | Pd/C then NiFe/TiO2 then HY then Ru/WO3-ZrO2 | Continuous flow, 180–270 °C, 100 bar H2 | Dimers formed |
2. Experimental section
2.1. Materials
All chemicals were used without further purification. Ruthenium(III) chloride hydrate (RuCl3·xH2O, ≥99.9%), palladium(II) chloride (PdCl2, 99%), chloroplatinic acid hexahydrate (H2PtCl6·6H2O, ≥37.5% Pt), nickel(II) chloride (NiCl2, 98%), cobalt(II) chloride hexahydrate (CoCl2·6H2O, 98%), GUA (C7H8O2, ≥98.0%), VANOL (C8H10O3, 98%), VAN (C8H8O3, 99%), 4-methyl cyclohexanol (C7H14O, 98%), activated charcoal, methyl cyclohexane (C7H14, ≥99%), and pyridine (C5H5N, 99.8%) were purchased from Sigma-Aldrich (Milwaukee, Wisconsin, USA). CRSOL (C8H10O2, ≥98%) and ammonium-form ZSM-5 zeolite (Si/Al = 50 mol mol−1) were purchased from Alfa Aesar (Ward Hill, Massachusetts, USA). n-Octane (C8H18, 97%) was purchased from Daejung Chemicals and Metals Co. Ltd (Siheung, Korea). Deionized (DI) water (18.2 MΩ⋅cm) in this study was prepared using an AquaMax Ultra 370 water purification system (Young In Chromass, Anyang, Korea). Nitrogen gas (N2, 99.999%), helium gas (He, 99.9999%), argon gas (Ar, 99.999%), hydrogen gas diluted with Ar (H2/Ar, 5% v/v), and carbon monoxide diluted with He (CO/He, 10% v/v) were purchased from Sinyang Medicine (Seoul, Korea).2.2. Catalyst preparation
Activated carbon-supported Ru catalysts (1, 3, and 5 wt% Ru/C) were prepared by incipient wetness impregnation. A solution of Ru precursor (RuCl3·xH2O) was prepared by dissolving the salt in DI water, using 5 mL of DI water per 10 g of support, based on total pore volume (0.5 cm3 g−1) of activated carbon support. The solution was slowly added to the activated carbon while stirring with a glass rod. The resulting mixture was dried at 105 °C for 16 h and then reduced under H2 flow (50 mL min−1) at 350 °C for 3 h. Catalysts containing 1 wt% of Pd, Pt, Ni, and Co (Pd/C, Pt/C, Ni/C, and Co/C) were prepared similarly using the respective metal chloride precursors (PdCl2, H2PtCl6·6H2O, NiCl2, and CoCl2·6H2O). The 3 wt% Ru/HZSM-5 catalyst was also prepared via the impregnation method.19 The ammonium form of ZSM-5 (Si/Al = 50 mol mol−1) was first calcined at 550 °C for 4 h to obtain the protonated form (HZSM-5). RuCl3·xH2O was dissolved in 100 mL of DI water, and HZSM-5 was added to the solution. After mixing for 3 h, the water was removed by vacuum evaporation. The resulting material was dried at 105 °C for 16 h and reduced under H2 flow (50 mL min−1) at 500 °C for 3 h.2.3. Catalyst characterization
N2 physisorption was performed using a Micrometrics (Norcross, Georgia, USA) ASAP2020 instrument. The Brunauer–Emmett–Teller (BET) surface area was determined using the BET model, while the micropore area was calculated through t-plot analysis. The average pore diameters and the Barrett–Joyner–Halenda (BJH) pore size distributions were obtained from the desorption branch of the N2 desorption isotherms. Carbon monoxide (CO) chemisorption measurements were performed using a BELCAT-M catalyst analyzer (MicrotracBEL, Osaka, Japan) equipped with a thermal conductivity detector (TCD). Prior to analysis, the catalyst (0.04–0.05 g) was reduced at 350 °C for 1 h under H2/Ar flow (5% v/v). After cooling to 50 °C, a CO/He mixture (10% v/v) was pulsed over the sample to quantify the amount of chemisorbed CO. Transmission electron microscopy (TEM) was performed using a Titan 80–300TM instrument (Thermo Fisher Scientific, Waltham, Massachusetts, USA) at the KIST Advanced Analysis Center (Seoul, Republic of Korea). X-ray diffraction (XRD) analysis was performed using a Rigaku (Tokyo, Japan) Dmax2500/PC X-ray diffractometer, scanning at 2° min−1 over a 2θ range of 10–90°. Pyridine-adsorbed Fourier transform infrared spectroscopy (Py-FTIR) was performed using a Nicolet iS20 FTIR spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). A catalyst pellet (0.02 g) was prepared and treated with He at 300 °C for 1 h to remove residual organics and moisture. Pyridine was adsorbed onto the catalyst for 30 min, and the sample was evacuated. FTIR spectra were recorded at 30 min intervals over 2 h. Quantitative analysis was performed using extinction coefficients of 1.13 cm μmol−1 for Brønsted acid sites and 1.28 cm μmol−1 for Lewis acid sites.2.4. Hydrogenation and condensation of VAN
Hydrogenation of VAN was performed in a 150 mL stainless-steel autoclave reactor. The reactor was charged with VAN (2 mmol), n-octane (15 mL), DI water (30 mL), and the required amount of catalyst. Prior to the reaction, the reactor was purged three times with H2 at 20 bar. The reactor was then heated in an electric furnace to the target temperature, with the reaction time counted from the moment the desired temperature was reached. Throughout the reaction, the reactor was stirred at 520 rpm. After the reaction, the reactor was immediately quenched using cold water (10 °C) to halt further reactions. Compounds in the n-octane and water layers were analyzed using gas chromatography–mass spectrometry (GC-MS) equipped with an HP-5MS column (60 m × 0.25 μm × 0.25 mm ID). Product quantification was performed using a gas chromatograph equipped with an HP-5 column (60 m × 0.25 μm × 0.25 mm ID) and a flame ionization detector (FID). To prevent column damage, 2 mL of the aqueous phase was extracted with 4 mL of ethyl acetate before analysis. n-Hexadecane was used as an internal standard for the quantification. The catalyst was recovered from the liquid products by vacuum filtration and dried in an oven at 105 °C for 16 h. The conversion of VAN and the yields of the products were calculated using the following equations: | (1) |
 | (2) |
The reusability of the spent catalyst was evaluated by recovering it through washing the remaining solid in the reactor with acetone after the reaction. The collected suspension was vacuum-filtered using a Whatman syringe filter (0.45 μm), and the recovered catalyst powder was dried at 105 °C for 16 h before being used in the subsequent reaction.
2.5. Condensation of VANOL and CRSOL
The condensation of VANOL and CRSOL was performed in a 150 mL stainless-steel autoclave reactor without a catalyst. VANOL and CRSOL (1 mmol each) were added to the reactor along with a mixed solvent of n-octane (15 mL) and DI water (30 mL). Prior to the reaction, the reactor was purged three times with N2 gas at 20 bar. The reactor was then pressurized to 50 bar with N2, and the temperature was incrementally adjusted to 50, 100, 150, or 200 °C. Once the target temperature was reached, the reaction was performed for 3 h. After the reaction, the reactor was immediately quenched using cold water (10 °C) to halt further reactions. Products in both the water and n-octane phases were analyzed using the same gas chromatography–mass spectrometry (GC-MS) and flame ionization detection (FID) methods described in section 2.4.2.6. Condensation of VAN, VANOL, and CRSOL
The condensation of VAN, VANOL, and CRSOL was conducted in a 150 mL stainless-steel autoclave reactor. When a catalyst was used, 1 wt% Ru/C (0.05 g) was added to the reactor. For all reactions, the reactants (2 mmol each) were introduced into a mixed solvent of n-octane (15 mL) andDI water (30 mL). For the mixture of VANOL and CRSOL, 1 mmol of each reactant was used. The reactor was purged three times with H2 before being pressurized to 50 bar with H2. The temperature was then raised to 200 °C, and the reaction was performed for 1 h. After completion, the reactor contents were immediately quenched with cold water (10 °C) to halt further reactions. Products from both the solid and liquid phases were analyzed using the methods detailed in section 2.4.2.7. Hydrodeoxygenation of condensed oxygenates
The hydrodeoxygenation was performed in a 150 mL stainless-steel autoclave reactor. VAN (2 mmol) and 1 wt% Ru/C catalyst (0.05 g) were added to the reactor containing a mixture of n-octane (15 mL) and DI water (30 mL). The system was purged three times with H2, after which the H2 pressure was set to 50 bar, and the temperature was increased to 150 °C. The reaction was performed for 3 h, followed by quenching with cold water (10 °C). Subsequently, 3 wt% Ru/HZSM-5 catalyst (0.3 g) was added to the reactor, and the system was again purged three times with H2. The temperature was then raised to 200 °C, and the reaction proceeded for 1 h. The reactor was quenched with cold water (10 °C) upon completion. Products from both the solid and liquid phases were analyzed using the methods detailed in section 2.4.2.8. Characterization of condensate
The reaction mixture was diluted with DI water and extracted with CH2Cl2. The organic layer was washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was purified by column chromatography on silica gel using a gradient of ethyl acetate/hexanes (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 → 1:6). Remaining impurities were removed via preparative thin-layer chromatography (ethyl acetate/hexanes = 1:8). NMR spectra were recorded on a Bruker (Tucson, Arizona, USA) 400 MHz spectrometer. 1H NMR chemical shifts were referenced to residual CDCl3 (δ = 7.26 ppm), while 13C NMR chemical shifts were referenced to residual CDCl3 (δ = 77.16 ppm). All chemical shift data are reported in δ (ppm). Abbreviations used for 1H NMR data include: s = singlet, d = doublet, and m = multiplet. The data for the isolated condensate are as follows:
:8 → 1:6). Remaining impurities were removed via preparative thin-layer chromatography (ethyl acetate/hexanes = 1:8). NMR spectra were recorded on a Bruker (Tucson, Arizona, USA) 400 MHz spectrometer. 1H NMR chemical shifts were referenced to residual CDCl3 (δ = 7.26 ppm), while 13C NMR chemical shifts were referenced to residual CDCl3 (δ = 77.16 ppm). All chemical shift data are reported in δ (ppm). Abbreviations used for 1H NMR data include: s = singlet, d = doublet, and m = multiplet. The data for the isolated condensate are as follows:
1H NMR (400 MHz, CDCl3) δ 6.82 (d, J = 8.5 Hz, 1H), 6.72–6.59 (m, 4H), 5.51–5.37 (m, 2H), 3.86 (s, 3H), 3.84–3.79 (m, 5H), 2.18 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 146.6, 144.8, 143.9, 143.5, 132.6, 132.2, 127.9, 121.5, 116.2, 114.3, 113.1, 111.4, 56.2, 56.0, 38.6, 19.4; Rf = 0.53 (ethylacetate/hexanes = 1/8).
2.9. Density functional theory calculation of product molecules
DFT calculations were conducted using Gaussian 16 software46 to explore the reaction mechanism and energy profile for the condensation of VANOL and CRSOL. All molecular geometries were fully optimized without constraints using the M06-2X functional with the 6-311++G(d,p) basis set. Geometry optimizations were performed with the 6-311++G(d,p) basis set. The optimization was performed with ‘opt = maxcycle = 1000’ to ensure convergence, followed by frequency calculations to confirm the nature of the stationary points and to obtain thermochemical corrections. The self-consistent field (SCF) procedure was controlled using ‘scf = (xqc,maxcycle = 1000)’ to enhance convergence reliability. All calculations were performed in the gas phase. Frequency calculations were used to confirm stationary points as minima (reactants, products, and intermediates) and to compute zero-point energy and thermal corrections.2.10. Density functional theory calculation of catalysts and reaction mechanism
To examine the changes in reaction mechanisms and product selectivity under adjusted reaction conditions, DFT calculations were performed using the Vienna Ab initio Simulation Package (VASP).47 The Bayesian error-estimation functional with van der Waals forces (BEEF-vdW) was employed to describe the exchange–correlation energy of electrons. Calculations utilized projector augmented wave (PAW) pseudopotentials, a plane wave energy cutoff of 460 eV, and the RMM-DIIS algorithm. Gaussian smearing with a width of 0.1 eV was applied, and all atomic positions were relaxed until the maximum force components were below 0.03 eV Å−1. A 2 × 2 × 1 Monkhorst–Pack grid was adopted for k-point sampling. Frequency calculations were performed to determine zero-point energy (ZPE) and thermal corrections. Reaction free energies (ΔG) were calculated as:| ΔG = ΔE + ΔZPE − TΔS | (3) |
3. Results
3.1. Catalytic hydrogenation of VAN
Hydrogenation and condensation of VAN were performed using 1 wt% Ru/C at 200 °C under 50 bar H2 (Fig. 1) based on the screening results of carbon-supported metal catalysts including Ru/C, Pd/C, Pt/C, Ni/C, and Co/C (Fig. S1†). Ru/C was selected because it exhibited the higher VAN conversion and the higher yield of phenolic dimer. For Ru/C, the product distribution varied with the amount of catalyst used, offering valuable insights into the reaction mechanism. Additionally, reaction temperature and pressure were systematically adjusted to evaluate their effects on catalytic performance. The optimal reaction conditions were determined based on these experimental results. | ||
| Fig. 1 Effects of catalyst-to-substrate ratio at (a) 150 °C and (b) 200 °C exhibiting products of CRSOL (2), GUA (3), dimer 1 (4), 2-methoxy-4-methyl-cyclohexanol (5), 4-methyl cyclohexanol (6), methylcyclohexane (7). (Reaction conditions: 2 mmol of VAN, a mixed solvent of 30 mL of DI water and 15 mL of n-octane, 1 h of reaction time, 0.03–0.15 g of 1 wt% Ru/C, 50 bar of H2 pressure measured at room temperature.) | ||
At 200 °C, CRSOL, GUA, 2-methoxy-4-methyl-cyclohexanol (the ring-saturated form of CRSOL), 4-methylcyclohexanol, and methylcyclohexane were produced, along with dimer 1 (Fig. 1(b) and Table S2†). At catalyst/substrate ratios ≥0.29 w/w, the formation of ring-saturated compounds, including methylcyclohexane, 4-methylcyclohexanol, and 2-methoxy-4-methylcyclohexanol, was predominant. In contrast, at catalyst/substrate ratios ≤0.25 w/w, partially hydrogenated phenolic compounds such as CRSOL, GUA, and dimer 1 were the major products.
The structure of dimer 1 was confirmed by 2D NMR spectroscopy, including COSY, HSQC, and HMBC analyses (Fig. 2; see ESI† for details). All proton and carbon signals were assigned based on these spectra. Notably, the HMBC result revealed key correlations: the proton at C7 exhibited 2J couplings with C5 and C8, and 3J couplings with C4, C6, C9, and C13. Additionally, the methyl protons at C14 exhibited 3J couplings with C3 and C5, and 2J couplings with C4, further supporting the proposed structure.
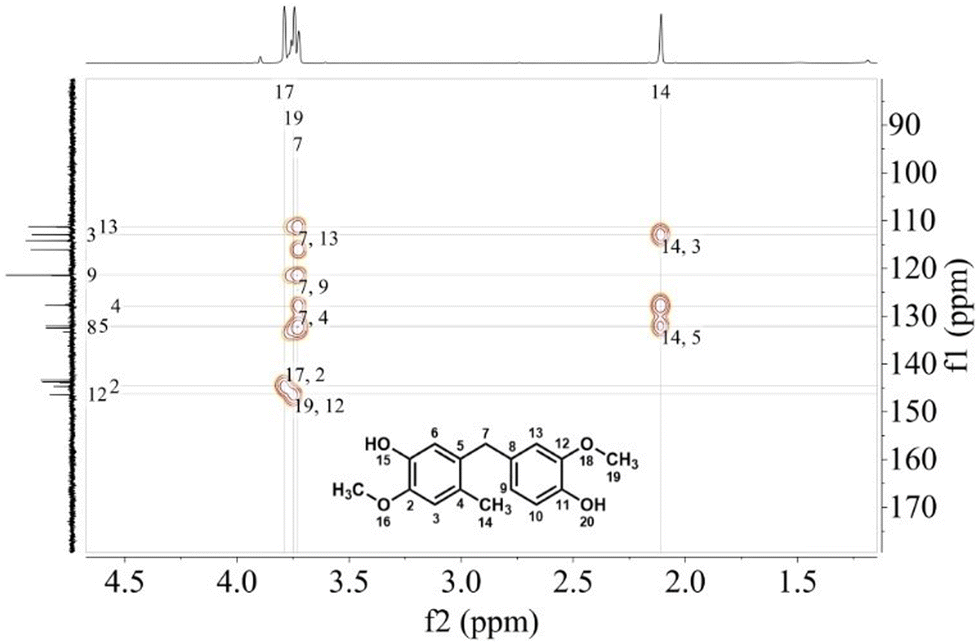 | ||
| Fig. 2 HMBC NMR result (400 MHz, CDCl3) of dimer 1. | ||
 | ||
| Fig. 3 Effects of Ru metal loading at (a) 150 °C and (b) 200 °C exhibiting products of CRSOL (2), GUA (3), dimer 1 (4), 2-methoxy-4-methyl-cyclohexanol (5), 4-methyl cyclohexanol (6), methyl cyclohexane (7). (Reaction conditions: 1–5 wt% Ru/C catalysts, 2 mmol of VAN in a mixed solvent of 30 mL of DI water and 15 mL of n-octane, 1 h of reaction time, 0.05 g of Ru/C catalyst, 50 bar of H2 pressure measured at room temperature.) | ||
The catalyst-to-substrate ratio was fixed at 0.16 w/w (<0.25 w/w, as discussed in Fig. 1), and the reaction time was set to 1 h. At 150 °C, VANOL, CRSOL, and dimer 1 were produced using 1 wt% Ru/C (Fig. 3(a) and Table S3†). However, with 3 wt% Ru loading, VANOL was not detected, and the yield of dimer 1 decreased. This indicates that VANOL was rapidly converted to CRSOL under these conditions, thereby suppressing the formation of dimer 1, which is typically obtained by alkylation between VANOL and CRSOL. At 5 wt% Ru loading, additional ring-saturated compounds derived from CRSOL were observed, indicating increased hydrogenation activity at higher Ru loading. At 200 °C, the production of ring-saturated oxygenated and deoxygenated cyclohexyl compounds increased with increasing Ru loading (1, 3, and 5 wt%), consistent with the trends observed at higher catalyst-to-substrate ratios. These results confirm that the formation of aromatic intermediates (VANOL, CRSOL, and aromatic condensates) during hydrogenation is favoured at lower catalyst loading. Based on the results in Fig. 1 and 2, a reaction temperature of 150 °C and a Ru loading of 1 wt% were identified as optimal for dimer 1 production.
 | ||
| Fig. 4 Effects of (a) H2 pressure measured at room temperature and (b) reaction temperature exhibiting products of VANOL (1), CRSOL (2), and dimer 1 (4). (Reaction conditions: 2 mmol of VAN in a mixed solvent of 30 mL of DI water and 15 mL of n-octane, 50–200 °C of reaction temperature, 1 h of reaction time, 0.05 g of 1 wt% Ru/C catalyst, catalyst/substrate = 0.17 w/w, 10–50 bar of H2 pressure measured at room temperature.) | ||
 | ||
| Fig. 5 Effects of reaction pressure and time on VAN hydrogenation and condensation for (a) 10 bar, (b) 30 bar, and (c) 50 bar of H2 pressure measured at room temperature. (Reaction conditions: 2 mmol of VAN in a mixed solvent of 30 mL of DI water and 15 mL of n-octane, 0.05 g of 1 wt% Ru/C catalyst, 150 °C of reaction temperature.) | ||
3.2. Non-catalytic condensation of VANOL and CRSOL
To further investigate the formation of dimer 1 through the condensation of VANOL and CRSOL, additional control experiments were performed (Fig. 6 and Table S9†). These reactions were performed without catalysts at 50 bar of N2 (measured at room temperature) for 3 h to prevent hydrogenation and promote reactant condensation. No dimer formation was observed at temperatures below 150 °C, whereas a 34% yield of dimer 1 was achieved at 200 °C. These results suggest that non-catalytic condensation requires a temperature as high as 200 °C. | ||
| Fig. 6 Product compositions of non-catalytic condensation of VANOL and CRSOL. Reaction conditions: 1 mmol of VANOL and CRSOL each, 30 mL of DI water, 15 mL of n-octane, 50 bar of N2 pressure (measured at room temperature), 3 h of reaction time. | ||
3.3. Condensation of VAN, VANOL, and CRSOL
To further understand the catalytic function and the reactivity of intermediates, control experiments were conducted (Table 2 and Fig. S3†). In the presence of a Ru/C catalyst, the reaction with VAN produced dimer 1, along with VANOL and CRSOL. VAN was first hydrogenated to VANOL and CRSOL, which subsequently condensed to form dimer 1. However, other combinations of reactants and intermediates did not undergo condensation in the presence of Ru/C. In a mixture of VANOL and CRSOL with Ru/C, no condensation occurred; Instead, hydrogenation produced CRSOL, 2-methoxy-4-methylcyclohexanol, and 4-methylcyclohexanol. This outcome is attributed to the presence of the Ru/C catalyst and high H2 pressure, which favoured hydrogenation over condensation. These findings suggest that the preferential hydrogenation of VANOL and CRSOL inhibits their condensation, indicating that optimal conditions, such as high H2 pressure and low catalyst-to-substrate ratios, are necessary to increase the yield of dimer 1.| Reactant | Catalyst | Formation of 1 |
|---|---|---|
| a Reaction conditions: 2 mmol of reactant (VAN, VANOL, or CRSOL) each, 30 mL of DI water, 15 mL of n-octane, 0 or 0.05 g of 1 wt% Ru/C, 50 bar of H2 pressure (measured at room temperature), 200 °C of reaction temperature, 1 h of reaction time. b Reactant: a mixture of 1 mmol VANOL and 1 mmol CRSOL. | ||
| VAN | Present | Formed |
| VANOL | Not formed | |
| VANOL, CRSOLb | Not formed | |
| CRSOL | Not formed | |
| VAN | Not present | Not formed |
| VANOL | Formed | |
| VANOL, CRSOLb | Formed | |
| CRSOL | Not formed | |
In the absence of catalysts, condensation to form dimer 1 was observed when the reaction was performed with VANOL or a mixture of VANOL and CRSOL. However, when VAN was used as the reactant, its hydrogenation to VANOL and CRSOL did not occur, and no subsequent condensation was observed. Similarly, no condensation was observed when CRSOL was used alone. Notably, condensation occurred in reactions involving VANOL or a mixture of VANOL and CRSOL, even under 50 bar of H2. These findings highlight that the formation of dimer 1 proceeds via condensation between VANOL and CRSOL.
In addition to all the catalysis results of Ru/C under the adjusted reaction conditions, the spent catalyst was used for the condensation of VAN. Although this study focuses more on the catalytic pathways than on the design of high-performance catalysts, the spent Ru/C exhibited a decrease in both VAN conversion and dimer 1 yield (Fig. S4†). Because the condensation between VANOL and CRSOL can occur without a catalyst, the decreased VAN conversion indicates that the selective hydrogenation of VAN to VANOL and subsequently to CRSOL became less active. This catalyst deactivation can be attributed to the well-known carbon deposition on the Ru metal surface induced by hydrogenation.48
3.4. DFT calculations for understanding the selective hydrogenation
Based on the Ru particle size (0.93–0.99 nm) measured using HAADF-STEM images as depicted in Fig. S5 and Table S5,† all mechanisms were modelled using a hexagonal close-packing (hcp)-based nanoparticle consisting of 57 Ru atoms (Ru57),49 corresponding to a particle size of approximately 0.5–1.1 nm (Fig. 7 and S5†). This model has been frequently used in the literature because of its small dimensions and representative structure.50,51 The Ru57 with an hcp structure has the (0001) surface on the top, which is considered the catalytically active phase, along with the (10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) and (12
0) and (12![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 0) surfaces on the sides. We calculated the relaxation of Ru cluster to obtain stabilized geometry prior to analysing the Ru surface interacting with molecules.
0) surfaces on the sides. We calculated the relaxation of Ru cluster to obtain stabilized geometry prior to analysing the Ru surface interacting with molecules.
 | ||
| Fig. 7 Energy diagram of VAN adsorbed on Ru surface covered with (a) 1 ML H* and (b1, b2 and b3) 0 ML H*. (p) denotes a physisorbed molecule. | ||
To investigate the effects of catalyst-to-substrate ratios on product formation, we assumed that variations in catalyst quantity at a fixed H2 concentration alter the H* (chemisorbed H atom) coverage per catalyst. Under this assumption, DFT calculations were performed for two limiting cases: 0 ML H* and 1 ML H* models. To reduce computational cost, the 1 ML H* model considered only the upper half of the Ru surface covered with H* (denoted as 1 ML H*/Ru, compared with 0 ML H*/Ru).
Prior to analysing the reaction mechanism, molecular adsorption of VAN on the 0 ML H*/Ru and 1 ML H*/Ru surfaces was examined, with geometric representations of the adsorbed configurations depicted in Fig. 7. On the 0 ML H*/Ru surface, VAN chemisorbs with the phenyl ring and aldehyde groups anchored to the Ru surface, while the methoxy and alcohol groups orient away from it. In contrast, on the 1 ML H*/Ru surface, the phenyl ring fails to chemisorb, and the aldehyde group bonds to the edge site of the Ru particle.
To elucidate the hydrogenation mechanism of VAN on these surfaces, free energy diagrams were generated via DFT calculations (Fig. 7 and S5†). On the 1 ML H*/Ru surface (Fig. 7(a)), the preferred conversion of VAN to CRSOL without the phenyl ring saturation was suggested. Chemisorption of VAN (C8H8O3 (g)) through the aldehyde group (A0) is more thermodynamically favourable than its physisorption (VP). The conversion of VAN (A0) to CRSOL (A5) proceeds via a main pathway involving the formation of a carbon monohydride intermediate (A2), while the formation of VANOL (A1 to A3 to C8H10O3(g)) constitutes a by-product pathway. Notably, the VANOL gas molecule can be easily chemisorbed to the Ru surface and subsequently converted through the main reaction pathway. Further hydrogenation of adsorbed CRSOL to B1 is hindered by a high energy barrier (ΔG = 1.63 eV), whereas CRSOL desorption (A5 to C8H10O2(g)) is more favourable (ΔG = 0.49 eV), indicating that desorption dominates over further hydrogenation.
On the 0 ML H*/Ru surface (Fig. 7(b1, b2 and b3)), selective hydrodeoxygenation of VAN to CRSOL, followed by phenyl ring saturation to 2-methoxy-4-methylcyclohexan-1-ol and subsequent hydrodeoxygenation to methylcyclohexane, was hypothesized based on the calculated reaction pathways. VAN strongly chemisorbs and is hydrogenated to 4-methyl cyclohexanol (VAN to B3). Similar to the 1 ML H*/Ru case, the reaction via a carbon monohydride intermediate (A0 to A1 to A2 to A4 to A5) represents the primary pathway, while the VANOL formation pathway (A0 to A3 to C8H10O3(g)) is less favourable. Hydrodeoxygenation of VAN to CRSOL (A0 to A5) is preferred over phenyl ring hydrogenation to 4-hydroxy-3-methoxycyclohexane-1-carbaldehyde (A0 to a1 to a2 to a3 to C8H14O3), because of the higher energy barrier (ΔG = 1.16 eV) associated with ring saturation. The phenyl ring of adsorbed CRSOL is further saturated to 2-methoxy-4-methylcyclohexan-1-ol (A5 to B1 to B2 to B3 to C8H16O2), which is then hydrodeoxygenated to methylcyclohexane (C8H16O2 to D1 to D2 to C7H14). The reaction pathway leading to 4-methylcyclohexanol (C2) may be favoured over the alternative higher-energy pathway to 1-methoxy-3-methylcyclohexane (C4).
Based on the calculated pathways, CRSOL production is favoured under the limiting case of θH = 1 or at lower catalyst-to-substrate ratios. The formed CRSOL can couple with VANOL to form dimer 1. In contrast, the production of ring-saturated compounds is preferred under the limiting case of θH = 0 or at higher catalyst-to-substrate ratios suppressing the formation of 1.
3.5. Hydrodeoxygenation of a mixed product containing dimer 1
High-carbon-number hydrocarbons containing two cyclohexyl groups were synthesized from dimer 1via a two-step catalytic hydrodeoxygenation process (Table 3 and Fig. S6†), which can be useful for the aviation fuels. In the first step, catalysis using 1 wt% Ru/C formed VANOL (10%) and CRSOL (43%), alongside the partial retention of dimer 1 (19%). In the subsequent step, 3 wt% Ru/HZSM-5 was introduced into the reaction mixture without removing the 1 wt% Ru/C catalyst. This led to the hydrodeoxygenation of dimer 1, producing 1-(cyclohexylmethyl)-2-methylcyclohexane (1sat) and mono-cyclohexyl derivatives, including 2-methoxy-4-methylcyclohexanol, 4-methylcyclohexanol, and methylcyclohexane. The lower yield of 1sat (5%) and the higher yield of mono-cyclohexyl derivatives (62%) compared to the first step results suggest the potential cracking of dimer 1 into mono-cyclohexyl products. The strong solid acids of crystalline ZSM-5 (Fig. S7†) cracked the hydrocarbons into smaller or gas-phase light hydrocarbons, decreasing the yield of 1sat (Table S10†), although high deoxygenation activity was still be achieved.52 Notably, the methylcyclohexane and 1sat obtained in this process show promise as components for aviation fuels. In the absence of the initial condensation step to form dimer 1, a one-step control reaction of VAN with 3 wt% Ru/HZSM-5 produced only mono-cyclohexyl derivatives and did not yield 1sat. This observation highlights the necessity of the condensation step for the formation of C14 deoxygenated hydrocarbons.| Product yielda (%) | |||||
|---|---|---|---|---|---|
| VANOL | CRSOL | 1 | 1sat | Mono-cyclohexylsb | |
| a Product yields of first and second steps were measured based on the quantity of VAN reactant. b Compounds including 2-methoxy-4-methyl cyclohexanol, 4-methyl cyclohexanol, and methyl cyclohexane. c The control one-step hydrogenation of VAN. Reaction conditions: 2 mmol of VAN, a mixed solvent of 30 mL of DI water and 15 mL of n-octane, 0.3 g of 3 wt% Ru/HZSM-5 catalyst, 50 bar of H2 pressure (measured at room temperature), 200 °C of reaction temperature, 3 h of reaction time. d The first step of two-step reaction is the hydrogenation and condensation of VAN. Reaction conditions: 2 mmol of VAN, a mixed solvent of 30 mL of DI water and 15 mL of n-octane, 0.05 g of 1 wt% Ru/C catalyst, 30 bar of H2 pressure (measured at room temperature), 150 °C of reaction temperature, 3 h of reaction time. e The second step of two-step reaction is the hydrodeoxygenation of the product of first step. Reaction conditions: a product mixture of first step as a reactant, 0.3 g of 3 wt% Ru/HZSM-5 catalyst, 50 bar of H2 pressure (measured at room temperature), 200 °C of reaction temperature, 3 h of reaction time. | |||||
| Control one-step using 3 wt% Ru/HZSM-5c | 73 | ||||
| Frist step using 1 wt% Ru/Cd | 10 | 43 | 19 | ||
| Second step using 3 wt% Ru/HZSM-5e | 5 | 62 | |||
4. Discussion
Based on the reaction results, a reaction pathway for the conversion of VAN to hydrogenated and condensed products is proposed (Fig. 8(a)). At a low catalyst-to-substrate ratio (≤0.25 w/w), the formation of GUA and CRSOL was observed. VANOL is initially produced from VAN through the reduction of the aldehyde group to a hydroxyl group,53 followed by its further conversion to CRSOL. Partial demethylation also occurred, leading to the formation of GUA. At a higher catalyst-to-substrate ratio (≥0.29 w/w), the ring saturation of CRSOL proceeded, leading to the formation of methylcyclohexane. The non-catalytic formation of dimer 1 requires the presence of both CRSOL and VANOL,20 and the further conversion of CRSOL must be suppressed to maximize the production of dimer 1. | ||
| Fig. 8 (a) Reaction pathway of hydrogenation and condensation of VAN using Ru/C and (b) its energy profile for the condensation of VANOL and CRSOL through the formation of intermediate (INT). | ||
To further investigate the possibility of non-catalytic condensation when VANOL and CRSOL coexist, DFT was employed to analyse the reaction energy profile. Building on our previous examination of dimer formation resulting in 1, we evaluated the feasibility of the reaction through dehydration and electrophilic substitution mechanisms. The computational results, illustrated in the energy diagram (Fig. 8(b)), confirm that the condensation reaction is both thermodynamically and kinetically favourable. The reaction profile depicts three key states: the initial state with separate VANOL and CRSOL molecules, an intermediate state (INT) likely involving the alignment of reactive groups, and the final product state representing dimer formation. The transition from the initial state to the intermediate involves an energy barrier of +9.6 kcal mol−1, indicating kinetic feasibility under experimental conditions (200 °C). The intermediate state is thermodynamically favourable, with an energy decrease of −3.9 kcal mol−1 relative to the initial state. The final step to dimer formation exhibits a significant energy decrease of −11.9 kcal mol−1, highlighting the strong thermodynamic favourability of the condensation product. Overall, the total energy change from reactants to the final product is −15.8 kcal mol−1, demonstrating that the entire condensation process is thermodynamically driven. These computational findings provide strong evidence that the condensation reaction between VANOL and CRSOL is both kinetically accessible and thermodynamically favourable, corroborating the experimental observations of condensation occurring in the absence of a catalyst when these compounds coexist.
5. Conclusion
In this study, a candidate for aviation fuel was prepared from lignin- or lignocellulose-derived reactants. Aviation fuel precursors, intended to be converted into high-carbon-number hydrocarbons (C14), were successfully synthesized from VAN using a two-step process involving both catalytic and non-catalytic reactions. The reaction mechanism was proposed based on DFT calculations and control experiments, which improved the understanding of phenolic compound condensation and suggested a strategy for enhancing the production of high-carbon-number hydrocarbons from lignin or lignocellulose. While the condensation of lignin derivatives has been reported in the literature (Table 1), this study focused on elucidating the reaction mechanism to improve the condensation of phenolic compounds.For the production of aviation fuel candidates, the first-step reaction using 1 wt% Ru/C was performed to selectively hydrogenate VAN into VANOL and CRSOL. The production of VANOL and CRSOL was critical for the formation of the VAN dimer, as confirmed by control experiments. The mixture of VANOL and CRSOL was subsequently noncatalytically condensed to form an aviation fuel precursor, achieving a 19% yield of dimer 1, the structure of which was confirmed via NMR analysis, without the formation of ring-saturated mono-cyclohexyl derivatives.
For the production of VANOL and CRSOL, selective conversion of VAN without ring saturation was required, which was successfully achieved by adjusting the catalyst (1 wt% Ru/C)-to-substrate (VAN) ratio to 0.25 w/w or lower in the batch reaction system. DFT calculations indicated that a high concentration of adsorbed hydrogen on the catalyst surface, facilitated by using less catalyst under constant H2 pressure, favoured the formation of VANOL and CRSOL over phenyl ring saturation leading to cyclohexyl derivatives.
Further deoxygenation to hydrocarbons was successfully performed via a second-step reaction using 3 wt% Ru/HZSM-5. Oxygen-free high-carbon-number hydrocarbons (5% yield of 1sat) from dimer 1 and mono-cyclohexyl derivatives (62% yield), not condensed to 1, were obtained from other compounds. The formation of these high-carbon-number hydrocarbons can improve the fuel properties of lignin- or lignocellulose-derived compounds for aviation applications.
The high-carbon-number hydrocarbons produced in the second step exhibit significant potential as bio-aviation fuels. This work establishes a viable approach for synthesizing aviation fuel precursors from biomass-derived VAN, emphasizing the critical role of intermediate selectivity in the reaction pathway. These findings provide a promising foundation for developing sustainable and environmentally friendly fuels from renewable resources. Future research will focus on optimizing system efficiency and expanding the approach to a broader range of biomass-derived feedstocks.
Abbreviations
| VAN | Vanillin (4-hydroxy-3-methoxybenzaldehyde) |
| VANOL | Vanillyl alcohol (4-(hydroxymethyl)-2-methoxyphenol) |
| CRSOL | Creosol (2-methoxy-4-methylphenol) |
| GUA | Guaiacol (2-methoyxl phenol) |
| 1 | 5-(4-Hydroxy-3-methoxybenzyl)-2-methoxy-4-methylphenol (oxygenated dimer) |
| 1sat | 1-(Cyclohexylmethyl)-2-methylcyclohexane |
Author contributions
Jina Eun: investigation, writing – original draft. Jeonghun Kim: software, writing – original draft. Han Byeol Kim: formal analysis. Do Heui Kim: validation. Jae-Wook Choi: data curation. Kwang Ho Kim: validation. Chun-Jae Yoo: validation. Seongmin Jin: validation. Kyeongsu Kim: software. Hyunjoo Lee: validation. Chang Soo Kim: validation. Kwan-Young Lee: supervision. Jong Suk Yoo: software, supervision, writing – review & editing. Seo-Jung Han: supervision, formal analysis, writing – review & editing. Keunhong Jeong: software, writing – review & editing. Jeong-Myeong Ha: conceptualization, supervision, writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the program of Development of Eco-friendly Chemicals as Alternative Raw Materials to Oil through the National Research Foundation (NRF) of Korea funded by the Ministry of Science and ICT (2022M3J5A1085250). This research was also supported by Korea Environment Industry & Technology Institute (KEITI) through Center of Plasma Process for Organic Material Recycling Program, funded by Korea Ministry of Environment (MOE) (2022003650001). J. S. Y. acknowledges the support from the Basic Study and Interdisciplinary R&D Foundation Fund of the University of Seoul (2021).References
- M. Prussi, U. Lee, M. Wang, R. Malina, H. Valin, F. Taheripour, C. Velarde, M. D. Staples, L. Lonza and J. I. Hileman, Renewable Sustainable Energy Rev., 2021, 150, 111398 CrossRef CAS.
- H. Wei, W. Liu, X. Chen, Q. Yang, J. Li and H. Chen, Fuel, 2019, 254, 115599 Search PubMed.
- R. Xing, A. V. Subrahmanyam, H. Olcay, W. Qi, G. P. van Walsum, H. Pendse and G. W. Huber, Green Chem., 2010, 12, 1933–1946 Search PubMed.
- G. Li, N. Li, Z. Wang, C. Li, A. Wang, X. Wang, Y. Cong and T. Zhang, ChemSusChem, 2012, 5, 1958–1966 CrossRef CAS PubMed.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 Search PubMed.
- F. F. Zormpa, A. G. Margellou, S. A. Karakoulia, E. Delli and K. S. Triantafyllidis, Catal. Today, 2024, 433, 114654 Search PubMed.
- Z. Zhang, X. Wang, C. Wang, Z. Yan, G. Zhuang, N. Ma and Q. Li, Chem. Eng. J., 2024, 483, 149367 CrossRef CAS.
- L. Faba, E. Díaz and S. Ordóñez, Appl. Catal., B, 2012, 113–114, 201–211 CrossRef CAS.
- J. Eun, R. Insyani, J.-W. Choi, D. J. Suh, K. Kim, H. Lee, K. H. Kim, C. S. Kim, K. Y. Lee, C.-J. Yoo and J.-M. Ha, Energy Convers. Manage., 2024, 314, 118696 CrossRef CAS.
- H. Gao, F. Han, G. Li, A. Wang, Y. Cong, Z. Li, W. Wang and N. Li, Sustainable Energy Fuels, 2022, 6, 1616–1624 RSC.
- M. S. Kollman, X. Jiang, R. Sun, X. Zhang, W. Li, H.-M. Chang and H. Jameel, Chem. Eng. J., 2023, 451, 138464 CrossRef CAS.
- D. Verma, H.-J. Chun, N. Karanwal, J. Choi, S. Oh, S. M. Kim, S. K. Kim and J. Kim, Chem. Eng. J., 2024, 490, 151420 CrossRef CAS.
- A. Kumar, A. Kumar, D. M. Santosa, H. Wang, P. Zuo, C. Wang, A. Mittal, R. Gieleciak, D. P. Klein, M. J. Manto and B. Yang, Appl. Catal., A, 2024, 676, 119649 CrossRef CAS.
- Y. Jian, X. Li, Y. Liu, X. Chen, Y. Zhang, S. Baroutian and Q. Yu, Fuel, 2023, 347, 128491 CrossRef CAS.
- Y. Zhu, Y. Liao, W. Lv, J. Liu, X. Song, L. Chen, C. Wang, B. F. Sels and L. Ma, ACS Sustainable Chem. Eng., 2020, 8, 2361–2374 CrossRef CAS.
- H. Fan, F. Qin, Q. Yuan, Z. Sun, H. Gu, W. Xu, H. Tang, S. Liu, Y. Wang, W. Chen, J. Li and H. Zhai, J. Mater. Chem. A, 2023, 11, 17560–17569 RSC.
- S. Govoni, A. Ventimiglia, C. P. Ferraz, I. Itabaiana Jr, A. Dufour, A. Pasc, R. Wojcieszak and N. Dimitratos, ChemCatChem, 2024, 16, e202400453 CrossRef CAS.
- X. Wang, Y. Zhang, J. Chen and Y. Xu, Catal. Lett., 2024, 154, 3195–3211 CrossRef CAS.
- H. Kim, S. Yang, Y. H. Lim, J.-M. Ha and D. H. Kim, J. Hazard. Mater., 2022, 423, 126525 CrossRef CAS PubMed.
- I. Yati, A. A. Dwiatmoko, J. S. Yoon, J.-W. Choi, D. J. Suh, J. Jae and J.-M. Ha, Appl. Catal., A, 2016, 524, 243–250 Search PubMed.
- J. H. Park, H. Kim, H. Jung, J.-M. Ha and D. H. Kim, Chem. Eng. J., 2024, 499, 155888 Search PubMed.
- H. Kim, J. H. Park, J.-M. Ha and D. H. Kim, ACS Catal., 2023, 13, 11857–11870 Search PubMed.
- H. Kim, J. Lee, Y. Kim, J.-M. Ha, Y.-K. Park, D. G. Vlachos, Y.-W. Suh and J. Jae, Chem. Eng. J., 2024, 481, 148328 CrossRef CAS.
- J. S. Reinhold, J. Pang, B. Zhang, F. E. Kühn and T. Zhang, Green Chem., 2024, 26, 10661–10686 RSC.
- J. Seo, J. S. Kwon, H. Choo, J.-W. Choi, J. Jae, D. J. Suh, S. Kim and J.-M. Ha, Chem. Eng. J., 2019, 377, 119985 Search PubMed.
- Y. An, Q. Wu, L. Niu, C. Zhang, Q. Liu, G. Bian and G. Bai, J. Catal., 2024, 429, 115271 Search PubMed.
- H. Kim, Y. H. Lim, J. H. Park, J.-M. Ha and D. H. Kim, Green Chem., 2024, 26, 2692–2704 RSC.
- A. B. Bindwal, A. H. Bari and P. D. Vaidya, Chem. Eng. J., 2012, 207–208, 725–733 Search PubMed.
- A. B. Jain and P. D. Vaidya, Energy Fuels, 2020, 34, 9963–9970 CrossRef CAS.
- A. Bjelić, M. Grilc and B. Likozar, Chem. Eng. J., 2018, 333, 240–259 CrossRef.
- A. B. Bindwal and P. D. Vaidya, Energy Fuels, 2014, 28, 3357–3362 CrossRef CAS.
- J. J. Musci, M. Montaña, A. B. Merlo, E. Rodríguez-Aguado, J. A. Cecilia, E. Rodríguez-Castellón, I. D. Lick and M. L. Casella, Catal. Today, 2022, 394–396, 81–93 CrossRef CAS.
- E. Lam and J. H. T. Luong, ACS Catal., 2014, 4, 3393–3410 CrossRef CAS.
- X. Wang, M. Arai, Q. Wu, C. Zhang and F. Zhao, Green Chem., 2020, 22, 8140–8168 RSC.
- M.-Y. Chen, Y.-B. Huang, H. Pang, X.-X. Liu and Y. Fu, Green Chem., 2015, 17, 1710–1717 RSC.
- S. Czernik and A. V. Bridgwater, Energy Fuels, 2004, 18, 590–598 CrossRef CAS.
- P. Hao, D. K. Schwartz and J. W. Medlin, ACS Catal., 2018, 8, 11165–11173 CrossRef CAS.
- Z. Zhu, H. Tan, J. Wang, S. Yu and K. Zhou, Green Chem., 2014, 16, 2636–2643 RSC.
- E. Aliu, A. Hart and J. Wood, Catal. Today, 2021, 379, 70–79 CrossRef CAS.
- R. Nie, H. Yang, H. Zhang, X. Yu, X. Lu, D. Zhou and Q. Xia, Green Chem., 2017, 19, 3126–3134 RSC.
- H. Xu and H. Li, J. Catal., 2023, 423, 105–117 CrossRef CAS.
- M. Liu, J. Zhang, L. Zheng, G. Fan, L. Yang and F. Li, ACS Sustainable Chem. Eng., 2020, 8, 6075–6089 CrossRef CAS.
- M. A. Dettori, P. Carta and D. Fabbri, Tetrahedron, 2024, 153, 133867 CrossRef CAS.
- X. Peng, S. Zeb, J. Zhao, M. Zhang, Y. Cui and G. Sun, R. Soc. Open Sci., 2020, 7, 200123 CrossRef CAS PubMed.
- A. A. Greish, A. P. Barkova, E. D. Finashina, T. O. Salmi and L. M. Kustov, Mol. Catal., 2021, 502, 111398 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, 2016 Search PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- I. Kim, A. A. Dwiatmoko, J.-W. Choi, D. J. Suh, J. Jae, J.-M. Ha and J.-K. Kim, J. Ind. Eng. Chem., 2017, 56, 74–81 CrossRef CAS.
- T. M. Soini, X. Ma, O. Üzengi Aktürk, S. Suthirakun, A. Genest and N. Rösch, Surf. Sci., 2016, 643, 156–163 CrossRef CAS.
- Y. Nanba, T. Ishimoto and M. Koyama, J. Phys. Chem. C, 2017, 121, 27445–27452 CrossRef CAS.
- D. S. Rivera Rocabado, M. Aizawa, T. G. Noguchi, M. Yamauchi and T. Ishimoto, Catalysts, 2022, 12, 331 CrossRef CAS.
- C. R. Lee, J. S. Yoon, Y.-W. Suh, J.-W. Choi, J.-M. Ha, D. J. Suh and Y.-K. Park, Catal. Commun., 2012, 17, 54–58 CrossRef CAS.
- J. L. Santos, M. Alda-Onggar, V. Fedorov, M. Peurla, K. Eränen, P. Mäki-Arvela, M. Á. Centeno and D. Y. Murzin, Appl. Catal., A, 2018, 561, 137–149 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5gc00281h |
| ‡ These authors equally contribute. |
| This journal is © The Royal Society of Chemistry 2025 |