
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMining waste as heterogeneous catalysts
Soo Lim
Kim†
a,
Heejin
Yang†
b,
Seonho
Lee
a,
Si-Kyung
Cho
c,
Chang-Gu
Lee
 *bd,
Seitkhan
Azat
*e and
Jechan
Lee
*af
*bd,
Seitkhan
Azat
*e and
Jechan
Lee
*af
aDepartment of Global Smart City, Sungkyunkwan University, Suwon 16419, South Korea. E-mail: jechanlee@skku.edu
bDepartment of Energy Systems Research, Ajou University, Suwon 16499, South Korea
cDepartment of Biological and Environmental Science, Dongguk University, Goyang 10326, South Korea
dDepartment of Environmental and Safety Engineering, Ajou University, Suwon 16499, South Korea. E-mail: changgu@ajou.ac.kr
eLaboratory of Engineering Profile, Satbayev University, Almaty 050013, Kazakhstan. E-mail: s.azat@satbayev.university
fSchool of Civil, Architectural Engineering, and Landscape Architecture, Sungkyunkwan University, Suwon 16419, South Korea
First published on 29th April 2025
Abstract
Mining activities generate significant waste that poses serious environmental challenges, emphasizing the urgent need for effective waste management strategies. Mining waste, such as tailings, pyritic materials, ore residues, and metallurgical by-products, is rich in metals and metal oxides (e.g., Mg, Fe, and Al species) that can serve as catalytic active sites or supports. This intrinsic property highlights its potential for application as heterogeneous catalysts. In recent years, there has been growing interest in utilizing mining waste for catalytic applications, sparking preliminary studies that explore its catalytic capacities and mechanistic roles across various processes. This review consolidates recent advancements in employing mining waste as catalysts, focusing on their characterization, preparation methods, and catalytic performance in diverse reactions. These include dry and steam reforming, wastewater treatment processes (e.g., Fenton, photo-Fenton, peroxymonosulfate activation, electrochemical methods, and ozonation), environmental remediation (e.g., denitrification, carbon monoxide oxidation, and carbon dioxide reduction), and other chemical transformations (e.g., esterification, acetylation, and hydrodeoxygenation). Furthermore, the review discusses key challenges and critical considerations for advancing research in mining waste-based catalysts.
Green foundationGreen chemistry is a set of practices that reduce or eliminate the use of hazardous chemicals in order to minimize pollution. Green chemistry can be applied to mining waste to reduce the environmental impact of the mining and relevant industries via the following points: use heterogeneous catalysts to minimize waste streams in chemical reactions; recycle mining waste to create new materials; use residual by-products as raw materials to create value-added products. In this regard, this review proposes the re-use of mining waste, one of the world's most generated types of waste, as heterogeneous catalysts. It not only evaluates the type and characteristics of mining waste that can be used for catalysts and relevant catalyst preparation methods, but also investigates applications of mining waste-derived catalysts for various chemical reactions (e.g., syngas/H2 production, wastewater treatment, environmental remediation, and various chemical conversion reactions). |
1. Introduction
Mining operations produce vast amounts of waste during the extraction and processing of mineral resources, including topsoil overburden, waste rock, and tailings.1 Appropriate management of these waste streams is critical for mitigating their harmful effects on the environment and human health.2 Mining waste is among the largest global waste streams, with billions of tons generated annually.3 For example, a survey of 107 mining companies estimated the global annual production of tailings to be approximately 13 billion tons, with at least 44.5 × 109 m3 of tailings stored at mine sites.4 The global mining waste management market size was estimated to be approximately 183 billion tons in 2023 and is projected to increase to ≈232 billion tons by 2032 at a compound annual growth rate of 2.7% between 2024 and 2032.5 The mining waste generated is often sent for recovery or neutralization for disposal in dedicated landfills that involve ground storage in neutralization tanks.6 However, it still raises the possibility of leakage into the ground, posing a threat to the surrounding area, and pretreatment for disposal generates additional waste.7 Moreover, current mining waste storage and treatment facilities are unprofitable,8 which might result in the disastrous spread of contaminants and increase mine waste-associated impacts.9 Consequently, there has been a surge in suitable mining waste treatment strategies.Mining waste may contain hazardous substances; however, not all mining waste poses environmental harm. In fact, a significant portion of mining waste holds valuable and reusable resources. For example, mining waste has been repurposed as construction materials10–16 and geopolymer17–20 products, offering sustainable alternatives in these industries. Furthermore, recoverable metals and minerals can often be extracted from mining waste, adding economic and environmental value.21–23 Some types of mining waste also contain metals and metal oxides that exhibit catalytic properties, making them suitable for use as catalysts in various chemical reactions. Although several studies have explored the use of mining waste in catalytic reactions, much of this research is fragmented and lacks a comprehensive framework. This gap highlights the need for a systematic review to thoroughly evaluate the potential of mining waste as catalytic materials. Accordingly, this review is organized to examine the diverse applications of mining waste as catalysts by consolidating and analyzing findings from recent studies. Specifically, section 2 classifies the types and characteristics of mining waste that can be used as catalysts. Section 3 introduces and compares the methods used to prepare mining waste-derived catalysts, examines their properties, and discusses their performance in various reactions. Section 4 identifies the current limitations of mining waste-based chemical processes and offers research recommendations to overcome these challenges. This study contributes novel insights in comparison with earlier reviews,24–30 as summarized in Table 1.
| Review | Way to reuse mining waste | Focus on |
|---|---|---|
| Lim and Alorro24 | Recovery of metal and mineral resources | • Technospheric mining |
| • Metal recovery techniques | ||
| Vitti and Arnold25 | Recovery of metal and mineral resources | • Extraction techniques for mineral & metal recovery mine tailings |
| Sarker et al.26 | Recovery of metal and mineral resources | • Characteristics of precious metals found in mining waste |
| • Precious metal recovery techniques | ||
| Abbadi et al.27 | Recovery of metal and mineral resources | • Processes for rare Earth metal recovery |
| Xiaolong et al.28 | Synthesis of geopolymers | • Methods for producing geopolymers from mining waste |
| • Pre-treatment of mining waste to enhance geopolymer reactivity | ||
| Carmignano et al.29 | Preparation of construction materials | • Applications relying solely on iron ore tailings |
| • Limited discussion on catalyst preparation and applications | ||
| Barraza et al.30 | Synthesis of nanomaterials | • Tailing reprocessing for nanomaterial synthesis |
| • Recovery of metal and metal oxide nanoparticles from mining tailings | ||
| • Application of the tailing-derived nanomaterials | ||
| This review | Use as heterogeneous catalysts for various reactions | • Components of mining waste with regard to catalytic activity |
| • Methods to prepare catalysts from different types of mining waste | ||
| • Chemical reactions facilitated by mining waste-derived catalysts |
2. Mining waste as raw materials for heterogeneous catalysts
Various types of mining wastes were used for heterogeneous catalyst preparation (Fig. 1 and Table 2). The mines presented in Fig. 1 produce bauxite, iron, flint kaolin, pumice, rare earth elements, etc.,31 which also generate mining waste consisting of a range of metals and metal oxides (Table 2). Most mining waste remains unused as a resource, leading to environmental and health issues. For example, the metallic substances in abandoned mining waste can cause environmental pollution through various pathways such as the leaching of heavy metals from mining and smelting and non-point source pollution from rainfall and atmospheric transport. This pollution has serious impacts on the environment and human health.32 Given that green chemistry is defined as the ‘Design of chemical products and processes that reduce or eliminate the use or generation of hazardous substances’, appropriate reuse of mining waste as heterogeneous catalysts substantially contributes to achieving green chemistry principles in the field of chemical production processes. | ||
| Fig. 1 Origins of various mining wastes used for heterogeneous catalyst preparation. | ||
| Mining waste | Components (content, %) | Ref. |
|---|---|---|
| Waste ore | • Fe3O4, Fe2O3, Co2O3, Co3O4, CuO, SiO2, MgO | 33 |
| Iron mining waste | • Fe2O3 (25), Fe3O4, FeOOH, FeO, CeO2 (8), Ce2O3, P2O3, SiO2, Al2O3, La2O3, CaO, BaO, TiO2, MnO, ZnO, Al(OH)3, MgO, MgFeAlO4, MgFe2O4, AlFe2O4 | 34, 35, 36, 37 and 38 |
| Magnetic mining waste | • Fe2O3 (73.72–75.85), TiO2 (9.48–10.09), P2O5 (3.58–3.92), SiO2 (2.92–6.3), CaO (2.18–2.57), MgO (0.85–2.12), MnO (0.91), Al2O3 (0.53–0.78), K2O (<0.05), Na2O (<0.05) | 39 and 40 |
| Tailings and slag originating from Serifos Island (Cyclades, Greece) | • FeO (97.42), MgO (0.67), Al2O3 (0.37), SiO2 (0.23), MnO (0.12) | 41 |
| Slag residue from metal mining | • MgFe2O4, MgFeAlO4, Fe3O4, Al2FeO4, NiO, MgO, MgO2 | 42 and 43 |
| Amethyst mining reject | • SiO2 (37.48), Fe2O3 (24.36), Al2O3 (13.56), CaO (12.44), TiO2 (6.17), MgO (2.08), K2O (1.23), P2O5 (1.18), SO3 (0.46), MnO (0.37), others (0.67) | 44 |
| Bauxite mining waste | • α-Fe2O3 & α-FeO(OH) (41), Al2O3 (17), SiO2 (10), TiO2 (9), CaO (9), Na2O (5) | 45 and 46 |
| Basalt mine tailings | • SiO2 (41.88), Fe2O3 (22.97), CaO (11.84), Al2O3 (11.74) | 47 |
| Ilmenite metallurgical residue | • Fe (31.26), Mg (17.49), Al (5.35), Ca (1.07), Mn (1.01), V (0.90), Ti (0.60), Cr (0.51), Na (0.17), Si (0.08), K (0.02), P (0.004), Zr (0.01), Zn (0.01) | 48 |
| Pyritic waste | • SiO2 (8.44), Al2O3 (2.55), CaO (0.12), K2O (0.19), Cr2O3 (0.04) | 49 |
| Pumice mining waste | • SiO2 (48.069), Al2O3 (8.275), K2O (4.312), Fe2O3 (1.591), Na2O (1.207), CaO (0.782), MgO (0.438), MnO (0.253), ZrO2 (0.052), P2O5 (0.044) | 50 |
| Rare Earth tailings | • Fe2O3 (27.67), CaO (27.2), SiO2 (11.86), MgO (3.31), CeO2 (3.01), MnO (1.96), Al2O3 (1.46), La2O3 (1.44), Nd2O3 (1.10), TiO2 (1), F (8.92), CaF2, Ce(CO3)F | 51, 52 and 53 |
| Kaolin waste | • SiO2 (39.43–42.30), Al2O3 (38.29–39.92), TiO2 (3.20–3.25), Fe2O3 (2.92–3.4) | 54 and 55 |
| Coal gangue | • SiO2 (8.38–58.3), Al2O3 (9.11–20.8), Fe2O3 (1.01–10), SO3 (0.05–3.97), K2O (0.05–4.75), MgO (0.98–2.11), CaO (0.02–1.59), TiO2 (0.51–0.98), Na2O (0.81), ZrO2 (0.01) | 56, 57 and 58 |
The composition of mining waste is closely related to the type of mined materials and resources as well as the mining methods employed.59Table 2 provides a summary of the compositions of several mining waste samples that have been utilized as heterogeneous catalysts in different chemical reactions. Mining waste typically contains a mixture of metal and metal oxide species, with major components including Fe (Fe2O3), Mg (MgO), Al (Al2O3), and Si (SiO2). These species are well-recognized as catalytic active sites or catalyst supports for numerous chemical reactions.60–66 Consequently, mining waste holds significant potential for use as heterogeneous catalysts. However, mining waste often contains impurities that may hinder its catalytic performance. For instance, while commercial kaolin consists primarily of aluminosilicate, kaolin mining waste also includes Fe and Ti impurities, which reduce kaolin brightness and decrease its crystallinity.67,68 Depending on the type of reaction, these impurities can significantly impair catalytic activity. As a result, effective pre- and post-treatment processes are often required to remove impurities, enhance crystallinity, and improve the material's properties as a solid support.69 In sections 3–5, a range of heterogeneous catalysts derived from mining waste and their applications in various chemical reactions are explored in detail.
3. Syngas and H2 production using mining waste-derived catalysts
3.1. Mining waste catalysts prepared for producing syngas and H2
As seen in section 2, mining waste can contain various metal oxides. Many of them are used either as catalyst supports or as catalytic active sites. When mining waste is used as the support to prepare catalysts for syngas or H2 production (sections 4.1 and 4.2), Ni is loaded on mining waste in most studies.34,42,48,56,70–73 Metals other than Ni, including Ru and Rh, have very rarely been tested for the reactions.74 Impregnation-based methods were most widely used to prepare mining waste-supported Ni catalysts using Ni nitrate (e.g., Ni(NO3)2·6H2O) as the Ni precursor. To make a pelletized form of a Ni/metallurgical residue catalyst, clay was mixed with the Ni precursor prior to impregnation.70 The form of Ni catalyst (powder or pellet) could affect the catalytic activity for dry reforming, as discussed in the following subsection.Table 3 shows the physicochemical properties of different Ni catalysts supported on mining waste. The mining waste-supported Ni catalysts tended to have lower surface areas (up to 10 m2 g−1), porosity (up to 0.03 cm3 g−1), and metal dispersion (<1%) than typical Ni catalysts supported on pure Al2O3 or SiO2 most likely due to the co-existence of different oxides, minerals, and impurities in mining waste (Table 2). The Ni loading on mining waste was found to be an important factor affecting the catalyst's physicochemical properties. For example, an increase in Ni loading on the Ni catalyst supported by iron-rich mining residue resulted in a reduction of the catalyst's surface area, pore volume, and pore size.34 However, the Ni loading did not significantly affect the crystal size of Ni spinel (NiFe2O4) present on the catalyst.
| Entry | Mining waste support (major components) | Properties | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Ni loading (%) | Surface area (m2 g−1) | Total pore volume (cm3 g−1) | Average pore diameter (nm) | Average particle size (nm) | Ni spinel crystallite size (nm) | Surface Ni sites | |||
| 1 | Ilmenite metallurgical residue (Fe, Mg, Al oxides) | 13.9 | 3.94 | 0.019 | — | 168 | — | — | 44 |
| 2 | Metallurgical residue | A form of powder | 4.1 | — | — | — | 29.4 | — | 48 |
| 3 | Metallurgical residue | A form of pellet (25% clay) | 0.4 | — | — | — | 56 | — | 48 |
| 4 | Iron-rich mining residue (Fe, Mg, Ce oxides) | 5–13 | 2.91–2.87 | 0.0134–0.0132 | 18.2–17.7 | — | 3.38–3.41 | — | 45 |
| 5 | Mining residue | 13 | 2.77 | 0.0177 | — | — | — | — | 49 |
| 6 | Slag residue from metal mining (Mg–Fe oxides) | 10 | 4.86 | 0.023 | 7.87 | — | — | 565.8 cc g−1 | 46 |
| 7 | Agglomerates of metallurgical residue | 12.5 | 10.3 | — | — | — | — | — | 50 |
| 8 | Agglomerates of metallurgical residue | 5 | 9.9 | 0.13 | 52.8 | 604.2 | — | Ni dispersion = 0.17% | 51 |
| 9 | Coal gangue (SiO2, Al2O3, Fe2O3) | 15 | 5.38 | 0.033 | 17.86 | — | — | — | 47 |
3.2. Syngas production
Synthesis gas (syngas) is a mixture primarily composed of H2 and CO in varying ratios. It serves as a critical intermediate for the production of ammonia,75 methanol,76 and hydrocarbon fuels.77 Syngas is also combustible, making it suitable for direct use as a fuel.78,79 Several studies have investigated syngas production using mining waste-derived catalysts via dry reforming and steam reforming, as summarized in Table 4 (entries 1–6).| Entry | Catalyst | Reaction | Reaction conditions | Performance | Ref. |
|---|---|---|---|---|---|
| T = temperature; P = pressure; t = time; Q = volumetric flow rate; SV = space velocity; GHSV = gas hourly space velocity. | |||||
| 1 | Ni/metallurgical residue | Dry reforming of CH4 | T = 810 °C; P = 1 atm; t = 4 h; Q = 15 mL min−1; SV = 2900 mL g−1 h−1; CH4/CO2 = 1 | CH4 conversion = 87%; CO yield = 84%; H2 yield = 70% | 48 |
| 2 | Ni/metallurgical residue (powder) | Dry reforming of CH4 | T = 800 °C; P = 5.5 atm; t = 4 h; GHSV = 810 L kg−1 h−1; CH4/CO2 = 0.5 | CH4 conversion = 82%; CO2 conversion = 53%; CO yield = 47%; H2 yield = 54% | 70 |
| 3 | Ni/metallurgical residue (pellet) | Dry reforming of CH4 | T = 800 °C; P = 5.5 atm; t = 4 h; GHSV = 810 L kg−1 h−1; CH4/CO2 = 0.5 | CH4 conversion = 85%; CO2 conversion = 67%; CO yield = 62%; H2 yield = 64% | 70 |
| 4 | Ni/iron-rich mining residue | Dry reforming of C2H4 | T = 650 °C; P = 1 atm; t = 2 h; Q = 40 mL min−1; GHSV = 4800 mL g−1 h−1; C2H4/CO2 = 3 | C2H4 conversion = 91.29%; CO2 conversion = 88.48%; H2 yield = 67.47% | 34 |
| 5 | Ni/slag residue | Steam reforming of CH4 | T = 900 °C; P = 1 atm; t = 168 h; Q = 15 mL min−1; SV = 3000 mL g−1 h−1; H2O/CH4 = 1.7 | CH4 conversion = 98%; CO yield = 91%; H2 yield = 92% | 43 |
| 6 | Ni/mining residue | Steam reforming of toluene | T = 800 °C; P = 1 atm; t = 24 h; WHSV = 4 h−1; C/H2O = 1 | Toluene conversion = 98.3 wt%; CO yield = 74%; H2 yield = 82% | 71 |
| 7 | Ni/iron-rich mining residue | Thermal decomposition of C2H4 | T = 750 °C; P = 1 atm; t = 2 h; Q = 40 mL min−1; GHSV = 4800 mL g−1 h−1; C2H4/Ar = 3 | Ethylene conversion = 92.24%; H2 yield = 74.46%; carbon yield = 76.25% | 34 |
| 8 | Ni/slag residue | Sequential non-catalytic pyrolysis–catalytic decomposition of (1) virgin HDPE, (2) used HDPE, and (3) plastic mixture | T = 700 °C (1st stage) & 650 °C (2nd stage); t = 2 h; Q (N2) = 0.03 SLPM; plastic feed rate = 0.33 g min−1 | (1) Gas yield = 33.13%; H2 yield = 75.62% | 42 |
| (2) Gas yield = 34.35%; H2 yield = 79.40% | |||||
| (3) Gas yield = 36.08%; H2 yield = 70.40% | |||||
| 9 | Ni/coal gangue | Pyrolysis of polyethylene | (1) 1st stage (non-catalytic): T = 500 °C (10 °C min−1); t = 20 min | H2 yield = 31.48 mmol g−1; H2 concentration = 66.48 vol% | 56 |
| (2) 2nd stage (catalyst bed): T = 750 °C; Q = 100 mL min−1 N2 | |||||
| 10 | Iron ore tailings | Thermal cracking of oleic acid | T = 450 °C; P = 1.25 MPa Ar; t = 3 h; oleic acid/catalyst = 1 (w/w) | Gas yield = 95 mol%; H2 selectivity = 68 mol% | 80 |
| 11 | Ni/metallurgical residue | Steam reforming of glycerol |
T = 580 °C; P = 1 atm; GHSV = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 966 cm3 (STP) g−1 h−1; water/glycerol = 9 966 cm3 (STP) g−1 h−1; water/glycerol = 9 |
Glycerol conversion = >99%; H2 yield = 80.7% | 73 |
| 12 | Rh/metallurgical residue | Steam reforming of glycerol |
T = 580 °C; P = 1 atm; GHSV = 10966 cm3 (STP) g−1 h−1; water/glycerol = 9 |
Glycerol conversion = >99%; H2 yield = 78% | 74 |
| 13 | Ru/metallurgical residue | Steam reforming of glycerol |
T = 580 °C; P = 1 atm; GHSV = 10966 cm3 (STP) g−1 h−1; water/glycerol = 9 |
Glycerol conversion = 94%; H2 yield = 71% | 74 |
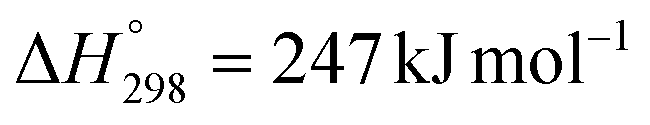 ) is a highly endothermic catalytic reaction that converts CH4 and CO2 into syngas.81 Ni-based catalysts are considered cost-effective, efficient, and stable catalysts for dry reforming of CH4.82 Maybe due to this, many studies have tried Ni loaded on mining waste support for dry reforming of CH4. For instance, Chamoumi et al. conducted dry reforming of CH4 using a Ni-loaded ilmenite metallurgical residue catalyst.48 The ilmenite metallurgical residue primarily consisted of CaO, MgO, and MnO. The reaction was performed at 810 °C under atmospheric pressure for 4 h with a CH4/CO2 molar ratio of 1. The catalyst showed a CH4 conversion of 87%, 84% CO yield, and 70% H2 yield (entry 1, Table 4). The catalytic performance of the Ni/metallurgical residue catalyst was superior to that of a spinel ferrite (NiFe2O4). For a 7 d run, however, the catalyst lost its activity as the CH4 conversion, CO yield and H2 yield were decreased by 22%, 19%, and 25%, respectively. Calcination at 900 °C restored the catalytic properties, confirming the regenerability of the Ni-loaded ilmenite metallurgical residue catalyst.
) is a highly endothermic catalytic reaction that converts CH4 and CO2 into syngas.81 Ni-based catalysts are considered cost-effective, efficient, and stable catalysts for dry reforming of CH4.82 Maybe due to this, many studies have tried Ni loaded on mining waste support for dry reforming of CH4. For instance, Chamoumi et al. conducted dry reforming of CH4 using a Ni-loaded ilmenite metallurgical residue catalyst.48 The ilmenite metallurgical residue primarily consisted of CaO, MgO, and MnO. The reaction was performed at 810 °C under atmospheric pressure for 4 h with a CH4/CO2 molar ratio of 1. The catalyst showed a CH4 conversion of 87%, 84% CO yield, and 70% H2 yield (entry 1, Table 4). The catalytic performance of the Ni/metallurgical residue catalyst was superior to that of a spinel ferrite (NiFe2O4). For a 7 d run, however, the catalyst lost its activity as the CH4 conversion, CO yield and H2 yield were decreased by 22%, 19%, and 25%, respectively. Calcination at 900 °C restored the catalytic properties, confirming the regenerability of the Ni-loaded ilmenite metallurgical residue catalyst.
Malik et al. prepared a Ni catalyst supported on metallurgical residue in both powder and pellet forms and compared their catalytic performance for dry reforming of CH4.70 The powder catalyst demonstrated superior performance under atmospheric pressure due to its higher surface area compared to the pelletized catalyst. However, under elevated pressures (at least 5.5 atm), the pelletized catalyst exhibited greater activity for syngas production (entries 2 and 3, Table 4). For instance, the pelletized catalyst achieved higher conversions of CH4 and CO2 and higher syngas yields than the powder catalyst within the pressure range of 5.5–6.5 atm. The improved catalytic performance of the pelletized catalyst under elevated pressures was attributed to increased coke oxidation rates, which mitigated coke deposition on the active sites. Nevertheless, a further increase in pressure from 6.5 to 10 atm resulted in reduced activity and selectivity of the pelletized catalyst, likely due to the formation of crystalline graphitic carbon, which caused the disintegration of the pellet particles.
Azara et al. prepared iron-rich mining residue-supported Ni catalysts with varying Ni loading for dry reforming of ethylene (C2H4 + 2CO2 → 4CO + 2H2,  ).34 Higher Ni loading improved the activity of the catalyst for H2 production. Specifically, the 13% Ni/iron-rich mining residue catalyst achieved a C2H4 conversion of 91.3%, a CO2 conversion of 88.5%, and an H2 yield of 67.5% under the following conditions: 650 °C, atmospheric pressure, a C2H4/CO2 molar ratio of 3, and a gas hourly space velocity (GHSV) of 4800 mL g−1 h−1 for 2 h time-on-stream (entry 4, Table 4).
).34 Higher Ni loading improved the activity of the catalyst for H2 production. Specifically, the 13% Ni/iron-rich mining residue catalyst achieved a C2H4 conversion of 91.3%, a CO2 conversion of 88.5%, and an H2 yield of 67.5% under the following conditions: 650 °C, atmospheric pressure, a C2H4/CO2 molar ratio of 3, and a gas hourly space velocity (GHSV) of 4800 mL g−1 h−1 for 2 h time-on-stream (entry 4, Table 4).
 ;83 entry 5, Table 4).43 The reaction was conducted at 900 °C with a CH4/H2O molar ratio of 0.59 and a space velocity of 3000 mL g−1 h−1. The catalyst achieved 98% CH4 conversion, remaining stable over 168 h time-on-stream, with 92% H2 yield and 91% CO yield (H2/CO ratio of approximately 3). The high catalytic activity under the reducing environment of the reforming reaction was attributed to two key factors: (1) the formation of Fe–Ni alloys such as FeNi, FeNi3, and Fe3Ni2, derived from the reduction of NiFe2O4 and FeNiAlO4, and (2) the solid solution of NiO–MgO, which provided an active Ni phase while preventing carbon deposition due to its alkalinity.
;83 entry 5, Table 4).43 The reaction was conducted at 900 °C with a CH4/H2O molar ratio of 0.59 and a space velocity of 3000 mL g−1 h−1. The catalyst achieved 98% CH4 conversion, remaining stable over 168 h time-on-stream, with 92% H2 yield and 91% CO yield (H2/CO ratio of approximately 3). The high catalytic activity under the reducing environment of the reforming reaction was attributed to two key factors: (1) the formation of Fe–Ni alloys such as FeNi, FeNi3, and Fe3Ni2, derived from the reduction of NiFe2O4 and FeNiAlO4, and (2) the solid solution of NiO–MgO, which provided an active Ni phase while preventing carbon deposition due to its alkalinity.
Belbessai et al. prepared a Ni catalyst supported on mining residue (entry 6, Table 4).71 It was observed that in this catalyst, metallic Ni was protected by MgO present in the NiO/MgO solid solution due to strong metal–support interactions. The metallic Ni particles were on the nanometer scale and highly uniformly dispersed, effectively suppressing coke formation.84 The catalyst's basicity, attributed to the presence of MgO, enhanced steam adsorption on the catalyst surface.85 Other promoters present in the mining residue, such as Ca, further contributed to the improved catalytic activity. The catalyst was tested for the steam reforming of toluene as a model reaction for tar upgrading. Under reaction conditions of 800 °C and 1-day time-on-stream, the toluene conversion reached 98.3% with an H2 yield of 82% and a CO yield of 74%.
3.3. H2 production
Hydrogen (H2) possesses a higher gravimetric energy density than conventional fuels such as natural gas, oil, and petrol.86 It is considered a promising alternative energy resource, serving as a clean and efficient energy carrier.87Table 4 (entries 7–13) summarizes studies on H2 production from various feedstocks on mining waste-derived catalysts via thermal decomposition and glycerol reforming. ). The reaction was conducted for 2 h under 1 atm with a C2H4/argon ratio of 3 at 750 °C and a GHSV of 4800 mL g−1 h−1. The catalyst yielded 74.5% H2 at C2H4 conversion of 92.2%. In addition to H2, thermal C2H4 cracking led to the formation of carbon deposited on the catalyst in 76.3% yield. Deposited carbon had a filamentous structure, which did not block the reactant's access to the catalyst's active sites. The iron species present on the mining residue support facilitated the growth of filamentous carbon, while Ni particles were responsible for cleaving C–C bonds in C2H4.
). The reaction was conducted for 2 h under 1 atm with a C2H4/argon ratio of 3 at 750 °C and a GHSV of 4800 mL g−1 h−1. The catalyst yielded 74.5% H2 at C2H4 conversion of 92.2%. In addition to H2, thermal C2H4 cracking led to the formation of carbon deposited on the catalyst in 76.3% yield. Deposited carbon had a filamentous structure, which did not block the reactant's access to the catalyst's active sites. The iron species present on the mining residue support facilitated the growth of filamentous carbon, while Ni particles were responsible for cleaving C–C bonds in C2H4.
A catalyst prepared by loading Ni onto a metal mining slag residue was utilized in a sequential process combining non-catalytic pyrolysis and catalytic decomposition of plastics to produce H2 as a plastic waste valorization strategy.42 In this system, the plastic feedstock was thermally decomposed at 700 °C in the first stage, evolving a range of hydrocarbon products. These hydrocarbon products were then passed through the catalyst bed at 650 °C in the second stage, where they were converted into lighter compounds. In the second-stage reactor, the cleavage of C–H and C–C bonds primarily occurred at Ni and Ni–Fe alloy active sites. Various plastic feedstocks were tested, including virgin high-density polyethylene (HDPE), used HDPE, and a plastic mixture comprising 80 wt% PE, 15 wt% polypropylene, 4 wt% polystyrene and polyethylene terephthalate (PET), and 1 wt% miscellaneous. Over the catalyst, the HDPE feedstocks achieved H2 yields of 75–79% with gas yields of 33–34 wt%, while the plastic mixture produced a H2 yield of 70.4% with a gas yield of 36 wt% under comparable reaction conditions (entry 8, Table 4).
Zhang et al. prepared Ni catalysts supported on coal gangue used for pyrolysis of polyethylene (PE) (entry 9, Table 4).56 Pyrolysis was carried out using a two-stage fixed bed reactor involving the 1st reactor that pyrolyzed PE at 500 °C and the 2nd reactor that catalytically decomposed pyrolytic volatiles evolved from PE. Different catalyst temperatures (700–900 °C) and Ni loadings (5–20%) were tested, showing that the 15% Ni/coal gangue catalyst led to the highest H2 yield of 31.5 mmol g−1 with 66.5 vol% concentration at 750 °C. The catalyst could be regenerated through oxidation, and it was reusable for at least five cycles, retaining a H2 yield of 27 mmol g−1 with 63 vol% concentration.
Luciano et al. used iron ore tailings rich in iron oxides and SiO2 for catalytic thermal decomposition of fatty acid (e.g., oleic acid) under a high pressure (entry 10, Table 4).80 The iron ore tailings were directly used as the catalyst with no treatment. In the presence of the catalyst, oleic acid was converted mainly into a gaseous product composed mainly of H2 at 450 °C under 1.25 MPa Ar for 3 h with an oleic acid/catalyst weight ratio of 1, achieving 95 mol% gaseous product yield and 68 mol% H2 selectivity.
The Ni catalyst was compared with Rh and Ru catalysts for H2 production from glycerol via steam reforming.74 The Ni catalyst demonstrated superior performance for hydrogen production compared to the Rh and Ru catalysts (entries 11–13, Table 4). Fig. 2 illustrates the cooperative interactions between the incorporated metal (Ni, Rh, or Ru) and the Fe/Mg-bearing species present on the mining waste support surface, which play a critical role in glycerol activation and promote its reforming. The regenerative mechanism (Fig. 2b) is CO conversion into CO2 by reacting with the oxygen produced by water dissociation or with the support's lattice oxygen.89 However, the presence of alkaline or alkaline-earth metals on the catalyst surface can promote an associative mechanism (Fig. 2a),90 which facilitates the conversion of CO and water. For Rh-based catalysts, the reaction pathway is believed to involve the formation of COO–formate species on FexOy species in close proximity to metallic Rh,91 promoting CO2 and H2 production. Similarly, an associative mechanism is also supported for reactions occurring on MgO.92 The results showed that the degree of interaction between the incorporated metal and MgO on the catalyst surface was strongly correlated with hydrogen production. The Ru catalyst exhibited the lowest H2 yield, which was attributed to its poor propensity for MgO–RuO2 interactions on the catalyst surface.
 | ||
| Fig. 2 Proposed mechanism of glycerol reforming over metallurgical waste-supported metal (M) catalysts through (a) associative and (b) regenerative mechanisms. Reprinted from Sahraei et al.,74 copyright (2021), with permission from Elsevier. | ||
4. Chemical conversion using mining waste-derived catalysts
4.1. Mining waste catalysts prepared for various chemical conversion reactions
Section 3 introduced that various mining waste could be used as a catalyst support for preparing supported Ni catalysts employed for the production of syngas and H2. In this section, various mining waste catalysts or mining waste-supported metal catalysts are discussed for different chemical conversion reactions such as esterification, hydrodeoxygenation (HDO), nitrogen oxides (NOx) removal, carbon monoxide (CO) oxidation, and carbon dioxide (CO2) reduction, as summarized in Table 5.| Entry | Mining waste (major components) | Treatment | Properties | Ref. | |||
|---|---|---|---|---|---|---|---|
| Surface area (m2 g−1) | Total pore volume (cm3 g−1) | Average pore diameter (nm) | Surface acid site (mmol g−1) | ||||
| CTAB = cetrimonium bromide. MPS = 3-mercaptopropyltrimethoxysilane. | |||||||
| 1 | Rare Earth tailings (Fe2O3, CaO, SiO2, MgO) | Ni and Cu loadings = 2% and 2.5%, respectively | 63.78 | 0.12 | 7.82 | 3.96 | 53 |
| 2 | Tailings and slags (FeO) | Au loading (1 wt%) | 63.8 | 0.076 | 1.6 | — | 41 |
| 3 | Coal gangue (SiO2, Al2O3, Fe2O3) | Ni loading (15 wt%) | 9.1 | 0.0126 | 56.03 | — | 57 |
| 4 | Iron mining tailings (Fe oxides) | H2SO4 treatment | — | — | — | 0.1211 | 35 |
| 5 | Kaolin waste (SiO2, Al2O3) | H2SO4 treatment; hydrothermal treatment at 110 °C in the presence of NaOH & CTAB; functionalization with MPS | 998–1016 | 0.78–0.80 | 3.14–3.25 | 5.93 | 54, 55 and 93 |
| 6 | Bauxite mining waste (Fe oxides, Al2O3, SiO2, TiO2) | A form of non-alkaline magnetic solid | — | — | — | — | 45 |
| 7 | Rare Earth tailings (Fe2O3, CaO, SiO2, MgO) | Na2CO3/Ca(OH)2 treatment | — | — | — | — | 51 |
| 8 | Mixture of rare Earth tailings and concentrate (1/1, w/w; Fe2O3, CaO, SiO2, MgO) | Na2CO3/Ca(OH)2 treatment, followed by HCl/citric acid treatment | 7.69 | 0.0403 | 17.44 | — | 52 |
For certain reactions (e.g., CO oxidation and CO2 reduction), supported metal catalysts (e.g., Cu, Ni, and Au) were prepared by an impregnation or precipitation method (entries 1–3, Table 5). Several mining wastes could be employed as catalysts themselves after pre-treatment (entries 4–8, Table 5). Pre-treatment involved acid and/or base treatment in order to activate catalytic sites and remove impurities. For example, iron mining waste (major mineral phase of hematite and quartz) treated with H2SO4 or ammonium sulfate ((NH4)2SO4) resulted in catalysts with varying surface acid densities—the H2SO4-treated catalyst exhibited a surface acid density approximately 4 times higher than the (NH4)2SO4-treated one.35 This difference is attributed to the enhanced solubilization of iron species in sulfuric acid, leading to the formation of highly dispersed iron sulfate groups upon calcination (Fig. 3). Treatment with Na2CO3 and Ca(OH)2 increased the Fe2O3 content in rare Earth tailings that acted as active sites for denitrification.51 The addition of HCl and citric acid to the Na2CO3/Ca(OH)2-treated catalyst modified the surface morphology of the mineral, increased the number of active sites, and enhanced the adsorption of NH3, which was beneficial for the selective catalytic reduction of NOx using NH3 as a reducing agent (NH3-SCR).52
 | ||
| Fig. 3 Schematic description of catalyst preparation from iron ore tailings for the esterification reaction. Reprinted from Prates et al.,35 copyright (2020), with permission from Elsevier. | ||
Beyond simple pretreatment with acid and base, Nascimento's group used kaolin waste to synthesize high-surface-area functionalized mesoporous aluminosilicates used for esterification reactions.54,55,93 The synthesis procedure involved acid leaching, hydrothermal treatment in the presence of NaOH and cetrimonium bromide, and then functionalization with 3-mercaptopropyltrimethoxysilane. The material exhibited a higher surface area of >900 m2 g−1 with the organic functional group (–SO3H) leading to a high density of surface acid sites, which gave high esterification activities for different feedstocks.
4.2. Esterification
Esterification of free fatty acids (FFAs) with alcohol using an acid catalyst is a key reaction in the production of biodiesel, a renewable alternative to petroleum-derived diesel fuels.94 A variety of solid acid catalysts have been used for esterification of FFAs, including acidic organic resins, zeolites, mesoporous silicate, heteropolyacids, sulfated metal oxides, and binary metal oxides.95 Some of the components contained in these solid acids (e.g., metal oxides) are also contained in mining waste (Table 2); thus, mining wastes with proper treatment/functionalization have been applied to esterifying FFAs (entries 1–4, Table 6). For instance, Prates et al. investigated the use of iron ore tailings-derived catalysts for the esterification of oleic acid as a surrogate reaction for biodiesel production using mining waste-derived catalysts.35 At 120 °C for 4 h of reaction, the H2SO4-impregnated catalyst achieved complete conversion (100%) of oleic acid into its corresponding ester under a methanol/oleic acid molar ratio of 15 and 5 wt% catalyst loading (entry 1, Table 6).| Entry | Catalyst | Reaction | Reaction conditions | Performance | Ref. |
|---|---|---|---|---|---|
| T = temperature; t = time; Q = volumetric gas flow rate; SV = space velocity; W/F = weight of catalyst/the total flow rate of the reactant gas. | |||||
| 1 | H2SO4-impregnated iron tailings | Esterification of oleic acid | T = 120 °C; t = 4 h; catalyst loading = 5 wt%; methanol/oleic acid molar ratio = 15 | Oleic acid conversion = 100% | 35 |
| 2 | SO3H-functionalized flint kaolin | Esterification of palm oil distillate | T = 130 °C; t = 2 h; catalyst loading = 4%; ethanol/palm oil distillate molar ratio = 30 | Palm oil distillate conversion = 95% | 93 |
| 3 | SO3H-functionalized flint kaolin | Acetylation of eugenol | T = 80 °C; t = 40 min; catalyst loading = 2%; eugenol/acetic anhydride molar ratio = 0.2 | Eugenol conversion = 99.9% | 55 |
| 4 | SO3H-functionalized kaolin waste | Esterification of palm oil deodorization waste | T = 130 °C; t = 2 h; catalyst loading = 5%; methanol/fatty acid molar ratio = 30 | Palm oil deodorization waste conversion = 98% | 54 |
| 5 | Reduced bauxite mining waste | Hydrodeoxygenation of levulinic acid | T = 365 °C; t = 4 h; 50 wt% aqueous solution; initial H2 pressure = 5.5 MPa catalyst loading = 9.1 wt% | C9 alkane and alkene yield = up to 76 wt% | 45 |
| 6 | Alkali/acid co-treated rare Earth tailings | Denitrification | T = 900 °C; CNO,i = 500 ppm; Q = 500 mL min−1; CO/NO ratio = 4 | NO removal efficiency = 96.2% | 51 |
| 7 | Alkali/acid co-treated rare Earth tailings | NH3-SCR |
T = 350 °C; Q = 100 mL min−1 (3% O2, SV = 25000 h−1); NH3/NO ratio = 1 |
NO removal efficiency = 82% | 52 |
| 8 | Ni–Cu/rare Earth tailings | NH3-SCR | T = 300 °C; Q = 100 mL min−1 (3% O2, SV = 8000 h−1); NH3/NO ratio = 1 | NO removal efficiency = 90%; N2 selectivity = 85% | 53 |
| 9 | Au/yellow hematite | CO oxidation | T = 62 °C; Q = 50 mL min−1; W/F = 0.36 g s mL−1; CO/O2 = 1/20 | CO conversion = 90% | 41 |
| 10 | Ni/coal gangue | CO2 methanation |
T = 450 °C; Q = 100 mL min−1; WHSV = 30000 mL g−1 h−1; CO2/H2 = 1/4 |
CO2 conversion = 73%; CH4 selectivity = 91% | 57 |
A by-product of kaolin extraction mining was utilized to synthesize a high-surface-area mesoporous aluminosilicate functionalized with –SO3H groups for catalytic applications, including esterification of palm oil distillate and acetylation of eugenol.93 The palm oil-derived feedstock contained 84% FFAs. During the esterification process, increasing the reaction temperature from 110 to 130 °C and extending the reaction time from 30 min to 2 h significantly improved the conversion of FFAs. The catalyst achieved up to 95% conversion of FFAs into fatty acid ethyl esters at 130 °C for 2 h under a molar ratio of ethanol to palm oil distillate of 30 and 4% catalyst loading (entry 2, Table 6). Additionally, the catalyst demonstrated reusability for at least four cycles, maintaining a conversion efficiency greater than 73%. The same catalyst was also applied to the acetylation of eugenol using acetic anhydride as the acetylating agent.55 The catalyst could convert 99.9% of eugenol into eugenyl acetate under optimized conditions (80 °C, 40 min, acetic anhydride/eugenol molar ratio of 5, and 2% catalyst loading; entry 3, Table 6). Successive reuse of the SO3H-functionalized aluminosilicate catalyst for the acetylation of eugenol resulted in a gradual decline in eugenol conversion, decreasing to 90% after 4 cycles. This reduction in efficiency is most likely due to active sites being occupied by molecules from reactants or products. A SO3H-functionalized aluminosilicate catalyst synthesized from kaolin waste was also employed for the esterification of industrial waste from palm oil deodorization (entry 4, Table 6).54 The esterification reaction was conducted at 130 °C for 2 h with a methanol/fatty acid molar ratio of 30 and 5% catalyst loading, achieving 98% conversion of palm oil deodorization waste into esters. In contrast, the non-catalytic reaction under identical conditions converted only 15% of the feedstock, highlighting the catalyst's effectiveness. After use, the catalyst was recovered and reused in the same reaction, yielding 81% conversion of the palm oil waste. This decline in performance indicates the need for further research to optimize the mining waste-derived catalyst, particularly in terms of improving its stability during successive reaction cycles.
4.3. Hydrodeoxygenation
HDO is a representative important reaction in biomass conversion and bio-oil upgrading, which reduces the oxygen content of biomass-derived products.96 Typically, bi-functional catalysts consisting of metal and solid acid sites have been employed for HDO of biomass feedstocks.97 One study could be found that was conducted by using a mining waste-derived catalyst for HDO.45 Bauxite mining waste, comprising a highly alkaline mixture of Fe2O3 (>60 wt%), TiO2, and complicated sodium alumino-silicates was employed as a catalyst that underwent in situ reduction at 350 °C. The reduced bauxite mining waste was found to be a non-alkaline magnetic solid. The catalyst was then used for HDO of levulinic acid (a well-known chemical derived from biomass) into a blend of C9 alkanes and alkenes with up to 76 wt% yield at 365 °C and an initial pressure of 5.5 MPa H2 (entry 5, Table 6). It could be re-used without loss of activity at least five times.4.4. NOx removal
NOx (e.g., NO and NO2) are a major component of kerbside pollution in large cities and in roadway microenvironments.98 Also, reprocessing spent nuclear fuel produces high-level radioactive liquid waste containing HNO3, which needs to be reduced to gas products by adding formaldehyde, resulting in the formation of NOx emitted via exhaust gas.99 In such applications, NOx removal is crucial. A variety of supported metal catalysts (e.g., Pt, Mn, Mo, V, Fe, Cu, and Ce) and binary metal oxides (e.g., CeO2/TiO2, MnOx/CeO2–ZrO2–Al2O3, and SnO2–MnOx–CeO2) have been used as NOx removal catalysts.100 There have been a few studies on using mining waste-derived catalysts for NOx removal.Wang et al. reported the denitrification performance of a catalyst derived from rare Earth tailings.51 To prepare the catalyst, rare Earth tailings underwent co-treatment with an alkali (calcium hydroxide and sodium carbonate) and an acid (HCl and citric acid) followed by calcination at 500 °C. The co-treatment process increased the Fe2O3 content in the catalyst, which played a crucial role in the denitrification reaction. Notably, the reduction temperature of the modified catalyst was higher than that of the original rare Earth tailings, indicating improved thermal stability. The catalyst achieved 96.2% efficiency for NO removal (initial NO concentration of 500 ppm, CO/NO = 4) at 900 °C (entry 6, Table 6). In the denitrification reaction over the catalyst, Fe2O3 is reduced to FeO by adsorbing CO molecules, while FeO is oxidized to Fe2O3 by adsorbing NO molecules (Fig. 4). Meanwhile, NO is reduced to N2. This redox capacity could enable continuous operation of the denitrification reaction.
 | ||
| Fig. 4 Schematic description of the catalytic denitrification reaction. Reprinted from Wang et al.51 and licensed under CC BY 4.0. | ||
NH3-SCR, considered one of the most efficient technologies for NOx removal from flue gas emitted by stationary and mobile sources,101 has been conducted using mining waste-derived catalysts.52,53 For example, a catalyst was synthesized from a mixture of rare Earth tailings and concentrate, containing cerium oxides and Fe–Ce composite oxides.52 The acid and alkali treatments increased the contents of Fe2+, Ce3+, and lattice oxygen, which significantly influenced the catalyst's NO degradation performance. Under controlled reaction conditions (entry 7, Table 6), the catalyst achieved 82% NO removal efficiency. A bimetallic Ni–Cu/rare Earth tailings catalyst was also applied for NH3-SCR (entry 8, Table 6).53 The addition of Ni and Cu introduced strong electron interactions between Fe, Ni and Cu, resulting in electron transfer between Ni–Fe and Cu–Fe and the subsequent formation and adsorption of nitrate species. Moreover, the formation of the NiFe2O4 spinel structure increased the number of surface-active sites, while the bimetallic Ni–Cu sites enhanced oxygen vacancies in CuO, improving the redox ability of the catalyst. The optimal composition of the bimetallic catalyst was found to be 2% Ni–2.5% Cu/rare Earth tailings, achieving a NO removal efficiency of 90% at 300 °C.
4.5. CO oxidation
CO gas, emitted from diverse sources, such as vehicle exhaust and industrial activities, poses significant risks to both the environment and human health.102 Thus, there have been continuous demands for the development of highly efficient and sustainable catalysts to mitigate CO emissions. CO oxidation is an attractive approach for eliminating CO from exhaust gas and in the fields of gas masks, gas sensors, and catalytic converters in fuel cells.103,104 In this regard, efforts were made to utilize mining waste originating from ore mines as CO oxidation catalysts. In a recent study by Mpiliou et al.,41 mesoporous iron oxide (e.g., hematite) derived from Serifos Fe-skarns mining waste was used as a catalyst support loaded with Au nanoparticles. The mining waste was calcined at 300 °C to remove impurities such as goethite, resulting in a more stable hematite support with a high surface area of up to 98.8 m2 g−1. Au nanoparticles were well-dispersed on the mining waste-derived hematite through a deposition–precipitation method. This catalyst achieved 90% CO conversion during the CO oxidation reaction at approximately 62 °C (entry 9, Table 6).4.6. CO2 methanation
Reducing CO2 emissions is an essential global challenge in the fight against climate change.105,106 CO2 methanation is considered a cost-effective method of CO2 reduction via the Sabatier reaction (CO2 + 4H2 → CH4 + 2H2O, ΔH° = −164 kJ mol−1).107 The Sabatier reaction is thermodynamically favorable at ambient temperature, but the production of CH4 is limited by the slow kinetics of this reaction. This necessitates the use of an active and selective catalyst.107 For CO2 methanation, Bahraminia and Anbia prepared Ni catalysts with different Ni loadings (5–20%) using coal gangue as the catalyst support.57 The coal gangue-supported Ni catalysts were used for the conversion of CO2 into CH4 at 450 °C and atmospheric pressure with 30000 mL g−1 h−1 of WHSV consisting of 60% H2/15% CO2/25% Ar (entry 10, Table 6). The mining waste-derived Ni catalyst with 15% Ni achieved 73% CO2 conversion and 91% CH4 selectivity. An increase in Ni loading from 5% to 15% increased the CO2 conversion and CH4 selectivity due to an increase in the number of active sites, but a further increase in Ni loading to 20% decreased the conversion and selectivity due to an increase in Ni particle size leading to blocking of the support cavities.
5. Wastewater treatment using mining waste-derived catalysts
5.1. Mining waste catalysts prepared for wastewater treatment
Mining waste-derived catalysts can also serve as effective materials for wastewater treatment.108 Recently, research has focused on recycling waste resources and modifying them into catalysts to efficiently remove pollutants from water. The preparation of mining waste-based catalysts for wastewater treatment commonly involves processes such as impurity removal, grinding, homogenization, and calcination. In some studies, additional salts and chemicals are added during synthesis to enhance catalytic properties.109 These catalysts have been successfully applied in Fenton, photo-Fenton, peroxymonosulfate (PMS) activation, electrochemical processes, and ozonation for pollutant removal from wastewater.In the wastewater treatment process, mining waste is converted into catalysts through various catalytic synthesis methods, as shown in Table 7. In some studies, untreated mining waste was used directly as a catalyst through grinding and milling (entries 5–7 and 9, Table 7). Conversely, in entries 2 and 3, HNO3 or CHBr3 was added to modify the mining waste. The addition of these chemicals enhances the purity110 of the modified catalyst by decomposing the mining waste through acid treatment or separating it via density separation.111 Furthermore, in most studies, the catalyst was modified using hydrothermal or thermal treatment (entries 1, 4, 8 and 10–15, Table 7). Thermal treatment removes volatile impurities from the mining waste and increases the number of active sites by altering the crystal structure.112 Catalysts modified through thermal treatment generally exhibited high specific surface areas, ranging from 20 to 63.48 m2 g−1. Various metal oxides and minerals, including Fe oxides (e.g., Fe2O3 and Fe3O4), SiO2, and Al2O3, were primarily present in the water treatment catalyst synthesized from mining waste. This composition closely resembles the mining waste components summarized in Table 2. Due to the presence of these metal-based compounds, some catalysts exhibited magnetic properties (entries 1, 10 and 14, Table 7). This magnetism enables easy catalyst separation after wastewater treatment, thereby improving catalyst reuse.
| Entry | Mining waste (major components) | Treatment | Properties | Ref. | ||||
|---|---|---|---|---|---|---|---|---|
| Surface area (m2 g−1) | Total pore volume (cm3 g−1) | Average pore diameter (nm) | Crystallite size (nm) | Saturation magnetization (Ms; emu g−1) | ||||
| 1 | Bauxite mining tailings (Fe oxides) | Fe3+ and Fe2+ molar ratio = 2:1 |
— | — | — | γ-Fe2O3 = 71.3; Fe3O4 = 61.3 | 9.8 | 46 |
| Hydrothermal treatment at 150 °C | ||||||||
| Thermal treatment at 400 °C | ||||||||
| 2 | Pyritic waste (FeS2, SiO2, Al2O3, CaO, K2O, Cr2O3) | CHBr3 purification | 2.37 | 0.0056 | 10900 |
— | — | 49 |
| 3 | Pyrite waste (SO3, Fe2O3, SiO2, Al2O3, Na2O3) | HNO3 treatment | 11.614 | 6.339 | 1.57 | — | — | 113 |
| 4 | Iron mining residue (SiO2, FeO) | Thermal treatment at 600 °C under an atmosphere of CH4 | 4.0 | — | — | — | — | 36 |
| 5 | Amethyst mining reject (SiO2, Fe2O3, Al2O3, CaO, TiO2) | — | 16.35 | 0.047 | 11.65 | — | — | 44 |
| 6 | Ilmenite mining residue (Fe oxides, FeTiO3, FeSi2, CaTiSiO5) | — | 0.6 | — | — | — | — | 114 |
| 7 | Iron mining residue (Fe2O3, Ce2O3) | — | 26 | — | — | — | — | 115 |
| 8 | Iron mining waste (Fe2O3, SiO2, CaO, B2O3, Al2O3) | Thermal treatment at 500 °C in air | 50.6 | 0.055 | 4.944 | — | — | 112 |
| 9 | Basalt mine tailings (SiO2, Fe2O3, CaO, Al2O3) | — | 13.43 | 0.033 | 9.73 | — | — | 47 |
| 10 | Waste ore (Fe3O4, Co3O4, SiO2, MgO) | Thermal treatment at 600 °C under an atmosphere of N2 | 11.26 | — | Meso = 4.18; macro = 61.87 | — | 7.42 | 33 |
| 11 | Iron mining waste (Fe oxides, g-C3N4, SiO2) | Suspension with melamine in H2O | 59.0 | — | — | — | — | 38 |
| Thermal treatment at 550 °C | ||||||||
| 12 | Coal gangue (Al2O3, SiO2, Fe2O3, SO3) | Suspension with melamine in H2O | 23.63 | — | 2–5 | — | — | 58 |
| Thermal treatment at 550 °C | ||||||||
| 13 | Pumice mining waste (SiO2, Al2O3, K2O) | Surface coating of nano-pumice onto carbon cloth | 63.48 | 0.117 | — | — | — | 50 |
| Thermal treatment at 160 °C | ||||||||
| 14 | Magnetic mining waste (Fe2O3, TiO2, SiO2, P2O5, CaO) | Polymerization with metakaolin at a ratio of 2.78 in the presence of NaOH and H2O | 10.4 | 1.05 | 40.26 | — | 18 | 39 |
| 15 | Magnetic mining waste (Fe2O3, TiO2, SiO2, P2O5, CaO) | Polymerization with metakaolin at a ratio of 0.96 in the presence of NaOH and H2O | 20.0 | 1.76 | 35.13 | — | — | 40 |
The synthesized catalyst generated reactive oxygen species (OH•, SO4˙−, O2˙−, 1O2, etc.) for pollutant decomposition in various water treatment processes. Additionally, the efficiency of reactive oxygen species generation and pollutant decomposition varied based on the catalyst characteristics, leading to an analysis of the optimal conditions and decomposition mechanisms for effective pollutant removal.
5.2. Fenton process
In the Fenton process, iron compounds such as Fe2O3, Fe3O4, FeO, and FeS2 included in catalysts play a pivotal role in determining the efficiency of pollutant degradation. These iron compounds react with hydrogen peroxide (H2O2), an oxidizing agent, leading to the oxidation of Fe2+ to Fe3+ and the subsequent formation of OH• radicals. Additionally, Fe3+ is reduced back to Fe2+ as it interacts with either the oxidizing agent or the pollutants, further enhancing radical generation.116 The OH• radicals produced through these reactions rapidly interact with pollutants, significantly improving the decomposition performance of the Fenton process.Santosa et al. synthesized magnetic catalysts via applying the hydrothermal method to the immobilized iron oxide precursor of Fe2+ and Fe3+ under alkaline conditions, followed by calcination.46 When the concentration of H2O2 was 1 mM and the pH was 7, these conditions proved to be optimal for tetracycline removal, achieving a decomposition rate of 89.7% (entry 1, Table 8). Additionally, the catalyst could be easily separated using its magnetic properties, and its practicality was further demonstrated by minimal crystal structure deformation after use. On the other hand, Cechinel et al. utilized bromoform for density separation to eliminate impurities from pyrite, which was subsequently used as a catalyst for organic dye discoloration.49 The use of bromoform effectively reduced impurities and enhanced the exposure of active sites by increasing access to the organic content in pyrite. As a result, the catalyst achieved a removal rate of 98% for reactive blue 21 dye under optimized conditions, with an H2O2 concentration of 2 g L−1 at pH 4.7 (entry 2, Table 8). Also, this high pollutant removal efficiency was attributed to the enhanced conductivity and electron transfer properties imparted by Si and S oxides, which contributed to increased radical generation via Fe oxide reactions.117 Mashayekh-Salehi et al. synthesized a catalyst from pyrite waste113 and improved its performance by eliminating surface impurities through HNO3 treatment, followed by milling. With a high FeS2 purity, the catalyst exhibited maximum tetracycline removal and catalytic efficiency of 96.1% under optimal conditions, specifically at a solution pH of 4.1 and an H2O2 concentration of 5 mM (entry 3, Table 8). Tetracycline removal reached approximately 96.1%, and after four consecutive reuse cycles of pyrite, the removal efficiency remained above 90%. This stability was due to high catalytic activity and low iron leaching, which ensured sustained performance even after multiple cycles. In the study by Augusto et al., iron mining residue was modified through calcination at 600 °C for 30 min under an atmosphere of methane. The catalyst indicated high thermal stability, presenting a negligible weight loss (<1%) in thermogravimetric analysis. The iron mining residue is primarily composed of SiO2, Fe2O3, and FeOOH, while calcination with CH4 transforms the catalyst into a reduced iron phase of FeO.118 The formation of Fe2+ enhances the removal efficiency to over 90%, as Fe2+ directly reacts with H2O2 at pH 7.2 and with 6.2 M of H2O2 (entry 4, Table 8).36 The higher efficiency compared to the raw catalyst is due to the faster H2O2 decomposition rate with Fe2+ and a higher radical generation rate. As illustrated in Fig. 5, the use of the catalyst in combination with H2O2 consistently resulted in higher removal efficiency compared to the use of H2O2 alone, while the extent of variation depends on the pH.
 | ||
| Fig. 5 Effect of pH on pollutant removal efficiency in the Fenton process. Reprinted from Mashayekh-Salehi et al.,113 copyright (2020), with permission from Elsevier. | ||
| Entry | Catalyst | Reaction | Reaction conditions | Performance | Ref. |
|---|---|---|---|---|---|
| T = temperature; t = time; CD = catalyst dosage; CA = concentration of A. | |||||
| 1 | γ-Fe2O3/Fe3O4-bauxite mining tailings | Fenton | T = 25 °C; t = 120 min; CTC = 20 mg L−1; CD = 1.2 g L−1; CH2O2 = 1.0 mM; pH = 7 | Tetracycline removal = 89.7% | 46 |
| 2 | Pyritic waste | Fenton | T = 20 °C; t = 210 min; CRB21 = 150 mg L−1; CD = 0.5 g L−1; CH2O2 = 2 g L−1; pH = 4.7 | Reactive blue 21 removal = 98% | 49 |
| 3 | Pyrite waste | Fenton | T = 40 °C; t = 60 min; CTC = 50 mg L−1; CD = 1 g L−1; CH2O2 = 5 mM; pH = 4.1 | Tetracycline removal = 96.1% | 113 |
| 4 | CH4–iron mining residue | Fenton | t = 150 min; Cpollutants = 10 mg L−1; CD = 1 g L−1; CH2O2 = 6.2 M; pH = 7.2 | Rhodamine b removal = >90%; paracetamol removal = >90% | 36 |
| 5 | Iron-bearing mining reject | Photo-Fenton | T = 25 °C; t = 180 min; Cphenol = 50 mg L−1; CD = 0.75 g L−1; CH2O2 = 7.5 mM; pH = 3; light flux = 52.5 W m−2 (400–1000 nm) | Phenol removal = 96.5% | 44 |
| 6 | Ilmenite mining residue | Photo-Fenton | T = 25 °C; t = 30 min; Cpollutants = 200 μg L−1; CD = 0.277 mg L−1; CH2O2 = 65.4 mg L−1; pH = 3.4; light flux = 6.8 W m−2 (350–400 nm) | Sulfamethoxazole removal = 89%; ciprofloxacin removal = 83%; tetracycline removal = 88% | 114 |
| 7 | Iron mining residue | Photo-Fenton | t = 60 min; CSTZ = 100 μg L−1; CD = 0.3 g L−1; CH2O2 = 1 mM; pH = 2.5; light flux = 18 W m−2 (320–400 nm) | Sulfathiazole removal = 96% | 115 |
| 8 | Iron mining waste | Photo-Fenton | T = 25 °C; t = 90 min; CSG dye = 10 mg L−1; CD = 0.15 g L−1; CNa2SO3 = 0.45 mM; pH = 3 | Sicomet green dye removal = 91.5% | 112 |
| 9 | Basalt mine tailings | Photo-Fenton | T = 25 °C; t = 60 min; CP4R = 50 mg L−1; CD = 0.75 g L−1; CH2O2 = 4.5 mM; pH = 3 | Ponceau 4R removal = 97.0% | 47 |
| 10 | Biochar-waste ore | PMS activation | t = 60 min; CTC = 20 mg L−1; CD = 0.9 g L−1; CPMS = 0.6 g L−1; pH = 5 | Tetracycline removal = 96.6% | 33 |
| 11 | Graphitic carbon nitride–iron mining waste | PMS activation | T = 25 °C; t = 60 min; CAPAP = 10 mg L−1; CD = 0.5 g L−1; CPMS = 0.6 mM; pH = 6.5 | Acetaminophen removal = 98% | 38 |
| 12 | Graphitic carbon nitride –coal gangue | PMS activation | T = 25 °C; t = 30 min; CBPA = 50 mg L−1; CD = 1.0 g L−1; CPMS = 0.27 mM; pH = 7 | Bisphenol A removal = 90% | 58 |
| 13 | Microbial fuel cells–pumice mine waste | Electrochemical |
T = 35 °C; t = 3 d; CCOD = 12000 mg L−1, BOD/COD = 0.4; electrode = 2 × 2 cm; catholyte = 50 mM (phosphate buffer); pH = 7 |
Generated voltages = 1.126 V; COD removal = 94% | 50 |
| 14 | Geopolymer–magnetic mining waste | Ozonation | T = 25 °C; t = 30 min; CD = 0.1 g L−1; pH = 4 | Ozone decomposition rate = 2.98 min−1 | 39 |
| 15 | Geopolymer–magnetic mining waste | Ozonation | T = 25 °C; t = 60 min; CTMP = 1 g L−1; CD = 0.1 g L−1; pH = 4 | Trimethoprim removal = 71% | 40 |
5.3. Photo-Fenton process
The photo-Fenton process closely resembles the conventional Fenton process, where iron oxides serve as activators for the oxidant reagent. In this process, Fe2+ reacts with H2O2 to generate OH• radicals while being oxidized to Fe3+. Among the iron oxides, Fe2O3 was identified as the primary component driving the reaction, highlighting its significance in the photo-Fenton mechanism. Unlike the standard Fenton process, where Fe3+ must be reduced back to Fe2+ through additional reagents, the photo-Fenton process utilizes light irradiation to rapidly reduce Fe3+ to Fe2+, ensuring a continuous cycle of radical generation.119 This self-sustaining reaction eliminates the need for an additional catalytic regeneration step, reducing catalyst consumption while maintaining high pollutant removal efficiency. Furthermore, the ability of light to regenerate iron oxides enables the reaction to proceed effectively in the presence of mining waste, regardless of the oxidation state of iron.Hollanda et al. synthesized a catalyst from amethyst mining reject, utilizing ball milling to reduce and homogenize its size, thereby increasing its surface area.44 In the pollutant removal experiment, a 96.5% removal efficiency was achieved using 0.75 g L−1 of catalyst, enhanced by the Fe2+/Fe3+ redox cycle under visible light (entry 5, Table 8).44 In the fourth cycle, 92.8% phenol degradation was achieved, demonstrating high stability and catalytic activity, with iron leaching (1.23 mg L−1) remaining well below the Brazilian regulatory limit of 15 mg L−1.120 da Silva et al.114 and Rojas-Mantilla et al.115 utilized ilmenite mining residue and iron mining residue as catalysts without additional modification. Despite its lower specific surface area than other catalysts,37 the ilmenite residue exhibited excellent pollutant removal efficiency, achieving 89% for sulfamethoxazole, 83% for ciprofloxacin, and 88% for tetracycline with 0.277 g L−1 of catalyst (entry 6, Table 8). Meanwhile, the iron mining residue-based catalyst achieved a 96% decomposition rate for sulfathiazole at a catalyst dose of 0.3 g L−1 (entry 7, Table 8). While both catalysts operated under UVA conditions, the iron mining residue, with a specific surface area more than four times higher, promoted greater radical generation, leading to an increased decomposition rate. Furthermore, the iron mining residue catalyst exhibited an electrical energy consumption of 1.3 kW h m−3 due to its rapid decomposition performance, highlighting its lower energy demands compared to other catalysts reported in the literature. When Kebir et al. calcined iron mining waste at 500 °C, the modified catalyst exhibited enhanced catalytic activity by increasing the active surface area and optimizing the distribution of active sites.112 Using 0.15 g L−1 of catalyst at pH 3 and 0.45 mM Na2SO3 under sunlight for 90 min, a dye removal rate of 91.5% was achieved (entry 8, Table 8). Despite Na2SO3 acting as an oxidizing agent, OH• and O2˙− radicals were generated through catalytic activation by light, with OH˙ radicals playing a dominant role in pollutant decomposition. Drumm et al. assessed the catalytic performance of basalt mine tailings sieved to a uniform size without additional modification.47 In dye removal experiments, the catalyst achieved 97% pollutant removal efficiency and a total organic carbon removal rate of 75.4% after 300 min (entry 9, Table 8). Light-reduced Fe2+ reacted with H2O2 to produce OH• radicals, which rapidly decomposed pollutants and efficiently mineralized them into CO2 and H2O. Fig. 6 indicates that the photo-Fenton process, like the traditional Fenton process, is more effective under low pH conditions. However, the photoreduction of Fe3+ to Fe2+ enhances the generation of OH•, thereby further accelerating the pollutant degradation rate.
 | ||
| Fig. 6 (a) Comparison of catalytic activities on pollutant removal and the effect of pH on the photo-Fenton process. Reprinted from Hollanda et al.,44 copyright (2024), with permission from Springer Nature. | ||
5.4. PMS activation
Unlike the traditional Fenton process, the PMS activation process utilizes PMS as an oxidizing agent, generating SO4˙− radicals, O2˙− radicals, and 1O2 as the primary reactive oxygen species for pollutant decomposition. In this process, various metals, including Fe, can participate, and PMS activation may also occur through carbon bonding.121 A detailed analysis was conducted to examine the active sites and radical generation involved in the PMS activation process.Wang et al. developed a PMS activation catalyst with waste ore and fine needle powder, mixed in a specific ratio and calcined at 600 °C to produce a metal composite-loaded biochar.33 Co2+, Fe2+, Co3+, and Fe3+ coexisted in the catalyst, actively contributing to the PMS activation reaction, as evidenced by the reduction in peak intensities. Under optimal reaction conditions, the catalyst degraded 96.6% of tetracycline within 60 min (entry 10, Table 8). PMS was decomposed by metal oxidation to form SO4˙− radicals and OH• radicals, but SO4˙− radicals mainly had a major influence on the decomposition of pollutants. Bicalho et al.38 and Zhang et al.58 synthesized catalysts by combining iron mining waste and coal gangue with graphitic carbon nitride. During the calcination process for graphitic carbon nitride synthesis, iron mining waste and metal gangue were incorporated in specific ratios, resulting in catalysts with various metal oxides distributed across the carbon nitride layer. The metals, dispersed over a large specific surface area, contributed to PMS activation for radical generation, while electron transfer at defect sites in carbon nitride further facilitated PMS activation. The iron mining waste-based catalyst achieved 98% acetaminophen removal efficiency (entry 11, Table 8), while the coal gangue-based catalyst demonstrated 90% bisphenol A removal efficiency (entry 12, Table 8). In both catalysts, various reactive oxygen species, including SO4˙− and OH• radicals, were generated; however, 1O2 and O2˙− radicals played a more significant role in pollutant decomposition. This suggests that the formation of O2˙− and 1O2 species was enhanced by the electron-deficient environment created due to differences in electronegativity.122
5.5. Electrochemical treatment
In the electrochemical process, the power generation capacity is influenced by anode characteristics such as potential and surface modification. Increased power generation enhances process efficiency while reducing energy consumption.123 With high chemical stability, mechanical strength, porosity, and specific surface area, mining waste serves as a cost-effective material that significantly boosts efficiency. Eslami et al. synthesized anode electrodes using pumice mining waste and applied them to electrochemical processes.50 The pumice was standardized in size through crushing and milling operations before being coated onto carbon cloth. A binder was added to secure the coating, and the assembly was calcined at 160 °C. With its high specific surface area, the pumice-coated electrode enhanced the adsorption of electrically generated microorganisms compared to uncoated electrodes. It also accelerated electron accumulation, resulting in a 47% increase at open circuit voltage. This electrode modification significantly improved the efficiency of COD decomposition in water, achieving over 40% improvement, with COD removal reaching 94% (entry 13, Table 8). Additionally, removal efficiencies for total suspended solids and total dissolved solids ranged from 70% to 80%. These improvements were attributed to enhanced electron transfer due to the accumulation of electrons in the electrode.124 Notably, the study, conducted with actual wastewater, demonstrated an energy efficiency improvement of 32%.5.6. Ozonation
The ozone process primarily focuses on evaluating the extent to which O3 decomposition is enhanced by the catalyst. In this process, the metal oxides present in the mining waste catalyst play a crucial role by promoting more efficient ion exchange and radical formation.39 This, in turn, leads to a significant increase in the ozone decomposition rate, enhancing the overall effectiveness of the process. Gier Della Rocca et al. synthesized an ozone generation catalyst using magnetic mining waste and investigated its application in pollutant decomposition via ozone oxidation.39,40 Magnetic mining waste was mixed with alkali activators (Na2SiO3, NaOH, and H2O) in specific ratios and reacted for 15 min at 65 °C to produce an inorganic binder, i.e., a geopolymer. Metakaolin, synthesized by calcining kaolin (Campina Grande, PB, Brazil), served as a control. Results revealed that the surface area of the catalyst decreased as the waste ratio increased. However, the catalytic performance improved, with the ozone decomposition rate peaking at waste ratios of 25–50 wt% (entry 14, Table 8). The application of the synthetic catalyst resulted in a 60% ozone decomposition rate and a 71% pollutant removal efficiency (entry 15, Table 8). Furthermore, mineralization, which was only 4% without the catalyst, increased significantly to 40% with its application.6. Summary and outlook
This review evaluated the performance of mining waste-derived catalysts across diverse fields of application. The catalytic properties of mining waste were examined in relation to their treatment procedures, highlighting their significant potential in various processes, including reforming, wastewater treatment, esterification, and environmental catalysis (e.g., denitrification, CO oxidation, and CO2 reduction). These findings demonstrate that mining waste can effectively replace costly and non-renewable conventional catalysts in several applications. Despite these promising results, there is still a need to further investigate the design of optimal mining waste-derived catalysts with enhanced activity and stability for different reactions. Continuous efforts in this field will not only improve the management of mining waste but also promote the sustainability of catalytic processes. To this end, several points need to be considered in future studies for further enhancement of the feasibility of mining waste-derived catalysts.First, various types of mining waste have been employed as heterogeneous catalysts, with characteristics such as composition, crystallite size (e.g., spinel), and impurities differing significantly based on their origin. However, inconsistencies in the characterization methods used across studies present a major challenge. For example, one study may report the content of metals measured via X-ray fluorescence, while another reports the content of metal oxides estimated via X-ray diffraction. These variations hinder the direct comparison of findings and complicate efforts to design more active and selective catalysts for specific reactions. Standardizing characterization techniques for mining waste is, therefore, essential to ensure uniformity and reproducibility in future research.
To enhance the catalytic properties of mining waste-derived catalysts—such as the type and number of active sites, surface area, porosity, and surface-active site density—greater efforts are needed to control the combined effects of key variables, including the type of mining waste and catalyst preparation conditions. However, little information is available on precise control over mining waste properties for catalytic applications. Further investigation into tailoring these properties is essential for designing active, selective, and stable mining waste-derived catalysts.
Although a few studies have reported data on equilibrium and kinetics for a limited range of reactions (e.g., reforming) conducted on mining waste-derived catalysts, more comprehensive studies are needed. Detailed investigations into chemical equilibrium, reaction kinetics, and thermodynamics for a broader array of reactions occurring over mining waste-derived catalysts are essential. These studies should also assess catalytic performance factors such as reaction rates, selectivity toward target products, and the extent of catalyst deactivation after reactions. This information is critical for process intensification and scaling up mining waste-based catalytic processes.
Various mining wastes have been modified and utilized as catalysts for wastewater treatment, demonstrating excellent pollutant removal performance. However, the range of pollutant types tested with these catalysts remains narrow, and their applicability to actual wastewater conditions is underexplored. Many studies lack comprehensive analyses of changes in catalyst performance when key parameters (e.g., temperature, scavengers, and anions) or water matrices (e.g., natural organic matter) are varied. These gaps make it challenging to evaluate the practicality and scalability of mining waste-derived catalysts in real-world applications. To address these issues, future research should focus on expanding the evaluation of these catalysts to include a broader spectrum of pollutants and perform correlation analyses with additional influencing factors.
In the studies reviewed, catalyst performance was primarily evaluated under controlled conditions using a single water treatment process, often achieving a removal efficiency of approximately 90%. While this performance is excellent, the presence of co-existing materials and background compounds in actual wastewater may significantly reduce the removal efficiency. To improve the practical applicability of mining waste-derived catalysts, further research should focus on optimizing catalyst performance through advanced reforming methods or by integrating multiple water treatment processes.
While mining waste-derived catalysts have shown great promise in various reactions, it is difficult to designate optimal reaction conditions for each of them because different studies have used different reaction conditions (e.g., temperature, pressure, time-on-stream, and more importantly, space velocity). Moreover, their stability and durability during long-term operations remain uncertain. Catalyst deactivation, particularly under high-temperature and high-pressure conditions, poses a significant challenge. Rigorous studies are required to optimize the reactions of mining waste catalysts and enhance the stability and durability of these catalysts, ensuring their ability to support continuous operations without significant performance degradation.
For mining waste to become a viable substitute for industrial catalysts, a stable and consistent supply of raw mining waste and residue is essential. Maintaining constant properties for large-scale catalyst production is critical for achieving industrial applicability. Addressing this challenge will require close cooperation between the chemical and mining industries, as well as local governments. To promote the integration of mining waste into catalyst production, tools such as techno-economic analysis (TEA) and life cycle assessment (LCA) should be employed. These methods can simulate the real-world application of mining waste-derived catalysts and highlight their contribution to sustainable mining waste management by demonstrating their potential to replace expensive and non-environmentally benign catalysts.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Research Foundation of Korea (NRF) grants funded by the Korean Government (Ministry of Science and ICT; grant no. RS-2023-00209044).References
- O. Agboola, D. E. Babatunde, O. S. I. Fayomi, E. R. Sadiku, P. Popoola, L. Moropeng, A. Yahaya and O. A. Mamudu, Results Eng., 2020, 8, 100181 CrossRef.
- J. Helser, E. Vassilieva and V. Cappuyns, J. Hazard. Mater., 2022, 424, 127313 CrossRef CAS PubMed.
- W. Haas, F. Krausmann, D. Wiedenhofer and M. Heinz, J. Ind. Ecol., 2015, 19, 765–777 CrossRef.
- D. M. Franks, M. Stringer, L. A. Torres-Cruz, E. Baker, R. Valenta, K. Thygesen, A. Matthews, J. Howchin and S. Barrie, Sci. Rep., 2021, 11, 5353 CrossRef CAS PubMed.
- Mining waste management market size, share & industry analysis, by source (surface mining and underground mining), by waste type (solid waste {waste rock, tailings, others} and liquid waste), by commodity (mineral fuels, iron, ferro alloys, industrial minerals, and others) and regional forecast, 2024–2032, Fortune Business Insights, Pune, Maharashtra, India, 2024.
- D. A. Vallero and G. Blight, Mine waste: a brief overview of origins, quantities, and methods of storage, in Waste, ed. T. M. Letcher and D. A. Vallero, Academic Press, Cambridge, MA, USA, 2nd edn, 2019, pp. 129–151, DOI:10.1016/B978-0-12-815060-3.00006-2.
- S. Kalisz, K. Kibort, J. Mioduska, M. Lieder and A. Małachowska, J. Environ. Manage., 2022, 304, 114239 CrossRef CAS PubMed.
- L. N. Bowker and D. M. Chambers, Earthwork Act, 2015, 24, 1–56.
- R. K. Valenta, É. Lèbre, C. Antonio, D. M. Franks, V. Jokovic, S. Micklethwaite, A. Parbhakar-Fox, K. Runge, E. Savinova, J. Segura-Salazar, M. Stringer, I. Verster and M. Yahyaei, Resour., Conserv. Recycl., 2023, 190, 106859 CrossRef CAS.
- P. Segui, A. e. M. Safhi, M. Amrani and M. Benzaazoua, Minerals, 2023, 13, 90 CrossRef CAS.
- S. Maruthupandian, A. Chaliasou and A. Kanellopoulos, Constr. Build. Mater., 2021, 312, 125333 CrossRef CAS.
- Z. Mkahal, Y. Mamindy-Pajany, W. Maherzii and N.-E. Abriak, Waste Biomass Valorization, 2022, 13, 667–687 CrossRef CAS.
- S. Yagüe, I. Sánchez, R. Vigil de la Villa, R. García-Giménez, A. Zapardiel and M. Frías, Minerals, 2018, 8, 46 CrossRef.
- Y. W. Choi, Y. J. Kim, O. Choi, K. M. Lee and M. Lachemi, Constr. Build. Mater., 2009, 23, 2481–2486 CrossRef.
- A. M. T. Simonsen, S. Solismaa, H. K. Hansen and P. E. Jensen, Waste Manage., 2020, 102, 710–721 CrossRef CAS PubMed.
- S. Maruthupandian, Upcycling mineral wastes: their characterisation and activation for use in sustainable cementitious binders, University of Hertfordshire, 2024, DOI:10.18745/th.28078.
- I. Capasso, S. Lirer, A. Flora, C. Ferone, R. Cioffi, D. Caputo and B. Liguori, J. Cleaner Prod., 2019, 220, 65–73 CrossRef CAS.
- S. M. A. Qaidi, B. A. Tayeh, A. M. Zeyad, A. R. G. de Azevedo, H. U. Ahmed and W. Emad, Case Stud. Constr. Mater., 2022, 16, e00933 Search PubMed.
- G. Lazorenko, A. Kasprzhitskii, F. Shaikh, R. S. Krishna and J. Mishra, Process Saf. Environ. Prot., 2021, 147, 559–577 CrossRef CAS.
- X. He, Z. Yuhua, S. Qaidi, H. F. Isleem, O. Zaid, F. Althoey and J. Ahmad, Ceram. Int., 2022, 48, 24192–24212 CrossRef CAS.
- L. D. Menéndez-Aguado, M. Marina Sánchez, M. A. Rodríguez, A. L. Coello Velázquez and J. M. Menéndez-Aguado, Materials, 2019, 12, 2047 CrossRef PubMed.
- A. J. Whitworth, E. Forbes, I. Verster, V. Jokovic, B. Awatey and A. Parbhakar-Fox, Cleaner Eng. Technol., 2022, 7, 100451 CrossRef.
- C. Falagán, B. M. Grail and D. B. Johnson, Miner. Eng., 2017, 106, 71–78 CrossRef.
- B. Lim and R. D. Alorro, Sustainable Chem., 2021, 2, 686–706 CrossRef.
- C. Vitti and B. J. Arnold, Min., Metall., Explor., 2022, 39, 49–54 Search PubMed.
- S. K. Sarker, N. Haque, M. Bhuiyan, W. Bruckard and B. K. Pramanik, J. Environ. Chem. Eng., 2022, 10, 107622 CrossRef CAS.
- A. Abbadi and G. Mucsi, J. Environ. Chem. Eng., 2024, 12, 113118 CrossRef CAS.
- Z. Xiaolong, Z. Shiyu, L. Hui and Z. Yingliang, J. Cleaner Prod., 2021, 284, 124756 CrossRef.
- O. R. Carmignano, S. S. Vieira, A. P. C. Teixeira, F. S. Lameiras, P. R. G. Brandão and R. M. Lago, J. Braz. Chem. Soc., 2021, 32, 1895–1911 CAS.
- F. P. Barraza, D. Thiyagarajan, A. Ramadoss, V. S. Manikandan, S. S. Dhanabalan, C. V. Abarzúa, P. S. Soloaga, J. C. Nazer, M. J. Morel and A. Thirumurugan, Renewable Sustainable Energy Rev., 2024, 202, 114665 CrossRef.
- V. Maus, S. Giljum, D. M. da Silva, J. Gutschlhofer, R. P. da Rosa, S. Luckeneder, S. L. B. Gass, M. Lieber and I. McCallum, Sci. Data, 2022, 9, 433 CrossRef PubMed.
- M. Jayakumar, U. Surendran, P. Raja, A. Kumar and V. Senapathi, Arabian J. Geosci., 2021, 14, 2156 CrossRef CAS.
- J. Wang, N. Huang, G. Wang, J. Yu, F. Wang, D. Zhang, F. Su, X. Jia, M. Wang, X. Meng, C. Kong, Z. Yang, T. Wang and H. Zhu, Colloids Surf., A, 2024, 685, 133249 CrossRef CAS.
- A. Azara, E.-H. Benyoussef, F. Mohellebi, M. Chamoumi, F. Gitzhofer and N. Abatzoglou, Catalysts, 2019, 9, 1069 CrossRef CAS.
- C. D. Prates, F. C. Ballotin, H. Limborço, J. D. Ardisson, R. M. Lago and A. P. d. C. Teixeira, Appl. Catal., A, 2020, 600, 117624 CrossRef CAS.
- T. d. M. Augusto, P. Chagas, D. L. Sangiorge, T. C. d. O. M. Leod, L. C. A. Oliveira and C. S. d. Castro, J. Environ. Chem. Eng., 2018, 6, 6545–6553 CrossRef CAS.
- S. C. Ayala-Durán, P. Hammer and R. F. P. Nogueira, Environ. Sci. Pollut. Res., 2020, 27, 1710–1720 CrossRef PubMed.
- H. A. Bicalho, R. D. F. Rios, I. Binatti, J. D. Ardisson, A. J. Howarth, R. M. Lago and A. P. C. Teixeira, J. Hazard. Mater., 2020, 400, 123310 CrossRef CAS PubMed.
- D. G. D. Rocca, F. A. S. e Sousa, J. D. Ardisson, R. A. Peralta, E. Rodríguez-Castellón and R. d. F. P. M. Moreira, Heliyon, 2023, 9, e17097 CrossRef PubMed.
- D. G. Della Rocca, A. De Noni Júnior, E. Rodríguez-Aguado, R. A. Peralta, E. Rodríguez-Castellón, G. L. Puma and R. F. P. M. Moreira, J. Environ. Chem. Eng., 2023, 11, 111163 CrossRef CAS.
- M. Mpiliou, K. Kappis, S. Tombros, G. Avgouropoulos, S. Kokkalas, P. Lampropoulou, S. Triantafyllidis, H. Li and J. Papavasiliou, Sustainable Chem. Environ., 2024, 5, 100067 CrossRef.
- S. Belbessai, E.-H. Benyoussef, F. Gitzhofer and N. Abatzoglou, Chem. Eng. J. Adv., 2022, 12, 100424 CrossRef CAS.
- M. Chamoumi and N. Abatzoglou, Catalysts, 2021, 11, 771 CrossRef CAS.
- L. R. Hollanda, J. A. B. de Souza, G. L. Dotto, E. L. Foletto and O. Chiavone-Filho, Environ. Sci. Pollut. Res., 2024, 31, 21291–21301 CrossRef CAS PubMed.
- E. Karimi, I. F. Teixeira, L. P. Ribeiro, A. Gomez, R. M. Lago, G. Penner, S. W. Kycia and M. Schlaf, Catal. Today, 2012, 190, 73–88 CrossRef CAS.
- E. D. A. Santosa, M. Tamyiz, S. Sagadevan, A. Hidayat, I. Fatimah and R.-a. Doong, Results Chem., 2022, 4, 100451 CrossRef.
- F. C. Drumm, P. Grassi, J. Georgin, D. P. Franco, D. Tonato, E. L. Foletto, G. L. Dotto and S. L. Jahn, Appl. Geochem., 2022, 136, 105136 CrossRef CAS.
- M. Chamoumi, N. Abatzoglou, J. Blanchard, M.-C. Iliuta and F. Larachi, Catal. Today, 2017, 291, 86–98 CrossRef CAS.
- M. A. P. Cechinel, T. de Oliveira Guidolin, A. R. da Silveira, J. dos Santos Tasca, O. R. K. Montedo and S. Arcaro, Sci. Total Environ., 2022, 807, 150823 CrossRef CAS PubMed.
- F. Eslami, K. Yaghmaeian, R. Shokoohi, R. Sajjadipoya, A. Rahmani, H. Askarpur, A. N. Baghani, H. J. Mansoorian and F. j. Ansari, Heliyon, 2024, 10, e40495 CrossRef CAS PubMed.
- Z. Wang, Y. Huang, H. Luo, Z. Gong, K. Zhang, N. Li and W. Wu, Green Process. Synth., 2019, 8, 865–872 CAS.
- J. Wang, C. Zhu, B. Li, Z. Gong, Z. Meng, G. Xu and W. Wu, Green Process. Synth., 2020, 9, 191–202 Search PubMed.
- L. Hou, Z. Li, J. Xu and W. Wu, React. Kinet., Mech. Catal., 2023, 136, 2499–2516 CrossRef CAS.
- E. T. L. Lima, L. S. Queiroz, L. H. de Oliveira Pires, R. S. Angélica, C. E. F. da Costa, J. R. Zamian, G. N. da Rocha Filho, R. Luque and L. A. S. d. Nascimento, ACS Sustainable Chem. Eng., 2019, 7, 7543–7551 CrossRef.
- A. d. N. de Oliveira, E. T. L. Lima, E. H. de Aguiar Andrade, J. R. Zamian, G. N. d. R. Filho, C. E. F. d. Costa, L. H. de Oliveira Pires, R. Luque and L. A. S. d. Nascimento, Catalysts, 2020, 10, 478 CrossRef CAS.
- Y. Zhang, W. Ma, Z. Zhang, Y. Chen, J. Sun, S. Yue, G. Wang and X. Wang, J. Environ. Manage., 2025, 373, 123557 CrossRef CAS PubMed.
- S. Bahraminia and M. Anbia, Int. J. Hydrogen Energy, 2024, 83, 842–855 CrossRef CAS.
- X. Zhang, R. Zhao, N. Zhang, Y. Su, Z. Liu, R. Gao and C. Du, Appl. Catal., B, 2020, 263, 118316 CrossRef CAS.
- É. Lèbre, G. D. Corder and A. Golev, Miner. Eng., 2017, 107, 34–42 CrossRef.
- R. Zhang, H. He, Y. Tang, Z. Zhang, H. Zhou, J. Yu, L. Zhang and B. Dai, ChemCatChem, 2024, 16, e202400396 CrossRef CAS.
- H. Lee, Y. Kim, H. K. Yu and J. Lee, Alexandria Eng. J., 2024, 91, 494–502 CrossRef.
- W. Yang, D. Choi, H. K. Yu, S. Jung and J. Lee, J. Environ. Manage., 2025, 373, 123564 CrossRef CAS PubMed.
- W. Yang, S. Jung, J. Lee, S. W. Lee, Y. T. Kim and E. E. Kwon, Environ. Pollut., 2023, 329, 121684 CrossRef CAS PubMed.
- H. S. Lee, S. Jung, S. W. Lee, Y. T. Kim and J. Lee, Korean J. Chem. Eng., 2023, 40, 2472–2479 CrossRef CAS PubMed.
- I. Riaz, O. A. Qamar, F. Jamil, M. Hussain, A. Inayat, L. Rocha-Meneses, P. Akhter, S. Musaddiq, M. R. A. Karim and Y. Park, Korean J. Chem. Eng., 2023, 40, 2683–2691 CrossRef CAS.
- I. Boz, M. S. Boroglu, Y. Zengin and B. Kaya, Korean J. Chem. Eng., 2023, 40, 1882–1891 CrossRef CAS.
- O. B. Al-Ameri, M. Alzuhairi, E. Bailón-García, F. Carrasco-Marín and J. Amaro-Gahete, Appl. Sci., 2024, 14, 9080 CrossRef CAS.
- Hartati, D. Prasetyoko, M. Santoso, I. Qoniah, W. L. Leaw, P. B. D. Firda and H. Nur, J. Chin. Chem. Soc., 2020, 67, 911–936 CrossRef CAS.
- É. Ujaczki, V. Feigl, M. Molnár, P. Cusack, T. Curtin, R. Courtney, L. O'Donoghue, P. Davris, C. Hugi, M. W. Evangelou, E. Balomenos and M. Lenz, J. Chem. Technol. Biotechnol., 2018, 93, 2498–2510 CrossRef PubMed.
- M. I. Malik, M. Rouabah, N. Abatzoglou and I. E. Achouri, Biofuels, Bioprod. Biorefin., 2024, 18, 1027–1046 CrossRef CAS.
- S. Belbessai, I. E. Achouri, E. H. Benyoussef, F. Gitzhofer and N. Abatzoglou, Catal. Today, 2021, 365, 111–121 CrossRef CAS.
- O. A. Z. Sahraei, F. Larachi, N. Abatzoglou and M. C. Iliuta, Appl. Catal., B, 2017, 219, 183–193 CrossRef.
- O. A. Sahraei, A. Desgagnés, F. Larachi and M. C. Iliuta, Appl. Catal., B, 2020, 279, 119330 CrossRef.
- O. A. Sahraei, A. Desgagnés, F. Larachi and M. C. Iliuta, Int. J. Hydrogen Energy, 2021, 46, 32017–32035 CrossRef.
- M. Farsi, Ammonia production from syngas: plant design and simulation, in Advances in Synthesis Gas: Methods, Technologies and Applications, ed. M. R. Rahimpour, M. A. Makarem and M. Meshksar, Elsevier, Amsterdam, Netherlands, 2023, vol. 4, pp. 381–399 Search PubMed.
- S. Sepahi and M. R. Rahimpour, Methanol production from syngas, in Advances in Synthesis Gas : Methods, Technologies and Applications, ed. M. R. Rahimpour, M. A. Makarem and M. Meshksar, Elsevier, Amsterdam, Netherlands, 2023, vol. 3, pp. 111–146 Search PubMed.
- S. Lee, J.-C. Seo, H.-J. Chun, S. Yang, E.-h. Sim, J. Lee and Y. T. Kim, Catal. Sci. Technol., 2022, 12, 5814–5828 RSC.
- J. Lee, K.-Y. A. Lin, S. Jung and E. E. Kwon, Chem. Eng. J., 2023, 452, 139218 CrossRef CAS.
- J. Lee, S. Kim, S. You and Y.-K. Park, Renewable Sustainable Energy Rev., 2023, 178, 113240 CrossRef CAS.
- V. A. Luciano, F. G. de Paula, P. S. Pinto, C. D. Prates, R. C. G. Pereira, J. D. Ardisson, M. G. Rosmaninho and A. P. C. Teixeira, Fuel, 2022, 310, 122290 CrossRef CAS.
- E. Benhelal, M. Hoseinpour, R. Karami, A. Mirvakili and M. I. Rashid, Energy and exergy analysis of blue hydrogen production and conversion, in Hydrogen Energy Conversion and Management, ed. M. M. K. Khan, A. K. Azad and A. M. T. Oo, Elsevier, Amsterdam, Netherlands, 2024, pp. 157–207, DOI:10.1016/B978-0-443-15329-7.00008-9.
- H. Zhu, H. Chen, M. Zhang, C. Liang and L. Duan, Catal. Sci. Technol., 2024, 14, 1712–1729 RSC.
- S. Wang, Z. Shen, A. Osatiashtiani, S. A. Nabavi and P. T. Clough, Chem. Eng. J., 2024, 486, 150170 CrossRef CAS.
- H. S. Bengaard, J. K. Nørskov, J. Sehested, B. S. Clausen, L. P. Nielsen, A. M. Molenbroek and J. R. Rostrup-Nielsen, J. Catal., 2002, 209, 365–384 CrossRef CAS.
- L. a. Garcia, R. French, S. Czernik and E. Chornet, Appl. Catal., A, 2000, 201, 225–239 CrossRef CAS.
- S. Mushtaq, J. Lee, F. Jamil, S. Imran, P. Akhter, M. Hussain and Y.-K. Park, Energy Environ., 2025, 36, 1063–1079 CrossRef.
- J. E. Lee, J. Lee, H. Jeong, Y.-K. Park and B.-S. Kim, Chem. Eng. J., 2023, 475, 146108 CrossRef CAS.
- A. Desgagnés and M. C. Iliuta, Chem. Eng. J., 2022, 429, 132278 CrossRef.
- C. Ratnasamy and J. P. Wagner, Catal. Rev., 2009, 51, 325–440 CrossRef CAS.
- A. Boudjemaa, C. Daniel, C. Mirodatos, M. Trari, A. Auroux and R. Bouarab, C. R. Chim., 2011, 14, 534–538 CrossRef CAS.
- L. Chen, C. K. S. Choong, Z. Zhong, L. Huang, T. P. Ang, L. Hong and J. Lin, J. Catal., 2010, 276, 197–200 CrossRef CAS.
- D. G. Rethwisch and J. A. Dumesic, Appl. Catal., 1986, 21, 97–109 CrossRef CAS.
- A. d. N. de Oliveira, I. M. Ferreira, D. E. Q. Jimenez, L. S. da Silva, A. A. F. da Costa, E. T. L. Lima, F. F. Costa, P. T. S. da Luz, G. N. d. R. Filho, S. M. Osman, R. Luque and L. A. S. d. Nascimento, Mol. Catal., 2022, 528, 112504 CrossRef CAS.
- J.-H. Kim, M. Kim, G. Park, E. Kim, H. Song, S. Jung, Y.-K. Park, Y. F. Tsang, J. Lee and E. E. Kwon, Biotechnol. Adv., 2024, 75, 108418 CrossRef CAS PubMed.
- D.-W. Lee and K.-Y. Lee, Catal. Surv. Asia, 2014, 18, 55–74 CrossRef CAS.
- S. Valizadeh, B. Valizadeh, J. Lee and Y.-K. Park, ChemCatChem, 2025, 17, e202401390 CrossRef CAS.
- S. Kim, E. E. Kwon, Y. T. Kim, S. Jung, H. J. Kim, G. W. Huber and J. Lee, Green Chem., 2019, 21, 3715–3743 RSC.
- P. Brimblecombe, M. Chu, C.-H. Liu, Y. Fu, P. Wei and Z. Ning, Atmos. Environ., 2023, 295, 119562 CrossRef CAS.
- S. Bhowmick, A. Badiwal and K. T. Shenoy, Chem. Eng. J. Adv., 2023, 15, 100511 CrossRef CAS.
- Y.-K. Park and B.-S. Kim, Chem. Eng. J., 2023, 461, 141958 CrossRef CAS.
- G. Li, B. Wang, H. Wang, J. Ma, W. Q. Xu, Y. Li, Y. Han and Q. Sun, Catal. Commun., 2018, 108, 82–87 CrossRef CAS.
- S. Xie, Y. Lu, K. Ye, W. Tan, S. Cao, C. Wang, D. Kim, X. Zhang, J. Loukusa, Y. Li, Y. Zhang, L. Ma, S. N. Ehrlich, N. S. Marinkovic, J. Deng, M. Flytzani-Stephanopoulos and F. Liu, Environ. Sci. Technol., 2024, 58, 12731–12741 CrossRef CAS PubMed.
- Q. Jiang, J. Zhang, H. Huang, Y. Wu and Z. Ao, J. Mater. Chem. A, 2020, 8, 287–295 RSC.
- S. Dey and G. C. Dhal, Mater. Sci. Energy Technol., 2020, 3, 6–24 CAS.
- S. Y. Lim, S. A. Younis, K.-H. Kim and J. Lee, Chem. Soc. Rev., 2024, 53, 9976–10011 RSC.
- A. Modak, P. Bhanja, S. Dutta, B. Chowdhury and A. Bhaumik, Green Chem., 2020, 22, 4002–4033 RSC.
- N. Czuma, K. Zarębska, M. Motak, M. E. Gálvez and P. Da Costa, Fuel, 2020, 267, 117139 CrossRef CAS.
- F. Rahimi, J. P. van der Hoek, S. Royer, A. Javid, A. Mashayekh-Salehi and M. J. Sani, J. Water Process Eng., 2021, 40, 101808 CrossRef.
- X. Guo, M. Zeng, H. Yu, F. Lin, J. Li, W. Wang and G. Chen, J. Cleaner Prod., 2024, 459, 142457 CrossRef CAS.
- J. Zhou, X. Ren, Z. Liu and S. Yuan, Mater. Today Sustainability, 2022, 20, 100225 CrossRef.
- C. M. Oliveira, C. M. Machado, G. W. Duarte and M. Peterson, J. Cleaner Prod., 2016, 139, 821–827 CrossRef CAS.
- M. Kebir, H. Tahraoui, I. K. Benramdane, N. Nasrallah, S. Toumi, J. Zhang and A. Amrane, Water Resour. Ind., 2024, 32, 100269 CrossRef CAS.
- A. Mashayekh-Salehi, K. Akbarmojeni, A. Roudbari, J. Peter van der Hoek, R. Nabizadeh, M. H. Dehghani and K. Yaghmaeian, J. Cleaner Prod., 2021, 291, 125235 CrossRef CAS.
- M. R. da Silva, D. L. Cunha, A. Kuznetsov, J. R. Araujo, A. Della-Flora, A. Dallegrave, C. Sirtori and E. M. Saggioro, J. Environ. Chem. Eng., 2024, 12, 111844 CrossRef CAS.
- H. D. Rojas-Mantilla, S. C. Ayala-Durán and R. F. P. Nogueira, J. Environ. Sci. Health, Part A, 2019, 54, 1277–1286 CrossRef CAS PubMed.
- F. Deng, H. Olvera-Vargas, M. Zhou, S. Qiu, I. Sirés and E. Brillas, Chem. Rev., 2023, 123, 4635–4662 CrossRef CAS PubMed.
- F. Cai, C. Sun, Z. Sun, Y. Lai and H. Ding, Appl. Surf. Sci., 2023, 623, 157044 CrossRef CAS.
- V. V. Figueiredo, R. P. F. Bonfim, M. A. Silva, C. R. Moreira, G. A. C. Pérez, W. A. Lourenço, B. R. Salles and V. M. M. Salim, Mater. Chem. Phys., 2025, 337, 130564 CrossRef CAS.
- H. Yang, S.-H. Jung, J. H. Lee, I. S. Cho, S.-J. Park and C.-G. Lee, J. Water Process Eng., 2024, 68, 106411 CrossRef.
- J. C. Mathews, F. P. da Costa Assunção, D. O. Pereira, J. C. C. da Silva, F. F. S. Almeida, A. C. P. Almeida, N. M. Mendonça, I. W. de Sousa Brandão, A. O. Menezes, L. E. P. Borges, J. F. H. Ferreira, J. A. R. Pereira and N. T. Machado, Sustainability, 2024, 16, 8370 CrossRef CAS.
- H. Yang, C.-G. Lee and J. Lee, J. Water Process Eng., 2023, 56, 104545 CrossRef.
- W.-D. Oh, A. Veksha, X. Chen, R. Adnan, J.-W. Lim, K.-H. Leong and T.-T. Lim, Chem. Eng. J., 2019, 374, 947–957 CrossRef CAS.
- Y. Liu, Y. Sun, M. Zhang, S. Guo, Z. Su, T. Ren and C. Li, J. Colloid Interface Sci., 2023, 629, 970–979 CrossRef CAS PubMed.
- J. S. Santos, M. Tarek, M. S. Sikora, S. Praserthdam and P. Praserthdam, J. Power Sources, 2023, 564, 232872 CrossRef CAS.
Footnote |
| † Co-first authors. |
| This journal is © The Royal Society of Chemistry 2025 |