
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImproved voltammetric discrimination of acetaminophen and uric acid in urine using CoO biochar nanocomposite†
Yihan
Zhang
a,
Yiliyasi
Baikeli
b,
Zehong
Gao
a,
Xamxikamar
Mamat
a and
Longyi
Chen
 *a
*a
aXinjiang Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Urumqi 830011, China. E-mail: chenly@ms.xjb.ac.cn
bCollege of Chemistry and Chemical Engineering, Dezhou University, Dezhou 253023, China
First published on 18th July 2024
Abstract
Overuse of acetaminophen (APAP) has become a severe societal burden in recent years. The rapid and reliable detection of urine APAP concentration can offer certain guidance for better management of APAP usage. This study explored the electrochemical sensing application of a novel electrocatalyst prepared from the biomass of Elaeagnus angustifolia gum. The biomass was first activated by ferric chloride to form a porous biomass carbon material (FBC). Then cobalt oxide (CoC) cracked nanoplate were synthesized by alkali precipitation and calcination and were then hybridized onto the biomass carbon via a simple sonication process. The electrocatalyst of CoO-FBC was characterized by scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDS), element mapping, transmission electron microscopy (TEM) and high resolution (HR-TEM), X-ray photoelectron spectroscopy (XPS), X-ray powder diffraction (XRD), thermogravimetric analysis (TGA), Raman spectroscopy, and nitrogen adsorption/desorption analysis. The CoO-FBC modified glassy carbon electrode (CoO-FBC/GCE) was characterized by various electrochemical methods including cyclic voltammetry (CV), electrochemical impedance spectroscopy (EIS) and differential pulse voltammetry (DPV). The CoO-FBC/GCE sensor was used to measure APAP in 0.1 M phosphate buffered saline (PBS) with a pH of 7.0, and with two linear sensing ranges from 1 μM to 10 μM and from 10 μM to 100 μM, with a sensitivity of 25.89 μA μM−1 cm−2 and 10.04 μA μM−1 cm−2, respectively, and a limit of detection of 0.46 μM. The unavoidable interference in measuring APAP is the inherent uric acid in urine. Uric acid and APAP exhibited adjacent and sometimes unseparable voltammetric peaks. This CoO-FBC/GCE sensor is capable of distinguishing APAP from uric acid and so APAP can be measured in human urine samples with good recoveries. This CoO-FBC/GCE sensor is a promising application for clinical diagnosis and environmental detection.
Keywords: Elaeagnus angustifolia gum; Ferric chloride; Polysaccharide biomass; Cobalt oxide nanoplate; Electrochemical sensing; Analgesic and antipyretic drug.
1 Introduction
Acetaminophen (N-acetyl-p-aminophenol, APAP, also commonly known as paracetamol) is a common ingredient used for analgesic and antipyretic effects. The excessive presence of APAP in human body fluids and contamination in environmental waters can heavily influence the human body and earth ecosphere.1–3 The U.S. Food and Drug Administration recommends that an adult dose of APAP is 4000 milligrams per day. The most notorious side effect of the overuse of APAP is acute liver damage. Besides hepatotoxicity, other potential risks may include pregnancy interference, rare skin reactions, and so on. Currently, multiple centralized laboratory methods can quantify APAP in various samples, e.g. high-performance liquid chromatography. However, such measurement requires use of expensive instrumentation and skilled personnel. Electrochemical sensing is promising in providing rapid, specific, inexpensive, internet integration, miniaturization and fully automated means for efficient clinical diagnoses and environmental detection.4,5Biomass represents a wide range of organic components, and sustainable and recyclable natural resources in the environment. Although more or less differences might exist in such huge biomass family, the major composition of biomass is carbon-based. Biomass derived carbon has found a multitude of applications in energy, sensing, biomedicine, the environment and catalysis.6–12Elaeagnus angustifolia gum is secreted in from tree trunks of Elaeagnus angustifolia L. and its major composition is polysaccharide. Our previous study revealed that the molecular weight of this polysaccharide is about 160 kDa. The E. angustifolia gum was originally used as a medicinal material. Here we explore the potential use of it as a biomass derived carbon material for electrochemical sensing applications. Biomass derived carbon has several characteristics including simple preparation, good conductance, high surface area and as a matrix for producing composites with nanomaterials, and so on.13,14
In order to achieve a larger surface area, several methods have been used to activate biomass carbon (BC) material, and a porous structure is usually formed after the activation. Traditional activation adopts a strong alkali etching approach, e.g. NaOH and KOH. In recent years, new activation chemicals were selected to activate biomass derived carbon material, including chloride salts like ZnCl2, FeCl3, and CaCl2, and so on. The intrinsic activating mechanism is complicated by the fact that the metal salts can form multiple materials, such as a metal hydroxide transforming into metal oxide by using a different calcination process. In addition to the chemical composition change and gas/liquid release, the activating agents may also expand or shrink and as a result, form a porous structure in the biomass material.15,16 For the FeCl3 activation, FeCl2, FeOCl, C3N4, and FeOOH, and so on, are formed during the calcination. The polysaccharide has a certain chemical reducibility and is able to interact with a variety of oxidative chemicals. The multiple iron species offer a complex reaction with polysaccharides which forms exquisite carbon structures.17
Transition metals have multiple orbital electrons and multiple valences which gives them a novel special catalytic function towards a mass of chemicals. Cobalt containing nanomaterials are one of the research topics for the degradation of various chemicals and sensing. Li and co-workers synthesized the Co–SnP composite, which can completely degrade APAP in Co–SnP/peroxymonosulfate system within 15 minutes. The catalyst of Co–SnP also exhibited excellent stability, reusability and universality.18 Kim and co-workers prepared a cobalt hexacyanoferrate incorporating a Fe-terephthalate metal organic framework as a robust and cost-effective acetaminophen sensing GCE modification material. The modified GCE sensor demonstrated excellent selectivity, sensitivity, stability, reproducibility.19 The Senthil Kumar group synthesized the highly uniform N-doped hollow carbon spheres with a uniform distribution of cobalt oxide materials and used them for the electrochemical sensing of APAP.20 Besides a cobalt nanomaterial for APAP sensing, the Mohamed group fabricated graphite carbon nickel nanoparticles with functionalized multiwalled carbon nanotubes for the detection of an immunomodulator drug, baricitinib, which showed a high precision in sensing baricitinib in plasma and medicinal formulations.21
Herein, we prepared the porous biomass carbon by ferric chloride activation of the gum derived from E. angustifolia using a simple pyrolysis procedure. To enhance the electrocatalytic property for use in measuring APAP concentrations, we synthesized cobalt hydroxide by alkali precipitation of Co2+ aqueous solution and calcined the cobalt hydroxide into a cobalt oxide cracked nanoplate. The sonication method was chosen to hybridize the cobalt oxide cracked nanoplate onto the porous biomass carbon material. This CoO-FBC/GCE sensor could sense APAP in a wide range of concentrations and was able to detect APAP in urine as well.
2 Results and discussion
2.1 Material characterization
Synthesized biomass derived carbon electrocatalyst materials of BC, FBC and CoO-FBC were checked using SEM, elemental mapping, TEM and HR-TEM techniques. As shown in Fig. 1a–c, the unactivated BC displays some mesopores on the surface, but a lot of solid parts remained. In comparison, the ferric chloride activated FBC shows pipe like etched micropores when compared to BC. A high degree of porous structure could be seen, and the inner parts are also highly porous. In the CoO-FBC SEM image, many cobalt oxide cracked nanoplates are attached onto the FBC's surface in random directions, demonstrating the successful incorporation of the CoO cracked nanoplate onto the FBC. In the magnified SEM image (Fig. S1†), irregular cracked holes are present in the hexagonal plate. These holes are assumed to come from the cobalt hydroxide shrinkage cracking during the calcination. These holes expose more cobalt atoms on the nanoplate surface. The hexagonal nanoplates have a side length of about 200 nm and a thickness of about 20 nm. In Fig. 1d–g a larger view of the electrocatalyst is shown for the elemental mapping. The CoO-FBC of (d) shows the presence of (e) the cobalt element, (f) the oxygen element and (g) the carbon element. The cobalt and oxygen elements ae mainly from the cobalt oxide cracked nanoplate. The inverse deep red carbon signal area reflects the biomass carbon material. The elemental mapping of BC and FBC, and the EDS spectra of all three biomass derived carbon materials can be found in Fig. S2.†Fig. 1h–j are the TEM images of BC, FBC and CoO-FBC, respectively. The BC TEM image shows the solid structure of the carbon material. Whereas the exterior part of the FBC is etched in an unorganized way as shown in Fig. 1i, the rich internal pipe-like hollow pore structure can still be clearly recognized. The CoO-FBC shows the successful hybridization of the CoO nanoplate on the FBC surface. The internal pipe-like hollow pores remained after the CoO decoration shown in Fig. 1j and the cracked nanoplate structure is clearly seen. The HR-TM image of the CoO in the CoO-FBC is shown in Fig. 1k, and the interplanar crystal spacing was measured and found to be 0.66 nm, which is in agreement with the (111) plane of the Co3O4 (PDF-#43-1003). The results of the XRD analysis are shown in Fig. 2c. | ||
| Fig. 1 SEM images of (a) BC; (b) FBC; (c) CoO-FBC; SEM image of (d) CoO-FBC and corresponding elemental mapping of (e) cobalt, (f) oxygen, (g) carbon; TEM images of (h) BC; (i) FBC; (j) CoO-FBC; (k) HR-TEM image of the enlarged CoO in CoO-FBC. Scale bar in (a–c) is 2 μm, in (d) is 10 μm, in (h–j) is 200 nm, and in (k) is 5 nm. | ||
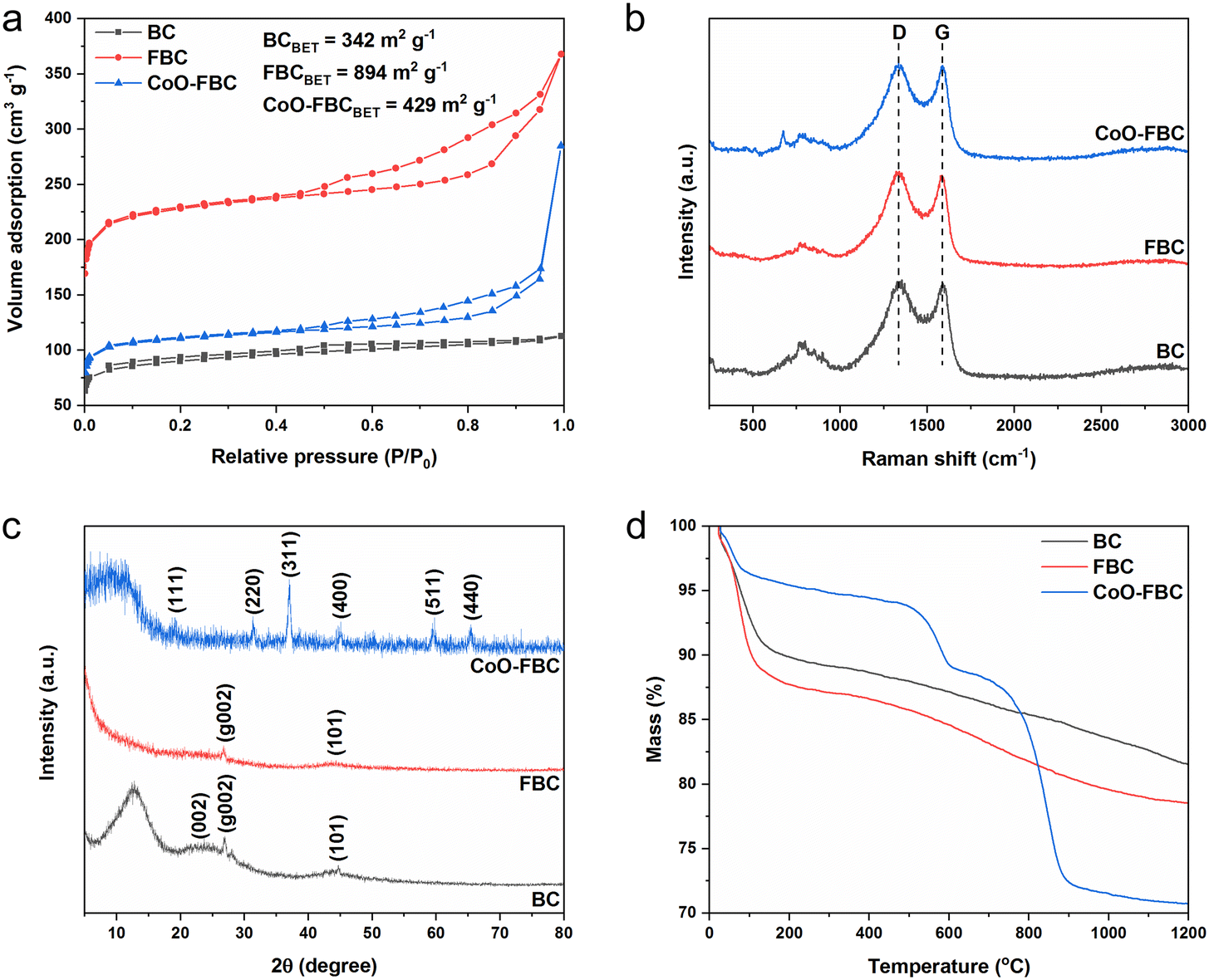 | ||
| Fig. 2 BC, FBC and CoO-FBC characterizations of (a) nitrogen adsorption/desorption curve and the calculated BET surface area; (b) Raman spectrum, with a laser wavelength of 532 nm; (c) XRD spectrum, Cu-Kα radiation, λ = 1.5406 Å; (d) TGA curves, 10 °C min−1, air flow rate of 30 mL min−1 combined with a nitrogen flow rate of 20 mL min−1. | ||
Fig. 2a shows the nitrogen adsorption/desorption curves of the prepared biomass derived materials. Obviously, all three carbon materials display characteristic features of type I and IV isotherms (according to IUPAC). The hysteresis loop is in the range of 0.45 to 1.0 (P/P0). The porous structure and the successful activation of the biomass carbon by the ferric chloride method are confirmed. The unactivated BC has the smallest calculated Brunauer–Emmett–Teller (BET) surface area of 342 m2 g−1, and the FBC has the largest calculated BET surface area of 894 m2 g−1, which is 2.6 times larger than the BC's surface area. After the use of sonication to incorporate the cobalt oxide onto the FBC, the CoO-FBC has a decreased calculated BET surface area of 429 m2 g−1. The decrease is assumed to come from the blocking by the cobalt oxide cracked nanoplate. From the measured pore size distribution (Fig. S3†), the mesopores with a size close to 17 nm are the major pores in the FBC and CoO-FBC, whereas the BC does not show any mesopores. After the nanoplate hybridization, the pore size near 17 nm is lowered.
Fig. 2b displays the Raman spectra of the biomass derived materials. The typical D band and G band of most carbon materials at around 1335 cm−1 and 1587 cm−1 are clearly seen. The D band signal represents the disordered carbon atom, originating from the carbon distortions and edges in the material structure. The G band signal represents the ordered graphitic carbon atom, originating from the vibration in the 2D hexagonal lattice of the sp2 carbon atom. Therefore, the band intensity ratio of ID/IG which reflects the disorderedness of the carbon atom in the carbon material. The ID/IG values of BC, FBC and CoO-FBC were 1.031, 1.036 and 1.026, respectively. After the activation, the ID/IG value of FBC increased slightly, and this came from the added etched porous inner pipeline holes and the outer surface. After the cobalt oxide nanoplate incorporation, the ID/IG value of CoO-FBC decreased to 1.026, which was smaller than that of BC. The diminution was speculated to come from the carbon edges forming physical and chemical bonds with the nanoplate, which reduce the carbon material's edges.22
Fig. 2c displays the XRD spectrum of the biomass derived materials. The BC shows the characteristic hard carbon diffraction peaks of (002), (g002) and (101) at 2θ of 22.7, 27.0 and 44.7, respectively. The (g002) is attributed to the crystallographic plane (002) reflection of graphitic carbon.23,24 As the BC has been calcined at 800 °C for 1 h, the polysaccharide may form certain sugar crystals and after calcination, transforms into graphitic carbon. The peak near the 2θ theta of 12.7 has rarely been seen reported in literature, and we only found one study about a cellulose biomass membrane that also exhibited this peak.25 We assume that this peak might be related to the sugar crystals formed during the fabrication procedure. The FBC still has the diffraction peaks of (g002) and (101), but the peak of (002) become much less apparent when compared to that of BC. The mesopores formed may cause this diminished peak. The CoO-FBC was discovered to agree well with the pattern of Co3O4 (PDF-#43-1003), showing the characteristic diffraction peaks of (111), (220), (311), (400), (511) and (440). The thermal stability of the biomass carbon material was checked using TGA, as shown in Fig. 2d. In order to resolve better of the material degradation process versus temperature increment, a total flow of 60% air and 40% nitrogen (protective gas) at the flow rate of 50 mL min−1 was used in the experiment. During the initial heating stage of 50 °C to 100 °C, the mass loss (about 10% loss) mainly came from the desorption of the physisorbed water. Starting from 200 °C, the BC and FBC decomposed slowly and stably until 1200 °C was reached, and about 80% of the mass still remained. This mass loss came from the burning of the carbon material, and the 40% protective gas slows down the burning speed. Rather than a quick and fast mass loss by using pure air, the adoption of partial protective gas resolved the thermal degradation curve better.
As can be seen from the TGA curve of the CoO-FBC, the mass loss explicitly requires detailed inspection. In the preparation of the CoO-FBC, an equal weight of FBC and CoO were added for the preparation. The CoO cracked nanoplate was calcined at 500 °C. Therefore, before 500 °C, the mass loss of FBC was about 2.5 times higher than the mass loss of CoO-FBC. For example, the mass loss of FBC at 200 °C, 300 °C and 400 °C was 12.28%, 12.91% and 13.41%, respectively, and the mass losses of CoO-FBC at 200 °C, 300 °C and 400 °C were 4.59%, 5.19% and 5.56%, respectively. At this range, the major mass loss came from the FBC part of the CoO-FBC. At the range from 500 °C to 600 °C, the CoO-FBC experienced a quick mass loss, and at the range from 700 °C to 900 °C, the CoO-FBC experienced another quick mass loss. The cobalt oxide is regarded as Co3O4 from the XRD spectrum, the composition of which is considered to be Co2+OCo23+O3. According to the mass loss calculation (Table S1†), the first mass loss is assumed to be from the reduction of the Co2+O component by the carbon, and the second mass loss is assumed from the reduction of the Co23+O3 component by the carbon. The reaction equation is described as:26
| C + Co2+OCo23+O3 → Co(s) + Cox(g) | (R1) |
The reaction is achieved at higher temperature (over 600 °C), but the nanomaterial condition of CoO may lower the reaction temperature to 600 °C. The separate reduction reaction may also be caused by the different reaction activation energy. The partial protective gas and flow rate may also lead to such a separate reduction procedure. At 1200 °C, the CoO-FBC has a mass percentage of about 70%.
The surface valence states of the characteristic elements of the biomass derived carbon materials were analysed using the XPS measurements. Fig. 3a shows the XPS survey scans of the biomass carbon materials, and all of them share the peaks of C 1s and O 1s at 284 eV and 530 eV, respectively. The CoO-FBC additionally displays the Co 2p peaks at 780 eV and 795 eV. The elemental contents measured from XPS are listed in Table S2,† and the minute N element is assigned to the inherent element in the E. angustifolia gum. Whereas the minute chloride element is supposed to be a residual from the hydrochloride washing. Fig. 3b–d show the XPS spectrum of CoO-FBC. In Fig. 3b, the two Co 2p peaks could be deconvoluted into two peaks. The two fitted peaks for Co 2p3/2 were assigned to Co3+ (779 eV) and Co2+ (781 eV). The peak of Co 2p1/2 was assigned to Co3+ (794 V) and Co2+ (796 eV). In the hydrothermal synthesis of cobalt hydroxide, the cobalt salt used is CoCl2, then after the calcination at 500 °C, the cobalt hydroxide was dehydrated and became partially oxidized into Co3O4. Therefore, the Co2+ and Co3+ valence states were detected. This result is also in accordance with the XRD spectrum and TGA analysis. There were two satellite peaks centred at approximately 788 eV and 804 eV. These satellite peaks belong to the Co2+ oxidation state, and thus confirm the chemical nature of the CoO.27
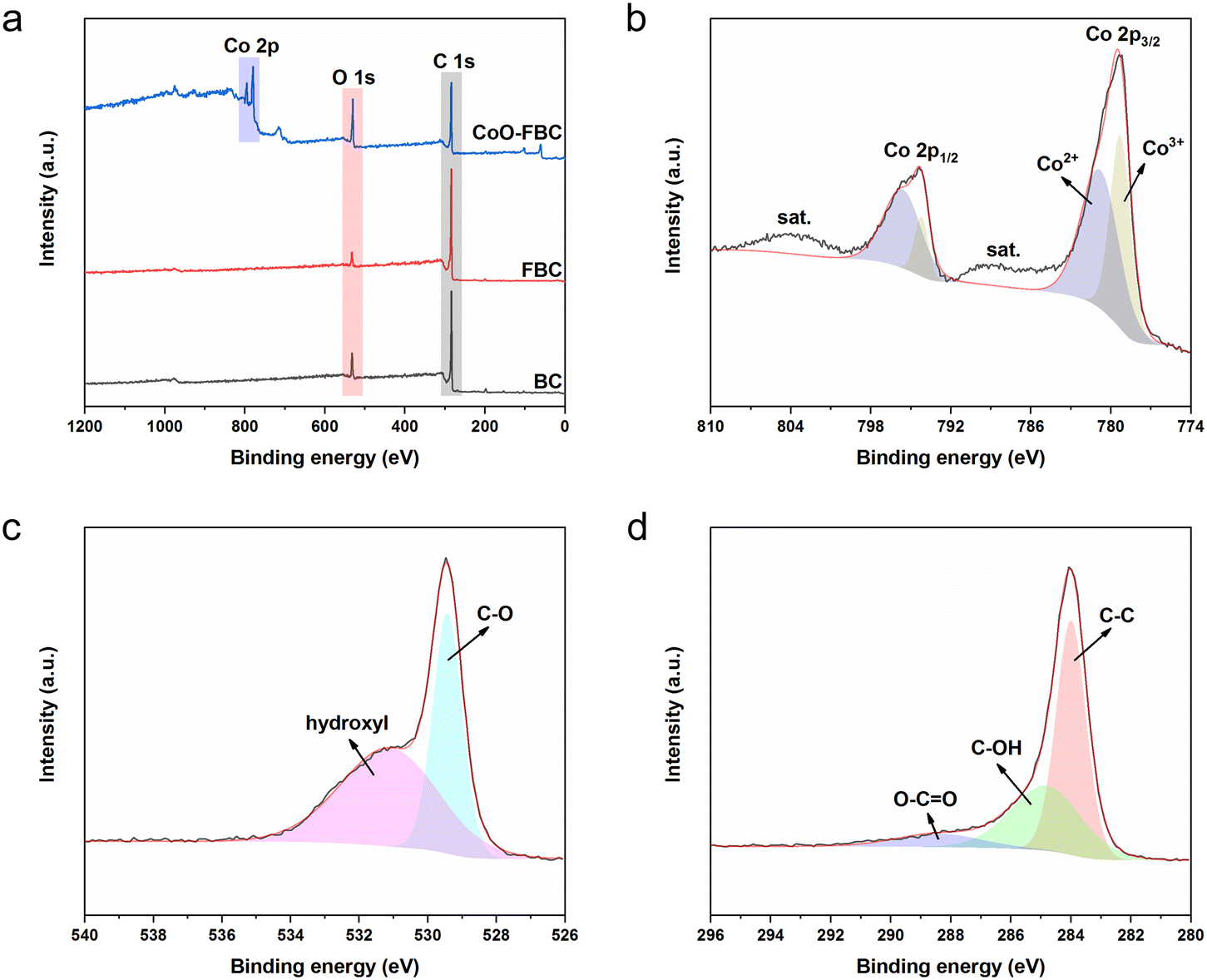 | ||
| Fig. 3 XPS (a) survey scan spectra of BC, FBC and CoO-FBC; (b) Co 2p scan of CoO-FBC; (c) O 1s scan of CoO-FBC; (d) C 1s scan of CoO-FBC. | ||
Fig. 3c displays the O 1s scan of CoO-FBC, where the peak is divided into two peaks at 529 eV and 531 eV. The 529 eV peak is related to the Co–O bond, whereas the 531 eV is related to the hydroxyl species. Fig. 3d shows the C 1s scan of the CoO-FBC sample, and the three deconvoluted peaks are at 284 eV, representing the C–C bond, at 284.9 eV, representing the C–OH bond, and at 288.6 eV, representing the O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond.28,29 It is assumed that after the sonication assisted incorporation of CoO onto FBC, some of the ethanol molecule may interact with the cobalt oxide cracked nanoplate and cause such a signal. Also, the calcination at 500 °C may also leave some cobalt hydroxide in the nanoplate.
O bond.28,29 It is assumed that after the sonication assisted incorporation of CoO onto FBC, some of the ethanol molecule may interact with the cobalt oxide cracked nanoplate and cause such a signal. Also, the calcination at 500 °C may also leave some cobalt hydroxide in the nanoplate.
2.2 Electrochemical characterization
The biomass derived carbon materials are fabricated onto a GCE for electrochemical analysis. Fig. 4a shows the CV spectra of bare GCE and the fabricated GCE in [Fe(CN)6]3−/4− solution. The Ipa for bare GCE, BC/GCE, FBC/GCE and CoO-FBC/GCE were 78.73 μA, 89.25 μA, 97.68 μA and 95.74 μA, respectively. The unactivated BC/GCE CV curve resembles the bare GCE CV curve, but with a slightly increasing Ipa. The FBC/GCE shows an apparently larger CV area and an increasing Ipa. The enlarged CV area originates from the porous structure and the high surface area, whereas the CoO-FBC/GCE has a slightly smaller Ipa than that of FBC/GCE. The reason for this may be due to the coverage of the FBC surface by the cobalt oxide cracked nanoplate and decreasing of the material surface area.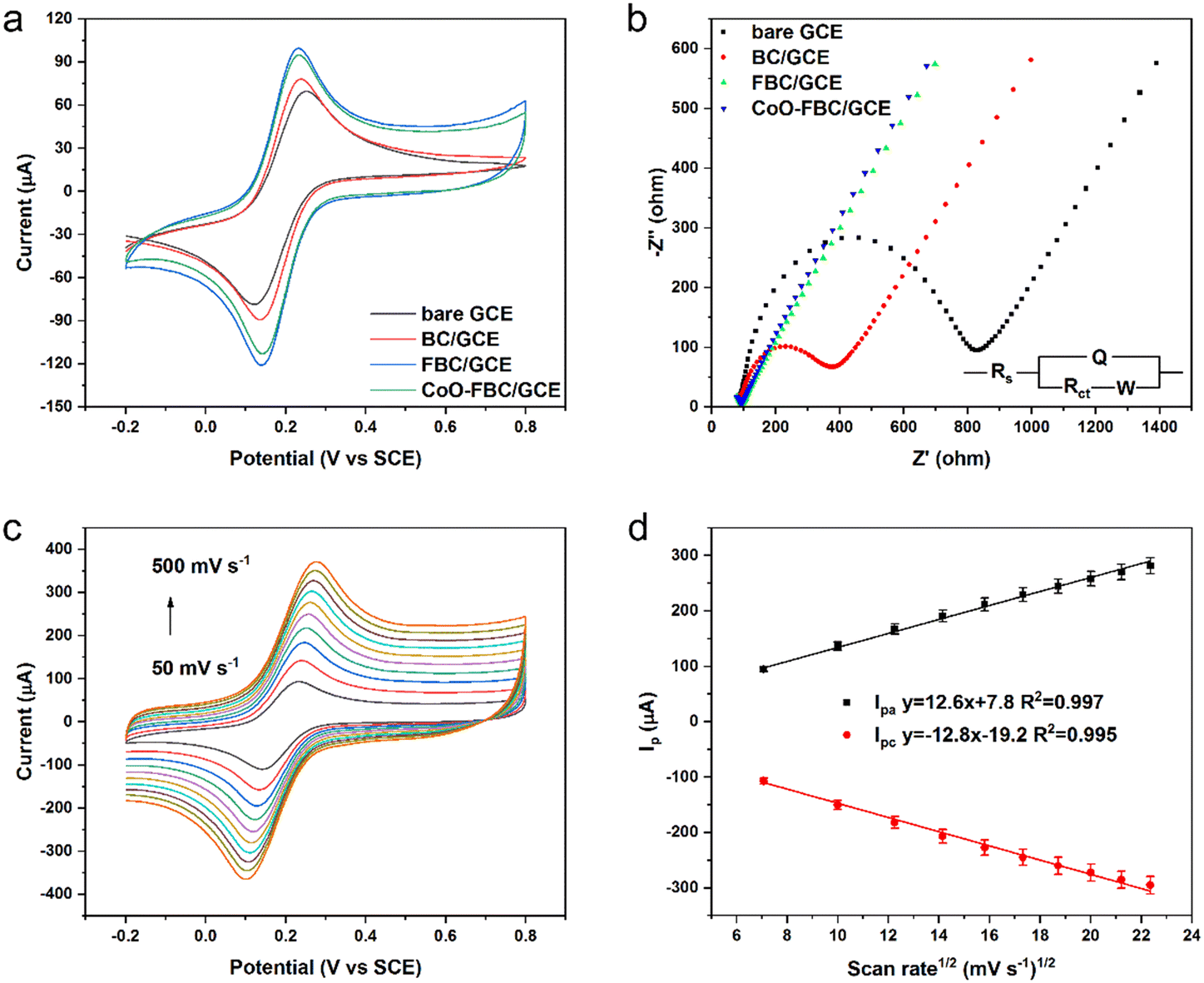 | ||
| Fig. 4 (a) CV spectra of the bare GCE and biomass derived carbon electrocatalyst modified GCE in 5 mM [Fe(CN)6]3−/4−, 0.1 M KCl, and a scan rate of 50 mV s−1; (b) EIS spectra of the bare GCE and biomass derived carbon electrocatalyst modified GCE in 5 mM [Fe(CN)6]3−/4−, 0.1 M KCl (frequency range from 100 kHz to 0.1 Hz); (c) the CoO-FBC/GCE CV spectrum in 5 mM [Fe(CN)6]3−/4−, 0.1 M KCl with a changing scan rate; (d) the linear fit of the Ipa and Ipc current versus the square root of the scan rate in (c). | ||
The corresponding GCEs were also tested using EIS analysis as shown in Fig. 4b. The high-frequency region is zoomed in and shown in Fig. S4.† The charge transfer resistance Rct of bare GCE, BC/GCE, FBC/GCE and CoO-FBC/GCE were simulated and calculated to be 710.6 ohm, 303.2 ohm, 8.1 ohm and 4.9 ohm, respectively. Although the BC is unactivated, the modification of the biomass carbon is still able to lower the interface resistance. The FBC/GCE and CoO-FBC/GCE have equivalent Rct and these are far smaller than those of the other two GCEs. The incorporation of the cobalt oxide cracked nanoplate did not affect the FBC's functionality too much. Although the nanoplate sits on the FBC surface, the hollow holes may still provide sufficient contact for the reactants in the solution when assessing the biomass carbon. As cobalt oxide is a semi-conductor material, it showed a limited influence on the conductance of the CoO-FBC biomass carbon.
In Fig. 4c, the CV spectra of the CoO-FBC/GCE was obtained with varying scan rates (100 mV s−1, 150 mV s−1, 200 mV s−1, 250 mV s−1, 300 mV s−1, 350 mV s−1, 400 mV s−1, 450 mV s−1 and 500 mV s−1). The redox pair [Fe(CN)6]3−/4− showed the redox peaks' potential positions to be at about 0.23–0.28 V and at 0.10–0.14 V. As the scan rate became faster, the CV spectrum showed a larger redox potential difference and a larger response current. Fig. 4d is the linear fit relationship between the Ipa and Ipc currents and the square root of the scan rate in Fig. 4c, indicated that the redox reaction of [Fe(CN)6]3−/4− electrocatalyzed by CoO-FBC was a diffusion controlled process. The linear fit R2 of both Ipa and Ipc were 0.997 and 0.994. The electrochemically active surface area of CoO-FBC was calculated by the Randles–Sevcik equation:30
| Ip = 2.69 × 105A·D1/2·n3/2·v1/2C | (1) |
The electrochemically active surface area of the bare GCE, BC/GCE, FBC/GCE and CoO-FBC/GCE sensor towards [Fe(CN)6]3−/4− were calculated by an average value using both Ipa and Ipc to give 0.104 cm2, 0.117 cm2, 0.135 cm2, 0.130 cm2, respectively. Compared to the bare GCE, the modified GCEs all had a larger electrochemically active surface area. In particular, the CoO-FBC/GCE had a slightly decreased value than that of FBC/GCE, which was caused by the coverage of the cobalt oxide cracked nanoplate on FBC.
2.3 Electrochemical analysis of APAP
As shown in Scheme 1, the chemical formula of APAP is C8H9NO2, and it can be electrochemically catalysed into N-acetyl-4-benzoquinone imine (C8H7NO2) at the oxidation potential near to the 0.3 V to 0.4 V range, together with 2 hydrogen atoms and 2 electrons were lost. This process is reversible with N-acetyl-4-benzoquinone imine reduced back to APAP in the potential near to the 0.2 V to 0.3 V region.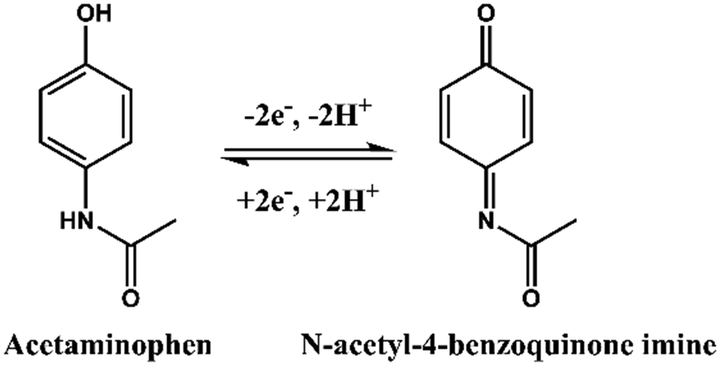 | ||
| Scheme 1 Electrochemical redox reaction of acetaminophen and N-acetyl-4-benzoquinone imine. | ||
To study the electrochemical response of APAP, the CV spectra of bare GCE and the fabricated GCE in APAP solution were measured as shown in Fig. 5a. Fig. S5† shows the CV spectra of the bare GCE and biomass derived carbon electrocatalyst modified GCE in 0.1 M PBS solution, at a scan rate of 50 mV s−1. The CV areas slowly increased in the sequence of bare GCE, BC/GCE, FBC/GCE and CoO-FBC/GCE. The Ipa for bare GCE, BC/GCE, FBC/GCE and CoO-FBC/GCE were 9.99 μA, 10.62 μA, 25.66 μA and 57.87 μA, respectively. The cobalt atom is considered to possess a specially enhanced electrocatalytic interaction towards the APAP molecule, which explains the intensified Ipa current response.31
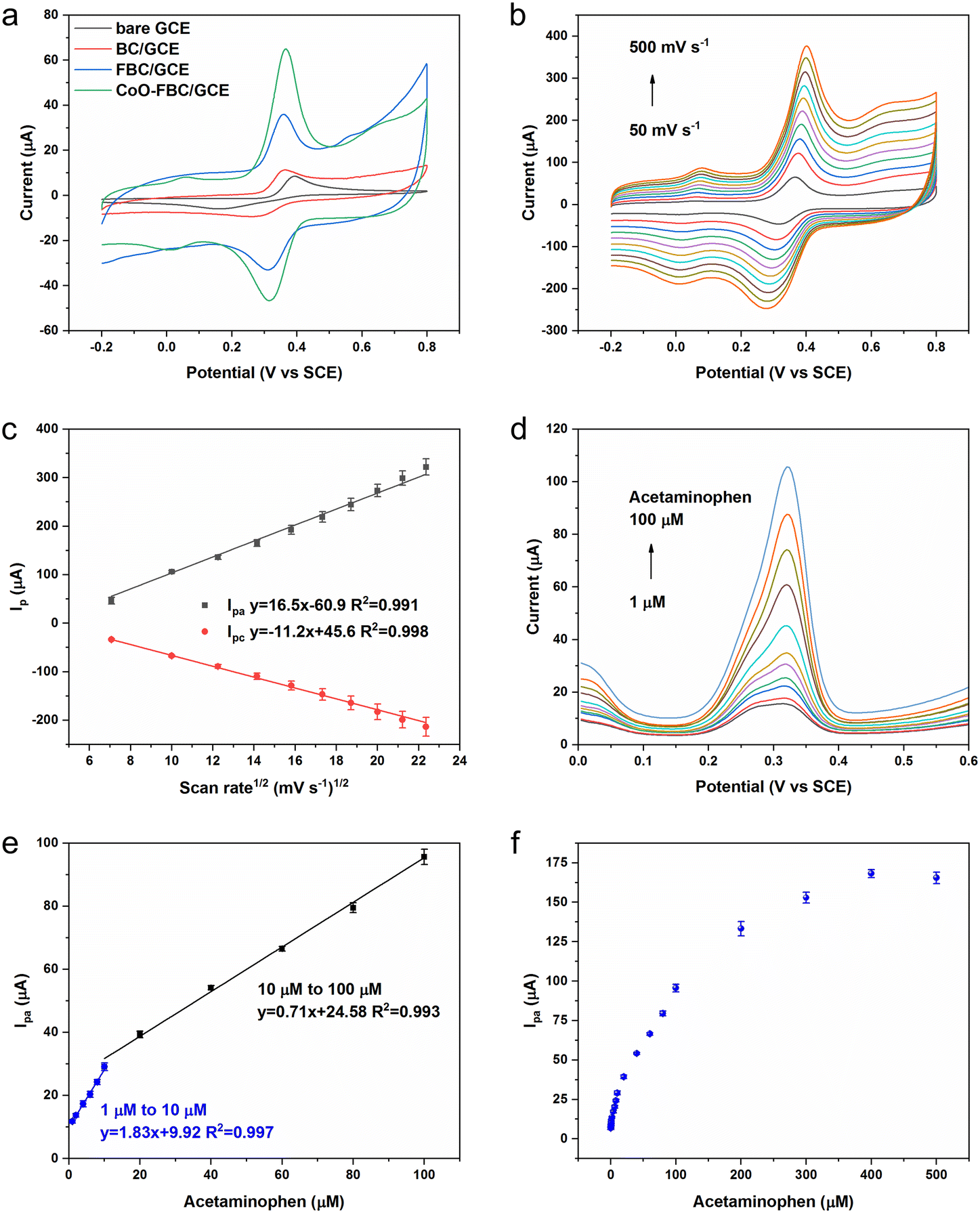 | ||
| Fig. 5 (a) CV spectra of the bare GCE and biomass derived carbon electrocatalyst modified GCE in APAP solution (500 μM in 0.1 M PBS, scan rate 50 mV s−1); (b) CoO-FBC/GCE CV spectrum in 500 μM APAP in 0.1 M PBS with a changing scan rate; (c) the linear fit of the Ipa and Ipc current versus the square root of the scan rate in (b); (d) DPV measurement of APAP in 0.1 M PBS with varying concentrations of APAP from 1 μM to 100 μM; (e) linear fit of DPV Ipa current versus the APAP concentrations in (d); (f) DPV Ipa current of APAP solution from 1 μM to 500 μM. | ||
The cracks in the nanoplate increase the surface cobalt atom exposure to the APAP molecule. The cobalt oxide is a semiconductor and is able to transfer the electrons efficiently to the biomass carbon matrix. All of these contribute to the almost doubled Ipa current response compared to the Ipa current response of FBC/GCE. Fig. 5b and c show the changing scan rate of the CoO-FBC/GCE CV spectra and the linear fit relationship between the Ipa and Ipc current and the square root of the scan rate in the APAP solution. Similar to the CoO-FBC/GCE CV scan in [Fe(CN)6]3−/4− solution, the CV scans of CoO-FBC/GCE in the APAP solution also show that the redox reaction of APAP electrocatalyzed by CoO-FBC is also a diffusion controlled process. Based on eqn (1), the electrochemically active surface area of the bare GCE, BC/GCE, FBC/GCE and CoO-FBC/GCE sensor towards APAP were calculated using the Ipa current response and found to be 0.013 cm2, 0.014 cm2, 0.033 cm2, 0.075 cm2, respectively. In Fig. 5b, there is a pair of redox peaks at about 0.01 V and 0.08 V, which are obvious at high scan rates. As discussed in the materials characterization section, the CoO is assumed to be Co3O4 (PDF-#43-1003). This redox peak is assumed to come from the oxidation and reduction from the Co2+ and Co3+ ions on the CoO nanoplate surface. There is also another oxidation peak of 0.65 V at high scan rates, which may be related to the Co2+/Co3+ redox process. The interaction of Co2+/Co3+ redox with the biochar may also form such peaks.32,33
Fig. 5d and e display the DPV measurement of CoO-FBC/GCE towards various concentrations of APAP solutions. Two linear ranges from 1 μM to 10 μM and 10 μM to 100 μM were found, with a sensitivity of 25.89 μA μM−1 cm−2 and 10.04 μA μM−1 cm−2, respectively, with a limit of detection of 0.46 μM. In the low APAP concentrations, the APAP oxidation peak of 0.32 V is deconvoluted into a mixture of peaks near 0.28 V to 0.32 V. We assumed that this might also come from the electrocatalyst multicomponent nanostructures. There were CoO nanoplates and biomass carbon present in the electrocatalyst surface. The APAP molecule might be oxidized at the nanoplate surface and the biomass carbon surface, thus resulting in the mixture of peaks around the range of 0.28 V to 0.32 V. Also different catalytic capabilities of the multicomponents existed and would also contribute to such a phenomenon. Fig. 5f shows the Ipa current of the DPV measurement. As the APAP concentration increases from 100 μM to 500 μM, the current response slowly became saturated and the 500 μM APAP signal shows a slightly decreasing value. This slight saturation trend may be caused by the overwhelming number of APAP molecules at the electrocatalyst surface, leading to the lowering of the electrocatalytic reaction.
As discussed in our previous study, the average male urine APAP concentration is recognized as 200 μM. The average male urine uric acid (UA) concentration is recognized to be 2000 μM. Uric acid is a frustrating interference when carrying out the electrochemical sensing of urine APAP concentrations.34 Because urine also contains a number of other biomolecules and inorganic components, the use of diluted urine should alleviate the potential sensing noise. We would use male urine diluted 10 times for the real sample measurement and based on the 10 times dilution, we would choose certain chemical concentrations. In the selectivity experiments, we chose the interfering chemicals to be uric acid, ascorbic acid, urea and glucose using equivalent concentrations of 200 μM. The average uric acid in the 10 times diluted urine was set as 20 μM. The average uric acid in the 10 times diluted urine was set as 200 μM. The selectivity experiment used the simulated 10 times diluted urine that was a much cleaner solution, and the APAP concentration was set as 10 μM.
Fig. 6a shows the DPV curves for the selected interferences. The ascorbic acid, urea and glucose show no response in the potential range. The APAP molecule is oxidized at the potential near 0.31 V, whereas the uric acid molecule is oxidized at the potential near 0.25 V. The uric acid peak and the APAP peak could be separated distinctly in the all the interference mixtures (APAP, uric acid, ascorbic acid, urea and glucose) and the mixture of APAP and uric acid solution. The current response may have been affected a little, but the deviation can be corrected by reconstruction under the designed conditions and applications. Fig. 6b displays the DPV curve of the diluted urine samples spiked by APAP. The diluted urine provides a current response near 0.24 V, which is probably coming from the uric acid. The spiked APAP (100 μM) urine sample exhibited two current responses, of which one is from the uric acid near 0.24 V, and the other one is from the APAP near 0.32 V. If the original urine APAP is in the range from 100 μM to 1 mM, the designed CoO-FBC/GCE would be capable of detecting the 10 times diluted urine APAP. Considering the situation of abuse of APAP intake, and considering the uric acid interference, the CoO-FBC/GCE sensor could fulfil the needs of clinical diagnosis. Further improvements can be done to enhance the CoO-FBC/GCE performance. For example, the biomass activation procedure, the cobalt oxide cracked nanoplate size and morphology, the sonication step and the biomass carbon to cobalt oxide ratio, co-doping with other transitional elements by alkali co-precipitation and so on could be all be considered for an in-depth study to achieve sensors with superior capability.
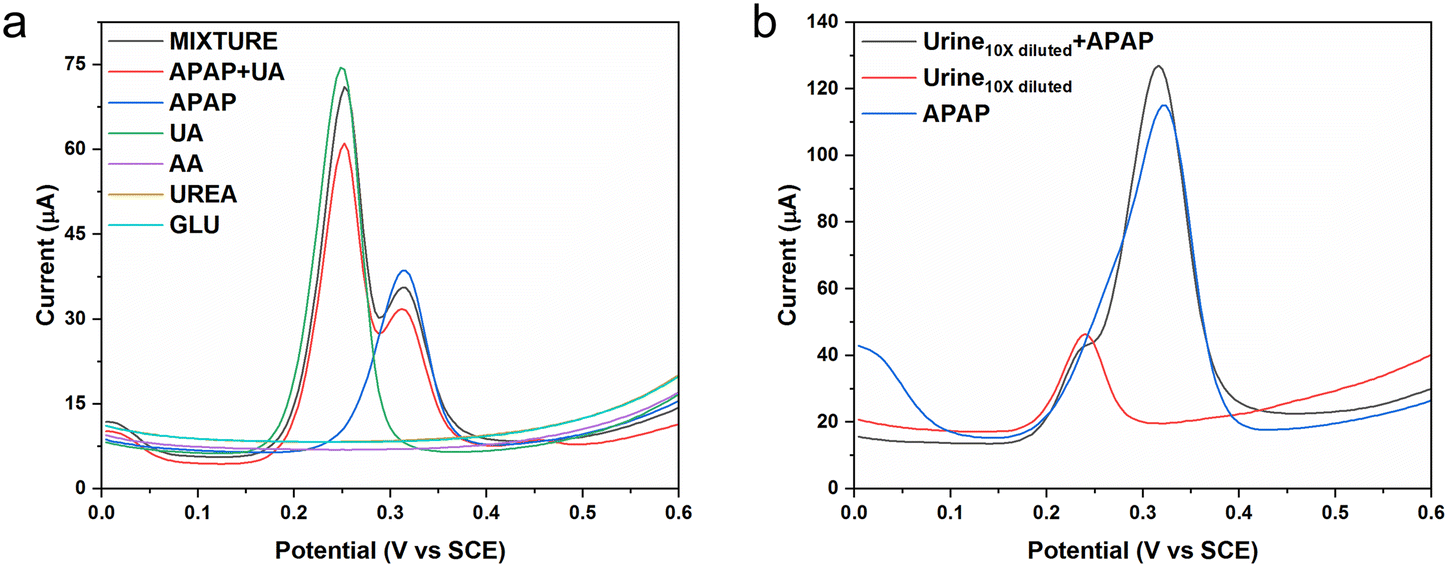 | ||
| Fig. 6 (a) The selectivity DPV measurement of the CoO-FBC/GCE against interferences of uric acid (UA), ascorbic acid (AA), urea (UREA) and glucose (GLU), all the interference solutions concentration are 200 μM in 0.1 M PBS, and the APAP concentration is 10 μM in 0.1 M PBS; (b) the DPV measurement of the urine sample, where the urine is diluted 10 times in 0.1 M PBS, and the APAP concentration is 100 μM in 0.1 M PBS. | ||
The cycling experiment result is shown in Fig. S6.† After 10 continuous CV cycles, the CoO-FBC/GCE still obtained about 82.1% of its first sensing signal. Also, the electrocatalyst was stored in a general room environment for over 3 months, there was no negative effect on the use of the sensor, and because the major composition of the electrocatalyst is cobalt oxide and carbonized biochar, the sensor remains very stable. The modified CoO-FBC/GCE could recover a similar current signal when compared with the results of experiments carried out 3 months previously. We think the CoO-FBC/GCE sensor has good stability, reproducibility, repeatability.
2.4 Comparison of other works
A survey of related research on nanomaterials' electrochemical sensing of APAP is summarized in Table 1. From the recent relevant studies, we can see various nanomaterials have been used, including a series of carbonaceous materials such as carbon nanotubes, graphene oxide, cellulose, porous carbon nanorods and so on. In addition, carbon containing materials from the pyrolysis of metal organic frameworks, and a series of nanomaterials using gold nanoparticles, zinc oxide nanoparticles, transition element co-doped nanomaterial, bi-metallic transition metal alloy, cobalt–zinc metal organic framework derived material, quantum dots and so on, have been hybridized and utilized for electrochemical catalytic sensing of APAP in a number of applications. Compared to the cited references, the biomass of E. angustifolia gum is a natural accessible abundant resource, and the hollowness of the cobalt oxide cracked nanoplate can be modulated by modifying the fabrication procedures. No complicated preparation steps are involved. The sensitivity of the CoO-FBC/GCE sensor is good. It is also worth mentioning that in this work, the uric acid response peak and the APAP response peak could be separated well and recognized in the rational physiological concentration range.| Electrocatalyst | Sensing range (μM) | Limit of detection (nM) | Sensitivity (μA μM−1 cm−2) | Applications | Ref. |
|---|---|---|---|---|---|
| COF@AuNPs@MWCNTs | 0.1 to 500, 500 to 1200 | 80 | 0.72, 0.30 | Human serum | 35 |
| Gd2ZnMnO6/ZnO | 0.090 to 900 | 25 | 2.75, 0.38 | Human urine, plasma and pharmaceutics | 36 |
| FeM@porous carbon nanorod | 0.001 to 0.8 | 0.59 | 540.70 | Pharmaceutics | 37 |
| ZnONPs/SBA-15 | 4 to 32 | 110 | 0.69 | Pharmaceutics | 38 |
| CoNC@rGO | 0.5 to 50 | 67 | 8.46 | Environmental water | 39 |
GO/MnO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :h-MoO3 :h-MoO3 |
0.06 to 10, 20 to 80 | 13.3 | 28.11, 18.27 | Pharmaceutics | 40 |
| Laser-induced rGO | 0.099 to 1987 | 5.2 | 2.73 | River water, pharmaceutics and human urine | 41 |
| MWCNT and poly(neutral red) | 2 to 70 | 15 | 56.40 | Pharmaceutics | 42 |
| Ag–Fe3O4/nanocellulose | 0.05 to 15 | 8 | 152.31 | Human blood and urine | 43 |
| Ti3C2 QDs/Fe-NC | 0.50 to 210 | 30 | 6.53 | Pharmaceutics and lake water | 44 |
| CoO-FBC | 1 to 10, 10 to 100 | 460 | 10.04, 25.89 | Human urine | This work |
3 Conclusions
We have developed a novel cobalt oxide cracked nanoplate decorated on biomass derived carbon electrocatalyst material for electrochemical sensing of APAP. The biomass derived carbon is prepared by the ferric chloride activation of E. angustifolia gum, a major composition of which is polysaccharides. The cobalt oxide cracked nanoplate is synthesized by alkali precipitation of cobalt chloride in aqueous solution and then by calcination. Simple sonication assisted hybridization of biomass carbon and cobalt oxide cracked nanoplate is adopted. The prepared CoO-FBC/GCE exhibits two linear sensing range from 1 μM to 10 μM and 10 μM to 100 μM, with sensitivities of 25.89 μA μM−1 cm−2 and 10.04 μA μM−1 cm−2, and a limit of detection of 0.46 μM. This CoO-FBC electrocatalyst is sufficient for sensing 10 times diluted APAP spiked urine samples and shows promise for integration into a disposable sensor for clinical diagnosis.4 Experimental
4.1 Chemicals
All the chemicals used were of analytical grade and used as received. Acetaminophen and phosphate buffered saline tablets are from Aladdin. The PBS solution used throughout the experiments is prepared using the PBS tablets. Ferric chloride hexahydrate (FeCl3·6H2O) and potassium ferricyanide (K3Fe(CN)6) are from the Tianjin Baishi Chemical Co., Ltd. The cobalt chloride (CoCl2) is from Alfa Aesar. Sodium hydroxide (NaOH) and hydrogen chloride (HCl) are from Tianjin Xinbote Chemical Co., Ltd. Urea is from the Tianjin Tianda Fine Chemical Plant. Potassium chloride is from Tianjin Bodi Chemical Co., Ltd. Glucose and potassium ferrocyanide trihydrate (K4Fe(CN)6·3H2O) are from the Tianjin Hedong District Hongyan Reagent Factory. Uric acid and ascorbic acid are from Shanghai Macklin Biochemical Technology Co., Ltd. The GCE, platinum wire electrode, platinum plate electrode, saturated calomel electrode (SCE) and aluminium slurry are from Wuhan Gaoss Union Technology Co., Ltd. All the aqueous solutions were prepared with water (18.2 MΩ cm) from a Millipore system. Dried E. angustifolia gum was provided by the Key Laboratory of Chemistry of Plant Resources in Arid Regions, and the State Key Laboratory Basis of Xinjiang Indigenous Medicinal Plants Resource Utilization. The human urine sample was provided by a healthy male volunteer in the laboratory, who did not consume any medicine containing APAP for at least a month before providing the urine.4.2 Preparation procedures
4.3 Fabrication of GCE
Before modification, the GCE (3 mm in diameter) was polished with 0.5 μm and 0.05 μm alumina slurries to obtain a mirror-like surface. The electrode was then cleaned in ethanol and water by sonication and dried in an ambient environment. The corresponding electrocatalyst aqueous solution (2 mg mL−1) was sonicated for 30 min to form a homogeneous solution, and then 5 μL of the solution was dropped onto the GCE surface and dried in an ambient environment.4.4 Instruments
A field-emission scanning electron microscope (Zeiss MERLIN VP Compact, Germany) equipped with an energy-dispersive X-ray spectrometer (Oxford Instruments X-Max, UK) was used to characterize the morphology, elemental composition and elemental mapping of the electrocatalyst. X-ray photoelectron spectroscopy (Thermo Scientific K-Alpha, USA) was used to perform the surface analysis of the electrocatalyst. Raman spectra of the samples were obtained using a spectrometer (Horiba LabRAM HR800, France). The thermogravimetric analysis was performed using a Netzsch STA 449 F5 Jupiter, Germany and a Thermo Fisher Scientific Nicolet iS50 FTIR spectrometer, USA. The X-ray powder diffraction measurements were recorded on a diffractometer (Bruker D8 Advance, Germany, Cu-Kα radiation). The nitrogen adsorption/desorption analysis was carried out using a Quantachrome Autosorb IQ, USA.An electrochemical workstation (CHI-760E, Shanghai, Chenhua, China) and a workstation (RST 5000C, Zhengzhou Shiruisi Instrument Technology Co., Ltd, Zhengzhou, China) were used in the electrochemical experiments. A three-electrode system was used for the CV and DPV scans, with an SCE, a platinum wire electrode, and GCE. For the EIS measurements, a three-electrode system of an SCE electrode, platinum plate electrode (10 mm × 10 mm × 0.1 mm), and a GCE were used.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Biological Resources Programme, Chinese Academy of Sciences (KFJ-BRP-007-011). Y. Z., Z. G. and L. C. acknowledge support from the Xinjiang Uygur Autonomous Region's Tianchi Talent Program.References
- S. van den Driesche, J. Macdonald, R. A. Anderson, Z. C. Johnston, T. Chetty, L. B. Smith, C. McKinnell, A. Dean, N. Z. Homer, A. Jorgensen, M. E. Camacho-Moll, R. M. Sharpe and R. T. Mitchell, Prolonged exposure to acetaminophen reduces testosterone production by the human fetal testis in a xenograft model, Sci. Transl. Med., 2015, 7, 288ra280 CrossRef.
- H. Jaeschke and A. Ramachandran, Acetaminophen hepatotoxicity: Paradigm for understanding mechanisms of drug-induced liver injury, Annu. Rev. Pathol.: Mech. Dis., 2024, 19, 453–478 CrossRef CAS.
- J.-l. Wu, Z.-h. Liu, Q.-g. Ma, L. Dai and Z. Dang, Occurrence, removal and risk evaluation of ibuprofen and acetaminophen in municipal wastewater treatment plants: A critical review, Sci. Total Environ., 2023, 891, 164600 CrossRef CAS PubMed.
- Q. Han, H. Wang and J. Wang, Multi-mode/signal biosensors: Electrochemical integrated sensing techniques, Adv. Funct. Mater., 2024, 2403122 CrossRef CAS.
- R. Singh, R. Gupta, D. Bansal, R. Bhateria and M. Sharma, A review on recent trends and future developments in electrochemical sensing, ACS Omega, 2024, 9, 7336–7356 CAS.
- R. S. Varma, Biomass-derived renewable carbonaceous materials for sustainable chemical and environmental applications, ACS Sustainable Chem. Eng., 2019, 7, 6458–6470 CrossRef CAS.
- Y. Yin, Q. Liu, Y. Zhao, T. Chen, J. Wang, L. Gui and C. Lu, Recent progress and future directions of biomass-derived hierarchical porous carbon: Designing, preparation, and supercapacitor applications, Energy Fuels, 2023, 37, 3523–3554 CrossRef CAS.
- L. Li, Y. Zhou, X. Xiao, Z. Chen, Z. Zhou, H. Deng, W. Deng, Y. Xu, G. Li, J. Zhang, X. Hu and Y. Wang, 3D biomass-derived carbon materials for electrochemical biosensors, Adv. Mater. Technol., 2023, 8, 2300666 CrossRef CAS.
- X. Xiao, L. Li, H. Deng, Y. Zhong, W. Deng, Y. Xu, Z. Chen, J. Zhang, X. Hu and Y. Wang, Biomass-derived 2D carbon materials: Structure, fabrication, and application in electrochemical sensors, J. Mater. Chem. B, 2023, 11, 10793–10821 RSC.
- S. Yu, J. He, Z. Zhang, Z. Sun, M. Xie, Y. Xu, X. Bie, Q. Li, Y. Zhang, M. Sevilla, M.-M. Titirici and H. Zhou, Towards negative emissions: Hydrothermal carbonization of biomass for sustainable carbon materials, Adv. Mater., 2024, 36, 2307412 CrossRef CAS PubMed.
- B. K. John, J. Mathew, S. K, R. E. K and B. Mathew, Biomass derived carbon quantum dots as a versatile platform for fluorescent sensing, catalytic reduction, fluorescent ink and anticancer agents, Mater. Today Sustain., 2024, 26, 100715 Search PubMed.
- Y.-C. E. Li, Sustainable biomass materials for biomedical applications, ACS Biomater. Sci. Eng., 2019, 5, 2079–2092 CrossRef CAS.
- Y. Wang, M. Zhang, X. Shen, H. Wang, H. Wang, K. Xia, Z. Yin and Y. Zhang, Biomass-derived carbon materials: Controllable preparation and versatile applications, Small, 2021, 17, 2008079 CrossRef CAS.
- H. Weldekidan, A. K. Mohanty and M. Misra, Upcycling of plastic wastes and biomass for sustainable graphitic carbon production: A critical review, ACS Environ. Au, 2022, 2, 510–522 CrossRef CAS PubMed.
- T. A. Saleh, A review on the technologies for converting biomass into carbon-based materials: Sustainability and economy, Bioresour. Technol. Rep., 2024, 25, 101771 CrossRef CAS.
- Z. Guo, X. Han, C. Zhang, S. He, K. Liu, J. Hu, W. Yang, S. Jian, S. Jiang and G. Duan, Activation of biomass-derived porous carbon for supercapacitors: A review, Chin. Chem. Lett., 2024, 35, 109007 CrossRef CAS.
- L. Hou, Z. Chen, Z. Zhao, X. Sun, J. Zhang and C. Yuan, Universal feCl3-activating strategy for green and scalable fabrication of sustainable biomass-derived hierarchical porous nitrogen-doped carbons for electrochemical supercapacitors, ACS Appl. Energy Mater., 2019, 2, 548–557 CrossRef CAS.
- S. Deng, Z. Shan, H. Zhang, J. Guo, C. Chen, Y. Li and Y. Lan, Peroxymonosulfate activation by tin (IV) phosphate supported cobalt composite for acetaminophen degradation: Performance and mechanism, J. Water Process Eng., 2024, 61, 105343 CrossRef.
- K. Theyagarajan, B. A. Lakshmi and Y.-J. Kim, Electrochemical sensing of acetaminophen in biofluids, pharmaceutical and environmental samples using cobalt hexacyanoferrate decorated iron terephthalate metal organic framework, Electrochim. Acta, 2024, 488, 144229 CrossRef CAS.
- V. Duraisamy, V. Sudha, V. Dharuman and S. M. Senthil Kumar, Highly efficient electrochemical sensing of acetaminophen by cobalt oxide-embedded nitrogen-doped hollow carbon spheres, ACS Biomater. Sci. Eng., 2023, 9, 1682–1693 CrossRef CAS PubMed.
- E. E. Abbas, A. S. Fayed, M. A. Hegazy, N. N. Salama and M. A. Mohamed, Toward an improved electrocatalytic determination of immunomodulator COVID medication baricitinib using multiwalled carbon nanotube nickel hybrid, ACS Appl. Bio Mater., 2024, 7, 3865–3876 CrossRef CAS.
- P. Veerakumar, A. Sangili, S.-M. Chen, A. Pandikumar and K.-C. Lin, Fabrication of platinum-rhenium nanoparticle-decorated porous carbons: Voltammetric sensing of furazolidone, ACS Sustainable Chem. Eng., 2020, 8, 3591–3605 CrossRef CAS.
- D. R. Lobato-Peralta, C. E. Arreola-Ramos, A. Ayala-Cortés, D. E. Pacheco-Catalán, M. Robles, A. Guillén-López, J. Muñiz, P. U. Okoye, H. I. Villafán-Vidales, C. A. Arancibia-Bulnes and A. K. Cuentas-Gallegos, Optimizing capacitance performance: Solar pyrolysis of lignocellulosic biomass for homogeneous porosity in carbon production, J. Cleaner Prod., 2024, 448, 141622 CrossRef CAS.
- X. Dou, I. Hasa, M. Hekmatfar, T. Diemant, R. J. Behm, D. Buchholz and S. Passerini, Pectin, hemicellulose, or lignin? Impact of the biowaste source on the performance of hard carbons for sodium-ion batteries, ChemSusChem, 2017, 10, 2668–2676 CrossRef CAS PubMed.
- H. Y. Nguyen Thi, S. Kim, B. T. Duy Nguyen, D. Lim, S. Kumar, H. Lee, G. Szekely and J. F. Kim, Closing the sustainable life cycle loop of membrane technology via a cellulose biomass platform, ACS Sustainable Chem. Eng., 2022, 10, 2532–2544 CrossRef CAS.
- S. Zallouz, B. Réty, L. Vidal, J.-M. Le Meins and C. Matei Ghimbeu, Co3O4 nanoparticles embedded in mesoporous carbon for supercapacitor applications, ACS Appl. Nano Mater., 2021, 4, 5022–5037 CrossRef CAS.
- W. Xu, F. Lyu, Y. Bai, A. Gao, J. Feng, Z. Cai and Y. Yin, Porous cobalt oxide nanoplates enriched with oxygen vacancies for oxygen evolution reaction, Nano Energy, 2018, 43, 110–116 CrossRef CAS.
- L. Zhang, J. Tang, J. Li, Y. Li, P. Yang, P. Zhao, J. Fei and Y. Xie, A novel dopamine electrochemical sensor based on 3D flake nickel oxide/cobalt oxide @ porous carbon nanosheets/carbon nanotubes/electrochemical reduced of graphene oxide composites modified glassy carbon electrode, Colloids Surf., A, 2023, 666, 131284 CrossRef CAS.
- P. Veerakumar, I. Panneer Muthuselvam, C.-T. Hung, K.-C. Lin, F.-C. Chou and S.-B. Liu, Biomass-derived activated carbon supported Fe3O4 nanoparticles as recyclable catalysts for reduction of nitroarenes, ACS Sustainable Chem. Eng., 2016, 4, 6772–6782 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical methods: fundamentals and applications, Wiley, New York, NY, USA, 2nd edn, 2011 Search PubMed.
- K. Wang, C. Wu, F. Wang, N. Jing and G. Jiang, Co/Co3O4 nanoparticles coupled with hollow nanoporous carbon polyhedrons for the enhanced electrochemical sensing of acetaminophen, ACS Sustainable Chem. Eng., 2019, 7, 18582–18592 CrossRef CAS.
- E. Watanabe, W. Zhao, A. Sugahara, B. Mortemard de Boisse, L. Lander, D. Asakura, Y. Okamoto, T. Mizokawa, M. Okubo and A. Yamada, Redox-driven spin transition in a layered battery cathode material, Chem. Mater., 2019, 31, 2358–2365 CrossRef CAS.
- E. Song, J. Moon, J. Y. Lee, C. O. Lee, W. S. Chi and J. T. Park, High-voltage solar energy conversion based on ZIF-67-derived binary redox-quasi-solid-state electrolyte, J. Electroanal. Chem., 2021, 893, 115264 CrossRef CAS.
- X. Mamat, Z. Gao and L. Chen, A Zn-doped Fe3O4 nanoparticle@N, S and P doped Elaeagnus angustifolia gum derived carbon hybrid electrocatalyst: Synthesis, characterization and electrochemical sensing of acetaminophen, Mater. Adv., 2023, 4, 4929–4942 RSC.
- Y. Liu, X. Yan, Y. Xing, P. Zhao, Y. Zhu, L. Li, N. Liu and Z. Zhang, Dispersed Au nanoparticles anchored on covalent organic frameworks/carbon nanotubes via self-reduction for electrochemical sensing of acetaminophen, ACS Appl. Nano Mater., 2024, 7, 4980–4988 CrossRef CAS.
- M. J. Tavakoli, M. Shabani-Nooshabadi and N. Ziaie, Application of Gd2ZnMnO6/ZnO nanocomposite for electrochemical measurement of acetaminophen, diphenhydramine, and phenylephrine, Anal. Chim. Acta, 2023, 1279, 341766 CrossRef CAS.
- H. Gu, X. Shui, Y. Zhang, T. Zeng, J. Yang, Z. Wu, X. Zhang and N. Yang, Porous carbon scaffolded Fe-based alloy nanoparticles for electrochemical quantification of acetaminophen and rutin, Carbon, 2024, 221, 118954 CrossRef CAS.
- L. A. Vomo, G. Deffo, C. G. Fotsop, L. G. Djemmoe, V. K. Tchieda, F. M. Eya'ane and E. Njanja, Synthesis of zinc oxide nanoparticles based on coffee husks embedded on mesoporous silica for the sensing of acetaminophen, ChemElectroChem, 2024, e202400088 CrossRef CAS.
- Y.-J. Shih, S.-K. Lin, Z.-L. Wu and W.-H. Chen, Cobalt and zinc imidazolate encapsulated in reduced graphene oxide and the derived nitrogen-enriched carbon frameworks (CoNC@rGO) for electrochemically sensing acetaminophen (APAP), Chem. Eng. J., 2024, 481, 148437 CrossRef CAS.
- Z. Dursun and M. Aktürk, A novel composite electrode based on graphene oxide/MnO2: h-MoO3 particles for square wave voltammetric determination of acetaminophen, Electroanalysis, 2024, e202400042 Search PubMed.
- K.-Y. Hwa, R. Murugan, S.-F. Tseng, A. Santhan and J.-Y. Lin, Laser-induced reduced graphene oxide for high-performance electrochemical sensors of antipyretic drug in real samples, Environ. Sci.: Nano, 2024, 11, 951–968 RSC.
- X. Liang, Y. Zhou, J. M. S. Almeida and C. M. A. Brett, A novel electrochemical acetaminophen sensor based on multiwalled carbon nanotube and poly(neutral red) modified electrodes with electropolymerization in ternary deep eutectic solvents, J. Electroanal. Chem., 2023, 936, 117366 CrossRef CAS.
- A. G. Alhamzani, A.-H. S. Mahdy, M. M. Abou-Krisha, T. A. Yousef and M. Abd-Elsabour, Eco-friendly synthesized silver-magnetic nanocomposite supported on nanocellulose modified glassy carbon electrode as an electrochemical sensor for simultaneous determination of dopamine and acetaminophen, Sens. Actuators, A, 2023, 364, 114810 CrossRef CAS.
- J. Zhang, S. Xu, W. Liu, Q. Wang and J. Qu, Detection of acetaminophen and P-aminophenol simultaneously by an electrochemical sensor based on Fe-NC derivatives attached with Ti3C2 QDs, Talanta, 2024, 275, 126192 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4im00069b |
| This journal is © Institute of Process Engineering of CAS 2025 |