
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReactive CO2 capture and mineralization of magnesium hydroxide to produce hydromagnesite with inherent solvent regeneration†
Xun Gaoa,
Peilong Lua,
Ivan Kuzmenkob,
Jan Ilavsky b and
Greeshma Gadikota*ac
b and
Greeshma Gadikota*ac
aSchool of Civil and Environmental Engineering, Cornell University, Ithaca, NY 14853, USA. E-mail: gg464@cornell.edu; Tel: +1 607 255 4796
bAdvanced Photon Source, Argonne National Laboratory, Lemont, IL 60439, USA
cSmith School of Chemical and Biological Engineering, Cornell University, Ithaca, NY 14853, USA
First published on 20th February 2025
Abstract
Valorization of multiple low value streams including CO2 emissions and magnesium-hydroxide bearing mine tailings to produce magnesium carbonate through reactive CO2 capture and mineralization provides a less explored opportunity to manage several gigatons of CO2 emissions. To resolve the feasibility of converting magnesium hydroxide to magnesium carbonate through reactive CO2 capture and mineralization, CO2 capture solvents such as sodium glycinate are harnessed to capture CO2 and react directly with Mg(OH)2 to produce hydromagnesite (Mg5[(CO3)4(OH)2]·4H2O). This approach eliminates the energy-intensive step of producing high purity CO2 associated with regenerating the solvent, and redissolving CO2 to produce magnesium carbonate. Interestingly, while temperatures below 50 °C facilitate CO2 capture, the mineralization kinetics are slow. However, at higher temperatures, accelerated carbon mineralization is favored by the faster kinetics of Mg(OH)2 dissolution and precipitation of magnesium carbonate. Reacting Mg(OH)2 at 90 °C with 15 wt% solids in the presence of 2.5 M sodium glycinate after 3 hours under well-stirred conditions results in an extent of carbon mineralization of 75.5%. The theoretical maximum extent of carbon mineralization when hydromagnesite is formed is 80%. Pre-loading CO2 on the solvent is also an effective approach to ensure that sufficient CO2 is available for reactive CO2 capture and mineralization, particularly when dilute CO2 and N2 mixtures are used. Higher extents of carbon mineralization are associated with an increase in the particle size and a reduction in the cumulative pore volume. These insights unlock the feasibility of harnessing reactive CO2 capture and mineralization as a pathway to convert magnesium-hydroxide bearing resources into industrially relevant magnesium carbonate products.
Keywords: Reactive CO2 capture and mineralization; Magnesium hydroxide; Magnesium carbonate; Regenerable CO2 capture solvents; Hydromagnesite.
1 Introduction
Harnessing multiple emissions such as CO2-bearing flue gas streams and magnesium hydroxide (or brucite) bearing tailings is emerging as a viable approach to capture and store CO2 emissions as stable magnesium carbonates, given the thermodynamically downhill nature of carbon mineralization pathways.1,2 The direct carbon mineralization of Mg(OH)2-bearing mine tailings with CO2 in ultra-dilute streams including air and flue gas is feasible but kinetically slow.1,3 Alternatively, bubbling highly concentrated or nearly pure CO2 through Mg(OH)2 is known to facilitate rapid carbon mineralization within two hours or less.4 However, CO2 capture technologies are needed to concentrate CO2 that can be used for subsequent carbon mineralization using this approach.5,6 To achieve fast kinetics of carbon mineralization while directly harnessing ultra-dilute sources of CO2 including flue gas streams or even air without additional steps to regenerate the solvents and solubilize CO2, reactive CO2 capture and mineralization pathways are being developed.7Reactive CO2 capture and mineralization pathways involve concentrating CO2 emissions to produce CO2 – loaded solvents. These CO2 – loaded solvents react directly with the alkaline source to produce the respective carbonates with concurrent regeneration.8 This approach is designed to circumvent: (i) the low solubility of CO2 by harnessing CO2 capture solvents with basic functionalities that enhance CO2 solubility at very low gas phase concentrations, (ii) energy needs associated with producing high purity CO2 for subsequent mineralization, and (iii) the additional unit operations associated with producing high purity CO2 which occurs conventionally via pressure – swing or thermal – swing sorption processes.
The efficacy of these pathways for mineralizing calcium – bearing resources, such as oxides and silicates, and heterogeneous resources, such as fly ash, has been verified by harnessing polyamines and water-lean solvents such as 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), branched amines such as 2-amino-2-methyl-1-propanol (AMP), monoamines such as monoethanolamine (MEA), and amino acid salts with varying alkyl functional groups such as sodium glycinate and sodium sarcosinate.9–14 These studies established that temperatures in range of 50–75 °C facilitate reactive CO2 capture and mineralization, with lower temperatures favouring CO2 capture but challenged by slow mineralization kinetics, whereas higher temperatures enable faster carbon mineralization but limit CO2 capture.
While the efficacy of reactive CO2 capture and mineralization of magnesium oxide was established using amine bearing solvents, the use of more environmentally benign solvents such as sodium glycinate has not been explored.15 Sodium glycinate is the simplest unit of amino acid salts. Establishing the efficacy of reactive CO2 capture and mineralization using this amino acid salt translates into the use of other amino acids with multiple amine groups, straight and branched alkyl groups, and varying positions of amine groups (e.g., alpha and beta positions) for this approach. Regardless of the placement of the amine functional groups, the key reactions associated with reactive CO2 capture and mineralization include: (i) CO2 hydration, (ii) dissolution of Mg(OH)2, and (iii) the formation of Mg-carbonate, which is shown in Fig. 1.16
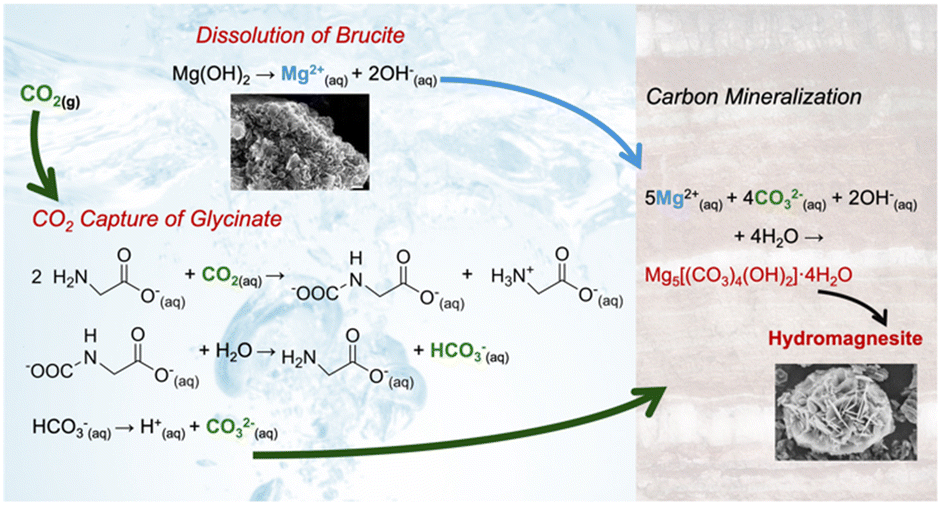 | ||
| Fig. 1 Schematic representation of reactive CO2 capture and carbon mineralization by harnessing Mg(OH)2 as the alkaline source and Na-glycinate as the CO2 capture solvent. | ||
Despite considerable advances made in reactive CO2 capture and mineralization,15 several outstanding research questions remain including: (i) is reactive CO2 capture and mineralization of Mg(OH)2 favoured at temperatures above or below 50 °C and why? (ii) How does the reactivity of Mg(OH)2 compare with that of Ca-bearing oxides and silicates for reactive CO2 capture and mineralization at comparable experimental conditions? (iii) Given that Mg2+ ions have a stronger hydration shell compared to Ca2+ ions,17 what is the influence of reactive CO2 capture and mineralization on the structural evolution of magnesium carbonate phases? For example, are metastable hydrated magnesium carbonate phases formed as opposed to stable magnesite forms? (iv) What is the influence of CO2 compositions in the range of 4–20 vol% on reactive CO2 capture and mineralization of Mg(OH)2? (v) What is the influence of the extent of carbon mineralization on the pore and particle morphological evolution of the Mg-bearing materials?
To address these questions, reactive CO2 capture and mineralization studies are conducted with 1 M and 2.5 M Na-glycinate contrasted with water at temperatures in the range of 25–90 °C with CO2 concentrations ranging from 4–100 vol%. The morphological features of the materials are characterized before and after carbon mineralization to determine the influence of carbon mineralization on the particle and pore morphologies of the products.
2 Results and discussion
2.1 Reactive CO2 capture and mineralization of magnesium hydroxide
Reactive CO2 capture and mineralization involves harnessing CO2 capture solvents to capture and concentrate CO2 in the aqueous phase and reacting these CO2-loaded solvents with an alkaline source to produce Mg-carbonate and regenerate the solvent. In this multiphase chemical reaction pathway, the solubility of CO2, reactivity of the alkaline source, and subsequent carbon mineralization behaviour are influenced by temperature, solvent concentration, and pH of the system. CO2 capture is aided at temperatures below 40 °C while mineralization kinetics are aided at higher temperatures. Furthermore, the solubility of magnesium carbonates decreases with temperature, thus favouring precipitation. To resolve the influence of temperature, reactive CO2 capture and mineralization behaviour with Mg(OH)2 is investigated at 25, 50, 75, and 90 °C. The reactivity of Mg(OH)2 is established with water, 1.0 M and 2.5 M sodium glycinate. Furthermore, the reactivity of Mg(OH)2 is compared with that of calcium oxide and calcium silicate at comparable experimental conditions to contrast their reactivity. These results are discussed in this section.To investigate the influence of temperature, experiments are conducted in a pure CO2 environment with 1.0 M and 2.5 M Na-glycinate and deionized water for comparison at 25, 50, 75, and 90 °C, respectively. As shown in Fig. 2, the extent of carbon mineralization increases from 17.1% at 25 °C to 75.5% at 90 °C when Mg(OH)2 is reacted in the presence of 2.5 M Na-glycinate. This enhancement in carbon mineralization which is more than three-fold indicates that the fast kinetics of carbon mineralization and lower solubility of Mg-carbonate at higher temperature of 90 °C dominate higher solubility of CO2 at lower temperatures.19–21 In the absence of the solvent, extents of carbon mineralization decreased from 26.9% at 25 °C to 22.2% at 75 °C and to 11.7% at 90 °C, respectively, due to the reduced solubility of CO2 with rising temperature which limits the availability of carbonate species needed for carbon mineralization.22 In contrast, calcium oxide is highly reactive, and carbon mineralization extents of 80% or higher are achieved at 25 °C or above. Interestingly, the reactivity of CaSiO3 increases to 31% at 75 °C and decreases at 90 °C. These results show that Ca-bearing solids are more reactive at temperatures below 90 °C. The significantly lower extents of carbon mineralization noted using Ca–silicate are attributed to the slow dissolution rate of silicate materials and the formation of silica passivation layer that may limit the diffusivity of the ions for carbon mineralization.23–25
 | ||
| Fig. 2 The extents of carbon mineralization of Mg(OH)2 with various Na-glycinate concentrations at different temperatures. All experiments are conducted at 1 atm of CO2. The solid products were analyzed by thermogravimetric analysis (TGA). Extents of carbon mineralization reported for CaO and CaSiO3 from Liu and coworkers are included for comparison.18 | ||
Investigations of the influence of 1 M and 2.5 M Na-glycinate on reactive CO2 capture and mineralization showed that higher extents of carbon mineralization are achieved with 2.5 M Na-glycinate compared to 1 M counterparts due to the greater availability of amine functional groups for reactive CO2 capture and mineralization. Specifically, extents of carbon mineralization achieved with Mg(OH)2 are 61.7% and 75.5% with 1 M and 2.5 M Na-glycinate, respectively, at 90 °C. It is important to note that the theoretical maximum extent of carbon mineralization for hydromagnesite is 80%, because the stoichiometric ratio of Mg(OH)2 to CO2 is determined to be 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 based on reaction 2. Assuming all Mg(OH)2 reacts with CO2, the EoC is calculated to be 80% based on eqn (3) and (4). As a result, the normalized extent of carbon mineralization is up to 94.3% for the 2.5 M Na-glycinate case at 90 °C (Fig. S1†). It is also worth noting that the extents of carbon mineralization of Mg(OH)2 at 25 °C in the presence of water, 1 M and 2.5 M Na-glycinate are 26.9%, 16.7%, and 17.1%, respectively. At 25 °C, the glycinate ligands can bind with the Mg2+ ions in the slurry to form Mg–glycinate complexes, which may be too stable to react with (bi)carbonate species to produce Mg-carbonates.26
:4 based on reaction 2. Assuming all Mg(OH)2 reacts with CO2, the EoC is calculated to be 80% based on eqn (3) and (4). As a result, the normalized extent of carbon mineralization is up to 94.3% for the 2.5 M Na-glycinate case at 90 °C (Fig. S1†). It is also worth noting that the extents of carbon mineralization of Mg(OH)2 at 25 °C in the presence of water, 1 M and 2.5 M Na-glycinate are 26.9%, 16.7%, and 17.1%, respectively. At 25 °C, the glycinate ligands can bind with the Mg2+ ions in the slurry to form Mg–glycinate complexes, which may be too stable to react with (bi)carbonate species to produce Mg-carbonates.26
While the efficacy of harnessing highly concentrated CO2 streams for the reactive CO2 capture and mineralization of Mg(OH)2 to produce the respective Mg-carbonate has been established, the influence of the dilute CO2 streams or flue gas which are representative of industrial operations remains less studied. Flue gas is the tail gas bearing nitrogen, carbon dioxide, and steam with trace amount of sulfur oxide or nitrogen oxide emerging from fuel combustion, industrial calcination or reduction processes.27,28 To simulate the conditions of industrial flue gases, a binary gas mixture of nitrogen and carbon dioxide is prepared and used to investigate reactive CO2 capture and mineralization with magnesium hydroxide.
As shown in the experimental section reactive CO2 capture and mineralization studies are conducted with binary mixture of N2 and CO2 inside a gas circulation system with CO2-preloaded solutions at 25 °C. A constant binary gas flow is injected into the Mg(OH)2 slurry as the CO2 source. Following the same calculation approach, the carbon mineralization extents from flue gas having varying CO2 levels with different pre-loading protocols are shown in Fig. 3. Generally, as the CO2 concentration in the binary gas flow increases from 4% to 20%, the extent of carbon mineralization increases from 3.4% to 31% in cases without CO2 pre-loading. The monotonically increasing correlation is aligned with Henry's law, by which CO2 solubility and dissolution rate are determined by the CO2 partial pressure, equivalent to the CO2 concentration in the flue gas.29 This observation indicates that increasing CO2 concentrations can linearly improve the extent of carbon mineralization by enhancing the amount of dissolved CO2. In contrast, CO2 pre-loading remarkably increases the extent of carbon mineralization compared to the cases without CO2 pre-loading, with increases of 15.2%, 19.9%, 26.8%, and 33.8% for 4, 10, 15, and 20 vol% CO2 at 25 °C, with associated extents of 18.6%, 35.4%, 50.1%, and 64.8%, respectively.
 | ||
| Fig. 3 The extents of carbon mineralization of Mg(OH)2 using binary flue gas with various CO2/N2 ratios. The carbonate content in the solid products is analyzed by thermogravimetric analysis (TGA). | ||
The significant increase in the extents of carbon mineralization with pre-loaded solvents are attributed to the ready availability of dissolved carbon for reacting with Mg(OH)2 to produce Mg-carbonate bearing species. This approach overcomes the challenge of the mass transfer of CO2 from the gas to the liquid phase. Pre-loading the solvent with CO2 also ensures that glycinate is effectively bound to CO2 as opposed to Mg2+ ions to produce Mg–glycinate complexes. When Na-glycinate is mixed with Mg(OH)2 before CO2 is introduced, the formation of Mg–glycinate complex over CO2-loaded Na-glycinate may limit CO2 loading and subsequent reactivity with Mg2+ ions to produce hydromagnesite.
It is interesting to note that the extents of carbon mineralization are 61.7% and 75.5% with 100 vol% CO2 without pre-loading 1.0 M and 2.5 M Na-glycinate solvent at 90 °C for a reaction time of 3 hours, respectively. In contrast, the extent of carbon mineralization is 64.4% with the pre-loaded 2.5 M Na-glycinate CO2 capture solvent when reacted at 25 °C for 3 hours. These results demonstrate that pre-loading the CO2 capture solvent is effective in enabling carbon mineralization at room temperature. Thus, these results show the feasibility of reactive CO2 capture and mineralization with Mg(OH)2 at 25 °C by harnessing pre-loaded CO2 capture solvents.
2.2 Chemical and structural evolution during reactive CO2 capture and mineralization
Evidence of carbonate formation in Mg-carbonate bearing products is established using attenuated total reflection Fourier transform-infrared (ATR FT-IR) spectroscopy and wide-angle X-ray scattering (WAXS). First, the formation of CO32− symmetric stretching vibration at 1119 cm−1 in the products as determined using ATR FT-IR measurements confirms solid carbonate formation (Fig. 4).30 The corresponding bending vibration of CO32− is also detected at 885 cm−1, 854 cm−1, and 798 cm−1, respectively.31 Besides, the CO32− asymmetric stretching vibration splits into two peaks at 1485 cm−1 and 1421 cm−1, respectively, which emerges in all the carbonate-bearing products, indicating the existence of bicarbonate in the carbonate-bearing solids.32 The identified infrared bands of different carbonate-bearing species and the corresponding assignments are listed in Table 1. The formation of hydromagnesite in the products is further confirmed through the wide-angle X-ray scattering (WAXS) measurements. | ||
| Fig. 4 Evidence of (bi) carbonate species as determined using attenuated total reflection-Fourier transform infrared (ATR-FTIR) spectra for carbonate – bearing solid products obtained on reacting in (a) DI water, (b) 1 M Na-glycinate, and (c) 2.5 M Na-glycinate solutions. The characteristic peaks in the dotted square areas indicate the formation of HCO3− and CO32− species. | ||
| Species | Band position (cm−1) | Assignment |
|---|---|---|
| CO32− | 1119 | Symmetric stretching |
| CO32− | 885 | Bending |
| 854 | ||
| 798 | ||
| HCO3− | 1485 | Asymmetric stretching |
| 1421 |
As shown in Fig. 5(a), the emerging peaks noted with black circles are observed after Mg(OH)2 mineralization in DI water. These well-defined characteristic peaks correspond to hydromagnesite (PDF 98-001-1519, noted with open circle). Products obtained after reacting in water do not have prominent hydromagnesite peaks compared to brucite (PDF 98-000-0130, noted with closed circle), indicating low extents of carbon mineralization. Additionally, the peak intensity of hydromagnesite showed negligible changes with temperature, demonstrating that carbon mineralization in DI water is not significantly influenced by temperature, which is consistent with the extents of carbon mineralization in Table 2.33,34
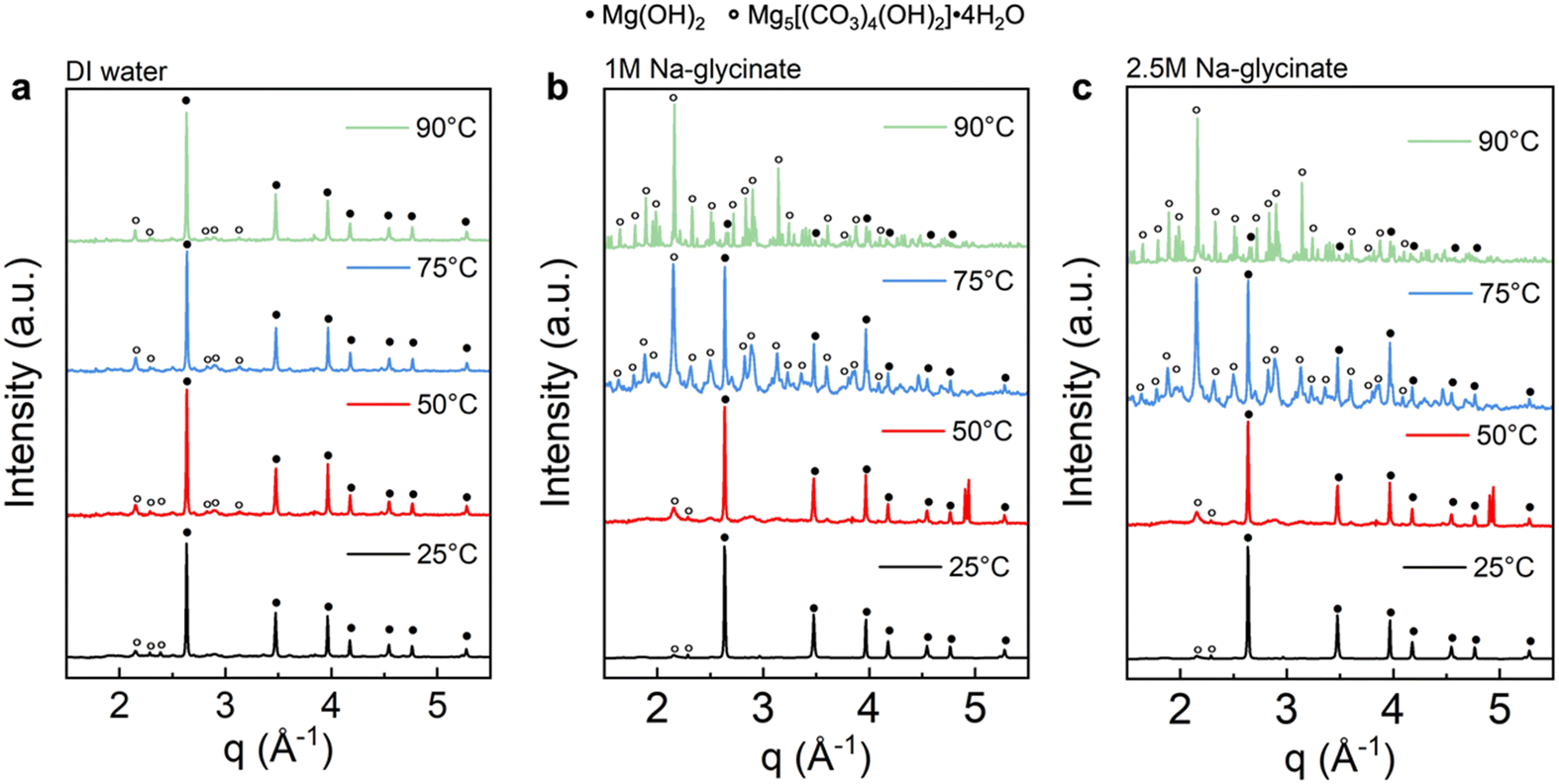 | ||
| Fig. 5 Evidence of hydromagnesite formation as determined using wide angle X-ray scattering (WAXS) patterns for the carbonate-bearing solid products obtained using (a) DI water, (b) 1 M Na-glycinate, and (c) 2.5 M Na-glycinate at different temperatures. Two types of crystal structures are observed: Mg(OH)2, originating from unreacted brucite, and Mg5[(CO3)4(OH)2]·4H2O, originating from the carbonate products. | ||
| Temperature (°C) | EoC (%) | Mean particle size (μm) | Pore volume (cc g−1) | |
|---|---|---|---|---|
| Mg(OH)2 | — | — | 7.85 | 0.047 |
| H2O | 25 | 26.90 ± 1.28 | 15.41 | 0.133 |
| 50 | 20.14 ± 1.88 | 16.97 | 0.218 | |
| 75 | 22.18 ± 0.93 | 19.85 | 0.118 | |
| 90 | 11.68 ± 0.85 | 15.63 | 0.101 | |
| 1 M Na-glycinate | 25 | 16.71 ± 3.88 | 7.77 | 0.066 |
| 50 | 29.97 ± 2.17 | 9.64 | 0.071 | |
| 75 | 36.71 ± 1.93 | 16.75 | 0.036 | |
| 90 | 61.69 ± 0.35 | 18.43 | 0.026 | |
| 2.5 M Na-glycinate | 25 | 17.08 ± 3.38 | 8.17 | 0.016 |
| 50 | 40.67 ± 4.25 | 9.63 | 0.02 | |
| 75 | 56.45 ± 0.72 | 20.56 | 0.011 | |
| 90 | 75.46 ± 0.77 | 19.30 | 0.008 |
In sharp contrast, the presence of Na-glycinate solution, particularly at elevated temperature, resulted in products with more prominent characteristic peaks of hydromagnesite with higher intensity. As shown in Fig. 5(b) and (c), magnesium hydroxide is the dominant phase at 25 °C, while hydromagnesite formation increases with temperature. At 90 °C, the dominance of hydromagnesite phase over Mg(OH)2 is noted.34 Prominent peaks corresponding to hydromagnesite emerge at 75 °C with 2.5 M Na-glycinate and at 90 °C with 1.0 M Na-glycinate. Furthermore, WAXS results in Fig. 5 confirm the formation of hydromagnesite, as the only Mg-carbonate bearing phase formed. Hydromagnesite formation is known to be dominant at 25–90 °C and in alkaline environments.35,36
2.3. Morphological changes during reactive CO2 capture and mineralization
Insights into the influence of carbon mineralization on the morphological evolution of Mg-carbonate are unlocked through particle and pore size analyses and microscopy, as discussed in the following sections. | ||
| Fig. 6 Morphologies of (a) unreacted brucite, and carbonated solid with 1 M Na-glycinate solution at (b) 50 °C and (c) at 90 °C, and carbonated solid with 2.5 M Na-glycinate solution at (d) 50 °C and (e) 90 °C, determined by scanning electron micrographs (SEM). | ||
 | ||
| Fig. 7 Particle size distributions of the carbonate – bearing solids obtained with (a) DI water, (b) 1 M Na-glycinate solution, and (c) 2.5 M Na-glycinate solution. The size distributions are measured by light scattering instrument (PSA) with volume – based normalization of the particle size distributions. | ||
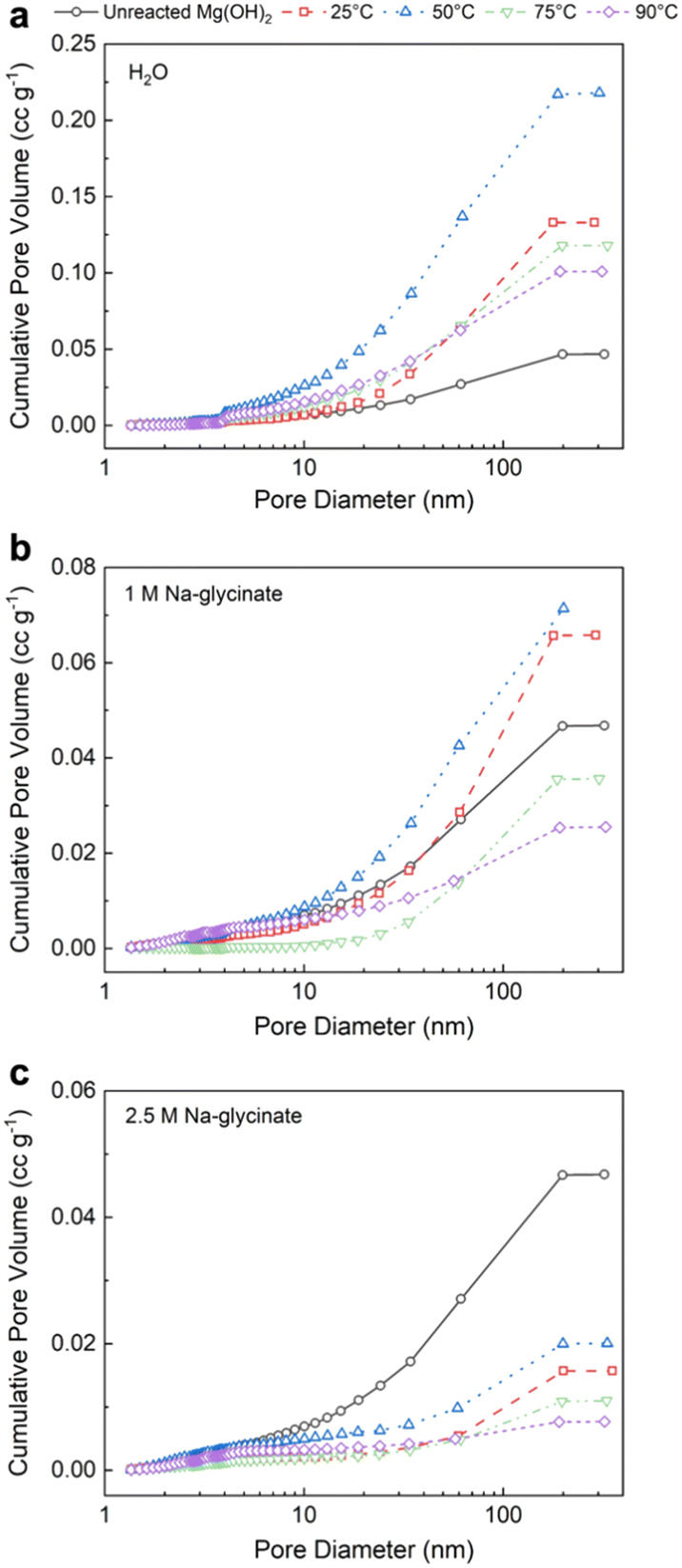 | ||
| Fig. 8 The cumulative pore volumes of various carbonate – bearing solids obtained using (a) DI water, (b) 1 M Na-glycinate solution, and (c) 2.5 M Na-glycinate solution. The pore volume distributions are determined using the Barrett, Joyner, Halenda (BJH) method with N2 adsorption data. | ||
It is interesting to note that with increasing temperature in the presence of higher concentration of 2.5 M Na-glycinate results in a lower cumulative pore volume at all temperatures of interest (Fig. 8(c)). The extensive formation of lamellar hydromagnesite (Fig. 6(d) and (e)) at higher temperatures in the presence of 2.5 M Na-glycinate corresponds to a 77% and 83% reduction in the pore volume at 75 °C and 90 °C, respectively (Table 2). Additionally, it is also worth noting that the highest pore volume and specific surface area are observed at 50 °C for all cases, indicating that hydromagnesite formation is not as extensive at these conditions to close the pore spaces (Fig. S3†). The increase in the pore volume at 50 °C facilitates greater diffusion of ions or mass transfer enabling carbon mineralization.
These studies show that hydromagnesite with varying morphologies can be produced by tuning the temperatures associated with reactive CO2 capture and mineralization with regenerable solvents such as Na-glycinate. Nest-like hydromagnesite at lower temperature is effective for flame retardant materials, while hydromagnesite with elongated lamellar particles can provide extra mechanical reinforcement as additives.41,42
Therefore, achieving tunable controls on the morphologies of the magnesium-carbonate bearing products unlocks new opportunities for introducing these products produced by harnessing CO2 emissions into markets for sustainable materials.
In contrast, Na-glycinate enhances the solubility of CO2 and provides sufficient concentration of (bi)carbonate species needed for carbon mineralization. While CO2 capture is aided at lower temperatures of 25 °C and 50 °C, the kinetics of Mg(OH)2 dissolution and the formation and precipitation of Mg-carbonate are aided at higher temperatures of 75 °C and 90 °C.19,44 Therefore, higher extents of carbon mineralization of Mg(OH)2 are noted at 75 °C and 90 °C relative to 25 °C and 50 °C. Interestingly, pre-loading the solvent with CO2 prior to reactive CO2 capture and mineralization overcomes the limitation of CO2 mass transfer from the gas to the liquid phase and provides a ready supply of carbonate species which enables rapid carbon mineralization as noted in Fig. 3. Higher concentrations of Na-glycinate of 2.5 M provide additional functional groups and enhanced alkalinity to enhance CO2 capture and mineralization compared to the 1 M Na-glycinate case (Fig. 2 and Table 2). The formation of hydromagnesite as the preferential magnesium carbonate phase is consistent with alkalinity, temperature, and ionic environments.45
Elevated temperature of 75 °C and 90 °C and enhanced alkalinity in the case of 2.5 M Na-glycinate facilitates the preferential formation of lamellar-shaped over nest-like hydromagnesite (Fig. 6). Carbon mineralization proceeds via the dissolution of particles smaller than 20 μm and the precipitation of Mg-carbonate particles with larger particle sizes (Fig. 7). The evolution of well-defined particle size distributions on extensive carbon mineralization is consistent with prior studies indicating that extensive carbonate formation in olivine ((Mg, Fe)2SiO4) results in distinct particle size distributions.46,47 The extensive formation of hydromagnesite phases and extents of carbon mineralization exceeding 60% are associated with >77% decrease in the cumulative pore volume (Fig. 8). These analyses establish the chemo-morphological mechanisms underlying reactive CO2 capture and mineralization using Mg(OH)2. These studies complement prior studies that discuss the feasibility of reactive CO2 capture and mineralization pathways by harnessing Ca – bearing alkaline sources.8–10,18,48–50
3 Conclusions
Reactive CO2 capture and mineralization of Mg(OH)2 to produce hydromagnesite (Mg5[(CO3)4(OH)2]·4H2O) by harnessing CO2 capture solvents such as Na-glycinate is established at concentrations of 1.0 M and 2.5 M and temperatures ranging from 25 to 90 °C. Reacting Mg(OH)2 at 90 °C with 15 wt% solids in the presence of 2.5 M sodium glycinate after 3 hours under well-stirred conditions results in an extent of carbon mineralization of 75.5%. In contrast, the extents of carbon mineralization in the presence of water without any solvent do not exceed 27% at all conditions of temperature. As a reference, the maximum possible extent of carbon mineralization is 80% when 100% hydromagnesite is produced. The coupled effects of enhanced kinetics of magnesium hydroxide dissolution and Mg-carbonate precipitation at elevated temperatures of 75 °C and 90 °C dominate favourable CO2 uptake by solvents at lower temperature of 25 °C. Well-defined particle size distributions and a significant decrease in the cumulative pore volume are noted with extensive hydromagnesite growth. Higher temperature and pH conditions favour the formation of lamellar-shaped versus nest-like hydromagnesite. Factors such as SOx and NOx in industrial flue gas, as well as operation optimization, need further investigation for practical applications. These findings unlock the potential for harnessing these reactive CO2 capture and mineralization for harnessing earth abundant magnesium hydroxide-rich tailings and anthropogenic CO2 emissions to produce Mg-carbonate bearing end products for a sustainable energy and environmental future.4 Experimental section
4.1 Chemicals
The reagents used in the study, magnesium hydroxide (Mg(OH)2, 99%), sodium hydroxide (NaOH, 98%), and glycine (NH2CH2COOH, 99%) are procured from Sigma Aldrich. Na-glycinate solutions with concentrations of 1 M and 2.5 M are prepared by adding calculated amounts of sodium hydroxide and glycine to deionized water (18.2 MΩ cm, Millipore) while stirring at 300 rpm for 1 hour at room temperature. The formation of Na-glycinate can be expressed by the following chemical equation (reaction 1):| NH2CH2COOH + NaOH ↔ NH2CH2COO− + Na+ + H2O | (1) |
4.2 Experimental approach
:17 or 15 wt%. CO2 gas (99.9% purity, Airgas Co., USA) is continuously injected into the reactor for five minutes to purge the residual air from the headspace. After the reactor is filled with CO2, the outlet valve is slowly closed until the internal CO2 pressure of 1 atm is reached. Next, the reactions are initiated by turning on stirrer with a set point of 300 rpm and applying a set of temperatures: 25 °C, 50 °C, 75 °C, and 90 °C, respectively. The reaction duration is set to 3 hours for each temperature. At the end of the reaction, the reactor is cooled and the solid carbonate-bearing products are collected by vacuum filtration, washed with deionized water, and dried at 80 °C for 24 hours. All experiments were replicated three times to calculate error bars.
 | ||
| Fig. 9 Schematic representation of the integrated CO2 capture and mineralization with (a) pure CO2 and (b) mixed gases composed of CO2 and N2. | ||
To investigate the influence of pre-loaded CO2 capture solvents on reactive CO2 capture and mineralization behaviour with Mg(OH)2, experiments are conducted for 3 hours in the presence of various CO2 and N2 mixtures. The hypothesis that the initial loading of CO2 overcomes the challenge of limited CO2 availability initially and enables faster kinetics of carbon mineralization, is investigated. The ratio of Mg(OH)2 to the solvent is 3:17 resulting in 15 wt% of solids in the system. The slurry is well-stirred at 300 rpm. The solid products are recovered through vacuum filtration, washed in deionized water, and dried at 80 °C for 48 hours before further characterization. All experiments were replicated three times to calculate error bars.
4.3 Determination of the extent of carbon mineralization
The extent of carbon mineralization defines CO2 mineralization conversion of Mg(OH)2 with respect to the theoretical maximum. In other words, this extent refers to the mole fraction of Mg(OH)2 that converts into solid carbonate after mineralization. In this study, hydromagnesite (Mg5[(CO3)4(OH)2]·4H2O), a hydrated form of magnesium carbonate is formed and is the only product of carbon mineralization, as determined by X-ray diffraction (XRD) and thermo-gravimetric analyses (TGA).51 The formation of hydromagnesite via carbon mineralization is shown in reaction 2.21 Accordingly, the extent of carbon mineralization can be defined as the ratio between the captured mole of CO2 and the theoretical CO2 mole capacity of the sorbent.52| 5Mg(OH)2 + 4CO2 = Mg5[(CO3)4(OH)2]·4H2O | (2) |
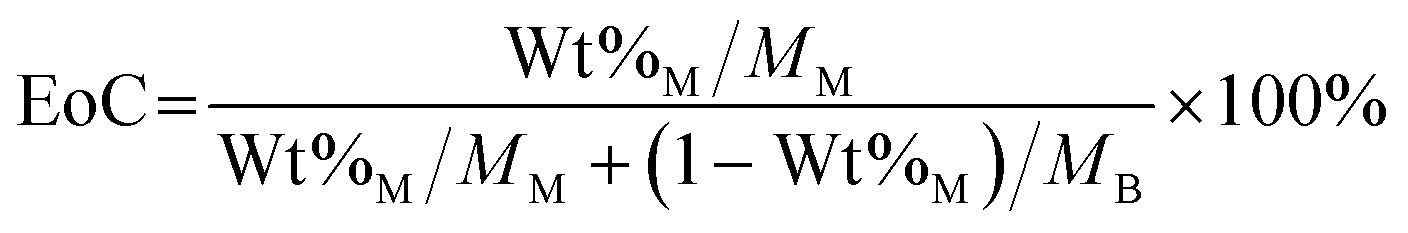 | (3) |
In the expression above, EoC refers to the extent of carbon mineralization, Wt%M refers to the weight percent of magnesium carbonate in the fully dried samples, while the MM and MB refer to the molecular weight of magnesium carbonate and brucite or magnesium hydroxide, respectively. Here, thermogravimetric analysis (TGA) is used to determine Wt%M. As shown in eqn (4), TGA is the actual weight loss of the fully dried samples in the temperature range 300–600 °C, while TGAM and TGAB refer to the theoretical weight loss of pure magnesium carbonate and magnesium hydroxide, respectively. Magnesium carbonate (MgCO3 → MgO + CO2) and magnesium hydroxide (Mg(OH)2 → MgO + H2O) decompose at 300–600 °C, the TGAM and TGAB values are calculated to be 52.20% and 30.89%, respectively.
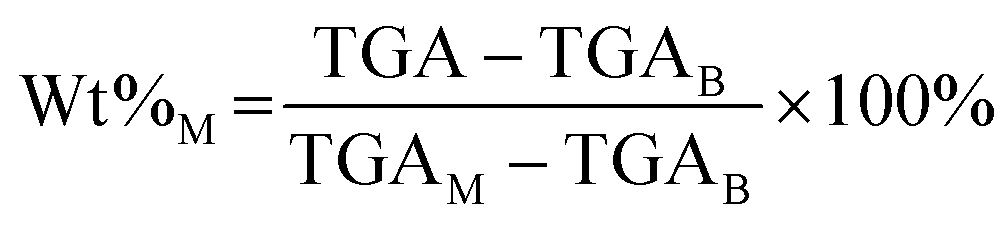 | (4) |
4.4 Characterization of the carbonate – bearing products
The chemical compositions and the morphological features of the carbonate-bearing products are analysed to provide insights into the mechanisms of reactive CO2 capture and mineralization. The CO2 content in the carbonate-bearing products is determined using thermogravimetric analysis (TGA, TA Instruments, SDT 650), in which the temperature is ramped from 25 °C to 1000 °C with a 2 °C min−1 ramping rate. Temperatures in the range of 300–500 °C are of particular interest since CO2 and water releases occur in this range. The magnesium carbonate phases present are determined using wide-angle X-ray scattering (WAXS) measurements. These measurements are conducted at Sector 9-ID-C at the Advanced Photon Source (APS) in Argonne National Laboratory (ANL). For these experiments, the X-ray energy is set to 21.0 keV, which corresponds to the wavelength of 0.59 Å. The total X-ray flux during the measurements is 1013 photons mm−2 s−1.53 Detailed information about the instrument is reported in prior publications.54,55 To further confirm the chemical bonds and functional groups in the carbonate-bearing products, attenuated total reflectance Fourier transform-infrared spectroscopy (ATR-FTIR, Nicolet FT-IR IS50, Thermo Scientific) analyses are performed. The morphological features and the particle size distributions are analysed using field emission scanning electron microscope (FESEM, LEO 1550 FESEM, Bruker) and the laser diffraction particle size analyzer (PSA, Anton Paar), respectively. The surface area and pore size distributions (PSD) before and after carbon mineralization are determined from N2 adsorption–desorption isotherms using the Brunauer–Emmett–Teller (BET) method and Barrett–Joyner–Halenda (BJH) method, respectively (Quantachrome Autosorb iQ Analyzer, Boynton Beach, FL). Prior to measuring the adsorption–desorption isotherms, the samples were outgassed at 120 °C for 18 hours.Data availability
The data supporting this article have been included in the main manuscript and as part of the ESI.†Author contributions
Greeshma Gadikota – supervision, conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, resources, software, validation, visualization, writing – original draft, writing – review & editing. Xun Gao – investigation, data curation, formal analysis, methodology, resources, software, validation, visualization, writing – original draft, writing – review & editing. Peilong Lu – validation, visualization, writing – review & editing. Ivan Kuzmenko – investigation. Jan Ilavsky – investigation.Conflicts of interest
Greeshma Gadikota is the co-founder of Carbon To Stone which is commercializing technologies for industrial decarbonization and carbon management. The other authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
G. G., X. G., and P. L.'s contributions are supported by the U. S. Department of Energy (DOE): DE-AC36-08GO28308. The authors acknowledge the use of shared facilities at the Cornell Center for Materials Research (CCMR). G. G. and P. L. gratefully acknowledge the support of the NSF Partnerships for Innovation (PFI) program (NSF Award #: 2141091). The authors also gratefully acknowledge the support of Ivan Kuzmenko and Jan Ilavsky at APS for assisting in this effort. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science user facility at Argonne National Laboratory and is based on research supported by the U.S. DOE Office of Science-Basic Energy Sciences, under Contract No. DE-AC02-06CH11357.References
- A. L. Harrison, I. M. Power and G. M. Dipple, Accelerated carbonation of brucite in mine tailings for carbon sequestration, Environ. Sci. Technol., 2013, 47, 126–134 CrossRef CAS PubMed.
- A. L. Harrison, G. M. Dipple, I. M. Power and K. U. Mayer, Influence of surface passivation and water content on mineral reactions in unsaturated porous media: Implications for brucite carbonation and CO2 sequestration, Geochim. Cosmochim. Acta, 2015, 148, 477–495 CrossRef CAS.
- I. M. Power, A. L. Harrison and G. M. Dipple, Accelerating mineral carbonation using carbonic anhydrase, Environ. Sci. Technol., 2016, 50, 2610–2618 CrossRef CAS PubMed.
- K. Rausis, A. Ćwik and I. Casanova, Phase evolution during accelerated CO2 mineralization of brucite under concentrated CO2 and simulated flue gas conditions, J. CO2 Util., 2020, 37, 122–133 CrossRef CAS.
- E. S. Sanz-Pérez, C. R. Murdock, S. A. Didas and C. W. Jones, Direct capture of CO2 from ambient air, Chem. Rev., 2016, 116, 11840–11876 CrossRef PubMed.
- X. Wang and C. Song, Carbon capture from flue gas and the atmosphere: A perspective, Front. Energy Res., 2020, 8, 560849 CrossRef.
- A. Sanna, M. Uibu, G. Caramanna, R. Kuusik and M. M. Maroto-Valer, A review of mineral carbonation technologies to sequester CO2, Chem. Soc. Rev., 2014, 43, 8049–8080 RSC.
- P. Ochonma, X. Gao and G. Gadikota, Tuning reactive crystallization pathways for integrated CO2 capture, conversion, and storage via mineralization, Acc. Chem. Res., 2024, 57, 267–274 CrossRef CAS PubMed.
- T. Yin, S. Yin, A. Srivastava and G. Gadikota, Regenerable solvents mediate accelerated low-temperature CO2 capture and carbon mineralization of ash and nano-scale calcium carbonate formation, Resour., Conserv. Recycl., 2022, 180, 106209 CrossRef CAS.
- M. Liu, A. Hohenshil and G. Gadikota, Integrated CO2 capture and removal via carbon mineralization with inherent regeneration of aqueous solvents, Energy Fuels, 2021, 35, 8051–8068 CrossRef CAS.
- X. Liu, S. Zhang, Q.-W. Song, X.-F. Liu, R. Ma and L.-N. He, Cooperative calcium-based catalysis with 1,8-diazabicyclo[5.4.0]-undec-7-ene for the cycloaddition of epoxides with CO2 at atmospheric pressure, Green Chem., 2016, 18, 2871–2876 RSC.
- J. M. Kang, et al., Energy-efficient chemical regeneration of AMP using calcium hydroxide for operating carbon dioxide capture process, Chem. Eng. J., 2018, 335, 338–344 CrossRef CAS.
- P. C. Chen and S.-Z. Lin, Optimization in the absorption and desorption of CO2 using sodium glycinate solution, Appl. Sci., 2018, 8, 2041 CrossRef CAS.
- F. M. Brethomé, N. J. Williams, C. A. Seipp, M. K. Kidder and R. Custelcean, Direct air capture of CO2 via aqueous-phase absorption and crystalline-phase release using concentrated solar power, Nat. Energy, 2018, 3, 553–559 CrossRef.
- M. Liu, H. Asgar, S. Seifert and G. Gadikota, Novel aqueous amine looping approach for the direct capture, conversion, and storage of CO2 to produce magnesium carbonate, Sustainable Energy Fuels, 2020, 4, 1265–1275 RSC.
- L. Zhao, L. Sang, C. Jun, J. Ji and H. H. Teng, Aqueous carbonation of natural brucite: Relevance to CO2 sequestration, Environ. Sci. Technol., 2010, 44, 406–411 CrossRef CAS PubMed.
- T. Dudev and C. Lim, Importance of metal hydration on the selectivity of Mg2+ versus Ca2+ in magnesium ion channels, J. Am. Chem. Soc., 2013, 135, 17200–17208 CrossRef CAS PubMed.
- M. Liu and G. Gadikota, Single-step, low-temperature and integrated CO2 capture and conversion using sodium glycinate to produce calcium carbonate, Fuel, 2020, 275, 117887 CrossRef CAS.
- P. Bénézeth, G. D. Saldi, J.-L. Dandurand and J. Schott, Experimental determination of the solubility product of magnesite at 50 to 200°C, Chem. Geol., 2011, 286, 21–31 CrossRef.
- D. Guo, et al., Amino acids as carbon capture solvents: Chemical kinetics and mechanism of the glycine + CO2 reaction, Energy Fuels, 2013, 27, 3898–3904 CrossRef CAS.
- Q. Gautier, P. Bénézeth, V. Mavromatis and J. Schott, Hydromagnesite solubility product and growth kinetics in aqueous solution from 25 to 75°C, Geochim. Cosmochim. Acta, 2014, 138, 1–20 CrossRef CAS.
- S. M. Klara, CO2 solubility in water and brine under reservoir conditions, Chem. Eng. Commun., 1990, 90, 23–33 CrossRef.
- P. Birkle and I. S. Torres-Alvarado, Water-Rock Interaction, CRC Press, 2010 Search PubMed.
- D. Daval, R. Hellmann, G. D. Saldi, R. Wirth and K. G. Knauss, Linking nm-scale measurements of the anisotropy of silicate surface reactivity to macroscopic dissolution rate laws: New insights based on diopside, Geochim. Cosmochim. Acta, 2013, 107, 121–134 CrossRef CAS.
- X. Gao, D. Prasad, M. A. Mahadik and G. Gadikota, Enhanced H2 recovery by coupling the water-gas shift reaction with in-situ CO2 capture and mineralization using earth-abundant Ca- and Mg-silicates and hydroxides, React. Chem. Eng., 2025 10.1039/D4RE00480A.
- C. Blaquiere and G. Berthon, Speciation studies in relation to magnesium bioavailability: Formation of Mg(II) complexes with glutamate, aspartate, glycinate, lactate, pyroglutamate, pyridoxine, and citrate, and appraisal of their potential significance towards magnesium gastrointestinal absorption, Inorg. Chim. Acta, 1987, 135, 179–189 CrossRef CAS.
- R. Weber, S. Orsino, N. Lallemant and A. Verlaan, Combustion of natural gas with high-temperature air and large quantities of flue gas, Proc. Combust. Inst., 2000, 28, 1315–1321 CrossRef CAS.
- F. Gholami, M. Tomas, Z. Gholami and M. Vakili, Technologies for the nitrogen oxides reduction from flue gas: A review, Sci. Total Environ., 2020, 714, 136712 CrossRef CAS PubMed.
- V. Majer, J. Sedlbauer and G. Bergin, Henry's law constant and related coefficients for aqueous hydrocarbons, CO2 and H2S over a wide range of temperature and pressure, Fluid Phase Equilib., 2008, 272, 65–74 CrossRef CAS.
- A. Botha and C. A. Strydom, DTA and FT-IR analysis of the rehydration of basic magnesium carbonate, J. Therm. Anal. Calorim., 2003, 71, 987–996 CrossRef CAS.
- C. M. Janet, B. Viswanathan, R. P. Viswanath and T. K. Varadarajan, Characterization and photoluminescence properties of MgO microtubes synthesized from hydromagnesite flowers, J. Phys. Chem. C, 2007, 111, 10267–10272 CrossRef CAS.
- T. Selvamani, A. Sinhamahapatra, D. Bhattacharjya and I. Mukhopadhyay, Rectangular MgO microsheets with strong catalytic activity, Mater. Chem. Phys., 2011, 129, 853–861 CrossRef CAS.
- R. Černý, V. Valvoda and M. Chládek, Empirical texture corrections for asymmetric diffraction and inclined textures, J. Appl. Crystallogr., 1995, 28, 247–253 CrossRef.
- M. Akao and S. Iwai, The hydrogen bonding of hydromagnesite, Acta Crystallogr., Sect. B, 1977, 33, 1273–1275 CrossRef.
- A. Abu Fara, M. R. Rayson, G. F. Brent, T. K. Oliver, M. Stockenhuber and E. M. Kennedy, Formation of magnesite and hydromagnesite from direct aqueous carbonation of thermally activated lizardite, Environ. Prog. Sustainable Energy, 2019, 38(3), e13244 CrossRef.
- Y. Wang, et al., Preparation, properties and phase transition of mesoporous hydromagnesite with various morphologies from natural magnesite, Powder Technol., 2020, 364, 822–830 CrossRef CAS.
- H. Pang, G. Ning, W. Gong, J. Ye and Y. Lin, Direct synthesis of hexagonal Mg(OH)2 nanoplates from natural brucite without dissolution procedure, Chem. Commun., 2011, 47, 6317–6319 RSC.
- D. Bhattacharjya, T. Selvamani and I. Mukhopadhyay, Thermal decomposition of hydromagnesite: Effect of morphology on the kinetic parameters, J. Therm. Anal. Calorim., 2012, 107, 439–445 CrossRef CAS.
- T. Selvamani, T. Yagyu, S. Kawasaki and I. Mukhopadhyay, Easy and effective synthesis of micrometer-sized rectangular MgO sheets with very high catalytic activity, Catal. Commun., 2010, 11, 537–541 CrossRef CAS.
- S. Guermech, J. Mocellin, L.-H. Tran, G. Mercier and L.-C. Pasquier, A study of hydromagnesite and nesquehonite precipitation in indirect aqueous carbonation of thermally activated serpentine in a batch mode, J. Cryst. Growth, 2022, 584, 126540 CrossRef CAS.
- H. Y. Atay and E. Çelik, Use of Turkish huntite/hydromagnesite mineral in plastic materials as a flame retardant, Polym. Compos., 2010, 31, 1692–1700 CrossRef CAS.
- A. A. Basfar and H. J. Bae, Influence of magnesium hydroxide and huntite hydromagnesite on mechanical properties of ethylene vinyl acetate compounds crosslinked by dicumyl peroxide and ionizing radiation, J. Fire Sci., 2010, 28, 161–180 CrossRef CAS.
- Q. Li, Y. Ding, G. Yu, C. Li, F. Li and Y. Qian, Fabrication of light-emitting porous hydromagnesite with rosette-like architecture, Solid State Commun., 2003, 125, 117–120 CrossRef CAS.
- D. A. Vermilyea, The dissolution of MgO and Mg(OH)2 in aqueous solutions, J. Electrochem. Soc., 1969, 116, 1179 CrossRef CAS.
- R. Hay, N. T. Dung, A. Lesimple, C. Unluer and K. Celik, Mechanical and microstructural changes in reactive magnesium oxide cement-based concrete mixes subjected to high temperatures, Cem. Concr. Compos., 2021, 118, 103955 CrossRef CAS.
- S. Katre, P. Ochonma, H. Asgar, A. M. Nair, K. Ravi and G. Gadikota, Mechanistic insights into the co-recovery of nickel and iron via integrated carbon mineralization of serpentinized peridotite by harnessing organic ligands, Phys. Chem. Chem. Phys., 2024, 26, 9264–9283 RSC.
- G. Gadikota, J. Matter, P. Kelemen and A. H. A. Park, Chemical and morphological changes during olivine carbonation for CO2 storage in the presence of NaCl and NaHCO3, Phys. Chem. Chem. Phys., 2014, 16, 4679–4693 RSC.
- G. Gadikota, Carbon mineralization pathways for carbon capture, storage, and utilization, Commun. Chem., 2021, 4, 23 CrossRef CAS PubMed.
- G. Gadikota, Multiphase carbon mineralization for the reactive separation of CO2 and directed synthesis of H2, Nat. Rev. Chem., 2020, 4, 78–89 CrossRef CAS PubMed.
- M. Liu and G. Gadikota, Integrated CO2 capture, conversion, and storage to produce calcium carbonate using an amine looping strategy, Energy Fuels, 2019, 33, 1722–1733 CrossRef CAS.
- L. A. Hollingbery and T. R. Hull, The thermal decomposition of huntite and hydromagnesite—A review, Thermochim. Acta, 2010, 509, 1–11 CrossRef CAS.
- A. A. Olajire, A review of mineral carbonation technology in sequestration of CO2, J. Pet. Sci. Eng., 2013, 109, 364–392 CrossRef CAS.
- X. Gao, H. Asgar, I. Kuzmenko and G. Gadikota, Architected mesoporous crystalline magnesium silicates with ordered pore structures, Microporous Mesoporous Mater., 2021, 327, 111381 CrossRef CAS.
- J. Ilavsky, F. Zhang, A. J. Allen, L. E. Levine, P. R. Jemian and G. G. Long, Ultra-small-angle X-ray scattering instrument at the Advanced Photon Source: History, recent development, and current status, Metall. Mater. Trans. A, 2013, 44, 68–76 CrossRef CAS.
- U. Bonse and M. Hart, Tailless X-ray single-crystal reflection curves obtained by multiple reflection, Appl. Phys. Lett., 1965, 7, 238–240 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4im00157e |
| This journal is © Institute of Process Engineering of CAS 2025 |