
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unlocking the potential of chemical-assisted water electrolysis for green hydrogen production†
Jiwoo Lee
a,
Sol A. Lee
ab,
Tae Hyung Lee
c and
Ho Won Jang
 *ad
*ad
aDepartment of Materials Science and Engineering, Research Institute of Advanced Materials, Seoul National University, Seoul 08826, Republic of Korea. E-mail: hwjang@snu.ac.kr
bDepartment of Applied Physics and Materials Science, California Institute of Technology, Pasadena 91126, USA
cSchool of Chemical and Biological Engineering, and Institute of Chemical Process, Seoul National University, Seoul 08826, Republic of Korea
dAdvanced Institute of Convergence Technology, Seoul National University, Suwon 16229, Republic of Korea
First published on 24th February 2025
Abstract
Despite global efforts to reduce the use of fossil fuels, carbon dioxide (CO2) emissions continue to rise. As the demand for clean energy grows, hydrogen (H2), which does not emit CO2 during combustion, is emerging as a promising energy resource. Among the various hydrogen production technologies, water electrolysis is attracting attention as a method for producing green hydrogen without carbon emissions. However, its high reaction overpotentials, due to complex reaction pathways, are a major factor limiting its energy efficiency. To address these issues, chemical-assisted water electrolysis is considered as an innovative alternative. This technology enables hydrogen production at lower voltages. Moreover, it can generate high-value products and remove pollutants, providing both environmental and energy benefits. In this review, we introduce various types of chemical-assisted water electrolysis and discuss the latest advances in catalyst design and reaction mechanisms aimed at reducing applied system voltage. Finally, we address the main challenges and prospects of chemical-assisted water electrolysis.
Keywords: Chemical-assisted water electrolysis; Hybrid water electrolysis; Overpotential; Hydrogen; Electrocatalyst; Value-added product.
 Jiwoo Lee | Jiwoo Lee is currently a PhD candidate under the supervision of Prof. Ho Won Jang at the Department of Materials Science and Engineering of Seoul National University (SNU). She received her BS degree in Materials Science and Engineering from Korea University in 2022. Her current research focuses on the design of metal-based catalysts and their application to the ammonia oxidation reaction. |
 Sol A Lee | Sol A Lee received her PhD degree from the Department of Materials Science and Engineering of Seoul National University in 2021 under the supervision of Prof. Ho Won Jang. She is currently a postdoctoral associate at the California Institute of Technology under the supervision of Prof. Harry Atwater. Her current research focuses on synthesizing electrocatalysts and semiconductors for energy conversion and designing unassisted tandem devices. |
 Tae Hyung Lee | Tae Hyung Lee received his PhD degree from the Department of Materials Science and Engineering of Seoul National University in 2021 under the supervision of Prof. Ho Won Jang. He is currently a postdoctoral associate at the Seoul National University under the supervision of Prof. Jungwon Park. His current research focuses on fabricating electrochemical catalysts for various energy conversion systems and their theoretical investigation through simulations. |
 Ho Won Jang | Ho Won Jang is a full professor at the Department of Materials Science and Engineering, Seoul National University. He received his PhD degree from the Department of Materials Science and Engineering, Pohang University of Science and Technology in 2004. He worked as a research associate at the University of Madison-Wisconsin from 2006 to 2009. He worked at the Institute of Science and Technology of Korea as a senior research scientist before joining Seoul National University. His research interests include the design of materials and device fabrication for (photo)electrocatalysis, thin film transistors, memristors, chemical sensors, and micro-light-emitting diodes. |
1 Introduction
Global carbon dioxide (CO2) emissions from fossil fuels were expected to reach 37.4 billion tons in 2024, reflecting a 0.8% increase compared to the previous year, according to recent data from the Global Carbon Project.1 This trend runs counter to global efforts to move toward a carbon-neutral society, highlighting the urgent need to reduce fossil fuel consumption and develop alternative clean energy technologies. Among these, hydrogen (H2) emerges as a critical resource for the transition to sustainable energy due to its ability to produce zero carbon dioxide (CO2) upon combustion.2 Hydrogen has a high energy density (140 MJ kg−1), which is significantly higher than that of conventional solid fuels (50 MJ kg−1).3 Moreover, hydrogen plays a key role in addressing the challenges of intermittent renewable energy supply, such as from solar and wind sources.4,5 By converting excess electricity into hydrogen and storing it, hydrogen offers a viable solution for energy storage and transportation. To fully realize this potential, it is crucial to produce hydrogen in a manner that does not emit carbon. Water electrolysis, a process that splits water (H2O) into hydrogen and oxygen (O2) using electricity, is the key technology for producing green hydrogen without carbon emissions.6,7 For this reason, water electrolysis is gaining increasing attention as a key technology for both energy transition and the realization of a sustainable society.The water electrolysis process consists of the following two half-cell reactions:
| Hydrogen Evolution Reaction (HER): 4H+ + 4e− → 2H2 |
| Oxygen Evolution Reaction (OER): 2H2O → O2 + 4H+ + 4e− |
| Overall Reaction: 2H2O → 2H2 + O2 |
Despite its great potential to produce clean hydrogen, water electrolysis still faces several challenges that must be addressed. While the HER is relatively simple, involving the transfer of only two electrons, the OER is far more complex, requiring the transfer of four electrons.8 The OER proceeds through a sequence of reaction intermediates: *OH → *O → *OOH → *O2. According to Man et al., the free energies required for the formation of these intermediates are theoretically the same, at 1.23 eV under ideal conditions.9 However, the *OH → *O and *O → *OOH transition steps require higher free energies in real systems. This results in the generation of an overpotential, requiring a higher applied potential to drive the reaction forward. Although significant efforts have been made to develop OER electrocatalysts with low overpotentials, current water electrolysis systems still face limitations in their efficiency for H2 production. One challenge is that O2 and H2 are produced simultaneously during the HER and OER processes, requiring complex systems for efficient gas separation and storage.10 In addition, O2 has limited economic value, underscoring the need for research aimed at producing value-added products at low potential.11
To address these limitations, chemical-assisted water electrolysis is emerging as a promising alternative to conventional water electrolysis.12 This approach replaces the anodic OER with alternative oxidation reactions that can occur at a lower voltage, thereby reducing the overall system voltage and improving energy efficiency. Various oxidation reactions, including the alcohol oxidation reaction, ammonia oxidation reaction, urea oxidation reaction, hydrazine oxidation reaction, and biomass oxidation reaction, can be employed at the anode in chemical-assisted water electrolysis.13–17 Fig. 1 illustrates the thermodynamic potentials of several chemical-assisted water electrolysis reactions. These reactions can be operated at lower voltages while generating high-value products or removing pollutants, depending on the operating conditions.18,19 This allows for both environmental improvement and energy generation. However, chemical-assisted water electrolysis faces challenges, including high overpotential, which arise from several reasons. To deal with the challenges and reduce the voltage for hydrogen generation, various strategies are being explored, such as facet engineering, nanostructuring, alloying, doping, and computational prediction.20–24
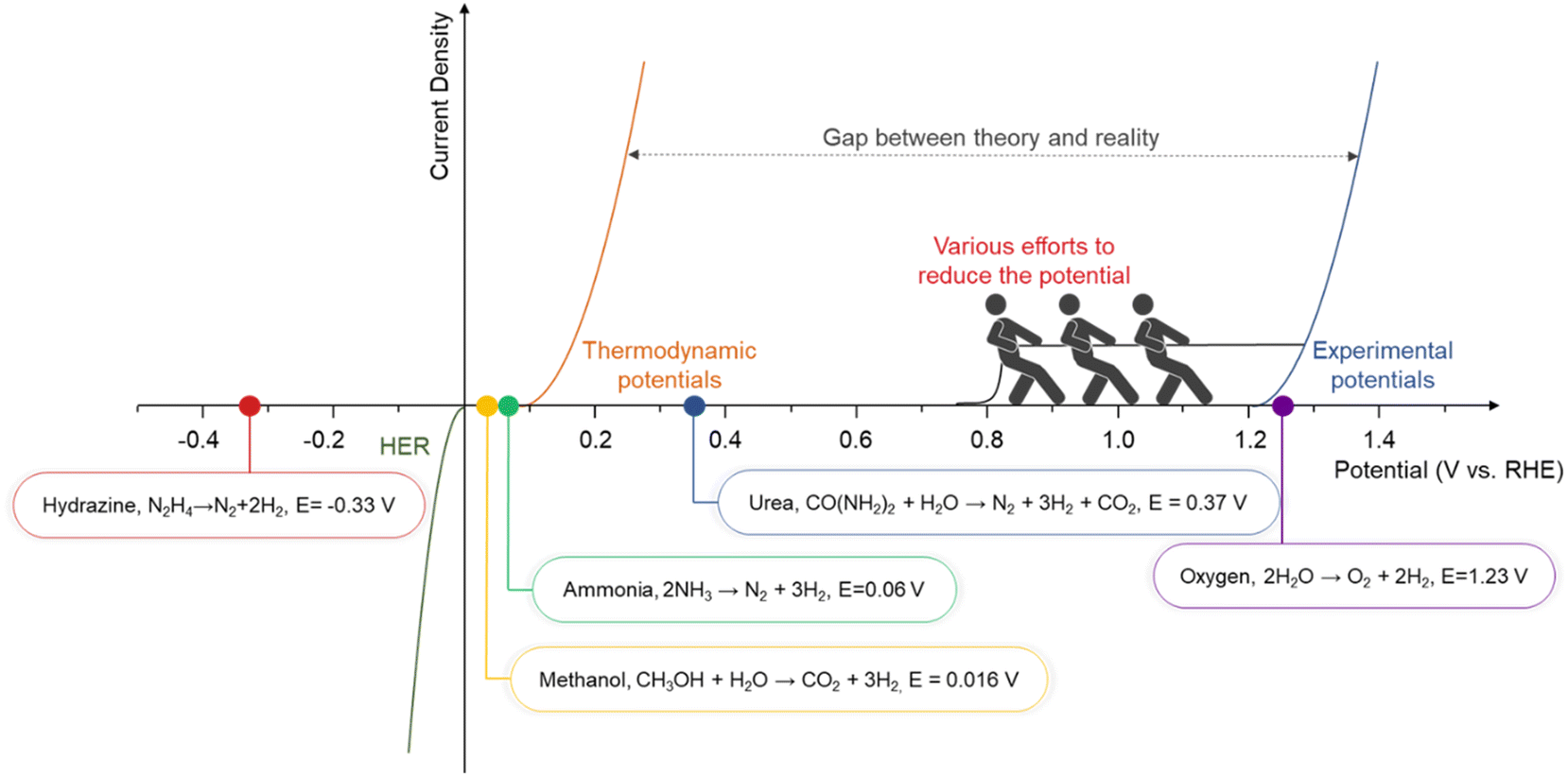 | ||
| Fig. 1 The gap between theoretical potential and practical application in various types of chemical-assisted water electrolysis. | ||
In this review, we examine various chemical-assisted water electrolysis systems—alcohol oxidation, ammonia oxidation, urea oxidation, hydrazine oxidation, and biomass oxidation—that enable hydrogen production at low voltages. As previously mentioned, the HER is relatively simple, and its overpotential does not vary significantly with the presence or absence of additives.25–27 Therefore, in this review, we primarily focus on anodic reactions, as they are critical in determining the overall energy efficiency and performance of chemical-assisted water electrolysis systems. Additionally, we explore the latest research trends in catalyst design and reaction mechanisms, focusing on the strategies to reduce the system voltage. Finally, we address the main challenges that chemical-assisted water electrolysis must overcome and provide insights into future prospects.
2 Strategies to reduce the overpotential of hybrid water electrolysis
2.1 Alcohol oxidation reaction
Methanol (CH3OH), as the simplest alcohol, has been extensively studied. In the methanol oxidation reaction (MOR), six electrons are involved in the complete oxidation of methanol to CO2. The thermodynamic potential of this process is 0.016 V, which is notably lower than that of the OER.31,32 The methanol oxidation reaction at the anode proceeds as follows:
| CH3OH + H2O → CO2 + 6H+ + 6e− (acidic electrolyte) |
| CH3OH + 6OH− → CO2 + 5H2O + 6e− (alkaline electrolyte) |
The MOR can proceed via two distinct pathways: the direct pathway and the indirect pathway, which differ in the formation of CO intermediates.31,36 In the direct pathway, methanol adsorbs on the catalyst, is gradually oxidized, and is eventually converted to CO2. In the indirect pathway, methanol adsorbs onto the catalyst surface, and its C–H bonds are sequentially broken to form COHx,ads intermediates and, finally, COads. However, strongly adsorbed COads species can deactivate the catalyst, resulting in a reduction in catalytic activity.
Platinum (Pt) is widely recognized as the most effective catalyst for the MOR. However, its application is hindered by slow reaction kinetics, which require an overpotential exceeding 0.4 V.37 This sluggish behavior is attributed to the complexity of the reaction mechanism, which involves a six-electron transfer. Furthermore, platinum is highly susceptible to poisoning by COads, limiting its long-term stability.38 To overcome these challenges, researchers have actively pursued the development and synthesis of advanced catalysts, employing various strategies to mitigate CO poisoning and enhance the performance of Pt-based electrocatalysts at low overpotentials.
Kim et al. proposed a single-atom-to-cluster (SAC) approach to form a monolayer of single Pt atoms on the surface of thiolated multiwalled carbon nanotubes (S-MWNTs) and then controlled the cluster size by heat treatment followed by slow quenching (q).39 Fig. 2a shows that the Pt cluster size varies with the heat treatment temperature (Th), and the particle size becomes larger and the size distribution becomes wider as Th increases. In situ X-ray diffraction (XRD) analysis proves that increasing Th enhances the crystallinity and enlarges the cluster size. In addition, the cyclic voltammetry (CV) patterns of Pth–q/MWNTs processed at different Th, shown in Fig. 2b, show that the onset potential of Pth–q/MWNTs decreases with smaller cluster size. This onset potential downshift is because the quantum-size effect becomes more pronounced as the Pt cluster size decreases, which leads to an increase in s–d mixing. X-ray absorption fine structure (EXAFS) and near edge structure (XANES) were used to quantitatively confirm the cluster size and electronic structure changes, and it was observed that the Pt–Pt bonding peak becomes more pronounced with increasing Th, and the electronic structure approaches the bulk state. These results demonstrate the transition of Pt from single atoms to clusters. Similarly, Chen et al. demonstrated the size effect of Pt-on-Au core–satellite nanocomposites by controlling the size of Pt particles from 2.1 to 7.6 nm using a seed-mediated growth method.40 Pt nanoparticles as small as 2.1 nm exhibited outstanding performance in the MOR, with a higher jf/jb ratio, indicating improved CO tolerance. This superior CO tolerance resulted in better long-term stability compared to nanocomposites with larger Pt nanoparticles.
 | ||
| Fig. 2 Methanol oxidation reaction (MOR). (a) Size distribution of Pth–q/MWNTs treated at various heating temperatures. (b) MOR CV graph of Pt-S-MWNTs and Pth–q/MWNTs treated at various heating temperatures and of Pt/CB in 0.5 M H2SO4 with 2 M CH3OH. (a and b) Adapted with permission.39 Copyright 2006, WILEY-VCH. (c) Scheme of the synergistic catalytic process between Co single atoms and adjacent Pt sites. (d) MOR LSV curves of Pt/C, N–C/Pt, and Co–N–C-x/Pt (x = 50, 100, and 300) in 1 M KOH with 3 M CH3OH. (c and d) Adapted with permission.41 Copyright 2022, Wiley-VCH GmbH. (e) MOR LSV curves of NiO, Fe2O3, RuO2, NiO/Fe2O3 mixture and Fe-NF-500 in 1 M KOH with 1 M CH3OH. (f) Charge density distribution of the Fe2O3/NiO heterojunction and plane-average electron difference diagram. (g) PDOS calculated for NiO and Fe2O3/NiO. (e–g) Adapted with permission.42 Copyright 2023, The Royal Society of Chemistry. | ||
In addition to particle size control, introducing other metals has become a key strategy in enhancing the performance of MOR catalysts. Ruan et al. proposed a novel approach by introducing Co single atoms on nitrogen-doped carbon (N–C) supports, followed by anchoring them to Pt nanoparticles.41 As shown in Fig. 2c, the Co single atoms modify the electronic structure of Pt by redistributing electrons on the support, thereby optimizing the activity of Pt nanoparticles. One of the significant contributions of Co single atoms is their oxophilic property, which promotes OH− adsorption and facilitates the oxidation of COads intermediates at the Pt active center. Fig. 2d shows the mass activity of Co-N–C-x/Pt in 1 M KOH with 3 M methanol, showing that the Co–N–C/Pt sample exhibited higher current density than N–C/Pt (1.8 A mgPt−1) and commercial Pt/C (1.6 A mgPt−1). Notably, Co–N–C-100/Pt reached a current density of about 6 A mgPt−1 and had a low onset potential. The effectiveness of the Co single atoms was demonstrated by CO-stripping measurements. The onset potential of the CO oxidation peak of Co–N–C-x/Pt was lower than that of N–C/Pt and commercial Pt/C, indicating that a smaller driving force is required to remove COads intermediates from the Pt surface. This indicates that the Co single atoms greatly enhance the CO tolerance of the Pt active sites. In addition, in the durability test, Co–N–C/Pt retained 60% of its initial current density after 4000 seconds, compared to only 25% for commercial Pt/C, demonstrating outstanding stability. Kuang et al. synthesized Pt/N-MoSe2@MHCS by applying N-doping and interfacial engineering to MoSe2 supported on mesoporous carbon spheres.43 N-doping with NH3 plasma enhanced the interaction between the support and Pt nanoparticles, leading to uniform dispersion. When Pt/N-MoSe2@MHCS is used as both an anode and a cathode in a two-electrode system, only 0.60 V is required to generate a current density of 10 mA cm−2 in 0.5 M H2SO4 with 1 M methanol. This is 1.07 V lower than that required for conventional water electrolysis, demonstrating that H2 production can be achieved with significantly lower energy input. Similarly, Zhou et al. synthesized Pt nanoparticles supported on CoSe/N-doped carbon nanospheres (Pt-CoSe/NCs) and utilized them as MOR catalysts, achieving a peak current density of 84.2 mA cm−2 in 0.5 M H2SO4 and 1 M methanol, a 3.1-fold increase over Pt/C.44 Pt-CoSe/NCs exhibited a low overpotential and high stability over 1000 cycles for HER performance. DFT calculations confirmed that CoSe/NCs enhanced the charge density of Pt, optimizing the H and CO adsorption energies. In the two-electrode system, Pt/C||Pt/C required 0.84 V to achieve 10 mA cm−2, whereas Pt-CoSe/NC-800||Pt-CoSe/NC-800 required only 0.67 V. Zhang et al. synthesized Pt–Ln/C catalysts (Ln = La, Ce, Pr, Nd) and explored how lanthanide elements induce electronic perturbations that influence catalytic activity.45 Alloying Pt with a Ln element modulates the electronic structure of Pt, significantly affecting CO adsorption and poisoning. DFT calculations revealed that the d–f bonding effect of Ce optimizes the d-band centers of Pt. This adjustment weakens the binding strength of intermediates such as COads. Furthermore, the electronic interaction between Pt and Ce redistributed the electron density on the Pt surface, increasing the energy barrier for CO formation while lowering the adsorption energy of COads. It significantly affects the onset potential, with Pt5Ce/C showing an onset potential of 0.17 V vs. RHE in 1 M KOH with 1 M methanol, which is 0.25 V lower than that of the Pt/C catalyst (0.42 V vs. RHE). These combined mechanisms inhibit the formation of COads, mitigate poisoning effects, and significantly enhance the durability of Pt catalysts.
The alloying of multiple elements ultimately leads to the fabrication of high entropy alloys (HEAs). Li et al. successfully synthesized Pt18Ni26Fe15Co14Cu27/C nanoparticles using a simple low-temperature oil phase synthesis method.46 This HEA exhibited exceptional MOR performance due to its multiple active sites and fast electron transfer ability. Partial projected density of states (PDOSs) analysis revealed that the strong electronic interactions between non-noble transition metals (Ni, Fe, Co, Cu) and Pt in the HEA structure shifted the d-band center of Pt closer to the Fermi level compared to commercial Pt/C. This modification in the electronic structure efficiently lowers the reaction energy barrier and enhances the structural stability of the catalyst, resulting in significant catalytic performance improvements. This also influences the onset potential, with the Pt18Ni26Fe15Co14Cu27/C nanoparticles exhibiting an onset potential of 412 mV vs. RHE, representing a significant 133 mV reduction compared to the commercial Pt/C catalyst. The HEA catalyst exhibited a peak mass activity of 15.04 A mgPt−1 and a peak area activity of 114.93 mA cm−2 in 1 M KOH with 1 M methanol, respectively, which were superior to those of the commercial Pt/C catalyst (1.45 A mgPt−1 and 27.48 mA cm−2, respectively). Moreover, durability tests revealed that the Pt18Ni26Fe15Co14Cu27/C catalyst retained 93.6% of its initial performance after 1000 cycles, demonstrating excellent stability compared to Pt/C, which showed 26.9% degradation.
Due to the problem of low stability caused by CO poisoning and the high cost of noble metals, non-noble metal-based MOR electrocatalysts, which are naturally abundant and theoretically exhibit excellent electrocatalytic activity, have recently attracted attention.47 Li et al. converted a Ni-metal organic framework (MOF) to Ni(OH)2-x through alkali treatment using different NaOH concentrations (x = 0.025, 0.25, and 0.5).48 Among these, the Ni(OH)2 treated with 0.25 M NaOH exhibited the best balance between structural stability and active sites, showing better activity compared to the other samples at both low (1.5 V vs. RHE) and high potential (1.7 V vs. RHE). Notably, at 1.5 V vs. RHE, Ni(OH)2-0.25 recorded 171.8 mA cm−2 in 1 M KOH with 1 M methanol, exceeding the performance of Ni(OH)2-0.025 and Ni(OH)2-0.5 by 1.5- and 2.5-fold, respectively. The alkali treatment converted the Ni-MOF to Ni(OH)2, enhancing the number of active sites and optimizing the electron transfer pathway. Furthermore, Ni(OH)2 promotes the MOR more effectively by increasing the surface oxygen vacancies and Ni3+/Ni2+ ratio, which are crucial for improving catalytic performance. The effect of oxygen vacancies (VO) has been investigated by Yang et al. in ultrathin-NiO nanosheets prepared by controlling the calcination reaction time.49 In 1 M KOH with 0.5 M methanol, VO-rich NiO achieved a current density of 85.3 mA cm−2 at 0.7 V vs. Ag/AgCl, which is 2.3 and 3.5 times higher activity than that of VO-poor and bulk NiO, respectively. In addition, the Tafel slope was reduced to 42 mV per decade on VO-rich NiO, compared to 80 mV per decade on VO-poor NiO and 111 mV per decade on bulk NiO, highlighting excellent reaction kinetics. VO-rich NiO also maintained a stable current density of approximately 25 mA cm−2 at 0.5 V vs. Ag/AgCl for 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s. These results indicate that oxygen defects in VO-rich NiO play a critical role in enhancing MOR catalytic performance. Hou et al. synthesized Ni NPs coated with ultrathin graphitic carbon shells by pyrolysis of Ni–Zn MOFs and utilized them as bifunctional catalysts for the HER and MOR.50 DFT calculations confirmed that the thin carbon shells optimized the electron transfer to Ni, ideally tuning the Gibbs free energy for H adsorption. Comparison of the LSV curves of the HER in 1 M KOH with and without 1 M methanol confirmed that the electrolyte does not interfere with the HER process. The two-electrode system using Ni1(Zn)@C as an anode and a cathode requires 1.63 V to achieve a current density of 10 mA cm−2 and shows good durability. Hao et al. investigated Fe2O3/NiO heterostructure catalysts synthesized via an ultrafast solution combustion strategy.42 Fe-NF-500 quickly reached an industrial-scale current density of 600 mA cm−2 at a low potential of 1.472 V vs. RHE in 1 M KOH with 1 M methanol, significantly outperforming other catalysts (Fig. 2e). The onset potential of Fe-NF-500 is close to the Ni2+/Ni3+ redox, indicating that Ni3+ was the actual active site in the MOR. The highly dispersed Fe–O–Ni interface promoted the electron excitation into the conduction band, improving the charge transport and diffusion ability. This accelerated the kinetics of the methanol oxidation process. DFT calculations also supported this electron transport mechanism. As shown in Fig. 2f, the plane-average electron difference diagram plotted along the direction of the heterojunction showed that electrons were transferred and accumulated from NiO to Fe2O3, and the NiO at the interface became electron-deficient. Additionally, the PDOS results in Fig. 2g showed that the d-band center of the Fe2O3/NiO heterojunction was significantly closer to the Fermi level compared to pure NiO. This shift enhances the anti-bonding state, which contributes to the strong coupling between the catalyst and methanol. Fig. 3 shows the stability time as a function of the potential applied to the MOR electrocatalyst. While noble metal-based catalysts exhibit shorter stability times, they facilitate reactions at relatively low potentials. However, non-noble metal-based catalysts operate at higher potentials, requiring more energy but demonstrating longer stability times.
000 s. These results indicate that oxygen defects in VO-rich NiO play a critical role in enhancing MOR catalytic performance. Hou et al. synthesized Ni NPs coated with ultrathin graphitic carbon shells by pyrolysis of Ni–Zn MOFs and utilized them as bifunctional catalysts for the HER and MOR.50 DFT calculations confirmed that the thin carbon shells optimized the electron transfer to Ni, ideally tuning the Gibbs free energy for H adsorption. Comparison of the LSV curves of the HER in 1 M KOH with and without 1 M methanol confirmed that the electrolyte does not interfere with the HER process. The two-electrode system using Ni1(Zn)@C as an anode and a cathode requires 1.63 V to achieve a current density of 10 mA cm−2 and shows good durability. Hao et al. investigated Fe2O3/NiO heterostructure catalysts synthesized via an ultrafast solution combustion strategy.42 Fe-NF-500 quickly reached an industrial-scale current density of 600 mA cm−2 at a low potential of 1.472 V vs. RHE in 1 M KOH with 1 M methanol, significantly outperforming other catalysts (Fig. 2e). The onset potential of Fe-NF-500 is close to the Ni2+/Ni3+ redox, indicating that Ni3+ was the actual active site in the MOR. The highly dispersed Fe–O–Ni interface promoted the electron excitation into the conduction band, improving the charge transport and diffusion ability. This accelerated the kinetics of the methanol oxidation process. DFT calculations also supported this electron transport mechanism. As shown in Fig. 2f, the plane-average electron difference diagram plotted along the direction of the heterojunction showed that electrons were transferred and accumulated from NiO to Fe2O3, and the NiO at the interface became electron-deficient. Additionally, the PDOS results in Fig. 2g showed that the d-band center of the Fe2O3/NiO heterojunction was significantly closer to the Fermi level compared to pure NiO. This shift enhances the anti-bonding state, which contributes to the strong coupling between the catalyst and methanol. Fig. 3 shows the stability time as a function of the potential applied to the MOR electrocatalyst. While noble metal-based catalysts exhibit shorter stability times, they facilitate reactions at relatively low potentials. However, non-noble metal-based catalysts operate at higher potentials, requiring more energy but demonstrating longer stability times.
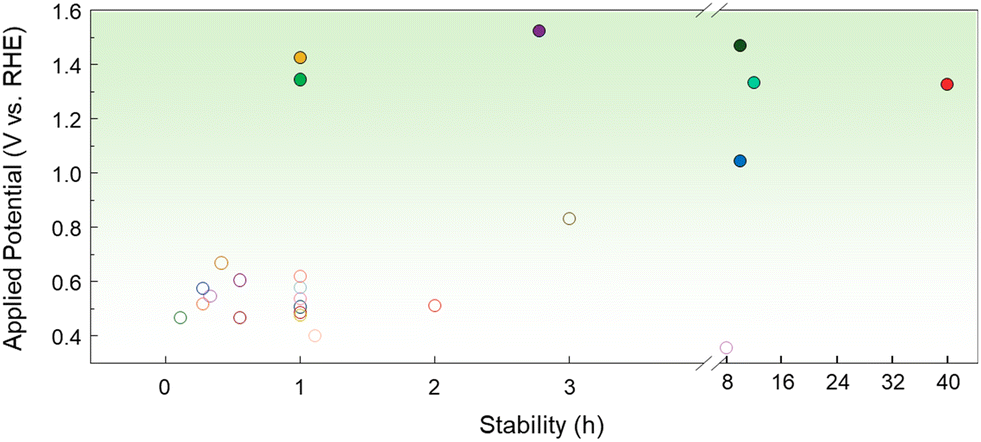 | ||
| Fig. 3 Comparison of the MOR performance of various electrocatalysts. Each potential was converted to RHE, considering the reference electrode and pH of the electrolyte. The detailed values are shown in the ESI.† | ||
| CH3CH2OH + H2O → 2CO2 + 12H+ + 12e− (acidic electrolyte) |
| CH3CH2OH + 12OH− → 2CO2 + 9H2O + 12e− (alkaline electrolyte) |
Similar to MOR catalysts, EOR catalysts also exhibit low overpotential in Pt-based materials, prompting numerous studies to enhance their efficiencies. Li et al. evaluated the MOR performance of Pt combined with hydroxyl-rich CeO2 and observed that the abundant OH groups and oxygen vacancies in the support optimized the electron transfer to Pt.55 This optimization reduced the adsorption of COads intermediates, resulting in high catalytic activity and durability. Wang et al. investigated how the surface structures of Pt influence the reaction pathway and product selectivity.56 Using transition-state (TS) searching methods and DFT calculations, they calculated the reaction barriers and adsorption energies of intermediates on Pt(111), Pt(211), and Pt(100) surfaces. Their findings revealed that Pt(100) is the most effective surface for the complete oxidation of ethanol to CO2. This efficiency arises from its ability to promote C–C bond cleavage through strongly adsorbed intermediates such as CH2CO or CHCO. In contrast, Pt(111) favors the formation of partial oxidation products such as CH3CHO and CH3COOH, showing that the selectivity of the EOR is highly sensitive to the surface structure. Pierozynski experimentally analyzed the effect of the surface crystal structure on the EOR in alkaline media using polycrystalline Pt, Pt(111), and Pt(100) single-crystal surfaces.57 The experimental results showed that Pt(111) exhibited higher catalytic activity and faster kinetics than Pt(100) and polycrystalline Pt. This is attributed to the separation of the underpotential deposition (UPD) H region at low potential and the OH− adsorption region at high potential. These distinct regions prevent competitive adsorption and allow for more efficient catalytic reactions. In contrast, on Pt(100), the overlap of the OH− and H adsorption regions causes OH− and H to be competitively adsorbed at the same site and reduces the catalytic reaction efficiency. The results show that Pt(111) promotes the oxidation of partial oxidation products such as CH3CHO and CH3COOH, while reducing the poisoning of intermediates such as COads, which can improve the reaction performance.
Extending this, researchers are focusing on facet modification to increase EOR efficiency. Liu et al. proposed a lattice expansion strategy for Pd@Pt core–shell nanoparticles to enhance the EOR performance.58 The lattice of the Pt shell was expanded by inserting H into the Pd core, thereby converting it to PdH0.43, which optimized the electronic structure of the catalyst and significantly improved the catalytic performance. Through high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images and fast Fourier transform (FFT) analysis, they confirmed that the core–shell structure was not destroyed and the lattice was expanded after H insertion. Notably, the Pt–Pt spacing increased during conversion to the PdH0.43 core, resulting in a lattice expansion of about 2.55% on the Pt(100) side. DFT calculations were conducted to determine the free energies of OH and CO adsorption on the Pt(100) and Pt(111) sides. The results demonstrated a reduction in the adsorption free energy of OH on the PdH0.43@Pt surface, facilitating OH adsorption. This effect is particularly pronounced on the (100) facet, which exhibits stronger Pt–OH and Pt–CO bonds due to its lower coordination number. Although the adsorption free energy of CO shows little change, the enhanced OH adsorption contributes to the more effective removal of CO poisoning. Consequently, the specific activity of PdH0.43@Pt was 14.86 A mgPt−1, representing a 25.19-fold improvement over commercial Pt/C in 1 M KOH with 1 M methanol.
Alloying is a strategy used to increase the efficiency of catalysts by modifying the structural and electronic properties of Pt. Yan et al. synthesized Pt3Ga-200/C catalysts by alloying Ga with Pt to improve the oxidation efficiency of the C2 pathway by tuning the electronic structure of Pt.59 The specific activity of Pt3Ga-200/C was 2.461 mA cm−2 at 0.56 V vs. SCE, which was more than four times higher than that of Pt/C, measured to be 0.584 mA cm−2 at 0.59 V vs. SCE in 0.1 M HClO4 with 0.1 M ethanol. According to the in situ Fourier transform infrared reflection (FTIR) spectroscopy results in Fig. 4a, compared to Pt3Ga-200/C, Pt/C showed a weaker CO2 signal and a distinct CO band caused by partial acetaldehyde dissociation. In contrast, Pt3Ga-200/C directly and continuously oxidized acetaldehyde to efficiently produce CO2. Fig. 4b shows the CO2 band area ratio calculated from the FTIR results, indicating that Pt3Ga-200/C achieved much higher CO2 selectivity than Pt/C. This highlights the critical role of Ga in enhancing catalytic efficiency. The CO stripping voltammetry results in Fig. 4c show that the Pt3Ga/C catalysts, including Pt3Ga-200/C, exhibit significantly better resistance to CO poisoning than Pt/C. This is attributed to the electronic modification of Ga, which reduces CO adsorption and promotes the rapid oxidation of CO. This effect arises from the shift of the d-band center of Pt atoms, which decreases CO adsorption, and the high hydrophilicity of the Ga surface facilitates the generation of more *O or *OH species, promoting CO oxidation at Pt sites. Wang et al. enhanced the EOR performance by incorporating monodispersed Ga atoms into Pt3Mn nanocrystals, forming Ga–O–Pt interfaces.60 The Ga–O–Pt3Mn nanocatalyst achieved a high activity of 4.71 mA cm−2 at 0.7 V vs. RHE in 0.1 M HClO4 with 0.5 M ethanol, outperforming Pt3Mn and commercial Pt/C by 3.68 and 8.41 fold, respectively. In situ FTIR spectroscopy revealed that CO2 generation occurred at 0.54 V vs. RHE on Ga–O–Pt3Mn, significantly lower than the 0.74 V vs. RHE required for Pt3Mn, demonstrating the inhibition of toxic CO intermediate formation. This result is consistent with the C1/C2 product ratio, indicating that Ga–O–Pt3Mn promotes C–C decomposition and follows a complete oxidation pathway. DFT calculations further confirmed that p–d orbital hybridization at the catalytic interface reduces both the intermediate formation and reaction energy barriers, facilitating efficient C–C bond cleavage. These results demonstrate that Ga doping enables effective ethanol oxidation at low potentials.
 | ||
| Fig. 4 Ethanol oxidation reaction (EOR). (a) In situ MSFTIR spectra of Pt3Ga-200/C and Pt/C for CO2 and HAc during the EOR. (b) The percentage of band area of CO2 (SCO2/(SCO2 + SHAc)). (c) CO-stripping voltammetry of catalysts in 0.1 M HClO4. (a–c) Adapted with permission.59 Copyright 2023, American Chemical Society. (d) LSV activation curve of Co(OH)2@Ni(OH)2. (e) Ni 2p XPS spectra before and after electrochemical activation. (f) EOR free energy diagram for CoOOH, NiOOH and CoOOH@NiOOH based on DFT calculations. (d–f) Adapted with permission.61 Copyright 2023, The Royal Society of Chemistry. | ||
In recent decades, significant efforts have been made to enhance the catalytic activity of Ni-based EOR catalysts through the rational design of active surfaces and interfaces.62 These improvements have been achieved by manipulating factors such as the atomic arrangement, electronic structure, and the incorporation of dopant elements.12 Chi et al. synthesized 2D MOF1-CDs@CC by combining a Ni-MOF with carbon dots (CDs) through a solvothermal method.63 The CDs enhanced the conductivity of the MOF, improving charge transfer efficiency and promoting ethanol adsorption. In 1 M KOH with 1 M ethanol, the optimized MOF1-CDs@CC demonstrated a high current density of 119 mA cm−2 at 1.6 V, with a significantly accelerated EOR onset compared to MOF1. This improvement is attributed to the increased surface area and high stability provided by the CDs, emphasizing the critical role of the support in catalytic performance. Metal doping can modify the electronic structure to increase catalytic activity. Wang et al. synthesized Cu-doped NiOOH by electrochemical reconstruction of a NiCu alloy.64 X-ray photoelectron spectroscopy (XPS) analysis revealed that the Ni3+/Ni2+ ratio in Cu-doped NiOOH is 72%, approximately twice that of NiOOH. This indicates that Cu doping increases the proportion of high-valence Ni species, which enhances the conductivity of NiOOH and facilitates higher charge transfer rates. In situ electrochemical Raman spectroscopy confirmed the production of acetaldehyde and acetate at different potentials during the EOR. However, 1H nuclear magnetic resonance (NMR) analysis detected no acetaldehyde signals, suggesting that acetaldehyde serves as a possible intermediate, subsequently converting to acetate during the EOR process. The Cu-doped NiOOH exhibited excellent stability, maintaining 73% of its initial current density for 32 hours at 1.7 V vs. RHE in 1 M KOH with 1 M ethanol. Li et al. synthesized a Co(OH)2@Ni(OH)2 heterostructure with a large specific surface area by a one-pot hydrothermal process using a Ni-doped ZIF-67 precursor.61 The in situ electrochemical activation process, as shown in Fig. 4d, demonstrated an enhancement in the EOR catalytic performance. XPS analysis in Fig. 4e revealed that the Ni 2p peak shifted by 1.4 eV to a higher binding energy after activation. In addition, an increase in Ni3+ species was observed, providing evidence for the conversion of Ni(OH)2 to NiOOH. The Co(OH)2@Ni(OH)2 heterostructure showed superior performance in terms of current density and stability compared to pure Ni(OH)2 and Co(OH)2 catalysts. In 1 M KOH with 1 M ethanol, the catalyst required a potential of 1.301 V vs. RHE to achieve a current density of 10 mA cm−2, which is 92 mV lower than that of Ni(OH)2. It also demonstrated excellent long-term stability, maintaining performance for over 100 hours at 50 mA cm−2. After the 20 h stability test, 1H NMR analysis showed a decrease in the ethanol signal and an increase in the acetic acid signal. Faradaic efficiency (FE) calculations confirmed a high potassium acetate selectivity of 97.9% up to 100 h, indicating improved reaction efficiency. Based on these results, the four-electron EOR pathway of *CH3CH2OH → *CH3CH2O → *CH3CHO → *CH3CO → *CH3COOH was proposed (Fig. 4f). During the EOR process, the second dehydrogenation step from *CH3CH2O to *CH3CHO was identified as the rate-determining step. The energy barrier for this step in CoOOH@NiOOH was 1.64 eV, which was significantly lower than that of CoOOH (2.21 eV) and NiOOH (2.76 eV). Furthermore, *CH3CHO was identified as the most unfavorable intermediate during the EOR process, theoretically explaining why the main product is acetic acid rather than acetaldehyde. Specifically, in the Pt/C||Co(OH)2@Ni(OH)2 two-electrode system, when methanol is added to 1 M KOH, 1.464 V is required to achieve 100 mA cm−2. This is a 200 mV decrease from the typical water splitting value.
2.2 Ammonia oxidation reaction
Ammonia (NH3) has high potential as an H2 carrier due to its high hydrogen storage capacity of 17.6 wt%. Unlike conventional hydrogen storage technologies, ammonia is easier and safer to handle, as it exists as a liquid at room temperature and pressure, making storage and transportation more practical.65,66 The ammonia oxidation reaction (AOR) is the reverse reaction of the Haber–Bosch process. It requires a low thermodynamic potential of 0.06 V, which reduces energy consumption by approximately 95% compared to the OER. Unlike methanol and ethanol oxidation reactions, which produce CO2, the ammonia oxidation reaction produces N2, a pollution-free substance.67,68 The production of N2 at the anode in an alkaline electrolyte proceeds as follows;| 2NH3 + 6OH− → N2 + 6H2O + 6e− |
| 2NH3 + 7OH− → NO2− + 5H2O + 6e− |
| 2NH3 + 9OH− → NO3− + 6H2O + 6e− |
The AOR involves the breaking of the N–H bond in the ammonia molecule and the release of N2 gas. Two primary mechanisms have been proposed for the AOR: the Oswin–Salomon mechanism and the Gerischer–Mauerer mechanism. In the Oswin–Salomon mechanism, NH3 undergoes dehydrogenation, sequentially producing *NH2, *NH, and *N intermediates, which then combine to form N2.71 In the Gerischer–Mauerer mechanism, NH3 is deprotonated to form an *NHx intermediate. The *NHx and *NHy intermediates subsequently combine to form a dimer (*N2Hx+y), which is converted to N2 through successive deprotonation steps.72 However, this process produces Nads, which can accumulate on the catalyst surface, leading to catalyst deactivation. The adsorption and desorption of these intermediates result in a relatively higher overpotential and limited durability. Therefore, catalysts that exhibit high performance and selectivity at low overpotentials, while maintaining stability, are critical for the ammonia oxidation reaction.73 To address these challenges, researchers are optimizing catalyst surface properties and electronic structure through various material design strategies.
Introducing other metals on Pt modifies the AOR performance by improving dehydrogenation ability, minimizing catalyst poisoning caused by Nads intermediates, lowering the onset potential, and modulating energy barriers.77,78 Pillai et al. utilized graph neural networks (GNNs) to predict catalytic activity, stability, and catalyst synthesizability descriptors, and then synthesized Pt3Ru–M (M: Fe, Co, Ni) alloy nanoparticles on reduced graphene oxide (rGO) and experimentally validated their catalytic performance.79 The onset potential of Pt3Ru1/2Co1/2 in 1 M KOH with 0.1 M ammonia is 0.42 V vs. RHE, which is much smaller than that of pure Pt (0.48 V vs. RHE) and Pt3Ru (0.46 V vs. RHE). In addition, the peak mass current activity of Pt3Ru1/2Co1/2 is 174.0 A gPt−1, which is more than two times larger than the 78.6 A gPt−1 and 80.2 A gPt−1 of pure Pt and Pt3Ru, respectively. The onset potential was observed to decrease as the electrolyte temperature increased from 25 °C to 80 °C, demonstrating that temperature affects the AOR performance. Ru and Co optimize the site by adjusting the electron density on the Pt surface. In particular, Co interacts with Pt–Ru to adjust the antibonding orbital to improve the electronic structure. Moreover, the introduction of Co slightly decreases the N2p orbital resonance energy of the adsorbate, weakening the adsorption strength to an appropriate level. This adjustment allows Pt3Ru1/2Co1/2 to exhibit optimal adsorption properties and show increased catalytic performance. Liu et al. improved the AOR performance by activating the inactive (220) facet of a PtMo alloy through single-atom Ni doping.80 Depending on the Ni doping ratio, Pt93Mo7, Ni7-Pt86Mo7, and Ni22-Pt71Mo7 catalysts were fabricated. Fig. 5a illustrates the onset potential and mass activity of each catalyst in 1 M KOH with 0.1 M ammonia. The onset potential of Ni7-Pt86Mo7 was measured to be 0.49 V vs. RHE, which is not significantly different from the control. However, Ni7-Pt86Mo7 achieved a peak current density of 94.96 A g−1, which is significantly higher than that of Pt93Mo7 (58.83 A g−1) and PtIr/C (20.31 A g−1). Fig. 5b presents the results of calculating the limiting potentials at the main crystal facets (111), (200), and (220) in the Pt93Mo7 and Ni7-Pt86Mo7 models. Unlike the Pt(200) facet, which is naturally the highest AOR active facet, the Pt(220) facet is generally considered an inactive face and has a high energy barrier of 0.22 eV, resulting in a low AOR contribution. However, Ni doping effectively reduces the energy barrier of the (220) facet to 0.13 eV, which is lower than that of the (200) facet. This finding indicates that the (220) facet has been successfully activated thanks to Ni doping. The electron density contour in Fig. 5c demonstrates that doping Ni on the (220) facet causes an obvious electron accumulation around the Pt atoms, resulting in a significant increase in electron density. This supports that Ni doping awakened the inactive facet and modified it into the most active surface.
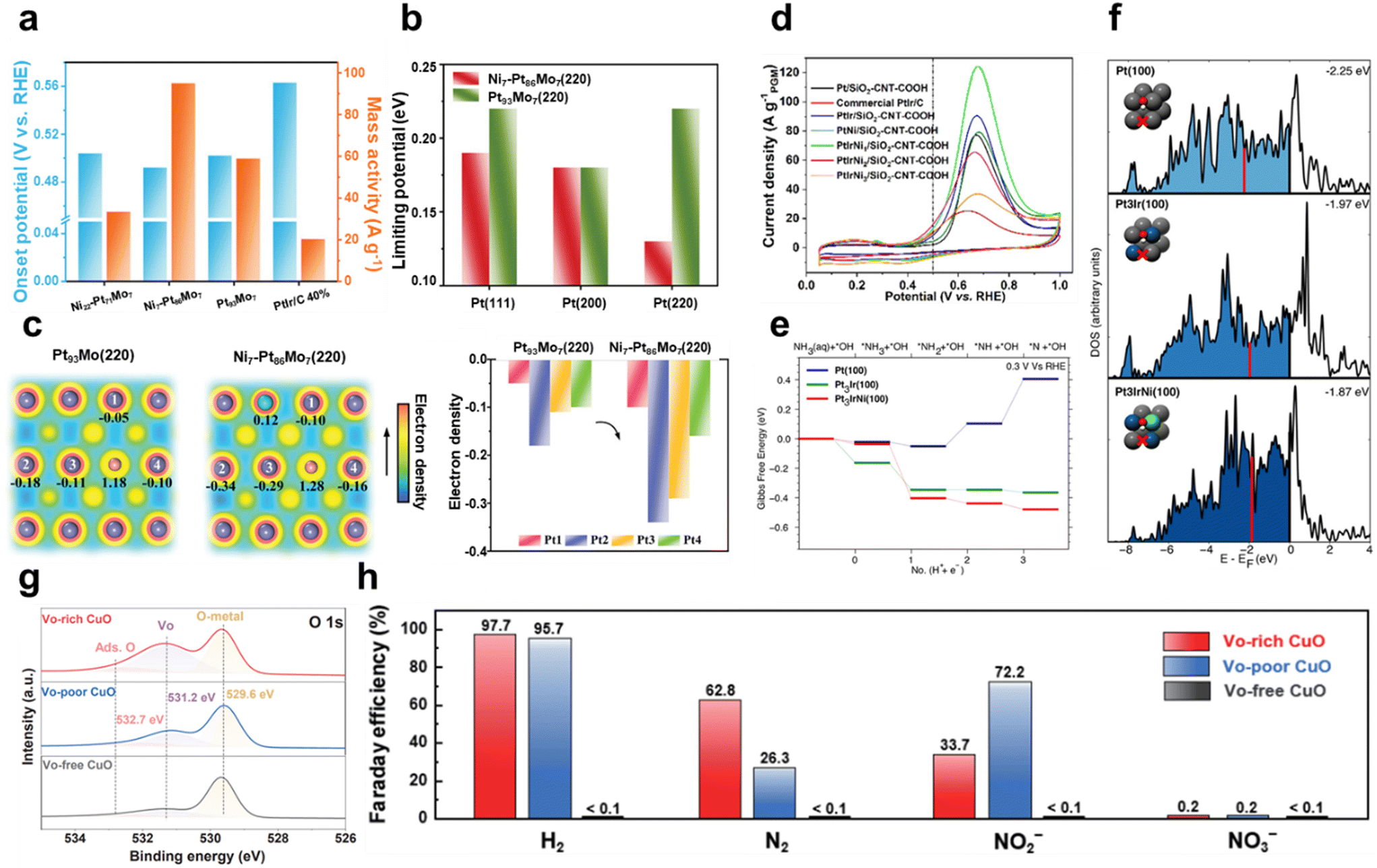 | ||
| Fig. 5 Ammonia oxidation reaction (AOR). (a) Comparison of onset potentials and mass activities for different Pt alloy catalysts in 1 M KOH with 0.1 M NH3. (b) Limiting potential of different facets in Ni7-Pt86Mo7 and Pt93Mo7. (c) Electron density contour map and electron densities of specific Pt atoms on the (220) facet of Ni7-Pt86Mo7 and Pt93Mo7. (a–c) Adapted with permission.80 Copyright 2023, Wiley-VCH GmbH. (d) CV curves for different Pt catalysts in 1 M KOH with 0.1 M NH3. (e) Gibbs free-energy pathway for the AOR through DFT calculations on Pt(100), Pt3Ir(100), and Pt3IrNi(100) at 0.3 V vs. RHE. (f) Group d-orbital density of states. Red lines indicate the d-band center. (d–f) Adapted with permission.81 Copyright 2020, American Chemical Society. (g) O 1s XPS spectra of VO-free, VO-poor and VO-rich CuO. (h) Faradaic efficiency of H2, N2, NO2− and NO3− for VO-free, VO-poor and VO-rich CuO at 0.6 V vs. Hg/HgO. (g and h) Adapted with permission.82 Copyright 2022, Springer Nature. | ||
In particular, the introduction of Ir lowers the onset potential by enhancing ammonia adsorption and promotes the dehydrogenation process.83 In Pt–Ir alloys, Ir facilitates the adsorption of *N and *NH species, making the vacant sites on Pt available to accept additional intermediates such as *NH2. Lin et al. introduced a unique PtIrCu AOR catalyst with hyperbranched concave octahedral nanodendrites (HCONDs).84 In 1 M KOH with 0.1 M ammonia, PtIrCu HCONDs exhibited an onset potential of 0.35 V vs. RHE, which was much lower than that of PtIr nanocubes (0.43 V vs. RHE), PtCu nanocubes (0.50 V vs. RHE), and commercial Pt/C (0.51 V vs. RHE). The peak current density also showed a notable difference, with PtIrCu HCONDs achieving 122.9 A gPGM−1, more than twice that of commercial Pt/C. PtIrCu HCONDs featured high-index facets such as (553), (331), and (221), providing a high density of active sites and enhancing the interaction with reactants. Furthermore, Cu and Ir atoms partially substitute Pt atoms, inducing a compressive strain of the lattice that weakens the oxygen adsorption and promotes the intermediate desorption. These properties lower the dehydrogenation potential and further accelerate the reaction kinetics, enhancing the AOR activity of PtIrCu HCONDs. Li et al. synthesized PtIrNi alloy nanoparticles on porous silicon dioxide (SiO2) and carboxyl-functionalized carbon nanotubes (SiO2–CNT–COOH).81 Porous SiO2 effectively provides OH−, while CNT–COOH has high electrical conductivity, thereby enhancing the catalytic activity of the alloy. The CV graph in Fig. 5d shows the catalytic performance of the alloys with SiO2–CNT–COOH as the support in 1 M KOH with 0.1 M ammonia. The onset potential of Pt/SiO2–CNT–COOH was rather high at 0.5 V vs. RHE, but it shifted negatively to 0.45 V vs. RHE after Ir introduction. This is due to the low NH3 adsorption energy of Ir, which facilitates NH3 adsorption. The individual introduction of Ni onto Pt has little effect on the onset potential, but the simultaneous introduction of Ir and Ni significantly influences the onset potential thanks to their synergistic effects. In particular, the onset potential of PtIrNi1/SiO2–CNT–COOH (molar ratio of Pt/Ir/Ni = 9:1:1) was significantly reduced to 0.40 V vs. RHE. Fig. 5e shows the free-energy pathway for the NH3 dehydrogenation step on Pt(100), Pt3Ir(100), and Pt3IrNi(100) surfaces at 0.3 V vs. RHE according to DFT calculations. The dehydrogenation of *NH2 to *NH on the Pt(100) surface has a thermodynamic barrier of 0.15 eV, while Pt3Ir(100) becomes thermoneutral, which reduces the reaction barrier. Furthermore, on Pt3IrNi(100) with Ni introduction, the dehydrogenation process becomes exergonic, leading to a predicted lower onset potential of 0.25 V vs. RHE. Fig. 5f shows the calculated density of states of the group d-orbital to analyze the changes in the electronic structure of the active site. The d-band center of Pt3Ir(100) is −1.97 eV, which shifts upward compared to −2.25 eV for Pt(100). This supports the previous results that the introduction of Ir enhances the adsorption of *NH intermediates. In Pt3IrNi(100) with Ni introduction, the d-band center further shifts upward to −1.87 eV, which further improves the catalytic performance and is consistent with the lower onset potential.
Huang et al. activated CuO by electrochemically introducing VO through CV scanning in an alkaline solution.82 In 1 M KOH with 0.1 M ammonia, VO-free CuO showed little AOR activity, but the current density progressively increased as the amount of VO increased. The onset potential of VO-poor CuO was 0.39 V vs. Hg/HgO, and it gradually shifted to 0.29 V vs. Hg/HgO as the number of CV cycles increased, resulting in VO-rich CuO. By analyzing the relative VO content to lattice oxygen using the O 1s XPS spectrum in Fig. 5g, the oxygen defect peak at 531.2 eV was found to have the largest area in VO-rich CuO. This confirms that the generation of VO is the key principle enhancing the electrochemical activity. DFT calculations revealed that VO shifts the d-band center of CuO closer to the Fermi level, which stabilizes the *NHx intermediate and improves the AOR performance. The FE results, shown in Fig. 5h, demonstrated that the N2 selectivity of VO-rich CuO was 62.8%, significantly higher than that of the control. These results were attributed to the introduction of VO, which changed the reaction energy of the dehydrogenation step, enabling selective oxidation to N2. Nagita et al. reported layered MnO2 thin films (NiCu/MnO2) co-intercalated with Ni2+ and Cu2+ ions coordinated with water molecules.91 While single-component Ni/MnO2 and Cu/MnO2 show little difference in current density before and after NH3 addition, NiCu/MnO2 shows a distinct difference. This shows that the intercalated Cu ions cannot directly oxidize NH3 at potentials lower than 0.6 V vs. Hg/HgO, but facilitate electron transfer when Ni2+ is oxidized to Ni3+. In 0.5 M NaOH and 55 mM NH4Cl, it selectively generated N2 with an FE of 97.4% at 0.6 V vs. Hg/HgO. In contrast, NiCu bimetal showed a lower N2 selectivity of 53.8% at 0.6 V vs. Hg/HgO. This is because the interlayer of NiCu/MnO2 promotes the formation of N–N bonds from partially dehydrogenated NH3 species before oxidation to NOx. Wang et al. introduced Ni1Cu1Co0.5-S-T/CP, a three-dimensional nanoflower structure, by co-doping Co and S onto NiCu.92 In 1 M NaOH and 0.2 M NH4Cl, the onset potential of Ni1Cu1Co0.5-S-T/CP was 1.241 V vs. RHE, which was lower than that of Ni1Cu1-T/CP (1.377 V vs. RHE) and Ni1Cu1-S-T/CP (1.374 V vs. RHE). S doping induces the surface reconfiguration of the electrode, exposing more active sites and improving the electrical conductivity. Meanwhile, Co doping modifies the NiCu electronic structure and increases the number of redox electron pairs, enhancing the catalytic performance. In addition, Co doping lowers the reaction energy barrier for N–H bond cleavage, which further facilitates the dehydrogenation process. As a result, the co-doping of Co and S accelerates the dehydrogenation reaction through the G–M mechanism, resulting in a high N2 selectivity of 81.21%. Kashale et al. synthesized boron-doped NiCu oxide (NiCuBO) and evaluated its AOR and OER performance.93 In the NiCuBO/NF-1||NiCuBO/NF-1 two-electrode system, 1.56 V is required to achieve 10 mA cm−2 in 1 M KOH with 0.5 M ammonia, representing a 110 mV decrease compared to water electrolysis. Furthermore, at 50 mA cm−2, the system demonstrates excellent stability for 24 h. Fig. 6 shows the N2 FE as a function of the potential applied to the AOR electrocatalyst. Noble metal-based catalysts exhibit high N2 FE at low potentials, while non-noble metal-based catalysts exhibit relatively low N2 FE at high potentials. Therefore, optimizing the operating potentials and controlling the reaction pathway of non-noble metal-based catalysts remain major challenges to improve N2 selectivity.
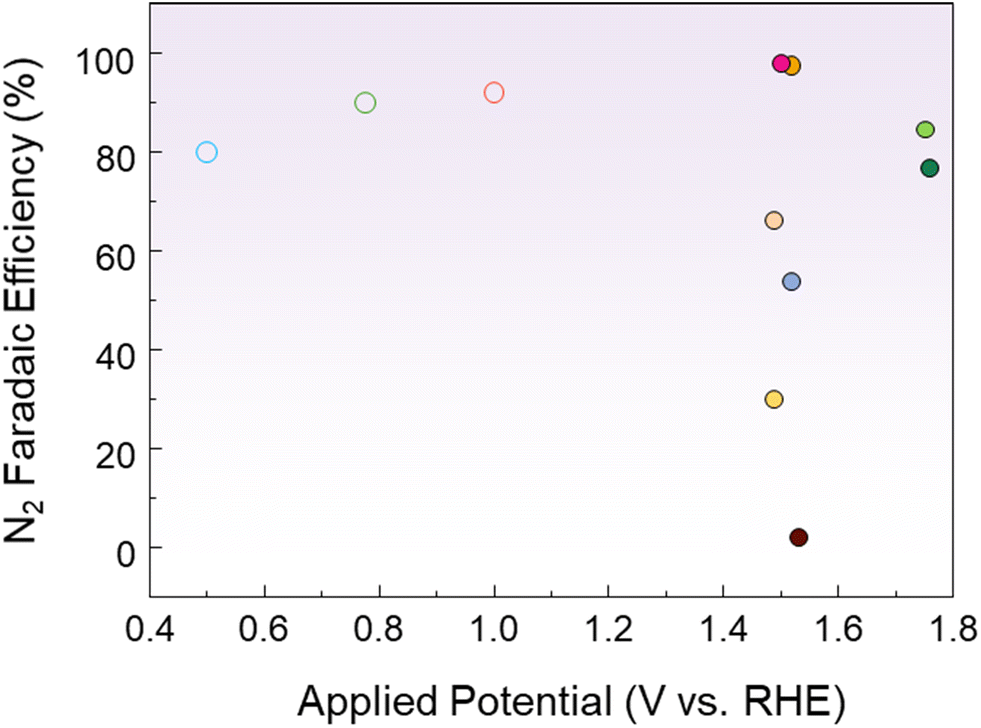 | ||
| Fig. 6 Comparison of the AOR performance of various electrocatalysts. Each potential was converted to RHE, considering the reference electrode and pH of the electrolyte. The detailed values are shown in the ESI.† | ||
2.3 Urea oxidation reaction
Large amounts of urine containing 2–2.5 wt% urea are produced by animals and humans, which requires urine-rich wastewater treatment to prevent producing toxic ammonia and further oxidized species.94 Electrocatalytic urea oxidation can be one of the environmentally-friendly technologies by converting urea into N2, CO2, and H2O using sustainable energy-derived electricity. The urea oxidation reaction at the anode requires a theoretical thermodynamic potential of −0.46 V vs. SHE (standard hydrogen electrode) and a cathode with a theoretical potential of −0.83 V vs. SHE for the HER. Therefore, the overall urea electrolysis requires 0.37 V.| Anode: CO(NH2)2 + 6OH− → CO2 + N2 + 5H2O + 6e− |
| Cathode: 6H2O + 6e− → 3H2 + 6OH− |
| Overall: CO(NH2)2 + 5H2O → CO2 + N2 + 3H2 |
DFT calculation methods provide a better understanding of the reaction mechanism for the UOR, which can undergo different pathways depending on the electrocatalysts. As a selected example, Zhang et al. showed that the NiOOH surface follows the pathway *CO(NH2)2 → *CONHNH2 → *CONHNH → *CONHN → *CON2 → *CO → *COOH → *COOHOH → *COO → CO2.95 In contrast, the reaction proceeding at NiOO with a higher oxidation surface shows *CO(NH2)2 → *CONHNH2 → *CONHNH → *VO–CONHN → *VO–NNH → *VO–NN → *VO → VO–OHOH → H2O. Although the theoretical voltage required for the UOR is significantly lower than that for the OER (1.23 V), it suffers from sluggish kinetics by the 6-electron transfer process, which requires the design of high-performance UOR electrocatalysts. In 1973, Yao et al. first demonstrated urea oxidation using a Pt anode.96 Sharing the same considerations for selecting electrocatalyst materials for water electrolysis, platinum group metal (Pt, Rh, Ir)-based materials are not ideal candidates for large-scale applications due to their high cost and scarcity.97 Non-noble metal-based electrocatalysts have gained attention due to their high performance in alkaline environments.98 Gao et al. summarized the three possible UOR pathways under alkaline conditions, depending on the adsorption of urea molecules.94 Considering that the predominantly used environment for the UOR is alkaline electrolytes, consuming the OH− at the anode sides, recent research has focused on Ni or Co-based hydroxides, alloys, sulfides, phosphides, selenides, oxides, and so on. Various strategies to develop efficient UOR catalysts include designing heterojunction catalysts, electrocatalysts having high oxidation states, and dual catalytically active sites by doping or mixing catalysts.99–101 Fig. 7 shows the comparison of urea-assisted water electrolysis using various electrocatalysts.
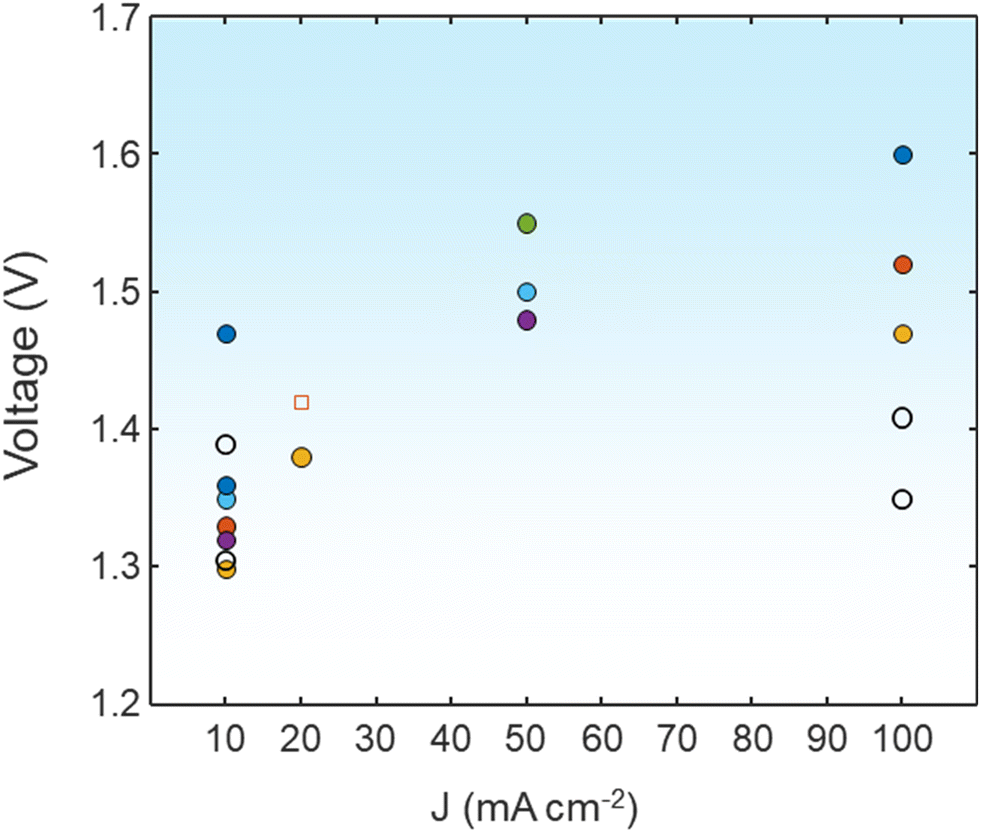 | ||
| Fig. 7 A comparison of the UOR||HER with various electrocatalysts in terms of cell voltage at various current densities. The detailed values are shown in the ESI.† | ||
Nickel hydroxide is one of the representative UOR electrocatalysts, which further reacts with OH− to form NiOOH active species. Sun et al. reported synthesized self-supported hierarchical Ni(OH)2-decorated CuO nanowires supported on Cu foam (Ni(OH)2/CuO NWs/CF) for the UOR.108 As shown in Fig. 8a, Ni(OH)2 was uniformly electrodeposited on CuO nanowires, which provides substantial active sites and high electrical conductivity for charge transfer. They used DFT calculations to unravel the UOR activity from the Ni(OH)2/CuO NWs/CF by comparing the adsorption energy of urea and OH− using (111) surface models. Both CuO and Ni(OH)2 have larger adsorption energy toward urea rather than OH−, compared to NiFe LDHs. As depicted in Fig. 8b, by comparing the adsorption energy of OH− on CuO (111), Ni(OH)2, and CuO (111)/Ni(OH)2, the intermediate adsorption behavior of the Ni(OH)2/CuO NWs/CF explains the highly catalytically active UOR performances. The polarization curves of CuO/CF and Ni(OH)2/CuO/CF show the preferential UOR over the OER in 1 M KOH with or without 0.5 M urea. Hierarchical Ni(OH)2/CuO/NF requires 1.334 V to generate the current density of 100 mA cm−2 for the UOR, which is 71 mV lower than that of CuO/NF (Fig. 8c). Urea electrolysis was performed by coupling Ni(OH)2/CuO NWs/CF as an anode and Pt/C/NF as a cathode at an applied current density of 20 mA cm−2, showing a linear increment of the H2 amount and nearly 100% faradaic efficiency (Fig. 8d). Chen et al. introduced nickel sulfoselenide (Ni–S–Se) on the Ni(OH)2 surface grown on Ni foam (Ni–S–Se/NF) as HER and UOR catalysts.109 The presence of S in Ni–S–Se/NF steers the HER with an optimal hydrogen free energy based on DFT calculations, and the in situ formed nickel oxyhydroxide (NiOOH) from sulfoselenide is an active species for the UOR.
 | ||
| Fig. 8 Urea oxidation reaction (UOR) using Ni-based electrocatalysts. (a) SEM image of Ni(OH)2/CuO NWs/CF. (b) Comparison of adsorption energies of urea of different surface models. (c) Polarization curves for the UOR and OER using CuO NWs/CF and Ni(OH)2/CuO NWs/CF. (d) Faradaic efficiency and generated amount of hydrogen during urea electrolysis. (a–d) Reprinted with permission.108 Copyright 2022, Wiley-VCH GmbH. (e) Schematic illustration of catalyst preparation of NF/NiMoO–Ar (UOR) and NF/NiMoO–H2 (HER). (f) Polarization curves of NF/NiMoO–Ar and NF/NiMoO–H2 for the UOR and OER. (g) Stability test of urea electrolysis (UOR||HER) using NF/NiMoO–Ar as a UOR catalyst and NF/NiMoO–H2 as a HER catalyst performed at a cell voltage of 1.58 V. (e–g) Reproduced with permission.110 Copyright 2018, The Royal Society of Chemistry. (h) SEM image of O–NiMoP/NF. (i) HER polarization curves of various electrocatalysts measured in 1.0 M KOH. (j) UOR polarization curves of various electrocatalysts in 1.0 M KOH with 0.5 M urea. (k) Comparison of cell voltage for water electrolysis and urea electrolysis at different current densities. (h–k) Reproduced with permission.111 Copyright 2021, Wiley-VCH GmbH. (l) XRD patterns of synthesized Ni2Fe(CN)6 catalysts. The inset shows the schematic illustration of Ni2Fe(CN)6. (m) Polarization curves of various catalysts in 1 M KOH with 0.33 M urea. (n) Comparison of LSV curves of the UOR||2e−-ORR and OER||2e−-ORR. (o) Faradaic efficiency and H2 production rate during urea electrolysis at different cell voltages. (l–o) Reprinted with permission.15 Copyright 2021, Springer Nature. | ||
Bimetallic nickel–molybdenum (NiMo)-based electrocatalysts have been considered as bifunctional catalysts for water electrolysis, especially showing catalytically active HER performance under alkaline conditions.112 Therefore, studies of NiMo-based electrocatalysts for urea electrolysis (UOR + HER) have been conducted by researchers. Yu et al. reported porous rod-like NiMoO4 with high oxidation states of the metal elements for an efficient UOR.110 As depicted in Fig. 8e, they first pre-synthesized NiMoO4·xH2O nanorods on Ni foam, which were subsequently annealed in an Ar atmosphere or reductive H2/Ar (5%/95%) mixture gas to obtain NF/NiMoO–Ar and NF/NiMoO–H2, respectively. Annealing of NiMo-based materials in different atmospheres affected UOR properties, as shown in Fig. 8f. NF/NiMoO–Ar showed a current density of 100 mA cm−2 at 1.42 V vs. RHE in 1 M KOH with 0.5 M urea, which is attributed to the easier oxidation of Ni2+ to Ni3+ by Mo6+ in NiMoO-Ar. However, when the sample was annealed under H2 conditions, NF/NiMoO–H2 showed mostly Ni0, Mo3+, and Mo4+, which makes it difficult to form Ni3+ active species. Instead, NF/NiMoO–H2 showed comparable catalytic activity to platinum in 1 M KOH, exhibiting low overpotentials of 11 mV to yield 10 mA cm−2 and 53 mV for the current density of 100 mA cm−2. By utilizing NF/NiMoO–Ar as an anode and NF/NiMoO–H2 as a cathode for the overall UOR, the potential of 1.38 V was required to reach the current density of 10 mA cm−2 and 1.55 V for 100 mA cm−2. Fig. 8g shows the stable urea electrolysis performed at a cell voltage of 1.58 V for 50 h, indicating potential for a noble-metal-free urea electrolyzer. Jiang et al. synthesized oxygen-incorporated NiMoP nanotube arrays, consisting of interconnected nanoparticles of 70 nm in diameter, as bifunctional electrocatalysts for the UOR and HER (Fig. 8h).111 The HER polarization curves exhibit that O–NiMoP/NF has a comparable overpotential of 54 mV (at J = 10 mA cm−2) to Pt/C/NF (Fig. 8i). In the case of the UOR, O–NiMoP/NF only requires 1.41 V for generating 100 mA cm−2, which is 340 mV lower than that of the OER. Compared to O–NiP/NF or conventional RuO2/NF, O–NiMoP/NF Mo, P, and O atoms modulated the electronic environment of catalytically active Ni sites, contributing to improved electrocatalytic activity and durability (Fig. 8j). The urea-assisted electrolysis system showed a cell voltage of 1.55 V at the current density of 50 mA cm−2 in 1 M KOH with 0.5 M urea, 0.3 V lower than that of water electrolysis (Fig. 8k). Liu et al. synthesized nickel phosphide nanoflake arrays on carbon cloth (Ni2P NF/CC) as a 3D catalyst electrode for the UOR, which requires 0.447 V to deliver the current density of 100 mA cm−2 in 1 M KOH with urea.113 Urea electrolysis using bifunctional Ni2P NF/CC||Ni2P NF/CC needs a cell voltage of 1.35 V to achieve 50 mA cm−2, 0.58 V lower than that of water electrolysis.
As a form of Ni-based framework electrocatalysts, Geng et al. demonstrated nickel ferrocyanide (Ni2Fe(CN)6) as a high-performance urea oxidation catalyst with high activity and stability.15 The structural analysis of Ni2Fe(CN)6 using XRD revealed that the Fe atom coordinates with carbon atoms in CN− and the Ni atoms coordinate with CN− species and are located in the center (Fig. 8l). Among catalysts having similar structures (Ni3[Co(CN)6]2 and Fe4[Fe(CN)6]3), Ni2Fe(CN)6 having Ni2+ and Fe2+ showed noticeable UOR properties in 1 M KOH with 0.33 M urea. It requires a potential of 1.35 V to generate a current density of 100 mA cm−2, 330 mV lower than that of the OER (Fig. 8m). As shown in Fig. 8n, urea electrolysis (UOR||HER) requires a cell voltage of 1.38 V to obtain the current density of 10 mA cm−2 and 1.5 V for 100 mA cm−2. This system was energy efficient compared to conventional water electrolysis, which requires 1.56 V and 1.85 V, respectively. The following H2 production rates increased as a function of cell voltage and the faradaic efficiency of urea electrolysis showed over 90% (Fig. 8o). By taking advantage of the structural diversity and porous features of MOFs, Li et al. reported a 2D MOF with mono-coordinated ferrocenecarboxylic acid (Fc) as a UOR catalyst.114 The NiFe-oxalate MOF, which was prepared by thermochemical treatment of Ni3Fe alloy foam in oxalic acid, showed a UOR current density of 500 mA cm−2 at 1.47 V vs. RHE in 1 M KOH with 0.33 M urea.115 The oxalate ligands induce the charge deficient Ni center and provide stable urea-O adsorption. Furthermore, the incorporated Fe enhances oxalate-O charge density and strengthens the H-bond with urea to overcome the rate-determining C–N cleavage of *CO(NH2). Metallic glasses (MGs) have a high Gibbs free energy state and a high density of low coordination states, which make them promising electrocatalyst candidates. Pei et al. prepared nanostructured Ni–P nanoglasses (NGs) with a heterogeneous microstructure, which showed high catalytic activity toward the UOR, OER, and HER. Ni–P-NG showed a low required potential for delivering 10 mA cm−2.116
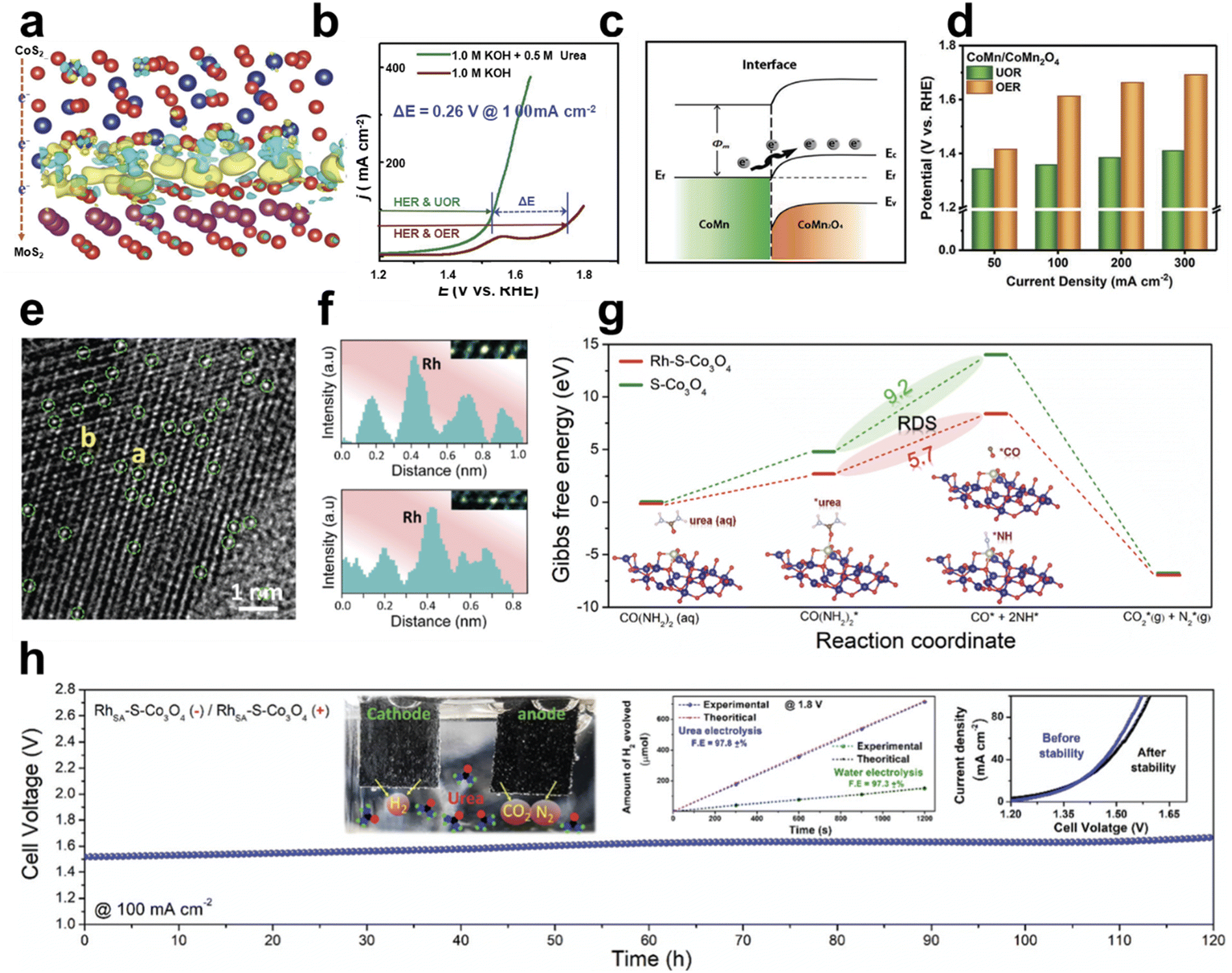 | ||
| Fig. 9 Urea oxidation reaction (UOR) using Co-based electrocatalysts. (a) The charge density distribution in the CoS2/MoS2 heterostructure. (b) Polarization curves of CoS2–MoS2 in 1 M KOH with or without 0.5 M urea. (a and b) Reprinted with permission.117 Copyright 2018, Wiley-VCH GmbH. (c) Energy band diagram of the CoMn/CoMn2O4 Schottky heterojunction. (d) Comparison of the UOR and OER performance at various current densities. (c and d) Reprinted with permission. Copyright 2020, Wiley-VCH GmbH. (e) TEM image of RhSA–S-Co3O4 with green circles indicating Rh single atom sites on S-Co3O4 and (f) line-scanning intensity profile at sites a and b. (g) Reaction free energy profiles of the UOR of S-Co3O4 with or without RhSA. (h) Stability test of urea electrolysis at a current density of 100 mA cm−2. The inset shows the comparison of H2 generation between urea and water electrolysis at a cell voltage of 1.8 V and the polarization curves before and after urea electrolysis. (e–h) Reprinted with permission.119 Copyright 2021, The Royal Society of Chemistry. | ||
Layered double hydroxides (LDHs) are a widely used form of electrocatalysts. Their 2D structures increase the active surface area and facilitate charge transfer in the oxygen evolution reaction. Song et al. prepared Co(OH)F and CoP-nanoarrays on Ni-foam, which have abundant active sites and facile pathways for ion/electron transport by taking advantage of their porous structure.120 Using Co–P/NF (cathode) and Co(OH)F/NF (anode), urea electrolysis requires a 1.42 V cell voltage for delivering a current density of 20 mA cm−2 in 1 M KOH with 0.7 M urea. Yu et al. reported hierarchical Fe-doped cobalt selenide coupled with FeCo LDH arrays designed for both the UOR and HER.121 Heterostructured Fe–Co0.85Se/FeCo LDH modulated the local interfacial bonding environment, facilitating the electron transfer and interface interactions at the interfacial Co–Se/O–Fe bonding. Free energy diagrams of the UOR through DFT calculation revealed that Fe–Co0.85Se/FeCo LDH can stabilize intermediates, resulting in high UOR catalytic activity. Wang et al. synthesized Cr-doped CoFe LDH on a Ni foam (NF) nanosheet array as an OER and UOR electrocatalyst, where the catalytic activity originates from the high-valence Cr6+ from the spontaneous oxidation of Cr-doped CoFe LDH acting as electron-withdrawing sites.122 Doping of Cr also enhances the extent of adsorption of the active sites for the H2O molecules. Urea electrolysis using Cr-doped CoFe LDH/NF (anode) and Pt–C/NF (cathode) required 1.305 V at the current density of 10 mA cm−2 in 1 M KOH with 0.33 M urea.
In a one-dimensional form, nanoparticles have been widely used for electrochemical systems due to their tunable size, composition, and nanoparticle–support interaction. Kang et al. formed ZIF-67-derived CoP nanoparticles confined in P, N, and Co-doped porous carbon (PNC) and attached to P-doped carbonized wood fibers (PCWF).123 With an enlarged surface area and high catalytic activity, CoP@PNC/PCWF required 1.321 V to generate a UOR current density of 50 mA cm−2, which is 257 mV lower than that of the OER. Single-atom catalysts (SACs) have shown great promise in energy conversion/applications due to their unique properties (electronic configuration, local coordination) and catalytic activities. However, the high surface energy of SACs causes agglomeration and results in low loading of SACs on the support. Kumar et al. introduced single-atom ruthenium (RhSA) on a tensile-strained Co3O4 (S-Co3O4) support prepared by liquid N2-quenching methods, achieving 2 times higher loading of RhSA for the UOR.119 As shown in Fig. 9e and f, the TEM image of RuSA–S-Co3O4 and the line scanning profile at sites a and b confirmed the stabilized distribution of RuSA on the S-Co3O4. The required potential to achieve the urea oxidation current density of 10 mA cm−2 of RuSA–S-Co3O4 was 1.28 V, 80 mV lower than that of S-Co3O4. RuSA–S-Co3O4 also showed 190 mV less potential (at 100 mA cm−2) for the UOR, compared to the OER. The reaction-free energy profiles for the UOR on RuSA–S-Co3O4 and S-Co3O4 were calculated to understand the effects of RuSA. As shown in Fig. 9g, *CO(NH2)2 to *CO + 2*NH is the rate-determining step (RDS) and RuSA stabilizes the *CO and *NH intermediates with a moderate urea adsorption energy (1.16 eV) compared to S-Co3O4 (1.72 eV), facilitating the adsorption and activation of urea. The urea electrolyzer using RhSA–S-Co3O4 for both the anode and cathode showed over 100 h stability at 100 mA cm−2, confirming the higher amount of hydrogen generation and faradaic efficiency compared to the water electrolysis at 1.8 V (Fig. 9h). Nguyen et al. introduced Rh single atoms in Co3S4/CoOx heterostructured nanosheet (NS)-assembled nanotube (NT) arrays on a 3D copper framework (3D-CF).124 Compared to the pure 3D-CF with low hydrophilicity behavior, Rh–Co3S4/CoOx NTs showed a super-hydrophilic surface, facilitating electrolyte infiltration and increasing the density of accessible active sites. Introducing Rh on Co3S4/CoOx NTs steered the UOR with a low overpotential of 1.32 V at 10 mA cm−2, lower than that of Co3S4/CoOx NTs (1.37 V), CoOx NTs (1.40 V), and CuO NRs (1.48 V). Furthermore, Rh showed small hydrogen adsorption free energy close to 0 from the DFT calculation, explaining its HER catalytic activity. Using Rh–Co3S4/CoOx NTs as an anode and a cathode, urea electrolysis required a cell voltage of 1.48 V to achieve a current density of 50 mA cm−2 and showed stable 50 h operation in 1 M KOH with 0.5 M urea.
2.4 Hydrazine oxidation reaction
Hydrazine (N2H4) is a small molecule, an important forming agent propellant and rocket fuel, a crucial electroanalytical target, and a useful reductant in organic synthesis.125 As a small-molecule oxidation reaction, hydrazine can be easily oxidized at lower potentials than the OER, decreasing significant energy consumption for hydrogen production. The hydrazine oxidation reaction (HzOR) has gained significant research attention due to its low thermodynamic potential (−0.33 V vs. RHE) and fast electro-oxidation kinetics.126| Anode: N2H4 + 4OH− → N2 + 4H2O + 4e− |
| Cathode: 4H2O + 4e− → 2H2 + 4OH− |
| Overall: N2H4 + H2O → N2 + 3H2 (E° = −0.33 V vs. RHE) |
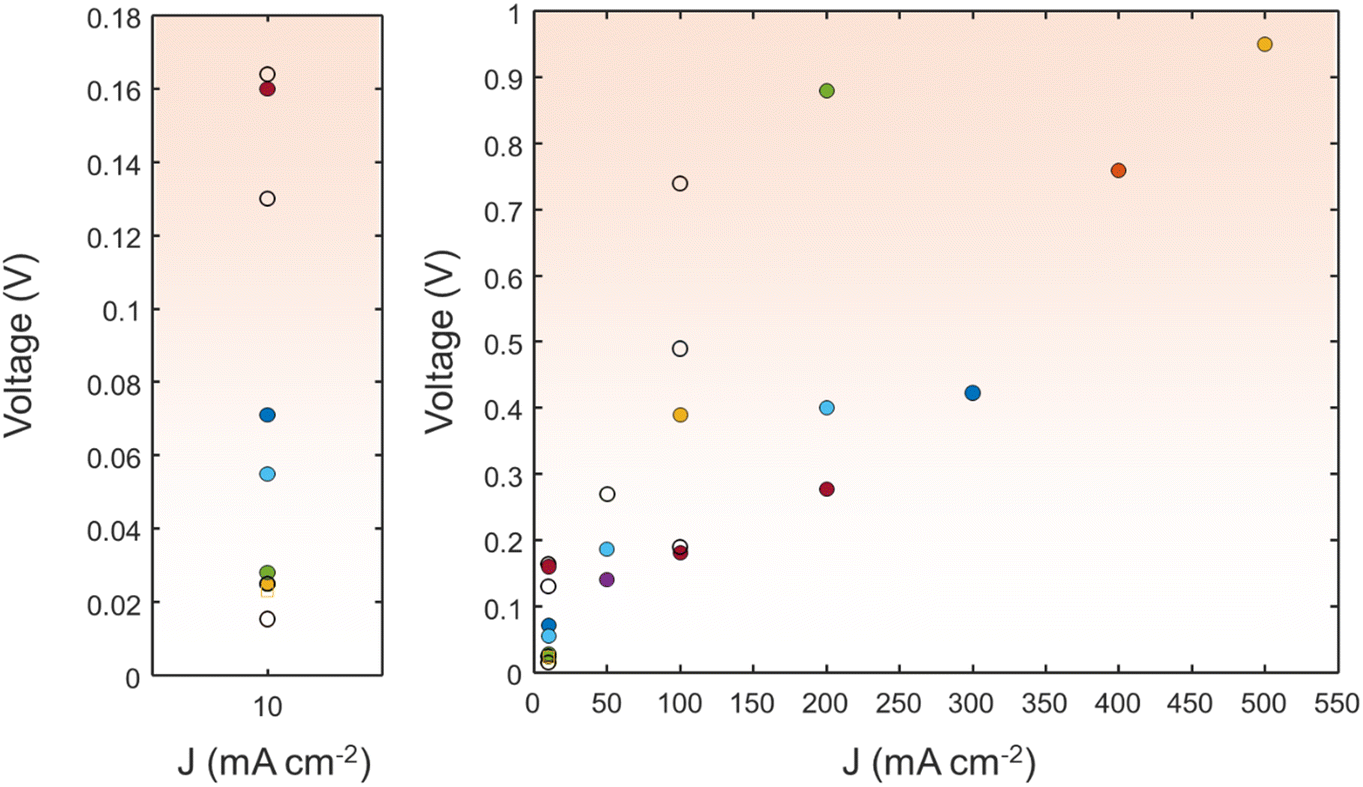 | ||
| Fig. 10 A comparison of the HzOR||HER with various electrocatalysts in terms of cell voltage at various current densities. The detailed values are shown in the ESI.† | ||
Metal phosphide-based hybrid structures have gained attention due to their Pt-like HER activity with lower metal loadings. As HzOR electrocatalysts, Zhu et al. synthesized 3D nickel cobalt phosphide heterostructures on Ni foam (Ni-Co-P/NF) with uniformly distributed CoP nanoparticles for both HER and HzOR electrocatalysts.16 The use of Ni-Co-P/NF catalysts as both an anode and a cathode requires 0.498 V to reach 500 mA cm−2. The high catalytic activity of the catalysts originated from the instantaneous recovery of metal phosphide active sites by N2H4, where the oxidized metal phosphide species could be reduced back to active metal phosphides. Li et al. designed RuP2-decorated N, P-doped carbon porous nanosheets (RP-CPM) for both HzOR and HER catalysts.127 A low cell voltage of 23 mV is required to achieve 10 mA cm−2 for HzOR-assisted water electrolysis. DFT calculation results supported the catalytic activity of RP-CPM: hydrogen adsorption on C-active sites is thermoneutral for the HER and favorable hydrazine dehydrogenation on active Ru sites.
Lattice strain engineering is one of the efficient strategies to improve the catalytic activity of various electrocatalysts. Modulating the lattice strain affects the adsorption strength of the reaction intermediates, facilitating the electrochemical reactions. Feng et al. manipulated the lattice strain of Ni2P nanosheets by dual cation (Cu and Co) co-doping by synthesizing Ni hydroxide precursors with dopants and subsequent phosphating of precursors to generate metal-doped Ni2P.128 As shown in Fig. 11a, synthesized metal-doped Ni2P shows porous two-dimensional structures with distinct lattice spacing toward the Ni2P (111) direction. Based on the lattice spacing of pristine Ni2P (111), doping of Co or Cu induces the tensile strain, while co-doping of Cu and Co formed the compression strain. The effects of strain engineering of Ni2P nanosheets are shown in the HzOR polarization curves (Fig. 11b). Co-doped Cu1Co2-Ni2P/NF showed the lowest required potential of −52 mV to achieve a current density of 10 mA cm−2. In comparison, those of tensile-strained Cu-Ni2P/NF and Co-Ni2P/NF were 18 mV and 28 mV, respectively. The compressive strain reduces the energy barrier of the *N2H4 to *N2H3dehydogenation step, contributing to the improved HzOR. With the optimized doping of Ni2P/NF, hydrazine electrolysis was conducted in 1 M KOH with 0.1 M N2H4 using Cu1Co2-Ni2P/NF as both an anode and a cathode. As shown in Fig. 11c, hydrazine-assisted water electrolysis required 0.16 V and 0.39 V to deliver the current density of 10 and 100 mA cm−2, respectively, which are significantly lower than those of the water electrolysis.
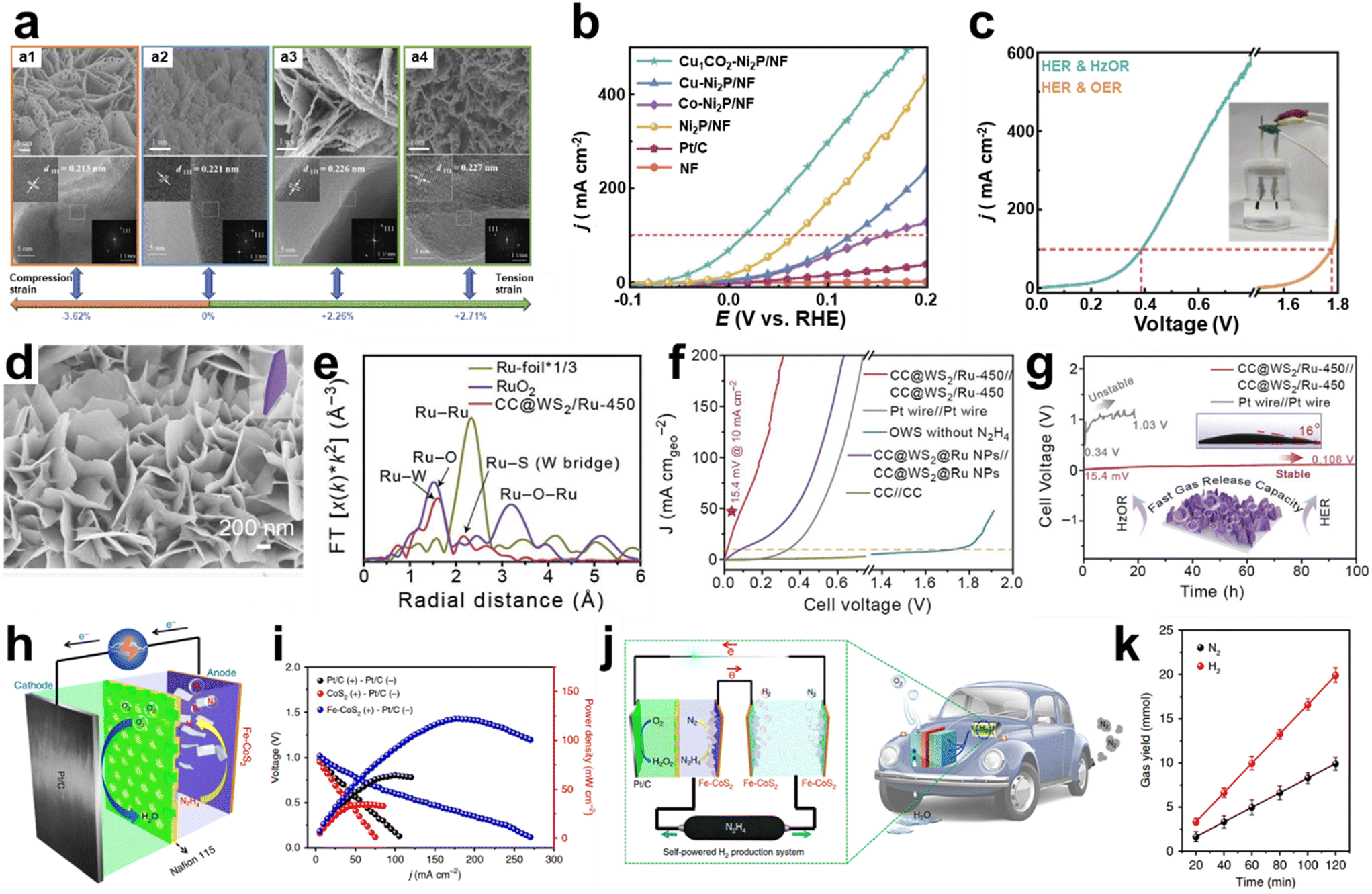 | ||
| Fig. 11 Hydrazine oxidation reaction (HzOR). (a) SEM (top) and HRTEM (bottom) images of catalysts. a1: Cu1Co2-Ni2P/NF, a2: Ni2P/NF, a3: Co-Ni2P/NF, and a4: Cu-Ni2P/NF. (b) Polarization curves of various catalysts in 1 M KOH with 0.1 M N2H4. (c) Polarization curves of Cu1Co2-Ni2P/NF as both an anode and a cathode in 1 M KOH with or without 0.1 M N2H4 (inset: image of the two-electrode electrolyzer). (a–c) Reprinted with permission.128 Copyright 2023, Wiley-VCH GmbH. (d) SEM image of CC@WS2/Ru-450. (e) FT-EXAFS spectra of CC@WS2/Ru-450, RuO2, and Ru-foil. (f) Polarization curves of CC@WS2/Ru-450 as both an anode and a cathode in 1 M KOH with or without 0.5 M N2H4. (g) Stability test of HzOR-assisted water electrolysis using CC@WS2/Ru-450||CC@WS2/Ru-450 and Pt wire||Pt wire (inset: contact angle test). (d–g) Reprinted with permission.129 Copyright 2022, Wiley-VCH GmbH. (h) Schematic illustration of DHzFCs using Fe-CoS2 nanosheets as the anode and commercial 40 wt% Pt/C as the cathode with O2 as the oxidizing reagent. (i) Current density–voltage profiles and current density–power density profiles for the DHzFC. (j) Schematic illustration of a self-powered H2 production system integrated with DHzFC and OHzS systems. (k) N2 and H2 generation in the system with 5.3 M hydrazine. (h–k) Reprinted with permission.130 Copyright 2018, Springer Nature. | ||
Although the platinum group metals are catalytically active, their high costs are challenging for large-scale applications. Therefore, it is required to reduce the amount of PGMs while maintaining their catalytic activity. Composition control of electrocatalysts is one well-known strategy to enhance catalytic activity and stability. Li et al. designed a hybrid structure composed of intermetallic CoPt3 and Pt-doped Co particles on the N-doped carbon skeleton (CoPt3/CoPt⊂PLNC) with a puff-like structure.131 Using CoPt3/CoPt⊂PLNC as both an anode and a cathode, the overall hydrazine splitting system requires low cell voltages of 0.13, 0.27, and 0.49 V, which are responsible for generating current densities of 10, 50 and 100 mA cm2. Feng et al. demonstrated the design of HEAs, which requires mixing of generally five or more elements equally or having relatively large proportions.132 HEA nanoclusters (NCs), having an average size of 1.48 nm, consist of Pt, Ru, Co, Mo, and Ni with optimized composition. Overall hydrazine oxidation-assisted splitting (OHzS) using HEANCs on carbon supports (C) as both an anode and a cathode requires 0.025 and 0.181 V for generating 10 and 100 mA cm−2.
Ruthenium (Ru) is a promising alternative to the Pt in Pt-group metals for the HER with similar hydrogen bond energy to Pt and electron-rich Ru sites. Ru can also benefit the HzOR because it can stabilize HzOR intermediates, as studied by DFT calculations in previous reports. Considering the overall hydrazine-assisted water electrolysis (HzOR/HER), utilizing Ru as catalytically active species for the HzOR has been studied. Guan et al. studied the role of Ru single atoms and nanoclusters prepared on a porous N-doped carbon (Ru/PNC) composite, which requires 1 and −20.4 mV to deliver 10 mA cm−2 for the HER and HzOR, respectively.133 DFT calculations revealed that the Ru SAs have optimal ad/desorption free energies for dehydrogenating N2H4 intermediates. Li et al. introduced Ru single atoms into the sulfur vacancies of tungsten disulfide nanosheets (WS2 NSs) using galvanostatic deposition grown on carbon cloth (CC@WS2/Ru SAs) (Fig. 11d), showing layers of the WS2 nanosheet structure without a morphology change.129 The presence of Ru SAs in WS2 is analyzed through extended EXAFS, where Ru K-edge Fourier-transformed (FT) EXAFS spectra show peaks at 1.63 Å, corresponding to Ru–W, and 2.2 Å, attributed to Ru–S (W bridge) interaction (Fig. 11e). The well-marked Ru–Ru peak at 2.33 Å and Ru–O–Ru peak at 3.19 Å didn't show up, which agrees with the presence of Ru SAs in WS2. Fig. 11f shows the polarization curves of HzOR-assisted water electrolysis with a distinct configuration, where the CC@WS2/Ru-450 (anode)||CC@WS2/Ru-450 (cathode) system showed the lowest potential of 15.4 mV to achieve 10 mA cm−2 in 1 M KOH with 0.5 M hydrazine. When the Ru NPs were used instead of Ru SAs, the potential of 86.6 mV was required to reach 10 mA cm−2. Furthermore, water electrolysis using CC@WS2/Ru-450||CC@WS2/Ru-450 required a higher voltage of 1.72 V, indicating a significant electric power saving through the HzOR-assisted water splitting reaction. The continuous operation over 100 h at the current density of 10 mA cm−2 revealed the durability of the electrolyzer, showing a slight increase of cell voltage to 0.108 V (Fig. 11g). Li et al. immobilized Ru SAs into the oxygen vacancies of urchin-like tungsten trioxide (VO–WO3) using galvanostatic methods for the HER and HzOR.134 They demonstrated the pH-universal HER and HzOR properties of VO–WO3: the HER performed in acid, alkaline, and neutral environments required the overpotentials of 17, 34, and 64 mV at 10 mA cm−2, and the HzOR performed in alkaline and neutral environments showed the overpotentials of −58 and 23 mV for achieving a current density of 10 mA cm−2, respectively. HzOR-assisted overall water splitting on CC@WO3/Ru-450||CC@WO3/Ru-450 delivers a current density of 10 mA cm−2 at 25 mV in 1 M KOH with 0.5 M hydrazine.
Metal sulfides and selenides are promising electrocatalysts due to their unique electronic structure and electrical conductivity. Zhang et al. demonstrated a bifunctional tubular cobalt per selenide (CoSe2) nanosheet electrode using a two-step hydrothermal method for the HzOR and HER.135 Hydrazine-assisted water electrolysis in 1 M KOH with 0.5 M hydrazine using CoSe2/CoSe2 required a cell voltage of 0.164 V at 10 mA cm−2. Liu et al. developed bifunctional Fe-doped CoS2 nanosheets for the HER and HzOR for direct hydrazine fuel cells (DHzFCs) and hydrazine-assisted water electrolyzers.130 They investigated the effect of Fe-doping on CoS2 by comparing the free energy profile of the HzOR using DFT calculations. Fe doping decreased the energy barrier of the rate-determining *N2H4 to *N2H3 step: 0.88 eV and 0.56 eV for CoS2 and Fe-CoS2, respectively. By taking advantage of the high catalytic activity and durability of the HzOR using Fe-CoS2, the DHzFC consisting of Fe-CoS2 (anode) and Pt/C (cathode) was operated using O2 (Fig. 11h). The maximum power density (pmax) of Fe-CoS2, CoS2, and Pt/C using the Pt/C cathode showed 125 mW cm−2, 36 mW cm−2, and 66 mW cm−2, respectively (Fig. 11i). They designed a self-powered H2 production system by integrating an overall hydrazine splitting (OHzS) unit and a DHzFC, as shown in Fig. 11j. The OHzS unit generates H2 and N2 at the anode and cathode using Fe-CoS2, where the generated amounts of H2 and N2 gases are measured using gas chromatography with a linear relationship as the measurement time increases (Fig. 11k). The integrated system showed an H2 evolution rate of 9.95 mmol h−1, a faradaic efficiency of 98%, and 20 h stability at room temperature and ambient pressure. This is a promising approach for H2 production without an external bias.
Aligning with the strategy of constructing heterojunction interfaces, nitride-based HzOR catalysts have gained attention due to their promising catalytic activity and physicochemical stability. Qian et al. reported hierarchical Ni3N–Co3N porous nanosheet arrays (Ni3N–Co3N PNAs/NF) as bifunctional catalysts for the HER and OER.136 The comparison of the most stable configurations between Ni3N, Co3N, and the Ni3N–Co3N heterostructure using DFT-calculated free energy diagrams of the HzOR revealed that the RDS for Ni3N–Co3N had a minimum free energy change, indicating higher HzOR activity than other catalysts. Adjustment of doping on metal nitrides was studied by Liu et al., where bifunctional P, W co-doped Co3N nanowire arrays (PW-Co3N NWA/NF) showed high catalytic activity toward the HzOR and HER, requiring −55 mV and −41 mV for achieving 10 mA cm−2, respectively.137 Low HzOR potentials of 27 and 127 mV vs. RHE are required to achieve high current densities of 200 and 600 mA cm−2 in 1 M KOH with 0.1 M hydrazine. Doping of P and W decreased the free energy changes for dehydrogenation of *N2H4 to *N2H3 and *N2H2 to *N2H. Mo-doped Ni3N and Ni heterostructure porous nanosheets grown on Ni foam (Mo-Ni3N/Ni/NF) showed the effects of interface engineering and doping, resulting in a cell voltage of 55 mV to deliver 10 mA cm−2 in 1 M KOH with 0.1 M hydrazine.138
2.5 Oxidation of higher carbon biomass-derived materials
Besides the utilization of methanol and ethanol, even higher carbon materials derived from biomass waste are also highly applied as an alternative to the OER for efficient hydrogen generation. The application of higher carbon material oxidation for anodic reactions can be advantageous when compared to methanol and ethanol oxidation. First, higher carbon materials have the potential to generate more valuable products, as they have various intermediates during the oxidation process.139,140 However, the oxidation of methanol and ethanol results in the production of low-value products like formaldehyde and acetaldehyde. In extreme cases, they can be fully oxidized to carbon dioxide, which is environmentally undesirable. For a similar reason, methanol or ethanol oxidation is more vulnerable to CO poisoning of the catalyst when compared to the oxidation reaction of higher carbon materials.141,142 Among various biomass-derived higher carbon materials, glycerol, 5-hydorxymethylfurfural (HMF) and glucose are intensively investigated due to their efficient coupling with the HER through the low oxidation overpotential and economical potential of their oxidation product. In this section, the reaction pathway of glycerol, HMF and glucose oxidation will be presented with their theoretical superiority over the OER. To focus on the comparative energy efficiency of the hybrid water splitting, studies on biomass oxidation coupled with the HER with a 2-electrode system will be briefly covered. Then, in addition to enhanced energy efficiency, research aimed at economic feasibility by enhanced selectivity for a single, high-value product will be presented.In Fig. 12a, the reaction pathway of the GEOR is presented. Starting from glycerol, each oxidation process can be categorized as C–OH oxidation, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O oxidation, and oxidative C–C cleavage. As the barriers for oxidation of each middle and terminal hydroxyl group are comparable, oxidation reactions of various pathways become inevitably competitive. The ability to control these competitive pathways is crucial for maximizing both efficiency and selectivity in the GEOR. To elucidate the GEOR mechanism, it is crucial to understand the stepwise transformations occurring during the process. The initial step typically involves the adsorption of glycerol onto the catalyst surface, where the interaction between hydroxyl groups and active sites determines the reaction pathway. Subsequent dehydrogenation leads to the formation of glyceraldehyde or dihydroxyacetone, depending on whether the terminal or middle hydroxyl group is oxidized. These intermediates can further undergo oxidation to yield carboxylic acids, such as glyceric acid and tartronic acid, or cleavage reactions, leading to smaller molecules, including formic acid and oxalic acid. The brief overview of recent studies on various GEOR products are provided in Table 1. According to the table, high faradaic efficiency and current density are achieved for formic acid while those of higher carbon products are relatively moderate. From the perspective of catalysts, transition metals are widely applied for the GEOR with formic acid generation. On the other hand, noble metals are featured to generate high carbon products, provoking the necessity for investigation on the reaction pathway and mechanism. Followed by such research trends, mechanisms underlying the GEOR have been thoroughly studied on noble metal catalysts. Noble metals, particularly Pt and Au, provide a well-defined platform for understanding the relationship between adsorption structures and reaction pathways. Garcia et al. presented how the glycerol adsorption structure can lead to different product selectivities.145 According to Fig. 12b, glycerol adsorbs on the Pt surface in different ways, depending on the surface facet of the Pt catalyst. Specifically, Pt (111) surfaces were shown to favor the oxidation of terminal hydroxyl groups, leading to the formation of glyceraldehyde and other terminal oxidation products. In contrast, Pt (110) and Pt(100) surfaces facilitated the oxidation of secondary hydroxyl groups, resulting in a higher yield of dihydroxyacetone. In addition to the catalyst structure, reaction conditions such as the reaction potential and pH play a significant role in the reaction pathways of the GEOR. Guschakowskin et al. investigated the varying behavior of the GEOR with Pt-based catalysts.146 According to Fig. 12c, the selectivity to various products at different applied potentials on Pt–Bi alloys is presented. At moderate potentials, the reaction pathway is dominated by the C3 products, optimizing selectivity towards these value-added chemicals. However, at higher potentials, further oxidation results in the oxidative C–C bond cleavage and increases the yields of C1 and C2 products. While moderate potentials are ideal for achieving high C3 selectivity, the conversion rate remains limited to about half of the higher potential. This indicates that for the practical application, the balanced potential that ensures both adequate selectivity and a sufficient conversion rate has to be acquired. The influence of pH on the GEOR is another critical factor determining reaction efficiency and selectivity. In addition to the potential, Hamada et al. presented that the GEOR kinetics and overpotential are closely correlated to pH.147 In acidic media, high overpotential was required for the large oxidation current, leading to forming formic acid with multiple C–C bond cleavage. On the other hand, in alkaline media, glycerol exhibited a much higher oxidation current density at low overpotential. The improved oxidation kinetics in alkaline media is contributed by the promotion of the deprotonation step in the basic environment. Kwon et al. further presented the product selectivity of the GEOR with noble metals at different pH values based on the new HPLC method.148 For the Pt catalyst, it sustains the catalytic activity under acidic conditions to basic conditions but lowered pH still significantly deactivates the GEOR, similar to the former study. The trend of major products changes from glyceric acid to formic acid along with the pH decrease. Compared to Pt, high oxidation potential is necessary for the GEOR on Au. In alkaline media, Au shows stronger catalytic activity at high potential than Pt, although the product mainly comprises formic acid. In contrast to its strong activity in alkaline media, Au completely loses its glycerol oxidation activity in acid. Verma et al. approached the Au-catalyzed GEOR theoretically to understand the origin of pH dependency.149 According to the DFT calculations, the GEOR activity and selectivity are controlled by the presence of a surface-bound hydroxyl group. With the environment of experimental onset potential under alkaline conditions, the Au surface has modest OH coverage, optimal for the formation of glyceric acid. Calculated thermodynamic reaction barriers indicate that the surface OH and clean Au site cooperatively facilitate the kinetics and thermodynamics of glyceric acid formation.
O oxidation, and oxidative C–C cleavage. As the barriers for oxidation of each middle and terminal hydroxyl group are comparable, oxidation reactions of various pathways become inevitably competitive. The ability to control these competitive pathways is crucial for maximizing both efficiency and selectivity in the GEOR. To elucidate the GEOR mechanism, it is crucial to understand the stepwise transformations occurring during the process. The initial step typically involves the adsorption of glycerol onto the catalyst surface, where the interaction between hydroxyl groups and active sites determines the reaction pathway. Subsequent dehydrogenation leads to the formation of glyceraldehyde or dihydroxyacetone, depending on whether the terminal or middle hydroxyl group is oxidized. These intermediates can further undergo oxidation to yield carboxylic acids, such as glyceric acid and tartronic acid, or cleavage reactions, leading to smaller molecules, including formic acid and oxalic acid. The brief overview of recent studies on various GEOR products are provided in Table 1. According to the table, high faradaic efficiency and current density are achieved for formic acid while those of higher carbon products are relatively moderate. From the perspective of catalysts, transition metals are widely applied for the GEOR with formic acid generation. On the other hand, noble metals are featured to generate high carbon products, provoking the necessity for investigation on the reaction pathway and mechanism. Followed by such research trends, mechanisms underlying the GEOR have been thoroughly studied on noble metal catalysts. Noble metals, particularly Pt and Au, provide a well-defined platform for understanding the relationship between adsorption structures and reaction pathways. Garcia et al. presented how the glycerol adsorption structure can lead to different product selectivities.145 According to Fig. 12b, glycerol adsorbs on the Pt surface in different ways, depending on the surface facet of the Pt catalyst. Specifically, Pt (111) surfaces were shown to favor the oxidation of terminal hydroxyl groups, leading to the formation of glyceraldehyde and other terminal oxidation products. In contrast, Pt (110) and Pt(100) surfaces facilitated the oxidation of secondary hydroxyl groups, resulting in a higher yield of dihydroxyacetone. In addition to the catalyst structure, reaction conditions such as the reaction potential and pH play a significant role in the reaction pathways of the GEOR. Guschakowskin et al. investigated the varying behavior of the GEOR with Pt-based catalysts.146 According to Fig. 12c, the selectivity to various products at different applied potentials on Pt–Bi alloys is presented. At moderate potentials, the reaction pathway is dominated by the C3 products, optimizing selectivity towards these value-added chemicals. However, at higher potentials, further oxidation results in the oxidative C–C bond cleavage and increases the yields of C1 and C2 products. While moderate potentials are ideal for achieving high C3 selectivity, the conversion rate remains limited to about half of the higher potential. This indicates that for the practical application, the balanced potential that ensures both adequate selectivity and a sufficient conversion rate has to be acquired. The influence of pH on the GEOR is another critical factor determining reaction efficiency and selectivity. In addition to the potential, Hamada et al. presented that the GEOR kinetics and overpotential are closely correlated to pH.147 In acidic media, high overpotential was required for the large oxidation current, leading to forming formic acid with multiple C–C bond cleavage. On the other hand, in alkaline media, glycerol exhibited a much higher oxidation current density at low overpotential. The improved oxidation kinetics in alkaline media is contributed by the promotion of the deprotonation step in the basic environment. Kwon et al. further presented the product selectivity of the GEOR with noble metals at different pH values based on the new HPLC method.148 For the Pt catalyst, it sustains the catalytic activity under acidic conditions to basic conditions but lowered pH still significantly deactivates the GEOR, similar to the former study. The trend of major products changes from glyceric acid to formic acid along with the pH decrease. Compared to Pt, high oxidation potential is necessary for the GEOR on Au. In alkaline media, Au shows stronger catalytic activity at high potential than Pt, although the product mainly comprises formic acid. In contrast to its strong activity in alkaline media, Au completely loses its glycerol oxidation activity in acid. Verma et al. approached the Au-catalyzed GEOR theoretically to understand the origin of pH dependency.149 According to the DFT calculations, the GEOR activity and selectivity are controlled by the presence of a surface-bound hydroxyl group. With the environment of experimental onset potential under alkaline conditions, the Au surface has modest OH coverage, optimal for the formation of glyceric acid. Calculated thermodynamic reaction barriers indicate that the surface OH and clean Au site cooperatively facilitate the kinetics and thermodynamics of glyceric acid formation.
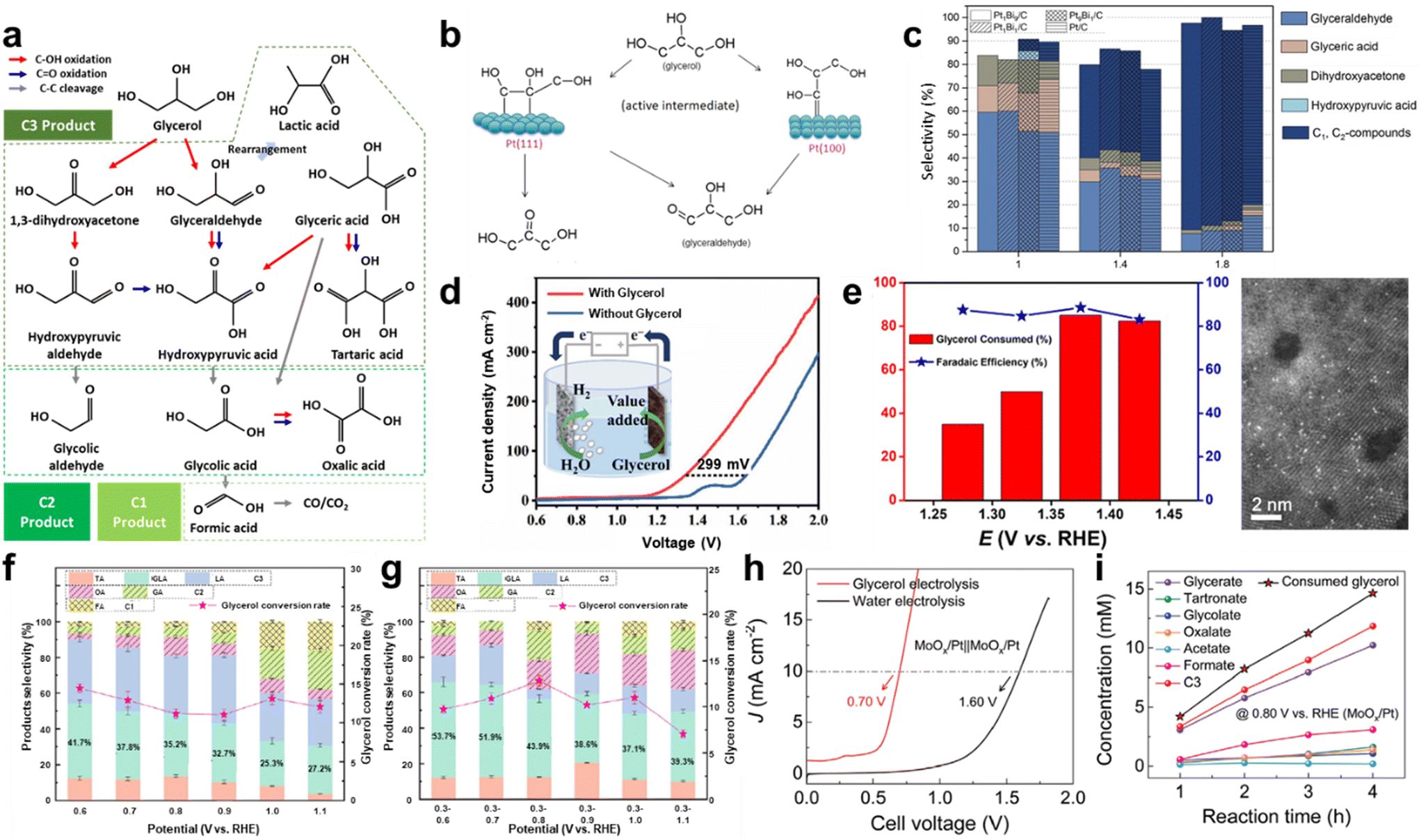 | ||
| Fig. 12 Glycerol oxidation reaction (GEOR). (a) Scheme of the GEOR pathway; (b) scheme of the reaction intermediate adsorption in the GEOR on Pt (100) and (111) surfaces. Reprinted with permission.145 Copyright 2016, American Chemical Society. (c) Product selectivity of direct electrochemical oxidation of 0.1 M glycerol in 0.5 M H2SO4 at different electrode potentials. Reprinted with permission.146 Copyright 2021, Wiley-VCH GmbH. (d) LSV curves (without iR correction) of the overall GOR||HER electrolyzer, which is depicted as an inset. Reprinted with permission.150 Copyright 2023, Wiley-VCH GmbH. (e) The glycerol conversion and faradaic efficiency of formic acid from PtSA–NiCo LDH at various potentials. Reprinted with permission.151 Copyright 2023, Elsevier. Product distribution and conversion rate of the GEOR in the electrolytic modes of (f) CE and (g) PE (EL= 0.3 V vs. RHE for 0.5 s and EH = 0.6–1.1 V vs. RHE for 0.5 s). (f and g) Reprinted with permission.152 Copyright 2024, Springer Nature. (h) Comparison of the two-electrode alkaline electrolyzer for the GEOR and water electrolysis. (i) Product concentration and glycerol conversion along with reaction time using the MoOx/Pt electrode applied at 0.8 V versus RHE based on HPLC analysis. (h and i) Reprinted with permission.153 Copyright 2021, Wiley-VCH GmbH. | ||
| Anode catalysts | Current density (mA cm−2) | Electrolytes (1 M KOH + xM glycerol) | Product (FE) | Ref. | |||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 10 | 20 | 100 | 150 | 300 | Glycerol (M) | |||
| NiCo2O4 | 1.42 | 0.1 | Formic acid (89.9% at 1.42 V) | 150 | |||||
| NiCo2O4/NF | 1.6 | 0.1 | Formic acid (97% at 1.6 V) | 154 | |||||
| NiOOH/Ni3S2/NF | 1.227 | 0.1 | Formic acid (97.7% at 1.4 V) | 155 | |||||
| PtSA–NiCo LDH/NF | 1.37 | 0.1 | Formic acid (88.7% at 1.375 V) | 156 | |||||
| CoMoO4 | 1.105 | 0.1 | Formic acid (90% at 1.26 V) | 157 | |||||
| CoNiCuMnMo NPs/CC | 1.25 | 0.1 | Formic acid (93.5% at 1.25 V) | 158 | |||||
| MnO2–CuO/CF | 1.3 | 0.1 | DHA (54% at 1.3 V) | 159 | |||||
| CoOx | 1.57 | 0.1 (1 M Na2B4O7) | DHA (49.4% at 1.5 V) | 160 | |||||
| NiOOH | 1.62 | 0.01 (0.75 M H2BO3 wt KOH) | DHA (44% at 1.52 V) | 161 | |||||
| Co–Bi/CF | 1.52 | 0.1 (0.2 M Na2B4O7) | DHA (35% at 1.52 V) | 162 | |||||
| MoOx/Pt | 0.7 | 0.1 | Glyceric acid (70% at 0.8 V) | 163 | |||||
| Pt/C–Al2O3 | 0.5 | 1 (1 M NaOH) | Lactic acid (31.3% at 0.5 V) | 164 | |||||
| Pt/NiF | 0.65 | 1 (3 M KOH) | Lactic acid (64% at 0.65 V) | 165 | |||||
Moving beyond the mechanistic studies on noble metal catalysts, recent research has increasingly focused on utilizing transition metal-based catalysts, based on a full system with the cathodic HER. Among transition metals, Ni, Co and their alloys have shown promising activity and stability under alkaline conditions. Luo et al. presented a NiCo2O4 catalyst deposited on Ni foam (NiCo2O4/NF) for a highly efficient GEOR.150 In Fig. 12d, the comparison of the two-electrode system LSV curves is presented. For the GEOR and HER coupled system, a 299 mV lower cell voltage (1.34 V) was required at 50 mA cm−2 compared to water splitting. High selectivity toward formic acid was also confirmed in the half-cell system, as the faradaic efficiency for formic acid was 89.9% at 1.42 V. The outstanding performance of the NiCo2O4/NF arises from the oxygen vacancy generation and formation of highly active NiIII–OOH/CoIII–OOH species, driven by the substitution of Ni for octahedral Co3+. Wu et al. further enhanced the activity of the NiCo2O4/NF catalyst by controlling the shape of synthesized NiCo2O4.166 With the hydrothermal approach, the synthesized NiCo2O4 has globular cactuses, fully covered with numerous nanowhiskers, which provide large reaction sites. With the hierarchical NiCo2O4 structure, a faradaic efficiency of 97% toward formate was achieved at 1.6 V. The overpotential in the two-electrode electrolyzer was reduced to 320 mV lower than that of the conventional water electrolysis system. To understand the exact reaction pathway on NiCo2O4, isotope-labelling NMR analysis was conducted. Two types of glycerol with isotope 13C in different positions were used for the GEOR, and the position of 13C was traced during the reaction pathway to formate. The oxidation route of glycerol was confirmed as glycerol → glyceraldehyde → glycerate → glycollate → formate, and C–C cleavage always occurs to C–C bonding next to the carboxyl group. The suggested reaction pathway is regarded as universal for the similar transition metal-based formate/formic acid generation, which was also observed through in situ electrochemical spectroscopy.167 In addition to the transition metal-based catalysts, Yu et al. applied a single atom catalyst to further maximize the energetic efficiency of the GEOR-coupled HER. Using NiCo layered double hydroxide (NiCo LDH) as a base structure, Pt single atoms are anchored at the oxygen vacancies, forming a Pt–O5 penta-coordination.151 The synthesized Pt single atom-decorated NiCo LDH (PtSA–NiCo LDH) has excellent catalytic properties not only for the GEOR, but also for the HER. Thus, a two-electrode system with the PtSA–NiCo LDH electrode as both the cathode and anode provides the current density of 100 mA cm−2 at only 1.37 V. As presented in Fig. 12e, even the faradaic efficiency toward formic acid has a competitive value of 88.7% at 1.37 V. From the DFT simulation, decorated single atom Pt sites are revealed to be a locally charge deficient region, providing HER active sites by lowering the d-band center.
While there have been numerous studies with low overpotential as a hybrid reaction for the HER, mostly their anodic products are limited to formic acid. Various research studies are striving to enhance selectivity for the relatively valuable C3 products, but most studies are still limited to the half-cell level, hardly considering the cathodic HER performance. The difficulty of achieving high selectivity for C3 products with enough current level for an efficient HER is mainly due to the continuous oxidation of GEOR intermediates to C1 products at oxidative potential. To ensure sufficient current level and C3 product selectivity, a strategy to prevent C–C cleavage through continuous oxidation is required. As part of that strategy, Chen et al. proposed a GEOR at pulsed potential.152 In Fig. 12f and g, the faradaic efficiency of a Pt–graphite (Pt@G) catalyst through constant-potential electrocatalysis (CE) and pulsed-potential electrocatalysis (PE) is presented. In the potential range, the selectivity for glyceric acid was enhanced and some of the formic acid decreased in the high potential region (1.0–1.1 V) with the application of PE. Here, PE is composed of a high potential section for the GEOR and a low potential section with open circuit potential (OCP). The PE electrolytic mode prevents the overaccumulation of intermediate species at the catalyst surface, releasing the intermediate as a product, and accelerates the re-adsorption of glycerol and hydroxyl groups under OCP for the repetitive GEOR. PE also enhances the current density for the GEOR in the high potential range by avoiding the poisoning of the catalyst surface with intermediates. Yu et al. presented a composite of MoOx nanosheets and Pt nanoparticles (MoOx/Pt) as a GEOR/HER bifunctional catalyst which operates at an extremely low cell voltage of 0.7 V with a current density of 10 mA cm−2.153 As shown in Fig. 12h, the GEOR of MoOx/Pt reduced the overpotential by 0.9 V compared to conventional water splitting. The point of the study is that such superior energetic efficiency was acquired while ensuring high C3 selectivity. From Fig. 12i, a high concentration of glycerate among various products, which is equivalent to a faradaic efficiency of ∼70%, can be confirmed. According to the DFT simulation, MoOx has the special role of attracting glycerol to the Pt interface, reducing the onset potential of the GEOR. Further, the electronic interaction between MoOx and Pt improves the reactivity of interfacial sites for both the GEOR and HER.
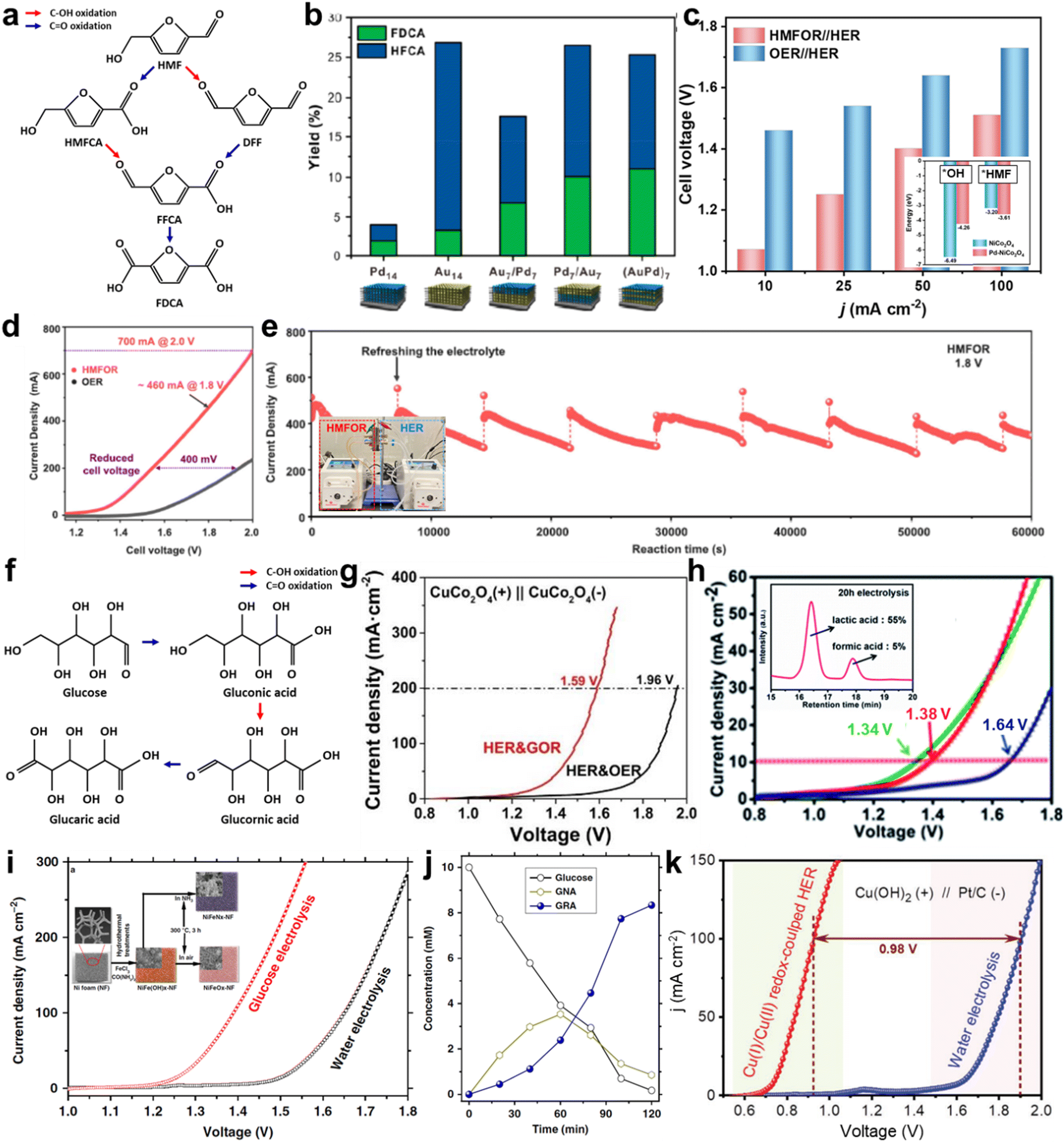 | ||
| Fig. 13 HMF and glucose oxidation reaction (HMFOR & GOR). (a) Scheme of the HOR pathway; (b) yield of FDCA from each electrode during the electrochemical oxidation. Reprinted with permission.168 Copyright 2020, American Chemical Society. (c) Comparison of the voltages required to achieve different current densities with *HMF and *OH adsorption energy on NiCo2O4 and Pd-NiCo2O4 as an inset. Reprinted with permission.169 Copyright 2024, Springer Nature. (d) The LSV curves of the A-Co-Ni2P catalyst in the MEA. (e) Stability measurement for the MEA electrolyzer at 1.8 V over A-Co-Ni2P in 1 M KOH containing 0.1 M HMF. (d and e) Reprinted with permission.170 Copyright 2024, Springer Nature. (f) Scheme of the GOR pathway. (g) LSV curves in a two-electrode system based on the bifunctional CuCoO4 electrode for glucose electrolysis and water electrolysis. Reprinted with permission.171 Copyright 2024, The Royal Society of Chemistry. (h) Comparison of polarization curves for water electrolysis, initial glucose electrolysis and after long-term electrolysis with the HPLC analysis result of the yield of oxidation products after 20 h electrolysis as an inset. Reprinted with permission.172 Copyright 2022, The Royal Society of Chemistry. (i) Comparison of glucose electrolysis and water electrolysis with the anodic NiFeOx-NF and cathodic NiFeNx-NF catalysts (scan rate: 5 mV s−1, glucose concentration: 100 mM, 1 M KOH). (j) Concentration of glucose and oxidation products as a function of time for the chronoamperometric test at 1.3 V vs. RHE. (i and j) Reprinted with permission.173 Copyright 2020, Springer Nature. (k) The LSV profiles in a two-electrode flow cell electrolyzer using Cu(OH)2 as the anodic electrode and Pt/C as the cathodic electrode. Reprinted with permission.174 Copyright 2021, Wiley-VCH GmbH. | ||
Various noble metals are applied for the HMFOR to achieve maximized FDCA selectivity. However, most noble metal catalysts presented inferior FDCA yields when compared to other transition metal-based catalysts. Pt and Ru catalysts are revealed to convert HMF to DFF, but further oxidation to FDCA is hardly observed.175,176 Au and Pd catalysts prefer to generate HMFCA, but still oxidation to FDCA is not favorable either.177,178 To overcome the inferior performance of monometallic noble metal catalysts, Park et al. proposed Au–Pd bimetallic electrodes and fabricated a HER/HMFOR full cell with an optimized cathode and anode.168 As presented in Fig. 13b, the electrochemical performances of three differently deposited Au–Pd bimetallic films are compared. The layer-by-layer structure of the bimetallic films is fabricated by dipping of ITO glass in Au/Pd-nanosized graphene oxide (nGO) precursor solution, which leads to Au or Pd layer formation through electrostatic interaction between noble metal and nGO. According to Fig. 13b, the FDCA yield apparently increased when the bimetallic film was applied. Particularly, the FDCA selectivity was increased to an ∼10% yield in Pd7/Au7 and (AuPd)7, while Au7/Pd7 has a lower value. Considering that the single Au electrode gives a high HFCA yield, HMF oxidation to HFCA is favorable in the Au site. Therefore, the outer Au region of Pd7/Au7 can generate HFCA to be transferred to the Pd layer for further oxidation to FDCA, leading to higher FDCA selectivity.
While noble metal-based catalysts only provide limited selectivity toward FDCA, most studies with transition metal-based catalysts provide excellent FDCA yields comparatively. To compare the intrinsic properties of various transition metal elements, Taitt et al. synthesized NiOOH, CoOOH, and FeOOH and investigated their catalytic properties.179 Among the three elements, NiOOH was the most efficient catalyst for the HMFOR, achieving 96% faradaic efficiency at 1.47 V vs. RHE. The current density for the HMFOR was also the highest with the NiOOH catalyst. In the case of the CoOOH catalyst, the initiation of the HMFOR was observed at a lower potential than NiOOH, which was confirmed in the indirect HMFOR mediated with Co(OH)2/CoOOH. However, the slow speed of Co(OH)2/CoOOH conversion leads to low current density, which is not practical for application. FeOOH barely showed any catalytic activity for the HMFOR below the onset potential for water oxidation. Wang et al. investigated a strategy to enhance the Ni catalyst for the HMFOR by controlling the different particle aggregation structures.180 With the electrodeposition of Ni on carbon paper at different current densities, various Ni nanoparticle assemblies of ordered nanoarray, disordered nanoarray and random aggregation were fabricated. Among various structures, the ordered nanoarray structure presented the best performance of 99.2% FDCA yield at a potential of 1.36 V. The high performance of the ordered nanoarray is closely related to the activation of Ni particles. The less agglomerated structure of the ordered nanoarray sample provides large interfacial areas between Ni and the aqueous electrolyte, leading to the facile conversion of surface Ni(OH)2 to NiOOH. The high coverage of catalytically active Ni(III) oxyhydroxide promotes the high yield of FDCA at comparatively low potential. Lu et al. proposed alloy catalysts with integrated catalytic sites for the HMFOR based on Co3O4.181 According to the DFT simulation, Co3O4 catalysts have higher activity for aldehyde oxidation than hydroxyl groups, while NiO has an opposite trend. Therefore, introducing Ni into the tetrahedral sites of Co3O4 can generate optimal integrated catalytic sites, favorable for both aldehyde and hydroxyl group oxidation. With the optimization of Ni composition, the Ni0.5Co2.5O4 catalyst achieved a faradaic efficiency of 90.3% for FDCA at 1.5 V vs. RHE. To further enhance the catalytic properties of Ni doped Co3O4, Jiang et al. applied a cascade structure of Pd-NiCo2O4.169 Based on the Ni-doped Co3O4, Pd was loaded by immersing NiCo2O4 in palladium chloride ethanol solution. The fabricated Pd-NiCo2O4 can be applied as a bifunctional catalyst for both the OER and HMFOR. As shown in Fig. 13c, Pd-NiCo2O4 demonstrated remarkable performance in a 2-electrode full cell system, achieving a current density of 100 mA cm−2 at a low cell voltage of 1.51 V. FDCA selectivity was also observed as an extremely high faradaic efficiency of 99.6% in the half cell system and ~80% with the full cell system. Both current density and faradaic efficiency were sustained for more than 14 hours for the full cell system. To understand the superior performance of Pd-NiCo2O4, the role of Pd was understood based on the DFT simulation. From the inset of Fig. 13c, the binding energies of the hydroxyl group and HMF were compared on NiCo2O4 and Pd-NiCo2O4. With the additional Pd on NiCo2O4, the binding energy of HMF increased, while the opposite was observed for the hydroxyl group. The enhanced adsorption behavior of HMF induces facile initiation of the HMFOR and is even beneficial to the desorption in subsequent steps.182,183 Chen et al. presented low-crystalline Co-doped nickel phosphide (A-Co-Ni2P), achieving industrial current density in a half cell system, which reaches the ampere level.170 Under the alkaline conditions of 1 M KOH, the current density of 1290 mA cm−2 was observed at a potential of 1.6 V, with nearly 100% FDCA yield. According to Fig. 13d, by applying the membrane electrode assembly (MEA) setup for the coupled HMFOR/HER, 200 mA cm−2 was acquired at 1.54 V even in a full cell system, which is 400 mV lower than that of the conventional OER coupled system. The operating stability of the HMFOR with MEA was evaluated at a voltage of 1.8 V, and stable electrolysis was maintained for 60000 s, as shown in Fig. 13e. The electrooxidation of HMF with A-Co-Ni2P was revealed to involve a coupling of electrochemical and non-electrochemical reactions through in situ ATR-FTIR. The electrochemical reaction goes through the dehydrogenation of the aldehyde group of HMF, followed by hydration and oxidation of the hydroxyl group, and finally oxidation to FDCA (HMF → HMFCA → FFCA → FDCA). The non-electrochemical reaction is related to the Cannizzaro reaction of DFF, which occurs under alkaline conditions. Without external potential, DFF rapidly converts to HMF and FFCA by the HMFOR.184,185
Similar to the GEOR, the glucose oxidation reaction (GOR) also can have various products during the electrooxidation process. Considering that glucose has a longer carbon chain than glycerol, the possible reaction pathway should be much more complex. Still, as it was the same in the GEOR, the products with a higher number of carbon chains are more desirable, and products without C–C cleavage are preferred. In this point, representative reaction pathways with preferred intermediates are presented in Fig. 13f. In particular, gluconic acid is considered as one of the most valuable products commercially. However, various studies which coupled the GOR with the HER mostly generate formic acid as the main product due to the difficulty of controlling continuous oxidation with C–C cleavage. Here, studies focused on energetically enhanced HER/GOR coupled systems will be discussed first, followed by approaches that aim for efficient hydrogen generation with valorized GOR products.
Lin et al. investigated the optimal catalyst based on spinel Co3O4, by introducing various transition metal elements.171 With a hydrothermal method, MCo2O4 (M = Co, Mn, Cu, Ni, and Zn) catalysts were synthesized on Ni foam and their performances for the GOR were compared. The trend of the catalytic activity was confirmed as Cu > Co > Zn > Ni > Mn and CuCo2O4 exhibited excellent GOR electrocatalytic activity, which requires 1.31 V vs. RHE for 200 mA cm−2 with 98% formic acid faradaic efficiency. According to Fig. 13g, fabricated CuCo2O4 also presented moderate activity for the HER, and 2-electrode HER/GOR performance with bifunctional CuCo2O4 could achieve 200 mA cm−2 at 1.59 V, which is 370 mV lower than that of the water electrolysis system. The GOR mechanism of CuCo2O4 was confirmed through in situ Raman spectroscopy and electrochemical impedance spectroscopy (EIS). When the oxidative potential was applied, the Co3O4 reconstruction to CoOOH was observed and its amount increased along with potential. However, as the potential entered the region of the GOR, some of the CoOOH remained unchanged, which indicates that reconstructed CoOOH is consumed in the indirect oxidation of glucose. To further enhance the catalytic properties of transition metal introduced Co3O4 catalysts, Zhang presented NiO/NiCo2O4 nanowires on nickel foam (NiO/NiCo2O4/NF) through a facile solvothermal reaction and calcination. From the DFT simulation, charge transfer from NiO to NiCo2O4 was observed, generating high oxidation state Ni at the interface, which can optimize the catalytic performance.186 In addition, the porous nanowire structure of NiO/NiCo2O4/NF generates enriched interfacial active sites and promotes the diffusion of active substances. Thanks to the morphology and synergetic interaction between NiO and NiCo2O4, remarkably low potentials of 0.212 V and 1.128 V vs. RHE for 100 mA cm−2 were achieved in the HER and GOR with a 72% yield of formic acid. Li et al. proposed a hybrid catalyst of cobalt nanoparticles supported on nitrogen doped porous carbon (Co@NFC) as a bifunctional HER/GOR catalyst and the generation of lactic acid as the main GOR product.187 The conversion of glucose to lactic acid can occur in alkaline aqueous solution, starting from the isomerization of glucose. With the Co@NFC bifunctional catalyst, a voltage of 1.56 V was applied to reach 10 mA cm−2, which is 180 mV lower than that of conventional water splitting. While the electrochemical performance is not much noticeable, the valorization of the GOR was achieved through a 45.4% yield of lactic acid. Wu et al. further improved the yield for lactic acid by applying a Co@CoO composite anchored on graphene.172 To maximize the catalytic activity, the Mott–Schottky heterojunction of Co@CoO was controlled by exploiting defective MOF precursors during the synthesis process. From Fig. 13h, the synthesized Co@CoO catalyst exhibited a 300 mV lowered GOR potential of 1.34 V for 10 mA cm−2 when compared to water electrolysis, and such performance was retained after 80 h of long-term operation. The product yield was observed through HPLC after 20 h of electrolysis, and lactic acid accounts for 55% of the product. Liu et al. fabricated a nanostructured NiFe oxide (NiFeOx) anode and NiFe nitride (NiFeNx) cathode assembly for efficient electrochemical glucaric acid and H2 production.173 The electrolyzer assembled with these two catalysts can deliver 100 mA cm−2 at 1.39 V, which is nearly 300 mV lower than that of water electrolysis as shown in Fig. 13i. The anode with NiFeOx was confirmed to be efficient for glucaric acid (GRA) generation. According to Fig. 13j, as the concentration of glucose decreases with oxidation, gluconic acid (GNA) is first generated and oxidized, which results in accumulation of GRA. Zhang et al. presented a glucose-assisted Cu(I)/Cu(II) redox-coupled hydrogen production system, which provided gluconic acid as the main product.174 The fabricated system was comprised of a Cu(OH)2 anode and a Pt/C cathode separated by an alkaline membrane. According to Fig. 13k, the glucose-assisted Cu(I)/Cu(II) redox-coupled HER exhibited a remarkable decrease of 0.98 V potential compared to water electrolysis for 100 mA cm−2. The liquid product at the anode was also analyzed by HPLC and only gluconic acid was detected, with a faradaic efficiency of 98.7%. The reaction mechanism of glucose on Cu(OH)2 was explained with Raman, XPS and DFT simulation. With the ex situ Raman and XPS, it is confirmed that the glucose is oxidized through dehydrogenation, reducing Cu(II) of Cu(OH)2 to Cu(I). The reduced Cu(I) is again oxidized to Cu(II) with oxidative potential, again ready to dehydrogenate glucose. The indirect oxidation of glucose through the Cu(I)/Cu(II) redox couple was compared with direct oxidation through DFT simulation. In the indirect oxidation, glucose dehydrogenation leads to H2O formation with the hydroxyl group of Cu(OH)2, leaving Cu2O, which is also confirmed in the experimental result. With the interaction with Cu, the reaction barrier is highly stabilized to 0.61 eV, while the dehydrogenation without Cu(OH)2 should overcome 1.76 eV.
3 Challenges and perspectives
Substituting the OER with a thermodynamically favorable alternative oxidation reaction is promising for producing H2 with reduced energy consumption. Several advantages of chemical-assisted water electrolysis include: (1) water electrolysis can potentially explode due to the crossover of produced H2 and O2; this explosion risk can be mitigated by replacing the OER with other oxidation reactions. (2) Oxidation of wastewater, which contains ammonia or urea, can effectively remove contaminants while generating H2; (3) organic alternative oxidation reactions can produce value-added products. Despite these advantages, there is still plenty of room to be considered for the industrial application of chemical-assisted water electrolysis as an alternative to conventional water electrolysis, as outlined below (Fig. 14).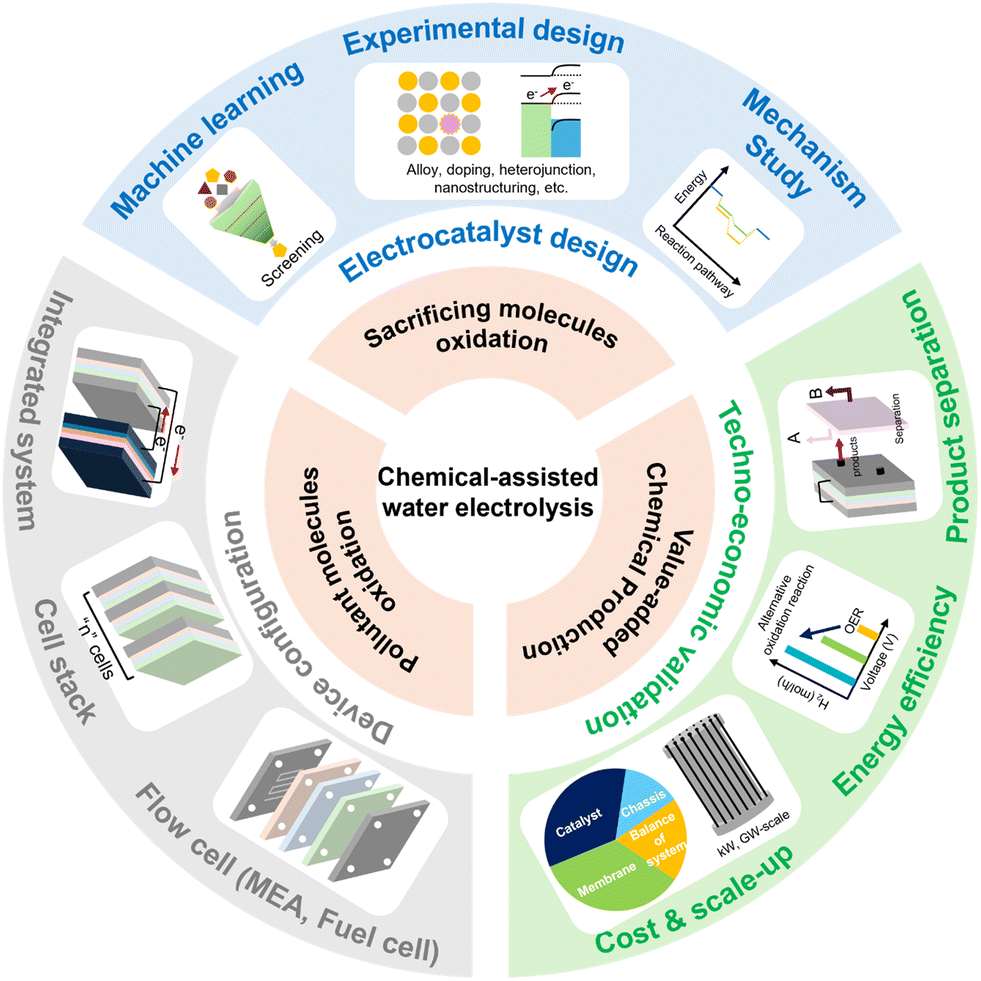 | ||
| Fig. 14 Schematic illustration of perspectives for chemical-assisted water electrolysis. | ||
Electrocatalyst design − securing the durability of chemical-assisted oxidation reactions with low overpotentials is a key area of interest. Extensive efforts have been made to develop cost-effective, catalytically active electrocatalysts for chemical-assisted water electrolysis, employing strategies such as alloying, nanostructuring, and doping. However, these reactions still require higher overpotentials than their thermodynamic reaction potentials. A notable issue in the AOR is that only Pt demonstrates selectivity toward direct N2 production at low voltage, but it suffers from deactivation due to surface poisoning by *N intermediates. One approach to mitigate this poisoning is operating the AOR with pulse cycling to facilitate the desorption of intermediates, although this is impractical for industrial operations. Another proposed solution is chronopotentiometry measurements at a lower operating voltage, which use partial catalytically active sites. However, this eventually results in complete deactivation due to poisoning of all active sites. Non-noble metal-based catalysts that selectively generate N2 have also been studied by several researchers. However, to date, no catalyst has been found that operates at voltages as low as Pt. Therefore, developing novel catalysts capable of operating at low voltages remains essential. Similarly, the alcohol oxidation reactions using Pt catalysts suffer from CO poisoning, which deteriorates the durability of the reaction. To address this, researchers explore non-precious metal-based catalysts, though these often require high operating potential. Recent research has focused on photoelectrochemical methanol oxidation to reduce energy consumption by harnessing solar energy.188,189
The fundamental investigation of chemical-assisted water oxidation reaction catalytic processes is pivotal to understanding the reaction mechanism of catalysts and addressing challenges such as strong adsorption of intermediates, catalyst dissolution, deactivation, and high energy barriers for the reaction. Thermodynamic-based DFT calculations have been instrumental in elucidating reaction mechanisms and identifying active catalytic surfaces. The rapid advancement of artificial intelligence has further enhanced catalyst design for electrochemical reactions by enabling data-driven screening of candidate materials for target reactions.190 Recent studies have shown the screening of electrocatalysts for chemical-assisted water oxidation reactions.79,132,191 Further systematic studies are required to screen electrocatalysts with multiple components for alternative water oxidation reactions, particularly multi-electron transfer reactions with ensured interpretability and reliability of machine learning models. In summary, combined high-throughput materials discovery, experimental catalyst development strategies, and materials characterization will pave the way for the design of catalytically active and durable chemical-assisted water oxidation electrocatalysts.
Device design − for industrial applications, achieving high current densities (scale of A cm−2) and long-term stability (>10000 h) using chemical-assisted water electrolysis is required. The development of MEA, a form of direct assembly of the anode, membrane, and cathode, has made it possible to reduce ohmic and mass transport losses, thereby achieving high operating current density. Studies have demonstrated ampere-scale electrochemical systems for chemical-assisted water electrolysis systems, suggesting a potential pathway toward industrial-scale applications.170,192,193 Additionally, fuel cell-type devices have been demonstrated, which may require elevated temperatures to achieve high performance.194,195 As an integrated system, the self-powered H2 production system has been demonstrated by combining direct DHzF and OHzS.137 However, the chemical-assisted water electrolysis system still requires advanced device design for large-scale, durable operation. To achieve this, further development of durable membranes with low product crossover, along with large-scale and uniform catalyst preparation methods, is required. The design of stack devices should also be followed by dynamic modeling and optimized operation environments.
Techno-economic aspects – meeting cost-effectiveness, energy efficiency, and scalability is crucial for chemical-assisted water electrolysis to serve as an alternative to conventional water electrolysis for hydrogen production at the system scale. Several factors must be considered for industrial applications, given the specific challenges of each reaction. The HzOR, which requires a very low voltage, represents a double-edged sword. On the positive side, the low voltage requirement enables significant savings in electrical energy consumption. However, from another perspective, it can be seen as hydrazine splitting rather than hydrazine-assisted water splitting. As pointed out by Badreldin et al., the electrochemical oxidation of N2H4 generates 2 moles of H2, but the N2H4 synthesis process consumes 3 moles of H2, leading to a net-negative production of 1 mol H2 per 1 mole of N2H4.196 In addition, sufficient N2H4, a key reactant for H2 production, should be supplied to the system, increasing overall costs. Therefore, it is necessary to balance the cost reduction from low energy consumption with the offsetting of the resource costs for N2H4. The concept of wastewater treatment or utilizing sacrificial reagents from wastewater should consider the use of low concentration reactants. To date, the use of actual wastewater for oxidation reactions has not been fully realized. In contrast to lab-scale research that uses high-purity, concentrated reactants (e.g., urea, ammonia), actual wastewater may introduce unforeseen issues, such as oxidation of pollutants, catalyst deactivation, stack failure due to impurities, and low operational current density from low reactant concentrations. This requires a pretreatment step and continuous supply of wastewater streams, which consume energy and incur additional costs. The categorization of chemical-assisted water oxidation may involve the generation of gaseous products from urea, ammonia, or hydrazine, or liquid value-added products from organic feedstocks. If multiple products are generated at the anode side, they must be sequestrated for further utilization. In terms of generating value-added products, complete oxidation of alcohols to CO2 has a low economic benefit. Glycerol oxidation reactions face challenges in controlling products due to their susceptibility to further oxidation, resulting in various products with small amounts. The process of separating multiple products also adds extra costs, highlighting the need for optimization of catalyst design, reaction conditions, and selective product formation to enhance economic viability.
In summary, we introduced 5 representative chemical-assisted oxidation reactions using agents (ammonia, alcohol, urea, hydrazine, and glycerol) as promising alternatives to the sluggish OER. In particular, we summarized the recent research on designing electrocatalysts for each reaction, explaining the catalyst design strategies and their effects on the catalytic activity. The various strategies were adjusted for electrocatalyst design, such as alloying, doping, heterostructure formation, control of dimensions from 3D to atomic scales, nanostructuring, etc. Overall, the chemical-assisted oxidation reaction and HER demonstrated promising systems with lower cell voltages compared to conventional water electrolysis (OER + HER). Considering the current state of the technology, future efforts should focus on integrating fundamental studies of catalytic reaction mechanisms, advancing electrocatalyst development, and conducting techno-economic assessments to meet the industrial performance requirements. With continued research and electrocatalyst development, chemical-assisted water electrolysis could become an environmentally friendly and energy-efficient technology, significantly contributing to the hydrogen economy.
Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
Jiwoo Lee, Sol A Lee, and Tae Hyung Lee contributed equally to this work. This research was supported by the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (RS-2024-00405016 and RS-2024-00421181).References
- The New York Times, https://www.nytimes.com/2023/12/04/climate/global-fossil-fuel-emissions.html, (accessed December 2024) Search PubMed.
- T. Capurso, M. Stefanizzi, M. Torresi and S. M. Camporeale, Perspective of the role of hydrogen in the 21st century energy transition, Energy Convers. Manage., 2022, 251, 114898 CrossRef CAS.
- A. Z. Arsad, M. A. Hannan, A. Q. Al-Shetwi, M. J. Hossain, R. A. Begum, P. J. Ker, F. Salehi and K. M. Muttaqi, Hydrogen electrolyser for sustainable energy production: A bibliometric analysis and future directions, Int. J. Hydrogen Energy, 2023, 48, 4960–4983 CrossRef CAS.
- S. G. Nnabuife, K. A. Quainoo, A. K. Hamzat, C. K. Darko and C. K. Agyemang, Innovative strategies for combining solar and wind energy with green hydrogen systems, Appl. Sci., 2024, 14, 9771 CrossRef CAS.
- Y. Gu, Q. Chen, J. Xue, Z. Tang, Y. Sun and Q. Wu, Comparative techno-economic study of solar energy integrated hydrogen supply pathways for hydrogen refueling stations in China, Energy Convers. Manage., 2020, 223, 113240 CrossRef CAS.
- J. H. Lim, K. Kim, J. H. Kang, K. C. Kwon and H. W. Jang, Tailored two-dimensional transition metal dichalcogenides for water electrolysis: Doping, defect, phase, and heterostructure, ChemElectroChem, 2024, 11, e202300614 CrossRef CAS.
- Z. Li, L. Sheng, R. Deng, Z. Zheng, P. Hou, M. Chen, Z. Ma, K. Sun, Y. Wang, Q. Liu, P. Xu, X. Ma and H. Chu, Regulating crystal phase in Ir-Ge interstitials for hydrogen evolution electrocatalysis, ACS Energy Lett., 2023, 8, 5136–5142 CrossRef CAS.
- H. Yang, M. Driess and P. W. Menezes, Self-supported electrocatalysts for practical water electrolysis, Adv. Energy Mater., 2021, 11, 2102074 CrossRef CAS.
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, Universality in oxygen evolution electrocatalysis on oxide surfaces, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- G. F. Swiegers, A. L. Hoang, A. Hodges, G. Tsekouras, C.-Y. Lee, K. Wagner and G. Wallace, Current status of membraneless water electrolysis cells, Curr. Opin. Electrochem., 2022, 32, 100881 CrossRef CAS.
- F. Arshad, T. ul Haq, I. Hussain and F. Sher, Recent advances in electrocatalysts toward alcohol-assisted, energy-saving hydrogen production, ACS Appl. Energy Mater., 2021, 4, 8685–8701 CrossRef CAS.
- K. Veeramani, G. Janani, J. Kim, S. Surendran, J. Lim, S. C. Jesudass, S. Mahadik, H. Lee, T.-H. Kim, J. K. Kim and U. Sim, Hydrogen and value-added products yield from hybrid water electrolysis: A critical review on recent developments, Renewable Sustainable Energy Rev., 2023, 177, 113227 CrossRef CAS.
- Y. Jun, J. Bang, S. Hong, S. Young Kim and S. Hyun Ahn, Unveiling facet engineering of ultrathin Pt overlayers for enhanced ammonia oxidation reaction, Chem. Eng. J., 2024, 498, 155433 CrossRef CAS.
- C. Bianchini and P. K. Shen, Palladium-based electrocatalysts for alcohol oxidation in half cells and in direct alcohol fuel cells, Chem. Rev., 2009, 109, 4183–4206 CrossRef CAS PubMed.
- S.-K. Geng, Y. Zheng, S.-Q. Li, H. Su, X. Zhao, J. Hu, H.-B. Shu, M. Jaroniec, P. Chen, Q.-H. Liu and S.-Z. Qiao, Nickel ferrocyanide as a high-performance urea oxidation electrocatalyst, Nat. Energy, 2021, 6, 904–912 CrossRef CAS.
- L. Zhu, J. Huang, G. Meng, T. Wu, C. Chen, H. Tian, Y. Chen, F. Kong, Z. Chang, X. Cui and J. Shi, Active site recovery and N-N bond breakage during hydrazine oxidation boosting the electrochemical hydrogen production, Nat. Commun., 2023, 14, 1997 CrossRef CAS PubMed.
- C. Chen, Z. Zhou, J. Liu, B. Zhu, H. Hu, Y. Yang, G. Chen, M. Gao and J. Zhang, Sustainable biomass upgrading coupled with H2 generation over in-situ oxidized Co3O4 electrocatalysts, Appl. Catal., B, 2022, 307, 121209 CrossRef CAS.
- J.-T. Ren, L. Chen, H.-Y. Wang, W.-W. Tian and Z.-Y. Yuan, Water electrolysis for hydrogen production: from hybrid systems to self-powered/catalyzed devices, Energy Environ. Sci., 2024, 17, 49–113 RSC.
- S. A. Lee, J. Bu, J. Lee and H. W. Jang, High-entropy nanomaterials for advanced electrocatalysis, Small Sci., 2023, 3, 2200109 CrossRef CAS.
- T.-G. Vo, C.-C. Kao, J.-L. Kuo, C. Chiu and C.-Y. Chiang, Unveiling the crystallographic facet dependence of the photoelectrochemical glycerol oxidation on bismuth vanadate, Appl. Catal., B, 2020, 278, 119303 CrossRef CAS.
- Y. Kuang, W. Qiao, S. Wang, F. Yang and L. Feng, Doping and interfacial engineering of MoSe2 nanosheets by NH3 plasma promoted Pt for methanol electrolysis, ACS Mater. Lett., 2024, 6, 1722–1731 CrossRef CAS.
- Z. Li, J. Li, Z. Zheng, K. Jiang, T. Zheng, D. Wang, H. Wei, Z. Shi, X. Li and H. Chu, Roles of hydroxyl and oxygen vacancy of CeO2·xH2O in Pd-catalyzed ethanol electro-oxidation, Sci. China: Chem., 2022, 65, 877–884 CrossRef CAS.
- Y. Kuang, M. Li, L. Fu and L. Feng, Deciphering promotion of MoP over MoC in Pt catalysts for methanol-assisted water splitting reaction, J. Colloid Interface Sci., 2025, 679, 921–929 CrossRef CAS PubMed.
- L. Fan, Y. Ji, G. Wang, J. Chen, K. Chen, X. Liu and Z. Wen, High entropy alloy electrocatalytic electrode toward alkaline glycerol valorization coupling with acidic hydrogen production, J. Am. Chem. Soc., 2022, 144, 7224–7235 CrossRef CAS PubMed.
- K. Hou, S. Zhang, P. Yin, T. Liu, Y. Cao, M. Wang, W. Zhan and L. Wu, Zinc vaporization induced formation of desired Ni nanoparticles coated with ultrathin carbon shells for efficient electrocatalytic H2 production coupling with methanol upgrading, ACS Appl. Mater. Interfaces, 2024, 16, 52326–52338 CrossRef CAS PubMed.
- F. Si, J. Liu, Y. Zhang, B. Zhao, Y. Liang, X. Wu, X. Kang, X. Yang, J. Zhang, X.-Z. Fu and J.-L. Luo, Surface spin enhanced high stable NiCo2S4 for energy-saving production of H2 from water/methanol coelectrolysis at high current density, Small, 2023, 19, 2205257 CrossRef CAS PubMed.
- M. He, C. Feng, T. Liao, S. Hu, H. Wu and Z. Sun, Low-cost Ni2P/Ni0.96S heterostructured bifunctional electrocatalyst toward highly efficient overall urea-water electrolysis, ACS Appl. Mater. Interfaces, 2020, 12, 2225–2233 CrossRef CAS PubMed.
- D. M. Fadzillah, S. K. Kamarudin, M. A. Zainoodin and M. S. Masdar, Critical challenges in the system development of direct alcohol fuel cells as portable power supplies: An overview, Int. J. Hydrogen Energy, 2019, 44, 3031–3054 CrossRef CAS.
- S. P. S. Badwal, S. Giddey, A. Kulkarni, J. Goel and S. Basu, Direct ethanol fuel cells for transport and stationary applications – A comprehensive review, Appl. Energy, 2015, 145, 80–103 CrossRef CAS.
- A. M. Sheikh, K. E.-A. Abd-Alftah and C. F. Malfatti, On reviewing the catalyst materials for direct alcohol fuel cells (DAFCs), J. Multidiscip. Eng. Sci. Technol., 2014, 1, N42350050 Search PubMed.
- X. Liu, Y. Han, Y. Guo, X. Zhao, D. Pan, K. Li and Z. Wen, Electrochemical hydrogen generation by oxygen evolution reaction-alternative anodic oxidation reactions, Adv. Energy Sustainability Res., 2022, 3, 2200005 CrossRef CAS.
- Z. Jusys, J. Kaiser and R. J. Behm, Methanol electrooxidation over Pt/C fuel cell catalysts: Dependence of product yields on catalyst loading, Langmuir, 2003, 19, 6759–6769 CrossRef CAS.
- D. S. Mekazni, R. M. Arán-Ais, A. Ferre-Vilaplana and E. Herrero, Why methanol electro-oxidation on platinum in water takes place only in the presence of adsorbed OH, ACS Catal., 2022, 12, 1965–1970 CrossRef CAS.
- B. Zhu, B. Dong, F. Wang, Q. Yang, Y. He, C. Zhang, P. Jin and L. Feng, Unraveling a bifunctional mechanism for methanol-to-formate electro-oxidation on nickel-based hydroxides, Nat. Commun., 2023, 14, 1686 CrossRef CAS PubMed.
- L. E. Heim, H. Konnerth and M. H. G. Prechtl, Future perspectives for formaldehyde: Pathways for reductive synthesis and energy storage, Green Chem., 2017, 19, 2347–2355 RSC.
- S. Jones, K. Tedsree, M. Sawangphruk, J. S. Foord, J. Fisher, D. Thompsett and S. C. E. Tsang, Promotion of direct methanol electro-oxidation by Ru terraces on Pt by using a reversed spillover mechanism, ChemCatChem, 2010, 2, 1089–1095 CrossRef CAS.
- G. Hou, J. Parrondo, V. Ramani and J. Prakash, Kinetic and mechanistic investigation of methanol oxidation on a smooth polycrystalline Pt surface, J. Electrochem. Soc., 2013, 161, F252 CrossRef.
- S. Li, Z. Fan, Y. Li, Y. Zhang, Y. Zhao, J. Zhao, J. Zhang and Z. Wang, Efficient catalysis for acidic methanol oxidation: Exploration of a low-platinum quaternary alloy catalyst via a two-step method, Chem. Eng. J., 2024, 500, 156355 CrossRef CAS.
- Y.-T. Kim, K. Ohshima, K. Higashimine, T. Uruga, M. Takata, H. Suematsu and T. Mitani, Fine size control of platinum on carbon nanotubes: From single atoms to clusters, Angew. Chem., Int. Ed., 2006, 45, 407–411 CrossRef CAS PubMed.
- X. Chen, M.-M. Liang, J. Xu, H.-L. Sun, C. Wang, J. Wei, H. Zhang, W.-M. Yang, Z.-L. Yang, J.-J. Sun, Z.-Q. Tian and J.-F. Li, Unveiling the size effect of Pt-on-Au nanostructures on CO and methanol electrooxidation by in situ electrochemical SERS, Nanoscale, 2020, 12, 5341–5346 RSC.
- J. Ruan, Y. Chen, G. Zhao, P. Li, B. Zhang, Y. Jiang, T. Ma, H. Pan, S. X. Dou and W. Sun, Cobalt single atoms enabling efficient methanol oxidation reaction on platinum anchored on nitrogen-doped carbon, Small, 2022, 18, 2107067 CrossRef CAS PubMed.
- Y. Hao, D. Yu, S. Zhu, C.-H. Kuo, Y.-M. Chang, L. Wang, H.-Y. Chen, M. Shao and S. Peng, Methanol upgrading coupled with hydrogen product at large current density promoted by strong interfacial interactions, Energy Environ. Sci., 2023, 16, 1100–1110 RSC.
- Y. Kuang, W. Qiao, S. Wang, F. Yang and L. Feng, Doping and interfacial engineering of MoSe2 nanosheets by NH3 plasma promoted Pt for methanol electrolysis, ACS Mater. Lett., 2024, 6, 1722–1731 CrossRef CAS.
- Y. Zhou, Q. Wang, X. Tian and L. Feng, Efficient bifunctional catalysts of CoSe/N-doped carbon nanospheres supported Pt nanoparticles for methanol electrolysis of hydrogen generation, Nano Res., 2022, 15, 8936–8945 CrossRef CAS.
- S. Zhang, Z. Zeng, Q. Li, B. Huang, X. Zhang, Y. Du and C.-H. Yan, Lanthanide electronic perturbation in Pt–Ln (La, Ce, Pr and Nd) alloys for enhanced methanol oxidation reaction activity, Energy Environ. Sci., 2021, 14, 5911–5918 RSC.
- H. Li, Y. Han, H. Zhao, W. Qi, D. Zhang, Y. Yu, W. Cai, S. Li, J. Lai, B. Huang and L. Wang, Fast site-to-site electron transfer of high-entropy alloy nanocatalyst driving redox electrocatalysis, Nat. Commun., 2020, 11, 5437 CrossRef CAS PubMed.
- D. Majumdar and S. K. Bhattacharya, Recent developments of methanol electrooxidation using nickel-based nanocatalysts, ChemistrySelect, 2022, 7, e202201807 CrossRef CAS.
- L. Li, W. Gao, M. Lei and D. Wen, Alkali-induced transformation of Ni-MOF into Ni(OH)2 nanostructured flowers for efficient electrocatalytic methanol oxidation reaction, Chem. – Eur. J., 2021, 27, 10966–10972 CrossRef CAS PubMed.
- W. Yang, X. Yang, J. Jia, C. Hou, H. Gao, Y. Mao, C. Wang, J. Lin and X. Luo, Oxygen vacancies confined in ultrathin nickel oxide nanosheets for enhanced electrocatalytic methanol oxidation, Appl. Catal., B, 2019, 244, 1096–1102 CrossRef CAS.
- K. Hou, S. Zhang, P. Yin, T. Liu, Y. Cao, M. Wang, W. Zhan and L. Wu, Zinc vaporization induced formation of desired Ni nanoparticles coated with ultrathin carbon shells for efficient electrocatalytic H2 production coupling with methanol upgrading, ACS Appl. Mater. Interfaces, 2024, 16, 52326–52338 CrossRef CAS PubMed.
- Y. Qin, H. Huang, W. Yu, H. Zhang, Z. Li, Z. Wang, J. Lai, L. Wang and S. Feng, Porous PdWM (M = Nb, Mo and Ta) trimetallene for high C1 selectivity in alkaline ethanol oxidation reaction, Adv. Sci., 2022, 9, 2103722 CrossRef CAS PubMed.
- C. Liang, R. Zhao, T. Chen, Y. Luo, J. Hu, P. Qi and W. Ding, Recent approaches for cleaving the C–C bond during ethanol electro-oxidation reaction, Adv. Sci., 2024, 11, 2308958 CrossRef CAS PubMed.
- S. C. S. Lai, S. E. F. Kleijn, F. T. Z. Öztürk, V. C. van Rees Vellinga, J. Koning, P. Rodriguez and M. T. M. Koper, Effects of electrolyte pH and composition on the ethanol electro-oxidation reaction, Catal. Today, 2010, 154, 92–104 CrossRef CAS.
- X. Fu, C. Wan, Y. Huang and X. Duan, Noble metal based electrocatalysts for alcohol oxidation reactions in alkaline media, Adv. Funct. Mater., 2022, 32, 2106401 CrossRef CAS.
- Z.-Y. Li, J. Zhou, L.-S. Tang, X.-P. Fu, H. Wei, M. Xue, Y.-L. Zhao, C.-J. Jia, X.-M. Li, H.-B. Chu and Y. Li, Hydroxyl-rich ceria hydrate nanoparticles enhancing the alcohol electrooxidation performance of Pt catalysts, J. Mater. Chem. A, 2018, 6, 2318–2326 RSC.
- H.-F. Wang and Z.-P. Liu, Comprehensive mechanism and structure-sensitivity of ethanol oxidation on platinum: New transition-state searching method for resolving the complex reaction network, J. Am. Chem. Soc., 2008, 130, 10996–11004 CrossRef CAS PubMed.
- B. Pierozynski, On the ethanol electrooxidation reaction on catalytic surfaces of Pt in 0.1 M NaOH, Int. J. Electrochem. Sci., 2012, 7, 4261–4271 CrossRef CAS.
- G. Liu, W. Zhou, Y. Ji, B. Chen, G. Fu, Q. Yun, S. Chen, Y. Lin, P.-F. Yin, X. Cui, J. Liu, F. Meng, Q. Zhang, L. Song, L. Gu and H. Zhang, Hydrogen-intercalation-induced lattice expansion of Pd@Pt core–shell nanoparticles for highly efficient electrocatalytic alcohol oxidation, J. Am. Chem. Soc., 2021, 143, 11262–11270 CrossRef CAS PubMed.
- W. Yan, G. Li, S. Cui, G.-S. Park, R. Oh, W. Chen, X. Cheng, J.-M. Zhang, W. Li, L.-F. Ji, O. Akdim, X. Huang, H. Lin, J. Yang, Y.-X. Jiang and S.-G. Sun, Ga-modification near-surface composition of Pt–Ga/C catalyst facilitates high-efficiency electrochemical ethanol oxidation through a C2 intermediate, J. Am. Chem. Soc., 2023, 145, 17220–17231 CrossRef CAS PubMed.
- Y. Wang, M. Zheng, Y. Li, C. Ye, J. Chen, J. Ye, Q. Zhang, J. Li, Z. Zhou, X.-Z. Fu, J. Wang, S.-G. Sun and D. Wang, p–d Orbital hybridization induced by a monodispersed Ga site on a Pt3Mn nanocatalyst boosts ethanol electrooxidation, Angew. Chem., Int. Ed., 2022, 61, e202115735 CrossRef CAS PubMed.
- Z. Li, S. Ning, J. Xu, J. Zhu, Z. Yuan, Y. Wu, J. Chen, F. Xie, Y. Jin, N. Wang, H. Meng and S. Sun, In situ electrochemical activation of Co(OH)2@Ni(OH)2 heterostructures for efficient ethanol electrooxidation reforming and innovative zinc–ethanol–air batteries, Energy Environ. Sci., 2022, 15, 5300–5312 RSC.
- X. Tan, S. Chen, D. Yan, R. Du, Q. Zhong, L. Liao, Z. Tang and F. Zeng, Recent advances in Ni-based catalysts for the electrochemical oxidation of ethanol, J. Energy Chem., 2024, 98, 588–614 CrossRef CAS.
- X. Chi, L. Gao, W. Zhou, Y. Zhang and T. Hu, Enhanced electrocatalytic performance of 2D Ni-MOF for ethanol oxidation reaction by loading carbon dots, J. Solid State Chem., 2022, 311, 123094 CrossRef CAS.
- H. Wang, A. Guan, J. Zhang, Y. Mi, S. Li, T. Yuan, C. Jing, L. Zhang, L. Zhang and G. Zheng, Copper-doped nickel oxyhydroxide for efficient electrocatalytic ethanol oxidation, Chin. J. Catal., 2022, 43, 1478–1484 CrossRef CAS.
- S. A. Lee, M. G. Lee and H. W. Jang, Catalysts for electrochemical ammonia oxidation: Trend, challenge, and promise, Sci. China Mater., 2022, 65, 3334–3352 CrossRef.
- C. Zhong, W. B. Hu and Y. F. Cheng, Recent advances in electrocatalysts for electro-oxidation of ammonia, J. Mater. Chem. A, 2013, 1, 3216–3238 RSC.
- H. Mashhadimoslem, M. Safarzadeh Khosrowshahi, M. Delpisheh, C. Convery, M. Rezakazemi, T. M. Aminabhavi, M. Kamkar and A. Elkamel, Green ammonia to hydrogen: Reduction and oxidation catalytic processes, Chem. Eng. J., 2023, 474, 145661 CrossRef CAS.
- Q. Qian, Y. Zhu, N. Ahmad, Y. Feng, H. Zhang, M. Cheng, H. Liu, C. Xiao, G. Zhang and Y. Xie, Recent advancements in electrochemical hydrogen production via hybrid water splitting, Adv. Mater., 2024, 36, 2306108 CrossRef CAS PubMed.
- H. Liu, X. Xu, D. Guan and Z. Shao, Minireview on the electrocatalytic ammonia oxidation reaction for hydrogen production and sewage treatment, Energy Fuels, 2024, 38, 919–931 CrossRef CAS.
- S. Cohen, S. Johnston, C. K. Nguyen, T. D. Nguyen, D. A. Hoogeveen, D. V. Zeil, S. Giddey, A. N. Simonov and D. R. MacFarlane, A CoOxHy/β-NiOOH electrocatalyst for robust ammonia oxidation to nitrite and nitrate, Green Chem., 2023, 25, 7157–7165 RSC.
- H. G. Oswin and M. Salomon, The anodic oxidation of ammonia at platinum black electrodes in aqueous KOH electrolyte, Can. J. Chem., 1963, 41, 1686–1694 CrossRef CAS.
- H. Gerischer and A. Mauerer, Untersuchungen zur anodischen oxidation von ammoniak an platin-elektroden, J. Electroanal. Chem. Interfacial Electrochem., 1970, 25, 421–433 CrossRef CAS.
- N. M. Adli, H. Zhang, S. Mukherjee and G. Wu, Review—Ammonia oxidation electrocatalysis for hydrogen generation and fuel cells, J. Electrochem. Soc., 2018, 165, J3130 CrossRef CAS.
- A. C. A. de Vooys, M. T. M. Koper, R. A. van Santen and J. A. R. van Veen, The role of adsorbates in the electrochemical oxidation of ammonia on noble and transition metal electrodes, J. Electroanal. Chem., 2001, 506, 127–137 CrossRef CAS.
- Y. Yang, J. Kim, H. Jo, A. Seong, M. Lee, H.-K. Min, M. Seo, Y. Choi and G. Kim, A rigorous electrochemical ammonia electrolysis protocol with in operando quantitative analysis, J. Mater. Chem. A, 2021, 9, 11571–11579 RSC.
- J. Liu, Z. Liu, H. Wang, B. Liu, N. Zhao, C. Zhong and W. Hu, Designing nanoporous coral-like Pt nanowires architecture for methanol and ammonia oxidation reactions, Adv. Funct. Mater., 2022, 32, 2110702 CrossRef CAS.
- F. J. Vidal-Iglesias, J. Solla-Gullón, V. Montiel, J. M. Feliu and A. Aldaz, Screening of electrocatalysts for direct ammonia fuel cell: Ammonia oxidation on PtMe (Me: Ir, Rh, Pd, Ru) and preferentially oriented Pt(100) nanoparticles, J. Power Sources, 2007, 171, 448–456 CrossRef CAS.
- M. H. M. T. Assumpção, R. M. Piasentin, P. Hammer, R. F. B. De Souza, G. S. Buzzo, M. C. Santos, E. V. Spinacé, A. O. Neto and J. C. M. Silva, Oxidation of ammonia using PtRh/C electrocatalysts: Fuel cell and electrochemical evaluation, Appl. Catal., B, 2015, 174–175, 136–144 CrossRef.
- H. S. Pillai, Y. Li, S.-H. Wang, N. Omidvar, Q. Mu, L. E. K. Achenie, F. Abild-Pedersen, J. Yang, G. Wu and H. Xin, Interpretable design of Ir-free trimetallic electrocatalysts for ammonia oxidation with graph neural networks, Nat. Commun., 2023, 14, 792 CrossRef CAS PubMed.
- S. Liu, Y. Jiang, M. Wang, Y. Huan, Y. He, Q. Cheng, Y. Cheng, J. Liu, X. Zhou, T. Qian and C. Yan, Awakening (220) as one more active facet of PtMo alloy via single-atom doping to boost ammonia electrooxidation in direct ammonia fuel cell, Adv. Funct. Mater., 2023, 33, 2306204 CrossRef CAS.
- Y. Li, X. Li, H. S. Pillai, J. Lattimer, N. Mohd Adli, S. Karakalos, M. Chen, L. Guo, H. Xu, J. Yang, D. Su, H. Xin and G. Wu, Ternary PtIrNi catalysts for efficient electrochemical ammonia oxidation, ACS Catal., 2020, 10, 3945–3957 CrossRef CAS.
- J. Huang, Z. Chen, J. Cai, Y. Jin, T. Wang and J. Wang, Activating copper oxide for stable electrocatalytic ammonia oxidation reaction via in-situ introducing oxygen vacancies, Nano Res., 2022, 15, 5987–5994 CrossRef CAS.
- H. Fang, C. Liao, Q. Cai, F. Zhong, L. Lin, C. Chen, Y. Luo and L. Jiang, Tuning surficial atomic configuration of Pt–Ir catalysts for efficient ammonia oxidation and low-temperature direct ammonia fuel cells, Chem. Eng. Sci., 2023, 280, 118836 CrossRef CAS.
- X. Lin, X. Zhang, Z. Wang, X. Zhu, J. Zhu, P. Chen, T. Lyu, C. Li, Z. Qun Tian and P. Kang Shen, Hyperbranched concave octahedron of PtIrCu nanocrystals with high-index facets for efficiently electrochemical ammonia oxidation reaction, J. Colloid Interface Sci., 2021, 601, 1–11 CrossRef CAS PubMed.
- H. Kim, W. Yang, W. H. Lee, M. H. Han, J. Moon, C. Jeon, D. Kim, S. G. Ji, K. H. Chae, K.-S. Lee, J. Seo, H.-S. Oh, H. Kim and C. H. Choi, Operando stability of platinum electrocatalysts in ammonia oxidation reactions, ACS Catal., 2020, 10, 11674–11684 CrossRef CAS.
- W. Xu, D. Du, R. Lan, J. Humphreys, D. N. Miller, M. Walker, Z. Wu, J. T. S. Irvine and S. Tao, Electrodeposited NiCu bimetal on carbon paper as stable non-noble anode for efficient electrooxidation of ammonia, Appl. Catal., B, 2018, 237, 1101–1109 CrossRef CAS.
- H. Shi, J. Tang, W. Yu, M. O. Tadé and Z. Shao, Advances in power generation from ammonia via electrocatalytic oxidation in direct ammonia fuel cells, Chem. Eng. J., 2024, 488, 150896 CrossRef CAS.
- S. He, Y. Chen, M. Wang, H. Nuomin, P. Novello, X. Li, S. Zhu and J. Liu, Metal nitride nanosheets enable highly efficient electrochemical oxidation of ammonia, Nano Energy, 2021, 80, 105528 CrossRef CAS.
- Y.-J. Shih and C.-H. Hsu, Kinetics and highly selective N2 conversion of direct electrochemical ammonia oxidation in an undivided cell using NiCo oxide nanoparticle as the anode and metallic Cu/Ni foam as the cathode, Chem. Eng. J., 2021, 409, 128024 CrossRef CAS.
- H. Zhang, H. Wang, L. Zhou, Q. Li, X. Yang, Y. Wang, M. Zhang and Z. Wu, Efficient and highly selective direct electrochemical oxidation of ammonia to dinitrogen facilitated by NiCu diatomic site catalysts, Appl. Catal., B, 2023, 328, 122544 CrossRef CAS.
- K. Nagita, Y. Yuhara, K. Fujii, Y. Katayama and M. Nakayama, Ni- and Cu-co-intercalated layered manganese oxide for highly efficient electro-oxidation of ammonia selective to nitrogen, ACS Appl. Mater. Interfaces, 2021, 13, 28098–28107 CrossRef CAS PubMed.
- H. Wang, X. Tong, L. Zhou, Y. Wang, L. Liao, S. Ouyang and H. Zhang, Unique three-dimensional nanoflower-like NiCu electrodes constructed by Co, S co-doping for efficient ammonia oxidation reaction, Sep. Purif. Technol., 2022, 303, 122293 CrossRef CAS.
- A. Ashok Kashale, C.-T. Wu, H.-F. Hsu and I.-W. Peter Chen, In-situ monitoring intermediate stages in ammonia oxidation reaction via high performance NiCuBOx-1/NF electrocatalysts, Chem. Eng. J., 2023, 474, 145907 CrossRef CAS.
- X. Gao, S. Zhang, P. Wang, M. Jaroniec, Y. Zheng and S.-Z. Qiao, Urea catalytic oxidation for energy and environmental applications, Chem. Soc. Rev., 2024, 53, 1552–1591 RSC.
- L. Zhang, L. Wang, H. Lin, Y. Liu, J. Ye, Y. Wen, A. Chen, L. Wang, F. Ni, Z. Zhou, S. Sun, Y. Li, B. Zhang and H. Peng, A lattice-oxygen-involved reaction pathway to boost urea oxidation, Angew. Chem., Int. Ed., 2019, 58, 16820–16825 CrossRef CAS PubMed.
- S. J. Yao, S. K. Wolfson, B. K. Ahn and C. C. Liu, Anodic oxidation of urea and an electrochemical approach to de-ureation, Nature, 1973, 241, 471–472 CrossRef CAS PubMed.
- Z.-P. Wu, X. F. Lu, S.-Q. Zang and X. W. Lou, Non-noble-metal-based electrocatalysts toward the oxygen evolution reaction, Adv. Funct. Mater., 2020, 30, 1910274 CrossRef CAS.
- S. Chen, J. Duan, A. Vasileff and S. Z. Qiao, Size fractionation of two-dimensional sub-nanometer thin manganese dioxide crystals towards superior urea electrocatalytic conversion, Angew. Chem., Int. Ed., 2016, 55, 3804–3808 CrossRef CAS PubMed.
- D. Zhu, H. Zhang, J. Miao, F. Hu, L. Wang, Y. Tang, M. Qiao and C. Guo, Strategies for designing more efficient electrocatalysts towards the urea oxidation reaction, J. Mater. Chem. A, 2022, 10, 3296–3313 RSC.
- C. Liu, P. Wang, B. Hu, X. Liu, R. Huang and G. Zhou, Asymmetric configuration activating lattice oxygen via weakening d-p orbital hybridization for efficient C/N separation in urea overall electrolysis, J. Energy Chem., 2024, 92, 233–239 CrossRef CAS.
- D. Zhu, C. Guo, J. Liu, L. Wang, Y. Du and S.-Z. Qiao, Two-dimensional metal–organic frameworks with high oxidation states for efficient electrocatalytic urea oxidation, Chem. Commun., 2017, 53, 10906–10909 RSC.
- B. K. Boggs, R. L. King and G. G. Botte, Urea electrolysis: Direct hydrogen production from urine, Chem. Commun., 2009, 4859 RSC.
- K. Yang, L. Hao, Y. Hou, J. Zhang and J.-H. Yang, Summary and application of Ni-based catalysts for electrocatalytic urea oxidation, Int. J. Hydrogen Energy, 2024, 51, 966–981 CrossRef CAS.
- Z. Ji, Y. Song, S. Zhao, Y. Li, J. Liu and W. Hu, Pathway manipulation via Ni, Co, and V ternary synergism to realize high efficiency for urea electrocatalytic oxidation, ACS Catal., 2022, 12, 569–579 CrossRef CAS.
- H. Jiang, J. Xia, L. Jiao, X. Meng, P. Wang, C.-S. Lee and W. Zhang, Ni single atoms anchored on N-doped carbon nanosheets as bifunctional electrocatalysts for urea-assisted rechargeable Zn-air batteries, Appl. Catal., B, 2022, 310, 121352 CrossRef CAS.
- G. Zhan, L. Hu, H. Li, J. Dai, L. Zhao, Q. Zheng, X. Zou, Y. Shi, J. Wang, W. Hou, Y. Yao and L. Zhang, Highly selective urea electrooxidation coupled with efficient hydrogen evolution, Nat. Commun., 2024, 15, 5918 CrossRef CAS PubMed.
- L. Wang, Y. Zhu, Y. Wen, S. Li, C. Cui, F. Ni, Y. Liu, H. Lin, Y. Li, H. Peng and B. Zhang, Regulating the local charge distribution of Ni active sites for the urea oxidation reaction, Angew. Chem., Int. Ed., 2021, 60, 10577–10582 CrossRef CAS PubMed.
- H. Sun, J. Liu, G. Chen, H. Kim, S. Kim, Z. Hu, J. Chen, S. Haw, F. Ciucci and W. Jung, Hierarchical structure of CuO nanowires decorated with Ni(OH)2 supported on Cu foam for hydrogen production via urea electrocatalysis, Small Methods, 2022, 6, 2101017 CrossRef CAS PubMed.
- N. Chen, Y.-X. Du, G. Zhang, W.-T. Lu and F.-F. Cao, Amorphous nickel sulfoselenide for efficient electrochemical urea-assisted hydrogen production in alkaline media, Nano Energy, 2021, 81, 105605 CrossRef CAS.
- Z.-Y. Yu, C.-C. Lang, M.-R. Gao, Y. Chen, Q.-Q. Fu, Y. Duan and S.-H. Yu, Ni–Mo–O nanorod-derived composite catalysts for efficient alkaline water-to-hydrogen conversion via urea electrolysis, Energy Environ. Sci., 2018, 11, 1890–1897 RSC.
- H. Jiang, M. Sun, S. Wu, B. Huang, C. Lee and W. Zhang, Oxygen-incorporated NiMoP nanotube arrays as efficient bifunctional electrocatalysts for urea-assisted energy-saving hydrogen production in alkaline electrolyte, Adv. Funct. Mater., 2021, 31, 2104951 CrossRef CAS.
- M. Fang, W. Gao, G. Dong, Z. Xia, S. Yip, Y. Qin, Y. Qu and J. C. Ho, Hierarchical NiMo-based 3D electrocatalysts for highly-efficient hydrogen evolution in alkaline conditions, Nano Energy, 2016, 27, 247–254 CrossRef CAS.
- D. Liu, T. Liu, L. Zhang, F. Qu, G. Du, A. M. Asiri and X. Sun, High-performance urea electrolysis towards less energy-intensive electrochemical hydrogen production using a bifunctional catalyst electrode, J. Mater. Chem. A, 2017, 5, 3208–3213 RSC.
- M. Li, H. Sun, J. Yang, M. Humayun, L. Li, X. Xu, X. Xue, A. Habibi-Yangjeh, K. Temst and C. Wang, Mono-coordinated metallocene ligands endow metal-organic frameworks with highly efficient oxygen evolution and urea electrolysis, Chem. Eng. J., 2022, 430, 132733 CrossRef CAS.
- J. Kim, M. Kim, S. S. Han and K. Cho, Accessible Ni-Fe-oxalate framework for electrochemical urea oxidation with radically enhanced kinetics, Adv. Funct. Mater., 2024, 34, 2315625 CrossRef CAS.
- C. Pei, S. Chen, T. Zhao, M. Li, Z. Cui, B. Sun, S. Hu, S. Lan, H. Hahn and T. Feng, Nanostructured metallic glass in a highly upgraded energy state contributing to efficient catalytic performance, Adv. Mater., 2022, 34, 2200850 CrossRef CAS PubMed.
- C. Li, Y. Liu, Z. Zhuo, H. Ju, D. Li, Y. Guo, X. Wu, H. Li and T. Zhai, Local charge distribution engineered by schottky heterojunctions toward urea electrolysis, Adv. Energy Mater., 2018, 8, 1801775 CrossRef.
- C. Wang, H. Lu, Z. Mao, C. Yan, G. Shen and X. Wang, Bimetal Schottky heterojunction boosting energy-saving hydrogen production from alkaline water via urea electrocatalysis, Adv. Funct. Mater., 2020, 30, 2000556 CrossRef CAS.
- A. Kumar, X. Liu, J. Lee, B. Debnath, A. R. Jadhav, X. Shao, V. Q. Bui, Y. Hwang, Y. Liu, M. G. Kim and H. Lee, Discovering ultrahigh loading of single-metal-atoms via surface tensile-strain for unprecedented urea electrolysis, Energy Environ. Sci., 2021, 14, 6494–6505 RSC.
- M. Song, Z. Zhang, Q. Li, W. Jin, Z. Wu, G. Fu and X. Liu, Ni-foam supported Co(OH)F and Co–P nanoarrays for energy-efficient hydrogen production via urea electrolysis, J. Mater. Chem. A, 2019, 7, 3697–3703 RSC.
- H. Yu, S. Zhu, Y. Hao, Y. Chang, L. Li, J. Ma, H. Chen, M. Shao and S. Peng, Modulating local interfacial bonding environment of heterostructures for energy-saving hydrogen production at high current densities, Adv. Funct. Mater., 2023, 33, 2212811 CrossRef CAS.
- Z. Wang, W. Liu, Y. Hu, M. Guan, L. Xu, H. Li, J. Bao and H. Li, Cr-doped CoFe layered double hydroxides: Highly efficient and robust bifunctional electrocatalyst for the oxidation of water and urea, Appl. Catal., B, 2020, 272, 118959 CrossRef CAS.
- J. Kang, F. Yang, C. Sheng, H. Xu, J. Wang, Y. Qing, Y. Wu and X. Lu, CoP nanoparticle confined in P, N Co-doped porous carbon anchored on P-doped carbonized wood fibers with tailored electronic structure for efficient urea electro-oxidation, Small, 2022, 18, 2200950 CrossRef CAS PubMed.
- D. C. Nguyen, T. L. L. Doan, S. Prabhakaran, D. H. Kim, N. H. Kim and J. H. Lee, Rh single atoms/clusters confined in metal sulfide/oxide nanotubes as advanced multifunctional catalysts for green and energy-saving hydrogen productions, Appl. Catal., B, 2022, 313, 121430 CrossRef CAS.
- T. Y. Burshtein, Y. Yasman, L. Muñoz-Moene, J. H. Zagal and D. Eisenberg, Hydrazine oxidation electrocatalysis, ACS Catal., 2024, 14, 2264–2283 CrossRef CAS.
- S. Li, Y. Hou, L. Jiang, G. Feng, Y. Ge and Z. Huang, Progress in hydrazine oxidation-assisted hydrogen production, Energy Rev., 2025, 4, 100105 CrossRef.
- Y. Li, J. Zhang, Y. Liu, Q. Qian, Z. Li, Y. Zhu and G. Zhang, Partially exposed RuP2 surface in hybrid structure endows its bifunctionality for hydrazine oxidation and hydrogen evolution catalysis, Sci. Adv., 2020, 6, eabb4197 CrossRef CAS PubMed.
- C. Feng, M. Lv, J. Shao, H. Wu, W. Zhou, S. Qi, C. Deng, X. Chai, H. Yang, Q. Hu and C. He, Lattice strain engineering of Ni2 P enables efficient catalytic hydrazine oxidation-assisted hydrogen production, Adv. Mater., 2023, 35, 2305598 CrossRef CAS PubMed.
- J. Li, Y. Li, J. Wang, C. Zhang, H. Ma, C. Zhu, D. Fan, Z. Guo, M. Xu, Y. Wang and H. Ma, Elucidating the critical role of ruthenium single atom sites in water dissociation and dehydrogenation behaviors for robust hydrazine oxidation-boosted alkaline hydrogen evolution, Adv. Funct. Mater., 2022, 32, 2109439 CrossRef CAS.
- X. Liu, J. He, S. Zhao, Y. Liu, Z. Zhao, J. Luo, G. Hu, X. Sun and Y. Ding, Self-powered H2 production with bifunctional hydrazine as sole consumable, Nat. Commun., 2018, 9, 4365 CrossRef PubMed.
- Z. Li, W. Wang, Q. Qian, Y. Zhu, Y. Feng, Y. Zhang, H. Zhang, M. Cheng and G. Zhang, Magic hybrid structure as multifunctional electrocatalyst surpassing benchmark Pt/C enables practical hydrazine fuel cell integrated with energy-saving H2 production, eScience, 2022, 2, 416–427 CrossRef.
- G. Feng, Y. Pan, D. Su and D. Xia, Constructing fully-active and ultra-active sites in high-entropy alloy nanoclusters for hydrazine oxidation-assisted electrolytic hydrogen production, Adv. Mater., 2024, 36, 2309715 CrossRef CAS PubMed.
- X. Guan, Q. Wu, H. Li, S. Zeng, Q. Yao, R. Li, H. Chen, Y. Zheng and K. Qu, Identifying the roles of Ru single atoms and nanoclusters for energy-efficient hydrogen production assisted by electrocatalytic hydrazine oxidation, Appl. Catal., B, 2023, 323, 122145 CrossRef CAS.
- J. Li, C. Zhang, C. Zhang, H. Ma, Y. Yang, Z. Guo, Y. Wang and H. Ma, Electronic configuration of single ruthenium atom immobilized in urchin-like tungsten trioxide towards hydrazine oxidation-assisted hydrogen evolution under wide pH media, Chem. Eng. J., 2022, 430, 132953 CrossRef CAS.
- J. Zhang, H. Wang, Y. Tian, Y. Yan, Q. Xue, T. He, H. Liu, C. Wang, Y. Chen and B. Y. Xia, Anodic hydrazine oxidation assists energy-efficient hydrogen evolution over a bifunctional cobalt perselenide nanosheet electrode, Angew. Chem., Int. Ed., 2018, 57, 7649–7653 CrossRef CAS PubMed.
- Q. Qian, J. Zhang, J. Li, Y. Li, X. Jin, Y. Zhu, Y. Liu, Z. Li, A. El-Harairy, C. Xiao, G. Zhang and Y. Xie, Artificial heterointerfaces achieve delicate reaction kinetics towards hydrogen evolution and hydrazine oxidation catalysis, Angew. Chem., Int. Ed., 2021, 60, 5984–5993 CrossRef CAS PubMed.
- Y. Liu, J. Zhang, Y. Li, Q. Qian, Z. Li, Y. Zhu and G. Zhang, Manipulating dehydrogenation kinetics through dual-doping Co3N electrode enables highly efficient hydrazine oxidation assisting self-powered H2 production, Nat. Commun., 2020, 11, 1853 CrossRef CAS PubMed.
- Y. Liu, J. Zhang, Y. Li, Q. Qian, Z. Li and G. Zhang, Realizing the synergy of interface engineering and chemical substitution for Ni3 N enables its bifunctionality toward hydrazine oxidation assisted energy-saving hydrogen production, Adv. Funct. Mater., 2021, 31, 2103673 CrossRef CAS.
- H. Luo, J. Barrio, N. Sunny, A. Li, L. Steier, N. Shah, I. E. L. Stephens and M. Titirici, Progress and perspectives in photo- and electrochemical-oxidation of biomass for sustainable chemicals and hydrogen production, Adv. Energy Mater., 2021, 11, 2101180 CrossRef CAS.
- Y. Li, Z. Dang and P. Gao, High-efficiency electrolysis of biomass and its derivatives: Advances in anodic oxidation reaction mechanism and transition metal-based electrocatalysts, Nano Sel., 2021, 2, 847–864 CrossRef CAS.
- Z. Wei, S. Yu and C. Li, Research development of anti-CO poisoning in electrocatalytic methanol oxidation processes: A review, Catal. Sci. Technol., 2024, 14, 5128–5142 RSC.
- J. Kaur, R. K. Gupta and A. Kumar, Electrocatalytic ethanol oxidation reaction: Recent progress, challenges, and future prospects, Discover Nano, 2024, 19, 137 CrossRef CAS PubMed.
- M. Kumar, B. Meena, A. Yu, C. Sun and S. Challapalli, Advancements in catalysts for glycerol oxidation via photo-/electrocatalysis: A comprehensive review of recent developments, Green Chem., 2023, 25, 8411–8443 RSC.
- N. Cai, C. Jin, C. Wan and R. Dong, Highly active PdAg/C catalysts for the electrooxidation of propan-1-ol, J. Electrochem. Soc., 2017, 164, H437 CrossRef CAS.
- A. C. Garcia, M. J. Kolb, C. Van Nierop, Y. Sanchez, J. Vos, Y. Y. Birdja, Y. Kwon, G. Tremiliosi-Filho and M. T. M. Koper, Strong impact of platinum surface structure on primary and secondary alcohol oxidation during electro-oxidation of glycerol, ACS Catal., 2016, 6, 4491–4500 CrossRef CAS.
- M. Guschakowski and U. Schröder, Direct and indirect electrooxidation of glycerol to value-added products, ChemSusChem, 2021, 14, 5216–5225 CrossRef CAS PubMed.
- T. Hamada, A. R. Circelli, H. Inoue, C. A. Randall and E. L. Clark, Investigating the origins of the pH-dependent oxidation of glycerol over platinum using differential electrochemical mass spectrometry, J. Phys. Chem. C, 2024, 128, 10790–10801 CrossRef CAS.
- Y. Kwon, K. J. P. Schouten and M. T. M. Koper, Mechanism of the catalytic oxidation of glycerol on polycrystalline gold and platinum electrodes, ChemCatChem, 2011, 3, 1176–1185 CrossRef CAS.
- A. M. Verma, L. Laverdure, M. M. Melander and K. Honkala, Mechanistic origins of the pH dependency in au-catalyzed glycerol electro-oxidation: Insight from first-principles calculations, ACS Catal., 2022, 12, 662–675 CrossRef CAS.
- W. Luo, H. Tian, Q. Li, G. Meng, Z. Chang, C. Chen, R. Shen, X. Yu, L. Zhu, F. Kong, X. Cui and J. Shi, Controllable electron distribution reconstruction of spinel NiCo2O4 boosting glycerol oxidation at elevated current density, Adv. Funct. Mater., 2024, 34, 2306995 CrossRef CAS.
- H. Yu, W. Wang, Q. Mao, K. Deng, Z. Wang, Y. Xu, X. Li, H. Wang and L. Wang, Pt single atom captured by oxygen vacancy-rich NiCo layered double hydroxides for coupling hydrogen evolution with selective oxidation of glycerol to formate, Appl. Catal., B, 2023, 330, 122617 CrossRef CAS.
- W. Chen, L. Zhang, L. Xu, Y. He, H. Pang, S. Wang and Y. Zou, Pulse potential mediated selectivity for the electrocatalytic oxidation of glycerol to glyceric acid, Nat. Commun., 2024, 15, 2420 CrossRef CAS PubMed.
- X. Yu, E. C. dos Santos, J. White, G. Salazar-Alvarez, L. G. M. Pettersson, A. Cornell and M. Johnsson, Electrocatalytic glycerol oxidation with concurrent hydrogen evolution utilizing an efficient MoO/Pt catalyst, Small, 2021, 17, 2104288 CrossRef CAS PubMed.
- G. Wu, X. Dong, J. Mao, G. Li, C. Zhu, S. Li, A. Chen, G. Feng, Y. Song, W. Chen and W. Wei, Anodic glycerol oxidation to formate facilitating cathodic hydrogen evolution with earth-abundant metal oxide catalysts, Chem. Eng. J., 2023, 468, 143640 CrossRef CAS.
- L. Xu, Y. Yang, C. Li, R. Ning, J. Ma, M. Yao, S. Geng and F. Liu, Unveiling the mechanism of electrocatalytic oxidation of glycerol by in-situ electrochemical spectroscopy, Chem. Eng. J., 2024, 481, 148304 CrossRef CAS.
- H. Yu, W. Wang, Q. Mao, K. Deng, Z. Wang, Y. Xu, X. Li, H. Wang and L. Wang, Pt single atom captured by oxygen vacancy-rich NiCo layered double hydroxides for coupling hydrogen evolution with selective oxidation of glycerol to formate, Appl. Catal., B, 2023, 330, 122617 CrossRef CAS.
- X. Yu, R. B. Araujo, Z. Qiu, E. Campos dos Santos, A. Anil, A. Cornell, L. G. M. Pettersson and M. Johnsson, Hydrogen evolution linked to selective oxidation of glycerol over CoMoO4—A theoretically predicted catalyst, Adv. Energy Mater., 2022, 12, 2103750 CrossRef CAS.
- L. Fan, Y. Ji, G. Wang, J. Chen, K. Chen, X. Liu and Z. Wen, High entropy alloy electrocatalytic electrode toward alkaline glycerol valorization coupling with acidic hydrogen production, J. Am. Chem. Soc., 2022, 144, 7224–7235 CrossRef CAS PubMed.
- Z. Huang, H. Ren, J. Guo, Y. Tang, D. Ye, J. Zhang and H. Zhao, High DHA selectivity and low-cost electrode for glycerol oxidation: CuO regulates MnO2 electron density to promote DHA desorption, Appl. Catal., B, 2024, 351, 123986 CrossRef CAS.
- T.-G. Vo, P.-Y. Ho and C.-Y. Chiang, Operando mechanistic studies of selective oxidation of glycerol to dihydroxyacetone over amorphous cobalt oxide, Appl. Catal., B, 2022, 300, 120723 CrossRef CAS.
- M. K. Goetz, M. T. Bender and K.-S. Choi, Predictive control of selective secondary alcohol oxidation of glycerol on NiOOH, Nat. Commun., 2022, 13, 5848 CrossRef CAS PubMed.
- X. Huang, Y. Zou and J. Jiang, Electrochemical oxidation of glycerol to dihydroxyacetone in borate buffer: Enhancing activity and selectivity by borate–polyol coordination chemistry, ACS Sustainable Chem. Eng., 2021, 9, 14470–14479 CrossRef CAS.
- X. Yu, E. C. dos Santos, J. White, G. Salazar-Alvarez, L. G. M. Pettersson, A. Cornell and M. Johnsson, Electrocatalytic glycerol oxidation with concurrent hydrogen evolution utilizing an efficient MoO/Pt catalyst, Small, 2021, 17, 2104288 CrossRef CAS PubMed.
- H. Luo, M. Xu, S. Liu, G. Tarantino, H. Ye, H. Yadegari, A. Y. Li, C. Hammond, G. Kastlunger, I. E. L. Stephens and M.-M. Titirici, Selective glycerol to lactic acid conversion via a tandem effect between platinum and metal oxides with abundant acid groups, EES Catal., 2025, 3, 87–96 RSC.
- S. Angizi, M. Nankali, A. Foroozan, J. Park, E. Yelekli Kirici, N. Noor, M. Fefer, Y. Terazono and D. Higgins, 3D bimetallic platinum-nickel electrodes for electro-oxidation of glycerol at ambient conditions, Adv. Funct. Mater., 2024, 2420622 CrossRef.
- G. Wu, X. Dong, J. Mao, G. Li, C. Zhu, S. Li, A. Chen, G. Feng, Y. Song, W. Chen and W. Wei, Anodic glycerol oxidation to formate facilitating cathodic hydrogen evolution with earth-abundant metal oxide catalysts, Chem. Eng. J., 2023, 468, 143640 CrossRef CAS.
- L. Xu, Y. Yang, C. Li, R. Ning, J. Ma, M. Yao, S. Geng and F. Liu, Unveiling the mechanism of electrocatalytic oxidation of glycerol by in-situ electrochemical spectroscopy, Chem. Eng. J., 2024, 481, 148304 CrossRef CAS.
- M. Park, M. Gu and B.-S. Kim, Tailorable electrocatalytic 5-hydroxymethylfurfural oxidation and H2 production: Architecture–performance relationship in bifunctional multilayer electrodes, ACS Nano, 2020, 14, 6812–6822 CrossRef CAS PubMed.
- X. Jiang, X. Ma, Y. Yang, Y. Liu, Y. Liu, L. Zhao, P. Wang, Y. Zhang, Y. Lin and Y. Wei, Enhancing the electrocatalytic oxidation of 5-hydroxymethylfurfural through cascade structure tuning for highly stable biomass upgrading, Nano-Micro Lett., 2024, 16, 275 CrossRef CAS PubMed.
- L. Chen, C. Yu, X. Song, J. Dong, J. Mu and J. Qiu, Integrated electrochemical and chemical system for ampere-level production of terephthalic acid alternatives and hydrogen, Nat. Commun., 2024, 15, 8072 CrossRef CAS PubMed.
- X. Lin, X. Xue and J. Du, Electrochemical glucose-to-formic acid conversion coupled with alkaline hydrogen production over nanostructured CuCo2O4 catalysts, J. Mater. Chem. A, 2024, 12, 32095–32103 RSC.
- M. Wu, J. Zhao, C. Li and R. Liu, Heterogeneity in a metal–organic framework in situ guides engineering Co@CoO heterojunction for electrocatalytic H2 production in tandem with glucose oxidation, J. Mater. Chem. A, 2022, 10, 4791–4799 RSC.
- W.-J. Liu, Z. Xu, D. Zhao, X.-Q. Pan, H.-C. Li, X. Hu, Z.-Y. Fan, W.-K. Wang, G.-H. Zhao, S. Jin, G. W. Huber and H.-Q. Yu, Efficient electrochemical production of glucaric acid and H2 via glucose electrolysis, Nat. Commun., 2020, 11, 265 CrossRef CAS PubMed.
- Y. Zhang, B. Zhou, Z. Wei, W. Zhou, D. Wang, J. Tian, T. Wang, S. Zhao, J. Liu, L. Tao and S. Wang, Coupling glucose-assisted Cu(I)/Cu(II) redox with electrochemical hydrogen production, Adv. Mater., 2021, 33, 2104791 CrossRef CAS PubMed.
- S. R. Kubota and K.-S. Choi, Electrochemical oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid (FDCA) in acidic media enabling spontaneous FDCA separation, ChemSusChem, 2018, 11, 2138–2145 CrossRef CAS PubMed.
- R. Latsuzbaia, R. Bisselink, A. Anastasopol, H. van der Meer, R. van Heck, M. S. Yagüe, M. Zijlstra, M. Roelands, M. Crockatt, E. Goetheer and E. Giling, Continuous electrochemical oxidation of biomass derived 5-(hydroxymethyl)furfural into 2,5-furandicarboxylic acid, J. Appl. Electrochem., 2018, 48, 611–626 CrossRef CAS.
- D. J. Chadderdon, L. Xin, J. Qi, Y. Qiu, P. Krishna, K. L. More and W. Li, Electrocatalytic oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid on supported Au and Pd bimetallic nanoparticles, Green Chem., 2014, 16, 3778–3786 RSC.
- N. Heidary and N. Kornienko, Operando Raman probing of electrocatalytic biomass oxidation on gold nanoparticle surfaces, Chem. Commun., 2019, 55, 11996–11999 RSC.
- B. J. Taitt, D.-H. Nam and K.-S. Choi, A comparative study of nickel, cobalt, and iron oxyhydroxide anodes for the electrochemical oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid, ACS Catal., 2019, 9, 660–670 CrossRef CAS.
- J. Wang, W. Zhao, H. Yu, W. Wang, Y. Xu, L.-L. Shen, G.-R. Zhang and D. Mei, Enhanced electrochemical oxidation of 5-hydroxymethylfurfural over tailored nickel nanoparticle assembly, Appl. Catal., B, 2024, 353, 124086 CrossRef CAS.
- Y. Lu, T. Liu, Y.-C. Huang, L. Zhou, Y. Li, W. Chen, L. Yang, B. Zhou, Y. Wu, Z. Kong, Z. Huang, Y. Li, C.-L. Dong, S. Wang and Y. Zou, Integrated catalytic sites for highly efficient electrochemical oxidation of the aldehyde and hydroxyl groups in 5-hydroxymethylfurfural, ACS Catal., 2022, 12, 4242–4251 CrossRef CAS.
- Z. Wang, S. Shen, Z. Lin, W. Tao, Q. Zhang, F. Meng, L. Gu and W. Zhong, Regulating the local spin state and band structure in Ni3S2 nanosheet for improved oxygen evolution activity, Adv. Funct. Mater., 2022, 32, 2112832 CrossRef CAS.
- Q. Xu, J. Zhang, H. Zhang, L. Zhang, L. Chen, Y. Hu, H. Jiang and C. Li, Atomic heterointerface engineering overcomes the activity limitation of electrocatalysts and promises highly-efficient alkaline water splitting, Energy Environ. Sci., 2021, 14, 5228–5259 RSC.
- B. You, N. Jiang, X. Liu and Y. Sun, Simultaneous H2 generation and biomass upgrading in water by an efficient noble-metal-free bifunctional electrocatalyst, Angew. Chem., Int. Ed., 2016, 55, 9913–9917 CrossRef CAS PubMed.
- B. You, X. Liu, N. Jiang and Y. Sun, A general strategy for decoupled hydrogen production from water splitting by integrating oxidative biomass valorization, J. Am. Chem. Soc., 2016, 138, 13639–13646 CrossRef CAS PubMed.
- Z. Zhang, X. Liang, J. Li, J. Qian, Y. Liu, S. Yang, Y. Wang, D. Gao and D. Xue, Interfacial engineering of NiO/NiCo2O4 porous nanofibers as efficient bifunctional catalysts for rechargeable zinc–air batteries, ACS Appl. Mater. Interfaces, 2020, 12, 21661–21669 CrossRef CAS PubMed.
- D. Li, Y. Huang, Z. Li, L. Zhong, C. Liu and X. Peng, Deep eutectic solvents derived carbon-based efficient electrocatalyst for boosting H2 production coupled with glucose oxidation, Chem. Eng. J., 2022, 430, 132783 CrossRef CAS.
- D. Paital, V. Thambi, M. S. Kutwal and S. Khatua, Porous plasmonic Au–Ag@Au nanostructures for photoelectrochemical methanol oxidation, ACS Appl. Nano Mater., 2022, 5, 13286–13294 CrossRef CAS.
- S. Mohajernia, S. Hejazi, P. Andryskova, G. Zoppellaro, O. Tomanec, R. Zboril and P. Schmuki, Conductive Cu-doped TiO2 nanotubes for enhanced photoelectrochemical methanol oxidation and concomitant hydrogen generation, ChemElectroChem, 2019, 6, 1244–1249 CrossRef CAS.
- R. Ding, J. Chen, Y. Chen, J. Liu, Y. Bando and X. Wang, Unlocking the potential: Machine learning applications in electrocatalyst design for electrochemical hydrogen energy transformation, Chem. Soc. Rev., 2024, 53, 11390–11461 RSC.
- H. Xin, T. Mou, H. S. Pillai, S.-H. Wang and Y. Huang, Interpretable machine learning for catalytic materials design toward sustainability, Acc. Mater. Res., 2024, 5, 22–34 CrossRef CAS.
- S. Li, S. Wang, Y. Wang, J. He, K. Li, Y. Xu, M. Wang, S. Zhao, X. Li, X. Zhong and J. Wang, Doped Mn enhanced NiS electrooxidation performance of HMF into FDCA at industrial-level current density, Adv. Funct. Mater., 2023, 33, 2214488 CrossRef CAS.
- Essential role of lattice oxygen in methanol electrochemical refinery toward formate, https://www.science.org/doi/10.1126/sciadv.adh9487, (accessed 20 January 2025).
- Y. Li, H. S. Pillai, T. Wang, S. Hwang, Y. Zhao, Z. Qiao, Q. Mu, S. Karakalos, M. Chen, J. Yang, D. Su, H. Xin, Y. Yan and G. Wu, High-performance ammonia oxidation catalysts for anion-exchange membrane direct ammonia fuel cells, Energy Environ. Sci., 2021, 14, 1449–1460 RSC.
- P. Wang, H. Cui and C. Wang, Ultrathin PtMo-CeO hybrid nanowire assemblies as high-performance multifunctional catalysts for methanol oxidation, oxygen reduction and hydrogen oxidation, Chem. Eng. J., 2022, 429, 132435 CrossRef CAS.
- A. Badreldin, E. Youssef, A. Djire, A. Abdala and A. Abdel-Wahab, A critical look at alternative oxidation reactions for hydrogen production from water electrolysis, Cell Rep. Phys. Sci., 2023, 4, 101427 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4im00163j |
| This journal is © Institute of Process Engineering of CAS 2025 |