
Development of a selective methodology for methylmercury quantification and evaluation of its accumulation in hippocampus†
Marcelo
Verdugo
 *ae,
Ferdinand
Ávila
a,
Jhoel
Ruiz
a,
Constanza
Vásquez
a,
Nicole
Roldán
b,
M. Gabriela
Lobos
ae and
Álvaro O.
Ardiles
cd
*ae,
Ferdinand
Ávila
a,
Jhoel
Ruiz
a,
Constanza
Vásquez
a,
Nicole
Roldán
b,
M. Gabriela
Lobos
ae and
Álvaro O.
Ardiles
cd
aLaboratorio de Química Analítica y Ambiental, Instituto de Química y Bioquímica, Facultad de Ciencias, Universidad de Valparaíso, Valparaíso, Chile. E-mail: marcelo.verdugo@uv.cl
bDepartamento de Cirugía, Escuela de Medicina, Facultad de Medicina, Universidad de Valparaíso, Valparaíso, Chile
cLaboratorio de Sinaptopatías, Escuela de Medicina, Facultad de Medicina, Universidad de Valparaíso, Valparaíso, Chile
dCentro de Investigación Traslacional en Neurofarmacología (CITNE), Universidad de Valparaíso, Valparaíso, Chile
eCentro de Investigación y Gestión de Recursos Naturales (CIGREN), Facultad de Ciencias, Universidad de Valparaíso, Valparaíso 2360102, Chile
First published on 21st November 2024
Abstract
Methylmercury (MeHg+) is a highly toxic compound with significant neurotoxic effects, necessitating precise and reliable quantification methods for its assessment in biological tissues. In this study, we developed and optimized a methodology combining Microwave-Assisted Extraction (MAE), derivatization by phenylation, and preconcentration through Liquid Phase Microextraction (LPME), coupled with Gas Chromatography-Pyrolysis-Atomic Fluorescence Spectrometry (GC-PYRO-AFS) for the selective quantification of MeHg+ in mouse brain tissue. The optimized method demonstrated high sensitivity and reproducibility, enabling the accurate detection of MeHg+ at trace levels without significant matrix effects. This methodological advancement is particularly important in the field of toxicology, as it addresses the limitations of traditional techniques by reducing analysis time and cost while improving accuracy. The ability to precisely quantify MeHg+ concentrations in biological tissues facilitates the study of toxicokinetic behaviors, the proposal of distribution mechanisms, and the evaluation of toxicological impacts, ultimately contributing to the development of biomarkers for human health risk assessment.
Introduction
Monomethylmercury (CH3Hg+; MeHg+) is an organic Hg species of significant toxicological interest due to its ability to bioaccumulate in the tissues of organisms, causing toxicity at both cellular and systemic levels.1–5 The central nervous system (CNS) is particularly vulnerable to MeHg+ exposure, with documented neurotoxic effects that have been widely investigated.6–11These studies show alterations in various brain areas, including the hippocampus.12–15 This area of the brain is associated with cognition, learning, and the initial generation and storage of new memories.16–19 Alterations in synaptic function and/or loss of synapses and neurons in the hippocampus have been linked to neurodegenerative diseases such as Parkinson's20,21 and Alzheimer's disease.22–25 Although these neurodegenerative diseases have multifactorial causes, some studies have considered exposure to MeHg+ as a potential risk factor.10,26–32 Therefore, understanding the exposure dose and its accumulation in the brain is crucial for elucidating the mechanisms of MeHg+ neurotoxicity.
The neurotoxic effects of MeHg+ in the brain and its relationship with exposure dose have been studied both in cell culture33–35 and in mice.36–39 It has been established that the implemented methods for the analysis of MeHg+ must be selective and also demonstrate a high sensitivity that permits the detection of trace levels of this contaminant in tissues that are hard to access, such as the brain.
Traditional techniques for the quantification of methylmercury (MeHg+) in biological tissues often face significant limitations, including time-consuming sample preparation, high costs, and challenges in achieving accurate measurements due to matrix effects.40,41 These limitations can hinder the ability to conduct precise toxicological assessments, which are crucial for understanding the toxicokinetic behavior of MeHg+ and its potential health impacts.42 Therefore, developing new methodologies that overcome these challenges is essential for advancing the field of toxicology, enabling more reliable quantification of MeHg+ in complex biological matrices, and supporting the proposal of distribution mechanisms and the identification of relevant biomarkers.43
Nevertheless, a significant limitation in current methods for estimating MeHg+ content in the brain is that they are often time-consuming and complex. These methods frequently rely on mass balance calculations based on the total Hg content in the matrix rather than providing direct quantification of MeHg+.44,45 Additionally, they often estimate MeHg+ content as equivalent to total Hg,38,39,46–48 overlooking the critical fact that this organic Hg species can be demethylated and converted into inorganic Hg (Hg2+). This oversight can lead to inaccurate assessments of MeHg+ levels and its associated neurotoxicity.
On the other hand, some selective analytical techniques exist that provide direct evidence of MeHg+ concentration in the matrix, but these methods are often limited by their high cost, the complexity of the procedures, and the need for sophisticated analytical equipment, such as gas chromatography-inductively coupled plasma-mass spectrometry (GC-ICP-MS),49 isotope dilution in combination with GC-ICP-MS,50 and liquid chromatography-inductively coupled plasma-mass spectrometry (LC-ICP-MS).51
For these reasons, there is a critical need for a methodology that allows for the selective and accurate determination of MeHg+ in the mouse brain. Such a methodology should not only provide precise results but also be user-friendly for the analyst, overcoming the complexity and high demands of current techniques. Additionally, it must be robust, reproducible, and sensitive, particularly when dealing with small samples where accuracy is crucial. Therefore, the development of our methodology is justified, as it is designed to address these exact requirements. It ensures that all stages, from sample treatment to the quantitative determination of the analyte, are performed with maximum efficiency and reliability, ultimately providing the best possible results in a more accessible and easier.
Given the above, the aim of this investigation was to develop and optimise a robust analytical methodology to directly quantify MeHg+ in mouse brain tissue exposed to distinct doses of MeHg+. Furthermore, the direct quantification of MeHg+ was determined in the hippocampus due to its relevance in the neuropathological process, providing direct evidence of its presence in this brain structure. Prior to the quantification of MeHg+ both in the brain and hippocampus, the samples were digested in alkaline medium and aliquots of extracts were derivatized to permit the preconcentration of MeHg+ in an organic medium.
In this line, alkaline digestion was performed to enable the extraction of MeHg+ from the matrix.52,53 Our search for derivatization reagents for MeHg+, applicable to mouse brain and hippocampus biological samples, led us to select sodium tetraphenylborate (NaBPh4) and phenylboronic acid (PhB(OH)2) as the derivatization reagents.54–56 These reagents were selected because they are easily accessible. Besides, the sample digestion, derivatization, and preconcentration stages were optimized through different experimental designs (DoE) to incorporate and study several factors simultaneously involved in each stage, thus obtaining the best experimental conditions.
Experimental
Reagents and stock standard solution
A stock standard solution of Hg2+ (1000 mg L−1, TraceCERT grade) and methylmercury chloride (Analytical Standard grade) were obtained from Merck. The methylmercury chloride was dissolved in Milli-Q water to prepare a standard solution, which was quantified using a Direct Mercury Analyzer (DMA-80, Milestone). All reagents and solutions were stored at 4 °C to ensure stability.Dichloroethane (C2H4Cl2, ≥99.5%, GC grade, EMPLURA®), dichloromethane (CH2Cl2, ≥99.8%, GC grade, SupraSolv), tetrachloroethylene (C2Cl4, ≥99%, GC grade, EMPLURA®), glacial acetic acid (CH3COOH, ≥99.7%, ACS grade), sodium hydroxide (NaOH, ≥99%, EMSURE®), sodium tetraphenylborate (NaBPh4, ≥99.5%, ACS grade), phenylboronic acid (PhB(OH)2, 95%), isoflurane (99.9%), hydrochloric acid (HCl, 37%, ACS grade), and nitric acid (HNO3, 65–67%, puriss. p.a. grade) were all procured from Merck.
Helium gas (≥99.999%) was employed as the carrier gas for GC. Nitrogen gas (≥99.999%) and argon gas (≥99.999%) were initially utilized as carrier gases in the PYRO-AFS system, with argon being the exclusive carrier gas in subsequent stages. Compressed air (≥99.999%) was used as the carrier gas in the DMA-80 system.
Method validation, including both the reference method for total Hg analysis (DMA-80, Milestone) and the optimized methodology, was conducted using the certified reference material ERM CE-464 (Tuna Fish), containing 5.5 mg MeHg per kg.
All glassware was meticulously cleaned using hydrochloric acid (37%, ACS grade) and nitric acid (65–67%, puriss. p.a. grade) to prevent contamination.
Animals and treatment
Two-month-old female C57BL/6 mice (17–23 g) were obtained from the Animal House Facility at the Faculty of Science, Universidad de Valparaíso. The animals were housed in polycarbonate cages (373 × 190 × 138 mm) in a temperature-controlled room (20 ± 1 °C) with a 12 hour light-dark cycle, and were provided food and water ad libitum. Animal maintenance and experimental procedures were conducted in accordance with the National Institute of Health (USA) guidelines for the use of experimental animals, with approval from the Institutional Animal Ethics Committee of the Universidad de Valparaíso.The 36 mice used in this study underwent an acclimation period of 4 days before being randomly divided into three groups: control group (n = 12), 0.5 mg L−1 MeHg+ group (n = 12), and 5 mg L−1 MeHg+ group (n = 12). The chosen concentrations were within a range that would not adversely affect the animals' health or alter their drinking behavior.37,38,57 The two exposure groups received methylmercury chloride (CH3HgCl) in filtered drinking water at the specified concentrations for 30 days, with the water being refreshed daily. The control group received only filtered water during the same period. Throughout the exposure period, the animals were monitored, and no signs of toxicity were observed.
Following the 30 day exposure period, the animals were anesthetized with isoflurane and sacrificed by cervical dislocation, followed by decapitation. The brains were promptly extracted and stored at −18 °C until analysis. Eighteen brains (n = 6 per group) were allocated for the determination of total Hg and MeHg+ using DMA-80 and GC-Pyro-AFS, respectively. The remaining eighteen brains were used to determine total Hg and MeHg+ concentrations in the hippocampus. The hippocampus was dissected following the method of Jaszczyk et al.58 which is optimized for molecular studies requiring contaminant-free tissues while preserving anatomical and molecular integrity.58 For hippocampal analysis, tissue samples from each group were pooled (n = 6 per group) for combined analysis.
Mercury ingestion calculation
To estimate the total mercury intake, it was assumed that each mouse consumed approximately 3 mL of water daily. Over the 30 day exposure period, this corresponds to a total consumption of about 540 mL per group (n = 6). For the group exposed to 0.5 mg L−1 of MeHg+, the estimated total mercury intake was approximately 270 mg per group, with each mouse receiving around 45 mg of mercury. Similarly, for the group exposed to 5 mg L−1 of MeHg+, the total mercury intake was approximately 2700 mg per group, with each mouse receiving around 450 mg of MeHg+. These calculations provide a quantitative assessment of the mercury dose administered to the animals, ensuring that the exposure levels are consistent with the intended experimental design.59,60Total mercury analysis
Total mercury in the samples was analyzed using a direct mercury analyzer (DMA-80, Milestone), which operates with a combustion dual cell detector, gold amalgamation, and atomic absorption. Hippocampus and brain samples were placed in nickel boats, weighed on an analytical balance (AUX220, Shimadzu), and then loaded into the autosampler tray. Calibration of the instrument was validated using the certified reference material ERM CE-464 (Tuna Fish). Each tissue analysis included a certified reference material, a method blank, and a duplicate sample to ensure accuracy and precision.GC-PYRO-AFS instrumentation
The separation and detection of MeHg+ were conducted using a GC-PYRO-AFS system. Gas chromatographic separation was performed on a TRACE 1300 GC (Thermo Scientific), where SLB-5MS and SLB-35 MS columns were evaluated during the optimization process. Ultimately, the SLB-5MS column was selected for further analysis. The outlet of the GC column was connected to the pyrolysis system via a T-connector (1/16′), with argon (Ar) serving as the carrier gas. The other end of the pyrolysis system was coupled to the atomic fluorescence detection system. The pyrolysis system was maintained at a working temperature of 800 °C. Detection was achieved using atomic fluorescence spectroscopy, with a Millennium System (P.S. Analytical).Derivatization reagents and LPME preconcentration factor (PF)
The derivatization of MeHg+ was evaluated using two reagents: sodium tetraphenylborate (NaBPh4) and phenylboronic acid (PhB(OH)2). A known amount of MeHg+ was added to a 15 mL centrifuge tube containing Milli-Q water, NaOH, and acetic acid (CH3COOH), forming a buffer with a total concentration of 0.4 M and a pH of 4.5. The tube was sealed and manually stirred before adding 0.2 mL of dichloroethane (C2H4Cl2) and the derivatization reagent at a final concentration of 0.3% w/v (n = 6). The tube was then sealed again and stirred at 700 rpm for 15 minutes using an orbital shaker (RS-OS-10, Phoenix). After stirring, the solution was centrifuged at 5000 rpm for 1 minute using a Hettich Universal 320R centrifuge. The resulting organic phase was separated and stored in a microcentrifuge tube at −18 °C until further analysis by GC-PYRO-AFS and DMA-80.The preconcentration factor (PF) was determined using synthetic samples as the ratio of the concentration of MeHg+ in the organic phase of derivatized samples to the concentration of MeHg+ in the aqueous phase of non-derivatized samples. This was performed (n = 3) using 0.2 mL of C2H4Cl2, CH2Cl2, or C2Cl4 as the organic solvent, along with an aqueous solution of 0.3% w/v NaBPh4 and acetate buffer (0.4 M, pH 4.5). The organic phase was then separated and stored in an Eppendorf tube at −18 °C until analysis by DMA-80. The preconcentration factor was calculated using the following equation:
| PF = [MeHg+]org/[MeHg+]ac |
Screening and optimization of the digestion, derivatization and preconcentration procedure
The microwave digestion procedure was optimized using a Central Composite Design (CCD). In this approach, power and digestion time were refined through 12 factorial experiments and 6 central points, using CRM ERM CE-464 (tuna fish) as the sample matrix (α = 0.05).The screening and optimization of the MeHg+ derivatization step and LPME preconcentration were assessed using a Plackett–Burman design for the initial screening, followed by a Box–Behnken design for further optimization, both with a 95% confidence level. Variables evaluated during the screening phase included the type of organic solvent, buffer pH, buffer concentration, percentage of derivatization reagent, agitation time, agitation speed, and LPME temperature. Both experimental designs (DoE) were carried out with 18 experiments using synthetic MeHg+ samples.
Statistical analysis and the development of experimental designs were conducted using Minitab® 21 statistical software.61 All DoEs were randomized to minimize the impact of uncontrollable factors on the response.
Evaluation of MeHg+ degradation using a focused single-mode microwave system
The recovery of MeHg+ during the extraction step was assessed to evaluate its potential degradation during microwave digestion. This evaluation was conducted by statistically comparing the chromatographic areas of MeHg+ in digested versus undigested synthetic samples. The digestion was performed using a focused single-mode microwave system (Discover, CEM), where MeHg+ was digested in a solution of 0.5% w/v NaOH in Milli-Q water for 6.8 minutes at 85 W (n = 4). These conditions were selected based on similar digestion protocols reported in previous studies.62 Following digestion, 1 mL of each digested synthetic sample was derivatized using the procedure previously described, with NaBPh4 as the derivatization reagent and C2H4Cl2 as the organic solvent. The resulting organic phases were separated and stored in microcentrifuge tubes at −18 °C. All samples were subsequently analyzed by GC-PYRO-AFS.Determination of MeHg+ in the hippocampus and mouse brain
Hippocampus and brain samples were digested using a focused single-mode microwave system (Discover, CEM) with an alkaline digestion method, employing 0.5% w/v NaOH in Milli-Q water. The analysis was performed under the optimized conditions for the complete methodology, which includes microwave digestion, LPME, and GC-PYRO-AFS. Additionally, total Hg in the samples was analyzed using a DMA-80 direct mercury analyzer.Statistical analysis
For the initial selection stage, a Plackett–Burman design of experiments (DOE) was employed to identify and reduce the number of significant factors. The statistical significance of variables such as agitation time, derivatizing agent percentage, pH, buffer concentration, temperature, mechanical shaker revolutions per minute, and organic solvent was evaluated based on the signal area of MeHg+ measured via GC-PYRO-AFS. Once the most relevant factors were identified, a response surface methodology (RSM) utilizing a Box–Behnken design was conducted to establish the optimal extraction conditions. All statistical analyses for optimization were performed using Minitab® 21 statistical software.61A one-way Brown–Forsythe ANOVA test with Welch's correction was used to compare total Hg and MeHg+ across different analysis groups. This test is suitable for experiments involving three or more groups with unpaired measurements, assuming a Gaussian distribution. Welch's correction was specifically applied to account for unequal variances among the groups.
For two-group comparisons, such as the assessment of matrix effects, an unpaired t-test with Welch's correction was employed. The normality of the data was assessed using the Shapiro–Wilk test. All analyses were conducted with a 95% confidence level using GraphPad Prism® 8 software.
OriginPro® 8 software was used to create the calibration curve graph and perform the associated linear regression analysis.
Results and discussion
Optimization of the chromatographic parameters of the hyphenated GC-PYRO-AFS system
The chromatographic parameters of the hyphenated GC-PYRO-AFS system were optimized to ensure reproducible, robust, sensitive, selective, and quantitative detection of MeHg+, specifically for application in mouse hippocampus and brain samples.Initially, the optimal conditions for the carrier gas in the PYRO-AFS system were evaluated. Synthetic MeHg+ samples were prepared and analyzed using the GC-PYRO-AFS system, employing either N2 or Ar as carrier gases. The use of Ar as the carrier gas resulted in a significantly higher mean peak area of 30 ± 2 mV min−1 with an RSD of 8.00%, compared to N2, which produced a much lower mean peak area of 1.4 ± 0.2 mV min−1 and a higher RSD of 14.80%. These findings are consistent with existing literature on the analysis of excited Hg0, where it has been demonstrated that N2 induces a more pronounced cross-quenching effect compared to Ar, likely due to increased Hg–N2 interactions under higher carrier gas pressure, which in turn diminishes Hg0 fluorescence.63 As a result, Ar was determined to be the optimal carrier gas for the PYRO-AFS system, enhancing both the sensitivity and reproducibility of MeHg+ detection in complex biological matrices.
The carrier gas pressure was subsequently optimized to maximize signal area while achieving an optimal asymmetry factor and minimizing tailing.64 Based on these criteria, the carrier gas pressure was set at 15 psi. The optimized operational conditions for the GC-PYRO-AFS system are detailed in Table 1.
| GC parameters | Condition |
|---|---|
| Oven program | 50/300 °C to 60 °C min−1 |
| Carrier gas pressure | He 15 psi |
| Pyrolysis oven temperature | 800 °C |
| GC injection mode | Splitless |
| GC injector temperature | 270 °C |
| Injection volume | 4 μL |
| GC column | (5%-Phenyl)-methylpolysiloxane (SLB-5MS 30 m × 0.25 mm × 0.25 μm) |
Derivatizing reagent and preconcentration factor
The LPME step was optimized by initially evaluating the derivatizing reagent. Synthetic MeHg+ samples containing 9.9 ng of MeHg+ were derivatized with either NaBPh4 or PhB(OH)2 and analyzed using DMA-80 and GC-PYRO-AFS. The results indicated that the preconcentration factor determined by DMA-80 was higher with NaBPh4 (34 ± 3) compared to PhB(OH)2 (29 ± 1), with both values showing statistically significant differences (p-value < 0.05). These results were corroborated by the chromatographic signals obtained via GC-PYRO-AFS, where the mean peak area for MeHg+ using NaBPh4 (6.1 ± 0.3 mV min−1) was significantly higher than that obtained with PhB(OH)2 (4.9 ± 0.3 mV min−1) (n = 5, p < 0.05). The comparison between these reagents is crucial because they play a key role in the derivatization process, which involves the transfer of MeHg+ from the aqueous phase to the organic phase during LPME.52,65,66 The differences between these derivatizing reagents are well-documented in the literature.67,68 Based on these findings, NaBPh4 was selected as the derivatization reagent for subsequent experiments.Following the determination of the optimal derivatization reagent, the ideal solvent for LPME was evaluated. Three solvents—dichloromethane (CH2Cl2), dichloroethane (C2H4Cl2), and tetrachloroethylene (C2Cl4)—were compared based on their preconcentration factors. Synthetic MeHg+ samples were derivatized with NaBPh4 and extracted using the evaluated organic solvents, with both the organic and aqueous phases analyzed by DMA-80. The preconcentration factors followed the order: CH2Cl2 > C2H4Cl2 > C2Cl4 (Fig. 1). Statistical analysis revealed significant differences between CH2Cl2 and C2H4Cl2 (p-value = 0.0067) and between CH2Cl2 and C2Cl4 (p-value = 0.0035), with no significant differences observed between C2Cl4 and C2H4Cl2. However, CH2Cl2 exhibited higher dispersion compared to the other solvents. This effect could be attributed to differences in boiling points and solvent volatilization during agitation in the LPME step, leading to reduced reproducibility and potential errors in calculating the preconcentration factor. The higher variability observed with CH2Cl2 suggests that its use could lead to inconsistent results, potentially compromising the precision of MeHg+ quantification in different samples. This increased dispersion could be particularly problematic when analyzing trace levels of MeHg+, where small variations in solvent behavior may lead to significant deviations in measured concentrations. As a result, CH2Cl2 and C2H4Cl2 were selected for further screening studies to determine the most suitable solvent for the method, with careful consideration of their impact on the reproducibility and accuracy of the LPME process.
 | ||
| Fig. 1 Comparison of preconcentration factors of MeHg+ LPME, obtained with different organic solvents. (a) Dichloromethane, (b) dichloroethane, (c) tetrachloroethylene (n = 3 each solvent). Significance parameter ** p-value < 0.01. | ||
Evaluation of MeHg+ degradation using a focused single-mode microwave system
The Microwave-Assisted Extraction (MAE) procedure, despite being considered a mild digestion process, has the potential to degrade MeHg+ during the irradiation phase. Therefore, prior to optimizing the extraction procedure using DoE, it was essential to assess the potential degradation of MeHg+. This was achieved by comparing undigested samples with those exposed to the MAE procedure under extreme conditions—specifically, extended exposure time (10 minutes) and high power (75 W). Synthetic samples containing 9.4 ng of MeHg+ were prepared and analyzed under non-optimized conditions using GC-PYRO-AFS. The chromatographic areas for the digested and undigested samples were 5.7 ± 0.5 and 5.7 ± 0.2 mV min−1, respectively. Statistical analysis via Analysis of Variance (ANOVA) indicated that the MAE procedure under these conditions does not cause significant degradation of MeHg+ (p-value = 0.807, α = 0.05). These findings align with those reported by Abrankó et al., who similarly observed no significant degradation of MeHg+ when using this mild digestion methodology.62To ensure the reliability of MAE in complex matrices, the absence of significant degradation under extreme conditions is crucial. This is particularly important for the accurate quantification of MeHg+ in biological matrices, where maintaining analyte integrity is essential for accurate results. The robustness of the MAE method under these conditions confirms its suitability for routine analysis, ensuring the extraction process does not compromise the quantification of MeHg+ in trace-level studies.
Screening and optimization of the digestion, derivatization and LPME procedure
| Factor | Condition |
|---|---|
| Agitation time (min) | 5–20 |
| % of derivatization reagent | 0.1–0.3 |
| pH | 4–5 |
| Buffer concentration (M) | 0.4–1 |
| Temperature (°C) | 20–35 |
| Agitation speed (rpm) | 100–700 |
| Solvent | CH2Cl2–C2H4Cl2 |
Additionally, the statistical significance of the independent variables' effects on the response was determined through an analysis of variance (ANOVA) of the regression, with the results visualized using a Pareto chart (Fig. 2). The analysis revealed that the type of organic solvent (p-value = 0.033) and pH (p-value = 0.001) were statistically significant, as both factors exceeded the calculated significance threshold (dotted line), indicating a substantial impact on the MeHg+ signal. The chart also depicted the nature of each factor's (positive or negative) on the response. Dichloroethane (C2H4Cl2) was identified as the solvent that generated higher peak areas, whereas increasing the pH had a negative effect, reducing the MeHg+ signal. The Pareto chart also displayed the variables with non-significant effects, distinguishing between those with positive (grey) and negative (white) impacts. To optimize the derivatization and LPME processes, the condition range of each non-significant factor was adjusted in subsequent experiments to enhance the MeHg+ signal.
 | ||
| Fig. 2 Pareto chart of the variables considered in the Placket Burman experimental design for the screening of derivatization and LPME process (α = 0.05). Positive effect: grey bars. Negative effect: white bars. | ||
| Factors | Level | |
|---|---|---|
| −1 | 1 | |
| pH | 3.5 | 4.5 |
| Agitation speed (rpm) | 400 | 800 |
| Agitation time (min) | 12.5 | 25 |
In relation to our results, the model demonstrated a significant relationship between the factors and the response (p-value = 0.011; R2 = 86.71). The statistical analysis confirmed that pH (B) retained its statistical significance and continued to exert a negative effect on the MeHg+ response (p-value = 0.001). Additionally, the interaction effect between agitation speed (A) and agitation time (C) was found to be significant, with a negative impact on the response. Specifically, an increase in the intensity of A generate a decrease in the effect of C (p-value = 0.033) (Fig. 3).
 | ||
| Fig. 3 Pareto chart of the variables considered in the Box–Behnken design for the optimization of derivatization and LPME process (α = 0.05). Positive effect: grey bars. Negative effect: white bars. | ||
The effect of the main factors and their interactions on the MeHg+. Signal, as predicted by quadratic modeling using the Box–Behnken design, enables the mathematical optimization of the process while maintaining a limited range of the independent variables. This limitation in the range of variables was necessary because it was not feasible to increase the solution's pH above 5.0 using the acetate buffer, as would have been required in a Central Composite Design. The change in the response is illustrated by the response surface plots of pH vs. agitation time, pH vs. agitation speed, and agitation speed vs. agitation time (S. Fig. 1). The optimization revealed that the highest MeHg+ signal area is achieved at a pH of 3.5, an agitation time of 25 minutes, and an agitation speed of 400 rpm, which are the optimal conditions for the derivatization and LPME processes to maximize the response.
| Factors | Conditions | ||||
|---|---|---|---|---|---|
| −α | −1 | 0 | 1 | α | |
| Microwave power (W) | 14.64 | 25 | 50 | 75 | 85.35 |
| Extraction time (min) | 1.17 | 2 | 4 | 6 | 6.80 |
The CCD model, which included a second-order term, was constructed with 4 axial points and 5 central points to estimate the variance of the experimental error. The results of the CCD yielded an R2 value of 80.37 and a p-value of 0.020, indicating a statistically significant correlation between the independent and dependent variables (α = 0.05). Additionally, the statistical analysis of the CCD revealed that the main factor, extraction time, was highly significant (p-value = 0.006). Moreover, the quadratic term for microwave power also showed significance (p-value = 0.025) (S. Fig. 2). The statistical significance of a quadratic term (AA) suggests that the response exhibits a notable curvature within the selected range for this factor. A response surface model (RSM) was developed to describe the behavior of the response across the tested range of independent variables, allowing for the selection of optimal conditions to achieve the highest MeHg+ signal. In this case, the optimal conditions were found at the extreme points of the applied range, specifically 85 W of microwave power and 6.8 minutes of exposure time, with an optimal desirability factor (D = 1). This desirability factor indicates that the effects of the evaluated variables are not antagonistic and work synergistically to maximize the response, a conclusion further supported by the surface plot graph (Fig. 4).
 | ||
| Fig. 4 Surface response plot for the optimization of MAE process by a central composite design. | ||
After the optimization of the derivatization-LPME and MAE processes, the optimal conditions for each variable were established and are presented in Table 5.
| Extraction MAE | Condition |
|---|---|
| Microwave power (W) | 85 |
| Extraction time (min) | 6.8 |
| NaOH (% w/v) | 0.5 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|
| Derivatization and LPME | |
| Agitation time (min) | 25 |
| Derivative reagent (%) | 0.3 |
| pH | 3.5 |
| Buffer concentration (M) | 0.4 |
| Temperature (°C) | 20 |
| Agitation speed (rpm) | 400 |
| Solvent | C2H4Cl2 |
Determination of analytical figures of merit and validation of GC-PYRO-AFS
The figures of merit calculated for the optimized methodology include the linearity limit (LOL), detection limit (LOD), and quantification limit (LOQ). The LOD and LOQ were determined using the formulas 3 × SD/slope and 10 × SD/slope, respectively, where SD corresponds to the standard deviation of blank signals from ten replicates. These values were calculated according to the general definitions provided by Eurachem, Demirkaya-Miloglu et al., and Ershadi et al.69–71 For the determination of the LOL, simple calibration curves were constructed, and an analysis of variance (ANOVA) test demonstrated a significant correlation (p-value < 0.0001) between the MeHg+ concentration and the area signal over a working range of 0.5 to 100 ng of MeHg+. The linearity of the calibration curve was assessed through linear regression, resulting in a correlation coefficient (r2) of 0.9999 and a linear equation given by y = 1.517x − 1.41. The calibration curve was then used to determine the LOD and LOQ, which were found to be 0.0045 and 0.113 μg L−1, respectively. Other parameters determined using C2H4Cl2, were preconcentration factor 35 ± 2, and extraction percentage 100 ± 6%.The results indicate that the sensitivity of the method is comparable to that reported in the literature, but with notable advantages in terms of speed and cost-effectiveness. While traditional methods often require labor-intensive sample preparation and the use of costly reagents, the optimized GC-PYRO-AFS system offers a streamlined process that reduces both time and expense, without compromising analytical performance. This reduction in time and cost is particularly advantageous for large-scale studies or routine analysis, where efficiency is crucial. The ability to achieve similar or better detection limits with a simpler and more cost-effective method could significantly influence the adoption of this methodology in future research and various applications. Moreover, the ease of implementation of this optimized method in laboratories with limited resources further highlights its potential for widespread use, particularly in regions where access to sophisticated analytical equipment and reagents may be constrained.
The LOD can be expressed as either a relative or absolute limit. The relative detection limit is defined as the minimum detectable analyte concentration (expressed in concentration units), while the absolute detection limit is defined as the minimum detectable analyte amount (expressed in mass units).72 In our study, the relative detection limit was 0.0045 μg L−1, while the absolute detection limit was 0.4 pg MeHg+ as Hg per injection volume. This distinction is important because LOD is reported in the literature in one or both formats. For instance, in studies using GC-PYRO-AFS systems for the determination of MeHg+ in various matrices, authors such as Gibičar et al., Ebdon et al., and Berzas Nevado et al. reported the LOD as an absolute detection limit, with values in the picogram range (0.22 pg and 1.8 pg MeHg+ as Hg, respectively).73–75 However, the enhanced sensitivity achieved by these authors contrasts with the time-consuming and labor-intensive sample preparation required for extracting MeHg+ and organomercurial compounds from certified reference materials (CRMs) and biological samples,73,74 which involves the use of highly volatile organic solvents like dichloromethane and hexane,74,75 and/or derivatizing agents recently discontinued in our country, such as sodium tetrapropylborate (NaBPr4)73 and sodium tetraethylborate (NaBEt4).75 Other authors, such as Mao et al., reported the LOD as a relative detection limit, obtaining a value of 0.03 ng L−1 for MeHg+ and EtHg+ through an aqueous phenylation process with NaBPh4, coupled to a purge-and-trap GC-PYRO-AFS system. However, the signals they obtained for both analytes were highly susceptible to parameters such as salt concentration in the aqueous medium, temperature, flow rate, and purge time, with a minimum purge time of 45 minutes being required.76
Given these considerations, our LOD is comparable to those reported in the literature. Our methodology is also faster, less time-consuming, and allows for straightforward sample processing at a lower cost, with NaBPh4 as a commercially accessible derivatizing agent compared to NaBPr4 and NaBEt4.
The matrix effect was also evaluated during the quantification of MeHg+ in mice brains. The analysis indicates that the sample matrix does not significantly affect the sensitivity of the method, ensuring accurate quantification with 95% confidence (p-value = 0.5279).
Regarding validation, the hyphenated GC-PYRO-AFS system, along with the MAE, derivatization, and LPME processes under optimal conditions, was validated through a statistical comparison at a 95% confidence level between the certified reference material ERM CE-464 (tuna fish) reported MeHg+ concentration (5.5 mg MeHg+ per kg) and the concentration obtained through GC-PYRO-AFS analysis (5.7 ± 0.3 mg MeHg+ per kg).
Selectivity of the hyphenated GC-PYRO-AFS system
After optimizing the chromatographic parameters and method procedures, the selectivity of the hyphenated GC-PYRO-AFS system was evaluated. Samples containing 10 ng of MeHg+, 10 ng of Hg2+, and blank samples were analyzed using the GC-PYRO-AFS system under identical conditions. The Hg2+ sample exhibited a low signal, similar to that of the blank, in contrast to the intense signal observed for the MeHg+ sample (Fig. 5). These results confirm that the method is selective for MeHg+ under the optimized conditions of the GC-PYRO-AFS system.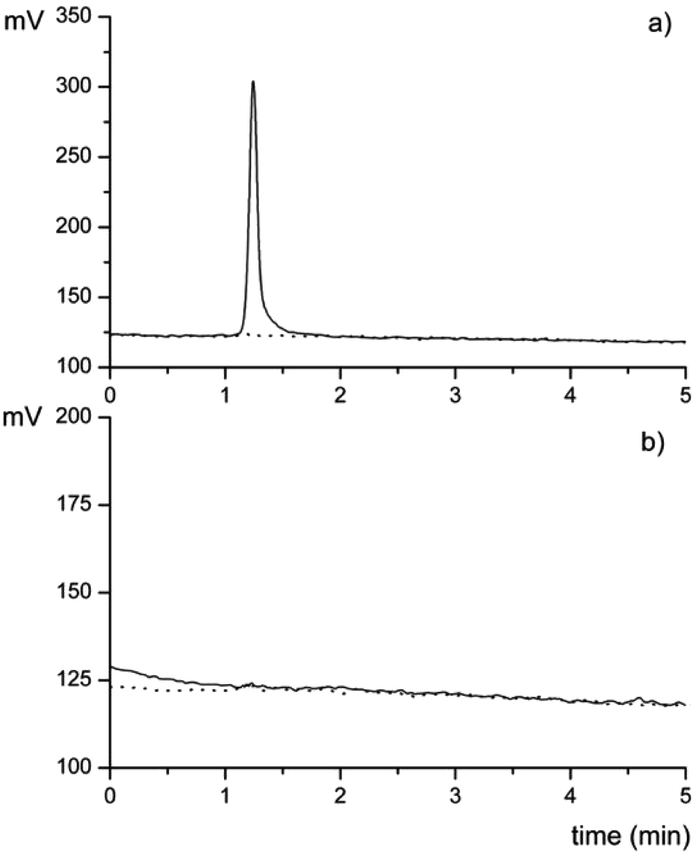 | ||
| Fig. 5 Chromatogram of (a) 10 ng of MeHg+ (solid line) and control (dotted line); (b) 10 ng Hg2+ (solid line) and control (dotted line), under optimal operating conditions (n = 3 each group). | ||
Biological application in mouse quantification and its importance in toxicology
 | ||
| Fig. 6 Total Hg and MeHg+ concentrations, along with their statistical significance, categorized by specific exposure conditions (measured in wet weight). Control group: white bars; 0.5 mg MeHg+ per L: grey bars; 5 mg MeHg+ per L: black and white bars. | ||
The results of MeHg+ accumulation in the brain correlate with the dose administered to each evaluated group. Additionally, the concentration of MeHg+ in the brain exhibits consistent ratios between groups. Specifically, the control and 0.5 mg MeHg+ per L group show a ratio of 22, while the 0.5 and 5 mg MeHg+ per L groups demonstrate a ratio of 25.
In the studies, it was observed that the hippocampus accumulates a higher amount of MeHg+ compared to the whole brain (p-value < 0.001 in all cases). This finding is significant because it indicates that the hippocampus may be more susceptible to the accumulation of this neurotoxin. Additionally, it was found that a portion of the mercury accumulated in these tissues is not in the form of MeHg+, and this difference is statistically significant (p-value < 0.0001 in all cases). This suggests that, in addition to MeHg+, other forms of mercury might be present, or that MeHg+ could be undergoing transformations within the tissue, which has important implications for understanding the mechanisms of toxicity and the risks associated with MeHg+ exposure. Therefore, the development of precise and specialized analytical techniques is essential for identifying and quantifying these different forms of mercury, which will allow for a better assessment of toxicological risk and the formulation of more effective mitigation strategies.
The development of new methodologies that are not only precise but also time and cost-efficient allows for improved detection and quantification of these compounds in various biological matrices. These methodologies enable the exact determination of toxicant concentrations in different tissues, which in turn helps propose detailed distribution mechanisms within the organism. Understanding these mechanisms is essential for linking toxicant distribution to observed toxicological effects, aiding in risk assessment and biomarker identification.
Regarding the entry and accumulation of MeHg+ in the brain, it is noted that while the precise molecular mechanism of distribution through the body remains unknown, the highly lipophilic MeHg+ can diffuse through cell membranes without the need for carrier proteins.78 It can also be transported across cell membranes due to the formation of thiol complexes, which enhance its water solubility and facilitate its circulation in the bloodstream. For instance, MeHg+ traverses the endothelial cells of the blood–brain barrier (BBB) conjugated with L-Cys via the L-type amino acid transporter 1 (LAT1) system.79 It has also been documented that exposure to MeHg+ can induce BBB damage due to the upregulation of genes that promote barrier leakage, a phenomenon predominantly observed in astrocytes.80 Similar effects have been noted in human brain microvascular endothelial cells, where MeHg+ exposure increased VEGF expression, leading to barrier leakage that could facilitate the entry of MeHg+ and other harmful substances.78
Given the complexities of MeHg+ transport and accumulation, the development of these studies is crucial for the future identification of biomarkers that can help detect these species in humans as accurately as possible.
Conclusions
The results obtained in this study demonstrate the successful development and optimization of a methodology that utilizes microwave-assisted extraction, derivatization by phenylation, and preconcentration through liquid phase microextraction for the selective quantification of MeHg+. The optimized methodology was effectively applied to the determination of MeHg+ in mice brain, enabling its quantification at trace levels without significant matrix effects. This methodology allowed for the determination of the accumulated concentration of MeHg+ in the mice brain, which correlated with the dose to which the mice were exposed through drinking water. Furthermore, it was observed that MeHg accumulates more extensively in the hippocampus compared to the entire brain, and that only a fraction of the total mercury remains as MeHg following ingestion.The high costs and complexities traditionally associated with MeHg+ quantification, often requiring mass balance calculations and the use of sophisticated equipment, have been effectively addressed by the methodology developed in this study. By integrating microwave-assisted extraction, phenylation derivatization, and preconcentration through liquid phase microextraction (LPME), this approach eliminates the need for mass balance assessments while offering a selective and sensitive quantification of MeHg+. The optimized methodology is not only cost-effective but also adaptable for use across a variety of matrices, enabling widespread application in both biological and environmental samples. This advancement permits for more accessible and accurate MeHg+ analysis, making it feasible to monitor exposure and accumulation in diverse contexts with minimal sample preparation and lower operational costs.
Moreover, the developed methodology not only enhances efficiency but also holds the potential to significantly improve the accuracy of MeHg+ quantification, which is crucial for toxicological risk assessment and biomarker development.
In the future, this selective quantification method for MeHg+ could be instrumental in establishing novel biological markers related to MeHg+ exposure, particularly in tissues that accumulate MeHg+ at trace levels, utilizing a small sample volume (0.25 g wet weight).
Abbreviations
| CNS | Central nervous system |
| DMA | Direct mercury analyzer |
| DoE | Design of experiments |
| EHC | Enterohepatic circulation |
| GC-PYRO-AFS | Gas chromatography with pyrolysis oven and atomic fluorescence spectroscopy |
| LOD | Limit of detection |
| LOL | Linearity limit |
| LOQ | Limit of quantification |
| LPME | Liquid-phase microextraction |
| MAE | Microwave-assisted extraction |
| MeHg+ | Monomethylmercury |
| NaBPh4 | Sodium tetraphenylborate |
| PhB(OH)2 | Phenylboronic acid |
| RSD | Relative standard deviation |
| VEGF | Vascular endothelial growth factor |
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Resources: Gabriela Lobos, Marcelo Verdugo. Supervision: Marcelo Verdugo. Project administration: Marcelo Verdugo. Conceptualization: Marcelo Verdugo. Writing – original draft: Marcelo Verdugo, Ferdinand Ávila, Jhoel Ruiz, Constanza Vásquez, Gabriela Lobos. Investigation: Marcelo Verdugo, Ferdinand Ávila, Jhoel Ruiz, Constanza Vásquez. Formal analysis: Marcelo Verdugo. Validation: Marcelo Verdugo, Nicole Roldán. Visualization: Marcelo Verdugo, Ferdinand Ávila, Jhoel Ruiz, Constanza Vásquez, Nicole Roldán. Writing – review & editing: Marcelo Verdugo, Ferdinand Ávila, Jhoel Ruiz, Constanza Vásquez, Nicole Roldán, M. Gabriela Lobos, Álvaro O. Ardiles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by ANID Fondecyt Iniciación 11190689 from the Government of Chile.References
- A. Antunes dos Santos, M. Appel Hort, M. Culbreth, C. López-Granero, M. Farina and J. B. T. Rocha, et al., Methylmercury and brain development: A review of recent literature, J. Trace Elem. Med. Biol., 2016, 38, 99–107 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S0946672X16300220.
- K. Eto, Minamata disease, Neuropathology, 2000, 20(s1), 14–19 CrossRef , https://pubmed.ncbi.nlm.nih.gov/11037181/.
- M. Farina, J. B. T. Rocha and M. Aschner, Mechanisms of methylmercury-induced neurotoxicity: Evidence from experimental studies, Life Sci., 2011, 89(15–16), 555–563, DOI:10.1016/j.lfs.2011.05.019.
- E. K. Tucker and R. A. Nowak, Methylmercury alters proliferation, migration, and antioxidant capacity in human HTR8/SV-neo trophoblast cells, Reprod. Toxicol., 2018, 78, 60–68, DOI:10.1016/j.reprotox.2018.03.008.
- Y. S. Hong, Y. M. Kim and K. E. Lee, Methylmercury Exposure and Health Effects, J. Prev. Med. Public Health, 2012, 45(6), 353–363, DOI:10.3961/jpmph.2012.45.6.353.
- G. d. P. Arrifano, M. Augusto-Oliveira, M. Sealey-Bright, J. Zainal, L. Imbiriba and L. M. P. Fernandes, et al., Contributing to understand the crosstalk between brain and periphery in methylmercury intoxication: Neurotoxicity and extracellular vesicles, Int. J. Mol. Sci., 2021, 22(19), 10855, DOI:10.3390/ijms221910855.
- K. Burke, Y. Cheng, B. Li, A. Petrov, P. Joshi and R. F. Berman, et al., Methylmercury elicits rapid inhibition of cell proliferation in the developing brain and decreases cell cycle regulator, cyclin E, NeuroToxicology, 2006, 27(6), 970–981, DOI:10.1016/j.neuro.2006.09.001.
- M. Fujimura, F. Usuki, J. Cheng and W. Zhao, Prenatal low-dose methylmercury exposure impairs neurite outgrowth and synaptic protein expression and suppresses TrkA pathway activity and eEF1A1 expression in the rat cerebellum, Toxicol. Appl. Pharmacol., 2016, 298, 1–8, DOI:10.1016/j.taap.2016.03.002.
- J. D. Mancini, D. M. Autio and W. D. Atchison, Continuous exposure to low concentrations of methylmercury impairs cerebellar granule cell migration in organotypic slice culture, Neurotoxicology, 2009, 30(2), 203–208, DOI:10.1016/j.neuro.2008.12.010.
- T. Toyama, T. Hoshi, T. Noguchi, Y. Saito, A. Matsuzawa and A. Naganuma, et al., Methylmercury induces neuronal cell death by inducing TNF-α expression through the ASK1/p38 signaling pathway in microglia, Sci. Rep., 2021, 11(1), 1–13 CrossRef , https://www.nature.com/articles/s41598-021-89210-7.
- T. Yorifuji, Lessons from an early-stage epidemiological study of minamata disease, J. Epidemiol., 2020, 30(1), 12–14, DOI:10.2188/jea.je20190089.
- R. M. Mellingen, L. S. Myrmel, K. K. Lie, J. D. Rasinger, L. Madsen and O. J. Nøstbakken, RNA sequencing and proteomic profiling reveal different alterations by dietary methylmercury in the hippocampal transcriptome and proteome in BALB/c mice, Metallomics, 2021, 13(5), mfab022, DOI:10.1093/mtomcs/mfab022.
- R. M. Mellingen, L. S. Myrmel, J. D. Rasinger, K. K. Lie, A. Bernhard and L. Madsen, et al., Dietary selenomethionine reduce mercury tissue levels and modulate methylmercury induced proteomic and transcriptomic alterations in hippocampi of adolescent BALB/c mice, Int. J. Mol. Sci., 2022, 23(20), 12242, DOI:10.3390/ijms232012242.
- T. S. Nascimento, D. V. Pinto, R. P. Dias, R. S. Raposo, P. I. G. Nunes and C. R. Roque, et al., Chronic methylmercury intoxication induces systemic inflammation, behavioral, and hippocampal amino acid changes in C57BL6J adult mice, Int. J. Mol. Sci., 2022, 23(22), 13837, DOI:10.3390/ijms232213837.
- A. Oguro, T. Fujiyama, Y. Ishihara, C. Kataoka, M. Yamamoto and K. Eto, et al., Maternal DHA intake in mice increased DHA metabolites in the pup brain and ameliorated MeHg-induced behavioral disorder, J. Lipid Res., 2023, 64(11), 100458, DOI:10.1016/j.jlr.2023.100458.
- O. Y. Chao, M. A. de Souza Silva, Y.-M. Yang and J. P. Huston, The medial prefrontal cortex - hippocampus circuit that integrates information of object, place and time to construct episodic memory in rodents: Behavioral, anatomical and neurochemical properties, Neurosci. Biobehav. Rev., 2020, 113, 373–407, DOI:10.1016/j.neubiorev.2020.04.007.
- E. Cherubini and R. Miles, The CA3 region of the hippocampus: how is it? What is it for? How does it do it?, Front. Cell. Neurosci., 2015, 9, 19, DOI:10.3389/fncel.2015.00019.
- J. Huijgen and S. Samson, The hippocampus: A central node in a large-scale brain network for memory, Rev. Neurol., 2015, 171(3), 204–216, DOI:10.1016/j.neurol.2015.01.557.
- Y. Yamagata, Y. Yanagawa and K. Imoto, Differential involvement of kinase activity of Ca2+/calmodulin-dependent protein kinase IIα in hippocampus- and amygdala-dependent memory revealed by kinase-dead knock-in mouse, eNeuro, 2018, 5(4), ENEURO.0133-18.2018, DOI:10.1523/eneuro.0133-18.2018.
- T. Das, J. J. Hwang and K. L. Poston, Episodic recognition memory and the hippocampus in Parkinson's disease: A review, Cortex, 2019, 113, 191–209, DOI:10.1016/j.cortex.2018.11.021.
- S. Villar-Conde, V. Astillero-Lopez, M. Gonzalez-Rodriguez, P. Villanueva-Anguita, D. Saiz-Sanchez and A. Martinez-Marcos, et al., The human hippocampus in Parkinson's disease: An integrative stereological and proteomic study, J. Parkinson's Dis., 2021, 11(3), 1345–1365, DOI:10.3233/jpd-202465.
- C. Boutet, M. Chupin, S. Lehéricy, L. Marrakchi-Kacem, S. Epelbaum and C. Poupon, et al., Detection of volume loss in hippocampal layers in Alzheimer's disease using 7 T MRI: A feasibility study, NeuroImage Clin., 2014, 5, 341–348, DOI:10.1016/j.nicl.2014.07.011.
- R. de Flores, R. La Joie and G. Chételat, Structural imaging of hippocampal subfields in healthy aging and Alzheimer's disease, Neuroscience, 2015, 309, 29–50, DOI:10.1016/j.neuroscience.2015.08.033.
- Y. L. Rao, B. Ganaraja, B. V. Murlimanju, T. Joy, A. Krishnamurthy and A. Agrawal, Hippocampus and its involvement in Alzheimer’s disease: a review, 3 Biotech, 2022, 12(2), 55, DOI:10.1007/s13205-022-03123-4.
- P. H. Reddy, X. Yin, M. Manczak, S. Kumar, J. A. Pradeepkiran and M. Vijayan, et al., Mutant APP and amyloid beta-induced defective autophagy, mitophagy, mitochondrial structural and functional changes and synaptic damage in hippocampal neurons from Alzheimer's disease, Hum. Mol. Genet., 2018, 27(14), 2502–2516, DOI:10.1093/hmg/ddy154.
- J. Azar, M. H. Yousef, H. A. N. El-Fawal and A. Abdelnaser, Mercury and Alzheimer's disease: a look at the links and evidence, Metab. Brain Dis., 2021, 36(3), 361–374, DOI:10.1007/s11011-020-00649-5.
- A. Antunes dos Santos, B. Ferrer, F. Marques Gonçalves, A. M. Tsatsakis, E. A. Renieri and A. V. Skalny, et al., Oxidative stress in methylmercury-induced cell toxicity, Toxics, 2018, 6(3), 47, DOI:10.3390/toxics6030047.
- D.-K. Kim, J.-D. Park and B.-S. Choi, Mercury-induced amyloid-beta (Aβ) accumulation in the brain is mediated by disruption of Aβ transport, J. Toxicol. Sci., 2014, 39(4), 625–635, DOI:10.2131/jts.39.625.
- F. Monnet-Tschudi, M.-G. Zurich, C. Boschat, A. Corbaz and P. Honegger, Involvement of environmental mercury and lead in the etiology of neurodegenerative diseases, Rev. Environ. Health, 2006, 21(2), 105–117, DOI:10.1515/reveh.2006.21.2.105.
- M. Ni, X. Li, J. B. T. Rocha, M. Farina and M. Aschner, Glia and methylmercury neurotoxicity, J. Toxicol. Environ. Health, Part A, 2012, 75(16–17), 1091–1101, DOI:10.1080/15287394.2012.697840.
- E. Paduraru, D. Iacob, V. Rarinca, A. Rusu, R. Jijie and O.-D. Ilie, et al., Comprehensive review regarding mercury poisoning and its complex involvement in Alzheimer's disease, Int. J. Mol. Sci., 2022, 23(4), 1992, DOI:10.3390/ijms23041992.
- J.-W. Song and B.-S. Choi, Mercury induced the accumulation of amyloid beta (Aβ) in PC12 cells: The role of production and degradation of Aβ, Toxicol. Res., 2013, 29(4), 235–240, DOI:10.5487/tr.2013.29.4.235.
- C. Sanfeliu, J. Sebastià and S. U. Kim, Methylmercury Neurotoxicity in Cultures of Human Neurons, Astrocytes, Neuroblastoma Cells, Neurotoxicology, 2001, 22(3), 317–327, DOI:10.1016/s0161-813x(01)00015-8.
- I. Vendrell, M. Carrascal, M.-T. Vilaró, J. Abián, E. Rodríguez-Farré and C. Suñol, Cell viability and proteomic analysis in cultured neurons exposed to methylmercury, Hum. Exp. Toxicol., 2007, 26(4), 263–272, DOI:10.1177/0960327106070455.
- B. Zimmer, S. Schildknecht, P. B. Kuegler, V. Tanavde, S. Kadereit and M. Leist, Sensitivity of Dopaminergic Neuron Differentiation from Stem Cells to Chronic Low-Dose Methylmercury Exposure, Toxicol. Sci., 2011, 121(2), 357–367, DOI:10.1093/toxsci/kfr054.
- S. Bellum, K. A. Thuett, B. Bawa and L. C. Abbott, The effect of methylmercury exposure on behavior and cerebellar granule cell physiology in aged mice, J. Appl. Toxicol., 2013, 33(9), 959–969, DOI:10.1002/jat.2786.
- B. Ferrer, T. V. Peres, A. A. d. Santos, J. Bornhorst, P. Morcillo and C. L. Gonçalves, et al., Methylmercury Affects the Expression of Hypothalamic Neuropeptides That Control Body Weight in C57BL/6J Mice, Toxicol. Sci., 2018, 163(2), 557–568, DOI:10.1093/toxsci/kfy052.
- M. Fujimura, F. Usuki, M. Sawada and A. Takashima, Methylmercury induces neuropathological changes with tau hyperphosphorylation mainly through the activation of the c-jun-N-terminal kinase pathway in the cerebral cortex, but not in the hippocampus of the mouse brain, Neurotoxicology, 2009, 30(6), 1000–1007, DOI:10.1016/j.neuro.2009.08.001.
- M. Iwai-Shimada, T. Takahashi, M.-S. Kim, M. Fujimura, H. Ito and T. Toyama, et al., Methylmercury induces the expression of TNF-α selectively in the brain of mice, Sci. Rep., 2016, 6, 38294 CrossRef CAS PubMed , http://www.ncbi.nlm.nih.gov/pubmed/27910896.
- H. Hintelmann, Organomercurials. Their formation and pathways in the environment, Met. Ions Life Sci., 2010, 7 Search PubMed , https://pubmed.ncbi.nlm.nih.gov/20877813/.
- C. H. Lamborg, C. R. Hammerschmidt, K. L. Bowman, G. J. Swarr, K. M. Munson and D. C. Ohnemus, et al., A global ocean inventory of anthropogenic mercury based on water column measurements, Nature, 2014, 512(7512), 65–68 CrossRef CAS PubMed , https://www.nature.com/articles/nature13563.
- T. W. Clarkson and L. Magos, The toxicology of mercury and its chemical compounds, Crit. Rev. Toxicol., 2006, 36(8), 609–662 CrossRef CAS PubMed , https://pubmed.ncbi.nlm.nih.gov/16973445/.
- D. Mergler, H. A. Anderson, L. H. M. Chan, K. R. Mahaffey, M. Murray and M. Sakamoto, et al., Methylmercury exposure and health effects in humans: A worldwide concern, Ambio, 2007, 36(1), 3–11 CrossRef CAS , https://pubmed.ncbi.nlm.nih.gov/17408186/.
- M. Vahter, N. K. Mottet, L. Friberg, B. Lind, D. D. Shen and T. Burbacher, Speciation of Mercury in the Primate Blood and Brain Following Long-Term Exposure to Methyl Mercury, Toxicol. Appl. Pharmacol., 1994, 124(2), 221–229, DOI:10.1006/taap.1994.1026.
- M. E. Vahter, N. K. Mottet, L. T. Friberg, S. B. Lind, J. S. Charleston and T. M. Burbacher, Demethylation of Methyl Mercury in Different Brain Sites of Macaca-fascicularis Monkeys during Long-Term Subclinical Methyl Mercury Exposure, Toxicol. Appl. Pharmacol., 1995, 134(2), 273–284, DOI:10.1006/taap.1995.1193.
- S. Bellum, K. A. Thuett, R. J. Taylor and L. C. Abbott, Assessment of Mercury Concentrations in Mouse Brain Using Different Routes of Administration and Different Tissue Preparations, Toxicol. Mech. Methods, 2007, 17(3), 165–173, DOI:10.1080/15376510601091368.
- H. Hiraoka, R. Nomura, N. Takasugi, R. Akai, T. Iwawaki and Y. Kumagai, et al., Spatiotemporal analysis of the UPR transition induced by methylmercury in the mouse brain, Arch. Toxicol., 2021, 95(4), 1241–1250, DOI:10.1007/s00204-021-02982-9.
- D. H. Roos, R. L. Puntel, T. H. Lugokenski, R. P. Ineu, D. Bohrer and M. E. Burger, et al., Complex Methylmercury–Cysteine Alters Mercury Accumulation in Different Tissues of Mice, Basic Clin. Pharmacol. Toxicol., 2010, 107(4), 789–792, DOI:10.1111/j.1742-7843.2010.00577.x.
- L. Yang, Z. Mester and R. E. Sturgeon, Determination of methylmercury in fish tissues by isotope dilution SPME-GC-ICP-MS, J. Anal. At. Spectrom., 2003, 18(5), 431–436 RSC , https://rsc.66557.net/en/content/articlehtml/2003/ja/b301299a.
- X. Ma, Y. G. Yin, J. B. Shi, J. F. Liu and G. B. Jiang, Species-specific isotope dilution-GC-ICP-MS for accurate and precise measurement of methylmercury in water, sediments and biological tissues, Anal. Methods, 2013, 6(1), 164–169 RSC , https://rsc.66557.net/en/content/articlehtml/2014/ay/c3ay41625a.
- J. Qvarnström, L. Lambertsson, S. Havarinasab, P. Hultman and W. Frech, Determination of methylmercury, ethylmercury, and inorganic mercury in mouse tissues, following administration of thimerosal, by species-specific isotope dilution GC-inductively coupled plasma-MS, Anal. Chem., 2003, 75(16), 4120–4124 CrossRef PubMed , https://pubmed.ncbi.nlm.nih.gov/14632125/.
- Z. Jókai, L. Abrankó and P. Fodor, SPME-GC-Pyrolysis-AFS determination of methylmercury in marine fish products by alkaline sample preparation and aqueous phase phenylation derivatization, J. Agric. Food Chem., 2005, 53(14), 5499–5505, DOI:10.1021/jf0501140.
- E. Ramalhosa, S. R. Segade, E. Pereira, A. Duarte and C. Vale, Microwave treatment of biological samples for methylmercury determination by high performance liquid chromatography–cold vapour atomic fluorescence spectrometry, Analyst, 2001, 126(9), 1583–1587, 10.1039/b104041n.
- H. E. L. Palmieri and L. V. Leonel, Determination of methylmercury in fish tissue by gas chromatography with microwave-induced plasma atomic emission spectrometry after derivatization with sodium tetraphenylborate, Fresenius. J. Anal. Chem., 2000, 366(5), 466–469, DOI:10.1007/s002160050094.
- R. Rodil, A. M. Carro, R. A. Lorenzo, M. Abuín and R. Cela, Methylmercury determination in biological samples by derivatization, solid-phase microextraction and gas chromatography with microwave-induced plasma atomic emission spectrometry, J. Chromatogr. A, 2002, 963(1–2), 313–323, DOI:10.1016/s0021-9673(02)00644-1.
- A. Sarafraz-Yazdi, E. Fatehyan and A. Amiri, Determination of Mercury in Real Water Samples Using in situ Derivatization Followed by Sol-Gel-Solid-Phase Microextraction with Gas Chromatography-Flame Ionization Detection, J. Chromatogr. Sci., 2014, 52(1), 81–87, DOI:10.1093/chromsci/bms208.
- B. Ferrer, L. M. Prince, A. A. Tinkov, A. Santamaria, M. Farina and J. B. Rocha, et al., Chronic exposure to methylmercury enhances the anorexigenic effects of leptin in C57BL/6J male mice, Food Chem. Toxicol., 2021, 147(111924), 111924, DOI:10.1016/j.fct.2020.111924.
- A. Jaszczyk, A. M. Stankiewicz and G. R. Juszczak, Dissection of mouse hippocampus with its dorsal, intermediate and ventral subdivisions combined with molecular validation, Brain Sci., 2022, 12(6), 799 CrossRef CAS PubMed , https://pubmed.ncbi.nlm.nih.gov/35741684/.
- N. Basu, A. Scheuhammer, N. Grochowina, K. Klenavic, D. Evans and M. O'Brien, et al., Effects of mercury on neurochemical receptors in wild river otters (Lontra canadensis), Environ. Sci. Technol., 2005, 39(10), 3585–3591 CrossRef CAS PubMed , https://pubmed.ncbi.nlm.nih.gov/15952362/.
- J. Bo Nielsen and O. Andersen, Oral mercuric chloride exposure in mice: effects of dose on intestinal absorption and relative organ distribution, Toxicology, 1989, 59(1), 1–10 CrossRef PubMed , https://pubmed.ncbi.nlm.nih.gov/2815096/.
- I. Minitab, Minitab 17 Statistical Software, State College, PA, 2010, https://www.minitab.com Search PubMed.
- L. Abrankó, Z. Jókai and P. Fodor, Investigation of the species-specific degradation behaviour of methylmercury and ethylmercury under microwave irradiation, Anal. Bioanal. Chem., 2005, 383(3), 448–453, DOI:10.1007/s00216-005-3395-x.
- W. T. Corns, L. C. Ebdon, S. J. Hill and P. B. Stockwell, Automated cold vapour flow-injection analysis of mercury at high concentrations, J. Anal. Methods Chem., 1991, 13(6), 267–271, DOI:10.1155/s1463924691000421.
- N. A. Katsanos, R. Thede and F. Roubani-Kalantzopoulou, Diffusion, adsorption and catalytic studies by gas chromatography, J. Chromatogr. A, 1998, 795(2), 133–184, DOI:10.1016/s0021-9673(97)00968-0.
- J. J. Berzas Nevado, R. C. R. Martín-Doimeadios, F. J. Guzmán Bernardo and M. Jiménez Moreno, Determination of mercury species in fish reference materials by gas chromatography-atomic fluorescence detection after closed-vessel microwave-assisted extraction, J. Chromatogr. A, 2005, 1093(1–2), 21–28 CrossRef CAS PubMed.
- M. Jiménez-Moreno, M. Á. Lominchar, M. J. Sierra, R. Millán and R. C. R. Martín-Doimeadios, Fast method for the simultaneous determination of monomethylmercury and inorganic mercury in rice and aquatic plants, Talanta, 2018, 176, 102–107 CrossRef PubMed , https://www.sciencedirect.com/science/article/pii/S0039914017308329.
- S. Díez and J. M. Bayona, Determination of Hg and organomercury species following SPME: A review, Talanta, 2008, 77(1), 21–27 CrossRef PubMed.
- A. Sarafraz Yazdi, S. Banihashemi and Z. Es'Haghi, Determination of Hg(II) in natural waters by diphenylation by single-drop microextraction: GC, Chromatographia, 2010, 71(11–12), 1049–1054, DOI:10.1365/s10337-010-1576-z.
- Eurachem, The Fitness for Purpose of Analytical Methods – A Laboratory Guide to Method Validation and Related Topics, ed. B. Magnusson and U. Örnemark, 2nd edn. 2014, https://www.eurachem.org Search PubMed.
- F. Demirkaya-Miloglu, M. E. Yaman and Y. Kadioglu, A new spectrofluorimetric method for determination of losartan potassium in rabbit plasma and its application to pharmacokinetic study, Luminescence, 2015, 30(1), 53–59 CrossRef CAS PubMed , https://pubmed.ncbi.nlm.nih.gov/24890844/.
- S. Ershadi and A. Shayanfar, Are LOD and LOQ reliable parameters for sensitivity evaluation of spectroscopic methods?, J. AOAC Int., 2018, 101(4), 1212–1213 CrossRef CAS PubMed , https://pubmed.ncbi.nlm.nih.gov/29566782/.
- N. Omenetto, G. A. Petrucci, P. Cavalli and J. D. Winefordner, Absolute and/or relative detection limits in laser-based analysis: the end justifies the means, Anal. Bioanal. Chem., 1996, 355(7–8), 878–882 CrossRef CAS PubMed , https://pubmed.ncbi.nlm.nih.gov/15045286/.
- D. Gibičar, M. Logar, N. Horvat, A. Marn-Pernat, R. Ponikvar and M. Horvat, Simultaneous determination of trace levels of ethylmercury and methylmercury in biological samples and vaccines using sodium tetra(n-propyl)borate as derivatizing agent, Anal. Bioanal. Chem., 2007, 388(2), 329–340, DOI:10.1007/s00216-007-1208-0.
- L. Ebdon, M. E. Foulkes, S. Le Roux and R. Muñoz-Olivas, Cold vapour atomic fluorescence spectrometry and gas chromatography-pyrolysis-atomic fluorescence spectrometry for routine determination of total and organometallic mercury in food samples, Analyst, 2002, 127(8), 1108–1114, 10.1039/b202927h.
- J. J. Berzas Nevado, R. C. Rodríguez Martín-Doimeadios, E. M. Krupp, F. J. Guzmán Bernardo, N. Rodríguez Fariñas and M. Jiménez Moreno, et al., Comparison of gas chromatographic hyphenated techniques for mercury speciation analysis, J. Chromatogr. A, 2011, 1218(28), 4545–4551 CrossRef CAS PubMed , https://pubmed.ncbi.nlm.nih.gov/21641604/.
- Y. Mao, G. Liu, G. Meichel, Y. Cai and G. Jiang, Simultaneous speciation of monomethylmercury and monoethylmercury by aqueous phenylation and purge-and-trap preconcentration followed by atomic spectrometry detection, Anal. Chem., 2008, 80(18), 7163–7168, DOI:10.1021/ac800908b.
- M. D. Rand, K. Conrad, E. Marvin, K. Harvey, D. Henderson and R. Tawil, et al., Developmental exposure to methylmercury and resultant muscle mercury accumulation and adult motor deficits in mice, Neurotoxicology, 2020, 81, 1–10 CrossRef CAS PubMed , https://pubmed.ncbi.nlm.nih.gov/32735808/.
- J. Korszun-Karbowniczak, Z. J. Krysiak, J. Saluk, M. Niemcewicz and R. Zdanowski, The progress in molecular transport and therapeutic development in human blood–brain barrier models in neurological disorders, Cell. Mol. Neurobiol., 2024, 44(1), 34, DOI:10.1007/s10571-024-01473-6.
- J. P. Novo, B. Martins, R. S. Raposo, F. C. Pereira, R. B. Oriá and J. O. Malva, et al., Cellular and molecular mechanisms mediating methylmercury neurotoxicity and neuroinflammation, Int. J. Mol. Sci., 2021, 22(6), 3101, DOI:10.3390/ijms22063101.
- T. Takahashi, M. Fujimura, M. Koyama, M. Kanazawa, F. Usuki and M. Nishizawa, et al., Methylmercury causes blood–brain barrier damage in rats via upregulation of vascular endothelial growth factor expression, PLoS One, 2017, 12(1), e0170623, DOI:10.1371/journal.pone.0170623.
Footnote |
| † Electronic supplementary information (ESI) available: The DoE experimental data and graphics. See DOI: https://doi.org/10.1039/d3ja00413a |
| This journal is © The Royal Society of Chemistry 2025 |