
A simple method for elemental analysis of liquids in sprayed microdroplets by laser-induced breakdown spectroscopy
Jun
Feng
ab,
Yihui
Yan†
c,
Yuzhu
Liu
 *ab and
Jozef
Lengyel
*c
*ab and
Jozef
Lengyel
*c
aState Key Laboratory Cultivation Base of Atmospheric Optoelectronic Detection and Information Fusion, Jiangsu Collaborative Innovation Center on Atmospheric Environment and Equipment Technology (CICAEET), Nanjing University of Information Science & Technology, Nanjing 210044, P. R. China. E-mail: yuzhu.liu@gmail.com; jozef.lengyel@tum.de
bJiangsu International Joint Laboratory on Meteorological Photonics and Optoelectronic Detection, Nanjing University of Information Science & Technology, Nanjing 210044, P. R. China
cLehrstuhl für Physikalische Chemie, TUM School of Natural Sciences, Technische Universität München, Lichtenbergstraße 4, 85748 Garching, Germany
First published on 30th October 2024
Abstract
The combination of paper spray with laser-induced breakdown spectroscopy (PS-LIBS) was used to analyze trace elements in aqueous solutions. The analysis of sprayed charged microdroplets eliminates the experimental challenges of bulk liquid analysis, such as splashing and surface ripples, which often affect LIBS sensitivity. In contrast to other electrospray techniques, PS is a simple and robust method that allows for the direct loading of liquid samples onto paper. Its performance is largely dependent on the quality of the spray plume, influenced by factors like paper shape, tip angle, spray voltage, and solvent composition. The cut angle of the paper tip significantly influences the spray plume and total ion current, with sharper tips being more efficient at generating sprayed microdroplets. The detection limits for Na, K, and Cr – 11.1 mg L−1, 28.9 mg L−1, and 67.9 mg L−1, respectively, – are in line with typical values from other LIBS experiments on liquid samples. These experimental results indicate that PS-LIBS is a promising technique for the in situ analysis of trace elements in liquid samples.
Introduction
Water is extensively utilized across various human activities, including agriculture, industry, and households, making it vulnerable to contamination, for example, by various toxic heavy metal contaminants. The most common sources of pollution are mining waste, municipal and industrial wastewater, leachates from landfills, and runoff from urban areas.1,2 Heavy metals are also introduced into the environment through natural events such as volcanic eruptions, weathering, and rock abrasion.3,4 Water pollution with heavy metals poses significant health risks owing to their high toxicity even at low concentrations.5 Minimal exposure levels often lead to their gradual accumulation in tissues, disrupting normal cellular functions and increasing disease risk. For example, long-term exposure to heavy metals in drinking water, such as arsenic, cadmium, and chromium, is associated with the development of cancer and tumor formation.6–8 Monitoring the elemental composition of water is thus essential for assessing environmental quality, detecting pollutants, and minimizing environmental impact through proper wastewater management. This is vital for ensuring compliance with regulatory standards for water released into the environment, particularly for protecting aquatic ecosystems and human health.Several techniques, including atomic absorption and emission spectroscopy as well as mass spectrometry, are capable of detecting metal contaminants in water. In atomic emission spectroscopy, both inductively coupled plasma optical emission spectroscopy (ICP-OES) and microwave plasma atomic emission spectroscopy (MP-AES) are well-established and commercially available techniques used for routine analysis of trace components in liquid samples.9,10 The liquid sample is nebulized into aerosols, introduced into a hot plasma, atomized, and subsequently excited to emit light at characteristic wavelengths for each element. An alternative approach that could also be considered is the application of a high-energy laser pulse, known as laser-induced breakdown spectroscopy (LIBS), which has found particular application in the analysis of the elemental composition of solid surfaces.11–15 Given its ability to directly analyze samples with minimal or no preparation and conduct rapid, simultaneous elemental analysis, it is not surprising that LIBS applications are expanding to analyze other forms of materials like liquids and aerosols.16–19
In contrast to conventional laboratory techniques like ICP-OES, LIBS enables the potential construction of small, portable devices for fast in situ and online measurements. Yet, analyzing liquid samples directly with LIBS faces notable experimental challenges, such as the formation of ripples on the liquid surface, liquid splashing upon laser irradiation, and the presence of vapors and aerosols above the liquid surface, which can lead to signal instability and thus affect the sensitivity of the technique.16,17,20–22 Therefore, particular attention has been paid to sample introduction and pretreatment, resulting in the development of novel approaches for LIBS experiments. These include performing experiments in bulk water,23 depositing liquid samples onto surfaces,24–28 freezing liquids,29,30 chemical derivatization,31–33 solid and liquid phase micro-extraction,34–37 injecting aqueous samples as liquid jets,38–40 or dispersing them as droplets and aerosols.35,41–43
The first LIBS experiments with sprayed droplets and aerosols used nebulizers, i.e., techniques adapted from ICP-OES.44 Later on, Lin and co-workers introduced electrospray (ES) to generate a spray of small microdroplets.41,43 In ES, a high voltage is applied to a liquid sample at the tip of a narrow capillary, resulting in the formation of a fine spray of charged droplets. These charged droplets further evaporate solvent molecules, thereby increasing the charge density and causing them to shrink into smaller charged particles. This enables precise monitoring of ion generation by measuring the ion current. The ion signal can be further adjusted by varying flow rates, spray voltages, distance between the capillary tip and laser spot, etc.45 Lin and co-workers determined the limit of detection for the ES-LIBS experiment to be 0.6 mg L−1 for Na, 2.7 mg L−1 for K, and 43 mg L−1 for Al.41,43 Coupling ES with separation techniques such as liquid chromatography or capillary electrophoresis can further improve its sensitivity in analyzing complex matrices.46 On the other hand, such experiments face challenges, as the tiny capillaries used in ES techniques are prone to clogging. As a result, substantial sample preparation is required, increasing the overall time for analysis.
To address these limitations, analytical mass spectrometry has undergone rapid development, focusing particularly on instrument miniaturization and direct sampling techniques, known as ambient ionization. Dozens of methods have been developed so far that allow for the direct injection of untreated samples.46 The present work builds upon the relatively limited studies on the combination of ES with LIBS and introduces paper spray as an alternative for generating charged microdroplets. In contrast to ES, PS was developed for direct MS injection with minimal sample preparation and has proven effective in the analysis of complex matrices, such as body fluids and food samples.47,48 This article describes the design of a PS-LIBS experiment and highlights key factors that influence the formation of charged microdroplets and the stability of the LIBS signal. The technique's capabilities are demonstrated by measuring the detection limits of selected metal ions in a model aqueous system. These results are compared to existing ES-LIBS data to evaluate whether PS is a viable alternative to ES for LIBS measurements.
Experimental
In all experiments, a piece of filter paper (VWR, Grade 413) with a circular base and a salient tip was cut out at five different angles (30°, 45°, 60°, 90°, and 120°) and attached to a copper clip, which was then connected to a high-voltage power supply. The paper cutoff geometry in our experiment follows the design by Cooks and coworkers.49 The paper substrate has two parts: a circular area for loading the sample solution and a salient part with a tip angle between 30 and 120°. The circular area is fixed and larger than the salient part to maintain consistent conditions, which allows for the comparison of sprays at different angles. After loading 500 μL of a methanol/water solution containing a sample onto the filter paper and applying a high voltage of 3.5 kV, the solution was sprayed from the tip of the paper with a pore size of 20–45 μm and guided towards the counter electrode (a copper plate) at a distance of about 8 mm. All PS measurements were conducted in positive ion mode, as negative ion mode usually needs a much higher onset voltage to achieve a stable spray.50 This assembly was held compactly on a 3D printed holder, shown in Fig. 1, which was mounted to the 3-axis translational stage (M7-600, Zolix with an MC600 controller) to ensure the spray and laser radiation are adjusted precisely. This allows us to measure LIBS signals at various distances from the tip, covering distinct spray modes.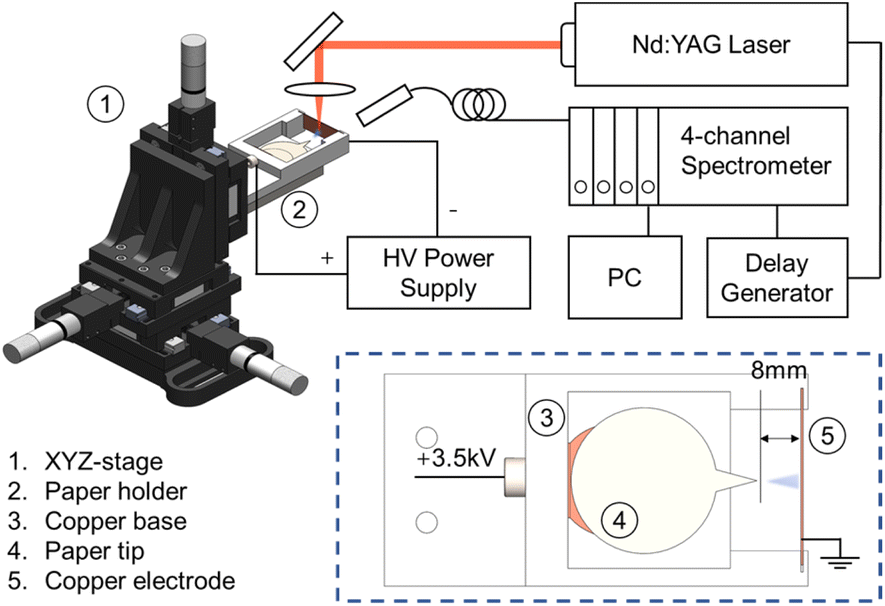 | ||
| Fig. 1 Scheme of the PS-LIBS experimental setup. | ||
The LIBS experiment was performed using a Q-switched nanosecond Nd:YAG pulse laser (Surelite II-10, Continuum Co., Ltd), and the signal was analyzed by a 4-channel spectrometer (AvaSpec ULS2048-4 Channel-usb2.0, Avantes) with a spectral window spanning 200 to 870 nm, as described elsewhere.51–53 The laser operated at a fundamental wavelength of 1064 nm, with a pulse duration of 6 ns and a repetition rate of 10 Hz. Three total reflective mirrors, designed for 1064 nm, were used to guide the excitation light and collect the emission light. A plano-concave lens with a 50 mm focal length was used to focus the pulsed laser onto the center of the spray. The probe was equipped with a collecting lens that directed the emission light into the spectrometer. The delay time of the signal spectrometer was optimized to 2.5 μs to eliminate bremsstrahlung noise. To ensure the detection of all trace elements in the LIBS signal, the integration time for all four channels of the optical spectrometer was set to 2 ms, consistent with our previous studies.54 This configuration enabled the capture of all effective spectral signals during the LIBS analysis, providing comprehensive data for accurate elemental analysis. Note that changes in integration time might have only a slight effect on plasma parameters.55 The emission lines of three elements (Fig. 2) were examined: Na I (588.995 nm and 589.592 nm), K I (766.490 nm and 769.896 nm), and Cr I (520.602 nm and 520.842 nm). Cr I emission lines near 520 nm were chosen over the more commonly reported ones at 425.43 nm due to the better signal-to-noise ratio. However, the analysis at both wavelengths yielded consistent LODs, indicating no significant difference in sensitivity.
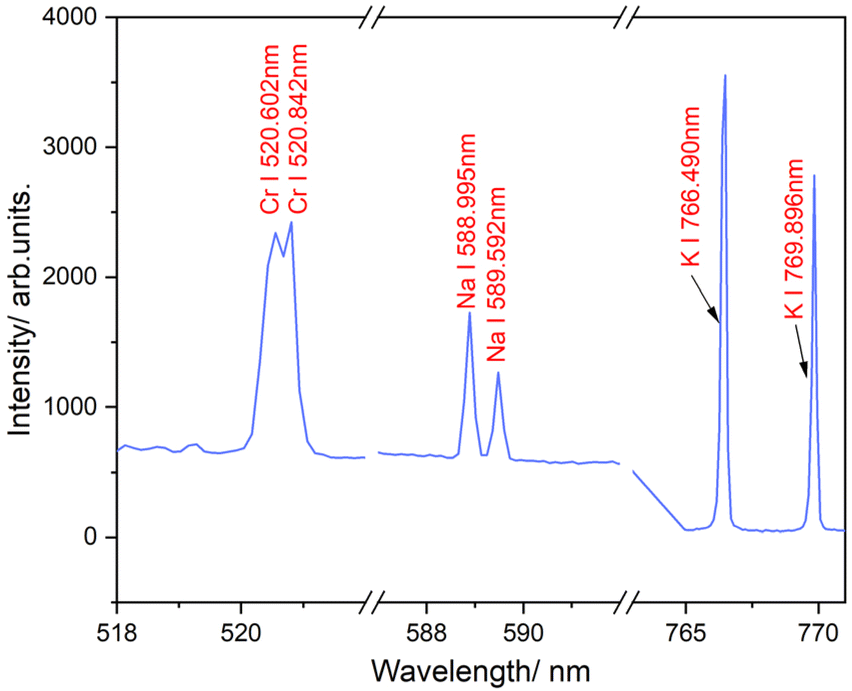 | ||
| Fig. 2 LIBS spectrum of selected trace metal elements. | ||
The experimental setup was optimized using a 1 mol NaCl solution in an H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH mixture (1:1, v/v). Methanol was added to reduce surface tension, which is essential for forming a stable spray at lower voltages. A small amount of Rhodamine B may be added to the solution for a quick visual assessment of spray quality. The LOD for sodium was determined by diluting the NaCl base solution, whereas chromium and potassium were quantitatively detected using a potassium chromate solution.
:MeOH mixture (1:1, v/v). Methanol was added to reduce surface tension, which is essential for forming a stable spray at lower voltages. A small amount of Rhodamine B may be added to the solution for a quick visual assessment of spray quality. The LOD for sodium was determined by diluting the NaCl base solution, whereas chromium and potassium were quantitatively detected using a potassium chromate solution.
As the final step, the ability of PS-LIBS to perform multi-element analysis in a real sample was evaluated using seawater collected from the coastal area of the Yellow sea near Rizhao city, Shandong province, China. For reference, the ICP-OES analysis, as described in ref. 56, was carried out at an external laboratory at the Institute of Geochemistry, Chinese Academy of Sciences, in Guiyang, Guizhou province, China. The analysis revealed that the sample contains Na (6036 ppm), Mg (977 ppm), Ca (327 ppm), K (395 ppm), and Pb (17 ppm), as the most abundant metal elements, with other metals detected at much lower concentrations, well below 10 ppm (e.g., trace amounts of Cr detected at 2 ppm).
Result and discussion
Characterization of the paper geometry and laser energy on the signal intensity
The main objective of this study is to systematically characterize the paper spray technique for integration with LIBS and identify the factors essential for achieving a high-quality spray, thereby improving the sensitivity of PS-LIBS analysis of liquids. The paper spray technique typically employs a piece of filter paper with specific porosity, positioned so that its apex faces the counter electrode. The sample is loaded directly onto the paper and transported via the microchannels to the tip. By applying a high voltage of about 3.0–3.5 kV, the spray is formed. Several factors can significantly influence the stability and shape of the spray, which ultimately impacts LIBS sensitivity. Among these factors, the most critical are the angle of the paper spray tip, the distance from the tip to the counter electrode, the location of laser focus in the spray, and the solvent composition.In this study, a constant distance was maintained between the paper spray tip and the counter electrode. The formation of the spray plume is driven by the potential difference between the paper tip and the electrode; at closer distances, lower voltages are usually required for spray formation. The addition of methanol is critical as it decreases the surface tension of the liquid, enabling the generation of a stable spray at lower voltages. In general, water needs a potential 1.8 times higher than methanol.57 It is important to note that using a sample dissolved in pure water requires the application of higher voltages, which can cause electrical discharges. These discharges might generate multiple spray jets and might damage the paper tip.
In electrospray techniques, high-polarity solvents are typically used to maintain strong conductivity in the sample solution. Electrospray is most effective when the analyte can easily be converted into its ionic form in solution. Adjusting the sample's pH and using buffers can further enhance ionization. Common polar solvents like water, methanol, acetonitrile, and isopropanol are often mixed with small amounts of electrolytes, such as acetic acid, ammonium formate, or ammonium acetate, to promote ion formation. However, this is not required for ionic compounds like NaCl, which spontaneously dissociate into Na+ and Cl− ions in aqueous solutions. The efficient generation of NaCl-enriched ionic particles by electrospray has been confirmed in various mass spectrometry studies.58
Fig. 3 illustrates the impact of varying paper angles on signal intensity. The absolute ion current was measured as a function of the applied potential for five different tip angles, namely 30°, 45°, 60°, 90°, and 120°. These measurements, reflecting the number of ions in the sprayed solution, enable a comparison of different paper angles to determine which one provides the best sensitivity for LIBS experiments. Yang et al. showed that spray occurs only at the sharp tips of the paper edge, so rounding the corners and choosing a specific apex determined the direction of the spray.49 The apex of the filter paper was then consistently positioned 8 mm from the counter electrode to ensure uniform conditions for all five tested paper geometries. As the spray voltage increased, different spray modes were observed. It is, therefore, essential to find the optimal voltage range by visual monitoring to ensure a stable spray. In our setup, this range was found to be between 3.0 to 3.5 kV.
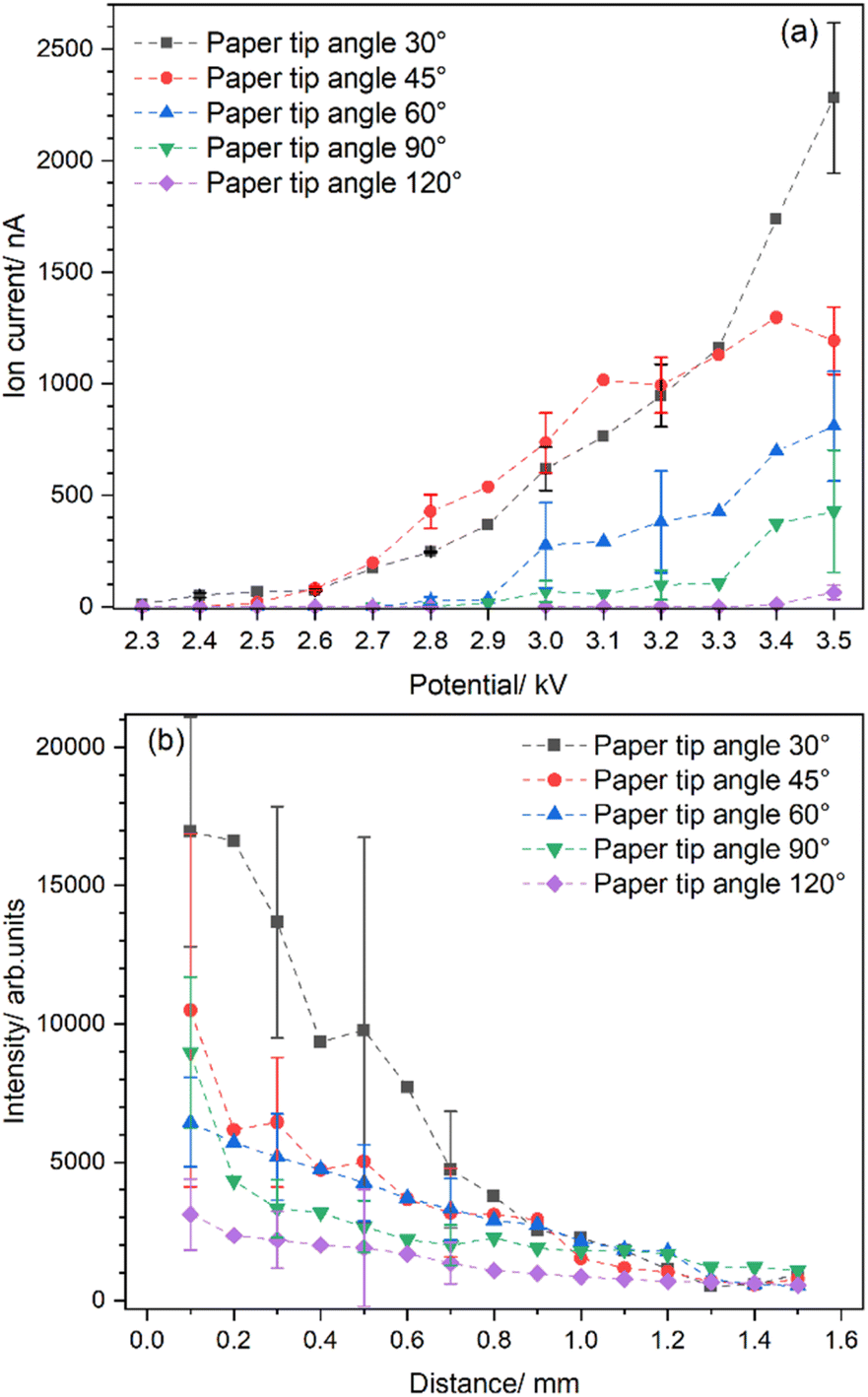 | ||
| Fig. 3 (a) Total ion current measured as a function of spray voltage for NaCl solution (1 mol L−1). (b) The intensity of the Na I (588.995 nm) emission line measured along the spray axis at a laser energy of 82 mJ. | ||
Overall, the measured ion current increases stepwise with rising voltages (Fig. 3a). Notably, sharper tips are also more efficient at producing sprays of charged microdroplets, due to their higher electric field densities. The maximum ion current was detected at paper tip angles of 30° and 45°, with the ion current decreasing as the angle increased beyond these points. The lowest ion currents, with barely any signal, were measured for a tip angle of 120°. This is likely caused by the shifted onset voltage of paper spray, which is usually higher with a larger tip angle. For example, Yang et al. found an onset potential of ≈3 kV for papers with 30° and 60° tips, and ≈4 kV for papers with 90° and 120° tips.49
Other essential factors are the particle and charge densities in the spray plume hit by the laser spot, which directly affects the intensity of the emitted spectral lines. As shown in Fig. 3a, the highest ion current is measured for the paper with a tip of 30° or 45° angle. However, the spray plume is getting wider as the distance from the paper tip increases, owing to the diverging electric field lines near the tip and the coulombic repulsion of the ions in the spray.59 Besides particle density, the droplet size distribution also decreases along the spray axis due to evaporation and Coulomb explosions of the charged microdroplets.50 A higher charge density indicates more analyte ions in the droplet, which should positively impact the LIBS intensity. On the other hand, the droplet quickly reaches the Rayleigh limit (i.e., the maximum charge a droplet can hold); at this point, electrostatic forces start to dominate, causing the droplet to disintegrate.
Fig. 3b shows that the highest signal intensity is observed when the laser is aimed almost at the tip, at a distance of 0.1 mm. At this point, the particle and charge densities are the highest, which can be seen in the intensity of the emission. The LIBS intensity from the spray plume generated by a 30° paper tip is by a factor of 1.6 stronger than that from a 45° tip, and more than five times stronger than that from a 120° tip. This difference is due to the amount of charged particles in the spray plume, as determined by the ion current measurements. As the distance between the paper tip and the laser spot increases, the intensity of the LIBS signal quickly drops. For example, with a paper tip angle of 30°, the LIBS intensity is reduced by half when the laser interacts with the spray at a distance of 0.6 mm from the tip, compared to 0.1 mm. Distances greater than 1 mm are characterized by low signal intensity and significant variation, regardless of the apex shape.
The sensitivity of LIBS in elemental analysis is further enhanced by the intensity of the laser pulse. Higher energy pulses generate more intense and stable plasma with a longer lifetime, enhancing both the qualitative analysis of elements and the overall sensitivity of LIBS. However, excessively high laser energy can elevate background signals, thereby reducing the signal-to-noise ratio.
Fig. 4 illustrates the intensity of the sodium emission lines as a function of laser energy. In this experiment, a sodium chloride solution was applied to filter paper with a tip angle of 30°. A voltage of 3.5 kV was used to generate a spray plume, and the laser beam was directed at a spot 1 mm from the tip of the paper. At low laser energies, there is almost no signal because of the low temperature of the generated plasma, which leads to its shorter lifetime. As the laser energy increases, the LIBS signal is significantly amplified, and the intensity gradually rises with the laser energy. The highest signal was observed at a laser power of 142 mJ, approximately double the signal measured at 130 mJ. However, the signals at high laser energies fluctuate significantly, resulting in large error bars. These error bars are based on 500 sets of spectra collected and analyzed for each energy level. High laser powers can generate background radiation, which affects the signal-to-noise ratio as surrounding material can also be excited.60–62 These fluctuations can also be explained by variations in the density of charged particles in the spray plume. This means that the number of charged particles in the laser focus varies, leading to strong fluctuations independent of the laser energy, as observed in our experiment.
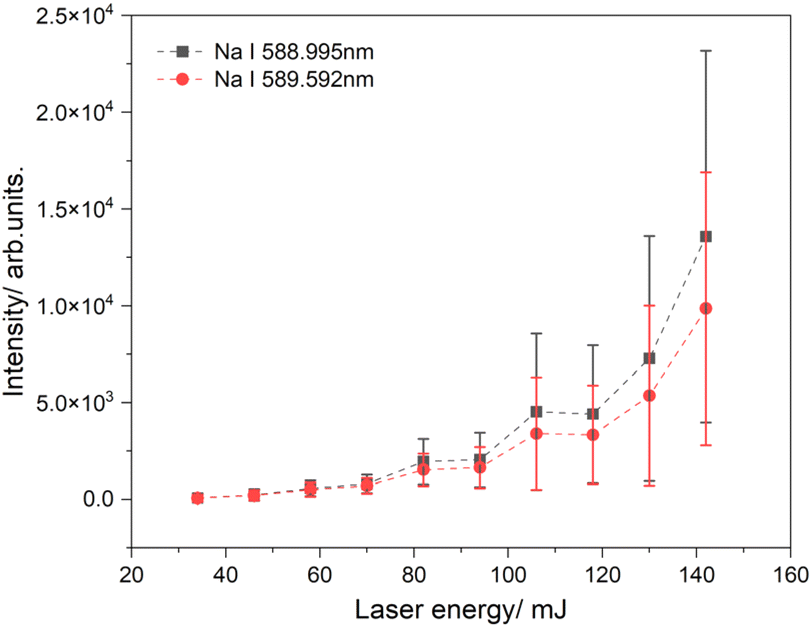 | ||
| Fig. 4 LIBS signal of Na I emission lines as a function of laser energy. | ||
Quantitative analysis
To evaluate the analytical capabilities of the PS-LIBS system, the limit of detection (LOD) for various analytes in model solutions was determined. This involved identifying the limit concentration of the analyte that can be safely distinguished by using this technique. Three metals were selected for this study: sodium (Na), potassium (K), and chromium (Cr), due to the extensive literature available on their LIBS analysis. Sodium was analyzed using a solution of sodium chloride, while potassium and chromium were analyzed in parallel using a solution of potassium dichromate. The spectral lines used for the analysis were: Na I 588.995 nm, Cr I 520.84 nm, and K I 766.49 nm. Spraying the NaCl solution should, in theory, allow for the detection of chlorine along with sodium. However, we did not observe any chlorine emission lines in our spectra. As shown in our previous measurements, chlorine, like other non-metallic elements such as sulfur, is particularly challenging to detect using LIBS due to its high excitation energy.63 For example, Bolshakov et al. reported limits of detection for sodium, potassium, and chromium in aqueous solutions under a rare gas atmosphere as 1.6, 11, and 5.8 mg kg−1, respectively, whereas the LOD for chlorine was orders of magnitude higher, at 5000 mg kg−1.64Calibration curves were plotted with the LIBS signal as a function of element concentration in the solution, and the experimental data were fitted using the least squares method,51 as shown in Fig. 5. The experimental conditions were kept constant for all measurements to prevent additional factors that might affect signal intensities. However, some deviations in the spectrum were observed. For example, as the sodium concentration increased, a slight discrepancy between the measured results and theoretical values appeared due to sodium self-absorption, which takes place at higher concentrations.17 The calibration curves in all three scenarios exhibit a strong linear dependence, with regression coefficients ranging from 0.96 to 0.98.
 | ||
| Fig. 5 Calibration curves illustrating the intensities of the emission lines for (a) sodium, (b) potassium, and (c) chromium as a function of analyte concentration. | ||
The limit of detection (LOD) can be determined using the formula LOD = 3σ/b, where σ represents the standard deviation of the LIBS signal from a sample with zero concentration of the analyte, and b is the slope of the calibration curve. The standard deviation of the background radiation, as measured closely around the LIBS signal for a particular element, served as the value for σ. For sodium, the LIBS signal was collected in the range of 584 to 587 nm. For chromium and potassium, the LIBS signal areas selected were 516–519 nm and 762–765 nm, respectively. By this method, LOD values for the PS-LIBS experiment were determined to be 11.1 mg L−1 for sodium, 28.9 mg L−1 for potassium, and 67.9 mg L−1 for chromium.
Table 1 compares the LOD measured by PS with other techniques based on liquid jets and sprayed droplets. Overall, PS shows slightly higher LOD than these techniques. On the other hand, even with the same approach, LOD can vary significantly, as demonstrated for liquid jets.17,65 For chromium, our experiments with paper produce LODs that are twice as high as those obtained with liquid jets and capillary measurements.17 For sodium, the difference ranges from a factor of 5 to 10, depending on the method used. This is primarily due to the differing number densities of analytes in each approach. In contrast to liquid jets and capillary measurements that essentially analyze bulk water, PS produces a plume of individual charged particles with far lower analyte number densities. However, spraying techniques can achieve LODs as low as around 1 ppm, as demonstrated in ES experiments.41
In addition to the LOD, practical considerations must also be taken into account to assess its suitability for liquid analysis in portable setups. For example, Zhang et al.17 recorded snapshots of laser pulses interacting with liquids and compared the images from the analysis of the liquid surface, the liquid jet, and a sample flowing through the capillary. They reported that, on the one hand, analysis in the closed capillary is more suitable than in the liquid jet, as the laser disrupts the jet and causes splashing, which can contaminate nearby optics. On the other hand, capillary experiments pose challenges, as precise alignment of the laser is crucial to prevent interaction with the capillary walls, which could lead to false positives due to ablation of the wall material. Hence, spraying liquids at rates of a few mL min−1 can be considered a feasible approach.
The primary advantage of PS over other spraying techniques is its simplicity. It is evident that even simple filter papers can lead to results comparable to those achieved with ES. Furthermore, handling filter paper is much easier than manipulating fragile ES emitters. This also applies to sampling aqueous solutions. For example, PS does not require a syringe pump to continuously introduce samples or to optimize the flow rate for stable spray formation. To date, PS has primarily demonstrated its potential in mass spectrometry, showing remarkable capability in the analysis of heterogeneous samples.40,48 It may enable the analysis of species separated on paper due to paper chromatography.66 It has now advanced to a stage that allows quantitative analysis with disposable sample cartridges, making it a promising option for in situ and online applications.47
The potential of the PS-LIBS technique to identify these elements can be further demonstrated through the test analysis of a seawater sample (for details, see the Experimental section). The LIBS spectrum of the seawater sample, shown in Fig. 6, reveals intense and well-resolved spectral lines for Na, Mg, Ca, and K, which largely correspond to the most abundant cations typically found in seawater. These metal elements are often reported in other LIBS studies focused on seawater analysis.67,68 The absence of Pb emission lines is likely due to a higher LOD. Likewise, other metals such as Cr, which were detected by ICP-OES at levels below 10 ppm, were not observed in the LIBS spectrum.
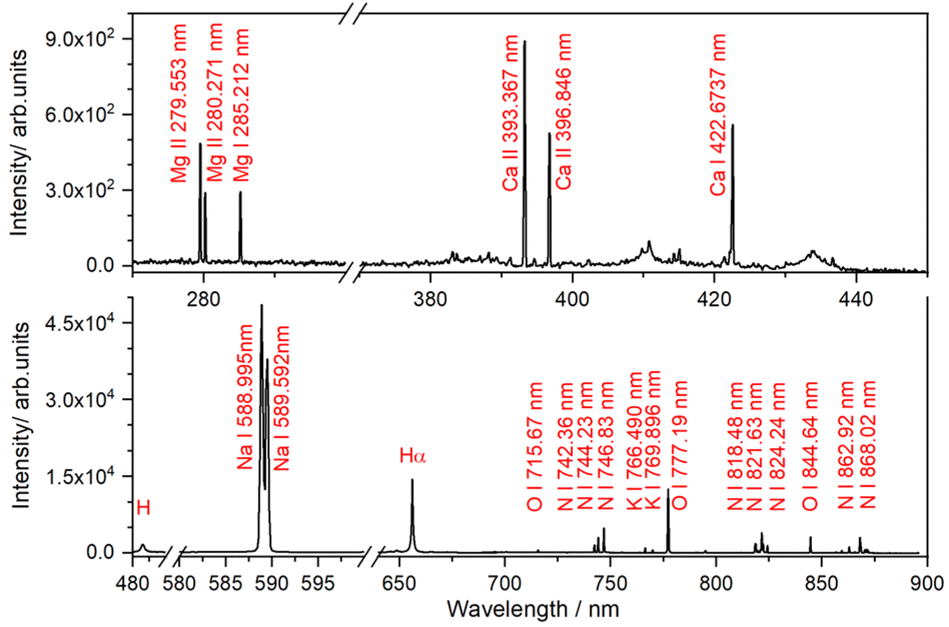 | ||
| Fig. 6 LIBS spectrum of the seawater sample. | ||
Overall, PS reduces the experimental challenges associated with liquid jet or spray techniques, simplifying the complexities of ES techniques while delivering comparable results. On the other hand, the key question remains the analytical capability of LIBS for quantitative analysis. While PS and other ES techniques can achieve detection limits at the ppm level, their sensitivity may not always be sufficient for monitoring contaminants in environmental samples.
Conclusions
The findings from this study demonstrate that integrating paper spray with the LIBS technique is an effective method for analyzing trace elements in liquids, particularly in detecting metals in water samples at concentrations as low as parts per million. However, the capability of this technique heavily relies on the quality of the spray, which can be improved by several factors. The most critical factor is the shape of the paper and, in particular, the tip angle, which determines the onset of spray formation—i.e., the minimum voltage needed for a stable spray. It can also enhance the number of charged particles present in the spray. Other factors to consider include the spray voltage, which impacts the spray mode and its stability, and the composition of the solvent. Mixing water samples with organic solvents lowers the surface tension, enabling spray formation at reduced voltages compared to pure water. A low voltage will prevent discharges and extend the lifespan of the paper substrate.Indeed, the characteristics of the LIBS signal can be influenced by the laser energy and other beam properties that affect the lifetime of the plasma. However, in PS and other ES techniques, the critical factor is the distance that charged particles must travel from the spray tip to interact with the laser radiation. Particles leaving the tip experience significant spreading due to the diverging electric field lines near the tip and repulsion between ions of the same charge. This spreading reduces the density of charged particles in a laser spot as the distance from the paper tip increases. Thus, fine-tuning the laser focus close to the paper tip in the spray solution is necessary.
The PS-LIBS combination produced strong emission lines for Na, K, and Cr in the model aqueous solution, allowing the determination of detection limits: 11.1 mg L−1 for Na, 28.9 mg L−1 for K, and 67.9 mg L−1 for Cr. This is comparable to the well-established approach employing electrospray with LIBS detection,41,43 while also being much more user-friendly with the potential for online measurements. The next step is to analyze heterogeneous samples.
Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.Author contributions
Jun Feng: investigation, formal analysis, visualization, writing – original draft. Yihui Yan: conceptualization, investigation, formal analysis, visualization, writing – review and editing. Yuzhu Liu: methodology, resources, writing – review and editing. Jozef Lengyel: methodology, resources, writing – original draft, review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation; project no.: 442679477) and National Natural Science Foundation of China (project no.: 62481540172, 62375136). Y. Y. acknowledges the support provided by the TUM Graduate School.Notes and references
- A. Hyder, J. A. Buledi, M. Nawaz, D. B. Rajpar, Z. U. H. Shah, Y. Orooji, M. L. Yola, H. Karimi-Maleh, H. J. Lin and A. R. Solangi, Environ. Res., 2022, 205, 112475 CrossRef CAS PubMed.
- K. H. H. Aziz, F. S. Mustafa, K. M. Omer, S. Hama, R. F. Hamarawf and K. O. Rahman, RSC Adv., 2023, 13, 17595–17610 RSC.
- J. O. Nriagu, Nature, 1989, 338, 47–49 CrossRef CAS.
- Z. L. L. He, X. E. Yang and P. J. Stoffella, J. Trace Elem. Med. Biol., 2005, 19, 125–140 CrossRef CAS PubMed.
- P. B. Tchounwou, C. G. Yedjou, A. K. Patlolla and D. J. Sutton, in Molecular, Clinical and Environmental Toxicology, ed. A. Luch, Springer, Basel, 2012, vol. 101 Search PubMed.
- P. Joseph, Toxicol. Appl. Pharmacol., 2009, 238, 272–279 CrossRef CAS.
- G. Bjorklund, J. Aaseth, S. Chirumbolo, M. A. Urbina and R. Uddin, Environ. Geochem. Health, 2018, 40, 955–965 CrossRef PubMed.
- Q. Y. Chen, T. DesMarais and M. Costa, Annu. Rev. Pharmacol., 2019, 59, 537–554 CrossRef CAS.
- N. Velitchkova, E. N. Pentcheva and N. Daskalova, Spectrochim. Acta, Part B, 2004, 59, 871–882 CrossRef.
- S. V. Smirnova, T. O. Samarina, D. V. Ilin and I. V. Pletnev, Anal. Chem., 2018, 90, 6323–6331 CrossRef CAS PubMed.
- P. Werheit, C. Fricke-Begemann, M. Gesing and R. Noll, J. Anal. At. Spectrom., 2011, 26, 2166–2174 RSC.
- S. Merk, C. Scholz, S. Florek and D. Mory, Spectrochim. Acta, Part B, 2015, 112, 10–15 CrossRef CAS.
- F. Boué-Bigne, Spectrochim. Acta, Part B, 2016, 119, 25–35 CrossRef.
- Y. J. Dai, C. Song, X. Gao, A. M. Chen, Z. Q. Hao and J. Q. Lin, J. Anal. At. Spectrom., 2021, 36, 1634–1642 RSC.
- M. Lanzinger, D. Huber, V. Merk, S. Kaufmann, M. Schuster and N. Ivleva, Spectrochim. Acta, Part B, 2023, 205, 106691 CrossRef CAS.
- X. D. Yu, Y. Li, X. F. Gu, J. M. Bao, H. Z. Yang and L. Sun, Environ. Monit. Assess., 2014, 186, 8969–8980 CrossRef PubMed.
- D. C. Zhang, Z. Q. Hu, Y. B. Su, B. Hai, X. L. Zhu, J. F. Zhu and X. Ma, Opt. Express, 2018, 26, 18794–18802 CrossRef CAS PubMed.
- X. Lu, Y. Z. Liu, Q. H. Zhang, Y. W. Chen and J. P. Yao, Opt. Laser Technol., 2022, 149, 107826 CrossRef CAS.
- Z. Y. Sun, C. Yu, J. Feng, J. Y. Zhu and Y. Z. Liu, J. Anal. At. Spectrom., 2024, 39, 1212–1224 RSC.
- V. Lazic, S. Jovicevic, R. Fantoni and F. Colao, Spectrochim. Acta, Part B, 2007, 62, 1433–1442 CrossRef.
- K. K. Kim, M. Roy, H. Kwon, J. K. Song and S. M. Park, J. Appl. Phys., 2015, 117, 074302 CrossRef.
- S. L. Zhong, Y. Lu, W. J. Kong, K. Cheng and R. Zheng, Front. Physiol., 2016, 11, 114202 CrossRef.
- B. Y. Xue, Y. You, I. Gornushkin, R. Zheng and J. Riedel, J. Anal. At. Spectrom., 2020, 35, 2901–2911 RSC.
- D. M. D. Pace, C. A. D'Angelo, D. Bertuccelli and G. Bertuccelli, Spectrochim. Acta, Part B, 2006, 61, 929–933 CrossRef.
- Z. J. Chen, H. K. Li, M. Liu and R. H. Li, Spectrochim. Acta, Part B, 2008, 63, 64–68 CrossRef.
- X. Wang, L. L. Shi, Q. Y. Lin, X. Q. Zhu and Y. X. Duan, J. Anal. At. Spectrom., 2014, 29, 1098–1104 RSC.
- N. Aras and S. Yalçin, Talanta, 2016, 149, 53–61 CrossRef CAS.
- L. Ripoll and M. Hidalgo, J. Anal. At. Spectrom., 2019, 34, 2016–2026 RSC.
- J. O. Cáceres, J. T. López, H. H. Telle and A. G. Ureña, Spectrochim. Acta, Part B, 2001, 56, 831–838 CrossRef.
- H. Sobral, R. Sanginés and A. Trujillo-Vázquez, Spectrochim. Acta, Part B, 2012, 78, 62–66 CrossRef CAS.
- S. Ünal and S. Yalçin, Spectrochim. Acta, Part B, 2010, 65, 750–757 CrossRef.
- J. B. Simeonsson and L. J. Williamson, Spectrochim. Acta, Part B, 2011, 66, 754–760 CrossRef CAS.
- S. Ü. Yesiller and S. Yalçin, Anal. Chim. Acta, 2013, 770, 7–17 CrossRef PubMed.
- M. A. Aguirre, S. Legnaioli, F. Almodóvar, M. Hidalgo, V. Palleschi and A. Canals, Spectrochim. Acta, Part B, 2013, 79–80, 88–93 CrossRef CAS.
- Q. Shi, G. H. Niu, Q. Y. Lin, X. Wang, J. Wang, F. Bian and Y. X. Duan, J. Anal. At. Spectrom., 2014, 29, 2302–2308 RSC.
- M. A. Aguirre, H. Nikolova, M. Hidalgo and A. Canals, Anal. Methods, 2015, 7, 877–883 RSC.
- M. A. Aguirre, E. J. Selva, M. Hidalgo and A. Canals, Talanta, 2015, 131, 348–353 CrossRef CAS PubMed.
- C. Haisch, J. Liermann, U. Panne and R. Niessner, Anal. Chim. Acta, 1997, 346, 23–35 CrossRef CAS.
- A. Kumar, F. Y. Yueh and J. P. Singh, Appl. Opt., 2003, 42, 6047–6051 CrossRef PubMed.
- Y. Feng, J. J. Yang, J. M. Fan, G. X. Yao, X. H. Ji, X. Y. Zhang, X. F. Zheng and Z. F. Cui, Appl. Opt., 2010, 49, C70–C74 CrossRef CAS.
- J. S. Huang, C. B. Ke, L. S. Huang and K. C. Lin, Spectrochim. Acta, Part B, 2002, 57, 35–48 CrossRef.
- N. Aras, S. Ü. Yesiller, D. A. Ates and S. Yalçin, Spectrochim. Acta, Part B, 2012, 74–75, 87–94 CrossRef CAS.
- J. S. Huang and K. C. Lin, J. Anal. At. Spectrom., 2005, 20, 53–59 RSC.
- L. J. Radziemski, T. R. Loree, D. A. Cremers and N. M. Hoffman, Anal. Chem., 1983, 55, 1246–1252 CrossRef CAS.
- Y. Yan, L. Schmitt, A. Khramchenkova and J. Lengyel, J. Mass Spectrom., 2023, 58, e4955 CrossRef CAS.
- J. H. Gross, Mass Spectrometry: A Textbook, Springer, Cham, 2017 Search PubMed.
- H. Wang, J. J. Liu, R. G. Cooks and Z. Ouyang, Angew. Chem., Int. Ed., 2010, 49, 877–880 CrossRef CAS PubMed.
- Z. P. Zhang, R. G. Cooks and Z. Ouyang, Analyst, 2012, 137, 2556–2558 RSC.
- Q. Yang, H. Wang, J. D. Maas, W. J. Chappell, N. E. Manicke, R. G. Cooks and Z. Ouyang, Int. J. Mass Spectrom., 2012, 312, 201–207 CrossRef CAS PubMed.
- R. D. Espy, A. R. Muliadi, Z. Ouyang and R. G. Cooks, Int. J. Mass Spectrom., 2012, 325, 167–171 CrossRef.
- Q. H. Zhang, Y. Chen and Y. Z. Liu, J. Anal. At. Spectrom., 2021, 36, 1028–1033 RSC.
- Z. Y. Zhou, Y. F. Ge, X. Y. Zhang, Y. P. Ye, M. L. Yang, Z. M. Sun and Y. Z. Liu, Spectrochim. Acta, Part B, 2024, 220, 107018 CrossRef CAS.
- Y. Z. Zhangcheng, Y. Z. Liu, Q. H. Zhang, Y. Chen, S. Saleem, Z. M. Zhuo and L. Li, Opt Laser. Eng., 2021, 142, 106586 CrossRef.
- Q. Zhang, Y. Liu, Y. Chen, Y. Zhangcheng, Z. Zhuo and L. Li, Opt. Express, 2020, 28, 22844–22855 CrossRef CAS PubMed.
- L. C. L. Borduchi, D. M. B. P. Milori and P. R. Villas-Boas, Spectrochim. Acta, Part B, 2022, 191, 106409 CrossRef CAS.
- C. Gong, H. Lu, K. Zhang, Y. Ding and L. Wang, Spectroscopy, 2024, 39, 6–17 Search PubMed.
- M. G. Ikonomou, A. T. Blades and P. Kebarle, J. Am. Soc. Mass Spectrom., 1991, 2, 497–505 CrossRef CAS PubMed.
- R. Juraschek, T. Dülcks and M. Karas, J. Am. Soc. Mass Spectrom., 1999, 10, 300–308 CrossRef CAS PubMed.
- M. Busman, J. Sunner and C. R. Vogel, J. Am. Soc. Mass Spectrom., 1991, 2, 1–10 CrossRef CAS PubMed.
- P. A. Benedetti, G. Cristoforetti, S. Legnaioli, V. Palleschi, L. Pardini, A. Salvetti and E. Tognoni, Spectrochim. Acta, Part B, 2005, 60, 1392–1401 CrossRef.
- E. Tognoni, G. Cristoforetti, S. Legnaioli and V. Palleschi, Spectrochim. Acta, Part B, 2010, 65, 1–14 CrossRef.
- D. W. Hahn and N. Omenetto, Appl. Spectrosc., 2012, 66, 347–419 CrossRef CAS PubMed.
- Q. H. Zhang, Y. Z. Liu, Y. Chen, Y. Z. Zhangcheng, Z. M. Zhuo and L. Li, Opt. Express, 2020, 28, 22844–22855 CrossRef CAS PubMed.
- A. A. Bol'shakov, S. J. Pandey, X. L. Mao and C. Y. Lou, Spectrochim. Acta, Part B, 2021, 179, 106094 CrossRef.
- N. H. Cheung and E. S. Yeung, Appl. Spectrosc., 1993, 47, 882–886 CrossRef CAS.
- J. J. Liu, H. Wang, N. E. Manicke, J. M. Lin, R. G. Cooks and Z. Ouyang, Anal. Chem., 2010, 82, 2463–2471 CrossRef CAS PubMed.
- H. M. Hou, Y. Tian, Y. Li and R. Zheng, J. Anal. At. Spectrom., 2014, 29, 169–175 RSC.
- J. J. Song, N. Li, Y. Tian, J. J. Guo and R. E. Zheng, J. Anal. At. Spectrom., 2020, 35, 2351–2357 RSC.
Footnote |
| † Same contribution as the first author. |
| This journal is © The Royal Society of Chemistry 2025 |