
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Real-time and regional analysis of the efficacy of anticancer drugs in a patient-derived intratumoral heterogeneous tumor microenvironment†
Ya-Hui
Lin‡
a,
Chiao-Min
Lin‡
a,
Kee-Ming
Man‡
 bcd,
Chih-Chiang
Hung
e,
Hsin-Ling
Hsu
f,
Yunching
Chen
g,
Hsuan-Yu
Mu
*a,
Tzu-Hung
Hsiao
*hij and
Jen-Huang
Huang
*a
bcd,
Chih-Chiang
Hung
e,
Hsin-Ling
Hsu
f,
Yunching
Chen
g,
Hsuan-Yu
Mu
*a,
Tzu-Hung
Hsiao
*hij and
Jen-Huang
Huang
*a
aDepartment of Chemical Engineering, National Tsing Hua University, Hsinchu 30013, Taiwan. E-mail: slevis185@gmail.com; jenhuang@mx.nthu.edu.tw
bDepartment of Anesthesiology, China Medical University Hsinchu Hospital, Hsinchu 30272, Taiwan
cDepartment of Life Sciences, National Chung Hsing University, Taichung 40227, Taiwan
dDepartment of Medicinal Botanicals and Health Applications, Da Yeh University, Changhua 51591, Taiwan
eDepartment of Surgery, Taichung Veterans General Hospital, Taichung 40705, Taiwan
fInstitute of Molecular and Genomic Medicine, National Health Research Institutes, Miaoli 35053, Taiwan
gInstitute of Biomedical Engineering, National Tsing Hua University, Hsinchu 30013, Taiwan
hDepartment of Medical Research, Taichung Veterans General Hospital, Taichung 40705, Taiwan. E-mail: thsiao@vghtc.gov.tw
iDepartment of Public Health, Fu Jen Catholic University, New Taipei 24205, Taiwan
jInstitute of Genomics and Bioinformatics, National Chung Hsing University, Taichung 40227, Taiwan
First published on 14th February 2025
Abstract
Preclinical evaluation of anticancer drug efficacy utilizes 2D cell culture systems, tumoroids or experimental animal models, but it suffers from limitations such as inaccurate simulation of tumor microenvironments in living tumors, difficulty in regional analysis, and low throughput. Therefore, in this study, we developed a system named tumor-microenvironment-on-chip (TMoC) comprising a 3D dynamic tumor tissue culture system, which recreated diverse and heterogeneous cellular tumor microenvironments. In addition to the culture with a dynamic circulation, TMoC allowed users to perform real-time regional analysis, independently assessing the drug response from the normoxic area to the hypoxic area in a gradient manner. Through cell composition analysis and gene analysis, we proved that TMoC has a tumor environment with close resemblance to the original tumor environment. By comparing 15 drug testing results with animal experiments, we proved that TMoC is 93% consistent with the response results of animal experiments. In addition, we confirmed that either mouse- or patient-derived tumor cell lines can be cultured and tested in TMoC, indicating its immense potential for all aspects of preclinical drug evaluation.
1. Introduction
After candidate drugs pass rigorous ex vivo screening and animal-based in vivo tests, they enter clinical trials, in which about 90% of them fail.1 In the field of anti-cancer medicine, accurately replicating the in vivo microenvironment is crucial for precise drug evaluation.2 The current widely adopted two-dimensional (2D) cell culture systems, three-dimensional (3D) tumoroids derived from cell lines or patients, and animal models, which are commonly used to evaluate the efficacy of therapies, fail to reproduce the complex tumor physiology or evaluate regional and spatial drug response in a tumor microenvironment (TME).3 Intratumoral heterogeneity (ITH) arises from intrinsic factors, such as genetic, transcriptional and protein level variations, as well as external factors like hypoxia, pH and cell–cell crosstalk within the TME.4 These complex interactions constantly alter the TME, providing tumors with dynamic spatial and temporal properties. Ignoring drug response variability due to ITH leads to over 80% of cancer drugs failing in phases 2 and 3 of clinical trials, primarily due to insufficient efficacy.5New therapy development requires the use of clinically relevant patient-derived xenograft (PDX) models to evaluate drugs, but the global trend of immunotherapy requires humanized mice or transplantation of PBMCs derived from healthy adults.6 Both PDX and humanized mouse models suffer from low throughput and high time consumption, which make them unsuitable for early drug development.7 Additionally, in recent years, driven by ethical trends and the FDA Modernization Act 2.0, animal reduction technology has flourished, with one prominent approach being organoid culture technology.8 Therefore, patient-derived tumoroids are significantly used in early drug development, and they provide data that are more relevant to clinical patient responses than 2D cell cultures. For the development of immunotherapy, many contract research organizations (CROs), and academic research institutions provide in vitro co-cultured tumoroids and PBMCs from healthy donors or paired patients for analyzing the effectiveness of the drug candidates, which provide good accuracy.9
Tumoroid culture involves seeding a mixture of hydrogels and cells onto a well plate, allowing the cells to aggregate into spherical structures. However, this method suffers from several challenges: (i) lack of dynamic circulation system. Tumoroid models are often cultured in static dishes, failing to represent the dynamic relationship between circulation and tumors.10 While rocker equipment can establish a medium flow, it provides less precision in assessing drug or immune cell infiltration. (ii) Difficulty in real-time analysis of drug response. Drug responses in tumors can change dynamically over time, such as through acquired resistance or immune cell chemotaxis.11 Even with a thorough initial tumor analysis, predicting real-time drug responses is difficult, highlighting the importance of continuous analysis methods. Continuous analysis of 3D tissues using a microscope is challenging because the dense tissue blocks the detection of core signals.12 Consequently, data can only be obtained on size changes of the tumoroid, ignoring the effects of the TME, such as drug resistance in hypoxic zones and their significant impact on drugs and immune cells.13 (iii) Difficulty in analyzing drug response in heterogeneous environments. Tumor responses to high environmental heterogeneity also need to be assessed. Independent analysis of IC50 and other parameters under various environmental conditions such as hypoxia, acidity, or desmoplasia can help in drug evaluation and further optimization of lead candidates.14
Recent advancements in organ-on-a-chip technology have enabled the miniaturization of tissues within microfluidic chips, allowing for the recapitulation of specific physiological functions relevant to disease modeling, drug development, and the replacement of animal models.15,16 We successfully employed this technology to identify the synergistic effects of chemo-immuno combination therapies, assessing five immunotherapy-enhancing drugs using a physiologically relevant tumor-microenvironment-on-chip (TMoC).17 Here we adapted this approach and integrated real-time visualization and ITH analysis, which enabled us to confirm the efficacy of chemotherapy drugs in real time across various tumor samples including those derived from cell lines, mouse-derived tumors, and patient-derived tumors.
2. Result
2.1. Design principle and operation of TMoC
The most used drug evaluation models are 2D cell cultures, 3D tumoroid cultures, and animal models. We aimed to compare these models with TMoC in terms of differences in gene expression, apoptosis induction, and physiological gradients including molecular diffusion and hypoxia. Through these comparisons, we seek to understand the advantages of TMoC in drug research compared to existing models (Fig. S1†). TMoC is designed with a dynamic culture medium circulation system, allowing precise control over the flow rate to regulate the infiltration of oxygen and drugs, thus accurately simulating the challenges of drug and immune cell infiltration into tumors (Fig. S2†). Its elongated culture area (2 cm) supports oxygen diffusion to mimic hypoxic conditions, while the thin 250 μm culture layer enables real-time microscopic observation of cancer cell apoptosis and tumor heterogeneity (Fig. 1A). The elongated tissue culture area further facilitates continuous analysis of tumor heterogeneity. To reconstruct the tumor microenvironment, we collected tumor tissues from mice or patients, dissociated them into single cells, and combined them with a tumor matrix. After implanting the sample into TMoC, we connected a pump to initiate the culture medium circulation. This setup allows for the introduction of drugs and immune cells into the circulation, simulating the process of infiltration from the bloodstream into the tumor. Real-time microscopic analysis was conducted thereafter to evaluate the functional characterization and responses of cells. The whole screening process can be finished within 72 h to realize the rapid drug evaluation process compared to other models (Fig. 1B).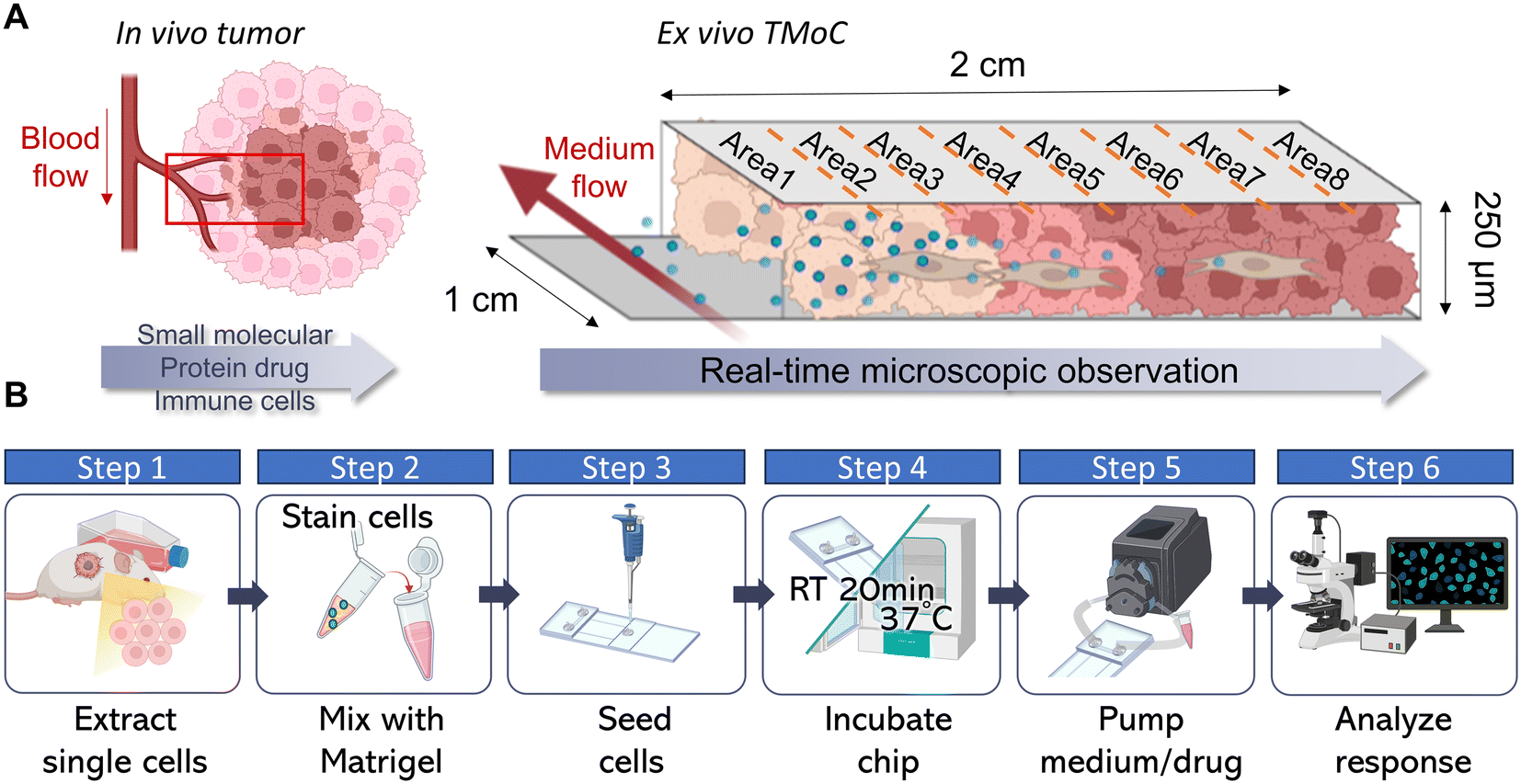 | ||
| Fig. 1 Drug evaluation using TMoC. (A) Tumors in vivo often exhibit molecular gradients owing to the rapid growth of cancer cells outpacing the formation of blood vessels penetrating deep into the tumor. This results in peripheral tumor tissues near blood vessels having higher concentrations of oxygen, drugs, and other molecules. TMoC utilizes an elongated-strip design to generate molecular gradients similar to those in vivo. The color changes of cancer cells on the chip demonstrate hypoxic cell characteristics, while the blue particles represent drugs delivered via the circulation system, showcasing the gradient properties within the chip. Further, the thin-layer design of TMoC allows users to easily observe drug responses in different areas using an inverted microscope. In subsequent experimental designs, the chip is divided into 8 areas; area 1 represents areas near the circulation system (oxygenated region), while area 8 represents the opposite. Additionally, the cultivation area on the chip is designed with dimensions of length × width × height = 2 cm × 1 cm × 250 μm. (B) Operation process of TMoC, including obtaining the cell line or tumor-derived cells, mixing them with the matrix, implanting into the TMoC, and waiting for 20 min at room temperature. Finally, TMoC is connected to the pump for drug analysis. | ||
2.2. Comparison of 2D, tumoroid and TMoC
We initially cultured 4T1 mouse breast cancer cells using three different methods: 2D, tumoroid, and TMoC. Live and dead assays indicated that the cells cultured on TMoC in area 1 and 8 maintained high viability for at least 72 hours (Fig. S3A†). We further monitored cell growth over a 72 hour period and analyzed RNA expression at the end of this period. KEGG analysis revealed that 4T1 cells cultured on TMoC showed a significant up-regulation of hypoxia-related genes (HIF-1 and glycolysis), while both TMoC and conventional tumoroid cultures showed significant down-regulation of cell proliferation and DNA replication mechanisms. This inhibition of replication rate is the key distinction between 2D and 3D cultures. These findings are consistent with growth status results, confirming that 4T1 cells cultured in conventional tumoroid or TMoC represent a 3D environment (Fig. S3B and C†).We further compared the differences between conventional tumoroid and TMoC. In the conventional tumoroid model, experimental results were limited to a uniform environment without distinct regional outcomes for molecular diffusion or drug efficacy (Fig. 2A–C). In contrast, TMoC allowed for the adjustment of flow rates to create different gradients of molecules such as oxygen, small molecule (e.g. fluorescein), and protein (e.g. green fluorescent protein, GFP), thereby simulating the challenges of molecular infiltration in the tumors. These molecules were selected to simulate the diffusion behavior of different drug molecules within the chip. Fluorescein, with a molecular weight of 343 Da, and GFP, with a molecular weight of 27 kDa, respectively represent small-molecule drugs (e.g., chemotherapy drugs) and large-molecule drugs (e.g., cytokines). Due to the dynamic culture system, increased convection flow in the interstitial fluid led to the diffusion of oxygen, small molecules, and proteins into deeper tumor regions (area 8 being the core area farthest from circulation), as indicated by hypoxia probes and fluorescence signals (Fig. 2D and E). In TMoC with a dynamic flow, the fluorescence intensity of the hypoxia probe demonstrated a clear gradient, whereas no significant differences were observed under static conditions. Additionally, the diffusion of small molecules in the dynamic flow model showed a concentration gradient more consistent with that reported in the literature.18
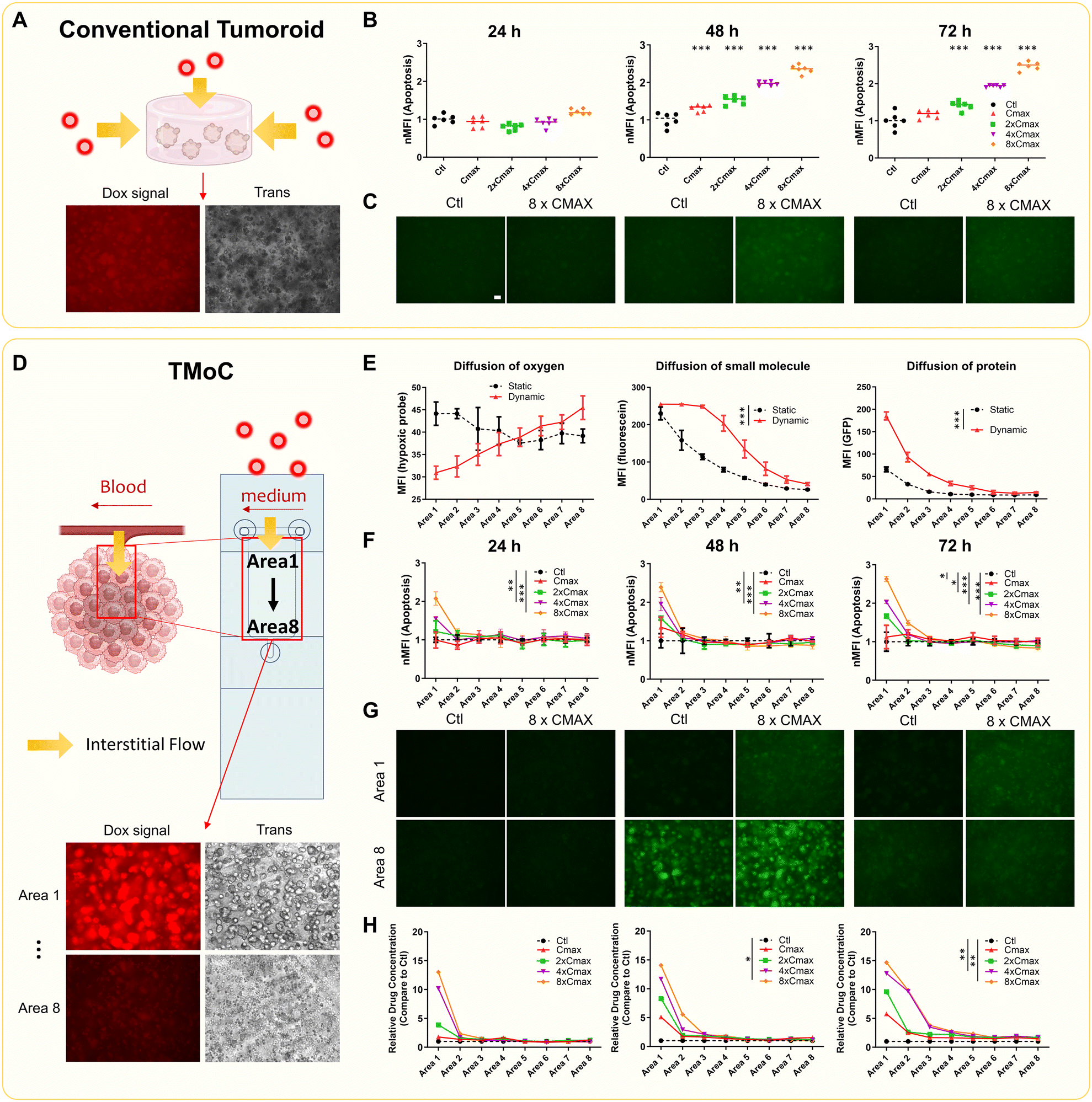 | ||
| Fig. 2 Comparison of drug treatment results in different models. (A) Schematic of drug molecule diffusion in the conventional tumoroids (using doxorubicin as an example). Red dots represent doxorubicin, and yellow arrows indicate its diffusion paths. The cells are suspended in a medium mixed with Matrigel to form the tumoroid, with the culture medium and molecules diffusing from all directions into the gel matrix, interacting with the tumoroid. The fluorescence image below shows doxorubicin signals in the tumoroid. (B) Tumoroids were treated with varying concentrations of doxorubicin. Apoptosis signals at 24, 48, and 72 h were normalized to control. MFI: mean fluorescence intensity. (C) Fluorescence images of tumoroids show increasing apoptosis signals over time and with higher doxorubicin concentrations. Scale = 100 μm. (D) Schematic of drug molecule diffusion in TMoC. Cells are suspended in a medium mixed with Matrigel and injected into the culture areas on the chip, forming a hydrogel layer with a thickness of only 200 μm to facilitate observation. Medium flow in TMoC system is designed to simulate the characteristics of blood flow. The culture medium and molecules diffuse from one side into the culture area, creating a physiological gradient. The fluorescence images below show doxorubicin signals from area 1 to area 8 in TMoC. (E) The small molecule fluorescein or the large-molecule GFP was added to the circulation system. Fluorescence signal intensity of fluorescein, GFP or hypoxia probes in different areas was analyzed after 24 h. Area 1 represents the region closest to the circulation, while area 8 represents the farthest region. (F) TMoC was treated with varying concentrations of doxorubicin, and apoptosis signals at 24, 48, and 72 h were normalized to control. (G) Fluorescence images of TMoC from area 1 show increasing apoptosis signals over time and with a higher doxorubicin concentration. In contrast, apoptosis in area 8 at high doxorubicin concentrations showed no significant difference compared to the control. Scale = 100 μm. Complete image data are provided in Fig. S4.† (H) TMoC was treated with varying concentration of doxorubicin, and doxorubicin signals at 24, 48, and 72 h were normalized to the control. Images from the conventional and TMoC models were adjusted for brightness and contrast equally. Statistical significance compared to the control group is denoted by *, with p-values less than 0.05, 0.01, and 0.001 indicated by one, two, and three stars, respectively. Cmax in this figure indicates maximum clinical serum dose. nMFI on the (B) and (F) represents the normalized mean fluorescence intensity. | ||
We also evaluated the effect of doxorubicin by treating 4T1 tumoroids and 4T1-derived TMoC with multiples of the maximum clinical serum dose (Cmax). The drug dosage was chosen to correspond to the actual serum concentrations observed in clinical patients, enabling a more accurate evaluation of the relationship between drug efficacy and clinical response. The 4T1 cells were pre-stained with a caspase 3/7 indicator to assess apoptosis induced by the drug. The analysis of apoptosis signals indicated that no significant apoptosis occurred at 24 h for any dose, but apoptosis signals appeared at 48 and 72 h, correlating positively with the dose (Fig. 2B and C). Notably, TMoC results showed dose-dependent apoptosis in areas 1 and 2 after 24 h due to dynamic circulation-derived convection. However, despite the presence of the doxorubicin signal in areas 6, 7, and 8, no significant apoptosis was observed in these regions at 48 and 72 h. Previous data indicated lower oxygen concentrations in areas 6, 7, and 8, affecting downstream drug resistance mechanisms and leading to drug resistance (Fig. 2F–H and S4†). This finding aligns with descriptions in other studies, where chemotherapy drugs face difficulty diffusing into tumor regions farther from the circulation system, resulting in a higher proliferation rate in these cancer tissues.19 These results demonstrate that TMoC differs from traditional tumoroids by enabling continuous analysis of regions with varying heterogeneous environments.
2.3. Comparison of in vivo tumor and TMoC models
Compared to genetically stable cell lines, the primary challenge of culturing patient-derived tumor tissues lies in the dynamic changes in tissue state after cultivation, making it difficult to accurately predict the original tumor's drug response. To address this, we transplanted the freshly obtained tumor tissues into TMoC and compared them with the original tumor in terms of the cell population, transcriptomic profile, and genomic profile using flow cytometry, bulk RNA sequencing (RNA-seq), and whole exome sequencing (WES), respectively. To determine whether culturing in TMoC or tumoroid formats alters the cell population, primary tumor tissue resected from mice was dissociated into single cells and subjected to flow cytometry analysis or cultured in TMoC and tumoroid conditions. After a 2-day culture period in either TMoC system or as tumoroids, flow cytometry was conducted again to characterize the tissues retrieved from TMoC and tumoroid cultures. In the triple-negative breast cancer (TNBC) model, compared to the significant depletion of certain immune cells (macrophages, CD8+ T cells, and CD4+ T cells) observed in the tumoroid culture system, the TMoC culture demonstrated a relatively mild decrease in these immune cells (Fig. 3A). For instance, 343 CD8+ T cells were counted in the primary tumor, while 262 and 191 cells were counted after TMoC and tumoroid culture, respectively. Moreover, the tumoroid culture significantly increased the population of cancer cells, whereas TNBC-derived TMoC was able to preserve the number of cancer cells. This ability of TMoC to maintain the cell population was also confirmed in the colorectal cancer (CRC) model (Fig. 3B).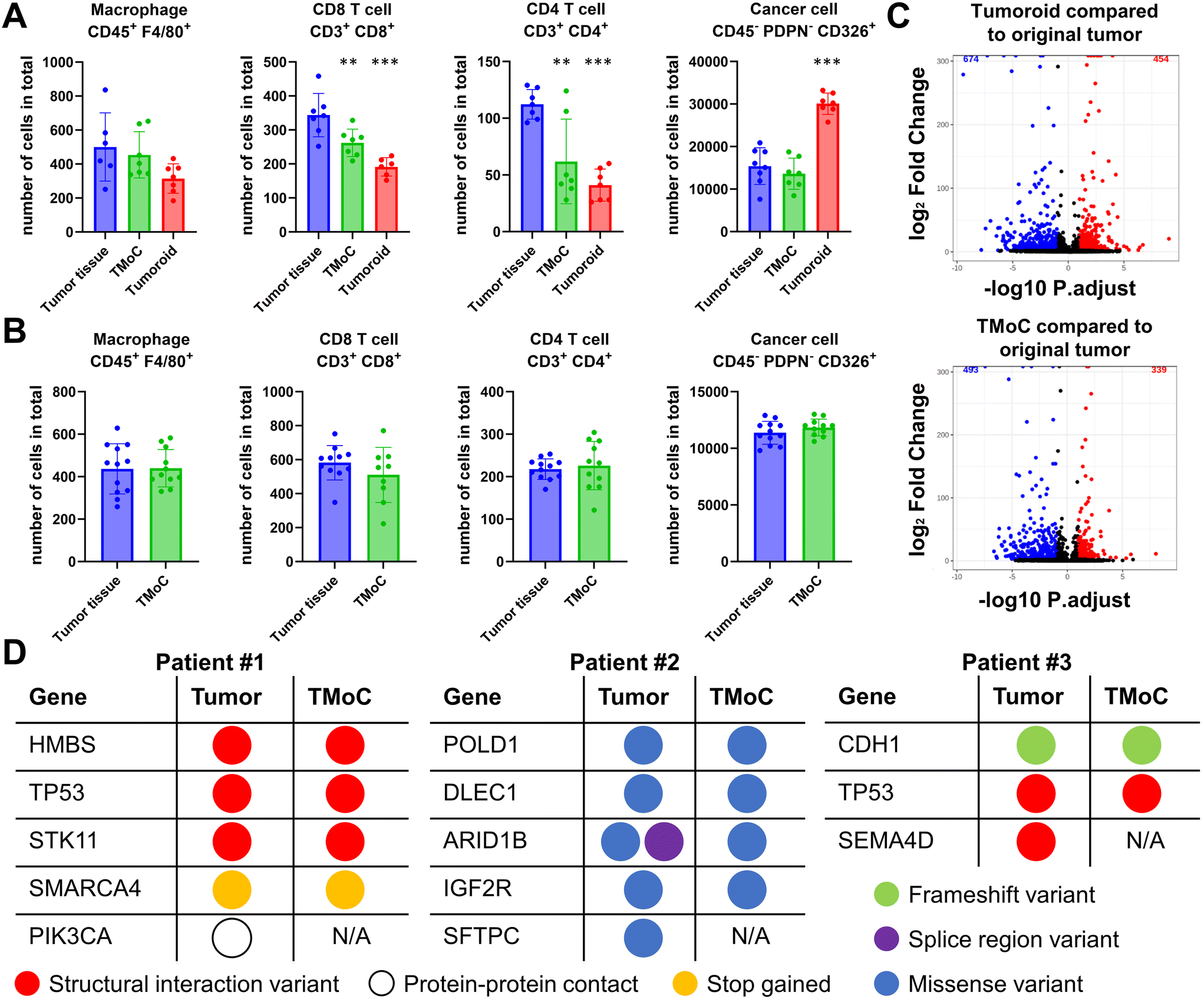 | ||
| Fig. 3 Profiling of TMoC, corresponding primary tumors, and tumoroids in terms of cell compositions, transcriptomics and genomics. (A) Number of macrophages (CD45+ F4/80+), CD8+ T cells (CD3+ CD8+), CD4+ T cells (CD4+ CD8+), and cancer cells (CD45+ PDPN− CD326+) in the TNBC primary tumor tissue, TMoC tissue, and tumoroid were quantified using flow cytometry. Notably, a depletion of immune cells (macrophages, CD8+ T cells, CD4+ T cells) was observed in both TMoC and tumoroid cultures of the TNBC model, whereas an increase in tumor cell numbers was detected in tumoroids. (B) In CRC primary tumor tissue and TMoC tissue, the number of macrophages (CD45+ F4/80+), CD8+ T cells (CD3+ CD8+), CD4+ T cells (CD4+ CD8+), and cancer cells (CD45+ PDPN− CD326+) were assessed via flow cytometry. The CRC-derived TMoC was able to preserve the populations of macrophage, CD8+ T cells, CD4+ T cells, and cancer cells. (C) Transcriptomic profiling of TMoC and tumoroids, compared to mouse CRC primary tumors. (D) Somatic mutations identified in TMoC and the corresponding primary tumors from patients. All data in (A) and (B) are expressed as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001. | ||
For transcriptomic analysis, RNA-seq was performed on primary tumor tissue resected from mice and corresponding tissue retrieved from TMoC or tumoroid cultures after 3 days. RNA samples from each tissue were isolated and subjected to bulk RNA-seq analysis. Compared to the transcriptomic profile of primary tumors, gene expression profiles in both TMoC and tumoroid models showed alterations. However, the TMoC system's gene expression profile was more similar to the primary tumor (493 differentially expressed genes (DEGs) downregulated; 339 DEGs upregulated) than the tumoroid culture (674 DEGs downregulated; 454 DEGs upregulated) (Fig. 3C). To further validate the ability of TMoC to preserve the genomic information of the corresponding patients' tumors, we conducted WES on tissues from a 3 day culture in TMoC and their corresponding primary tumors. Focusing on highly relevant oncology-associated genes listed for individual patients, TMoC was found to retain 77% of the mutations from the primary tumor, in terms of both the mutated genes and the types of mutations (Fig. 3D). In summary, TMoC exhibited a strong ability to preserve the TME in terms of the cell population, transcriptomic profile, and genomic profile, making it a suitable model for precise drug evaluation.
2.4. Drug response of in vivo tumor and TMoC models
To evaluate TMoC as a preclinical screening tool bridging the gap between traditional in vitro testing and in vivo studies—and thereby reducing reliance on animal models—we aimed to reduce the time required for drug efficacy testing. Compared to the 15–45 days typically needed for animal model studies, TMoC requires only 5–7 days, making it particularly suitable for early-stage drug development. To validate TMoC's ability to accurately replicate in vivo tumor responses, we established disease models for three different cancers—pancreatic cancer (PDAC), CRC, and TNBC—on TMoC and assessed their responses to various drugs (Fig. 4A). Additionally, the results of TMoC drug efficacy testing were compared with those from animal experiments (Table S1†).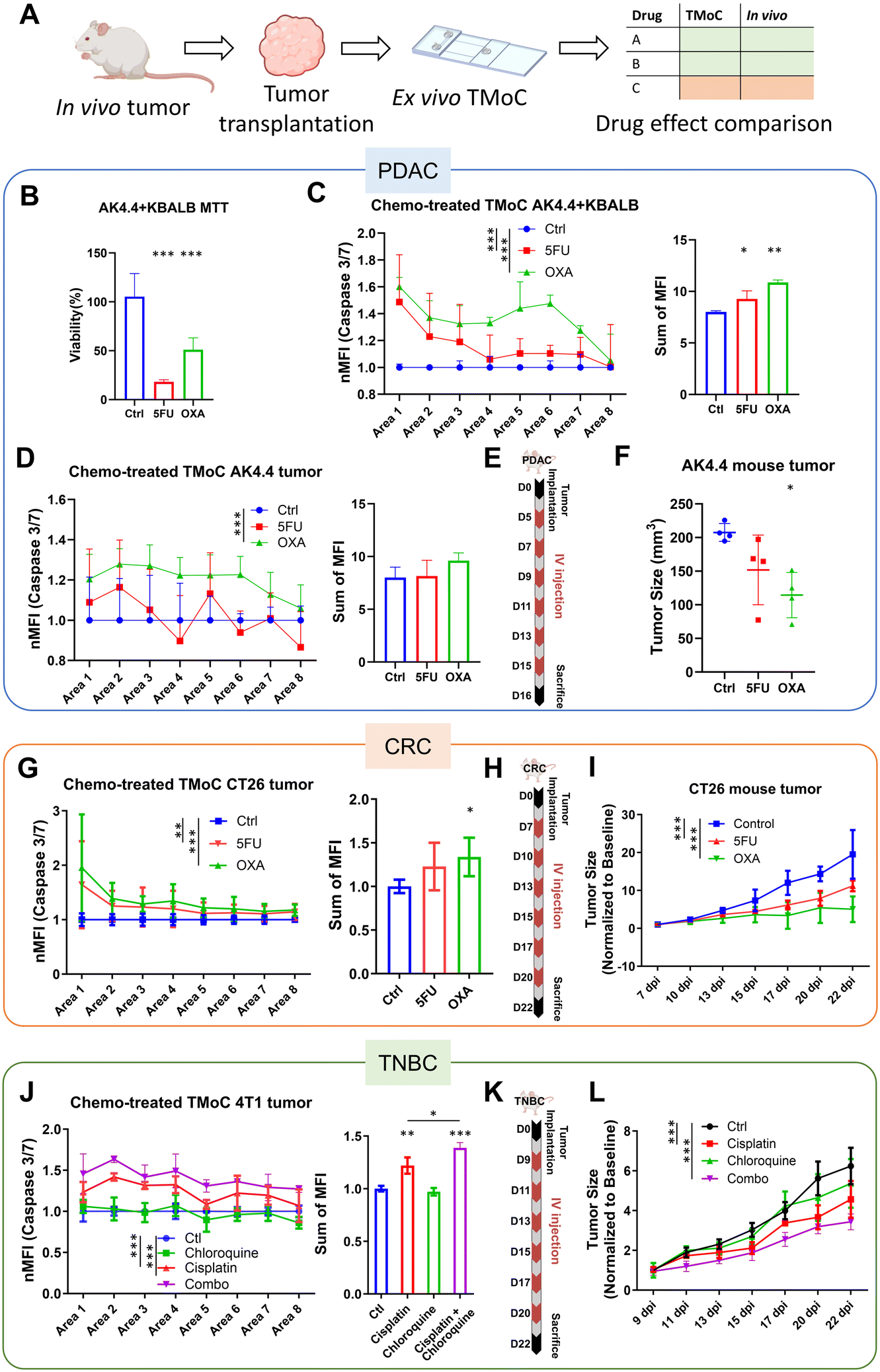 | ||
| Fig. 4 Comparison of drug evaluation results in 2D, TMoC and animal models. (A) After establishing mouse tumor models, the tumors were transplanted into TMoC for drug testing. The same drugs were also directly administered to the mouse models, and their efficacy results were compared. The comparison involved testing more than one drug to evaluate whether the efficacy ranking of the same drugs in the in vivo tumor treatment matched the results observed in ex vivo TMoC. (B) Viability assessment of AK4.4 and KBALB cell line co-culture post-treatment with 5-FU and oxaliplatin. (C) Apoptosis intensity induced by 5-FU and oxaliplatin in different regions of TMoC using the AK4.4 + KBALB cell line (left) and the cumulative intensity (right), measured via caspase 3/7 signaling to determine the drug efficacy in cancer cell eradication. Higher intensity indicates greater drug effectiveness. (D) Apoptosis intensity induced by 5-FU and oxaliplatin in different regions of TMoC using murine PDAC tumor tissue (left) and the cumulative intensity (right). (E) Chemotherapy treatment protocol for PDAC involved intravenous injections on days 5, 7, 9, 11, 13, and 15 post-tumor implantation. (F) Anti-tumor effect evaluation, where PDAC mice received chemotherapy, and the tumor size was measured on day 16 post-tumor implantation. (G) Apoptosis intensity induced by 5-FU and oxaliplatin in different regions of TMoC using murine CRC tumor tissue (left) and the cumulative intensity (right). (H) Chemotherapy treatment protocol for CRC involved intravenous injections on days 7, 10, 13, 15, 17 and 20. (I) Anti-tumor effect evaluation, where CRC mice received chemotherapy, and the tumor size was measured on day 22 post-tumor implantation. (J) Apoptosis intensity induced by cisplatin and chloroquine in different regions of TMoC using murine TNBC tumor tissues. (K) Chemotherapy treatment protocol for TNBC involved intravenous injections on days 9, 11, 13, 15, 17 and 20. (L) Anti-tumor effect evaluation, where TNBC mice received chemotherapy, and the tumor size was measured on day 22 post-tumor implantation. * denotes statistical significance compared to the control group. p-Values less than 0.05, 0.01, and 0.001 were considered statistically significant for one, two, and three stars, respectively. nMFI in (C), (D), (G), and (J) represents the normalized mean fluorescence intensity. | ||
First, widely administered drugs, 5-FU and oxaliplatin, were tested in the PDAC model. The co-cultures of the AK4.4 PDAC cell line and fibroblast cell line KBALB were conducted in both 2D and TMoC models. The two models showed contrasting results; in the 2D cell culture model, 5-FU demonstrated significantly better apoptosis induction (Fig. 4B), while in TMoC, oxaliplatin showed significantly better efficacy (Fig. 4C). To construct a more physiologically relevant in vitro model, PDAC mouse tumor tissue-derived cells were cultured on TMoC for further drug screening, and the results were similar to those of the cell line TMoC, with oxaliplatin showing superior efficacy (Fig. 4D). Finally, animal experiments were conducted for validation using the same drugs and concentrations as the maximum serum concentrations reported in the clinical literature.20 After six rounds of intravenous injection therapy, tumor size analysis was performed on day 16 (Fig. 4E). The group treated with oxaliplatin showed a 45% reduction in tumor size compared to the control group, while the 5-FU group only showed a 27% reduction, indicating significantly better results with oxaliplatin treatment (Fig. 4F). These animal experiment results aligned with the TMoC drug screening findings.
Next, we conducted drug screening with the same two drugs, 5-FU and oxaliplatin, in the CRC model. Using the maximum reported serum drug concentrations in the circulation system, a mouse CRC model was established using the CT26 CRC cell line. Drug screening was performed on TMoC and traditional tumoroids using cells derived from the tumor tissue. Oxaliplatin exhibited significantly better efficacy in inducing apoptosis of CRC tumor-associated cells (Fig. 4G), consistent with the findings in animal experiments. However, no significant difference between each group was observed in conventional tumoroid cultures (Fig. S5†). After six rounds of intravenous chemotherapy (Fig. 4H), the mouse group treated with oxaliplatin showed 74% reduction in tumor size compared to the control group, while 5-FU reduced the tumor size by 42% (Fig. 4I). Consistent with the predictions of TMoC drug screening, oxaliplatin demonstrated significantly better therapeutic efficacy.
Further, we established a mouse TNBC tumor model using the 4T1 cell line for drug screening. The candidate treatment strategies included cisplatin, chloroquine, and combination therapy of the two drugs, all administered at the maximum serum concentrations. Using cells derived from mouse tumor tissues, TMoC was established for drug screening. The results indicated that combination therapy significantly outperformed single-drug treatments (Fig. 4J). In animal experiments, after six rounds of treatment in each group (Fig. 4K), the tumor size reduction was 45% for the combination therapy group, 27% for cisplatin, and only 13% for chloroquine, with no significant difference compared to the control group (Fig. 4L). These results were consistent with the TMoC drug screening outcomes.
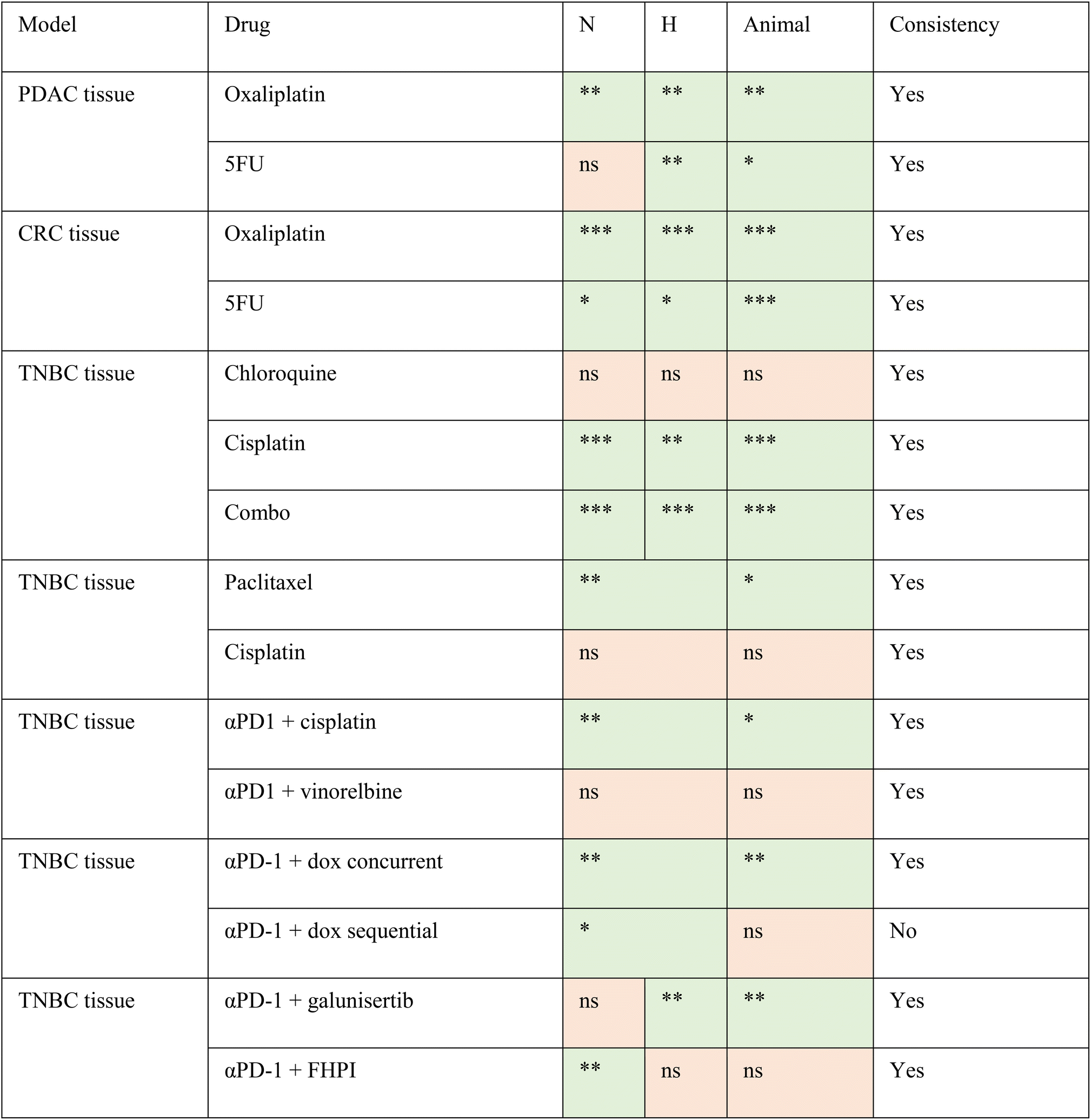
The TMoC platform was used to evaluate the efficacy of treatment across various cancer types (TNBC, CRC, PDAC) and treatment strategies (chemotherapy, targeted therapy, targeted immunotherapy, immunotherapy, and chemo-immunotherapy). The results indicated a high correspondence between TMoC and animal models with 93% (14/15) agreement across different treatments (Table 1 and Fig. S6†). In the study of TNBC treated with targeted immunotherapy (αPD-1 combined with galunisertib or FHPI), besides the overall treatment efficacy, TMoC revealed the regional hypoxia-dependent synergistic effect of the treatment, consistent with the response observed in primary tumors resected from mice.17 Based on the statistical significance of treatment groups and controls, we indicated in the table whether each group showed significant differences (p-value < 0.05), marking significant results in green and non-significant results in orange. These findings underscore TMoC's utility as a rapid, reliable preclinical screening tool with high predictive accuracy for in vivo outcomes.
| Statistical significance for each group at different time points is indicated as follows: ** denotes p-value < 0.01, * denotes p-value < 0.05, and ns denotes not significant. ‘Consistency’ refers to the alignment between the significance of results obtained from TMoC and animal experiments. |
|---|
|
2.5. Real-time regional response of ex vivo patient-derived cultures in TMoC
After establishing the role of TMoC in preclinical drug development, we sought to explore whether a platform closer to clinical patient tissue could be used to evaluate the suitability of new drugs for clinical trials. Here, we obtained post-surgery tumor tissues from clinical breast cancer patients to confirm the feasibility of real-time regional drug evaluation in TMoC. The tissues were treated with doxorubicin and Paclitaxel alone or in combination. Due to the automatic generation of molecular gradients and dynamic environments in TMoC, the treated tissues were analyzed in real time. In the case of patient #4, doxorubicin exhibited better efficacy in both proximal and distal blood vessel areas (Fig. 5). However, after 72 hours, the efficacy of doxorubicin began to decline, showing no significant difference in apoptosis from the control group. This suggests that the drug's effectiveness is diminishing. Overall, doxorubicin was superior to Paclitaxel in this patient. For patient #5, treatment with doxorubicin, Paclitaxel, or their combination showed better efficacy in the proximal vascular area, while the distal vascular area showed no significant effect. After 72 hours, neither doxorubicin nor Paclitaxel alone differed from the control group. Only the combination therapy maintained excellent efficacy, indicating that combination therapy is superior for extended treatment durations. Conversely, for patients #6 and #7, neither doxorubicin, nor paclitaxel, nor their combination showed any effect on the patient's tissue at any time point or in any area within TMoC, indicating the resistance to these drugs.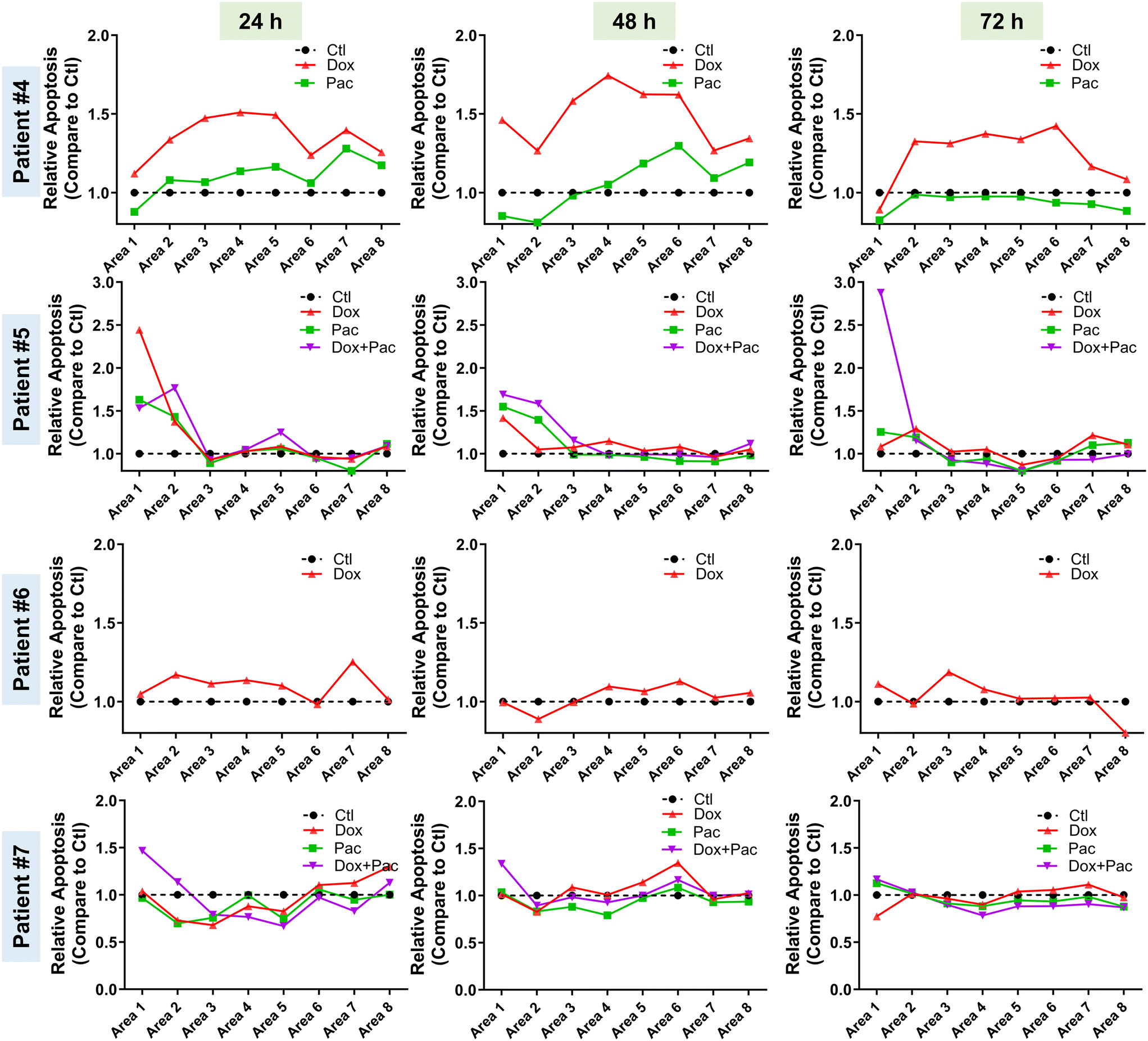 | ||
| Fig. 5 Real-time regional analysis of drugs in patient-derived TMoC. Tumors from four breast cancer patients were collected and digested. Following the establishment of TMoC, doxorubicin or paclitaxel was introduced into the circulation at doses equivalent to the clinical Cmax. The regional apoptosis signal was recorded at 24, 48, and 72 h. | ||
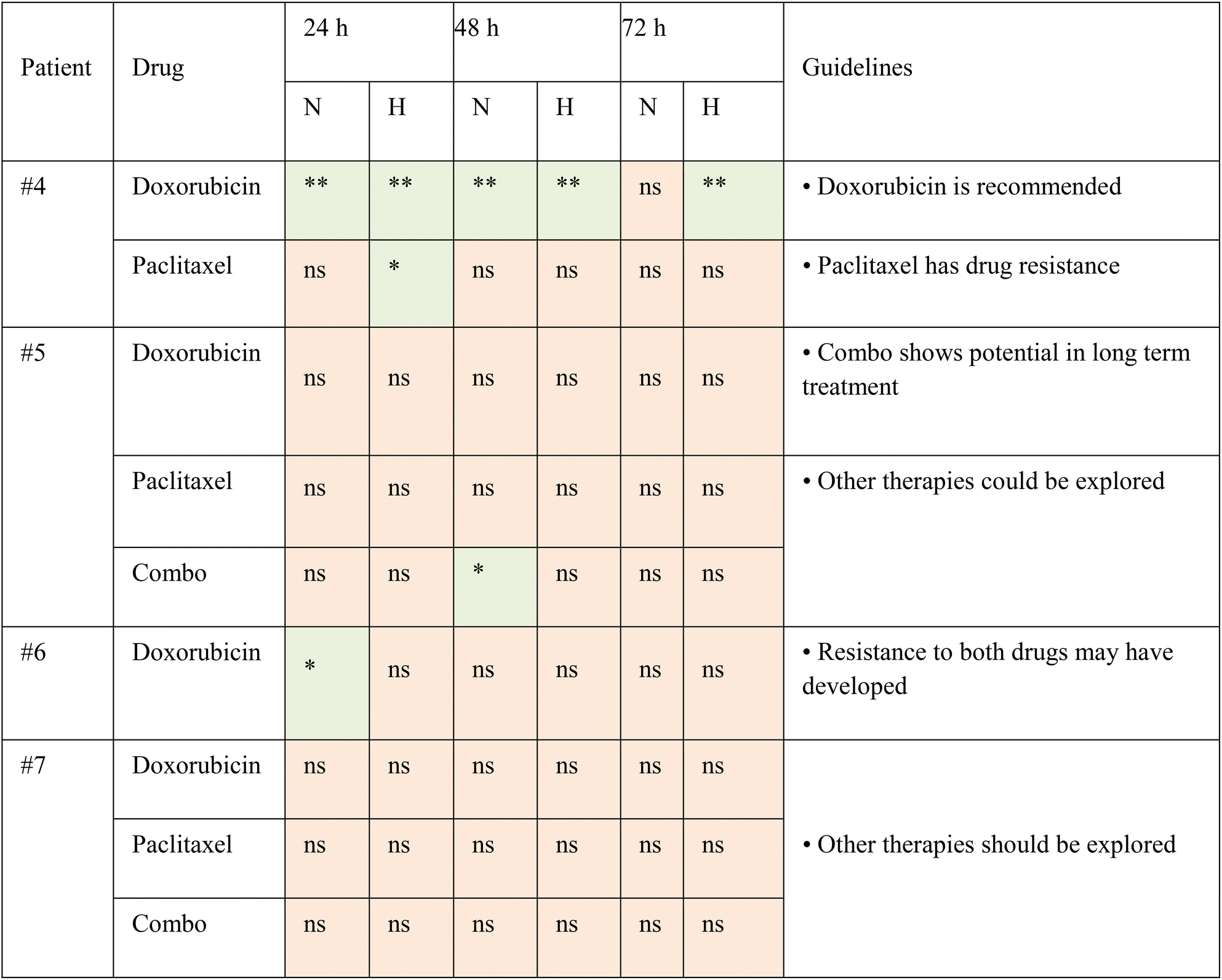
We presented these findings in statistical results to demonstrate the capability of TMoC to evaluate drug responses at different times and in various regions (Table 2). Based on the data, treatment guidelines were developed, recommending candidate drugs that demonstrated significant efficacy compared to the control group for patient use. In cases where no significant differences were observed in the experimental group, it was inferred that the patient exhibited resistance to the tested drugs, and alternative treatment regimens were recommended. Overall, these findings underscore the feasibility and significance of TMoC in preclinical drug evaluation. TMoC offers flexibility in culturing cell lines, mouse-derived tumors, or patient-derived tumors, tailored to the specific needs of the users.
| Statistical significance for each group at different time points is indicated as follows: ** denotes p-value < 0.01, * denotes p-value < 0.05, and ns denotes not significant. Based on the drug screening results, corresponding treatment guidelines were suggested. |
|---|
|
3. Discussion
In this study, we developed a 3D tumor tissue culture platform for dynamic and combinatorial drug screening to help study and model real-time regional responses of human tumors to drugs. Our tissue culture platform enables experimental analysis of drug response, cellular composition, and genetics in various samples including cell lines, mouse-derived tumors, and patient-derived tumors, while accommodating screening conditions for combined treatments. The advantage of this technology is that traditional 2D monolayer cell lines cannot reflect the heterogeneous structure of in vivo tissues. Recently developed 3D model culture methods such as organoid systems and tumoroids can overcome these limitations, but they face the inability to observe in real time. Limitations include regional drug responses and the inability to analyze drug infiltration due to the static environment. Therefore, we believe that TMoC can serve as a complement to tumoroids and living animal models and has the potential to be used in preclinical drug evaluation in the future together with traditional methods.TMoC can be used in the lead compound optimization and experimental animal validation stages of preclinical drug evaluation. For example, under the global trend of animal reduction, preclinical dose range finding, formulation development, and combination drug selection can all be tested using TMoC first, and the results can be confirmed using the smallest number of experimental animals. Even for precision medicine, TMoC could be a companion diagnostic tool. For example, there are many new immunotherapies related to cell infiltration and function, such as CAR-T cell and T cell-engaging therapies (BiTE).21,22 Before patients receive these therapies, TMoC could be used to evaluate whether immune cells are affected by the tumor environment and cause failure, thus avoiding unnecessary waste of patients' lives and resources. In addition, TMoC has the potential to retain the intact tissue pattern and retrieve the tissue after drug treatment is completed. Subsequently, technology that can analyze spatial biology and the tumor microenvironment can be applied to evaluate the gene expression changes of individual cells, coupled with dynamic tumor apoptosis analysis, providing users with a more complete ex vivo tumor analysis of drug efficacy.23 Furthermore, different types of dyes can be replaced or used in combination in TMoC testing. For example, to investigate a wider range of cancer cell death mechanisms, pre-stained cell tracker cancer cells were used instead of caspase dyes to evaluate non-caspase-dependent cancer cell death (Fig. S7†). Preliminary results demonstrated the feasibility of dye replacement. Additionally, other indicator dyes such as hypoxia markers, pH indicators, ferroptosis markers, and ROS dyes have the potential to be employed to represent the response changes of cancer tissues after treatment.
TMoC has demonstrated exceptional accuracy, with 93% of the results aligning with animal studies (Table 1). The only discrepancy arose during sequential drug administration, highlighting the potential future applications of TMoC. In TMoC experiments, chemotherapy was administered for one day, followed by anti-PD-1 treatment the next day, whereas in animal studies, three doses of chemotherapy were followed by three doses of anti-PD-1.17 This discrepancy underscores the inherent challenge of replicating in vivo pharmacokinetics within an in vitro system, where drug retention, metabolism, and clearance differ significantly.24 Moving forward, bridging the gap between in vivo and in vitro systems will involve comparing pharmacokinetics across models, optimizing dosing schedules, selecting appropriate dosages, and potentially developing predictive models to forecast animal study outcomes. These advancements could significantly increase the potential for replacing animal models in research. Additionally, TMoC has demonstrated the capability to reconstruct the tumor microenvironment using patient-derived tissues, retaining 77% of the mutation characteristics, which highlights its potential in personalized medicine (Fig. 3D). We are closely monitoring the treatment outcomes of the patients from whom these samples were obtained, and TMoC drug screening results could potentially guide clinical decision-making for personalized treatment regimens (Table 2).
Like other tumor-on-a-chip models, TMoC has inherent limitations in fully replicating the in vivo tumor microenvironment. For example, TMoC currently does not account for the role of vascular endothelial cells in drug infiltration and immune cell activation. However, by optimizing and fine-tuning the pump flow rate, we can approximate drug infiltration closer to the actual tumor concentration gradient. While a universal tumor matrix cannot perfectly simulate the tumor environment, the inclusion of specialized growth factors (e.g. FGF and EGF) and other matrix components (e.g. collagen IV) is a potential solution to enhance the simulation accuracy.25,26 Adopting synthetic matrices instead of animal-derived Matrigel offers additional benefits such as improved stability and batch-to-batch consistency.
Moreover, the diversity of tumor types and stages presents a challenge for a one-size-fits-all chip design. Early-stage tumors with low hypoxia, for example, may require a shorter chip length due to the proximity between blood vessels and the tumor core.27 From the perspective of tumor tissue characteristics, the current TMoC design faces challenges in replicating the highly heterogeneous TMEs observed between patients, including fibrotic tumors or those with low hypoxia in highly vascularized organs. These challenges can be addressed through material modifications (e.g., replacing PET with highly oxygen-permeable PDMS) or structural adjustments, such as introducing fibroblast co-cultures into specific compartments while maintaining the existing TMoC framework.
TMoC serves as a foundational platform for creating tumor gradient environments and can be further customized to generate specialized derivative chips tailored for different tumor types. For instance, the integration of tissue-specific matrices or engineered stromal cell populations could enhance its ability to mimic tumor heterogeneity. This adaptability expands the utility of TMoC in modeling various tumor conditions. Additionally, scalability remains a key challenge for the widespread application of tumor-on-chip technology. To address this, we have recently implemented a thin-film pump system capable of driving multiple TMoC units simultaneously (up to 12 independent chips), thus meeting the high-throughput demands of drug screening.28 For large-scale industrial manufacturing, TMoC's simplified framework facilitates the use of injection-molded chips, which are cost-effective, easy to produce, and compatible with single-use applications. This design not only aligns with the goal of reducing animal use but also enhances reproducibility in preclinical drug studies, fulfilling critical requirements for drug developers. Despite these challenges, TMoC remains a valuable and versatile tool, particularly in the absence of more sophisticated alternatives. Ongoing advancements in materials science, chip architecture, and system scalability will further address these limitations, solidifying TMoC's critical role in preclinical drug evaluation and cancer research.
4. Conclusion
In conclusion, TMoC represents a drug testing platform that, despite its current limitations, holds substantial promise for enhancing drug evaluation across various stages of development. Our study demonstrates that TMoC provides a more comprehensive and dynamic approach to drug testing compared to traditional tumoroid cultures, enabling real-time regional analysis and improved preservation of gene expression and cellular composition. The incorporation of dynamic circulation within TMoC significantly enhances the assessment of drug infiltration, more accurately mimicking in vivo conditions. The validation of TMoC responses against animal experimental data further supports its accuracy and efficacy. With further refinement, TMoC has the potential to become a crucial tool for predicting in vivo drug performance, supporting drug development and precision medicine, and ultimately reducing the reliance on experimental animal models. Furthermore, the successful application of TMoC to patient-derived tissues highlights its versatility and potential for wide-ranging clinical applications.5. Experimental section
Chip fabrication
The chip design continues from the previously published content, featuring a culture area with a length-to-width ratio of 2/1 and a long rectangular channel on one side of the short edge of the culture area. The primary material of the chip is polymethyl methacrylate (PMMA), manufactured via CNC machining (MDX-40A, Roland, USA). At the bottom of the chip, a gas-impermeable PET sheet with a thickness of 0.1 mm serves as the substrate. It is fabricated by cutting a 0.1 mm-thick PET sheet pre-laminated with biocompatible adhesive tape (9122, 3M Company, USA) on one side using a CO2 laser cutting machine (PLS6.75, Universal Laser System, USA). After cleaning with alcohol and removing the adhesive corresponding to the culture area, it is attached to the bottom of the chip. Once assembled, the entire chip is sterilized by exposure to ozone and ultraviolet (UV) light for at least 20 min before cell culture experiments. The entire chip material is gas-impermeable to create oxygen gradients, while the PMMA portion of the chip can be reused.28TMoC establishment
To establish TMoC, the cells were prepared and loaded on the chip, as described previously. In brief, cell lines or cells isolated from mouse-derived or human-derived tumors, optionally with dead cell removal carried out with using a MACS annexin V magnetic separation system (Miltenyi Biotec, 130-090-201, USA), were suspended in 40 μL culture medium per 2 × 106 cells. The cell suspension was subsequently mixed with 40 μL Matrigel (Corning, 356255, USA) and introduced into the TMoC 3D culture area (70 μL for 20 × 10 × 0.25 mm in L × W × H, or 7 μL for 20 × 1 × 0.25 mm in L × W × H) via the cell loading hole. The gelation was achieved by 10 min of incubation at room temperature and the following 10 min of incubation at 37 °C. The sterilized silicon tubes (ID = 1/16′′, 8ARN7, Tygon, USA) and medium reservoir were connected to the automation platform, followed by rinsing with sterilized PBS. The chips of complete gelation were placed and fixed to the automation system, and the culture medium with designated treatment was introduced to the individual medium reservoirs. The automation system enabled the automated connection between chips and the adapters, coupled with a dynamic culture powered by a 12-channel peristaltic pump (AM17HDB4, Moons', China) and a gear reducer (PLF42A-L1-10-S2-P2, Nanyang Transmission, China) at a medium flow rate of 30 μL min−1. The flow rate selection for TMoC system has been addressed in previous studies,17 where a flow rate of 30 μL min−1 was shown to generate moderate shear stress (e.g. 5.5 dyne per cm2) in the culture area, aligning with the range of shear stress observed in tumor microvasculature. Additionally, this flow rate does not compromise the structural integrity of the hydrogel and was consistently applied across all tests to replicate the physiological conditions of blood flow.Tumoroid culture
Cell lines were prepared and suspended at a density of 2 × 104 cells per μL in the culture medium. The cell suspension was then mixed with Matrigel at a 1/1 ratio, resulting in a final concentration of 1 × 104 cells per μL in the matrix mixture. Subsequently, 10 μL of the mixture was aliquoted into each well of a 12-well plate to form a dome-shaped gel at the bottom of the well, with each well containing four domes. After seeding, the plates were inverted and incubated at 37 °C for 15 min to allow gelation. Following gelation, 1 mL of medium, with or without drug treatment, was added to each well. The plates were then incubated under standard culture conditions for subsequent analyses.Cell lines and primary cell culture
The murine TNBC cell line 4T1, fibroblast KBALB, murine CRC cell line CT26, and human TNBC cell line MDA-MB-231 were purchased from the Bioresource Collection and Research Centre (Taiwan Hsinchu, Taiwan). The 4T1, AK4.4, MDA-MB-231 and primary murine tissue-derived cells were cultured in high-glucose Dulbecco's modified Eagle's medium (DMEM, SH30022.01, HyClone, GE Healthcare Life Sciences, USA) supplemented with 10% fetal bovine serum (SH30084.03, HyClone, GE Healthcare Life Sciences, USA) and 1% penicillin/streptomycin solution (SV30010, HyClone, GE Healthcare Life Sciences, USA). KBALB cells were cultured in 10% bovine calf serum (SH30073.03, HyClone, GE Healthcare Life Sciences, USA), high-glucose Dulbecco's modified Eagle's medium, and 1% penicillin/streptomycin. CT26 was cultured in an RPMI-1640 medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin solution. Patient-derived cells were cultured in Cancer Cell Line Medium XF (Promocell, C-28077, USA). All the cells were cultured at 37 °C in an incubator (Forma 370, Thermo Fisher Scientific, USA) in an atmosphere of 5% CO2.Human tumor samples acquisition
Human tissue samples in this study were obtained from Taichung Veterans General Hospital and China Medical University Hsinchu Hospital. The procedure followed in this study involved patient tumor tissues, which was approved by the Institutional Review Boards (IRB) of participating hospitals, including Taichung Veterans General Hospital (IRB number: CF23147B-1) and China Medical University Hsinchu Hospital (IRB number: CMUH111-REC3-179). The written informed consent was obtained from patients before the surgery to agree with the use of individual tissues in this study.Animals and orthotopic tumor models
Female BALB/cByJ, CB17 and FVB mice (BioLASCO, Taiwan) were purchased to establish heterotopic colon cancer models, and orthotopic TNBC models and orthotopic PDAC models. All animals received humane care in compliance with the “Guide for the Care and Use of Laboratory Animals”, published by the National Academy of Sciences. All study procedures and protocols were approved by the Institutional Animal Care and Use Committee (IACUC) at National Tsing Hua University (Hsinchu, Taiwan) with approval numbers listed as follows: 112017 (TNBC); 111083 (CRC); 112055 (PDAC); 111026 (human TNBC).CRC:29 murine colon cancer CT26 cells were subcutaneously implanted on the 6–8 week-old BALB/cByJ female mice. Briefly, 2 × 106 CT26 cells were suspended in 50 μL PBS–Matrigel (Corning 356255, USA) solution in a volume ratio of 1/1, and syringes with 25 G needle were used to inject the cells subcutaneously after the removal of mouse fur. The successful injection without leakage from the injection spot was confirmed through visual inspection. TNBC: murine TNBC 4T1 cells were orthotopically inoculated under the nipple of 6–8 week-old BALB/cByJ female mice. Concisely, 1 × 106 4T1 cells suspended in 40 μL PBS were injected under the nipple using syringes equipped with 25 G needles, after the mouse fur was removed with depilatory cream. The successful injection without leakage from the injection spot was confirmed through visual inspection. PDAC:30 1.0 × 105 AK4.4 cells were orthotopically implanted into the pancreas of 5 to 6 weeks old female FVB mice. First, a 1 cm longitudinal cut was made in the left upper quadrant of the abdomen. The spleen was gently squeeze out from the body, and the pancreas was dragged with a sterilized cotton swab. The AK4.4 and matrix gel mixture was injected into the end of the pancreas (20 μL) using a 28 G needle. In this experiment, 20 μL of matrix gel mixture contained 1 × 105 AK4.4 cells in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 solution of matrix and culture medium. The success of the implantation was evaluated via a direct contact of sterile cotton swab with the pancreas. If there was no leakage matrix, it was considered as successful implantation.
:1 solution of matrix and culture medium. The success of the implantation was evaluated via a direct contact of sterile cotton swab with the pancreas. If there was no leakage matrix, it was considered as successful implantation.
Human TNBC: human TNBC cancer cells were subcutaneously implanted into 6–8 week-old CB17/Icr-Prkdc SCID female mice. Specifically, 1 × 106 MDA-MB-231 cells were suspended in 50 μL PBS–Matrigel solution (Corning 356255, USA) in a volume ratio of 1/1. The cell suspension was subcutaneously injected using a syringe with a 25 G needle after carefully removing the fur at the injection site. Successful injection, without leakage from the injection site, was confirmed through visual inspection.
Fluorescence staining for the assessment of hypoxia gradient
Image-iT™ green Hypoxia Reagent (I14833, Invitrogen, USA) was used to evaluate the hypoxic gradient in area 1 to area 8 on the chip. Then 5 μL of 5 mM reagent was mixed with 2 × 106 4T1 cancer cells in 0.5 mL of culture medium, followed by 30 min of incubation at 37 °C. The cells were washed with PBS once and mixed with a tumor matrix, as indicated previously. After 24 h of on-chip incubation, an image was captured using a fluorescence microscope at ex/em = 490/520 nm. The hypoxia-positive or negative photos were counted in randomly selected visual fields.Molecular diffusion testing
We tested the diffusion of small molecules, proteins, and oxygen. The oxygen diffusion was described in the previous section (see fluorescence staining for the assessment of hypoxia gradient). The concentration changes on the chip were observed to evaluate the diffusion model. For small molecules, we used 1 mg mL−1 fluorescein (46955, Sigma, USA), and for proteins, we used 1 mM GFP protein (Assenzyme, Taiwan). Both molecules were prepared in a complete culture medium and introduced into the pre-established 4T1-TMoC chip independently. Following the 24 hour on-chip incubation, an image was captured using a fluorescence microscope at excitation/emission wavelengths of 490/520 nm. The fluorescence intensity under the microscope was used to identify the proportion of diffusion into different regions of the chip.Fluorescence staining for the assessment of cell apoptosis and cell death
According to the supplier protocol, the apoptosis assay was performed using a Cell Meter™ Live Cell Caspase 3/7 Imaging Kit (20130, AAT Bioquest, Inc., USA). Briefly, 1 μL of 200 × caspase 3/7 substrate solution was mixed with 2 × 106 cancer cells/fibroblasts. The cells were seeded on the chip, as described above. After at least 24 h of incubation, the images were captured using a fluorescence microscope at ex/em = 490/520 nm. For caspase-independent death in the chip, we evaluated by staining with the Red Cell Tracking Dye Kit Cytopainter (ab138893, Abcam, USA). Cancer cells were collected and stained with a cytopainter solution according to the manufacturer's protocol. Briefly, 1 × 106 cancer cells were suspended in DMEM (0.5 mL) and mixed with 5 μL of cytopainter. After incubation in the incubator for 60 min, cells were washed once with PBS. Then, cells were inoculated into TMoC, as described previously. For data processing and normalization, the fluorescence images were first analyzed using the ImageJ software (version 1.53, NIH, USA) to obtain the fluorescence intensity.31 The data from the treatment group were then normalized by subtracting the average intensity of the control group, resulting in the normalized MFI, recorded as nMFI.Live and dead assay
After 72 h of incubation, the upper layer of TMoC was separated from the lower layer to allow for further staining. The cells in the matrix gel were stained with 60 μL of 40 μM calcein AM and ethidium homodimer-1 (L3224, Thermo Fisher Scientific, USA) in PBS for 15 min in the darkness in an incubator. A fluorescence microscope (iRiSTM Digital Cell Imaging System, Logos Biosystems, Korea) was used to observe the positive signal of ethidium homodimer-1 (ex/em = 490/610) and calcein AM (ex/em = 490/520).Compounds
All drugs used for combination therapy evaluation via chemotherapy screening were purchased from MedChemExpress, USA, and are listed as follows: cisplatin (HY-17394), paclitaxel (HY-B0015), 5-FU (HY-90006), doxorubicin (HY-15142), oxaliplatin (HY-17371), chloroquine (HY-17589), and talazoparib (HY-16106). The doses of all chemotherapy drugs are indicated in figure legends.Dosage
When conducting drug screening in PDAC, CRC, and TNBC models, we used doses reflecting the clinical dosage applied in mouse serum, simulating real-world usage conditions. The doses of 5FU, oxaliplatin, cisplatin, and chloroquine used in TMoC and animal models are listed in Table S1.†Drug treatment
In the drug treatment evaluations on the chip, a total volume of 3 mL of culture medium was used, to which the respective drugs were added. We dissolved the drug powder in DMSO to prepare a stock solution and adjusted the concentration to ensure that the final culture medium contained no more than 1% DMSO, minimizing its impact on treatment outcomes.Tissue dissociation and primary tumor cell collection
(a) Mouse tumor tissue: the primary mouse tumor cells were isolated from tumor tissue using a Tumor Dissociation Kit for mouse (MACS, 130-096-730, USA), according to the manufacturer's instructions. In brief, the mouse tumor tissues were collected and cut into small pieces and placed in a gentleMACS C tube containing the enzyme mix. The subsequent dissociation and incubation procedures were performed using the soft/medium program for CT26 mouse tumors, or the Tough program for 4T1 and PDAC mouse tumors, according to the manufacturer's instructions. (b) Human tumor tissue: the primary human tumor cells were isolated from tumor tissue using a Tumor Dissociation Kit for human (MACS, 130-095-929, USA), according to the manufacturer's instructions. In brief, the human tumor tissues were collected and cut into small pieces, and placed in a gentleMACS C tube containing the enzyme mix. The subsequent dissociation and incubation procedures were performed according to the manufacturer's instructions.Flow cytometry analysis
After 72 h of dynamic incubation, the chips were collected. The bottom layer of TMoC was separated from the main body for flow cytometry. Tumor tissues were collected and digested with collagenase (1.5 mg mL−1) and hyaluronidase (1.5 mg mL−1) in a culture medium at 37 °C for 30 min. We then used a 40 μm filter to separate the undigested tissues. The single cells were washed and resuspended in a cold buffer (1% BSA in PBS). The cell suspensions were then stained with antibodies. To evaluate immune population, cells were stained with CD3, CD4 and CD8 antibodies for T cell population, stained with CD45 and F4/80 for macrophage, and CD326, PDPN, and CD45 for cancer cells. Flow cytometry data were obtained using a BD FACSAria III flow cytometer (Becton Dickinson, USA) and analyzed using the FACSDiva™ software (BD, USA). The following antibodies were used for flow cytometry analysis: APC-CD3 (100235), PE-Cy7-CD8 (100721), PE-CD4 (100407), FITC-CD45 (110706), PE-F4/80 (123109), APC-CD326 (118213), and PE-PDPN (127407), all of which were purchased from BioLegend, USA.MTT assay
AK4.4 and KBALB cell lines were plated in a 96-well plate in a ratio of 1/1. Following a 48 hour incubation period with chemotherapeutic drugs, including 5-FU and oxaliplatin, the culture medium was replaced with PBS containing 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, SERVA, Germany) at a concentration of 5 mg mL−1. Subsequently, the cells were incubated for an additional 2 hours. Finally, the MTT solution was replaced with 50 μL of DMSO, and the absorbance was measured at 570 nm using a UV spectrophotometer (Multiskan, Thermo Scientific, Rockford, IL), following the manufacturer's protocol.Total RNA collection
After 72 h of incubation, TMoC, tumoroids, or 2D cells were harvested. For TMoC, the upper layer of the chip was separated from the lower layer for RNA extraction. Total RNA was extracted using the TRIzol™ reagent (Invitrogen, 15596018, USA), according to the manufacturer's protocol. In short, 400 μL of TRIzol™ Reagent was mixed with the cells and matrix to collect total RNA, and RNA was extracted using 150 μL of 1-bromo-3-chloropropane. Total RNA was re-dissolved in RNAse-free water and kept at −80 °C once RNA was prepared.Bulk RNA sequencing
The 4T1 cancer cell line, tumor derived cells, tumoroids or TMoC were collected separately to extract total RNA for investigating the transcriptome changes. In particular, 1 μg total RNA per sample was used as the input material for RNA sample preparation. Sequencing libraries were generated using the KAPA mRNA HyperPrep Kit (KAPA Biosystems, Roche, Switzerland), following the manufacturer's protocol, and index codes were added to attribute sequences to each sample. Briefly, mRNA was purified from total RNA using magnetic oligo-dT beads. The captured mRNA was fragmented by incubation at a high temperature in the presence of magnesium in the KAPA fragment, prime, and elute buffer. First-strand cDNA was synthesized using random hexamer priming. Combined 2nd-strand synthesis and A-tailing converted the cDNA:RNA hybrid to double-stranded cDNA (dscDNA), incorporated dUTP into the second cDNA strand, and added dAMP to the 3′ ends of the resulting dscDNA. The dsDNA adapter with 3′dTMP overhangs was ligated to the library insert fragments to generate library fragments carrying the adapters. To select cDNA fragments of preferentially 300–400 bp in length, the library fragments were purified using a KAPA Pure Beads system (KAPA Biosystems, Roche, Switzerland). The library carrying appropriate adapter sequences at both ends was amplified using a KAPA HiFi HotStart ReadyMix (KAPA Biosystems, Roche, Switzerland) and library amplification primers. Finally, PCR products were purified using a KAPA Pure Beads system, and the library quality was assessed using a Qsep 100 DNA/RNA Analyzer (BiOptic Inc., Taiwan), followed by RNA sequencing. The original data obtained by high-throughput sequencing (Illumina NovaSeq 6000 platform, USA) were transformed into raw sequenced reads by CASAVA base calling and stored in the FASTQ format. The obtained high-quality data were used for subsequent analyses. Read pairs from each sample were aligned to the reference genome using the HISAT2 software. Feature counts were used to count the read numbers mapped to individual genes.Whole-exome sequencing (WES)
To perform WES for evaluating the changes in mutations with original dissociated cells, 2 × 106 breast cancer patient-derived cells were collected from TMoC. We used a total DNA collection kit according to the manufacturer's protocol for further WES analyses. WES was performed using Illumina. For sequence and SNV analysis, suboptimal quality raw sequencing data underwent preprocessing to remove adapter sequences and low-quality reads using Trimmomatic. The resulting clean reads were used for subsequent analyses. The quality assessment of the cleaned data was performed using FastQC. The alignment of clean reads to the human reference genome (UCSC_hg19/GRCh37) was conducted using the Burrows-Wheeler Aligner (BWA). Germline SNV/INDEL analysis used the HaplotypeCaller workflow within the Genome Analysis Toolkit (GATK). Somatic SNV/INDEL analysis employed the MuTect2 workflow within GATK. Subsequent annotation and analysis were performed using SnpEff. The distribution and types of variants across the genome were elucidated, with variant types being statistically analyzed. In addition to SNVs and Indels, genomic variations encompass SVs and CNVs, representing large-scale alterations in the genome structure and copy number, respectively. Manta and Control-FREEC tools were employed for the analysis of SVs and CNVs, respectively. The annotation and integration of SNV/INDEL variants used ANNOVAR, InterVar, and SnpEff. CNVs and SVs were annotated using AnnotSV. For SNVs and Indels, ANNOVAR was employed to annotate: genomic position, variant type, and associated genes. Pathogenicity prediction was made using various software tools like SIFT, PolyPhen2, CADD, and REVEL. In addition, we screened out the genes with the highest disease-causing impact on patients and compared the types of mutations to evaluate whether mutation characteristics were retained before and after TMoC culture.Statistics and reproducibility
Statistical analyses were performed using the GraphPad Prism 9 software (GraphPad, USA). One-way ANOVA was used to compare the differences between single groups. Two-way ANOVA was used to evaluate the statistics with more than 2 parameters, such as tumor growth and regional response. Data are presented graphically as mean ± SD. The values were normally distributed. P-Values less than 0.05, 0.01, and 0.001 were considered statistically significant for one, two, and three stars, respectively.Data availability
The data supporting the findings of this study are available within the article and its ESI.†Author contributions
Ya-Hui Lin: data curation, formal analysis, methodology, writing – original draft. Chiao-Min Lin: data curation, validation, visualization, writing – original draft. Kee-Ming Man: conceptualization, funding acquisition, methodology, resources, writing – review & editing. Chih-Chiang Hung: methodology, resources, validation. Hsin-Ling Hsu: funding acquisition, supervision, validation. Yunching Chen: conceptualization, supporting, methodology, resources. Hsuan-Yu Mu: conceptualization, data curation, investigation, supervision, writing – original draft, writing – review & editing. Tzu-Hung Hsiao: conceptualization, funding acquisition, resources, supervision, writing – review & editing. Jen-Huang Huang: conceptualization, funding acquisition, methodology, resources, validation, visualization, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Science and Technology Council (NSTC), Taiwan under grant numbers 111-2628-E-007-002-MY2, 113-2221-E-007-015-MY3, and 113-2823-8-007-002, National Health Research Institutes (NHRI) under grant number NHRI-EX113-11319BI, Veterans General Hospitals and University System of Taiwan Joint Research Program under grant numbers VGHUST112-G2-3-2 and VGHUST113-G2-3-2, and National Tsing Hua University/China Medical University Hsinchu Hospital under grant numbers NTHU-CMUHH-112Q2527E1 and NTHU-CMUHH-113QF043E1. C.M. Lin acknowledges the support of the Emmanuel O Salawu Scholarship for this research.References
- S. Yamaguchi, M. Kaneko and M. Narukawa, Clin. Transl. Sci., 2021, 14, 1113–1122 CrossRef PubMed.
- X. Xu, M. C. Farach-Carson and X. Jia, Biotechnol. Adv., 2014, 32, 1256–1268 CrossRef CAS PubMed.
- J. Rodrigues, M. A. Heinrich, L. M. Teixeira and J. Prakash, Trends Cancer, 2021, 7, 249–264 CrossRef CAS PubMed.
- A. Zhang, K. Miao, H. Sun and C.-X. Deng, Int. J. Biol. Sci., 2022, 18, 3019 CrossRef CAS PubMed.
- D. Sun, W. Gao, H. Hu and S. Zhou, Acta Pharm. Sin. B, 2022, 12, 3049–3062 CrossRef CAS PubMed.
- K. T. Jin, W. L. Du, H. R. Lan, Y. Y. Liu, C. S. Mao, J. L. Du and X. Z. Mou, Cancer Sci., 2021, 112, 2592–2606 CrossRef CAS PubMed.
- S. Abdolahi, Z. Ghazvinian, S. Muhammadnejad, M. Saleh, H. Asadzadeh Aghdaei and K. Baghaei, J. Transl. Med., 2022, 20, 206 CrossRef PubMed.
- P.-J. H. Zushin, S. Mukherjee and J. C. Wu, J. Clin. Invest., 2023, 133, e175824 CrossRef CAS PubMed.
- H. Gao, J. M. Korn, S. Ferretti, J. E. Monahan, Y. Wang, M. Singh, C. Zhang, C. Schnell, G. Yang and Y. Zhang, Nat. Med., 2015, 21, 1318–1325 CrossRef CAS PubMed.
- L. Ferreira, V. Gaspar and J. Mano, Acta Biomater., 2018, 75, 11–34 CrossRef CAS PubMed.
- J. M. Greene, D. Levy, S. P. Herrada, M. M. Gottesman and O. Lavi, Cancer Res., 2016, 76, 2882–2890 CrossRef CAS PubMed.
- I. Abd El-Sadek, L. T.-W. Shen, T. Mori, S. Makita, P. Mukherjee, A. Lichtenegger, S. Matsusaka and Y. Yasuno, Sci. Rep., 2023, 13, 15377 CrossRef CAS PubMed.
- E. Yokota, M. Iwai, T. Yukawa, M. Yoshida, Y. Naomoto, M. Haisa, Y. Monobe, N. Takigawa, M. Guo and Y. Maeda, npj Precis. Oncol., 2021, 5, 29 Search PubMed.
- H.-C. Huang, Y.-C. Sung, C.-P. Li, D. Wan, P.-H. Chao, Y.-T. Tseng, B.-W. Liao, H.-T. Cheng, F.-F. Hsu and C.-C. Huang, Gut, 2022, 71, 1843–1855 CrossRef CAS PubMed.
- G. Vunjak-Novakovic, K. Ronaldson-Bouchard and M. Radisic, Cell, 2021, 184, 4597–4611 CrossRef CAS PubMed.
- D. E. Ingber, Nat. Rev. Genet., 2022, 23, 467–491 CrossRef CAS PubMed.
- H. Y. Mu, C. M. Lin, L. A. Chu, Y. H. Lin, J. Li, C. Y. Liu, H. C. Huang, S. L. Cheng, T. Y. Lee and H. M. Lee, Adv. Healthcare Mater., 2024, 13, 2302268 CrossRef CAS PubMed.
- A. J. Primeau, A. Rendon, D. Hedley, L. Lilge and I. F. Tannock, Clin. Cancer Res., 2005, 11, 8782–8788 CrossRef CAS PubMed.
- A. I. Minchinton and I. F. Tannock, Nat. Rev. Cancer, 2006, 6, 583–592 CrossRef CAS PubMed.
- D. R. Liston and M. Davis, Clin. Cancer Res., 2017, 23, 3489–3498 CrossRef CAS PubMed.
- R. C. Sterner and R. M. Sterner, Blood Cancer J., 2021, 11, 69 CrossRef PubMed.
- M.-E. Goebeler and R. C. Bargou, Nat. Rev. Clin. Oncol., 2020, 17, 418–434 CrossRef PubMed.
- B. Hu, M. Sajid, R. Lv, L. Liu and C. Sun, Front. Immunol., 2022, 13, 996721 CrossRef CAS PubMed.
- M. Bouhaddou, L. J. Yu, S. Lunardi, S. K. Stamatelos, F. Mack, J. M. Gallo, M. R. Birtwistle and A. C. Walz, Clin. Transl. Sci., 2020, 13, 419–429 CrossRef CAS PubMed.
- J. S. Di Martino, A. R. Nobre, C. Mondal, I. Taha, E. F. Farias, E. J. Fertig, A. Naba, J. A. Aguirre-Ghiso and J. J. Bravo-Cordero, Nat. Cancer, 2022, 3, 90–107 CrossRef CAS PubMed.
- C. Mazouni, B. Arun, F. Andre, M. Ayers, S. Krishnamurthy, B. Wang, G. Hortobagyi, A. Buzdar and L. Pusztai, Br. J. Cancer, 2008, 99, 68–71 CrossRef CAS PubMed.
- S. McKeown, Br. J. Radiol., 2014, 87, 20130676 CrossRef CAS PubMed.
- W.-H. Lai, H. Y. Mu, Y. L. Lu, H. Chen, J.-W. Wen, H.-J. Wu, C.-M. Cheng and J. H. Huang, Chem. Eng. Sci., 2022, 253, 117555 CrossRef CAS.
- J. C. Castle, M. Loewer, S. Boegel, J. de Graaf, C. Bender, A. D. Tadmor, V. Boisguerin, T. Bukur, P. Sorn and C. Paret, BMC Genomics, 2014, 15, 1–12 CrossRef PubMed.
- H. Y. Mu, Y. N. N. Ta, M. J. R. Tham, F. F. Hsu, Y. C. Lin, H. C. Huang, Y. C. Sung, C. I. Huang, C. L. Wu and C. H. Chang, Adv. Funct. Mater., 2024, 34, 2303033 CrossRef CAS.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lc00990h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |