DOI:
10.1039/D5LC00040H
(Paper)
Lab Chip, 2025, Advance Article
Denaturation methods for reusable magnetic biosensors†
Received
13th January 2025
, Accepted 21st May 2025
First published on 23rd May 2025
Abstract
Nanoscale biosensors for sensitive DNA detection require advanced and precise fabrication techniques, which make them highly expensive and result in low yield rates. For such DNA biosensors, sensor regeneration is highly desirable. In this study, we investigated the effectiveness of various denaturants, including ultrapure water, urea solution, tris-ethylenediaminetetraacetic acid buffer, and dimethyl sulfoxide (DMSO), for the denaturation of target DNAs hybridized to probe DNAs on sensors. We used giant magnetoresistive (GMR) biosensors equipped with a temperature control unit in conjunction with magnetic nanoparticles. To examine the effect of DNA sequence on denaturation efficiency, 14 orthogonal DNA pairs were designed and tested. Furthermore, to maintain a consistent sensitivity in subsequent measurements, we evaluated the integrity of the probe DNAs on the sensors after denaturation. Among all the denaturants tested, 40% DMSO demonstrated excellent performance in the denaturation of probe DNAs covalently bonded to the sensors, without any heating process. This optimal denaturant can be applied to other planar DNA biosensor systems; moreover, GMR biosensors can facilitate the evaluation of newly developed denaturants.
Introduction
Sensitivity is one of the most critical factors that researchers strive to enhance for better diagnostics because DNA biosensors with higher sensitivities can significantly reduce the preparation time needed for amplifying target copies using techniques such as polymerase chain reaction (PCR),1,2 loop-mediated isothermal amplification (LAMP),3 and recombinase polymerase amplification (RPA).4 Therefore, considerable efforts have been made to improve sensitivity through the development of various biosensors utilizing nanowires,5,6 field-effect transistors (FETs),7,8 and magnetic sensors.9,10 However, owing to their high manufacturing costs and low yields, there have been growing needs for regenerating these biosensors.
Conventional approaches for DNA detection using planar biosensors involve immobilizing probe DNAs on biosensor surfaces and subsequently introducing target DNAs for hybridization. Regenerating the DNA biosensors presents practical challenges, including the complete detachment of target DNAs hybridized with the probes while retaining the probe DNAs on the surfaces and preserving their reactivity for subsequent measurements. Additionally, to ensure their multiplexing capability, the unhybridized probe DNAs on different sensors for different targets must remain intact throughout the regeneration process for accurate measurements.
Several denaturation methods have been reported to address these issues,11,12 including heating,13 treatment with chemical agents,11,14 and their combination to disrupt the hydrogen bonds between nucleic bases, ultimately breaking double-stranded DNA (dsDNA) into two single-stranded DNAs (ssDNA). However, these studies primarily focused on the denaturation phenomenon itself and did not address the integrity of the probe DNAs post-denaturation, which is a crucial factor for reusability. Additionally, previous studies have predominantly investigated solution-phase hybridization and denaturation, which differ from solid-phase reactions involving surface-bound probe DNAs.
As multiple reactions can occur in parallel, planar biosensors have been developed to achieve superior multiplexing capabilities and high throughput. One of the most sensitive planar biosensors is the giant magnetoresistive (GMR) biosensor, which employs the quantum-mechanical effect of giant magnetoresistance to detect minute stray magnetic fields from the magnetic nanoparticles (MNPs).15–19 GMR biosensors have demonstrated the ability to detect multiple DNAs in clinical samples;10,20,21 however, they have not been reused to measure multiple DNA samples using a single device. In this study, we investigated the effectiveness of four denaturants – ultrapure water (UPW), urea solution, tris-ethylenediaminetetraacetic acid (TE) buffer, and dimethyl sulfoxide (DMSO) – by utilizing the real-time monitoring capability of GMR biosensors under both heating and non-heating conditions. To examine the effect of denaturation on different nucleic acid sequences, 14 orthogonal DNA pairs were designed with different sequences but similar thermodynamic properties. We proposed a reliable and straightforward approach to regenerate planar GMR biosensors and validated the method through multiple cycles of hybridization and denaturation, demonstrating its effectiveness in denaturation and its ability to preserve probe DNAs. This approach can be used to screen newly developed denaturants and evaluate their effectiveness in real time using GMR biosensors, which may also be applicable to other types of planar biosensors.
Experimental
Materials and instruments
All ssDNAs including probes and targets (pairs P1-1 to 4, P2-1 to 4, P3-1 to 4, P4-1, and P4-2), were sourced from Integrated DNA Technologies (USA). DMSO, 8 M urea solution, poly(ethylene-alt-maleic anhydride), poly(allylamine hydrochloride), Tween-20, and N-hydroxysuccinimide (NHS) were purchased from Sigma-Aldrich. Bovine serum albumin (BSA), biotinylated BSA, phosphate buffered saline (PBS), 20× saline-sodium citrate (SSC), 0.5 M ethylenediaminetetraacetic acid (EDTA) at pH 8.0, 1 M Tris-HCl (pH 8.0), and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) were obtained from Thermo Fisher (USA). UPW was obtained using a Direct-Q3 UV water purification system (Merck, Germany). The streptavidin-coated MNPs with multiple superparamagnetic cores were purchased from Miltenyi Biotec (Germany).
Preparation of GMR biosensor chips
The GMR biosensor chip was fabricated to contain 80 GMR biosensors based on a spin-valve structure, as previously described.22 The chips were washed twice with acetone, methanol, and isopropanol. Then, each probe DNA diluted to 20 μM in 1× SSC, 1% BSA (negative control), and 10 μg mL−1 biotinylated BSA (positive control) were deposited on different GMR biosensors in quadruplicate using a robotic spotter (sciFlexArrayer S3; Scienion, Germany). The GMR biosensor chip was then kept in a humid chamber at 4 °C overnight. After overnight incubation, the chip was assembled with a custom-designed cartridge to create a reaction well over the chip and to control the temperature of the sensors with an embedded temperature-control module, as previously described.23 The assembled chip was washed with washing buffer (0.05% Tween-20 and 0.1% BSA in PBS) and blocked using 1% BSA for 1 h at room temperature in a shaker. The chip was then washed sequentially with washing buffer and 1× SSC buffer. Subsequently, a mixture of target DNAs containing eight targets (P2-1, P2-2, P2-3, P2-4, P3-1, P3-2, P3-3, and P3-4) at 0.5 μM each in 1× SSC buffer was added to the chip and hybridized with their respective probes on the GMR biosensors for 1 h at 25 °C in a temperature-controlled shaker. The chip was washed with 1× SSC buffer and inserted into a custom-designed reader station with the initial temperature set at 25 °C. After recording the baseline signals without any MNPs for 1 min, the 1× SSC buffer in the reaction well was removed, and 60 μL of streptavidin-coated MNPs were added. Upon the binding of the MNPs to the DNA hybrids on the sensors, signals were generated from the GMR sensors underneath.
Denaturation with UPW, urea solution, and TE buffer
After the signals from the GMR biosensors were saturated, the MNP solution in the reaction well was replaced with denaturants, such as UPW, urea solution, and TE buffer. The TE buffer contained 10 mM Tris-HCl (pH 8.0) and 0.1 mM EDTA. Tween-20 was mixed with each denaturant at a final concentration of 0.5%. In the first set of denaturation experiments, the temperature of the chips was maintained at 25 °C after each denaturant was added, and the experiments were terminated 30 min later. In the second set, the chips were heated to approximately 55 °C at a heating rate of ∼0.25 °C s−1 while connected to the reader stations to monitor real-time denaturation signals. To accelerate denaturation, the cartridges were placed in a 90 °C oven for different durations (10, 30, and 60 min) in the third set of experiments. To measure the remaining MNPs on the sensors after denaturation in the oven, the chips were rinsed outside the oven and incubated with biotinylated BSA (10 μg mL−1 in 1% BSA) for 30 min. The chips were then rinsed again and blocked with 1% BSA for 30 min. After washing and insertion into the reader stations, 60 μL of streptavidin-coated MNPs were added while signals were being recorded. To reuse the chips with different target DNAs, they were rinsed sequentially with washing buffer and 1× SSC buffer. The chips were then incubated for 1 h with another mixture of target DNAs containing eight targets (P1-1, P1-2, P1-3, P1-4, P3-1, P3-2, P3-3, and P3-4) at 0.5 μM each in 1× SSC buffer. After hybridization, 60 μL of streptavidin-coated MNPs were added to the chips on the reader stations to obtain signals.
Denaturation with DMSO
DMSO was added to another set of prepared GMR biosensor chips on the stations at 25 °C after washing away unbound MNPs. DMSO was diluted to different concentrations (20%, 30%, 40%, 60%, and 80%) using UPW, and each concentration was added to different chips while signals were recorded. After obtaining real-time denaturation signals induced by DMSO, the chips were unplugged from the stations and washed with a washing buffer. The chips were then blocked with 1% BSA for 30 min and washed sequentially with washing buffer and 1× SSC buffer. A mixture of target DNAs (eight targets, P1-1, P1-2, P1-3, P1-4, P3-1, P3-2, P3-3, and P3-4) was then incubated with the chips for 1 h in a temperature-controlled shaker at 25 °C to test the reusability of the chips. The chips were plugged back into the reader stations, and 60 μL of streptavidin-coated MNPs were added.
Regeneration of GMR biosensor chips
To validate regeneration, GMR biosensor chips were used to measure the same mixture of target DNAs (P2-1, P2-2, P2-3, P2-4, P3-1, P3-2, P3-3, and P3-4) five times by repeatedly performing hybridization and denaturation on the same chips. For denaturation, treatment with TE buffer in a 90 °C oven for 10 min or the addition of 40% DMSO at 25 °C was performed. Signal acquisition with MNPs, along with blocking after denaturation, treatment with denaturants, and acquisition of denaturation signals followed the same procedures outlined above for TE and DMSO.
Surface treatment for covalent bonding
To covalently attach the probe DNAs to the sensors, the chip was cleaned using an oxygen plasma generator (PDC-002, Harrick Plasma) for 2 min after sequential washing with acetone, methanol, and isopropanol. Next, the chip was assembled using a cartridge and incubated with 1% poly(allylamine hydrochloride) in UPW for 10 min, followed by washing with UPW. The chip was then baked for 1 h on a hot plate at 120 °C. It was then incubated with 1% poly(ethylene-alt-maleic anhydride) in UPW for 10 min, washed again with UPW, and incubated with an aqueous mixture of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) and N-hydroxysuccinimide (NHS) for 1 h. The aqueous mixture was prepared by dissolving 50 mg each of NHS and EDC in 0.6 mL of UPW. Each probe DNA was deposited on the activated surfaces of different sensors, and the chip was incubated overnight in a humid chamber. The remaining hybridization and denaturation steps were the same as those described above for DMSO denaturation.
Results and discussion
As the GMR biosensor chip contains 80 GMR sensors, individual sensors can be used to simultaneously test the hybridization and denaturation of multiple pairs of DNAs with different sequences (Fig. 1a). To ensure independent testing by each sensor, orthogonal pairs of probes and targets were obtained to accommodate a mixture of targets on the chip during hybridization. Using previously reported orthogonal 25-mer oligonucleotides,24 we designed 14 orthogonal pairs of 35-mers, including a poly-thymine spacer attached to the 5′-terminus. In this work, we focused on 25-mer oligonucleotides for hybridization motifs because they have been widely used in DNA microarrays and other biosensors.25,26 These 35-mer probes and their complementary targets were thoroughly screened to avoid any potential cross-hybridization, hairpin formation, and homodimerization, following previously reported thermodynamic criteria.27 The criteria for the orthogonal pairs are detailed in the ESI.† The GMR biosensors on the chip were divided into four sections (quadrants P1, P2, P3, and P4), and the probes of the 14 orthogonal pairs were immobilized on different sections and named according to their location (Fig. 1a). For P1, P2, and P3, four orthogonal pairs were allocated, whereas two orthogonal pairs were included in P4. For example, the four orthogonal pairs tested for P1 were P1-1, P1-2, P1-3, and P1-4. Detailed information on these pairs is presented in Table S1.† Along with the probes, BSA and biotin-BSA were deposited on different sensors to serve as negative and positive controls, respectively (Fig. 1b). Subsequently, the biotinylated target DNAs were added to the chip as a mixture and hybridized with the respective probe DNAs immobilized on the sensors (Fig. 1c). After hybridization, streptavidin-coated MNPs were introduced onto the chip and attached to the hybrids on the sensors (Fig. 1d). The sensors underneath generated signals that are proportional to the number of MNPs near the surface. Thus, the signals of GMR biosensors were presented as changes in the magnetoresistance ratio (ΔMR) induced by the presence of MNPs. Upon adding denaturants to the chip, the target DNAs conjugated with MNPs detached from their respective probes on the sensors, resulting in a decrease in sensor signals (Fig. 1e).
 |
| | Fig. 1 Schematics of denaturation using GMR biosensors and the effectiveness of different denaturants. (a) The 80 GMR sensors were divided into four sections, P1, P2, P3, and P4 (outlined with green, purple, red, and yellow dashed lines, respectively). The blue arrow indicates an individual sensor, while the red arrow indicates a sensor covered with probe DNAs. A GMR biosensor chip was assembled with a custom-made cartridge, creating a reaction well over the chip (black arrow). The scale bar is 500 μm. (b) All probe DNAs, BSA, and biotin-BSA were deposited on different sensors. (c) Biotinylated target DNAs were added to the chip and hybridized with their respective probe DNAs. (d) Streptavidin-coated MNPs were introduced into the reaction well and bound to the hybrids via streptavidin–biotin interactions. (e) Hybrids were denatured upon the addition of a denaturant to the chip. Probe DNAs immobilized on the sensors remained on the surface whereas the target DNAs conjugated with MNPs detached and diffused away from the sensors. Real-time signals were obtained from individual sensors treated with (f) UPW, (g) urea solution, and (h) TE buffer while the chip was maintained at 25 °C. Real-time signals from sensors treated with (i) UPW, (j) urea solution, and (k) TE buffer when the chips were gradually heated up to 55 °C, starting at 13 min. Blue shading represents the labeling of hybrids with MNPs (added to the chips at 1 min; not shown), and the red shading indicates the treatment with the denaturant. | |
Denaturation using UPW, urea solution, and TE buffer
For denaturation of the DNA hybrids, we selected three extensively researched denaturants for initial tests: UPW, urea solution, and TE buffer. UPW has been used to destabilize DNA structures by removing cations around the hybrids, as cations can attenuate the repulsive coulombic interaction of the phosphates, thus stabilizing the hybrids.28 Secondly, urea is known for its ability to destabilize natural structures and induce the denaturation of DNA molecules. Urea denaturation occurs through the disruption of intramolecular hydrogen bonding interactions or the weakening of intermolecular interactions.29 Lastly, alkaline solutions can denature DNA hybrids by removing protons that contribute to hydrogen bonding using abundant hydroxide ions.11,30 Therefore, we included a TE buffer at pH 8.0, which is a readily available reagent used in DNA experiments. To assess the effectiveness of each denaturant, the DNA pairs in P2 and P3 were hybridized, whereas the probes in P1 and P4 remained unhybridized for later use and evaluation of any physical detachment of the probes caused by the denaturant. After hybridization, each denaturant was added to different chips with the same hybridization patterns while maintaining the chip temperature at 25 °C. No significant denaturation signals were observed during the first 15 min after the addition of UPW (Fig. 1f and S1†), urea solution (Fig. 1g), or TE buffer (Fig. 1h). Although slight decreases in signals were detected upon adding the denaturant, these were attributed to partial breaks between biotin and streptavidin, as similar reductions were observed in the sensors coated with biotin-BSA or from minor physical detachment during washing and exchanging buffers. Although the experiments lasted for 30 min, no further denaturation signals were detected. Therefore, all denaturants tested were confirmed to be ineffective in denaturing the hybrids at room temperature. Next, the temperature dependency of each denaturant was investigated by gradually heating the chip to 55 °C in the presence of each denaturant. Notably, weak denaturation (10% signal drops) was initiated in the UPW at approximately 53 °C (Fig. 1i). Although this was not observed in the temperature range tested, denaturation can be further accelerated at higher temperatures as previously reported.31 Unlike UPW, urea solution and TE buffer revealed clear and strong denaturation signals starting at around 40 and 42 °C, respectively, with all hybrids significantly denatured at 55 °C (Fig. 1j and k). Their melting temperatures (TM) were determined to be 45 and 48 °C, respectively. The urea solution induced strong denaturation at lower temperatures than the TE buffer, but it could not completely denature the hybrids. The large drops in signals were solely due to the denaturation of the hybrids, as the denaturants did not disrupt the binding between streptavidin and biotin. Signals from sensors coated with biotin-BSA remained constant, indicating that biotin–streptavidin interaction was intact. To achieve complete denaturation, the chips would need to be treated with denaturants at higher temperatures; however, our reader station was not compatible with higher temperatures and thus could not monitor the denaturation signals in real time.
Denaturation at elevated temperatures
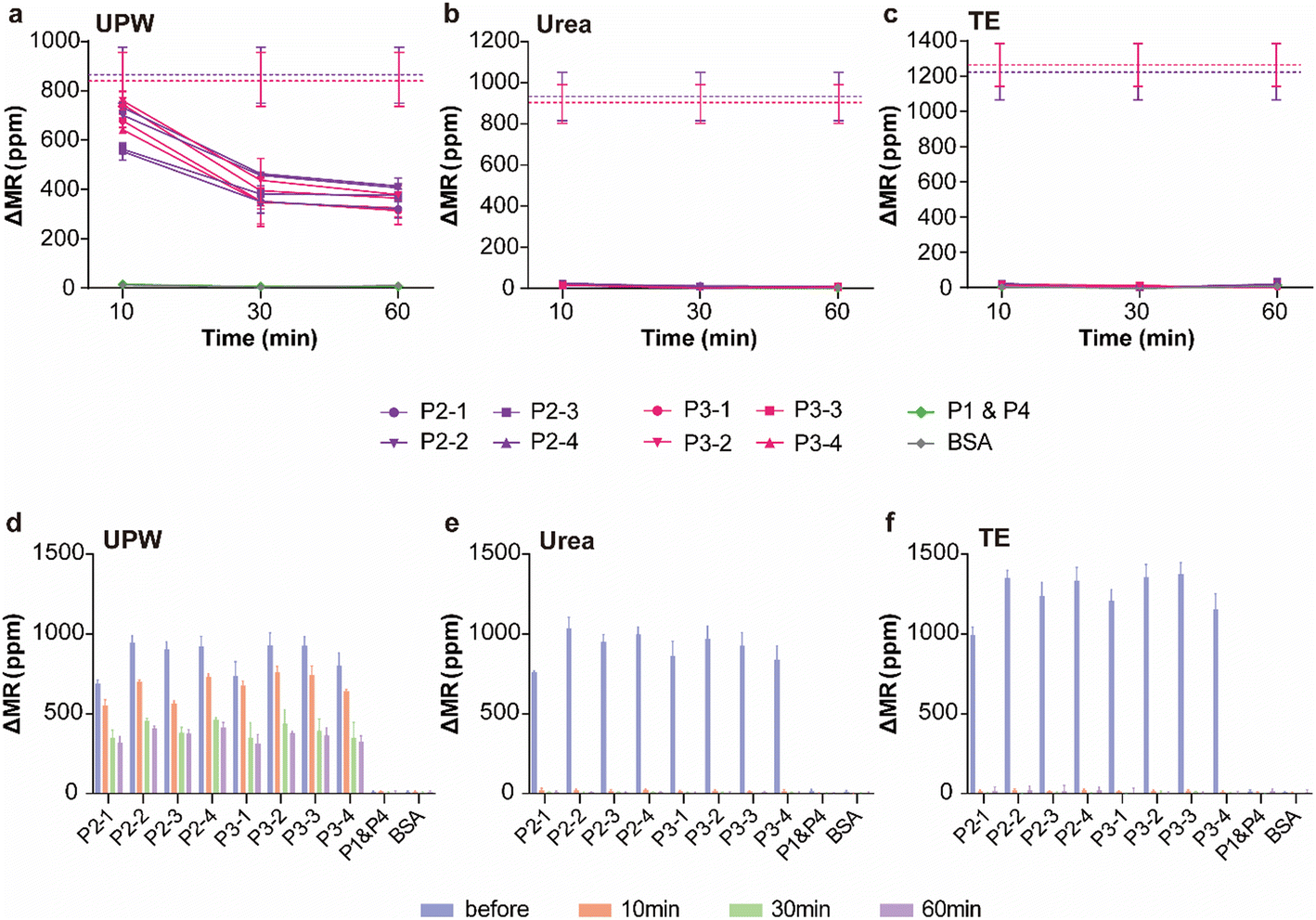
To achieve complete denaturation with each denaturant, the chips were heated to 90 °C in an oven. This denaturation condition was selected because lower temperatures (50 to 70 °C) were insufficient to achieve complete denaturation (Fig. S2†). While the chips were in the oven, they were not connected to the reader stations; therefore, real-time denaturation signals were not recorded during the 90 °C denaturation process. Instead, we counted the number of remaining MNPs (or DNA hybrids conjugated with MNPs) after denaturation by attaching additional MNPs to the remaining MNPs on the sensor surface using biotinylated BSA (Fig. S3a†). The amount of intact dsDNA was inferred from the number of remaining MNPs, which was estimated based on the magnitude of the signals from the additional MNPs (Fig. S3b†). Additionally, different denaturation durations of 10, 30, and 60 min at 90 °C were tested to determine the kinetics of each denaturant. For UPW, complete denaturation was not achieved even after 60 min (Fig. 2a), whereas both urea and TE buffer effectively denatured the dsDNA within 10 min at 90 °C (Fig. 2b and c). No significant differences in denaturation were observed across the different sequences when treated with UPW (Fig. 2d), urea (Fig. 2e), or TE buffer (Fig. 2f), indicating that these denaturants have a universal effect (Fig. S4†).
 |
| | Fig. 2 GMR biosensor signals representing remaining DNA hybrids after denaturation with (a) UPW, (b) urea solution, and (c) TE buffer at 90 °C for different durations of 10, 30, or 60 min. The error bars represent the standard deviation (SD) of four identical sensors with the same probe in P2 and P3 (n = 4) or all sensors in P1 and P4. The dashed lines indicate the average hybridization signals from the sensors in each quadrant (purple: P2 and red: P3) before denaturation. The GMR biosensor signals are displayed for different denaturation times for each probe DNA. No significant differences were observed across the different sequences with (d) UPW, (e) urea solution, and (f) TE buffer. | |
Estimation of probe DNA detachment from the surface
Although the urea solution and TE buffer demonstrated their effectiveness in removing target DNAs, their ability to preserve probe DNAs for subsequent measurements had not been fully investigated. Therefore, we reused the chips that had been treated with denaturants to measure a different set of targets and subsequently evaluated the integrity of the probe DNAs. Since the target groups of P2 (including P2-1, P2-2, P2-3, and P2-4) and P3 were probed by the sensors and denatured, the target groups of P1 and P3 were measured using the regenerated chips to estimate the integrity of the never-used probes (target group P1) and second-used probes (target group P3). The hybridization signals of targets P1 from the chip treated with UPW for 10 or 30 min were comparable to those obtained from new chips without any denaturation step, indicating that treatment with UPW at 90 °C for up to 30 min does not cause significant detachment of unused probes (Fig. 3a). However, the 60 min treatment with UPW clearly showed attenuated hybridization signals from the unused probes (Fig. 3a). This reduction may be due to the detachment of the probes from the sensor surface because the immobilization of probe DNAs involves physisorption and charge interactions. For the probes used after denaturation with UPW, the hybridization signals were much lower than those from the new chips. This is because denaturation with UPW did not completely remove the hybridized targets on these probes (Fig. 2a); thus, the second hybridization with the target group P3 was disrupted by the remaining targets. For urea and TE, no remaining targets were observed, as they were completely denatured. However, both denaturants detached both used and unused probes (Fig. 3b and c). Treatment with the urea solution for only 10 min significantly reduced the integrity of both used and unused probes, and the effect became more severe with longer denaturation times. The effect of denaturation time on probe detachment was more pronounced in the TE buffer. However, since treatment with TE buffer at 90 °C for 10 min induced minimal physical detachment, this denaturation method was used for subsequent comparisons. The overall performance of each denaturant is summarized in Table S2.†
 |
| | Fig. 3 Hybridization signals from the chips regenerated using (a) UPW, (b) urea solution, and (c) TE buffer at 90 °C for different denaturation times (10, 30, and 60 min). After regeneration, the chips were incubated with target groups P1 and P3. The dashed lines indicate the average hybridization signals from new chips without any regeneration (green: P1 and red: P3). The error bars represent the SD of four identical sensors (n = 4). | |
Denaturation with DMSO
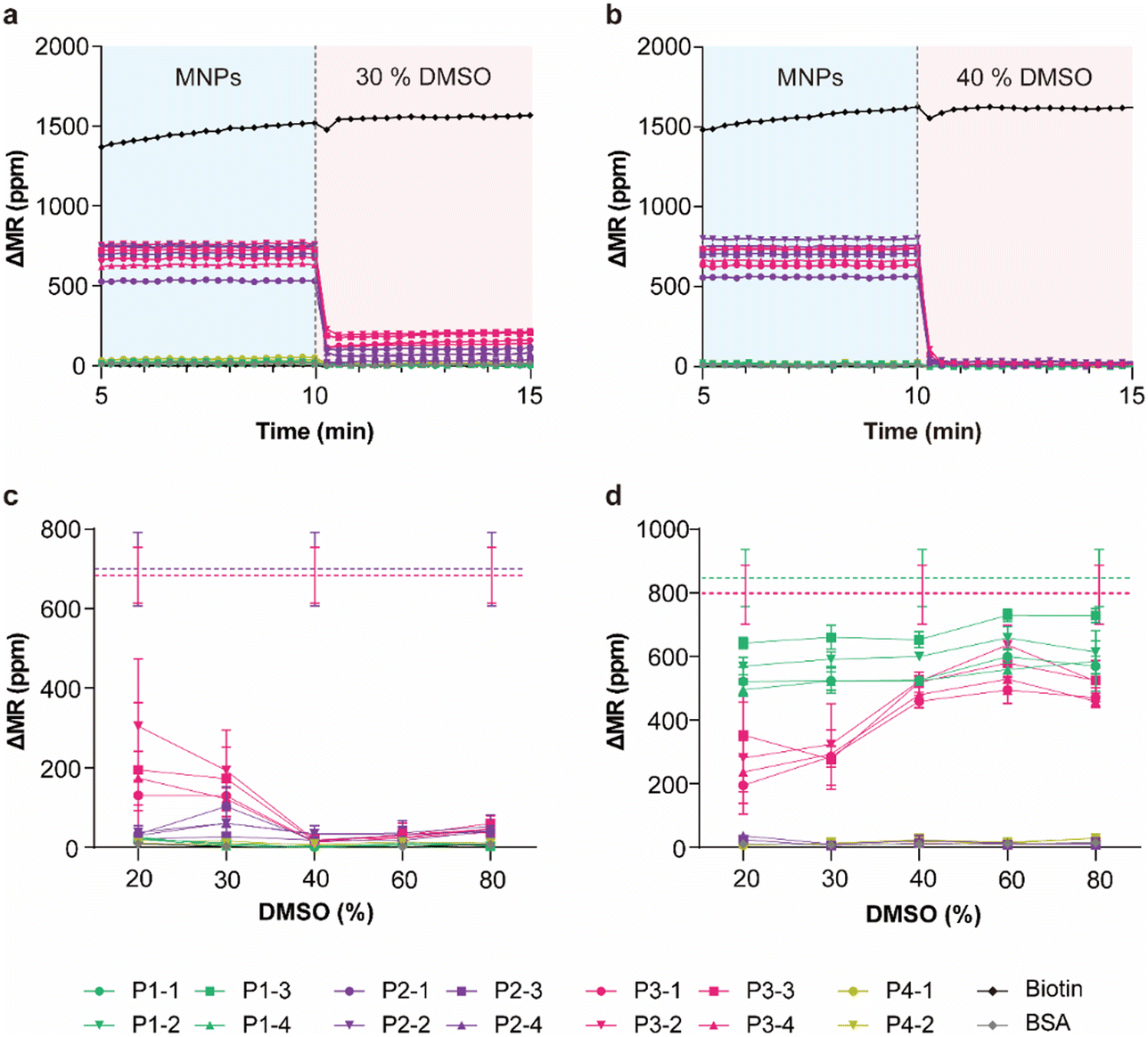
Although the urea solution and TE buffer can effectively denature target DNAs at high temperature, they may detach the probe DNAs during extended treatment. Furthermore, using denaturants at high temperatures or heating the system may not be feasible in resource-limited settings or could cause damage to the peripherals of biosensing systems or the biosensors themselves. A previous study reported that the addition of DMSO can lower the TM of DNA hybrids by 1.25 °C per percent increase in DMSO concentration.32 Therefore, we investigated the effectiveness of different DMSO concentrations for denaturing DNA hybrids on planar biosensors. After incubating the GMR biosensor microarrays with the target groups P2 and P3, hybridization signals were acquired from the reader system. While the chips were inserted into the reader system, 20%, 30%, 40%, 60%, and 80% DMSO at room temperature were added to each chip, which were predicted to lower the TM by 25.0, 37.5, 50.0, 75.0, and 100.0 °C, respectively (detailed calculations in Table S3†). Based on the calculation, we anticipated that 40% DMSO could reduce all the TM values of the hybrids to approximately 10 °C below the experimental temperature. As predicted, 30% DMSO exhibited incomplete denaturation because the TM lowered by this concentration was approximately room temperature (Fig. 4a), whereas 40% DMSO clearly detached the targets from the probes on the surface (Fig. 4b). Since the signals from biotin-BSA-coated biosensors did not show any decrease, the reduction in signals after adding DMSO was attributed primarily to denaturation between probes and targets, and not to breaks between biotin and streptavidin. Interestingly, denaturation with DMSO was completed within 20 s, indicating relatively high denaturation rates. Due to the TM dependency on the nucleotide sequence, the remaining signals after denaturation with 20% DMSO showed more variation and indicated incomplete denaturation compared to 30% DMSO (Fig. 4c). DMSO at 60% and 80% seemed to completely denature the hybrids but exhibited relatively higher remaining signals compared to 40% DMSO (Fig. 4c). Since 80% DMSO can disrupt the biotin–streptavidin interactions (Fig. S5†), we selected 40% DMSO as the most effective concentration for subsequent denaturation experiments. To determine whether DMSO disengages the probes or the entire hybrid from the surface, the chips were reused to measure the target groups P1 and P3. Unused probes (P1) showed similar minor physical detachment across all DMSO concentrations, while the used probes showed reduced signals for 20% and 30% DMSO, which were again disrupted by incomplete denaturation (Fig. 4d). Therefore, DMSO was demonstrated to effectively denature DNA hybrids on planar biosensors without heating and to preserve relatively intact probes. For clarity, the regeneration performance at different DMSO concentrations is summarized in Table S4.†
 |
| | Fig. 4 Real-time denaturation signals upon introducing (a) 30% and (b) 40% DMSO. (c) The remaining signals after denaturation with different DMSO concentrations. The dashed lines indicate the average hybridization signals (purple: P2 and red: P3) before denaturation. (d) Hybridization signals with targets P1 and P3 were obtained from reused chips after regeneration with different DMSO concentrations. The dashed lines indicate the average hybridization signals obtained from new chips (green: P1 and red: P3). The error bars represent the SD of four identical sensors (n = 4). | |
Regeneration of GMR biosensor chips
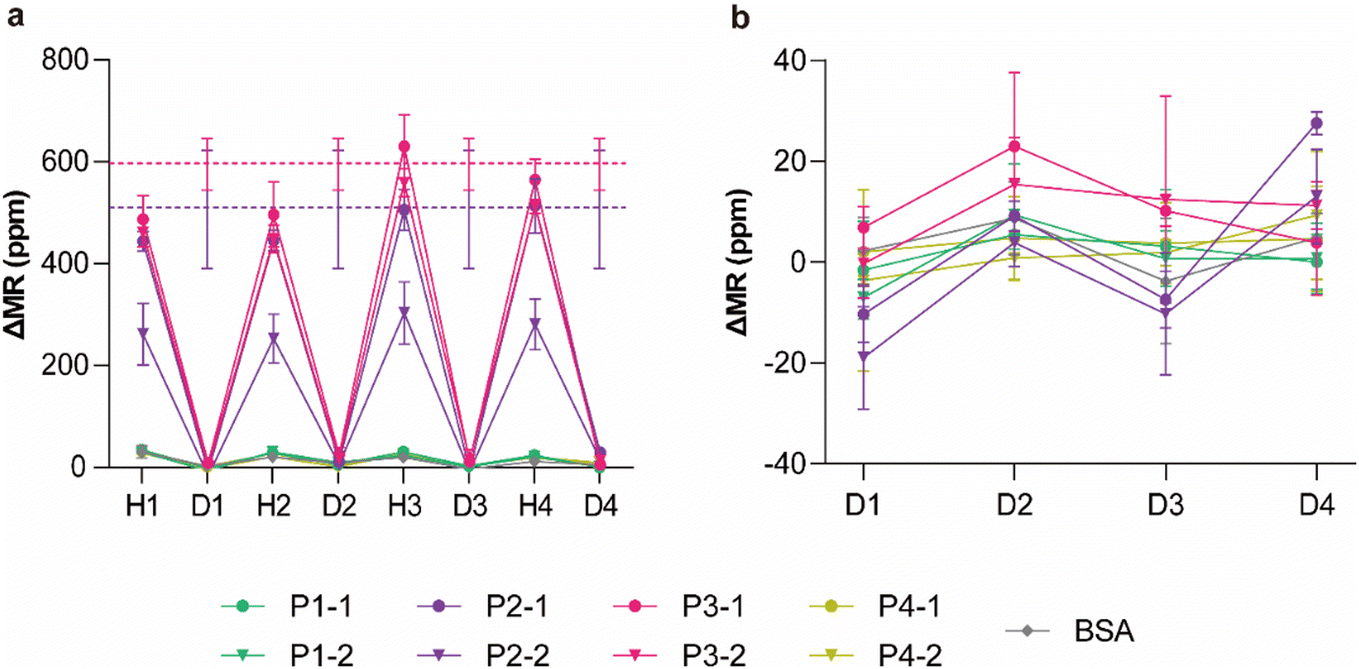
Using TE buffer at 90 °C and 40% DMSO, we conducted regeneration experiments with five cycles of hybridization and denaturation using the same chips to demonstrate the reliability of each denaturant. The same sample solution containing the target groups P2 and P3 was used repeatedly for hybridization, and each denaturant was applied to regenerate each identical chip. As observed previously, even a single regeneration cycle with each denaturant reduced the integrity of the probes, resulting in fewer signals for the same sample (Fig. 5a and b). The hybridization signals from the first cycle were considered the baseline level achieved by a fresh chip and are displayed as dashed lines. As the remaining signals after each denaturation were close to the baselines (Fig. 5c and d), complete denaturation was confirmed, and the reduced hybridization signals in subsequent cycles were attributed primarily to the loss of probes during denaturation. Representative real-time signals of 40% DMSO are provided in Fig. S6.† To further verify that the signal drops were induced by complete denaturation of the target–MNP complexes from the surface-bound probes, additional experiments were conducted to confirm the complete removal of target DNAs and the preservation of the majority of the probes (Fig. S7†). Overall, DMSO exhibited superior regeneration performance and nearly complete denaturation compared to TE buffer (Fig. 5c and d). Additionally, denaturation with DMSO can be performed at room temperature, eliminating the need for heating. However, to improve measurement repeatability, the degradation of probe integrity after denaturation must be addressed. Since the hybridization signals decreased significantly in the first few cycles of regeneration and then stabilized, we hypothesized that relatively weakly immobilized probes detached from the sensor surface upon treatment with the denaturant, while strongly immobilized probes remained in subsequent cycles. This is because probe immobilization involves physisorption and charge interactions. Washing the chips with DMSO before use could produce consistent results with strongly immobilized probes in subsequent measurements; however, this approach may reduce sensitivity.
 |
| | Fig. 5 Regeneration of chips for repeated measurements of target groups P2 and P3 using (a) TE buffer at 90 °C for 10 min and (b) 40% DMSO. H and D indicate the signals obtained after hybridization and denaturation in each cycle, respectively. The dashed lines represent the average hybridization signals (purple: P2 and red: P3) obtained before regeneration. Remaining signals after each denaturation cycle using (c) TE buffer and (d) DMSO. The error bars represent the SD of four identical sensors (n = 4). | |
Regeneration using covalently bonded probe DNAs
To improve the integrity of the probe DNAs tethered to the surface during denaturation, we employed covalent bonding for immobilizing probe DNAs using the amine groups on their 5′ ends. Using the NHS–EDC chemistry method previously suggested,33 the probe DNAs were immobilized on the sensor surface via covalent bonding. The same regeneration experiment was conducted with hybridization using a new sample containing the target groups P2 and P3, but only 40% DMSO was used for denaturation, as DMSO demonstrated superior regeneration performance compared to TE buffer and, more importantly, enables efficient denaturation at room temperature. Across multiple regeneration cycles, consistent hybridization signals were obtained, and their levels were comparable to those obtained using a fresh chip (Fig. 6a), with real-time signals shown in Fig. S8.† However, the surface chemistry resulted in slightly higher non-specific binding signals for the non-hybridized and BSA-coated sensors compared to those from the chip without covalent bonding surface chemistry. This increase is likely due to enhanced probe orientation achieved by covalent attachment through the amino modification and spacer at the 5′-terminus. The enhanced orientation may have increased the affinity of the probes not only for their targets but also for off-target oligonucleotides, resulting in reversible non-specific interactions. Nearly complete denaturation was again achieved in each cycle for the covalently bound probes, as the signals dropped to the baseline after denaturation (Fig. 6b). Therefore, among the denaturation conditions evaluated in this study, the combination of 40% DMSO and covalently bound probe DNAs is the most effective and reliable method for facilitating the regeneration of magnetic biosensors or similar platforms for DNA detection.
 |
| | Fig. 6 (a) Regeneration of chips for repeated measurements of target groups P2 and P3 using 40% DMSO. H and D indicate the signals obtained after hybridization and denaturation in each cycle, respectively. The dashed lines represent the average hybridization signals (purple: P2 and red: P3) obtained before regeneration. (b) Remaining signals after each denaturation cycle using 40% DMSO. The error bars represent the SD of four identical sensors (n = 4). | |
Conclusions
We observed that 40% DMSO exhibited superior denaturation capability with minimal physical detachment of the probe DNAs on the sensors compared to other common denaturants such as UPW, urea solution, and TE buffer. Unlike other denaturants, DMSO can denature hybridized DNAs at room temperature, which is particularly desirable for point-of-care (POC) or resource-limited settings. Additionally, the GMR biosensor platform demonstrated the capability to evaluate the effectiveness of a denaturant, indicating that this platform can serve as an evaluation testbed for newly developed denaturants. The TE buffer also revealed good denaturation performance at elevated temperatures and thus required an additional system to heat the chips; however, it can be used for applications where mild chemicals are preferred. Among the four denaturants tested, no significant effect of DNA sequence was observed on denaturation, indicating that they are universal denaturants. However, the probe DNAs used in this study were all 25-mers for hybridization motifs, which are common lengths employed in DNA microarrays and other biosensors,25,26 and were designed to feature similar GC contents. Therefore, their hybridization affinities to their target DNAs were comparable, and no significant differences were observed across the different pairs. When longer probe DNAs with varying GC contents such as circulating tumor DNA or DNA isolates from patients are measured, the DMSO concentration should be tailored to achieve optimal results. This method is not limited to magnetic biosensors and can be applied to other sensing platforms with covalently bound probes to obtain the best outcomes. More importantly, the regeneration scheme can significantly reduce manufacturing costs for high volumes of single-use chips, especially if their yields are relatively low.
Data availability
The data supporting the findings of this study have been included in this article and the ESI.†
Author contributions
Suhyeon Park: methodology, validation, formal analysis, visualization, writing – original draft. Songeun Kim: methodology, software, visualization. Shan X. Wang: supervision, funding acquisition, resources, writing – review and editing. Jung-Rok Lee: conceptualization, supervision, funding acquisition, resources, writing – original draft.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Korea Health Technology R&D Project grants through the Korea Health Industry Development Institute (KHIDI) funded by the Ministry of Health & Welfare (RS-2023-00266554), the National Research Foundation of Korea (NRF) grants funded by the Korean government (NRF-2020R1C1C1005416 and NRF-2023R1A2C1006611), and the National Institutes of Health (NIH) grants (1R01CA257843 and 1R21DK131776).
References
- Z. Li, S. Zou, S. Wu, X. Miao and D.-L. Ma, Talanta, 2021, 221, 121661 Search PubMed.
- Y. Zhang, S. Li, J. Tian, K. Li, Z. Du and W. Xu, Talanta, 2021, 222, 121575 CrossRef CAS PubMed.
- L. L. Poon, B. W. Wong, E. H. Ma, K. H. Chan, L. M. Chow, W. Abeyewickreme, N. Tangpukdee, K. Y. Yuen, Y. Guan, S. Looareesuwan and J. M. Peiris, Clin. Chem., 2006, 52, 303–306 Search PubMed.
- I. M. Lobato and C. K. O'Sullivan, TrAC, Trends Anal. Chem., 2018, 98, 19–35 Search PubMed.
- W. C. Maki, N. N. Mishra, E. G. Cameron, B. Filanoski, S. K. Rastogi and G. K. Maki, Biosens. Bioelectron., 2008, 23, 780–787 Search PubMed.
- T. Adam and U. Hashim, Biosens. Bioelectron., 2015, 67, 656–661 CrossRef CAS PubMed.
- D.-S. Kim, Y.-T. Jeong, H.-J. Park, J.-K. Shin, P. Choi, J.-H. Lee and G. Lim, Biosens. Bioelectron., 2004, 20, 69–74 CrossRef CAS PubMed.
- J. Ping, R. Vishnubhotla, A. Vrudhula and A. C. Johnson, ACS Nano, 2016, 10, 8700–8704 CrossRef CAS PubMed.
- J.-R. Lee, N. Sato, D. J. Bechstein, S. J. Osterfeld, J. Wang, A. W. Gani, D. A. Hall and S. X. Wang, Sci. Rep., 2016, 6, 18692 CrossRef CAS PubMed.
- J. C. Nesvet, K. A. Antilla, D. S. Pancirer, A. X. Lozano, J. S. Preiss, W. Ma, A. Fu, S.-M. Park, S. S. Gambhir, A. C. Fan, J. W. Neal, S. K. Padda, M. Das, T. Li, H. A. Wakelee and S. X. Wang, Clin. Chem., 2021, 67, 534–542 Search PubMed.
- X. Wang, H. J. Lim and A. Son, Environ. Health Toxicol., 2014, 29, e2014007 CrossRef PubMed.
- J. Esteban, N. Alonso-Rodriguez, G. del-Prado, A. Ortiz-Pérez, D. Molina-Manso, J. Cordero-Ampuero, E. Sandoval, R. Fernández-Roblas and E. Gómez-Barrena, Acta Orthop., 2012, 83, 299–304 Search PubMed.
- G. Rizzi, M. Dufva and M. F. Hansen, Lab Chip, 2017, 17, 2256–2263 Search PubMed.
- M. Xu, T. Dai, Y. Wang and G. Yang, RSC Adv., 2022, 12, 23356–23365 RSC.
- J.-R. Lee, D. J. Bechstein, C. C. Ooi, A. Patel, R. S. Gaster, E. Ng, L. C. Gonzalez and S. X. Wang, Nat. Commun., 2016, 7, 12220 CrossRef CAS PubMed.
- J. Im, S. Kim, S. Park, S. X. Wang and J.-R. Lee, Biosens. Bioelectron., 2024, 249, 116017 CrossRef CAS PubMed.
- R. S. Gaster, D. A. Hall, C. H. Nielsen, S. J. Osterfeld, H. Yu, K. E. Mach, R. J. Wilson, B. Murmann, J. C. Liao, S. S. Gambhir and S. X. Wang, Nat. Med., 2009, 15, 1327–1332 Search PubMed.
- R. S. Gaster, D. A. Hall and S. X. Wang, Lab Chip, 2011, 11, 950–956 RSC.
- Y. Lee, S. Kim, T.-J. Song, S. X. Wang and J.-R. Lee, Anal. Chim. Acta, 2024, 1331, 343347 CrossRef CAS PubMed.
- D. L. Cortade, J. Markovits, D. Spiegel and S. X. Wang, J. Mol. Diagn., 2023, 25, 197–210 CrossRef CAS PubMed.
- V. D. Krishna, K. Wu, A. M. Perez and J.-P. Wang, Front. Microbiol., 2016, 7, 400 Search PubMed.
- S. Kim, J. Kim, J. Im, M. Kim, T. Kim, S. X. Wang, D. Kim and J.-R. Lee, Microchim. Acta, 2022, 189, 256 Search PubMed.
- S. Kim, S. X. Wang and J.-R. Lee, Biosens. Bioelectron.: X, 2023, 14, 100356 CAS.
- Q. Xu, M. R. Schlabach, G. J. Hannon and S. J. Elledge, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2289–2294 Search PubMed.
- J. G. Hacia, Nat. Genet., 1999, 21, 42–47 CrossRef CAS PubMed.
- K. Glynou, P. C. Ioannou, T. K. Christopoulos and V. Syriopoulou, Anal. Chem., 2003, 75, 4155–4160 Search PubMed.
- S. Kim, J. Im, S. X. Wang and J.-R. Lee, Anal. Chem., 2024, 96, 19447–19455 Search PubMed.
- Z.-J. Tan and S.-J. Chen, Biophys. J., 2006, 90, 1175–1190 CrossRef CAS PubMed.
- S. Raghunathan, T. Jaganade and U. D. Priyakumar, Biophys. Rev., 2020, 12, 65–84 CrossRef CAS PubMed.
- M. Ageno, E. Dore and C. Frontali, Biophys. J., 1969, 9, 1281–1311 CrossRef CAS PubMed.
- J. M. Sturtevant and E. P. Geiduschek, J. Am. Chem. Soc., 1958, 80, 2911 CrossRef CAS.
- S. A. Markarian, A. M. Asatryan, K. R. Grigoryan and H. R. Sargsyan, Biopolymers, 2006, 82, 1–5 CrossRef CAS PubMed.
- J.-R. Lee, D. J. Haddon, N. Gupta, J. V. Price, G. M. Credo, V. K. Diep, K. Kim, D. A. Hall, E. C. Baechler, M. Petri, M. Varma, P. J. Utz and S. X. Wang, ACS Nano, 2016, 10, 10652–10660 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab