
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Compartmentalized perfusion for temporal control of the chemical microenvironment of iPSC-derived cardiac cells†
Kaisa Tornberg *,
Wolfram Grötsch,
Niina Ritari,
Saara Haikka,
Lassi Sukki,
Katriina Aalto-Setälä,
Mari Pekkanen-Mattila and
Pasi Kallio
*,
Wolfram Grötsch,
Niina Ritari,
Saara Haikka,
Lassi Sukki,
Katriina Aalto-Setälä,
Mari Pekkanen-Mattila and
Pasi Kallio
Faculty of Medicine and Health Technology, Tampere University, Tampere, Finland. E-mail: kaisa.tornberg@tuni.fi
First published on 15th May 2025
Abstract
Organ-on-chip structures are predicted to have a significant influence in drug research. In these structures, perfusion can provide cells a more controllable environment to receive signaling molecules. In many current organ-on-chip applications, perfusion is used for shear stress stimulus for the cells, but it can also provide a more precise way of controlling the chemical microenvironment around the cells. In this paper, we propose an open-top organ-on-chip structure with compartment-specific perfusion to introduce stimulating molecules to cells with only minimal extra unspecific stimulus. Using numerical simulations, we show that shear stress sensed by the cells within the structure is low. We further validated the flow profile experimentally. We showed that the hiPSC-CMs accommodate to the flow environment where the shear stress is kept below 0.035 mPa. We also show that the beating rate of hiPSC-CMs increases due to the stimulation provided by chemical stimulant molecules introduced through the flow.
Introduction
In 2022, the FDA Modernization Act 2.0 created opportunities for organ-on-chip systems to be used in drug development and safety testing.1 Organ-on-chip technology combines microfluidic channel systems with live cells to model normal physiology, diseases, complex tissue architectures and electrical, mechanical and biochemical cues that cells face within the human body. Hence, this technology provides better tools for drug research.2 Microfluidic channel architectures in these systems go beyond just perfusion channels. These systems can capture the complex interactions taking place in the cellular microenvironments, containing different cell types, extracellular matrix (ECM) molecules and physiological cues. By including a flow of fluids actively controlled with pumps in the cell culture compartments, researchers have means to modify the cellular microenvironment and have control over the compounds the cells are exposed to. Additionally, introduction of flow enables control over cell-secreted substances.The in vivo formation and homeostasis of tissues and organs is orchestrated by a dynamic microenvironment, with blood flow serving as an essential mediator.3 In soft tissues, slow interstitial flow provides necessary nutrients, removes metabolic waste, and delivers signalling molecules that guide tissue and organ development and homeostasis.4 Correspondingly, adding perfusion to a cell culture compartment enables researchers to manipulate the chemical microenvironment of cells, allowing for the precise delivery of growth factors, cytokines, drugs and other chemicals, as opposed to manual medium exchange. When researchers currently add or remove culture medium containing stimulants, cells experience a sudden change in concentration with completely fresh medium, instead of the dose- and time-dependent exposure seen in in vivo pharmacokinetics.5–7 In addition, adding and removing medium disturbs cells by introducing a change in temperature, pH and shear stress.8 The changes in cell behaviour caused by these parameters can overlap with the cells' response to the stimulant and interfere with measurements.
While perfusion helps replicate an in vivo-like environment for cells, it does have its limitations. Culturing cells within closed perfusion channels still presents challenges in accurately positioning cells in their designated locations, despite efforts to optimize cell entrapment structures.9 These cell-seeding challenges can be avoided by open-top designs. Additionally, very complex microfluidic designs are not easily scalable, further hindering the throughput of these systems.10 The need for specialized equipment such as pumps for handling fluid flows increases costs.11 Avoiding the use of pumps and other fluid handling equipment creates additional challenges, such as maintaining the stability of flow for long-term studies. For example, gravity-driven flow is transient and rocker-assisted flow moves in two directions by nature.12 Engineering solutions to address this include optimized microfluidic structures to be used with 3D-tilting systems13 and slow-tilting tables14 to enable one-directional flow. Additional challenges in perfusion systems include interfacing issues11 and spontaneous bubble formation.15 Replicating in vivo flow conditions and shear stresses within chip platforms remains complex, as each tissue in the body possesses unique flow characteristics.16
Despite the limitations, perfusion has its substantial advantages in cell culture. Perfusion enhances the mass transport of nutrients and oxygen, improving the viability and growth especially with highly metabolically active cells and 3D cultures.9,17 Additionally, in some applications, it is the effect of the shear stress generated by the flow that is desired. For example, shear stress has been shown to enhance differentiation of gut cells,18,19 endothelial cells,20 and osteoblasts21 and guide more in vivo-like tissue barrier formation including those in the intestine,22 endothelial layers23,24 and blood–brain barrier.25 With numerical models, the detailed fluid dynamics can be resolved and further apply the results to observe the effect exerted on cells.22,26,27 In contrast to high shear stress applications, Wei et al. used low shear stress as a guiding principle in their perfusion system, as it was intended to be used with cardiomyocytes and other excitable cells.17 The reasoning was that in human physiology, cardiomyocytes are protected from the shear stress as vasculature mediates the blood flow. The device improved cell viability and was suitable for live-cell imaging, but it lacked features to promote direct cell–cell interactions.17 Liu et al. utilized another type of perfusion system enabling cell–cell contacts between cardiac cells, fibroblasts and endothelial cells. According to them, these cell–cell interaction promoted cardiac cells to have more in vivo-like responses.28 Both of these applications utilized perfusion to create a more in vivo-like environment for the cells. However, they did not utilize perfusion in more in vivo-like pharmacokinetic studies, even though perfusion could provide highly precise temporal control of drug concentrations.
Perfusion facilitates the transition from organ-on-chip devices to body-on-chip systems. In this approach, various cell types are linked together via perfusion or using microstructures enabling cell-to-cell contacts. Perfusion facilitates downstream signaling and exchange of metabolites.29,30 Multi-organ systems are compelling for drug studies as they more closely model the whole-body drug responses.2 However, these advancements do not come without limitations. Picollet-D'hahan et al.31 list challenges for these multi-organ platforms. One challenge is the common medium circulation. It is challenging to find a culture medium that can support the growth, viability and functions of various cell types.31 Even as the tissues are allowed to mature and differentiate within organ-specific media in their individual tissue modules, challenges arise after their integration and shared medium perfusion.32 Another challenge to consider is how to ensure that different cells remain within their in vivo-like shear stress environment when multiple organs are connected via a common medium flow.31
Advancing compartment-specific perfusion is crucial to meet the needs of multi-organ cultures and ensure that each organ experiences its physiological flow environment. To categorize compartmentalized perfusion systems, Zhang et al. have divided them roughly into two approaches: vertical approach and horizontal approach.33 In the vertical approach, two channels34–36 or other perfusable compartments22,37 are separated from each other by porous membranes, allowing cells to be in contact via this planar plane of pores. In the horizontal approach, micropillars,38,39 phase guides40–42 or surface treatments43 constrict hydrogels and other 3D matrixes to a central compartment creating side walls to flanking channels, allowing for the creation of multiple such compartments in a scalable manner44,45 and with varying geometries.46 When positioning compartments in parallel, cell–cell interactions can be observed in a single horizontal plane, enabling real-time imaging. As the side channels are most often separated from each other by hydrogels or similar biomaterials, they do not completely restrict solutes from diffusing from one compartment to another, even though cellular barriers are established within these interfaces.47 Therefore, the compartments cannot completely separate chemical environments from each other. Additionally, the formulated 3D barriers are subjected to mechanical forces introduced by the flow. In particular, ECM mimicking materials are not particularly durable and possess some degree of biodegradability;48 thus the degradation of barriers changes the fluidic behavior within these platforms. Further approaches developed for compartmentalized perfusion, while maintaining cell–cell interactions between the compartments, include microfeatures such as microchannels and microgrooves in rigid transparent materials, such as polydimethylsiloxane (PDMS)49 or glass.50
To enhance the physiological relevance of organ-on-chip models and to improve their utility for drug toxicity and efficacy testing, human-induced pluripotent stem cell (hiPSC)-derived cells are used.2 As an example, hiPSC-derived cardiomyocytes (hiPSC-CMs) hold the potential to predict the cardiotoxic effects of drugs and other stimulants. Toxicity and drug efficiency studies have been carried out in perfusion platforms either containing cardiomyocytes alone or in combination with other tissue types. The flow is introduced either by gravity within the chip51,52 or produced outside the chip using a syringe53,54 or peristaltic pumps.55 The most common methods to evaluate drug effects and responses with hiPSC-CM models include recording electrophysiological activity, calcium imaging for measuring changes in the excitation–contraction coupling and contractility assays utilizing optical methods to measure the contractile properties reflecting the mechanical functionality of cardiomyocytes.56 Combination of such measurements with precise spatiotemporal control of drug molecules allows researchers to study drug mechanisms and interactions at a detailed level using human-based cell models.
In this paper, we introduce an organ-on-chip device with the following attributes: the device features an open-top design, enabling cell seeding from the top rather than exclusively relying on the perfusion channel. For co-culturing purposes, the device contains three separate culture compartments connected via microtunnels, arranged in planar configuration for real-time imaging of cells. Perfusion is compartmentalized to one of these compartments, making it the only compartment exposed to flow and drug stimulation. A perfusion channel is established within this defined compartment to deliver stimulating molecules at flow velocities similar to those in soft tissue interstitial space.4 The device enables observing the cells' live response to temporary changes in their chemical environment without additional interventions. We characterize the device and the established perfusion function in detail and show that relatively high flow rates can be used in the structure while the cells are protected from high shear stress. We resolve the fluid dynamics using numerical simulations in COMSOL and characterize the key flow features in laboratory experiments. We accommodate the structure with human-based iPSC-CMs and analyze changes in their beating behavior when exposed to low shear stress flow and temporary adrenaline stimulation.
Results and discussion
Chip structure
In order to have control over the chemical microenvironment of the hiPSC-CMs, a perfusion channel was established to a previously published 3-compartment chip57 through a middle cell culture compartment (Fig. 1a and S1†). The inlet and the outlet of the chip are connected to a 17 mm long and 200 μm high perfusion channel guiding the flow through a 2.2 mm high cell culture compartment. The perfusion channel is separated from the cell culture area (5 mm × 4 mm) by an array of equilateral triangular pillars with a side length of 0.3 mm and spacing of 0.15 mm (ESI† Fig. S1). The pillars are included to restrict the cell growth solely inside the cultivation area. An array of microtunnels with 100 μm spacing and dimensions of 250 μm long, 10 μm wide and 3.5 μm high lead from each side of the middle cell culture compartment to the side compartments, providing fluidically restricted connection (ESI† Fig. S1). This allows compartmentalization of different cell types within the same platform, with chemical microenvironment tailored for each cell type.58 | ||
| Fig. 1 The chip structure and the system setup for observing the beating of cardiomyocytes under continuous perfusion. (a) The base of the chip (at the left and right lower corners) includes the cell culture compartments and the perfusion channel, which can be accessed through the openings on top. These openings enable cell seeding and provide access to medium reservoirs (left top corner) during stationary experiments. The medium reservoirs increase the volume of the cell culture medium, further supporting cell culture in these compartments. Once the chip is used for perfusion experiments, the medium compartment is replaced with a sheet of PDMS (top right corner) to seal the openings and to form a closed channel. (b) The inlet side of the perfusion setup consists of two syringe pumps; one filled with culture medium and the other with culture medium including 1 μM adrenaline, both connected to a recirculation valve. The valve injects adrenaline to the inlet stream of the chip. The chip is placed into an incubator box, where temperature and 19% oxygen and 5% carbon dioxide levels are maintained constant throughout the experiments. A third syringe pump withdraws medium from the chip aiding the medium supply through the chip. Beating of the human-induced pluripotent stem cell-derived cardiomyocytes is recorded in different time points with a camera attached to an optical microscope. Created in BioRender. Tornberg, K. (2025) https://BioRender.com/b05j377. | ||
Chip design principles and the system setup
The following requirements were defined for the chip design: (1) accessible from above to facilitate cell seeding, (2) possibility for co-culture, (3) live imaging and (4) introduction of active flow to cells with low shear stress.In order to meet the first two requirements, we utilized the 3-compartment chip used for neuron–cardiomyocyte58 co-culture and introduced perfusion modification. By plasma-bonding the chip on top of a cover slip, the cells are easily observed and imaged live with a microscope. The chip structure consists of two layers. The plasma-bonded base contains the cell culture compartments and the perfusion channel, remaining unchanged throughout the experiments (Fig. 1a). For perfusion experiments, the top layer can be exchanged for a polydimethylsiloxane (PDMS) sheet in place of medium reservoirs. Within the base, directly above the square-shaped cell culture compartments, there are openings designed to facilitate cell seeding. Placing oval-shaped medium reservoirs on top increases the culture medium volume, supporting the cell culture in a stationary setup and further ensuring that the cells adhere properly to the cell culture areas. For perfusion experiments, the openings in the cell culture compartments need to be sealed to create a closed channel, with access limited through the inlet and outlet. To achieve this, we utilized the reversible bonding property of PDMS and replaced the medium reservoirs with a rectangular PDMS sheet, sealing the openings of the cell culture compartments (Fig. 1a).
The chip is placed into an incubator box of a microscope system and cell medium flow of 5 μl min−1 is introduced using a syringe pump. In the inlet side of the chip, the system includes two pumps connected to a microfluidic recirculation valve, which controls whether a pure culture medium or a culture medium containing 1 μM adrenaline is supplied to the chip. When adrenaline is used, a 15-minute adrenaline injection is followed by flushing out with the culture medium. A third syringe pump is connected to the chip outlet to aid in pulling the medium through the chip (Fig. 1b). The flow conditions sensed by the hiPSC-CMs are studied with numerical simulations.
Chip characterization
 | ||
| Fig. 2 Shear stress values resolved with numerical simulation along the perfusion channel. (a) Schematic of the bottom of the chip showing the fluid channel geometry. The red line highlights the positions along the channel, where shear stress values are resolved using numerical simulations and depicted in (b). (b) When shear stress at a flow rate of 5 μl min−1 along the perfusion channel is compared with and without triangular pillars, the only visible difference is noticed in the areas between the pillars approximately at 5.9 mm and 11.1 mm from the beginning of the inlet. | ||
 | ||
| Fig. 3 Numerical simulations and measurements reveal details of the flow within the perfusion channel. In all panels, simulated 5 μl min−1 flow is from left to right. (a) Shear stresses within the channel and cell culture compartment along the ZX-plane (top) and YZ-plane (bottom). The shear stress is higher at the start and end where the channel is narrower. Towards the cell culture compartment, it decreases, reaching its lowest value in the cell culture area due to its height. The shear stress peaks around the triangular pillars, but this does not affect the shear stress within the cell culture area. (b) Streamlines of flow through the cell compartment. (c) Arrow field showing the relative velocities of simulated particles flowing through the cell compartment. (d) Particle tracking result showing relative velocities of measured particles to proof the arc-shaped flow profile and height-dependent velocity profile. | ||
The detrimental effects of perfusion occur when modelling organ systems, where tissues are protected from shear forces by the surrounding vasculature, such as the brain. It has been shown that shear stress below 500 mPa has a minimal effect on neuronal viability and differentiation,60,61 but shear stress above 1000 mPa has been shown to decrease the viability of cortical neurons, especially when applied for more than 12 hours.61 In contrast, a shear stress value of 1000 mPa has been used to induce differentiation of cardiomyocytes from mesenchymal stem cells.62–64 Correspondingly, cardiac cells derived from hiPS cells and other sources are known to benefit from mechanical65,66 and electrical stimulation.67,68 To enhance cell differentiation using shear stress, the stimulus is typically applied long term.
In the short term, for example, when monitoring live cell responses to drugs, shear stress should be kept below values that are known to induce mechanosensitive responses in cells. Figallo et al.69 considered 100 mPa as the threshold below which shear stress does not affect cells. They reported a chip configuration with shear stress values as low as 0.1 mPa.69 The shear stress values reported by Figallo et al. resemble the values of the present study (Fig. 3a). However, Figallo et al. used a flow rate as low as 0.3 μl min−1 to achieve the desired shear stress and it was much lower than the flow rates used in this study.69 At very low flow rates, the dead volume of a chip system and its tubing significantly affects the speed at which signalling molecules can be flushed in and out of the chip. In other words, when using low flow rates in systems with high dead volume, the delivery of supplied molecules is slow. Within the chip proposed here, the flow rate can be increased up to 280 μl min−1, while shear stress still remains low, under 100 mPa (ESI† Fig. S3).
The low shear stress values in the cell culture compartment (Fig. 3a) are related to the development of the flow within the perfusion channel structure. As the flow passes the pillars, the flow profile changes and the stream lines are no longer parallel to bottom of the channel (Fig. 3b). In the cell culture compartment, the flow profile follows an arc where the flow velocities are lowest at the top and bottom and highest in the middle (Fig. 3c). A flow rate of 5 μl min−1 results in velocities of ∼14.5 μm s−1 in the centre of the chip at 1.1 mm height (Fig. 3c). Similarly, in the centre of the chip, at 200 μm height, the velocity is only 5 μm s−1 (Fig. 3c). Thus, the velocity varies along the height of the cell culture compartment. Moreover, close to the inlet and the outlet the velocity can get as high as 130 μm s−1 (Fig. 3c). Since this arc shaped flow and its height dependent velocity is crucial for the function of the chip, it was experimentally validated.
Validating the simulated flow profile and velocities
To confirm the simulated arc-shaped flow pattern within the cell compartment, we added particles to the simulation model (Fig. 3c). To measure the same phenomenon in the laboratory, we supplied 6 μm diameter polystyrene particles through the compartment at a flow rate of 5 μl min−1. Video recordings verified the arc-shaped flow and its height dependent velocity profile (ESI† Video S1). Particle tracking software was used to visualize the movement of the polystyrene particles (Fig. 3d). The results show that the shape of the arrow field and the relative velocities of the particles closely match with the simulation (Fig. 3c and d).Supply of stimulating molecules to the cells
To study how the supplied stimulant molecules reach the cells, we estimated the dead volume in the tubing system and calculated the time required for the molecules to arrive at the chip. This was done by 15 minutes of fluorescein injection and observing the pillars on the inlet side. The dead volume of the tubing is estimated to be between 210 and 230 μl. With a flow rate of 5 μl min−1, the estimated time of arrival for the molecules is between 42 and 45 minutes. Based on acquired fluorescent images, it takes 42 minutes for the fluorescein molecules to reach the pillar area (Fig. 4a) which corresponds to calculations. The fluorescein signal reaches its peak intensity at a time point of 58 minutes. From the fluorescein measurements, we observe that after 80 minutes, all molecules are completely flushed out from the pillar area (Fig. 4a). This proves that the supply of molecules is transient and molecules are available to the cells longer than at the initial injection time of 15 minutes. | ||
| Fig. 4 Study of the arrival of molecules provided by the flow to the chip and the cells adhering to the cell culture area. (a) Fluorescent images at four time points where fluorescein reaches the chip inlet pillars after a 15-minute injection followed by flushing. Measurements were conducted to determine the time delay between switching the valve and arrival of a substance at the cell culture compartment. At 42 minutes after switching the valve, the first fluorescein molecules arrive at the pillar area, indicated by a higher intensity, higher than that in the reference image (the 10-minute time point). The highest fluorescein intensity is seen at 58 minutes, and by 80 minutes, all fluorescein molecules are flushed out. A 15-minute injection of 1 μM adrenaline followed by a flush was simulated and the average adrenaline concentration over time in three regions as shown in (b) were studied. The regions include the inlet (blue) and outlet (magenta) regions with a 150 μm × 150 μm area and the middle region (yellow) with a 2 mm × 2 mm area. (c) Plot of the simulated arrival of adrenaline molecules in the inlet, middle and outlet regions, and (d) a comparison of the simulated arrival of adrenaline molecules (blue) at the inlet region with the measured arrival of fluorescein molecules shown as normalized intensity (green) over time. | ||
We simulated a constant 5 μl min−1 flow within the chip structure with 15-minute injections of 1 μM adrenaline through the inlet boundary at time point 0, following a flush out. We plotted the changes in adrenaline concentration in regions from both the inlet and the outlet sides within a 150 μm × 150 μm area as well as in the middle region within a 2 mm × 2 mm area (Fig. 4b). The inlet region reaches a concentration of 1 μM in 6.5 minutes, maintains this level until 15 minutes and approaches zero by 21 minutes (Fig. 4c). The middle region reaches its peak concentration at 14.5 minutes, displaying a more transient nature, and starts to decrease immediately afterward. By 30 minutes, the concentration approaches zero. In the outlet region, the molecules are diluted, resulting in a peak concentration below 1 μM which is reached in 18.5 minutes. The concentration approaches zero by 43 minutes. The simulations indicate an 8-minute time difference between the inlet and the middle regions in reaching their concentration peaks, and a 12-minute difference between the inlet and the outlet regions (Fig. 4c).
When comparing simulation data with the measured fluorescent intensity data, we can observe that the measured intensities spread over a wider time window due to Taylor dispersion (Fig. 4d). Taylor dispersion states that an injected pulse of molecules is dispersed along the flow direction in the solvent flow and it is caused by both advection and diffusion.70 Taylor dispersion occurring in the tubing leading to the chip is not included in the simulation, as it primarily takes place outside the chip, while the simulations focus on the dynamics of the chip itself. Additional assumptions for the system include constant temperature and atmospheric pressure. In addition, water is used as the solvent instead of cell culture medium and no evaporation, leakage or cells are included in the model. The inflowing and outflowing stimulant concentrations are considered to be equal, meaning that concentration-reducing effects such as cellular uptake or absorption of stimulant molecules by PDMS are neglected. Nevertheless, the model allowed us to study the chip dynamics in a greater detail than sole flow experiments. Taylor dispersion also states that high diffusion coefficients lead to smaller dispersion than low diffusion coefficients. Since fluorescein (diffusion coefficient 0.42 × 10−5 cm2 s−1)71 has a lower diffusion coefficient than adrenaline (diffusion coefficient 1.03 × 10−5 cm2 s−1),72 it disperses more widely, broadening the solute pulse and offering a longer time window for its presence in the system compared to adrenaline. Thus, it makes a good estimator for arrival and flush out of the adrenaline. We address the importance of the diffusion and recognize that the system is partially diffusion dependent. We took this into account within the dynamics study. Therefore, to study the adrenaline supply to the cells within this system, initial flow simulation was expanded by a transport of diluted species module which allows for the simulation of the movement of a solute based on diffusion governed by Fick's first law (eqn (1)).
Due to the arc-shaped flow profile, most of the adrenaline will flow above the cells, as shown in the simulated results in Fig. 5a. We further simulated how varying diffusion coefficients of stimulant molecules and different flow rates affect their arrival times in the defined region in the middle of the cell culture area (the region depicted in Fig. 4b). The injection duration of adrenaline was kept constant at 15 minutes. Fig. 5b illustrates the simulation results and shows simulated concentration profiles for two diffusion coefficients at different flow rates. The higher the flow rate, the more step-like the change in the signalling molecule concentration response becomes (Fig. 5b). At the flow rate of 1 μl min−1, the 1 μM concentration is not achieved since the total volume supplied within the 15-minute injection time is only half of the cell culture compartment volume diluting the signalling molecule. This represents a different peak concentration system and was omitted from the follow-up analysis. When comparing cases where the 1 μM concentration is reached, the areas under the curve (AUCs) of the two molecules in all three flow rates (5 μl min−1, 25 μl min−1 and 280 μl min−1) differ from each other by less than 1% and are close to a value of 0.9. Thus, the total exposures of the molecules are very close to each other in each case (Fig. 5c). The diffusion dependency of the system is best illustrated by comparing the rise times – the time required for the concentration to increase from 10% to 90% of its maximum value – between two molecules with different diffusion coefficients at the same flow rate. The fluorescein molecule exhibits a slightly longer rise time than adrenaline across all studied flow rates due to its smaller diffusion coefficient. This demonstrates that the system is partially diffusion-dependent and thus influenced by the diffusion coefficient of the supplied molecule. However, it is notable that the difference between the rise times becomes smaller as the flow rate increases (Fig. 5d). Thus, the higher the flow rate within the chip, the less dependent it is on the diffusion coefficient of the stimulant molecule. The downside is that the shear stress within the chip increases linearly when the flow rate is increased (ESI† Fig. S3).
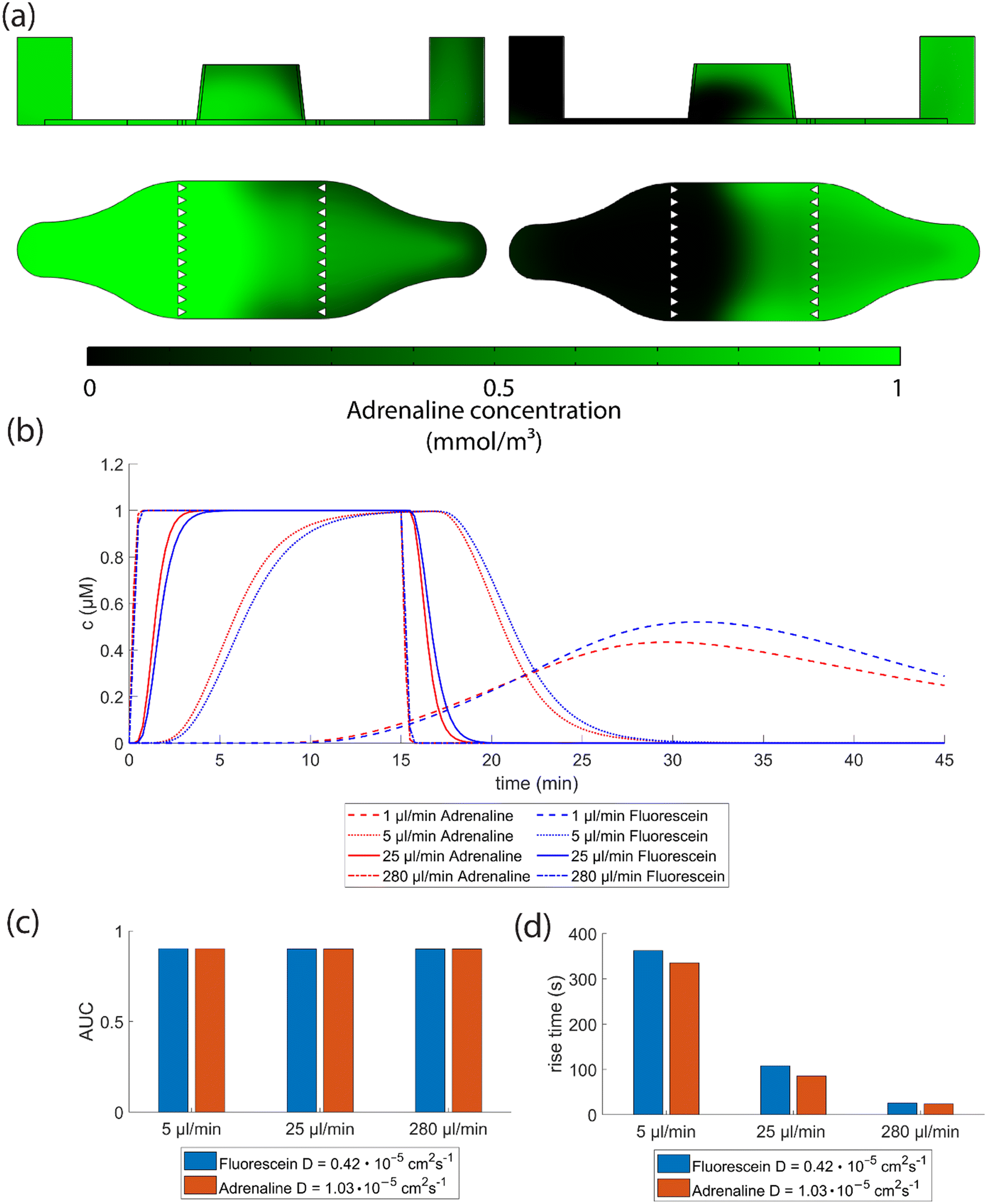 | ||
| Fig. 5 Simulations reveal that diffusion and diffusion coefficients of the molecules within the perfusion system contribute to the system dynamics. (a) Simulations illustrate the concentration of adrenaline in the YZ and ZX plane at two time points: arrival at 10 minutes (left) and flush out at 22.5 minutes (right). The figures highlight the interaction between the arc-shaped flow and diffusion. The simulated arrival of a 15-minute injection of fluorescein (diffusion coefficient of 0.42 × 10−5 cm2 s−1) and adrenaline (diffusion coefficient of 1.03 × 10−5 cm2 s−1) molecules into a defined area within the cell culture compartment is plotted in (b), depicting the concentration profile over time with varying flow rates. (c) The area under the curve (AUC) calculations for both fluorescein and adrenaline molecules show that at flow rates where a maximum concentration of 1 μM is achieved, the AUC is similar across different flow rates. (d) Comparing the effect of the diffusion coefficients of fluorescein and adrenaline molecules on the concentration rise times reveals that the difference due to different diffusion coefficients diminishes as the flow rate increases. | ||
Cell responses to the flow
Human iPSC-CMs were plated onto gelatin-coated chips for cell stimulation experiments. Immunocytochemistry using the cardiac marker myosin binding protein C (MyBPC3) confirmed that the plated cells were cardiomyocytes, exhibiting sarcomere structures (Fig. 6a). The cells were seeded in such a density that they were either single cells or clusters of a few cells, allowing confinement of their own regions of interest (ROIs) for analysis (Fig. 6b). As the hiPSC-CMs within the chip were not in contact with each other, they did not exhibit synchronized beating behaviour; instead, each cell/cell cluster had a distinct beat rate (ESI† Fig. S6). We studied how the cardiomyocytes respond to the flow by analysing their beating frequency. We studied the beating rates across multiple chips and ROIs by normalizing the beating rates to the values that were recorded before the start of the flow. Normalized values showed an increase within the beating frequency at the start of flow, but the beating rate levels out after 60 minutes (see ESI† Video S2). Thus, as presented in Fig. 6c, cells accommodate to their new perfusion environment within 60 minutes after the start of the flow, and once the accommodation period is complete, the stimulation experiment can begin. | ||
| Fig. 6 Human-induced pluripotent stem cell-derived cardiomyocyte response to the flow and adrenaline molecules carried within the flow. (a) The plated hiPSC-CMs express the cardiac specific marker MYBPC3 (red). Nuclei were stained with DAPI (blue). (b) Individual ROIs cropped from the phase contrast images containing beating cell clusters. (c) Depicts the change in the beating frequency during the time period of 110 minutes and depicts that after 60 minutes the beating rate is stabilized between 1 and 1.5. (d) Simulated results fitted to the dataset of measured beating rates of hiPSC-CMs during the stimulation experiment. The blue line depicts the arrival of adrenaline molecules to hiPSC-CMs. Increase in the beating frequency can be seen simultaneously with the arrival of adrenaline molecules. The beating frequency doubles and remains elevated at least 8 minutes after the concentration peak. The increase in the beating frequency is temporal as 18 minutes after the peak concentration the normalized beating frequency returns to the 1–1.5 range. In all figures, the beat rate is normalized to the values before the start of the flow and represent averages ± standard error of mean (SEM) across three separate chips (where a total of six ROIs were analyzed (n = 6)). | ||
Beating response to the stimulant molecule supplied with the flow
To demonstrate how molecules carried within the flow stimulate cardiomyocytes, we choose adrenaline as the stimulating agent for hiPSC-CMs. As the arrival of the adrenaline molecules onto the cell culture area cannot be experimentally measured, we used fluorescein molecules to estimate the arrival of adrenaline molecules onto the chip, as discussed earlier. We plotted the simulated change in the adrenaline concentration together with normalized hiPSC-CM beating rates to see if we can increase the beating rate of hiPSC-CMs by supplying adrenaline into the flow stream (Fig. 6d).Fig. 6d shows that once the adrenaline concentration starts to increase the normalized beating rate of hiPSC-CMs increases gradually. Here, the arrival of the adrenaline molecules was estimated by fitting the simulation data so that the positions of the centre of the peaks in the measured inlet area and simulated inlet area are aligned (Fig. 4d). After the alignment, the simulated change in the adrenaline concentration in the middle region is plotted into Fig. 6d together with normalized beating rates, as it is corresponding to the position of the measured hiPSC-CMs. Recognizing the influence of Taylor dispersion in our experimental measurement system, adrenaline is expected to be available for cells sooner than simulations suggest. Fig. 6d shows that once the system reaches peak concentration, the normalized beating rate is elevated and remains elevated until the 60-minute time point, which is approximately 8 minutes after the peak concentration is achieved (see ESI† Video S3). By the 70-minute time point, as the adrenaline molecules are flushed from the cells, the beating frequency returns approximately back to the initial level. Our system reaches the peak concentration in 14.5 minutes, which resembles time constants that are measured from patient samples collected after a subcutaneous administration of adrenaline.73 In order to model drug delivery methods where more rapid peak concentrations are achieved, for example, intravenous administration,74 the peak concentration should be achieved within a minute. To achieve concentration rise times within a minute scale, the flow rate can be increased up to 25 μl min−1 (Fig. 5d). This naturally affects the shear forces experienced by cells, but the maximum shear stress remains under 10 mPa (ESI† Fig. S3). Modelling this in static in vitro cultures, where the desired stimulant concentration must be pipetted onto the cells and then replaced with culture medium to flush the molecules from the cell environment, the proposed system better captures in vivo-like pharmacokinetics of the stimulants, providing the capability for continuous monitoring of cell responses in real time. In static systems, cells are exposed twice to a step change in their environment along with shear stress forces resulting from manual pipetting. Thus, in static systems, the response to a stimulant molecule is hard to distinguish from the stress induced by medium exchange. The proposed flow-based system enables the delivery of molecules into a flow environment to which the cells have already adapted, allowing the collection of more detailed data of the cell responses to the studied molecules.
Materials and methods
Chip fabrication
In the chip fabrication, an SU-8 mold was produced using a multilayer method. First, the channel designs were created in SolidWorks (Dassault Systèmes, Vélizy-Villacoublay, France) and finalized in AutoCAD (Autodesk, California, USA) to a format compatible with the direct-writer photolithography system μPG 501 (Heidelberg Instruments Mikrotechnik GmbH, Heidelberg, Germany). Two chrome-on-glass masks were fabricated such that the first one contained an array of 10 μm wide microtunnels and the second one contained the cell culture areas and the perfusion channel including pillars. To fabricate the microtunnels utilizing the first mask, SU-8 5 was spin coated to a thickness of 3.5 μm and developed on a silicon wafer. SU-8 3050 was then spin coated to a thickness of 200 μm and developed utilizing the second mask. Before spin-coating, the wafer was treated using oxygen plasma at 30 W for 2 minutes in Vision 320 Mk II RIE (Advanced Vacuum, Malmö, Sweden). The cell culture compartments were 3D-printed similarly as previously.58Trichloro(1H,1H,2H,2H-perfluorooctyl)silane was used to coat the mold to improve demoulding properties (note that storing and working with trichloro(1H,1H,2H,2H-perfluorooctyl)silane involves serious hazards and risks, see ESI† section 6). Polydimethylsiloxane (PDMS) was mixed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 cross linker:polymer ratio and poured on the fabricated mold. PDMS was cured at 60 °C for 10 h and cut after demoulding. The cell culture compartments were punched with 3 mm and 2.5 mm biopsy punches and inlets and outlets with a modified 18G needle. The medium reservoirs were fabricated using laser cutting as previously published58 with the following modification to the protocol; the PDMS sheets were covered with a thin layer of dish soap to prevent laser cut debris adhering on the component surfaces.
:10 cross linker:polymer ratio and poured on the fabricated mold. PDMS was cured at 60 °C for 10 h and cut after demoulding. The cell culture compartments were punched with 3 mm and 2.5 mm biopsy punches and inlets and outlets with a modified 18G needle. The medium reservoirs were fabricated using laser cutting as previously published58 with the following modification to the protocol; the PDMS sheets were covered with a thin layer of dish soap to prevent laser cut debris adhering on the component surfaces.
Numerical model
Shear stress, flow profile, particle transport and drug dynamics in the cell culture compartment were studied using convection and diffusion. The numerical models (ESI† Fig. S2) were implemented in 3D using a commercial finite-element modelling tool, COMSOL Multiphysics® Version 6.1. All models utilized a mesh based on COMSOL's predefined ‘fine’ mesh size. Both the stationary and time-dependent studies were solved using the MUMPS solver.Flow velocities and shear stress were simulated using a Laminar Flow node applicable due to the low flow velocities and small geometry. It was run using a stationary solver governed by the Navier–Stokes equations for incompressible water flow.
The particle simulation was implemented using the particle tracing in fluid flow module. It was run using a time-dependent simulation with a duration of 90 min and a timestep of 30 s. Particles were continuously released along the inlet boundary. The particles were spherical with a 6 μm diameter and used polystyrene as material. Particles were set to “bounce” off the wall. The module was coupled to the resulting velocity field of the laminar flow node with the Oseen correction as the applied drag law.
Stimulant transport was modelled using the transport of diluted species which used Fick's first law as the governing equation for diffusion:
 | (1) |
| R = ∇J + u × ∇c, | (2) |
The results were exported from COMSOL either to MATLAB (The MathWorks, Massachusetts, USA) or to Excel (Microsoft, Washington, USA) for plotting. For the AUC calculations, a built-in trapezoidal approximation function was used in MATLAB. For the rise time calculations, the built-in MATLAB functions were used.
Flow characterization
To enable visualization of the particles within the cell compartment chip, we modified a plasma-bonded chip by cutting it along the edge of the cell compartment in the flow direction. The chip was then re-sealed by plasma-bonding a clear 400 μm thick membrane onto the cut surface and the glass plate. The chip was placed horizontally on an XY table and imaged 22.66 frames per second from the side through the PDMS sheet using an IDS CP3370CP-C-HQ color camera (IDS Imaging Development Systems GmbH, Obersulm, Germany). A mixture of 4 ml DI water and 10 μl of 6 μm polystyrene particles (DRP6000, Lab261, California, USA) was prepared in a 5 ml syringe and pumped through the chip using a syringe pump at a flow rate of 5 μl min−1. For particle tracking analysis, two frames (ESI† Fig. S5) 12 frames apart were cropped and extracted from the acquired video and analysed with particle image velocimetry (PIV) code by A. F. Forughi (March, 2014)75 within MATLAB. For visualization purposes, the acquired video was further edited in Adobe Premiere Pro (Adobe, California, USA) by cropping and adjusting the brightness and contrast while speeding up the real-time video to eight times faster (ESI† Video S1).The system and system characterization
The chip was placed in an incubator box of an inverted microscope system (DMi8 Leica Microsystems, Wetzlar, Germany) and the flow at 5 μl min−1 was introduced by syringe pumps (AL-1000, World precision instruments, Florida, USA). The pumps providing liquids into the chip were connected to a microfluidic recirculation valve (MUX Recirculation 6/2, Elvesys, Paris, France) with Tygon (I.D. 0.030′′ O.D.0.090′′) tubing (Saint-Gobain, Paris, France). Two pumps, one containing pure medium and one containing medium and 1 μM adrenaline, pushed liquid into the chip and one syringe pump was connected to the outlet to aid in pulling the liquids through the chip.For characterization purposes, 1 μM fluorescein sodium salt (Sigma Aldrich, Missouri, USA) was perfused through the system at a 5 μl min−1 flow rate. Before measurement, all the tubing was primed with culture medium. At the start of the measurement, the valve system directed fluorescein to the tubing that led to the chip. After 15 minutes, the valve was turned to flush out the fluorescein with culture medium. During the measurement, fluorescence images from the cell and pillar regions were captured with a 2-minute interval using a Leica DMi8 microscope. Acquired images were analysed in Fiji ImageJ software (1.53f51, NIH, USA) using a rectangular (200 pixels × 200 pixels) area to measure the intensity. The intensities were normalized by scaling each value to the maximum measured intensity, resulting in a range from 0 to 1 to show the dynamics of how fluorescein reaches the chip and is flushed out.
Chip preparation and cell culture
The human iPS cell line used in the present study was UTA.14802.WT produced by reprogramming peripheral blood mononuclear cells collected from a healthy donor. The iPS cell line used was acquired from a voluntary subject who had given written and informed consent. The iPS cell line was reprogrammed at Tampere University by Prof. Aalto-Setälä's research group, which has a supportive statement from the ethics committee of Pirkanmaa Hospital District, Finland, to generate iPSCs from human donor cells (R08070 and R12123) and all experiments were performed with approval in accordance with relevant guidelines and regulations. The pluripotency of the line was confirmed regularly, and the cultures maintained normal karyotypes and were free of mycoplasma. The iPSC-CMs were differentiated and plated similarly as described earlier.58Chips and coverslips were sterilized with 70% ethanol and left to dry over nigh in a laminar hood. Chips were bonded to cover slips with air plasma (Pico, Diener Electronic GmbH + Co. KG, Ebhausen, Germany). Chips were left to bond for 10 minutes and immediately after 0.1% gelatin was pipetted to the channels. Chips were left with the gelatin solution overnight in an incubator. Before cell plating the solution was removed and replaced with cell culture medium and incubated for a minimum of 30 minutes in the incubator. 8000 cells were seeded into the middle cell culture compartment and 4000 cells were seeded into the side cell culture compartment to represent the coculture aspect. Cells were cultured in KnockOut-DMEM (KO-DMEM Gibco) containing 20% fetal bovine serum (FBS; Sigma-Aldrich, Saint Louis, MO, USA) 1% non-essential amino acid (NEAA; Euroclone, Pero, Italy), 1% GlutaMAX (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) and penicillin/streptomycin (Pen/Strep; Euroclone, Pero, Italy). Cells were left to attach for four days and after that the cell culture medium was changed every 2–3 days only in the medium compartment. The cells were cultured for a minimum of 7 days within the chip until exposed to the perfusion environment.
Cardiomyocyte imaging and analysis
Cell responses were captured in phase contrast images of cardiomyocytes with the Leica DMi8 microscope 5× objective. The iPSC-CMs were imaged every 10 minutes during the flow experiment without adding any stimulants and every 10 minutes before and after the adrenaline stimulation. Upon arrival of the adrenaline molecules, the iPSC-CMs were imaged every 2 minutes. At each time point, images were acquired for 2 minutes with a frame rate of 12 frames per second. 1–3 regions of interest (ROIs) were chosen from each chip for further analysis. These ROIs were exported as AVI files and analysed with MUSCLEMOTION on the ImageJ software.76,77 The software automatically detected each beat and the beating rate of each ROI was calculated. Mean beating rates and standard error of the mean across six ROIs were calculated in MATLAB excluding outliers with a detected beat rate of zero. All normalized beat rates across whole measurements in all ROIs are plotted in ESI† Fig. S7. ESI† Videos S2 and S3 were exported from the cropped microscope files at a frame rate of 12 frames per second.Immunocytochemistry
Cells were fixed in 4% paraformaldehyde after perfusion experiments. Samples were blocked with 10% normal donkey serum (NDS; Biowest, Riverside, Newry and Mourne, UK) for 45 minutes. Myosin binding protein C3 was stained against mouse monoclonal IgG MYBPC3 (1:200, Santa Cruz Biotechnology, Dallas, TX, US) at 4 °C overnight. Donkey anti-mouse Alexa Fluor 568 (1:400, Thermo Fisher Scientific, Waltham, MA, USA) was used as secondary antibody and incubated for 1 h. Finally, Vectashield including DAPI (Vector Laboratories Newark, CA, USA) was added to the cell culture compartments to stain the nuclei. Cells were imaged with an Olympus IX 51 fluorescence (Olympus, Tokyo, Japan) microscope. Images were edited with Fiji ImageJ software.
Conclusions
In this paper, we introduced compartment-specific perfusion to an organ-on-chip platform. As all the compartments have open tops, uniform seeding of cells within all cell culture compartments, including the perfusion channel, is straightforward. We characterized the shear stress values across the chip and demonstrated that the shear stress experienced by the cells is low as desired to represent interstitial flow in soft tissues. The low shear stress within the cell culture area of the chip is a result of the flow profile in the cell culture compartment. This was validated through both simulations and laboratory experiments. Due to the flow profile, the system is partially dependent on the diffusion coefficient of the supplied molecules. In the future, this established numerical model can be used to precisely estimate the time it takes for studied molecules to reach the cells and further reveal details of the cell responses. The studied structure can be easily combined with micro electrode arrays to record the electrophysiological responses. Combining knowledge of the detailed molecular arrival at the cells with the recorded electrophysiological data could reveal even more precise details on the cell responses, allowing more precise pharmacokinetic studies, for example. Furthermore, using hiPSC-CMs, we demonstrated that the proposed chip is suitable for chemical stimulation of cells. We showed that hiPSC-CMs adapt to the introduced flow environment and respond to adrenaline stimulation carried out within the flow. We have shown that the platform is suitable for optical microscopy, so further analysis such as calcium imaging can be applied. Our previous studies have shown that other cell types can be cultured in the compartments within their own microenvironment. However, the current design allows for the co-culture of cells, where only one compartment is exposed to flow and the stimulation it brings. In the future, perfusion is intended to be implemented in all compartments, allowing the establishment of multi-tissue models with a modifiable microenvironment regulated by flow.The current version of the structure still faces challenges with interfacing, as Taylor dispersion was seen to disturb the direct comparison of the numerical model with the real-life measurements. Additionally, while the use of syringe pumps allows for the establishment of steady flow conditions within the chip and, together with the valve system, offers precise temporal control of the cellular microenvironment, these pieces of equipment may pose limitations for certain studies. The chip, combined with human-based cells, can be used for drug studies to gain a better understanding of the dynamics, effects and responses of drugs in target tissue within the human body.
Data availability
Data sets, CAD models and numerical models generated and analysed for this study are available from the corresponding author on request. In addition to that, ESI† is available for this paper.Author contributions
K. Tornberg: conceptualization, methodology, investigation, validation, data curation, formal analysis, visualization, project administration, writing – original draft. W. Grötsch: methodology, data curation, formal analysis, conceptualization of perfusion designs, investigation, visualization, writing – original draft. N. Ritari: investigation, data curation, formal analysis. S. Haikka: methodology – fabrication of chips, investigation, writing – review & editing. L. Sukki: methodology – fabrication of molds, visualization, writing – review & editing. K. Aalto-Setälä: conceptualization, funding acquisition, supervision, resources, writing – review & editing. M. Pekkanen-Mattila: conceptualization, project administration, formal analysis, supervision, writing – review & editing. P. Kallio: conceptualization, funding acquisition, supervision, resources, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Research Council of Finland is acknowledged for the funding (Centre of Excellence in Body-On-Chip Research grant number 353174 and 353175). K. Tornberg would like to acknowledge the Finnish Cultural Foundation, the Pirkanmaa Regional Fund (grant number 50231683) and the doctoral school of the Faculty of Medicine and Health Technology, Tampere University, for a personal grant. Chat GPT and copilot were used for proofreading of this manuscript.References
- U.S. Congress. FDA Modernization Act 2.0, https://www.congress.gov/bill/117th-congress/senate-bill/5002/text, (accessed April 29, 2024).
- D. E. Ingber, Nat. Rev. Genet., 2022, 23, 467–491 CrossRef CAS PubMed.
- Y. Wang, P. Wang and J. Qin, Acc. Chem. Res., 2021, 54, 3550–3562 CrossRef CAS PubMed.
- M. A. Swartz and M. E. Fleury, Annu. Rev. Biomed. Eng., 2007, 9, 229–256 CrossRef CAS PubMed.
- P. Erickson, G. Jetley, P. Amin, A. Mejevdiwala, A. Patel, K. Cheng and B. Parekkadan, Toxicol. In Vitro, 2023, 91, 105623 CrossRef CAS PubMed.
- Y. A. Guerrero, D. Desai, C. Sullivan, E. Kindt, M. E. Spilker, T. S. Maurer, D. E. Solomon and D. W. Bartlett, AAPS J., 2020, 22, 53 CrossRef CAS PubMed.
- D. Singh, S. P. Deosarkar, E. Cadogan, V. Flemington, A. Bray, J. Zhang, R. S. Reiserer, D. K. Schaffer, G. B. Gerken, C. M. Britt, E. M. Werner, F. D. Gibbons, T. Kostrzewski, C. E. Chambers, E. J. Davies, A. R. Montoya, J. H. L. Fok, D. Hughes, K. Fabre, M. P. Wagoner, J. P. Wikswo and C. W. Scott, PLoS Biol., 2022, 20, e3001624 CrossRef CAS PubMed.
- P. Erickson, T. Houwayek, A. Burr, M. Teryek and B. Parekkadan, Anal. Biochem., 2021, 625, 114213 CrossRef CAS PubMed.
- E. L. Jackson-Holmes, A. W. Schaefer, T. C. McDevitt and H. Lu, Analyst, 2020, 145, 4815–4826 RSC.
- H. Azizgolshani, J. R. Coppeta, E. M. Vedula, E. E. Marr, B. P. Cain, R. J. Luu, M. P. Lech, S. H. Kann, T. J. Mulhern, V. Tandon, K. Tan, N. J. Haroutunian, P. Keegan, M. Rogers, A. L. Gard, K. B. Baldwin, J. C. de Souza, B. C. Hoefler, S. S. Bale, L. B. Kratchman, A. Zorn, A. Patterson, E. S. Kim, T. A. Petrie, E. L. Wiellette, C. Williams, B. C. Isenberg and J. L. Charest, Lab Chip, 2021, 21, 1454–1474 RSC.
- D. Kumar, R. Nadda and R. Repaka, Med. Biol. Eng. Comput., 2024, 62, 1925–1957 CrossRef PubMed.
- K. S. Nitsche, I. Müller, S. Malcomber, P. L. Carmichael and H. Bouwmeester, Arch. Toxicol., 2022, 96, 711–741 Search PubMed.
- M. Busek, A. Aizenshtadt, T. Koch, A. Frank, L. Delon, M. Amirola Martinez, A. Golovin, C. Dumas, J. Stokowiec, S. Gruenzner, E. Melum and S. Krauss, Lab Chip, 2023, 23, 591–608 RSC.
- N. Limjanthong, Y. Tohbaru, T. Okamoto, R. Okajima, Y. Kusama, H. Kojima, A. Fujimura, T. Miyazaki, T. Kanamori, S. Sugiura and K. Ohnuma, J. Biosci. Bioeng., 2023, 135, 151–159 Search PubMed.
- A. Tajeddin and N. Mustafaoglu, Micromachines, 2021, 12, 1443 CrossRef PubMed.
- H. Chen, Z. Yu, S. Bai, H. Lu, D. Xu, C. Chen, D. Liu and Y. Zhu, TrAC, Trends Anal. Chem., 2019, 117, 186–199 CrossRef CAS.
- L. Wei, W. Li, E. Entcheva and Z. Li, Lab Chip, 2020, 20, 4031–4042 Search PubMed.
- H. J. Kim and D. E. Ingber, Integr. Biol., 2013, 5, 1130–1140 Search PubMed.
- C. A. M. Fois, A. Schindeler, P. Valtchev and F. Dehghani, Biomed. Microdevices, 2021, 23, 55 CrossRef CAS PubMed.
- H. Wang, G. M. Riha, S. Yan, M. Li, H. Chai, H. Yang, Q. Yao and C. Chen, Arterioscler., Thromb., Vasc. Biol., 2005, 25, 1817–1823 CrossRef CAS PubMed.
- S. Kapur, D. J. Baylink and K.-H. William Lau, Bone, 2003, 32, 241–251 CrossRef CAS PubMed.
- M. Lindner, A. Laporte, S. Block, L. Elomaa and M. Weinhart, Cell, 2021, 10, 2062 CrossRef CAS PubMed.
- W. J. Polacheck, M. L. Kutys, J. Yang, J. Eyckmans, Y. Wu, H. Vasavada, K. K. Hirschi and C. S. Chen, Nature, 2017, 552, 258–262 Search PubMed.
- J. S. Gnecco, V. Pensabene, D. J. Li, T. Ding, E. E. Hui, K. L. Bruner-Tran and K. G. Osteen, Ann. Biomed. Eng., 2017, 45, 1758–1769 CrossRef PubMed.
- L. Cucullo, M. Hossain, V. Puvenna, N. Marchi and D. Janigro, BMC Neurosci., 2011, 12, 40 CrossRef CAS PubMed.
- T. Wongpakham, T. Chunfong, W. Jeamsaksiri, K. Chessadangkul, S. Bhanpattanakul, W. Kallayanathum, T. Tharasanit and A. Pimpin, Cell, 2024, 13, 2132 CrossRef CAS PubMed.
- R. Lu, J. Li, Z. Guo, Z. Wang, J. J. Feng and Y. Sui, Biomech. Model. Mechanobiol., 2025, 24, 489–506 Search PubMed.
- Y. Liu, R. Kamran, X. Han, M. Wang, Q. Li, D. Lai, K. Naruse and K. Takahashi, Sci. Rep., 2024, 14, 18063 Search PubMed.
- Y. Baert, I. Ruetschle, W. Cools, A. Oehme, A. Lorenz, U. Marx, E. Goossens and I. Maschmeyer, Hum. Reprod., 2020, 35, 1029–1044 Search PubMed.
- S. Bauer, C. Wennberg Huldt, K. P. Kanebratt, I. Durieux, D. Gunne, S. Andersson, L. Ewart, W. G. Haynes, I. Maschmeyer, A. Winter, C. Ämmälä, U. Marx and T. B. Andersson, Sci. Rep., 2017, 7, 14620 CrossRef PubMed.
- N. Picollet-D'hahan, A. Zuchowska, I. Lemeunier and S. Le Gac, Trends Biotechnol., 2021, 39, 788–810 CrossRef PubMed.
- P. Loskill, S. G. Marcus, A. Mathur, W. M. Reese and K. E. Healy, PLoS One, 2015, 10, e0139587 Search PubMed.
- X. Zhang, M. Karim, M. M. Hasan, J. Hooper, R. Wahab, S. Roy and T. A. Al-Hilal, Cancers, 2022, 14, 648 Search PubMed.
- D. Huh, H. Fujioka, Y.-C. Tung, N. Futai, R. Paine, J. B. Grotberg and S. Takayama, Proc. Natl. Acad. Sci., 2007, 104, 18886–18891 Search PubMed.
- D. Huh, B. D. Matthews, A. Mammoto, M. Montoya-Zavala, H. Y. Hsin and D. E. Ingber, Science, 2010, 328, 1662–1668 Search PubMed.
- B. Jing, Y. Luo, B. Lin, J. Li, Z. A. Wang and Y. Du, RSC Adv., 2019, 9, 17137–17147 RSC.
- D. W. Lee, S. K. Ha, I. Choi and J. H. Sung, Biomed. Microdevices, 2017, 19, 100 CrossRef PubMed.
- Y.-C. Toh, T. C. Lim, D. Tai, G. Xiao, D. van Noort and H. Yu, Lab Chip, 2009, 9, 2026–2035 Search PubMed.
- V. Vickerman, J. Blundo, S. Chung and R. Kamm, Lab Chip, 2008, 8, 1468–1477 RSC.
- G. S. Offeddu, K. Haase, M. R. Gillrie, R. Li, O. Morozova, D. Hickman, C. G. Knutson and R. D. Kamm, Biomaterials, 2019, 212, 115–125 CrossRef CAS PubMed.
- H. Ehlers, A. Nicolas, F. Schavemaker, J. P. M. Heijmans, M. Bulst, S. J. Trietsch and L. J. van den Broek, Front. Immunol., 2023, 1118624 CrossRef CAS PubMed.
- A. Yrjänäinen, E. Mesiä, E. Lampela, J. Kreutzer, J. Vihinen, K. Tornberg, H. Vuorenpää, S. Miettinen, P. Kallio and A.-J. Mäki, Sci. Rep., 2024, 14, 22916 CrossRef PubMed.
- C. Olaizola-Rodrigo, S. Palma-Florez, T. Ranđelović, C. Bayona, M. Ashrafi, J. Samitier, A. Lagunas, M. Mir, M. Doblaré, I. Ochoa, R. Monge and S. Oliván, Lab Chip, 2024, 24, 2094–2106 RSC.
- M. B. Chen, J. A. Whisler, J. Fröse, C. Yu, Y. Shin and R. D. Kamm, Nat. Protoc., 2017, 12, 865–880 Search PubMed.
- Y. Shin, S. Han, J. S. Jeon, K. Yamamoto, I. K. Zervantonakis, R. Sudo, R. D. Kamm and S. Chung, Nat. Protoc., 2012, 7, 1247–1259 CrossRef CAS PubMed.
- T. Liu, B. Lin and J. Qin, Lab Chip, 2010, 10, 1671–1677 RSC.
- S. W. Chen, A. Blazeski, S. Zhang, S. E. Shelton, G. S. Offeddu and R. D. Kamm, Lab Chip, 2023, 23, 4552–4564 Search PubMed.
- L. Sun, F. Bian, D. Xu, Y. Luo, Y. Wang and Y. Zhao, Mater. Horiz., 2023, 10, 4724–4745 RSC.
- Y. Gao, J. Broussard, A. Haque, A. Revzin and T. Lin, Microsyst. Nanoeng., 2016, 2, 15045 CrossRef CAS PubMed.
- J. Yeste, M. García-Ramírez, X. Illa, A. Guimerà, C. Hernández, R. Simó and R. Villa, Lab Chip, 2018, 18, 95–105 RSC.
- C. Oleaga, C. Bernabini, A. S. T. Smith, B. Srinivasan, M. Jackson, W. McLamb, V. Platt, R. Bridges, Y. Cai, N. Santhanam, B. Berry, S. Najjar, N. Akanda, X. Guo, C. Martin, G. Ekman, M. B. Esch, J. Langer, G. Ouedraogo, J. Cotovio, L. Breton, M. L. Shuler and J. J. Hickman, Sci. Rep., 2016, 6, 20030 CrossRef CAS PubMed.
- K.-C. Weng, Y. K. Kurokawa, B. S. Hajek, J. A. Paladin, V. S. Shirure and S. C. George, Tissue Eng., Part C, 2020, 26, 44–55 CrossRef CAS PubMed.
- O. Schneider, L. Zeifang, S. Fuchs, C. Sailer and P. Loskill, Tissue Eng., Part A, 2019, 25, 786–798 CrossRef CAS PubMed.
- G. Bergström, J. Christoffersson, K. Schwanke, R. Zweigerdt and C.-F. Mandenius, Lab Chip, 2015, 15, 3242–3249 RSC.
- A. Skardal, S. V. Murphy, M. Devarasetty, I. Mead, H.-W. Kang, Y.-J. Seol, Y. Shrike Zhang, S.-R. Shin, L. Zhao, J. Aleman, A. R. Hall, T. D. Shupe, A. Kleensang, M. R. Dokmeci, S. Jin Lee, J. D. Jackson, J. J. Yoo, T. Hartung, A. Khademhosseini, S. Soker, C. E. Bishop and A. Atala, Sci. Rep., 2017, 7, 8837 CrossRef PubMed.
- K. Andrysiak, J. Stępniewski and J. Dulak, Pflugers Arch., 2021, 473, 1061–1085 CrossRef CAS PubMed.
- M. Ristola, L. Sukki, M. M. Azevedo, A. I. Seixas, J. B. Relvas, S. Narkilahti and P. Kallio, J. Micromech. Microeng., 2019, 29, 065009E CrossRef.
- M. Häkli, S. Jäntti, T. Joki, L. Sukki, K. Tornberg, K. Aalto-Setälä, P. Kallio, M. Pekkanen-Mattila and S. Narkilahti, Int. J. Mol. Sci., 2022, 3148 CrossRef PubMed.
- H. J. Kim, D. Huh, G. Hamilton and D. E. Ingber, Lab Chip, 2012, 12, 2165–2174 RSC.
- B. Wahlberg, H. Ghuman, J. R. Liu and M. Modo, Sci. Rep., 2018, 8, 9194 CrossRef PubMed.
- M. Liu, W. Song, P. Li, Y. Huang, X. Gong, G. Zhou, X. Jia, L. Zheng and Y. Fan, PLoS One, 2013, 8, e63473 CrossRef PubMed.
- Y. Huang, X. Jia, K. Bai, X. Gong and Y. Fan, Arch. Med. Res., 2010, 41, 497–505 CrossRef PubMed.
- P. Izadpanah, A. Golchin, T. Firuzyar, M. Najafi, A. Jangjou and S. Hashemi, Mol. Biol. Rep., 2022, 49, 3167–3175 CrossRef CAS PubMed.
- S. A. Vaez, S. Ebrahimi-Barough, M. Soleimani, S. Kolivand, S. Farzamfar, S. H. Ahmadi Tafti, M. Azami, F. Noorbakhsh and J. Ai, EXCLI J., 2018, 17, 762–774 Search PubMed.
- J. Kreutzer, M. Viehrig, R.-P. Pölönen, F. Zhao, M. Ojala, K. Aalto-Setälä and P. Kallio, Biomech. Model. Mechanobiol., 2020, 19, 291–303 Search PubMed.
- W. Dou, L. Wang, M. Malhi, H. Liu, Q. Zhao, J. Plakhotnik, Z. Xu, Z. Huang, C. A. Simmons, J. T. Maynes and Y. Sun, Biosens. Bioelectron., 2021, 175, 112875 Search PubMed.
- M. Radisic, H. Park, H. Shing, T. Consi, F. J. Schoen, R. Langer, L. E. Freed and G. Vunjak-Novakovic, Proc. Natl. Acad. Sci., 2004, 101, 18129–18134 CrossRef CAS PubMed.
- S. S. Nunes, J. W. Miklas, J. Liu, R. Aschar-Sobbi, Y. Xiao, B. Zhang, J. Jiang, S. Massé, M. Gagliardi, A. Hsieh, N. Thavandiran, M. A. Laflamme, K. Nanthakumar, G. J. Gross, P. H. Backx, G. Keller and M. Radisic, Nat. Methods, 2013, 10, 781–787 CrossRef CAS PubMed.
- E. Figallo, C. Cannizzaro, S. Gerecht, J. A. Burdick, R. Langer, N. Elvassore and G. Vunjak-Novakovic, Lab Chip, 2007, 7, 710–719 Search PubMed.
- E. L. Cussler, Diffusion: Mass Transfer in Fluid Systems, Cambridge University Press, 2009 Search PubMed.
- T. Casalini, M. Salvalaglio, G. Perale, M. Masi and C. Cavallotti, J. Phys. Chem. B, 2011, 115, 12896–12904 CrossRef CAS PubMed.
- A. Sethu Madhavan, J. Kakkaraparambil Vijayan and L. Rajith, ChemistrySelect, 2022, 7, e202203252 CrossRef CAS.
- S. Dreborg and H. Kim, Allergy Asthma Clin. Immunol., 2021, 17, 25 CrossRef CAS PubMed.
- B. E. Heradstveit, G. A. Sunde, H. Asbjørnsen, R. Aalvik, T. Wentzel-Larsen and J.-K. Heltne, Resuscitation, 2023, 193, 110025 CrossRef PubMed.
- GitHub - forughi/PIV: A Particle Image Velocimetry (PIV) code in Python and Matlab, https://github.com/forughi/PIV?tab=readme-ov-file#readme, (accessed May 8, 2024).
- L. Sala, B. J. van Meer, L. G. J. Tertoolen, J. Bakkers, M. Bellin, R. P. Davis, C. Denning, M. A. E. Dieben, T. Eschenhagen, E. Giacomelli, C. Grandela, A. Hansen, E. R. Holman, M. R. M. Jongbloed, S. M. Kamel, C. D. Koopman, Q. Lachaud, I. Mannhardt, M. P. H. Mol, D. Mosqueira, V. V. Orlova, R. Passier, M. C. Ribeiro, U. Saleem, G. L. Smith, F. L. Burton and C. L. Mummery, Circ. Res., 2018, 122, e5–e16 Search PubMed.
- B. J. van Meer, L. Sala, L. G. J. Tertoolen, G. L. Smith, F. L. Burton and C. L. Mummery, Curr. Protoc. Hum. Genet., 2018, 99, e67 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5lc00072f |
| This journal is © The Royal Society of Chemistry 2025 |