
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhancing cancer cell immunocapture on orientation-controlled nanoimprinted microcone arrays in microgap channels†
Yuhei
Saito
a,
Natsumi
Shimmyo
a,
Shuhei
Aoyama
 b,
Rie
Utoh
a,
Minoru
Seki
a and
Masumi
Yamada
*a
b,
Rie
Utoh
a,
Minoru
Seki
a and
Masumi
Yamada
*a
aDepartment of Applied Chemistry and Biotechnology, Graduate School of Engineering, Chiba University, 1-33 Yayoi-cho, Inage-ku, Chiba 263-8522, Japan. E-mail: m-yamada@faculty.chiba-u.jp; Tel: +81 43 290 3398
bDenka Innovation Center, Denka Co., Ltd., 3-5-1 Asahi-machi, Machida, Tokyo 194-8560, Japan
First published on 28th May 2025
Abstract
Cancer detection through circulating tumor cell (CTC)-based liquid biopsy has been expected to be a new modality for less-invasive, next-generation diagnosis. However, due to limitations such as the complexity of the cell capture devices, as well as the cost and reproducibility of their fabrication and surface functionalization, these methods are not yet practical for clinical use. In this study, we propose a new strategy for affinity-based selective capture of cancer cells using a microfluidic system integrating nanoengineered polymeric structures. Polycarbonate (PC) sheets with closely assembled microcone arrays were prepared using thermal nanoimprint lithography (T-NIL). These structures are suitable for mass production and can strongly absorb antibodies without the need for chemical linkers. Microgap channels incorporating the microcones were formed by simply sandwiching the sheet between two plates, which were highly functional in capturing cancer cells from blood samples. In this study, we clarified the effect of the orientation angle of the microcone array in a closely packed hexagonal pattern to ensure high capture efficiency even under high flow-rate conditions. The feasibility of detecting cancer cells through post-capture processing was also demonstrated. The cell capture structures proposed in this study are simple and reproducible in their fabrication, highly productive, and practical, and may become a new tool for cell-based cancer diagnosis.
Introduction
Over the past decade, liquid biopsy based on circulating tumor cells (CTCs) as micrometastasis biomarkers has shown great promise as a minimally invasive tool for the early detection, monitoring, and prognosis of various cancers.1 The selective capture and enrichment of CTCs from patient blood samples is a crucial step in CTC processing, as the population of CTCs is extremely low compared to other blood cells.2 Since the emergence of microfluidic technologies as a powerful platform for biochemical affinity-based capture and subsequent detection of CTCs in blood samples, researchers have devoted considerable effort to pursuing efficient micro/nanoengineered structures.3–5 Microfluidic devices modified with antibodies6–8 and aptamers9–11 have been proven highly effective for selective capture of target rare cell populations. Incorporating cell-sized micro/nanoengineered structures into microfluidic platforms not only enhances CTC capture efficiency but also enables seamless downstream characterization, including single-cell molecular analysis.12,13 Such information on cancer heterogeneity is expected to be highly useful for tailoring cancer therapies to individual patients. Previous studies have demonstrated early phenotyping of CTCs through 100- to 1000-fold enrichment using microfluidic systems.14–16 In this context, the need for simple and versatile capture devices and processes for CTCs is still increasing, and expectations for practical devices for cell-based cancer detection are growing.17Effective immobilization of capture molecules on substrate surfaces is crucial for the successful construction of such microfluidic immunocapturers. Generally, it is not trivial to introduce capture molecules, mostly antibodies, onto a specific surface area inside a preassembled and closed microchannel system. In particular, PDMS microfluidic devices are cost-effective and highly reproducible, but a multistep chemical modification process is needed. Because of the chemical inertness of PDMS, multiple linkers based on silane coupling and/or biotin–avidin interaction18–22 are often employed. The resultant complexity and low controllability in the conventional modification process may compromise the reproducibility and reliability of device fabrication and performances. Additionally, existing CTC capture devices have limited productivity in fabricating microstructures, which may pose a significant obstacle to their practical application. While numerous studies have been conducted and clinical applications have been eagerly anticipated, the use of CTC-based cancer cell assays in real-world medical practice remains limited.23
Substrates that can easily and strongly adsorb antibodies have been widely used in biotechnology and biochemical research fields. For example, nitrocellulose membranes are often employed as the base material for lateral-flow immunoassay (LFIA) devices,24,25 while PVDF membranes are employed for Western blotting.26 Antibodies can be spontaneously and firmly adsorbed onto the surface of these materials, due to hydrophilic/hydrophobic and electrostatic interactions.27,28 We have recently reported that nanoengineered polymeric sheets prepared by thermal nanoimprint lithography (T-NIL) can be a new material for antibody immobilization,29,30 which were employed as substrates for LFIA. Among several polymer materials, polycarbonate (PC) showed high ability for antibody adsorption and immobilization.29 The closely packed pattern of the micrometer structures on the surface with nanometer-sized surface topographies, along with the chemical properties of PC, contributed to the autonomous adsorption of antibodies without the need for any chemical crosslinking protocols. The microfabrication process with T-NIL is also amenable to the highly reproducible, mass production of identical structures. Adoption of such material as a new substrate to the selective capture of rare cells would therefore facilitate the immunological capture of CTCs for highly potent liquid biopsy with dramatically simplified fabrication and surface modification processes.
The key to realizing such a concept is the development of a facile strategy for integrating nanoengineered polymer structures into microfluidic platforms. Such systems allow us to continuously introduce biological fluids with controlled flow profiles and enhanced cell–substrate interactions. Generally, pre-assembled microchannels have been employed for capturing rare cells; however, creating such channels with nanoengineered structures using thermoelastic polymers via thermal nanoimprint lithography (T-NIL) is challenging. Additionally, it is non-trivial to seamlessly integrate and immobilize pre-patterned antibody-functionalized nanoengineered sheets into specific regions of microchannels without introducing any gaps, steps, or defects. In fact, to the best of our knowledge, such processes have yet to be developed.
From these perspectives, here we propose a unique strategy to utilize the inter-structural space on the nanoengineered polymeric sheet as a “microgap” channel, as shown in Fig. 1, for effectively immunocapturing blood cancer cells. The device is formed by physically sandwiching the nanoengineered PC sheet with pre-coated antibody molecules between a solid plate (e.g., glass slide) as the bottom and an elastic plate (e.g., PDMS plate) as the top substrate. A closely packed array of cell-sized microcones with nanometer-sized surface roughness was formed on the sheet by T-NIL and utilized. To assemble the nanoengineered PC sheet into a seamless microfluidic format, the microcones in the periphery of the sheet were embedded in a hydrophobic sealing material. In this configuration, the gap between the microcones functions as a flow channel, in which the target cells are immunologically captured on the surface of the cones. In the experiment, we fabricated immunocapture devices and characterized their performances. Particularly, the orientation of the hexagonally patterned microcone array against the flow direction, which is denoted as the orientation angle, was investigated to maximize the capture performance of several types of cancer cells spiked into blood samples.
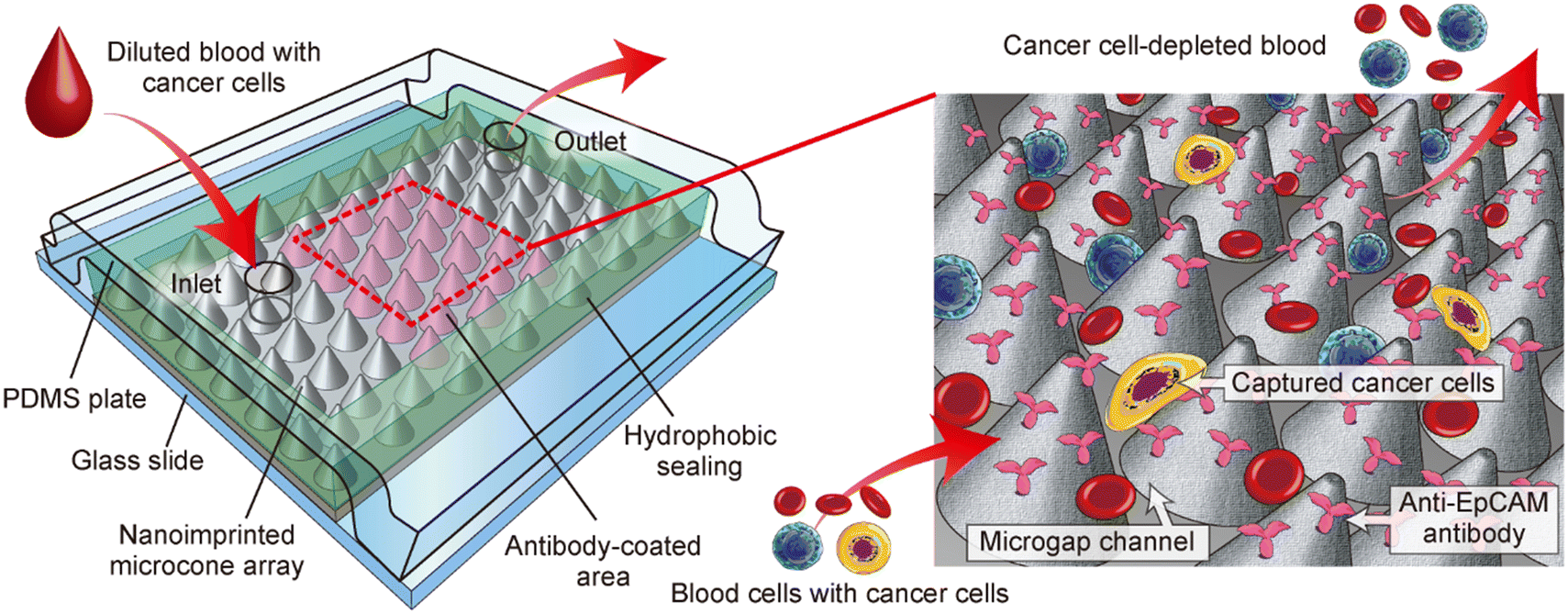 | ||
| Fig. 1 Schematic diagram showing a microgap channel incorporating a microcone array structure, enabling the selective capture of cancer cells. | ||
Materials and methods
Preparation of microfluidic devices with microcone arrays
The fabrication process of the device with the microgap channel is shown in Fig. 2(a). A PC sheet (250 μm thick) with microcone array structures was prepared by T-NIL using an aluminum mold with the laser-ablated concave structures, as described before.30 Both the diameter and height of the microcone were 30 μm, and the cone structures were packed most tightly into a hexagonal pattern. Nanoimprinted sheets were cut into small pieces, 10 mm × 15 mm in size. The periphery region of the cut sheet was coated with hydrophobic water repellent (A-PAP Pen, Daido Sangyo, Tokyo, Japan). The coating operation was repeated three times to completely fill the microcones in the peripheral region with the hydrophobic repellent, leaving a 6 mm × 12 mm area uncoated in the center of the sheet (Fig. 2(b)). A 10 μL drop of mouse anti-human EpCAM antibody (final conc. of 50 μg mL−1, sc-59906, Santa Cruz Biotechnology, CA, USA) in 50 mM Tris–HCl solution containing 2% (w/v) trehalose (Fujifilm Wako Pure Chemical, Osaka, Japan) was applied to the central 6 mm × 6 mm area (antibody-bound area) of the sheet. After incubating at room temperature for 1 h, the sheet was dried in a convection oven at 40 °C for 1 h. | ||
| Fig. 2 (a) Fabrication process of the microdevice incorporating the microgap channel and the T-NIL PC sheet with antibody-coated microcone structures. (b and c) Schematic images showing (b) the design of the microgap channel and (c) the orientation angle of the microcone array against the flow. | ||
To fabricate the microgap channels, the imprinted PC sheet partially bound with the antibody was sandwiched between two plates. An elastic PDMS plate with a thickness of 4–5 mm was prepared from PDMS prepolymer (Silpot 184, Dow Corning Toray, Tokyo, Japan), and inlet/outlet ports were made by punching. This PDMS plate and a glass slide (S1112, Matsunami, Osaka, Japan) were treated with O2 plasma using a plasma reactor (PR500, Yamato Scientific, Tokyo, Japan) and these two plates were bonded while sandwiching the antibody-bound sheet. Silicone tubes were attached to the inlet and outlet ports and then glued. In this study, three types of microgap channels with different orientation angles of the microcone array were prepared, as shown in Fig. 2(c), to investigate the effect of the microcone arrangement on the capture behaviors of the cancer cells. In the 0° device the packed row of the microcones was aligned parallel to the flow direction, whereas in the 15° and 30° devices the rows of the microcones were tilted at 15° and 30°, respectively.
Cell culture and preparation
MCF-7, A549, and HeLa were used as model cells for CTCs. These cells were kindly provided by Riken BRC, Ibaraki, Japan. Cells were cultured in DMEM supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, MA, USA) and 1% penicillin/streptomycin (Sigma-Aldrich) in a CO2 incubator at 37 °C with 5% CO2. Cells were collected from the dish by trypsin/EDTA treatment, washed with phosphate buffered saline (PBS). After removing large cell aggregates using a cell strainer (mesh size of 40 μm, Corning, NY, USA) cells were used for capture experiments.Capture of cancer cells spiked in human blood samples
All experiments using human blood samples were conducted in accordance with the Ethical Guidelines for Medical and Biological Research Involving Human Subjects established by the Ministry of Education, Culture, Sports, Science and Technology and the Ministry of Health, Labour and Welfare of Japan. The study protocol was approved by the Ethics Committee of the School of Engineering, Chiba University (Approval No. R4-10). Informed consent was obtained from all participants involved in this study. A small amount of blood was collected from healthy adult volunteers by finger pricking. The collected blood was diluted 10-fold with PBS containing 0.1% EDTA-2Na (Dojindo, Kumamoto, Japan), 0.5% BSA (Rockland Immunochemicals, PA, USA), and 2.5% anti-adhesion reagent (Through Path Plus 40x, On-chip Biotechnologies, Tokyo, Japan). When visualizing the capture behaviors of the cancer cells, cells were stained before use in experiments. Cancer cells at approximately 80% confluency on the culture dish were labeled with a red fluorescent dye (Vybrant DiI, Thermo Fisher) according to the manufacturer's protocol. Labeled cancer cells were recovered from the culture dish and added to the diluted blood sample at a final concentration of 3 × 103 cells per mL. To evaluate the capture efficiency with high accuracy, the CTC concentration in this sample was set to be >100 times higher than the CTC concentration in blood samples. Hoechst 33342 (final conc. of 10 μg mL−1, Thermo Fisher) was added to this sample to stain the nuclei of the cancer cells and leukocytes. When in situ staining of cancer cells was performed, non-labeled cancer cells were used.The diluted blood sample with the spiked cancer cells (volume of 100 μL, theoretically with 300 cancer cells) was loaded into a Teflon tube (inner φ of 1.0 mm) and one end of this tube was connected to a plastic syringe containing a washing buffer (PBS with 2.5% of the anti-adhesion reagent). The other end of the tube was then connected to the inlet silicone tube of the microgap channel, and the sample was pumped into the channel at a flow rate of 20–200 μL min−1 using a syringe pump (KDS200, KD Scientific, MA, USA). After introducing 100 μL of the blood sample, 500 μL of the washing buffer was serially introduced to wash out cells remaining inside the tube and the device. After washing, CTCs and leukocytes trapped in the microgap channel were observed and counted using a fluorescence microscope (IX73, Olympus, Tokyo, Japan) and a CCD camera (DP74, Olympus).
As indices showing the capture behaviors of cancer cells, we quantitatively evaluated the following two values: capture efficiency and selectivity. The capture efficiency (capture yield) was defined as the ratio of the cancer cells captured in the antibody-coated area to the introduced cancer cells (300 cells). The capture selectivity (capture purity) was defined as the ratio of cancer cells to the nucleated cells (both leukocytes and cancer cells) captured in the antibody-bound area. Each device was used only once, and the experiments were repeated four times under each condition using different devices to obtain the data.
Visualization of flow profiles by micro PIV
Confocal micro particle image velocimetry (micro PIV) was performed to investigate the flow profiles in the microgap channels with different orientation angles of the microcone array. Green fluorescent microparticles (ϕ of 1.1 μm, G0100, Thermo Fisher) were used as a tracer for visualizing the flow profiles in the microgap channels. These particles were suspended in PBS containing 0.5% tween 20 at a concentration of 1.0 × 108 particles per mL. The particle suspension was introduced into the microgap channel using a syringe pump, and the flow behaviors of particles were observed using a high-speed confocal micro PIV system (Confocal Scanning Micro PIV System, Seika Corp., Tokyo, Japan) in an x-y plane at the z position of 15 μm from the base of the microcone array. Particle motion was analyzed using an image processing software (Image J, NIH, MD, USA).Immunohistological staining
To confirm the expression of EpCAM on the cancer cells investigated in this study, immunostaining was performed. Cells were seeded in a 12-well plate and cultured until they reached approximately 50–60% confluency. The cells were washed with PBS and fixed using cold methanol at −15 °C, followed by blocking with PBST containing 10% goat serum (Vector Laboratories, CA, USA). Cells were subsequently treated with PBST containing a primary antibody (rabbit anti-EpCAM antibody, Abcam, Cambridge, UK; final conc. of 5 μg mL−1) for 1 h and then with PBST containing a secondary antibody (Alexa 594 goat anti-rabbit IgG, Thermo Fisher; final conc. of 2 μg mL−1) for 30 min. Finally, cell nuclei were stained blue using PBST containing Hoechst 33342. Fluorescence images were captured using the fluorescence microscope and the CCD camera.Additionally, we performed in situ post-capture immunostaining of non-stained cancer cells in the microgap channel. After capturing and washing the cells in the channel, the following staining and washing solutions were introduced stepwise at a flow rate of 30 μL min−1 for 10 min and then incubated at stopped-flow conditions for 10–60 min; (i) 10% neutral buffered formalin (Fujifilm Wako) for 15 min, (ii) PBST containing 10% donkey serum (ImmunoBioScience, WA, USA) for 30 min, (iii) PBST containing the primary antibody for EpCAM (conc. of 5 μg mL−1) for 60 min, (iv) PBST containing a secondary antibody for rabbit IgG (donkey anti-rabbit IgG H&L, Alexa Fluor 594, Thermo Fisher; final conc. of 4 μg mL−1) for 30 min, and (v) PBST containing Hoechst 33342 for 10 min. Between steps PBST was introduced for 10 min for washing. Consequently, EpCAM-positive cells (cancer cells) were stained red and blue, whereas leukocytes were stained only blue.
Statistical analysis
Statistical analyses were performed using IBM SPSS Statistics 22 software (IBM Japan, Tokyo, Japan). Statistical significance was examined by one-way analysis of variance (ANOVA) with Tukey or Bonferroni post hoc test. Differences were considered significant with *p < 0.05 and **p < 0.01.Results and discussion
Characterization of the microfluidic device
One prerequisite for successful CTC capture in this study is that the microstructure can easily and efficiently absorb and immobilize antibodies. In a previous study, both the chemical properties of the material of the polymer sheet and the morphological property of the nanometer-sized bumps were reported to be important for antibody adsorption.30 From the observation of the surface morphology of the microcones by SEM, we confirmed that the surface of the microcone was not smooth but had nanometer-sized bumps (Fig. 3(a)). This roughness is considered to be derived from the aluminum mold used for T-NIL. In this study, we prepared microcone structures with a base diameter of 30 μm and a height of 30 μm, which were closely packed with a distance between the tops of the neighboring microcones of 30 μm. As a result, nanometer-sized bumps were also observed over the entire area of the nanoimprinted PC sheet. | ||
| Fig. 3 (a) SEM image showing the microcone structure prepared by T-NIL on a PC sheet. (b) Fluorescence microscopic image showing the boundary between the coated and non-coated areas with fluorescence-labeled antibody. (c) Photograph of the assembled microfluidic device with the microgap channel. | ||
We evaluated whether antibody adsorption and immobilization could be achieved simply by dropping an antibody solution onto the microcone. A fluorescence micrograph of the boundary between the antibody-coated and uncoated areas is shown in Fig. 3(b). Even after washing the microcones with a buffer solution by ultrasonication, strong signal of the FITC-labelled antibody was observed on the coated region. This result indicated that the antibody was firmly adsorbed on the microcone surface. SEM observations revealed that the microcone slopes exhibited rougher surfaces compared to the apexes and intercone spaces. This roughness likely contributed to the relatively higher fluorescence signals observed in the slope regions. While the fluorescence signal distribution across the microcone surface was not perfectly uniform, it was not deemed a significant obstacle for capturing CTCs with an average size of approximately 15 μm.31 The significantly lower fluorescence intensity observed in the uncoated area indicated that antibody adsorption was effectively confined to the coated region. It is notable that the microcone sheet without antibody coating was stable and showed no decrease in antibody adsorption capacity even after long-term storage (approximately 1 year).
Next, we attempted to implement the nanoimprinted sheet with microcones into a microfluidic platform. Introducing the sheet into pre-assembled microchannels with controlled arrangements was anticipated to be challenging. In this regard, we proposed a facile but effective process, that is, sandwiching the antibody-coated nanoimprinted sheet between a flexible flat PDMS plate and a rigid glass slide, as shown in Fig. 2(a). This process effectively creates a gap between the top of the microcones on the sheet and the PDMS plate, which function as a fluidic channel facilitating the continuous introduction of blood samples.
A photograph of the fabricated device is shown in Fig. 3(c). The channel structure was formed at the center of the imprinted sheet, and the periphery of the sheet was coated with a hydrophobic repellent to prevent fluid leakage. Despite the manual process, the thickness of the hydrophobic area could be controlled by varying the number of coatings, and in this experiment, it was approximately 40 μm. This value was set to be slightly greater than the cone height (30 μm) to ensure that the microcones in the peripheral region were completely covered by the hydrophobic repellent. This consequently means that there was a gap of approximately 10 μm between the PDMS plate and the microcone apexes. By using the nanoimprinted sheet with the convex microcone array structure, we were able to fabricate the gap channels by a simple sandwiching process without employing any conventional techniques for microchannel fabrication. The device was tested for water introduction using a pressure control device. As a result, a relatively high flow rate of approximately 5 mL min−1 was obtained at an applied pressure of 10 kPa, and no liquid leakage was observed.
Selective capture of MCF-7 cells in blood samples
We then verified whether the prepared devices could actually function as a new modality for selectively capturing cancer cells and investigated factors affecting the capture performances. First, MCF-7 cells, a commonly used cell line as a model for CTCs, were spiked into diluted human blood samples and introduced into the device. To quantitatively analyze the capture efficiency and selectivity of the rare cell populations, 100 μL of diluted human blood containing 300 MCF-7 cells was introduced. In this experiment, the membrane and nuclei of MCF-7 cells were pre-stained with red and blue dyes, respectively, to distinguish cancer cells from leukocytes, which were stained only with blue dye.The capture behaviors of MCF-7 cells in the microgap channel with the orientation angle of 30° at a flow rate of 20 μL min−1 are shown in Fig. 4(a) and ESI† Movie S1. In this experiment the nuclei of the MCF-7 cells only were stained blue. The MCF-7 cells flowed through the microgap channel in a zigzag pattern, at an average flow velocity of approximately 1.4 mm s−1. Although there was a small gap between the top of the microcones and the channel ceiling, most of the MCF-7 cells flowed through the intercone space near the bottom of the channel, which was possibly assisted by gravity-driven cell precipitation onto the bottom. Cells were rolling while flowing through the intercone spaces, but no clear regularity or periodicity in their rotation was observed. Consequently, MCF-7 cells collided with the microcones and were subsequently captured on their surfaces. Fig. 4(b) shows an SEM image of the captured MCF-7 cells. MCF-7 cells were captured in the intercone region, not on the surface of the single cones, suggesting that the closely packed microcone arrangement was effective for capturing cancer cells. Fig. 4(c) shows fluorescence images of antibody-bound regions in the upstream, middle, and downstream sections after blood sample introduction and washing. These micrographs indicate that MCF-7 cells were effectively captured, along with a small number of leukocytes. The number of captured cells in the upstream region was larger than that in the central and downstream regions. In particular, MCF-7 cells were primarily captured in the upstream region of the antibody-coated area, with 70–80% of these cells detected within approximately 2 mm from the entrance of the antibody-bound region. These results suggest that cell capture occurs with a certain probability for the event of microcone-cell collision. Under this flow-rate condition, approximately 95% of the introduced MCF-7 cells were captured. Hence, the size of the antibody-coated area (6 mm × 6 mm) was considered sufficient for capturing cancer cells. In contrast to the cancer cells, the ratio of captured leukocytes was limited, accounting for less than 0.1%. A small number of erythrocytes were also observed on the microcones via SEM observation, but their population was extremely low (fewer than 100 erythrocytes per device). We have not investigated the capture behaviors of platelets, but their influence on CTC capture was negligible because the flows of the cell samples were stably maintained through the CTC capture experiments.
 | ||
| Fig. 4 (a) Time-laps images showing the capture behaviors of cancer cells (MCF-7 cells) in diluted blood on the microcone array in the microgap channel with the orientation angle of 30°. The nuclei of the MCF-7 cells were selectively stained blue with Hoechst 33342. (b) SEM image showing a captured cancer cell; the cell is pseudocolored red. (c) Fluorescence micrographs showing cells captured and remained after washing in the upstream, central, and downstream regions of the microgap channel. Leukocytes are stained blue and indicated with blue arrows, whereas MCF-7 cells are stained both blue and red. | ||
Furthermore, to validate if the capture of cancer cells was actually caused by the antibodies, we performed a control experiment using microcones without antibody coating. As a result, approximately 25% of MCF-7 cells were captured but this value was significantly smaller than the antibody-coated microcones. The number of captured leukocytes was still very low, less than 0.1%. From these results the effect of antibody on the selective capture of cancer cells was clarified. The small amount of trapped MCF-7 cells was possibly due to the physically captured of the cancer cells in the narrow space between the microcones especially near the channel bottom; leukocytes, which exhibit more deformable nature than cancer cells,31,32 were not captured. Although the obtained result indicates the possibility of nonspecific capture of cancer cells, the low capture efficiency of leukocytes suggests that this is not a significant issue for the application in cancer diagnosis using blood samples.
Many parameters should significantly affect the cell capture performances, and hence, we conducted capture experiments by varying the two important parameters, namely, the orientation angle of the microcone array and the flow rate. Notably, if CTCs can be efficiently captured even under high flow-rate conditions, the processing volume would increase, reducing detection time. Fig. 5(a) shows the capture ratio of MCF-7 cells for the three types of devices with different orientation angles of the microcones, under different flow rates. In the 0° device, more than 90% of MCF-7 cells were captured at a flow rate of 20 μL min−1, whereas the capture ratio decreased at flow rates higher than 50 μL min−1. This may be because the direction of the closest row of the microcones was parallel to the direction of the flow in this device, so once cells flow through the intercone spaces, they continued to flow straight through the microgap channel. In the 15° and 30° devices, more than 90% of MCF-7 cells were captured even at a flow rate of 100 μL min−1. This may be because the arrangement of the closest rows of cones was tilted against the flow, so that cells were more likely to collide with the microcones at a certain frequency regardless of their initial position. There was no statistical difference in the capture ratios of cancer cells between the 15° and 30° devices. These results indicate that the microcone array arrangement and flow rate significantly impact cell capture efficiency, and that tilting the orientation angle of the microcone array was crucial for enhancing capture performance. Fig. 5(b) shows the capture selectivity of MCF-7 cells. The selectivity was approximately 70% under all conditions, but this value tended to decrease slightly with the 30° device, especially under high flow conditions. However, considering that more than 99% of leukocytes were removed under all conditions, efficient removal of leukocytes was possible regardless of the flow rate conditions and the arrangement of the microcone array. In previous studies on antibody-based capture of CTCs in microchannels, experiments were performed mostly under flow-rate conditions typically in the range of 10–30 μL min−1.3–6,33–36 In addition, it was reported that the capture efficiencies dramatically decreased as the flow rate increased. In comparison, in the present study, highly efficient capture was achieved even at relatively high flow rate conditions even with using a relatively small antibody-bound region (6 mm × 6 mm), which suggests that the 3D microcone geometry was a key to successful capture of cancer cells. We anticipate that using microgap channels with a wider antibody-bound region will effectively enhance the throughput of CTC capture.
 | ||
| Fig. 5 Effects of the flow rate and the orientation angle of the microcone array on (a) capture efficiency and (b) selectivity of MCF-7 cells. Each data shows the mean ± SD of 4 experimental results. *p < 0.05, **p < 0.01. | ||
Visualization of the flow profiles through microcone arrays
To obtain direct evidence of the effect of the orientation angle of the microcone array on the cell capture efficiency, we analyzed in detail the flow profiles in the microgap channel using confocal high-speed micro PIV. Green fluorescent microparticles with average diameter of 1.1 μm were used as a tracer, and the particle movement in the x–y plane at the z = 15 μm position from the base of the microcone was observed. The particle suspension was introduced at a flow rate of 50 μL min−1 for the 0°, 15°, and 30° devices, and the images were captured at a speed of 250 fps.The positions of the tracer particles flowing through the intercone spaces were observed as shown in ESI† Movie S2, and their trajectories were visualized by plotting the particle positions and connecting them with lines. The results are shown in Fig. 6(a). In the 0° device, the flow speed of each particle was relatively uniform, and the variation in the flow velocity of each particle was not large; particles flowing near the microcone surface tended to constantly flow slower, while those far from the cones flowed faster. In the 15° and 30° devices, on the other hand, particles exhibited repeated acceleration and deceleration in accordance with the microcone array period; when particles flowed in the proximity of the microcones, they were decelerated. This result indicates that by tilting the arrangement of the microcone array against the flow direction, cells would have an equal opportunity to collide with microcones regardless of their initial position.
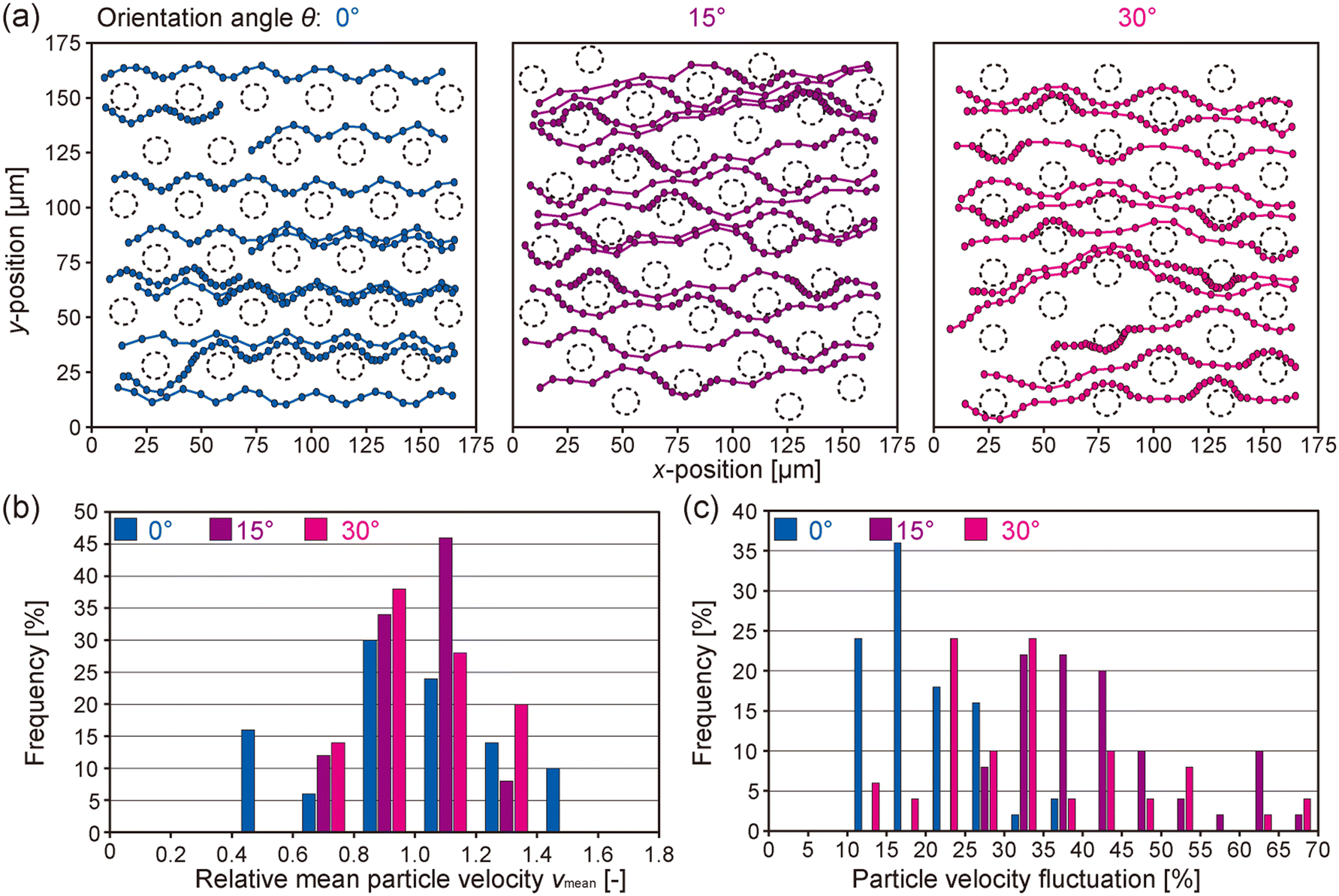 | ||
| Fig. 6 Visualization of flow profiles through the microgap channels. (a) Confocal micro PIV analysis of the flow profile using fluorescent microparticles (ϕ of 1.1 μm) as a tracer. Flow patterns of representative particles are illustrated, in an x–y plane at z = 15 μm from the base. The outlines of the microcones at this z-position are indicated by dotted lines. The time interval between each dot representing particle movement is 20 ms. Three types of microgap channels with different microcone arrangements were investigated. (b and c) Distributions of (b) relative mean particle velocity and (c) particle velocity fluctuation. | ||
To quantitatively evaluate the differences in the flow profiles depending on the microcone arrangement, we defined and analyzed two indices: (1) the variation in the mean flow velocity (vmean) of a single particle and (2) the variation in the velocity fluctuation of a single particle. The velocity fluctuation was defined as the standard deviation of velocities of a particle over a 20 ms interval, normalized by the mean flow velocity. The variation of vmean is shown in Fig. 6(b). For the device with the orientation angle of 0°, the distribution of relative vmean was broadened and its coefficient of variation (CV) was 27.6%. In contrast, for the devices with the orientation angles of 15° and 30°, the distributions of relative vmean were narrower, with CVs of 15.6% and 18.2%, respectively. In addition, the differences in vmean between 15° and 30° devices were small. Fig. 6(c) shows the variation in the particle velocity fluctuation. In the 0° device, most particles showed small changes in velocity, while there are many particles with large velocity fluctuations in the 15° and 30° devices. These results were consistent with the observation of flow profiles described above, that is, particles in the 0° device tended to keep their initial flow velocities. It could be therefore suggested that in the 0° device, once cells flowed through the intercone space, they flowed directly down the channel without contacting the downstream microcones. On the other hand, the 15° and 30° devices caused efficient collision of cells with the cones, resulting in a high capture efficiency even under the relatively high flow-rate conditions. Our findings clearly indicate that the arrangement of microcones is a key factor dominating the affinity-based cancer cell capture.
Capture of different cancer cell types
It is naturally anticipated that the expression levels of the surface marker are a critical factor dominating the capture efficiency of the target cells.37 We therefore investigated the capture behaviors of several types of cancer cells; in addition to MCF-7 cells, we employed A549 cells, a human lung cancer cell line, and HeLa cells, a human cervical cancer cell line. According to previous studies, MCF-7 cells express the highest amount of EpCAM, A549 cells moderately, and HeLa cells do not express EpCAM.38,39Before performing capture experiments, the expression of EpCAM was examined for these cells by immunohistochemical staining. Fluorescence micrographs of the stained cells are shown in Fig. 7(a). MCF-7 cells exhibited strong fluorescence signals, showing that these cells are highly EpCAM positive. The expression of EpCAM for the A549 cells was also confirmed, but its level was lower compared to MCF-7 cells and was nonuniform. HeLa cells were not positive for EpCAM.
 | ||
| Fig. 7 (a) Immunohistochemical analysis of EpCAM for the three types of cancer cells used. (b and c) Capture efficiency and selectivity of three types of cancer cells using the microgap channel, at the flow rate of 20 μL min−1. | ||
Based on this result, the capture efficiencies of these cells from diluted blood samples were investigated at a flow rate of 20 μL min−1 in the devices with the orientation angle of 30°. The results are shown in Fig. 7(b and c). Compared to the MCF-7 cells, the capture efficiency of the A549 cells was slightly decreased but the difference was not statistically significant; more than 90% of cells were captured. The capture selectivity of the A549 cells was comparable to the MCF-7 cells. The expression of EpCAM in A549 cells was lower and not uniform. It has been previously reported that the capture efficiency of A549 cells using only the anti-EpCAM antibody was relatively low.40 Our approach, on the other hand, achieved high capture efficiency for A549 cells, possibly due to the closely packed microcone configuration along with the enhanced antibody immobilization on the rough surface. In contrast, the capture efficiency of HeLa cells was low, which was less than 20%. From the immunohistochemical observation, HeLa cells do not express EpCAM, which was the main reason for this decreased capture efficiency. The small amount of the HeLa cells captured would be attributed to the non-specific trapping, as in the MCF-7 cells captured on the non-antibody-coated microcones described above. In all of these experiments the ratios of the removed leukocytes were higher than 99%, resulting in higher selectivities especially for MCF-7 and A549 cells. In previous studies, it was suggested that using a single surface marker was not sufficient for cancer cell detection and characterization,41 and cell capture using multiple affinity molecules is effective. For cancer cells with low expression level of EpCAM, other markers, such as HER2, CD44, and IGF1R42–44 could be simultaneously used for clinical diagnosis of cancer, as reported by previous studies.45–48 We expect that using microgap channels with multiple types of antibodies in series will enable us to target multiple cancer cell types with diverse phenotypes.
Post capture visualization and identification of cancer cells
In applying the presented approach to the actual clinical diagnostic, it is necessary to validate its ability for in situ post-capture identification of cancer cells on the microcone array. For this purpose, additionally, we should detect and characterize the captured cells by specific staining or by DNA/RNA analysis following cell lysis and nucleic acid extraction. In this experiment, to demonstrate the post-capture detection, in situ immunostaining was performed by multistep introduction of reagent solutions into the gap channels after capturing CTCs.Fig. 8 shows microscopic images of the cells captured in the gap channel and immunohistologically stained. During the staining process with several introductions of reagent solutions and washing buffers, cells stably remained in the gap channels without flowing out of the channel. After the staining process, MCF-7 cells were double-stained in red and blue, whereas leukocytes were stained blue. Because of the transparency of the PDMS plate on top of the nanoimprinted sheet, fluorescence observation was easily possible. These results indicated the ability of the post-capture staining of the non-prelabeled cancer cells for in situ identification, showing the clinical applicability and versatility of the presented system.
 | ||
| Fig. 8 In situ post-capture staining of MCF-7 cells in the microgap channel. Cell nuclei and the target antigen (EpCAM) were stained blue with Hoechst 33342 and red with anti-EpCAM antibody, respectively. These images were captured from the top PDMS plate using fluorescence microscopy. | ||
Conclusions
Here, we successfully developed a new approach for the efficient and selective capture of cancer cells from blood samples. The system consisted of a microcone array on a PC sheet and flat glass and PDMS plates, which were easily assembled into closed microgap channels without the need for complex fabrication procedures. Since the physicochemical nature of the nanoengineered PC sheets eliminates the need for multistep chemical functionalization protocols for antibody immobilization on the substrate surface, this system is feasible for clinical cancer diagnostics based on liquid biopsy. Additionally, the microcone structures could be prepared using a simple T-NIL process, which is suitable for rapid and large-scale production with high reproducibility.The most remarkable advantage of the system is the high trapping efficiency of cancer cells on microcone surfaces. This can be attributed to the three-dimensional “cone” morphology of the structure, that is, the cell-sized intercone spaces generate irregular flow patterns, which were highly functional in immunoaffinity-based cell capture. We further revealed that controlling the microcone arrangement, i.e., tilting the microcone array against the flow direction, further enhanced the contact between the cells and microcones. As a result, we achieved high capture efficiency and selectivity even at relatively high flow rates for the devices with the orientation angle of 15° and 30°. The applicability to several types of cancer cells and post-capture identification was also demonstrated. In the future, the clinical relevance of the presented approach will be investigated using blood samples from cancer patients with a smaller number of CTCs. Multiplexing capture strategies based on multiple antigens and further increases in the processing flow rate should also be pursued, as well as CFD simulations of the flow profiles in the microgap channels to maximize the capture performances.
Data availability
Raw data supporting the findings of this study are available from the corresponding author upon reasonable request.Author contributions
Conceptualization: SA, MY. Investigation: YS, NS, SA, RU. Methodology: YS, NS, RU, MY. Formal analysis: SA. Supervision: MS. Writing – original draft: YS, MY. Writing review & editing: YS, NS, SA, RU, MS, MY.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported in part by Grants-in-aids for Scientific Research (23 K17343, 23 K23143, and 25 K01585) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan.References
- S. N. Lone, S. Nisar, T. Masoodi, M. Singh, A. Rizwan, S. Hashem, W. El-Rifai, D. Bedognetti, S. K. Batra, M. Haris, A. A. Bhat and M. A. Macha, Mol. Cancer, 2022, 21, 79 CrossRef PubMed
.
- P. Augustsson, C. Magnusson, M. Nordin, H. Lilja and T. Laurell, Anal. Chem., 2012, 84, 7954–7962 CrossRef PubMed
- S. Nagrath, L. V. Sequist, S. Maheswaran, D. W. Bell, D. Irimia, L. Ulkus, M. R. Smith, E. L. Kwak, S. Digumarthy, A. Muzikansky, R. B. J. Settleman, J. W. Tompkins, D. A. Haber and M. Toner, Nature, 2007, 450, 1235–1239 CrossRef PubMed
- A. Abdulla, Z. Zhang, K. Z. Ahmad, A. R. Warden, H. Li and X. Ding, Biosens. Bioelectron., 2022, 201, 113965 CrossRef PubMed
- W. Shi, S. Wang, A. Maarouf, C. G. Uhl, R. He, D. Yunusb and Y. Liu, Lab Chip, 2017, 17, 3291–3299 Search PubMed
- S. Wang, K. Liu, J. Liu, Z. T. Yu, X. Xu, L. Zhao, T. Lee, E. K. Lee, J. Reiss, Y. K. Lee, J. C. Lee and J. H. Park, Angew. Chem., Int. Ed., 2011, 50, 3084–3088 CrossRef PubMed
- Y. Chikaishi, K. Yoneda, T. Ohnaga and F. Tanaka, Oncol. Rep., 2017, 37, 77–82 CrossRef PubMed
- Q. Wu, L. Zhu, X. Wei, M. Zhang, C. Zhang, Z. You, S. Zhang, Y. Song, D. Liu and C. Yang, Sens. Actuators, B, 2023, 379, 133274 CrossRef
- Q. Shen, L. Xu, L. Zhao, D. Wu, Y. Fan, Y. Zhou, W. H. Ouyang, X. Xu, Z. Zhang, M. Song and M. Lee, Adv. Mater., 2013, 25, 2368–2373 CrossRef PubMed
- Y. Song, Y. Shi, M. Huang, W. Wang, Y. Wang, J. Cheng, Z. Lei, Z. Zhu and C. Yang, Angew. Chem., Int. Ed., 2019, 58, 2236–2240 CrossRef PubMed
- S. Fukuyama, S. Kumamoto, S. Nagano, S. Hitotsuya, K. Yasuda, Y. Kitamura, M. Iwatsuki, H. Baba, T. Ihara, Y. Nakanishi and Y. Nakashima, Talanta, 2021, 228, 122239 CrossRef CAS
- S. Maheswaran, L. V. Sequist, S. Nagrath, L. Ulkus, B. Brannigan, C. V. Collura, E. Inserra, S. Diederichs, A. J. Iafrate, D. W. Bell, R. B. J. Settleman, J. W. Tompkins, D. A. Haber and M. Toner, N. Engl. J. Med., 2008, 359, 366–377 CrossRef CAS PubMed
- Y. K. Wang, M. Wang, S. B. Cheng, Y. J. Chen, C. W. Li, M. Xie and W. H. Huang, View, 2022, 4, 20220054 CrossRef
- L. Zhu, H. Lin, S. Wan, X. Chen, L. Wu, Z. Zhu, Y. Song, B. Hu and C. Yang, Anal. Chem., 2020, 92, 15229–15235 CrossRef CAS
- S. Gao, S. Chen, Y. Liu, H. Mao and Q. Lu, ACS Appl. Mater. Interfaces, 2021, 13, 19603–19612 CrossRef CAS PubMed
- X. Zhang, Y. Ma, Y. Wang, Z. Liang, X. Zhang, Y. Chen, Q. Wang, H. Qin and J. Wang, Micromachines, 2024, 15, 542 CrossRef PubMed
- L. Descamps, D. Le Roy and A. L. Deman, Int. J. Mol. Sci., 2022, 23, 1981 CrossRef CAS
- N. S. K. Gunda, M. Singh, L. Norman, K. Kaur and S. K. Mitra, Appl. Surf. Sci., 2014, 305, 522–530 CrossRef CAS
- P. Xue, Y. Wu, J. Guo and Y. Kang, Biomed. Microdevices, 2015, 17, 39 CrossRef
- S. B. Cheng, M. M. Chen, Y. K. Wang, Z. H. Sun, Y. Qin, S. Tian, W. G. Dong, M. Xie and W. H. Huang, Anal. Chem., 2021, 93, 7102–7109 CrossRef CAS
- A. Shakeri, S. Khan and T. F. Didar, Lab Chip, 2021, 21, 3053–3075 RSC
- Q. Zeng, B. Xu, C. Qian, N. Li, Z. Guo and S. Wu, Biomater. Sci., 2024, 12, 2369–2380 RSC
- R. Lawrence, M. Watters, C. R. Davies, K. Pantel and Y. J. Lu, Nat. Rev. Clin. Oncol., 2023, 20, 487–500 CrossRef PubMed
- D. Quesada-Gonzalez and A. Merkoci, Biosens. Bioelectron., 2015, 73, 47–63 CrossRef PubMed
- Z. Chen, Z. Zhang, X. Zhai, Y. Li, L. Lin, H. Zhao, L. Bian, P. Li, L. Yu, Y. Wu and G. Lin, Anal. Chem., 2020, 92, 7226–7231 Search PubMed
- B. Liu, L. Huang, C. Sihlbom, A. Burlingame and J. D. Marks, J. Mol. Biol., 2002, 315, 1063–1073 Search PubMed
- Y. Jung, J. Y. Jeong and B. H. Chung, Analyst, 2008, 133, 697–701 RSC
- N. G. Welch, J. A. Scoble, B. W. Muir and P. J. Pigram, Biointerphases, 2017, 12, 02D301 CrossRef
- S. Aoyama, Y. Akiyama, K. Monden, M. Yamada and M. Seki, Analyst, 2019, 144, 1519–1526 RSC
- S. Aoyama, K. Monden, Y. Akiyama, M. Yamada and M. Seki, Anal. Chem., 2019, 91, 13377–13382 CrossRef PubMed
- N. Shimmyo, M. Furuhata, M. Yamada, R. Utoh and M. Seki, Analyst, 2022, 147, 1622–1630 RSC
- Y. Chen, C. Ni, X. Zhang, Z. Ni and N. Xiang, Small Methods, 2024, 8, e2301195 CrossRef PubMed
- C. Wang, M. Ye, L. Cheng, R. Li, W. Zhu, Z. Shi, C. Fan, J. He, J. Liu and Z. Liu, Biomaterials, 2015, 54, 55–62 CrossRef
- S. Yan, X. Zhang, X. Dai, X. Feng, W. Du and B. F. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 33457–33463 CrossRef
- M. G. Ahmed, M. F. Abate, Y. Song, Z. Zhu, F. Yan, Y. Xu, X. Wang, Q. Li and C. Yang, Angew. Chem., Int. Ed., 2017, 56, 10681–10685 CrossRef PubMed
- J. Chen, C. Y. Liu, X. Wang, E. Sweet, N. Liu, X. Gong and L. Lin, Biosens. Bioelectron., 2020, 150, 111900 CrossRef PubMed
- Z. Svobodova, J. Kucerova, J. Autebert, D. Horak, L. Bruckova, J. L. Viovy and Z. Bilkova, Electrophoresis, 2014, 35, 323–329 CrossRef
- M. Czaplicka, K. Nicinski, A. Nowicka, T. Szymborski, I. Chmielewska, J. Trzcinska-Danielewicz, A. Girstun and A. Kaminska, Cancers, 2020, 12, 3315 CrossRef
- Y. Zhou, X. Wang, Z. Luo, X. Liu, J. Hou and S. Zhou, Small Sci., 2022, 2, 2200061 CrossRef
- S. He, P. Liu, Y. Wu, M. Ø. Agerbæk, A. Salanti, L. W. M. M. Terstappen, P. Jonkheijm and M. Stevens, Int. J. Mol. Sci., 2024, 25, 9816 CrossRef
- D. Lin, L. Shen, M. Luo, K. Zhang, J. Li, Q. Yang, F. Zhu, D. Zhou, S. Zheng, Y. Chen and J. Zhou, Signal Transduction Targeted Ther., 2021, 6, 404 CrossRef
- J. Peng, Q. Zhao, W. Zheng, W. Li, P. Li, L. Zhu, X. Liu, B. Shao, H. Li, C. Wang and Y. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 18423–18428 CrossRef
- R. E. Wilson, Jr., R. O'Connor, C. E. Gallops, E. A. Kwizera, B. Noroozi, B. I. Morshed, Y. Wang and X. Huang, ACS Appl. Mater. Interfaces, 2020, 12, 47220–47232 CrossRef
- X. Li, T. Cui, W. Zhang, Z. Zhai, F. Wu, Y. Zhang, M. Yang, W. Zhong and W. Yue, Colloids Surf., B, 2020, 196, 111281 CrossRef PubMed
- Y.-T. Kang, Y. J. Kim, J. Bu, S. Chen, Y.-H. Cho, H. M. Lee, C. J. Ryu, Y. Lim and S.-W. Han, Sens. Actuators, B, 2018, 260, 320–330 CrossRef
- G. Galletti, M. S. Sung, L. T. Vahdat, M. A. Shah, S. M. Santana, G. Altavilla, B. J. Kirbyd and P. Giannakakou, Lab Chip, 2014, 14, 147–156 RSC
- X. Wang, L. Deng and B. T. Gjertsen, Anal. Chim. Acta, 2020, 1129, 1–11 CrossRef PubMed
- J. Cha, H. Cho, J. S. Chung, J. S. Park and K. H. Han, Cancers, 2023, 15, 2825 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: Movie S1 showing the capture behaviors of cancer cells and Movie S2 showing the flow visualization in the microgap channels. See DOI: https://doi.org/10.1039/d5lc00143a |
| This journal is © The Royal Society of Chemistry 2025 |