
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectropolishing Fe-based biodegradable metals for vascular applications: impact on surface properties, corrosion and cell viability
Letícia Marin
de Andrade
a,
Carlo
Paternoster
a,
Pascale
Chevallier
a,
Sofia
Gambaro
ab,
Francesco
Copes
a,
Vinicius Fidelis
de Oliveira Sales
a and
Diego
Mantovani
 *a
*a
aLaboratory for Biomaterials and Bioengineering (LBB), Canada Research Chair Tier I in Biomaterials and Bioengineering for the Innovation in Surgery, Department of Mining, Metallurgy, and Materials Engineering, Laval University, Quebec City, G1V 0A6, Canada. E-mail: diego.mantovani@gmn.ulaval.ca
bNational Research Council, Institute of Condensed Matter Chemistry and Technologies for Energy (CNR-ICMATE), Genoa, Italy
First published on 5th December 2024
Abstract
Biodegradable metals constitute a new class of materials for medical application. By breaking the paradigm that a metallic biomaterial to be implanted in the body must be corrosion resistant, biodegradable metals advance surgery allowing clinicians to dispose of temporary devices. Among them, Fe–Mn–C steel has emerged due to its outstanding mechanical properties, while its degradation rate must be carefully controlled. For this purpose, especially for medical devices, the surface finishing plays a pivotal role and influences both the corrosion behavior and biological response of these materials. Therefore, this research investigated the impact of electropolishing (EP) processes on the Fe–Mn–C alloy surface finishing in terms of composition, morphology, topography, and wettability. Three electrolytes were carefully selected and used in this study: EP1 (ethanol, perchloric acid, and glycerol), EP2 (perchloric acid, acetic acid, and glycerol), and an ionic liquid EP3 (choline chloride and ethylene glycol). Corrosion behavior and cell viability were investigated and compared with those obtained on mechanically polished (MP) samples. The results displayed that electropolishing was governed by two mechanisms: 1) controlled mass transport for EP1 and EP2, and 2) an adsorption mechanism for EP3. Among the tested conditions, EP2 emerged as a promising overall EP process. It promoted the smoothest and most hydrophilic passivated surface (Ra ∼ 10 nm, WCA = 53°, respectively) and the highest ratio of metal oxides to metallic elements. In addition, EP2 exhibited appropriate corrosion behavior suitable for biodegradable metal devices by inducing the formation of a protective oxide layer. Furthermore, cell viability with EP2 was comparable to that observed with MP. These findings emphasize the potential of electropolishing for enhancing the properties of Fe–Mn–C alloys, paving their applicability, especially in cardiovascular devices.
1. Introduction
Biodegradable metals represent a significant advance in cardiovascular applications,1 for example for temporary stents, where complete degradation is expected to occur between 6 and 24 months.2 Despite several candidates such as Fe, Mg, and Zn alloys, only one Mg-based stent is currently available on the market.3 However, its low mechanical properties impose a larger structure design, much larger than the gold standard conventional stents made of Co–Cr alloys, or AISI 316 L.4 Fe–Mn–C has therefore emerged as a promising solution due to its exceptional mechanical properties, notably its high ductility (strain at break, A ≈ 20%) and strength (yield strength, σY ≈ 620 MPa), much closer than those of Co–Cr alloys, thus enabling thin stents, thereby reducing the risks of complications.5,6 Furthermore, in vitro assays with endothelial cells have exhibited satisfactory cytocompatibility levels, depending on the Mn content within Fe–Mn–C substrates.7,8 Metallic bare metal devices, if used as stents for vascular applications, must have a very smooth surface finish, in the range of a few nanometers.9,10 While this requirement is easily addressed with an electropolishing step in Cr–Co or SS316L corrosion-resistant metals, it represents a major challenge for biodegradable metals, mainly because it has the potential to initiate corrosion, thus leading to degradation. Moreover, smoother surfaces are less prone to cellular hyperplasia and inflammation when in contact with the artery wall,11 and hydrophilic surfaces reduce platelet adhesion, promoting early vascular healing, and minimize late tissue response.12 Regarding corrosion behavior, for instance, increasing surface roughness induces a higher degradation rate.13In the light of these results, researchers in academia and industry agree that there is an urgent need to develop an efficient surface finishing process for Fe–Mn–C alloys susceptible to be used in vascular applications. A stable engineered surface that compromises corrosion behavior and improves biological response in the early stages of alloy–tissue interactions is required. To achieve this goal, recent studies have focused on the development of new surface modification processes, such as thermo-mechanical treatments,14,15 acid pickling,16,17 anodic oxidation,18 sandblasting19 and laser texturing,20 which have been explored to treat Fe-based and Fe–Mn–C-based alloys.
Mechanical polishing (MP) is a common step performed before preliminary surface finishing tests on metallic materials.21–25 This process uses abrasives to remove microscopic chips from metallic surfaces; the process is composed of several steps: the first ones foresee the use of coarse grain abrasives, whose size is progressively reduced until the final stages, which are executed with micron-size or less abrasives, buffing to achieve a mirror-like surface.26 This considerably hinders its application on devices with a complex geometry and reduced thickness, such as those found in stents.16 In addition, residual stress after MP can smear the surface or even reduce the corrosion resistance of the substrate.27,28
To overcome these drawbacks, electropolishing (EP) has been adopted as a standard process for surface finishing of corrosion-resistant metallic implants with complex shapes.29 This technique is employed to promote material smoothening and to increase its corrosion resistance by tuning, respectively, the surface roughness and chemical composition.30 EP is a surface finishing process wherein the anodic dissolution of the workpiece (anode) occurs within a suitable electrolyte in an electrochemical cell.31–33 Controlling the current density and voltage, the outermost material from the workpiece surface is removed.34 Subsequently, mass transport phenomena promote surface passivation through the formation of a nanometric oxide layer on the metallic surface. This technique can be used for the precise control of surface features, finally imparting a bright appearance to the material.35
Several EP protocols were developed for corrosion-resistant alloys for biomedical practice, for example stainless steel,36–41 Co–Cr,42,43 and nitinol,44 generally based on strong acids, such as sulfuric and ortho-phosphoric acids as electrolytes.45 However, as far as EP processes for biodegradable metals are concerned, to our knowledge, only a few have been described in the literature, but none for Fe–Mn–C alloys. In fact, electrolytes composed of C2H5OH, C4H9OH, AlCl3·6H2O, ZnCl2 and distilled water held at 21–23 °C for 90 s were used for an EP pure Zn stent, resulting in the formation of a chemically homogeneous and smooth surface.46 Another EP process was applied to a commonly studied magnesium alloy, WE43, featuring an electrolytic mixture of HNO3 and C2H5NO3.47 The EP process effectively smoothens the surface and maintains high cell viability, comparable to that of untreated surfaces.
In parallel, recent research has been focusing on the viability of employing organic acids or ionic liquids (ILs) as alternatives to water-based solutions.48 IL solvents are a large class of environmentally-conductive solvents which have the advantages of not being based on a water solution and of presenting high current efficiencies with negligible gas evolution at the anode/solution interface.49 ILs were successfully applied to the EP of stainless steel,50 pure silver,51 and aluminum- and nickel-based alloys.52 The most commonly used electrolyte, composed of choline chloride (ChCl) and ethylene glycol (EG), features a unique EP mechanism, distinct from traditional aqueous acid solutions – the ChCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EG mixture shows slower and more potential-dependent oxide removal, shifting to a mass transport-limited process after the oxide layer is removed.53 In all the cases, the use of this electrolyte results in bright surfaces and a noticeable reduction in surface roughness. Furthermore, while electropolishing with water-acid solutions lasts for 1 to 15 minutes at a temperature of 30 to 90 °C, IL solutions take 10 to 120 minutes, depending on their chemical composition.36 Despite this longer duration, electropolishing with IL solutions is carried out at a lower temperature, ranging from room temperature to 50 °C.54
:EG mixture shows slower and more potential-dependent oxide removal, shifting to a mass transport-limited process after the oxide layer is removed.53 In all the cases, the use of this electrolyte results in bright surfaces and a noticeable reduction in surface roughness. Furthermore, while electropolishing with water-acid solutions lasts for 1 to 15 minutes at a temperature of 30 to 90 °C, IL solutions take 10 to 120 minutes, depending on their chemical composition.36 Despite this longer duration, electropolishing with IL solutions is carried out at a lower temperature, ranging from room temperature to 50 °C.54
Therefore, the aim of this research was to develop an electropolishing process as surface finishing for Fe–Mn–C biodegradable alloys in order to achieve effective passivation, homogeneous chemical composition and surface roughness. The effect on surface features of EP process parameters among three different solutions controlling the potential (V), current density (A cm−2), time (min) and electrolyte bath temperature (°C) within the electrochemical cell was investigated. In addition, the relationship between these surface characteristics and their influence on corrosion behavior was evaluated by comparing electropolished and mechanically polished samples. Finally, the impact of the surface finish on vascular cells was assessed in order to validate the potential of these treatments for vascular applications.
2. Materials and methods
2.1. Sample preparation
Square samples (10 mm × 10 mm × 1 mm) of an Fe–13Mn–1.2C alloy (ISO GX120Mn13), commercially known as Hadfield steel, were supplied by Polstar Metals Inc. (Canada). The amount of Fe and Mn in both alloys was determined by atomic absorption spectroscopy (AAS). The carbon content was analyzed by employing a pyrolytic carbon/sulfur detector (LECO CS200, USA). The experimental composition of the alloy is reported in Table 1.| Element | C | Mn | Si | S | P | Fe |
|---|---|---|---|---|---|---|
| Composition (wt%) | 0.99 ± 0.02 | 12.80 ± 0.50 | <0.4 | <0.02 | <0.035 | Bal. |
The sample surface was subjected to two surface finishings, that is, MP and EP, as detailed in Table 2. First, as a pre-treatment, all samples were mechanically polished using 80–240 grit SiC paper (AS-REC condition). The MP surface finishing was obtained by applying complete mechanical polishing until a mirror-like surface finishing was achieved for the pretreated samples. Following the MP treatment, a thorough cleaning step with an industrial non-acidic cleanser (DiverseyTM, Canada) and deionized water was performed to remove any accumulated impurities on the specimen surface. Finally, samples were immersed in an ultrasonic bath containing 10 mL of pure acetone for 5 minutes, then extracted and dried with filtered compressed air. Subsequently, they were sonicated in a similar way with methanol before drying with filtered compressed air. This exact cleaning step was also employed prior to EP treatment.
| Treatment | Conditions | SiC abrasive paper (grit) | Cleaning after each stage | Diamond suspension (μm) |
|---|---|---|---|---|
| 1. Grinding (pre-treatment) | All samples | 80–240 | Sonication in acetone and methanol + final compressed air drying | – |
| 2. Mechanical polishing | MP | 320–800 | Sonication in acetone and methanol + final compressed air drying | 6, 3, 1 |
| Treatment | Conditions | Electrolyte composition (* % vol/vol † % w/w) | Potential (V) | Current density (A cm−2) | Time (min) | Initial electrolyte bath temperature (°C) |
|---|---|---|---|---|---|---|
| 3. Electropolishing | EP1 | *70% ethanol | 60–90 | 0.5 | 2.5 | 24 ± 0.5 |
| 20% HClO4 | ||||||
| 10% glycerol | ||||||
| EP2 | *94.8% CH3COOH | 65–100 | 0.5 | 2.5 | 24 ± 0.5 | |
| 4.7% HClO4 | ||||||
| 0.5% glycerol | ||||||
| EP3 | †69% choline chloride | 6 | 0.25 | 10 | 40 ± 0.6 | |
| *31% ethylene glycol |
For the EP finishing, three electrolytes were selected: two acid solutions55 and one ionic liquid solution described by Abbott et al.54 The chemical composition of electrolytes and operating parameters are described in Table 2. All the acids (CH3COOH, 99.5 vol% and HClO4, 80 vol%) used in the present work were commercial ACS grade solutions (Lab Mat, Canada). A volume of 150 mL of freshly prepared solution was used for each test. Square samples with the same composition as the substrate alloy were used as the cathode. The distance between the electrodes was 6 cm. The current density in this electrochemical cell setup was controlled using a Sorensen DC power supply (Ametek, USA). After the electropolishing procedure, all the samples were sonicated in acetone and methanol, as described above, and then dried with a jet of filtered compressed air.
2.2. Surface characterization
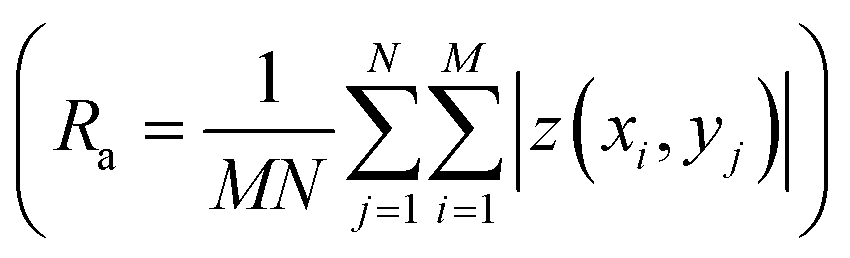 and root mean roughness
and root mean roughness 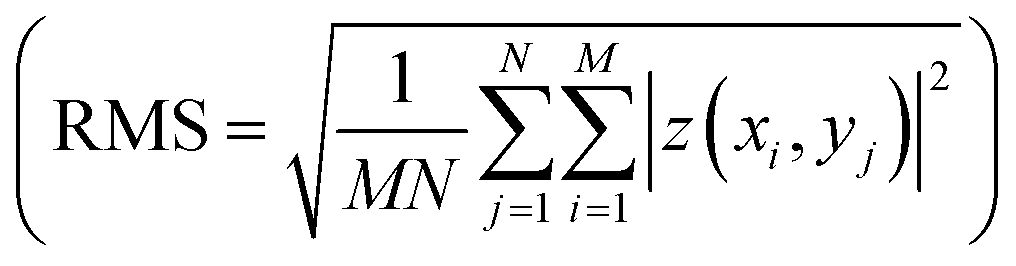 were calculated using WSxM software,56 where Ra is the arithmetic average of profile height deviations from the mean line and RMS is the quadratic mean or root mean square average of profile height deviations from the mean line.57 In addition, a first derivative was applied to the 20 μm × 20 μm images to enhance the contrast of features such as grain boundary steps.
were calculated using WSxM software,56 where Ra is the arithmetic average of profile height deviations from the mean line and RMS is the quadratic mean or root mean square average of profile height deviations from the mean line.57 In addition, a first derivative was applied to the 20 μm × 20 μm images to enhance the contrast of features such as grain boundary steps.
2.3. Electrochemical surface characterization
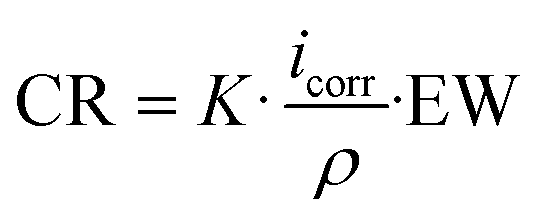 | (1) |
The impedance spectra were recorded using an AC amplitude with a root mean square (RMS) of ±10 mV using a frequency range from 100 kHz to 10 mHz. The electrochemical behavior of samples was studied in modified Hanks' solution60 and M199 culture medium (Gibco, Invitrogen Corporation, Canada). Table 3 presents the ionic concentration of both solutions compared to human plasma.
2.4. Biological tests
:3 or used for experiments. For the experiment reported here, HUVECs have been used at passage 5.
| Supplement | HUVEC medium |
|---|---|
| a Gibco, Invitrogen Corporation, Canada. b Life Sciences, USA. c Sigma Aldrich, Canada. | |
| Fetal bovine serum (FBS)a | 5%;1 10%2 |
| Penicillin–streptomycin (P/S)a | 1% |
| Human fibroblast growth factor-basic (FGF)b | 2 ng mL−1 |
| Human epidermal growth factor (EGF)b | 0.5 ng mL−1 |
| Ascorbic acidc | 1 μg mL−1 |
| Heparinc | 90 μg mL−1 |
| Hydrocortisonec | 1 μg mL−1 |
000 cells per cm2 and incubated at 37 °C and in 5 vol% CO2 for 24 hours in 100 μL per well of complete M199. The day after, the medium was removed, and 100 μL of the extracts were added to the well containing the HUVECs and incubated for 24 h. The extracts were then removed, and 100 μL of 1% solution of resazurin sodium salt in complete M199 medium was added to the cells and incubated for 4 hours at 37 °C and in 5 vol% CO2. After the incubation, the solutions containing the now reduced resorufin product were collected and the fluorescence intensity at a 545 nmex/590 nmem wavelength was measured with a SpectraMax i3x multi-mode plate reader (Molecular Devices, USA). Fluorescence intensity is proportional to cell viability.
2.5. Statistical analysis
All numerical data were analyzed by ANOVA and followed by Tukey's test for specific comparisons between mean values. Results were expressed as the mean value ± standard deviation (SD). A statistically significant difference was considered, if the probability values for a set of data were found to be <0.05 (p < 0.05).3. Results
3.1. Diffusion-limited current plateau
Three scan rates were applied to determine the key parameters for EP, specifically the applied voltage and current density range. Additionally, the corresponding voltage–current curves were used to evaluate the mass transport mechanism occurring at the solution–sample interface for each electrolyte. The schematic LSV curve in Fig. 1a shows the four characteristic regions (A, B, C and D). In brief, region A corresponds to the anodic dissolution of the metal, and therefore to the highest corrosion rate at critical current density (Icrit). Region B is associated with the formation of a passive layer, followed by region C, where mass transport controls both dissolution and diffusion of anions through the passive layer. Finally, region D represents the transpassive region where oxygen gas is produced, leading eventually to pitting.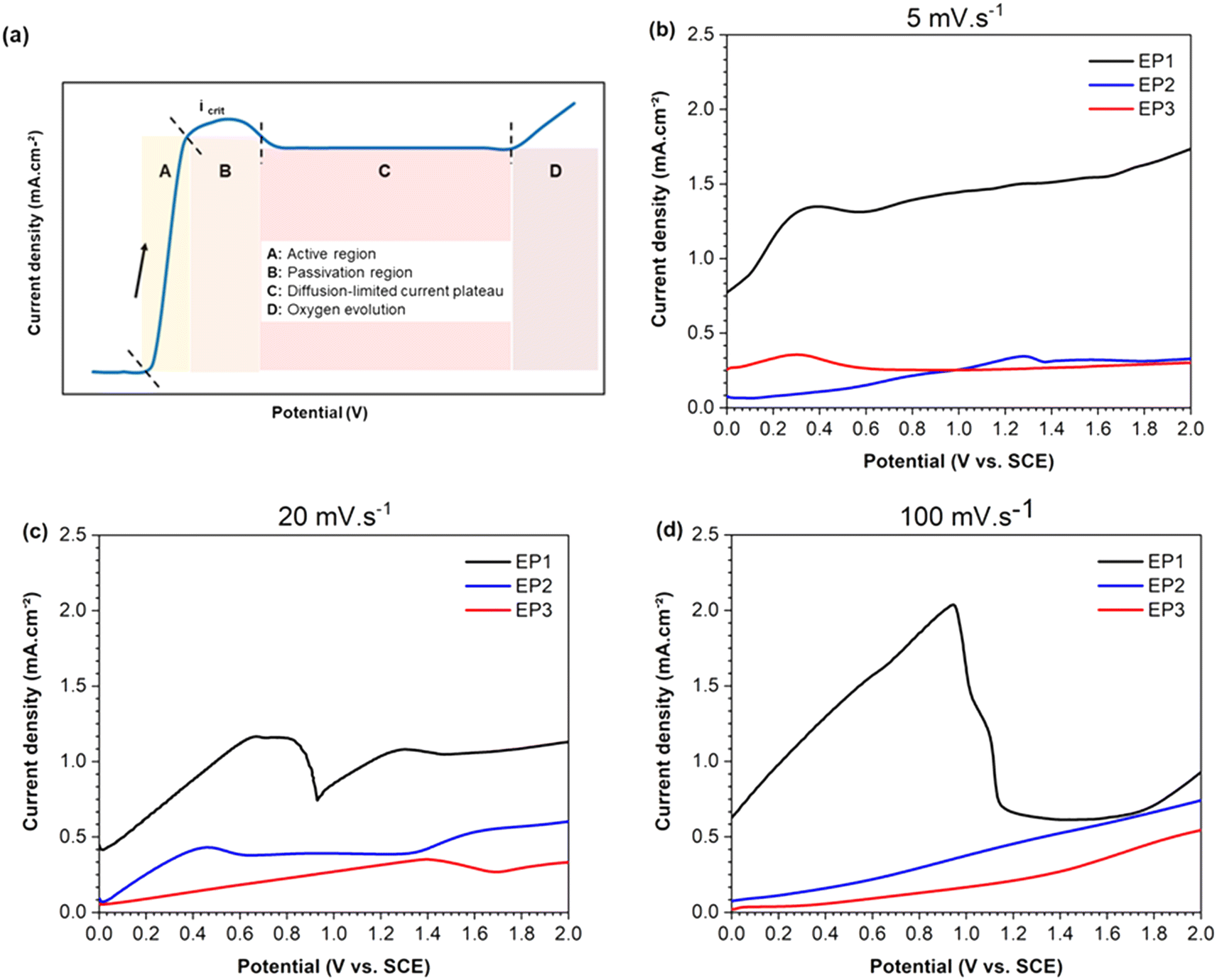 | ||
| Fig. 1 (a) Schematic illustration of a reference LSV plot, and curves for Hadfield samples in EP1 (perchloric acid, ethanol and glycerol), EP2 (acetic acid, perchloric acid and glycerol) and EP3 (choline chloride and ethylene glycol) electrolytes. All experiments were conducted at 21.0 ± 0.5 °C with scan rates of (b) 5 mV s−1, (c) 20 mV s−1 and (d) 100 mV s−1. | ||
Fig. 1b–d show the linear sweep voltammograms, acquired from 0 to 2 V, measuring the anodic behavior of the Hadfield alloy immersed in the three electrolyte compositions at different scan rates. When a scan rate of 5 mV s−1 was adopted, samples tested in EP1 solution exhibited a more positive anodic current (black line of Fig. 1b) compared to those obtained for samples tested in EP2 and EP3 solutions (blue and red lines, respectively). However, EP1 conditions displayed a continuous progressive curve within the studied potential range without reaching a maximum current peak. This observation may be correlated to the absence of a passive layer formation on the sample surface. For EP2 and EP3 electrolytes, a tiny maximum peak current (Icrit ∼ 0.35–0.36 mA) was reached, followed by a plateau of diffusion for potentials higher than 0.6 V. The rising of a low current density for samples treated with EP2 and EP3 electrolytes showed a limited mass transport of the dissolving ions to the electrolyte–sample interface, which could be associated with the slow scan rate used (5 mV s−1).
When the intermediate scan rate of 20 mV s−1 (Fig. 1c) was applied, EP1 conditions still exhibited the highest positive anodic current above all conditions, with a maximum current peak at Icrit ∼ 0.68 mA. The active dissolution region of EP1 was not immediately followed by a steady passive region plateau (represented by the C section of Fig. 1a). Still, the current density increased immediately until 1.5 V followed by a slight but not significant increase. In contrast, samples tested in EP2 solution at 20 mV s−1, showed an Icrit ∼ 0.46 mA (for low potentials, <0.6 V) and a passive region from 0.6 to 1.35 V. After this potential range, the current still increased, indicating oxygen evolution. The EP3 voltammogram displayed an increasing linear trend reaching a value of 0.4 mA at 1.4 V, followed by a small decrease until 1.7 V. Then a continuous slight rise until the final studied potential (2 V) was registered.
At the highest scan rate of 100 mV s−1 (Fig. 1d), LSV curves showed an Icrit peak followed by a plateau region only for samples tested in EP1 solution, suggesting that no passive film occurs on sample surfaces.
Based on these results, the current density was settled at 0.5 A cm−2 for EP1 and EP2 electrolytes while 0.25 A cm−2 for EP3. The potential range for each electrolyte was calculated based on the exposed area of the samples used in the following experiments, as indicated in Table 2.
3.2. Microstructure and morphology
The morphology, microstructure, and detailed patterns of the EP sample surfaces were analyzed by means of SEM, and compared to the MP surface (Fig. 2). | ||
| Fig. 2 Secondary electron mode SEM images for EP1 (a), EP2 (b), EP3 (c), and MP (d) samples. | ||
After EP and MP processes, samples exhibited a mirror-like surface; no noticeable features were observed with the naked eye. For EP1 conditions (Fig. 2a), several pits (small black points) across the surface were noticed, while for EP2, negligible pitting corrosion was observed (Fig. 2b). Additionally, the formation of a thin brown layer was observed during the EP1 and EP2 electropolishing processes. This layer was easily removed with methanol during the rinsing step. It is likely associated with a viscous layer that forms at the metal–electrolyte interface due to anodic dissolution facilitated by the diffusion–adsorption process during the EP process.66
From Fig. 2b, the microstructure observed is associated with austenite (γ) and martensite (α′ or ε) phases which are commonly observed in the type of steel used in the present study.67 The EP3 conditions showed a heterogeneous surface (Fig. 2c); the microstructure was evidenced, but the grain boundaries were not distinctive as for EP1 and EP2 conditions. Different phases and grains exhibited distinct crystallographic orientations, resulting in uneven dissolution rates as observed in the AFM images of the EP2 conditions (Fig. 3). In Fig. 3a, a line was drawn through two regions (A and B) touching prominent boundaries to assess if height profilometry could qualitatively depict the dissolution phenomena. As depicted in Fig. 3b, a height difference of nearly 20 nm was observed between regions A and B, with region B showing smaller height peaks. Additionally, Fig. 3c indicates that the phase in region B is predominantly formed of small particles (less than 10 nm), contributing to a more pronounced surface smoothening compared to region A. Moreover, the amplitude mode from the same image delineated distinct grain boundaries. In fact, Landolt31 explained this phenomenon as the anodization of polycrystals with random orientations which leads to the formation of crystallographic facets, revealing different patterns on crystal planes. These observations indicate that the two main phenomena observed during EP, i.e. macro- and micro-smoothening, are influenced by the microstructure itself and the electrolyte chemical composition.66 However, anhydrous solutions used in EP treatments may lead to the formation of complex oxides on the surface, which could be attributed to the adsorption of organic products originating from the viscous organic solution of choline chloride and ethylene glycol, as well as from perchloric reactions.36
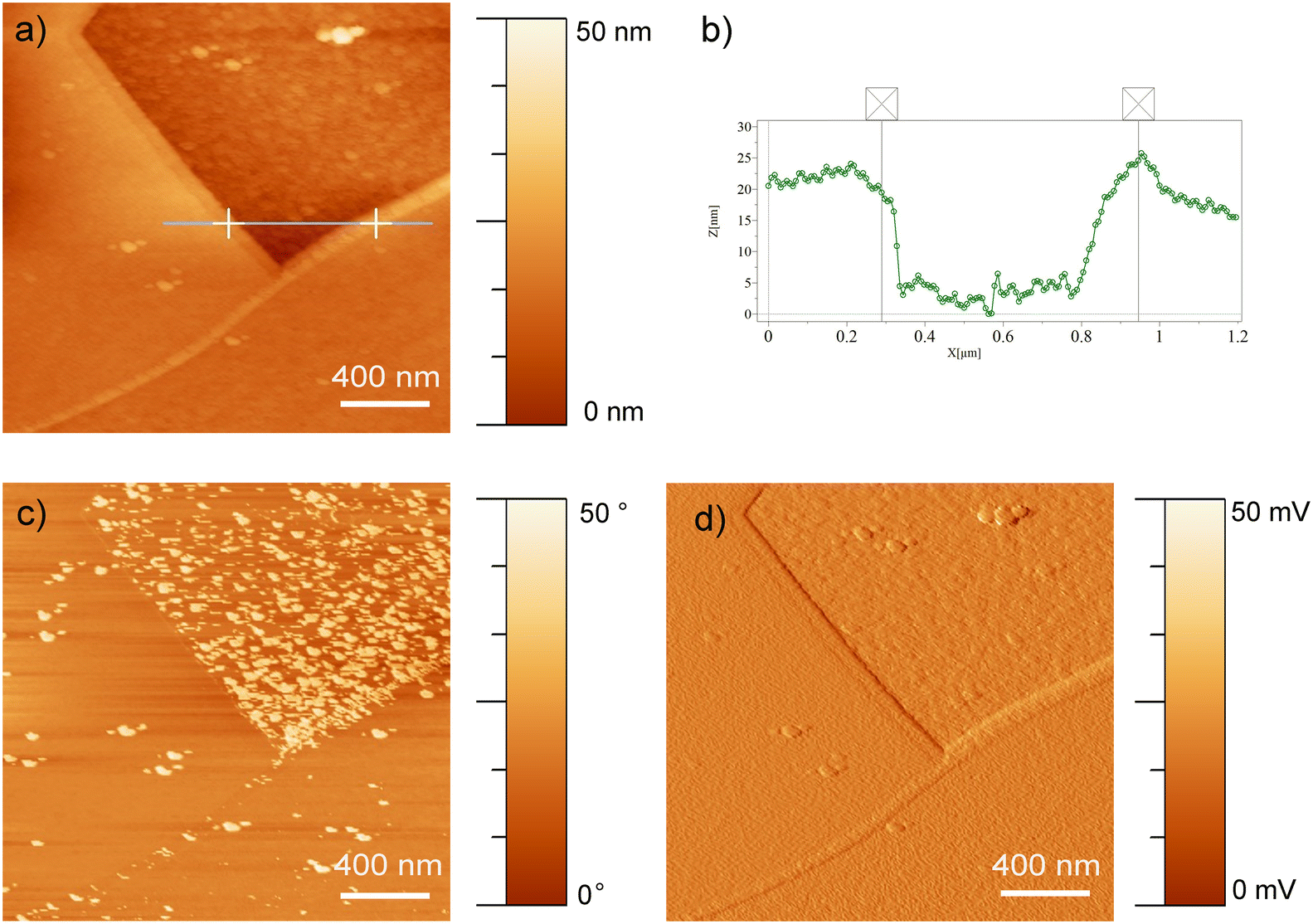 | ||
| Fig. 3 AFM measurements of EP1 conditions show the a) height image and the b) height profile of the line passing through grain boundaries. The c) phase, and d) amplitude images are also presented. | ||
Finally, MP samples (Fig. 2d) showed parallel lines associated with the grooves formed during the mechanical polishing process. No other features, such as grain boundaries, were observed for these conditions contrary to the tested electropolishing conditions.
3.3. Surface roughness, topography, and wettability
The RMS and Ra roughness values of EP1, EP2, EP3, and MP samples were evaluated for an area of 20 μm × 20 μm (Fig. 4a) and 2 μm × 2 μm (Fig. 4b).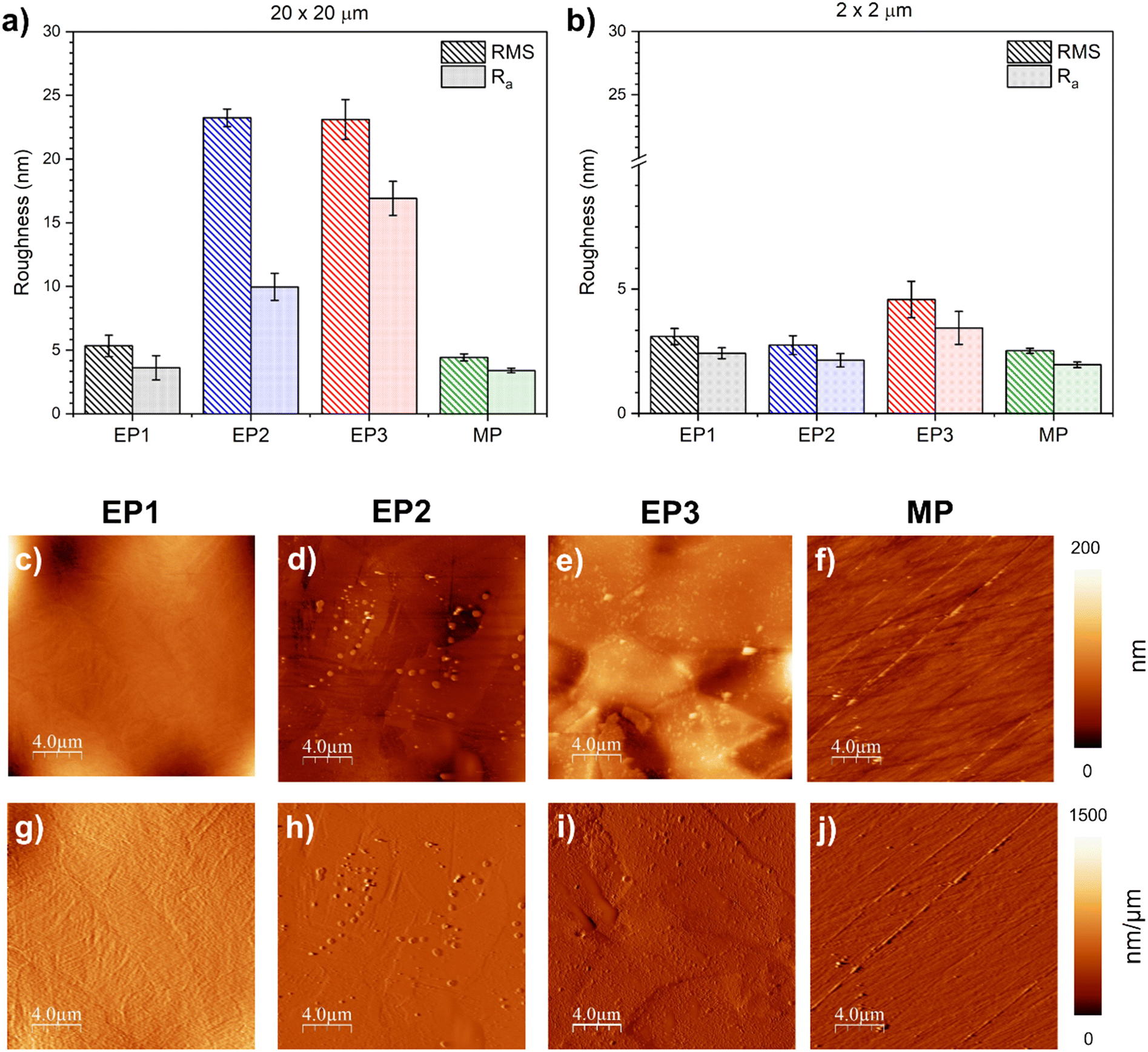 | ||
| Fig. 4 Surface roughness values (Ra and RMS) were measured for two scan sizes, (a) 20 μm × 20 μm and (b) 2 μm × 2 μm. Figures (c–f) present AFM images and their respective first derivative images (g–j) for EP1, EP2, EP3 and MP conditions. | ||
For the large scan area (images of 20 μm × 20 μm), EP1 and MP treatments appeared to produce a smoother surface (RMSEP1 = 5.3 ± 0.8 nm and RMSMP = 4.4 ± 0.3 nm; Ra, EP1 = 3.6 ± 0.9 nm and Ra, MP = 3.4 ± 0.2 nm, respectively), whereas EP2 and EP3 samples exhibited a rougher surface (RMSEP2 = 23.2 ± 2.0 nm and RMSEP3 = 23.1 ± 1.6 nm; Ra, EP2 = 10.0 ± 1.1 nm and Ra, EP3 16.9 ± 1.3 nm, respectively). When the analyses were carried out on a smaller area (2 μm × 2 μm), surface roughness decreased, as expected, with no significant differences between EP1, EP2, and MP conditions. However, EP3 exhibited the highest calculated surface roughness (4.6 ± 0.7 μm) which displayed a significant difference in both RMS and Ra calculations when compared to the others. As suggested by Sautebin et al.,68 it is possible to achieve microsmoothening without macrosmoothening. Both these definitions are linked to local current densities on a rough surface and the limitation of mass transport, respectively.66,69 Furthermore, the first derivative images (Fig. 4g–j), revealed that grain boundaries form steps along two adjacent grains, a phenomenon primarily correlated to the dissolution process during electropolishing. For EP1, the average height step was about 20 nm. EP2 conditions displayed a similar average height of 9 nm (Fig. 4h), and EP3 conditions exhibited a maximum average height step of ∼32 nm, with the highest variation among the three EP conditions. In the case of MP, the fine grooves across the surface showed an approximate average of ∼16 nm from the edge to the substrate.
Finally, the surface wettability for all the studied conditions was assessed by static contact angle (WCA) measurements. All of them showed a hydrophilic behavior; even though EP1 and MP showed the lowest roughness values compared to EP2 and EP3, the surface wettability did not follow the same trends. In fact, the EP2 surface displayed the lowest WCAEP2 value, 53 ± 2°, among all treatments. EP1 showed an intermediate behavior (WCAEP1 = 64 ± 2°), whereas EP3 and MP presented no significantly different CA values, WCAEP3 = 72 ± 5° and WCAMP = 76 ± 3°, respectively, compared to the other conditions.
3.4. Chemical composition after EP and MP
The chemical surface composition (at%) for all the conditions was evaluated by XPS survey analysis (Table 5).| Sample | % Fe | % Mn | % C | % O | %(Fe + Mn) | Ometala/(Fe + Mn) | R Fe/Mn (R0b = 6.7) |
|---|---|---|---|---|---|---|---|
| a Ometal corresponds to the proportion of O1 area per absolute oxygen (Table 6). b R 0 is the ratio between Fe and Mn in the base material. Cl and N traces were found to be less than 0.1 at% under all conditions. | |||||||
| EP1 | 11.0 ± 1.5 | 2.9 ± 0.1 | 40.9 ± 2.0 | 43.4 ± 1.9 | 13.8 | 0.5 | 3.8 |
| EP2 | 12.7 ± 1.4 | 1.2 ± 0.4 | 40.7 ± 1.1 | 44.9 ± 1.4 | 13.8 | 1.3 | 10.6 |
| EP3 | 16.1 ± 1.3 | 2.5 ± 0.1 | 39.4 ± 1.2 | 39.8 ± 0.9 | 18.7 | 1.1 | 6.4 |
| MP | 13.3 ± 0.2 | 1.8 ± 0.7 | 44.1 ± 1.2 | 37.2 ± 1.4 | 15.1 | 1.0 | 7.4 |
The chemical composition of each condition appeared to be homogeneous all over the surface, with a standard deviation of less than 2% for all elements detected. The Fe amount for EP1 and EP2 conditions was similar to those mechanically polished (∼12 at%), while EP3 conditions exhibited the highest amount of Fe concentration (16.1 ± 1.3 at%). EP1 samples presented the highest Mn content (2.9 ± 0.1 at%) followed by EP3 (2.5 at%), MP (1.8 ± 0.7 at%), and finally, EP2 (1.2 ± 0.4 at%). The overall metallic percentage (Fe + Mn) at% found on each sample surface was calculated (Table 5). EP3 and MP samples revealed a more metallic surface, with 18.7% and 15.1%, respectively, than EP1 and EP2 samples with 13.8%. That said, EP3 and MP samples displayed a low oxygen amount, ∼40 at%, while they had the highest metallic concentration. By opposite, the highest oxygen percentage was obtained for EP1 and EP2 conditions, ∼44 at%, with a lower metallic percentage than MP and EP3. Finally, the amount of carbon was similar for all EP samples, ∼40 at%, while MP had the highest C value of 44 at%. The high-resolution spectra of the different elements present on the surface were obtained (Table 6). The deconvolution of the O 1s high-resolution spectra evidenced differences. O1 (ref. 17) referred to metal oxide (Me–Ox), and O2 was related to the formation of hydroxides (Me–OHn), while O3 and O4 were associated with physio-adsorbed water.
| Sample | % area (O 1s peaks) | |||
|---|---|---|---|---|
| O1 | O2 | O3 | O4 | |
| 529.7–530.1 eV | 531.2–531.5 eV | 532.3–532.5 eV | 533.5–533.9 eV | |
| EP1 | 16.5 ± 0.8 | 40.0 ± 8.2 | 33.0 ± 9.7 | 10.4 ± 2.3 |
| EP2 | 41.1 ± 6.7 | 43.8 ± 3.4 | 15.1 ± 8.9 | – |
| EP3 | 53.4 ± 2.0 | 30.2 ± 0.7 | 13.8 ± 1.3 | 2.6 ± 1.4 |
| MP | 41.5 ± 4.7 | 37.6 ± 4.1 | 17.8 ± 4.6 | 3.1 ± 0.5 |
The oxygen species present on the surface were strongly correlated to the treatment used (Table 6). The ones found for EP1 conditions were significantly different from those present on the surface of the other conditions. Indeed, they displayed the highest O3 (33.0%) and O4 (10.4%) contributions, a relevant presence of O2 (40.0%) and the lowest O1 percentage (16.5%). In opposite, the EP3 sample exhibited the highest contribution of metal oxides (O1) (53.4%), when compared to the other conditions, and the lowest area associated with O3 and O4, respectively, 13.8% and 2.6%. The EP2 and MP samples presented a similar composition of the O band, with an O1 contribution of ∼41%, and an O2 one of the same amount as O1. The oxidation state of the surface given by the ratio Ometal/(Fe + Mn) was similar for the EP2, EP3 and MP samples, ranging from 1 to 1.3, while the EP1 conditions displayed the lowest value (0.5). The latter presented a higher amount of O bound to hydroxides than to metal oxides. The EP2 conditions displayed the highest metallic oxide contribution, with an O/metal ratio of 1.3, and no O4 contribution, as well as the lowest WCA value, WCAEP2 = 53 ± 2°, among all the conditions. These conditions also led to the highest RFe/Mn compared to the other conditions, with visible grain boundary features (∼9 nm step height). Furthermore, although the EP1 and EP3 conditions presented a mirror-like surface, EP1 showed numerous corrosion pits (Fig. 2a), while for the EP3 conditions, adsorption of organic residues from the viscous organic solution was observed. These two patterns can mislead electrochemical analysis, resulting in a falsely elevated corrosion rate due to the high presence of pits and slower dissolution, in the case of EP3. Therefore, EP2 exhibited the most promising chemistry and surface properties and will be used for further electrochemical and cytotoxicity assays compared to the reference sample, the MP conditions.
3.5. Electrochemical characterization
The results of PDP and EIS analyses of the EP2 and MP samples in both media, modified Hanks' solution and M199, are reported in Fig. 5. The pH of both solutions remained stable after electrochemical tests, consistently remaining within a range of 7.4 and 7.3; the detailed ionic composition of the solutions are presented in Table 3. The corrosion potential (Ecorr), current density (icorr), and corrosion rate (CR) were extracted from PDP curves (Fig. 5a). Regardless of the solution used, MP exhibited similar corrosion potentials (Ecorr, MP = −740 mV). However, EP2 showed a significant difference in corrosion potentials when analyzed in M199 (Ecorr, EP2 = −710 mV) and in Hanks' (Ecorr, EP2 = −750 mV) solutions. Therefore, the EP and MP samples displayed significantly different corrosion current densities. Indeed, when analyzed in Hanks' solution (Fig. 5b, solid lines), the MP surfaces showed a lower corrosion current density (icorr, MP = 7.2 ± 1.3 μA cm−2) than EP2 (icorr, EP2 = 11.2 ± 1.7 μA cm−2). While similar trends for cathodic branches were observed, the anodic dissolution one differed depending on the conditions. In fact, the EP2 ones displayed a faster anodic dissolution when compared to the MP ones, showing an overall corrosion resistance. Indeed, for Hanks' solution, the EP2 corrosion rate (138 ± 25 μm per year) was twice as high as that of the MP samples (45 ± 24 μm per year). Electrochemical measurements performed in M199 medium (dashed curves, Fig. 5b) showed an opposite trend to that observed for samples tested in Hanks' solution. It is noteworthy that the corrosion rate of MP showed no significant difference regardless of the solution.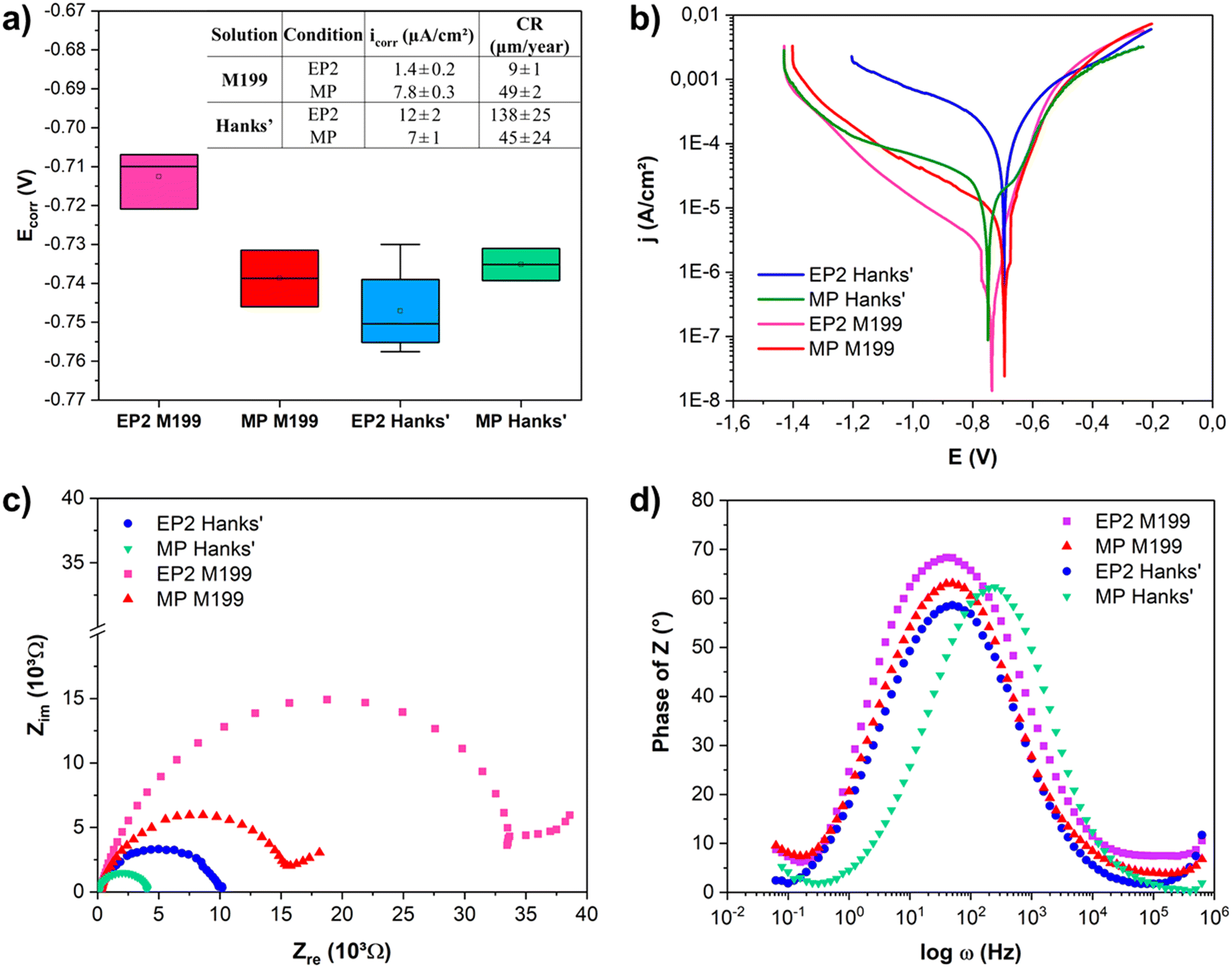 | ||
| Fig. 5 Electrochemical measurements of samples after EP2 and MP treatments: a) OCP and kinetic parameters of PDP measurements for EP2 and MP conditions in Hanks' solution and M199, b) PDP curves, c) Nyquist plots, and d) Bode plots as a function of phase angle in modified Hanks' solution (green) and M199 medium (blue). | ||
Fig. 5c presents the Nyquist plots. Regardless of the medium, the EP2 surface displayed a more capacitive behavior than that of the MP sample in both solutions; in other words, the impedance values of EP2 reached higher values than those obtained for the MP samples. This behavior could be related to the adsorption of inorganic molecules, such as HPO42− or other phosphate-based anions, forming a film at the surface that acts as a barrier against the corrosion process.70 Analyzing the Bode plots (Fig. 5d), all the conditions presented a time constant for the medium frequency region (∼100 Hz). The time constant in this region was attributed to localized corrosion of the substrate and the formation of corrosion products; the indication of another time constant at lower frequencies (∼0.1 Hz) was associated with species adsorption on the metallic substrate.71 Comparing the Bode plots, the time constant of the MP samples shifted to higher frequencies (around 500 Hz) when Hanks' solution was used. This shift indicates a faster corrosion process compared to the other conditions as seen for a Mg-based system.72
Electrochemical results were fitted using an electric equivalent circuit (EEC) to quantify changes in impedance values (Fig. 6a). The EEC was composed of Rs, the electrolyte resistance, with R1 and Q1 (Q as the constant phase element, or CPE) representing the ohmic resistance of the oxide/corrosion product interface and the capacitance of this layer, respectively. The additional (R2 and Q2) components represent the resistance of the corrosion products adsorbed when in contact with the studied solutions, as well as the capacitance of the electric double layer. All fitted electric parameters are shown in Fig. 6. The lowest values of Rs were found for the MP conditions, with Rs, MP = 10 Ω for Hanks' solution and Rs, MP = 8 Ω for M199. Both conditions exhibited a higher outer layer resistance R1 than the corresponding R2 (resistance of the inner passive oxide layer). For the EP2 conditions, the exponential n1, representative of the homogeneity of the system interface, was lower than 0.75 for both solutions. For the MP conditions, the same parameter presented a higher value when in contact with Hanks' solution (>0.7) than for M199 (0.2–0.5). Both conditions presented a deviation from the ideal capacitor which is related to the materials' surface chemical heterogeneities and roughness.73
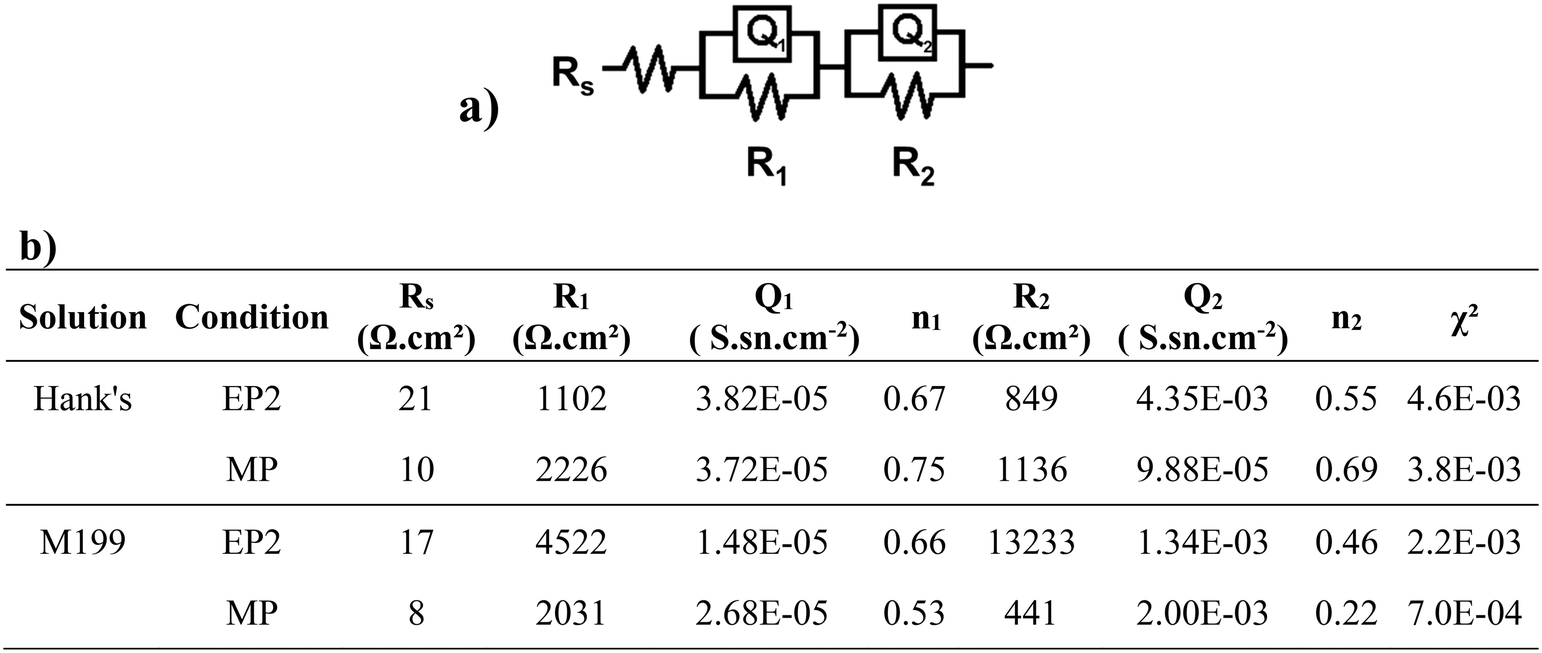 | ||
| Fig. 6 Electrical equivalent circuit attributed to this study (a) and the fitting parameters of EP2 and MP samples in contact with Hanks' and M199 solutions (b) where Rs: the electrolyte resistance, R1: resistance of the oxide/corrosion product layer, Q1: capacitance of the oxide/corrosion product layer, n1: dimensionless coefficient associated with Q1, R2: resistance of the inner oxide layer, Q2: capacitance of the inner oxide/metal layer, and n2: dimensionless coefficient associated with Q2. | ||
Although a different EP2 behavior was observed after Hanks' or M199 exposure, these conditions showed the highest charge transfer resistances, suggesting that their conductivity was low, whereas MP-treated samples exhibited lower corrosion resistance for both solutions.
XPS analyses were performed for EP2 and MP surfaces after exposure to both solutions as shown in Fig. 7. A lower C content, that is ∼30 at%, was observed when compared to that of pristine conditions, which was around 40 at% (Table 5). Compared to the chemical composition before corrosion assays, the oxygen content increased for all conditions (from 40 to 50 at%) except for EP2 analyzed in M199 solution. The MP conditions displayed the highest Fe amount on the surface after immersion in Hanks' solution, that is 20 ± 3 at% versus 5.8 ± 0.7 from EP2 analyzed in the same solution. Several elements present in both solutions, such as P and Mg, were found on EP2 and MP corroded surfaces.
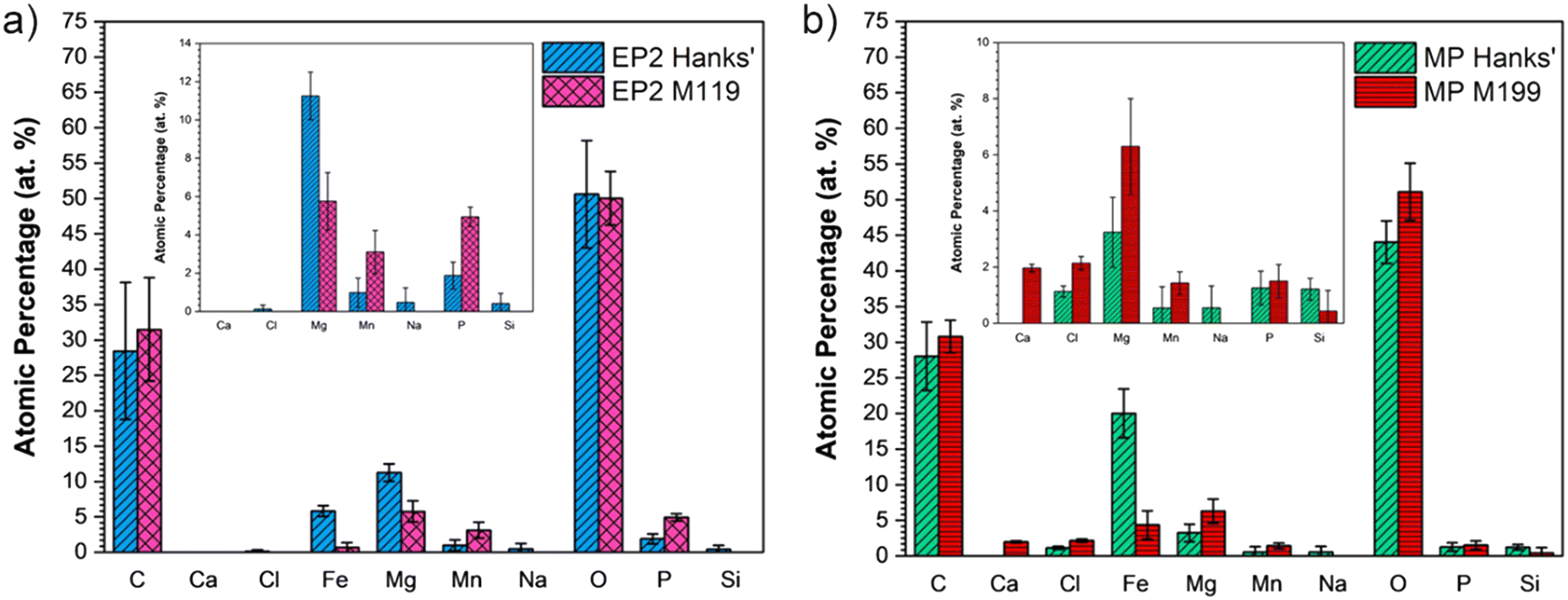 | ||
| Fig. 7 Chemical composition (at%) of a) EP2 and b) MP conditions analyzed after the PDP test in modified Hanks' solution and M199 medium. | ||
As seen in Fig. 7, Cl, Na, and Si were found in lower amounts (<2 at%) under EP2 conditions when analyzed in modified Hanks' solution and in either solution for MP samples. On the other hand, Fe and Mn were detected under both conditions in either solution. No statistical difference was observed for Mn detected amounts and all samples presented a lower Fe presence, except for MP in modified Hanks' solution, where the values increased compared with the initial conditions. Interestingly, no Ca was detected under either conditions in modified Hanks' solution, despite both electrolytes containing Ca2+ (as shown in Table 3, Ca2+Modified Hanks': 35 mg L−1 and Ca2+M199: 80 mg L−1). Ca was only detected on the MP surfaces immersed in the M199 solution. Moreover, the presence of Mg and P was detected under all corroded conditions since these elements were also present in both chemical compositions of solutions (see Table 3).
Overall, the Fe/Mn ratio decreased after corrosion tests, except for the MP conditions in modified Hanks' solution, which presented a ratio RFe/Mn: 37 (at%/at%), which means five times higher than the initial conditions, i.e. MP. This is mainly attributed to the increase of Fe content since no significant statistical changes were observed for Mn amounts in MP samples before and after the PDP test in Hanks' solution. The low Mn content may be attributed to a preferential pickling of the Mn compounds when in contact with both solutions.17 Moreover, the increase of oxygen may be attributed to the oxidation and adsorption of degradation products originating from Hanks' solution, which often contains carbonate, phosphate, and hydroxides.74,75 Regarding the Cl content found in MP samples analyzed in both solutions, it might be related to the higher amount of metallic species under the pristine conditions (% Fe + Mn: 15.1 at%) leading to the formation of metallic chloride (FeCl2) in the innermost layer at the surface, since Cl− ions present a high ionic volume (approximately 23.3 cm3 mol−1).76 Moreover, the higher Fe content observed on MP surfaces exposed to Hanks' solution might explain the less capacitive behavior evident in Nyquist plots. This could indeed be attributed to the dissolution of metallic elements from the bulk to the oxide interface since the resistance R2 (inner oxide layer) was lower than R1 (resistance of the oxide/corrosion product layer). Since the metallic ions encounter more resistance, they remain in place while the cathodic reaction occurs at the outer interface, increasing the oxygen content.
3.6. Cell viability assays
The relative cell viability of EP2 and MP conditions was compared with that of the control and SS316 samples for 1, 3 and 7 days at different eluate concentrations (100%, 10% and 1%).Regarding the viability of cells with 100% elution (pure extract), the SS316L conditions did not alter the HUVEC viability over time compared with the control. For the Fe-13Mn-1.2C alloy, both MP and EP2 conditions induced a decrease in cell viability, with no statistical difference between them. After 3 and 7 days, a decrease in cell viability was observed. Indeed, this behavior was expected, and it was related to the concentration of the degradation products of the medium in contact with the cells; longer cell contact (7 days vs. 1 day) with high-concentration degradation products (100% vs. 1%) was generally responsible for the lower viability21,77 for the HUVEC assays.
SS316L diluted extracts again exerted no effect on cell viability (Fig. 8b and c): in fact, cells showed viability comparable with the control conditions. As for the EP2 and MP samples, once again, their extracts induced cytotoxic effects on the treated HUVECs. However, an inverse correlation between extract concentrations and cytotoxic effects can be observed: increasing the dilution factor enhances the viability of the treated HUVECs. Moreover, for both conditions, no statistical difference was observed for EP2 and MP after 3 and 7 days in contact with the extract.
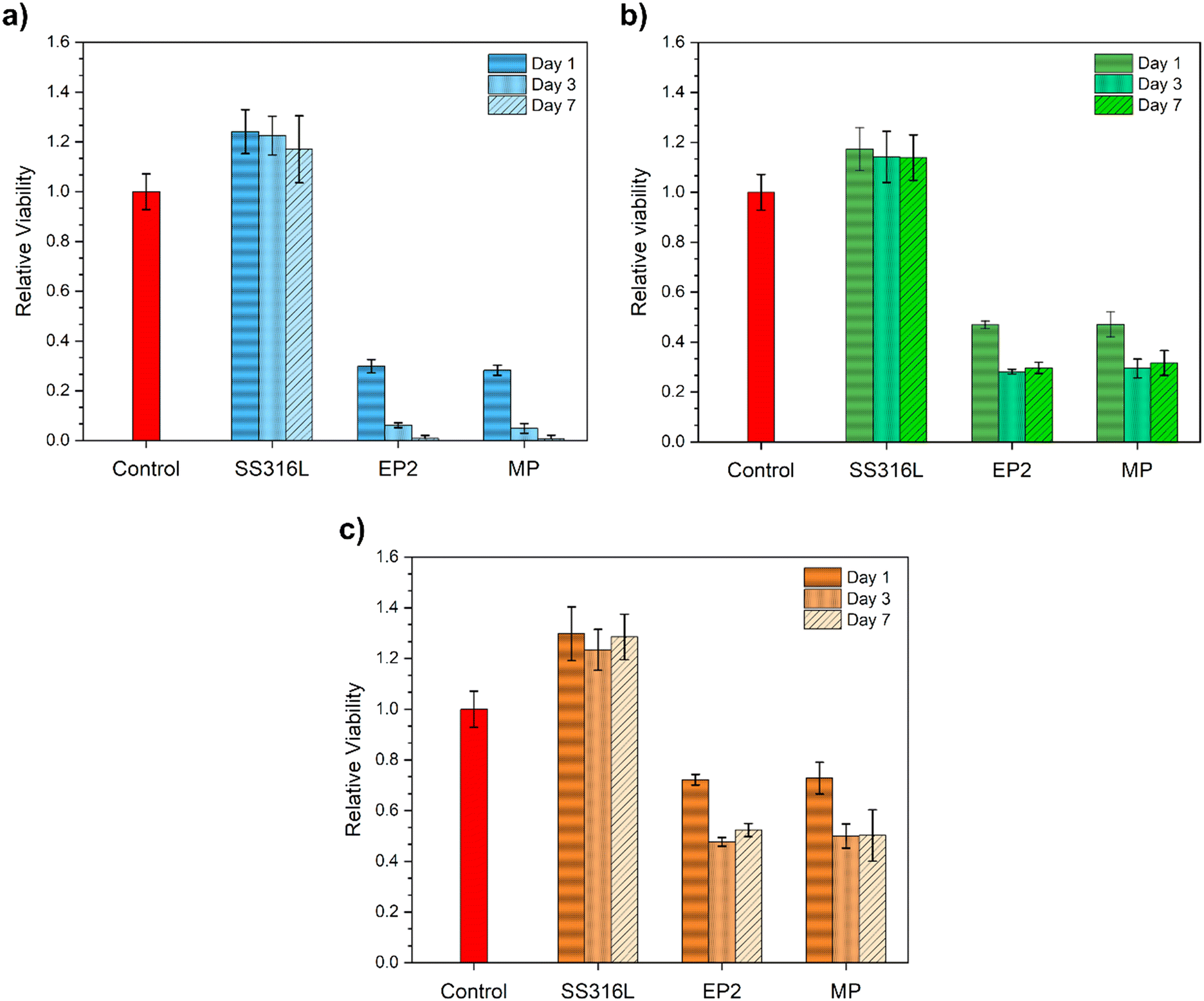 | ||
| Fig. 8 Cell viability of Fe-13Mn-1.2C at different eluate concentrations: (a) 100%, (b) 10% and (c) 1%. The extracts from EP2 and MP samples, upon contact with HUVECs, were analyzed and compared to the standard culture medium (control) and extracts from SS 316 L. Cell viability was measured after 1 day of incubation by means of a resazurin salt solution assay. The graph shows the mean fluorescence ± standard deviation, recorded from HUVECs treated with the different experimental conditions. Results have been normalized against the control conditions. | ||
4. Discussion
Although degradable metallic materials are designed for gradual dissolution, their surface properties play a critical role in regulating corrosion and eliciting a host response at the initial interface between the medical implant and the biological environment.78,79 Natural and induced oxide layers have been demonstrated to enhance surface corrosion resistance and improve cell compatibility on biodegradable materials.80 However, achieving control over the oxidation of biodegradable materials becomes a challenging endeavor when striving for simultaneous degradation and avoidance of cytotoxic effects in the human body.81 The passivation mechanism through electropolishing has undergone extensive investigation across various steel systems. The nature of electropolishing treatments on biodegradable steel surfaces is elucidated based on the chemical composition of the electrolyte, surface morphology, and topography, as well as the cytotoxicity effects and electrochemical analyses for each EP and MP treatment studied in this research.4.1. The influence of anodic dissolution on the electropolishing parameters and surface features
EP parameters were mainly related to the voltage–current curve where the limiting current region in LSV plots can be affected by, among other things, the electrolyte composition and the distribution and/or the concentration of electroactive species.82,83 The rate and reversibility of a reaction may be associated with variations in the peak observed in the current–potential graph. Indeed, samples tested in EP1 solution presented, at all scan rates, the highest current density peak (Icrit), even though no plateau was observed at 5 mV s−1 (Fig. 1b). The sharp increase in current density by increasing the scan rate from 20 to 100 mV s−1 could be associated with a fast reaction between the sample and the electrolyte due to the high concentration of acid in the solution, i.e., perchloric acid.84 At 100 mV s−1, the development of a plateau confirms the formation of an oxide layer on the steel surface. Indeed, the main mechanism proposed for this electrolyte is based on the formation of a salt film by the ions extracted from the substrate (Fe2+, Mn2/4+) which triggers the formation of pits. Polar groups, such as ethanol, may cover the surface and not allow metallic ions such as Fe2+ and Mn4+ to dissolve in the bulk solution, leading to a heterogeneous electropolishing process and an undefined plateau (region C, Fig. 1a).For EP2, perchlorate (ClO4−) and acetate (CH3COO−) anions can create a film through an adsorption process. Two hypothetical reactions of acetic acid and acetate ions with a divalent metallic element (Me2+) can occur:
| Me + 2CH3CO2H → Me(CH3CO2)2 + H2(g) | (2) |
| (CH3COO)2O + 3Me − 2e− → MeO + Me2+ + (CH3COO)2Me | (3) |
Samples treated in ChCl:EG solution (EP3) at room temperature and scan rates higher than 5 mV s−1 did not show a limiting current plateau, suggesting slow anodic dissolution, mainly controlled by an adsorption mechanism.
For all electropolishing conditions, the SEM images highlighted visible grain boundaries and a wavy surface morphology (Fig. 2). Indeed, a free-etched surface is expected after EP treatment.35 The etching mechanism occurs in the active dissolution region before reaching a diffusion-limited current (Fig. 1, region A). The crystal orientation influences the dissolution, resulting in distinct etch patterns,31 as observed in Fig. 3. The etching process stops at the formation of a passive oxide layer (Fig. 1, region B).69 However, grain boundaries have already been observed in the case of electropolished pure Zn (ref. 46) and pure Fe.87 While the observation of grain boundaries is generally considered a nonconformity in the electropolishing process, there is ongoing controversy in the literature regarding this aspect.30,49,88,89 Additionally, due to the limited number of published studies on the EP of bioabsorbable metallic alloys,46,87 specific mechanisms for this class of materials remain elusive.
4.2. Effect of the electropolishing process on the surface roughness and wettability
After the dissolution of the native oxide layer, a new surface with improved chemical homogeneity is expected to be formed.90 The MP conditions exhibit low roughness values similar to the EP1 conditions, and both are smoother compared to the EP2 and EP3 conditions (Fig. 4a). In fact, the roughness is dependent on the mechanism by which the material is removed, dissolved, or treated.91 For abrasive mechanical polishing, the effect of surface roughness is determined by the particle size of the abrasive element and the normal load applied on the surface during the process.26 For electropolishing, however, material removal is known to occur by two different mechanisms, which are anodic leveling (macro-smoothening) and micro-smoothening.31,92 The first is a function of the local charge distribution at the surface, where protuberances are preferentially dissolved. Micro-smoothening is induced by the suppression of the influence of surface defects (or crystallographic orientation) due to ionic salt formation, resulting in a diffusion-controlled process. However, the difference observed among the analyzed areas for EP2 and EP3 could be attributed to a microscale pattern and nanotopography. Liu et al.93 observed that nanotopography can stimulate the initial osteoblast cell adhesion and spreading compared to the outer most surface chemistry. In fact, the authors concluded that the nanotopography and the chemical composition synergetically are the main mechanisms of cell differentiation. Therefore, distinct electrolytes produce different ionic salts, and thus, different surface roughness.36 The presence of glycerol in EP1 and EP2 solutions promoted a significant decrease in surface roughness, Ra (Fig. 4a), and the formation of steps between adjacent grains with different orientations (Fig. 3g and h) compared to the EP3 sample (Fig. 4i). In the literature, similar trends have been observed on electropolished pure iron specimens where significant surface roughness variations were found by adding deionized water to the electrolyte composition.87The impact of micro-roughness on cell attachment is known,94 but it must also be correlated with wettability and nano-topography,95e.g., cell adhesion is enhanced on more hydrophilic surfaces. In this work, a range of water contact angles from 53° to 76° was found for all treated samples. Although samples treated in EP1 exhibited the smoothest surface among other conditions, the most hydrophilic character was obtained for EP2 (WCAEP2 = 53° ± 2°). For stenting applications, a slightly hydrophilic surface (between 40°and 60°) has been suggested for improving the adhesion of endothelial cells (ECs).96 In fact, highly hydrophilic surfaces bind proteins by weak forces and can lead to cell detachment, whereas hydrophobic surfaces promote EC adsorption but in a rigid and denatured state, resulting in conformational protein changes and reducing accessible sites to cell attachment.90,95,97
4.3. Chemical composition after passivation: metallic oxides
EP2 and EP1 conditions displayed a higher contribution of the O2 band, associated with the presence of hydroxides, than the MP one, that is ∼44%, ∼40%, and ∼38%, respectively. These hydroxides increased the surface wettability. Indeed, the EP2 surface displayed a more hydrophilic behavior than EP1 followed by MP, with WCAEP2 = 53 ± 2° < WCAEP2 = 64 ± 2° < WCAMP = 76 ± 3°, respectively. Therefore, the hydrophilic behavior seems to be related to the OH moieties found on the surface, corresponding to the O2 peak, without neglecting the contributions of O3 and O4 peaks, associated with physio-adsorbed water (Table 6). In contrast, the EP3 sample showed the highest contribution of the O1 peak (Me–O) and the lowest for the O2 peak, which may explain its low hydrophilicity character, WCAEP372 ± 5°, while exhibiting the lowest roughness values. Furthermore, EP3 displayed the highest concentration of metallic elements (Fe, Mn), with 18.7 at%, compared to the other conditions which exhibited a concentration range from 14–15% (Table 5). Finally, effective surface passivation was correlated with a metallic oxide enrichment, a high Ometal/metal ratio.4.4. The effect of surface treatments on the corrosion behavior and biological performances
As illustrated in Fig. 8, cell viability was influenced by the extract concentrations, with the most dilute solution (Fig. 8c) leading to a twofold increase in cell viability by mitigating the concentration level, as observed in other studies.74,98,99 In addition, surprisingly, there was no statistical difference in HUVEC viability between the two conditions, even with varying eluate concentrations (Fig. 8). Indeed, if we consider that the samples present a different corrosion rate (section 3.5), the amount of Fe and Mn released should therefore be different and potentially lead to a cytotoxic effect. The corrosion rate of EP2 in M199 was 9 ± 1 μm per year, which is ∼5 times less than that of MP (Fig. 5a, inset table). This difference in the corrosion rate, estimated by PDP tests, may be explained by the fact that EP2 degradation products formed on the surface act as “protective” barriers against corrosion compared to those of MP. Indeed, XPS analyses (Fig. 7e and f) show that the phosphorus content is higher in EP2 than in MP, suggesting that EP2 is more prone to adsorbing phosphates, as corroborated by its high resistance to charge transfer observed in Nyquist diagrams (Fig. 5c).The correlation between the lower corrosion resistance and the lower corrosion rate in MP-treated samples suggests a complex interaction between surface characteristics from the treatment and corrosion kinetics. While the treatment may not prevent corrosion initiation effectively, it could alter corrosion mechanisms or retard corrosion propagation once initiated. XPS analysis revealing a higher iron content implies greater permeability, mitigated by an anodically formed outer surface that restricts significant corrosion rate escalation. Thus, MP substrates show susceptibility to corrosion, albeit at a slower rate compared to EP2 conditions.
These findings underscore the importance of considering dynamic physiological conditions and larger sample volumes in toxicity assessments and corrosion studies for a more accurate representation of the material behavior in the human body. Indeed, assessing toxicity levels for soluble iron and manganese ions in static in vitro tests may be prone to overestimation, given the limited volume of the used medium (approximately 20 to 100 mL) compared to the total volume of human blood (6 L). In fact, toxic levels of metallic ions differ from in vitro to in vivo studies; therefore, concentrations of 3.5–5 μg mL−1 for Fe and 0.03–0.06 μg mL−1 for Mn in blood are known as critical concentrations.100 The absence of dynamic flow, mimicking the conditions within the human body, further complicates the accuracy of such estimations.101
5. Conclusions
This study systematically investigated the influence of three distinct electrolytes and electropolishing conditions on the surface features of Fe-13Mn-1.2C biodegradable steel suitable for applications in a cardiovascular context. Mechanically polished samples were used as reference conditions, to compare the electropolishing effect of two acidic solutions and an ionic liquid-based one. Dissolution mechanisms occurring at the cathode/solution interface were discussed, as well as their effect on the properties of the treated surface.1. Electropolishing was regulated mainly by two main mechanisms: a) a controlled mass transport observed for EP1 and EP2 electrolytes and b) an adsorption mechanism evident for the EP3 electrolyte. These findings suggest that EP1 and EP2 anodic dissolution was faster than that for EP3; EP1 showed the fastest anodic dissolution. Although surfaces subjected to EP1 and EP2 conditions had similar surface properties, those treated in EP2 displayed the highest ratio (1.3) of metal-bound O to metallic elements (Fe and Mn).
2. EP2 showed a passivated surface with reduced roughness (Ra ∼ 10 nm), a moderately hydrophilic behavior (WCAEP2 = 53°), a chemical composition combining a substantial proportion of metallic oxides (41.4% area of the O1 peak) with the highest contribution of metallic hydroxides (43.8% area of O2), and the absence of pitting and scattered degradation product heaps on the surface.
3. The significant corrosion resistance of EP2 samples in both modified Hanks' and M199 media could be attributed to the formation of a homogeneous oxide layer, hindering the diffusion of ions toward the material/medium interface.
4. No significant difference was observed in terms of cell viability studied for EP2 and MP conditions.
Further work should focus on the development of this process for tubular devices and on improving in vitro tests to more accurately simulate an endothelialization process.
Data Availability
OSF.io registration: https://osf.io/dashboard. Electropolishing Fe-based biodegradable metals for vascular applications: impact on surface properties, corrosion and cell viability https://osf.io/9dnve/?view_only=56104f08bb8b4d08bd64f54da56fdc33.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by the Natural Sciences and Engineering Research Council of Canada (Discovery and Alliance Programs), and the Government of Quebec Ministry of Economy, Innovation and Energy (PRIMA). L. M. d. A. acknowledges the support of Nathalie Moisan and Maxime Therrien from Laval University during the sample preparation for this work.References
- S. Hyuk, Y. Jung and S. Hyun, Current status and future direction of biodegradable metallic and polymeric vascular scaffolds for next-generation stents, Acta Biomater., 2017, 60, 3–22 CrossRef PubMed.
- H. H. Seop, S. Loffredo, I. Jun, J. Edwards, K. Y. Chan and S. H. Kwang, et al., Current status and outlook on the clinical translation of biodegradable metals, Mater. Today, 2018, 23, 57–71 Search PubMed.
- S. Verheye, A. Wlodarczak, P. Montorsi, J. Bennett, J. Torzewski and M. Haude, et al., Twelve-month outcomes of 400 patients treated with a resorbable metal scaffold: Insights from the BIOSOLVE-IV registry, EuroIntervention, 2020, 15, E1383–E1386 CrossRef PubMed.
- P. Poncin and J. Proft, Stent tubing: Understanding the desired attributes, Med. Device Mater., Proc. Mater. Processes Med. Devices Conf., 2003, 2003, 253–259 Search PubMed.
- M. Schinhammer, A. C. Hänzi, J. F. Löffler and P. J. Uggowitzer, Design strategy for biodegradable Fe-based alloys for medical applications, Acta Biomater., 2010, 6, 1705–1713 CrossRef CAS PubMed.
- G. Gąsior, J. Szczepański and A. Radtke, Biodegradable iron-based materials—what was done and what more can be done?, Materials, 2021, 14, 1–25 CrossRef PubMed.
- M. Caligari Conti, D. Aquilina, C. Paternoster, D. Vella, E. Sinagra and D. Mantovani, et al., Influence of cold rolling on in vitro cytotoxicity and electrochemical behaviour of an Fe–Mn–C biodegradable alloy in physiological solutions, Heliyon, 2018, 4, 1–32 CrossRef PubMed.
- P. Li, C. Schille, E. Schweizer, F. Rupp, A. Heiss and C. Legner, et al., Mechanical characteristics, in vitro degradation, cytotoxicity, and antibacterial evaluation of Zn-4.0Ag alloy as a biodegradable material, Int. J. Mol. Sci., 2018, 19, 755 CrossRef PubMed.
- E. Scarcello and D. Lison, Are Fe-based stenting materials biocompatible? A critical review of in vitro and in vivo studies, J. Funct. Biomater., 2020, 11, 1–16 Search PubMed.
- Y. Su, C. Luo, Z. Zhang, H. Hermawan, D. Zhu and J. Huang, et al., Bioinspired surface functionalization of metallic biomaterials, J. Mech. Behav. Biomed. Mater., 2018, 77, 90–105 CrossRef CAS PubMed.
- A. S. Hoffman, Modification of Material Surfaces to Affect How They Interact with Blood, Ann. N. Y. Acad. Sci., 1987, 516, 1–685 CrossRef PubMed.
- V. Milleret, A. Ziogas, M. Ehrbar, P. M. Markham, A. R. Tzafriri and D. L. Bhatt, Ultra-hydrophilic stent platforms promote early vascular healing and minimise late tissue response: a potential alternative to second-generation drug-eluting stents, EuroIntervention, 2019, 12, 2148–2156 Search PubMed.
- J. Chen, J. Dai, J. Qian, W. Li, R. Li and D. Pang, et al., Influence of Surface Roughness on Biodegradability and Cytocompatibility of High-Purity Magnesium, Materials, 2022, 15, 1–17 Search PubMed.
- C. Sunday Obayi, R. Tolouei, C. Paternoster, S. Turgeon, B. Adeleh and D. Oray, et al., Influence of cross-rolling on the micro-texture and biodegradation of pure iron as biodegradable material for medical implants, Acta Biomater., 2015, 17, 68–77 CrossRef PubMed.
- S. Loffredo, C. Paternoster, N. Giguère, G. Barucca, M. Vedani and D. Mantovani, The addition of silver affects the deformation mechanism of a twinning- induced plasticity steel: Potential for thinner degradable stents, Acta Biomater., 2019, 98, 103–113 CrossRef CAS PubMed.
- H. Hermawan and D. Mantovani, Process of prototyping coronary stents from biodegradable Fe-Mn alloys, Acta Biomater., 2013, 9, 8585–8592 CrossRef CAS PubMed.
- L. M. de Andrade, C. Paternoster, P. Chevallier, S. Gambaro, P. Mengucci and D. Mantovani, Surface processing for iron-based degradable alloys: A preliminary study on the importance of acid pickling, Bioact. Mater., 2022, 11, 166–180 CAS.
- Y. Yang, J. Zhou, R. Detsch, N. Taccardi, S. Heise and S. Virtanen, et al., Biodegradable nanostructures: Degradation process and biocompatibility of iron oxide nanostructured arrays, Mater. Sci. Eng., C, 2018, 85, 203–213 CrossRef CAS PubMed.
- J. Zhou, Y. Yang, M. Alonso Frank, R. Detsch, A. R. Boccaccini and S. Virtanen, Accelerated Degradation Behavior and Cytocompatibility of Pure Iron Treated with Sandblasting, ACS Appl. Mater. Interfaces, 2016, 8, 26482–26492 CrossRef CAS PubMed.
- J. Fiocchi, C. A. Biffi, S. Gambaro, C. Paternoster, D. Mantovani and A. Tuissi, Effect of laser welding on the mechanical and degradation behaviour of Fe-20Mn-0.6C bioabsorbable alloy, J. Mater. Res. Technol., 2020, 9, 13474–13482 CrossRef CAS.
- S. Loffredo, S. Gambaro, F. Copes, C. Paternoster, N. Giguère and M. Vedani, et al., Effect of silver in thermal treatments of Fe–Mn–C degradable metals: Implications for stent processing, Bioact. Mater., 2022, 12, 30–41 CAS.
- R. Xu, X. Yang, X. Zhang, M. Wang, P. Li and Y. Zhao, et al., Effects of carbon dioxide plasma immersion ion implantation on the electrochemical properties of AZ31 magnesium alloy in physiological environment, Appl. Surf. Sci., 2013, 286, 257–260 CrossRef CAS.
- N. Shevchenko, M. Pham and M. F. Maitz, Studies of surface modified NiTi alloy, Appl. Surf. Sci., 2004, 235, 126–131 CrossRef CAS.
- T. Huang, Y. Cheng and Y. Zheng, In vitro studies on silver implanted pure iron by metal vapor vacuum arc technique, Colloids Surf., B, 2016, 142, 20–29 CrossRef CAS PubMed.
- Q. Feng, D. Zhang and C. Xin, Characterization and in vivo evaluation of a bio-corrodible nitrided iron stent, J. Mater. Sci.:Mater. Med., 2013, 24, 713–724 CrossRef CAS PubMed.
- K. Geels, K. W. Ulrich and M. Rückert, Mechanical Surface Preparation—Polishing, in Metallographic and Materialographic Specimen Preparation, Light Microscopy, Image Analysis and Hardness Testing, ASTM International, West Conshohochen, 1st edn, 2007, pp. 138–173 Search PubMed.
- L. R. Hilbert, D. Bagge-Ravn, J. Kold and L. Gram, Influence of surface roughness of stainless steel on microbial adhesion and corrosion resistance, Int. Biodeterior. Biodegrad., 2003, 52, 175–185 CrossRef CAS.
- G. Yang, B. Wang, K. Tawfiq, H. Wei, S. Zhou and G. Chen, Electropolishing of surfaces: theory and applications, Surf. Eng., 2017, 33, 149–166 CrossRef CAS.
- I. De Scheerder, J. Sohier, L. Froyen, J. Van Humbeeck and E. Verbeken, Biocompatibility of Coronary Stent Materials: Effect of Electrochemical Polishing, Materwiss. Werksttech., 2001, 32, 142–148 CrossRef CAS.
- M. Haïdopoulos, S. Turgeon, C. Sarra-Bournet, G. Laroche and D. Mantovani, Development of an optimized electrochemical process for subsequent coating of 316 stainless steel for stent applications, J. Mater. Sci.: Mater. Med., 2006, 17, 647–657 CrossRef PubMed.
- D. Landolt, Fundamental aspects of electropolishing, Electrochim. Acta, 1987, 32, 1–11 CrossRef CAS.
- S. J. Lee and J. J. Lai, The effects of electropolishing (EP) process parameters on corrosion resistance of 316L stainless steel, J. Mater. Process. Technol., 2003, 140, 206–210 CrossRef CAS.
- E. S. Lee, Machining characteristics of the electropolishing of stainless steel (STS316L), Int. J. Adv. Manuf. Technol., 2000, 16, 591–599 CrossRef.
- D. K. Das and G. Freedman, Electropolishing of surfaces: theory and applications, United States Patent Office, US3352667A, 1964, vol. 14, pp. 131–134.
- W. Han and F. Fang, Fundamental aspects and recent developments in electropolishing, Int. J. Mach. Tools Manuf., 2019, 139, 1–23 CrossRef.
- E. Łyczkowska-Widłak, P. Lochyński and G. Nawrat, Electrochemical polishing of austenitic stainless steels, Materials, 2020, 13, 1–25 CrossRef PubMed.
- P. Sojitra, C. Engineer, D. Kothwala, A. Raval, H. Kotadia and G. Mehta, Electropolishing of 316LVM stainless steel cardiovascular stents: An investigation of material removal, surface roughness and corrosion behaviour, Trends Biomater. Artif. Organs, 2010, 23, 115–121 Search PubMed.
- C. C. Lin and C. C. Hu, Electropolishing of 304 stainless steel: Surface roughness control using experimental design strategies and a summarized electropolishing model, Electrochim. Acta, 2008, 53, 3356–3363 CrossRef CAS.
- C. C. Lin, C. C. Hu and T. C. Lee, Electropolishing of 304 stainless steel: Interactive effects of glycerol content, bath temperature, and current density on surface roughness and morphology, Surf. Coat. Technol., 2009, 204, 448–454 CrossRef CAS.
- S. Mohan, D. Kanagaraj, R. Sindhuja, S. Vijayalakshmi and N. G. Renganathan, Electropolishing of stainless steel - A review, Trans. Inst. Met. Finish., 2001, 79, 140–142 CrossRef CAS.
- C. C. Irving, Electropolishing Stainless Steel Implants, ASTM Spec. Tech. Publ., 1985, 136–143 CAS.
- J. A. Sandoval-Robles, C. A. Rodríguez and E. García-López, Laser surface texturing and electropolishing of CoCr and Ti6Al4V-ELI alloys for biomedical applications, Materials, 2020, 13, 1–20 CrossRef PubMed.
- H. Aihara, Surface and biocompatibility study of electropolished Co-Cr alloy L605, San Jose State University, 2009 Search PubMed.
- E. S. Lee and T. H. Shin, An evaluation of the machinability of nitinol shape memory alloy by electrochemical polishing, J. Mech. Sci. Technol., 2011, 25, 963–969 CrossRef.
- R. I. M. Asri, W. S. W. Harun, M. Samykano, N. A. C. Lah, S. A. C. Ghani and F. Tarlochan, et al., Corrosion and surface modification on biocompatible metals: A review, Mater. Sci. Eng., C, 2017, 77, 1261–1274 CrossRef CAS PubMed.
- R. J. Guillory, M. Sikora-Jasinska, J. W. Drelich and J. Goldman, In Vitro Corrosion and in Vivo Response to Zinc Implants with Electropolished and Anodized Surfaces, ACS Appl. Mater. Interfaces, 2019, 11, 19884–198893 CrossRef CAS PubMed.
- K. P. Liu, J. L. You, S. Y. Jian, Y. H. Chang, C. C. Tseng and M. D. Ger, Effect of electropolishing parameters of WE43 magnesium alloy on corrosion resistance of artificial plasma, J. Mater. Res. Technol., 2023, 26, 4989–5000 CrossRef CAS.
- A. P. Abbott, G. Capper, K. J. McKenzie, A. Glidle and K. S. Ryder, Electropolishing of stainless steels in a choline chloride based ionic liquid: an electrochemical study with surface characterisation using SEM and atomic force microscopy, Phys. Chem. Chem. Phys., 2006, 8, 4214–4221 RSC.
- T. M. Abdel-Fattah and J. D. Loftis, Comparison of Electropolishing of Aluminum in a Deep Eutectic Medium and Acidic Electrolyte, Molecules, 2020, 25, 1–8 CrossRef PubMed.
- A. P. Abbott, G. Capper, B. G. Swain and D. A. Wheeler, Electropolishing of stainless steel in an ionic liquid, Trans. IMF, 2005, 83, 51–53 CrossRef CAS.
- J. D. Loftis and T. M. Abdel-Fattah, Nanoscale electropolishing of high-purity silver with a deep eutectic solvent, Colloids Surf., A, 2016, 511, 113–119 CrossRef CAS.
- A. J. Goddard, R. C. Harris, S. Saleem, M. Azam, C. Hood and D. Clark, et al., Electropolishing and electrolytic etching of Ni-based HIP consolidated aerospace forms: a comparison between deep eutectic solvents and aqueous electrolytes, Trans. Inst. Met. Finish., 2017, 95, 137–146 CrossRef CAS.
- A. P. Abbott, G. Frisch, J. Hartley, W. O. Karim and K. S. Ryder, Anodic dissolution of metals in ionic liquids, Prog. Nat. Sci.: Mater. Int., 2015, 25, 595–602 CrossRef CAS.
- A. P. Abbott, G. Capper, K. J. McKenzie and K. S. Ryder, Voltammetric and impedance studies of the electropolishing of type 316 stainless steel in a choline chloride based ionic liquid, Electrochim. Acta, 2006, 51, 4420–4425 CrossRef CAS.
- ASTM International, ASTM E1558-09: Standard guide for electrolytic polishing of metallographic specimens, Annu. Book ASTM Stand., 2009, 09, 1–13 Search PubMed.
- I. Horcas, R. Fernández, J. M. Gómez-Rodríguez, J. Colchero, J. Gómez-Herrero and A. M. Baro, WSXM: A software for scanning probe microscopy and a tool for nanotechnology, Rev. Sci. Instrum., 2007, 78, 1–8 CrossRef PubMed.
- E. S. Gadelmawla, M. M. Koura, T. M. A. Maksoud, I. M. Elewa and H. H. Soliman, Roughness parameters, J. Mater. Process. Technol., 2002, 123, 133–145 CrossRef.
- ASTM International, ASTM D7334-08: Standard practice for surface wettability of coatings, substrates and pigments by advancing contact angle measurement: active standard, Annu. Book ASTM Stand., 2013, 08, 1–3 Search PubMed.
- ASTM International, ASTM G102 A: Standard Practice for from Electrochemical Measurements, Annu. Book ASTM Stand., 2018, 89, 1–7 Search PubMed.
- J. Lévesque, H. Hermawan, D. Dubé and D. Mantovani, Design of a pseudo-physiological test bench specific to the development of biodegradable metallic biomaterials, Acta Biomater., 2008, 4, 284–295 CrossRef PubMed.
- Thermofisher Scientific, M199-Media Formulation [Internet]. Technical ressources, 2024, [cited 2024 Jun 16], Available from: https://www.thermofisher.com/ca/en/home/technical-resources/media-formulation.86.html.
- J. D. Bronzino, The Biomedical Engineering Handbook: Biomedical Engineering Fundamentals, Clavier Companion, CRC - Taylor & Francis, Boca Raton, 3rd edn, 2006, vol. 8, pp. 1–1569 Search PubMed.
- C. Loy, S. Meghezi, L. Lévesque, D. Pezzoli, H. Kumra and D. Reinhardt, et al., A planar model of the vessel wall from cellularized-collagen scaffolds: focus on cell – matrix interactions in mono-, bi- and tri-culture models, Biomater. Sci., 2017, 5, 153–162 RSC.
- ISO 10993-5, Biological evaluation of medical devices - Part 5: tests for in vitro cytotoxicity, International Organization for Standardization, 2009 Search PubMed.
- C. S. Campelo, P. Chevallier, C. Loy, R. S. Vieira and D. Mantovani, Development , Validation , and Performance of Chitosan-Based Coatings Using Catechol Coupling, Macromol. Biosci., 2020, 20, 1–11 Search PubMed.
- M. Matlosz, Modeling of impedance mechanisms in electropolishing, Electrochim. Acta, 1995, 40, 393–401 CrossRef CAS.
- S. Allain, J. P. Chateau, O. Bouaziz, S. Migot and N. Guelton, Correlations between the calculated stacking fault energy and the plasticity mechanisms in Fe–Mn–C alloys, Mater. Sci. Eng., A, 2004, 387–389 Search PubMed.
- R. Sautebin, H. Froidevaux and D. Landolt, Theoretical and Experimental Modeling of Surface Leveling in ECM under Primary Current Distribution Conditions, J. Electrochem. Soc., 1980, 127, 1096–1100 CrossRef CAS.
- T. P. Hoar and J. Mowat, Mechanism of electropolishing, Nature, 1950, 165, 64–65 CrossRef CAS.
- H. Yang, K. Yang and B. Zhang, Pitting corrosion resistance of La added 316L stainless steel in simulated body fluids, Mater. Lett., 2007, 61, 1154–1157 CrossRef CAS.
- J. Huang, A. Gonzalez Orive, J. T. Krüger, K. P. Hoyer, A. Keller and G. Grundmeier, Influence of proteins on the corrosion of a conventional and selective laser beam melted FeMn alloy in physiological electrolytes, Corros. Sci., 2022, 200, 110186 CrossRef CAS.
- S. Gambaro, M. L. Nascimento, M. Shekargoftar, S. Ravanbakhsh, V. Sales and C. Paternoster, et al., Characterization of a Magnesium Fluoride Conversion Coating on Mg-2Y-1Mn-1Zn Screws for Biomedical Applications, Materials, 2022, 15, 1–18 CrossRef PubMed.
- L. Wang, D. Snihirova, M. Deng, C. Wang, B. Vaghefinazari and S. V. Lamaka, Insight into physical interpretation of high frequency time constant in electrochemical impedance spectra of Mg, Corros. Sci., 2021, 187, 1–13 Search PubMed.
- S. Loffredo, S. Gambaro, L. Marin De Andrade, C. Paternoster, R. Casati and N. Giguère, et al., Six-Month Long in Vitro Degradation Tests of Biodegradable Twinning-Induced Plasticity Steels Alloyed with Ag for Stent Applications, ACS Biomater. Sci. Eng., 2021, 7, 3669–3682 CrossRef CAS PubMed.
- E. Mouzou, C. Paternoster, R. Tolouei, A. Purnama, P. Chevallier and D. Dubé, et al., In vitro degradation behavior of Fe-20Mn-1.2C alloy in three different pseudo-physiological solutions, Mater. Sci. Eng., C, 2016, 61, 564–573 CrossRef CAS PubMed.
- Y. Marcus, Properties of individual ions in solution, in Solution Chemistry Research Progress, ed. D. V. Bostrelli, Nova science Publishers, Inc., New York, 2008, pp. 51–68 Search PubMed.
- E. Mouzou, PhD Thesis, Laval University, 2017 Search PubMed.
- S. Gambaro, C. Paternoster, B. Occhionero, J. Fiocchi, C. A. Biffi and A. Tuissi, et al., Mechanical and degradation behavior of three Fe–Mn–C alloys for potential biomedical applications, Mater. Today Commun., 2021, 27, 1–12 Search PubMed.
- Y. Shen, M. Gao, Y. Ma, H. Yu, C. F. Zhai and H. Gregersen, et al., Effect of surface chemistry on the integrin induced pathway in regulating vascular endothelial cells migration, Colloids Surf., B, 2015, 126, 188–197 CrossRef CAS PubMed.
- S. Zhu, N. Huang, H. Shu, Y. Wu and L. Xu, Corrosion resistance and blood compatibility of lanthanum ion implanted pure iron by MEVVA, Appl. Surf. Sci., 2009, 256, 99–104 CrossRef CAS.
- N. S. Fagali, C. A. Grillo, S. Puntarulo and M. A. Fernández Lorenzo de Mele, Is there any difference in the biological impact of soluble and insoluble degradation products of iron-containing biomaterials?, Colloids Surf., B, 2017, 160, 238–246 CrossRef CAS PubMed.
- G. Yang, B. Wang, K. Tawfiq, H. Wei, S. Zhou and G. Chen, Electropolishing of surfaces: theory and applications, Surf. Eng., 2017, 33, 149–166 CrossRef CAS.
- C. Ponce-De-León, C. T. J. Low, G. Kear and F. C. Walsh, Strategies for the determination of the convective-diffusion limiting current from steady state linear sweep voltammetry, J. Appl. Electrochem., 2007, 37, 1261–1270 CrossRef.
- D. Pourjafari Nokandeh, D. I. Martínez Delgado, A. Vázquez Dimas and I. Gómez De la Fuente, The effect of changing the electrolyte species volume ratio on the electropolishing of aluminum foil, Química Hoy Chemistry Sciences, 2012, 2, 9–12 Search PubMed.
- A. Kahyarian, A. Schumaker, B. Brown and S. Nesic, Acidic corrosion of mild steel in the presence of acetic acid: Mechanism and prediction, Electrochim. Acta, 2017, 258, 639–652 CrossRef CAS.
- D. Yan, M. Z. Bazant, P. M. Biesheuvel, M. C. Pugh and F. P. Dawson, Theory of linear sweep voltammetry with diffuse charge: Unsupported electrolytes, thin films, and leaky membranes, Phys. Rev. E, 2017, 95, 1–20 CrossRef PubMed.
- V. O. F. Sales, Presented in part at 17th Enemet, Sao Paulo, 2017 Search PubMed.
- G. Han, Z. Lu, X. Ru, J. Chen, Q. Xiao and Y. Tian, Improving the oxidation resistance of 316L stainless steel in simulated pressurized water reactor primary water by electropolishing treatment, J. Nucl. Mater., 2015, 467, 194–204 CrossRef CAS.
- S. Diaz-Rodriguez, C. Loy, P. Chevallier, C. Noël, G. Caligiuri and L. Houssiau, et al., Comparison of the linking arm effect on the biological performance of a CD31 agonist directly grafted on L605 CoCr alloy by a plasma-based multistep strategy, Biointerphases, 2019, 14, 051009–051024 CrossRef PubMed.
- H. U. Sajid and R. Kiran, Influence of corrosion and surface roughness on wettability of ASTM A36 steels, J. Constr. Steel Res., 2018, 144, 310–326 CrossRef.
- P. Pendyala, M. S. Bobji and G. Madras, Evolution of surface roughness during electropolishing, Tribol. Lett., 2014, 55, 93–101 CrossRef CAS.
- J. Edwards, The Mechanism of Electropolishing of Copper in Phosphoric Acid Solutions, J. Electrochem. Soc., 1953, 100, 189–194C CrossRef.
- X. Liu, Y. Xie, S. Shi, Q. Feng, A. Bachhuka and X. Guo, et al., The co-effect of surface topography gradient fabricated via immobilization of gold nanoparticles and surface chemistry via deposition of plasma polymerized film of allylamine/acrylic acid on osteoblast-like cell behavior, Appl. Surf. Sci., 2019, 473, 838–847 CrossRef CAS.
- M. Rahmati, E. A. Silva, J. E. Reseland, C. A. Heyward and H. J. Haugen, Biological responses to physicochemical properties of biomaterial surface, Chem. Soc. Rev., 2020, 49, 5178–5224 RSC.
- L. Bacakova, E. Filova, M. Parizek, T. Ruml and V. Svorcik, Modulation of cell adhesion, proliferation and differentiation on materials designed for body implants, Biotechnol. Adv., 2011, 29, 739–767 CrossRef CAS PubMed.
- Y. Arima and H. Iwata, Effect of wettability and surface functional groups on protein adsorption and cell adhesion using well-defined mixed self-assembled monolayers, Biomaterials, 2007, 28, 3074–3082 CrossRef CAS PubMed.
- J. Drelich, E. Chibowski, D. D. Meng and K. Terpilowski, Hydrophilic and superhydrophilic surfaces and materials, Soft Matter, 2011, 7, 9804–9828 RSC.
- M. Schinhammer, I. Gerber, A. C. Hänzi and P. J. Uggowitzer, On the cytocompatibility of biodegradable Fe-based alloys, Mater. Sci. Eng., C, 2013, 33, 782–789 CrossRef CAS PubMed.
- M. Peuster, P. Wohlsein, M. Brügmann, M. Ehlerding, K. Seidler and C. Fink, et al., A novel approach to temporary stenting: degradable cardiovascular stents produced from corrodible metal-results 6-18 months after implantation into New Zealand white rabbits, Heart, 2001, 86, 563–569 CrossRef CAS PubMed.
- H. Hermawan, A. Purnama, D. Dube, J. Couet and D. Mantovani, Fe-Mn alloys for metallic biodegradable stents: Degradation and cell viability studies, Acta Biomater., 2010, 6, 1852–1860 CrossRef CAS PubMed.
- B. Paul, A. Lode, A. M. Placht, A. Voß, S. Pilz and U. Wolff, et al., Cell-Material Interactions in Direct Contact Culture of Endothelial Cells on Biodegradable Iron-Based Stents Fabricated by Laser Powder Bed Fusion and Impact of Ion Release, ACS Appl. Mater. Interfaces, 2022, 14, 439–451 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |