
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective adsorption and separation of C6 hydrocarbons: the role of structural flexibility and functionalization in zeolitic imidazolate frameworks†
Kevin
Dedecker
 *,
Martin
Drobek
* and
Anne
Julbe
*,
Martin
Drobek
* and
Anne
Julbe
Institut Européen des Membranes (IEM), CNRS, ENSCM, Univ Montpellier, Place Eugène Bataillon, 34095 Montpellier, France. E-mail: kevin.dedecker@umontpellier.fr; martin.drobek@umontpellier.fr
First published on 19th December 2024
Abstract
This study investigates the selective adsorption and separation of C6 hydrocarbons (benzene, cyclohexane, and n-hexane) by zeolitic imidazolate frameworks (ZIFs), focusing on their structural flexibility and functionalization. ZIF-8_CH3 and ZIF-8_Br were synthesized and compared, indicating distinct adsorption behaviors. ZIF-8_CH3 showed higher uptake for benzene (9.5 molecules per unit cell) and n-hexane (8.0 mlc uc−1) compared to cyclohexane (1.0 mlc uc−1). In contrast, ZIF-8_Br exhibited enhanced adsorption for cyclohexane (5.0 mlc uc−1) and reduced n-hexane uptake (0.5 mlc uc−1). Computational simulations supported these findings, identifying the involved host–guest interactions. Ideal adsorbed solution theory analysis confirmed that ZIF-8_CH3 demonstrated virtually zero uptake of cyclohexane from binary mixtures containing either n-hexane or benzene, while ZIF-8_Br exhibited negligible adsorption of n-hexane from its mixtures with cyclohexane or benzene. It was concluded that bromine functionalization in ZIF-8_Br increased structural rigidity and selectivity for aromatic compounds. These results highlight the crucial role of functionalization and gate-opening phenomena in ZIFs to achieve efficient volatile organic compound capture and separation where traditional adsorbents may not be effective.
Introduction
In recent decades, the issue of airborne pollution has emerged as a critical concern, leading to the development of environmental policies aimed at reducing emissions, detecting hazardous gases, and removing contaminants. Among these pollutants, volatile organic compounds (VOCs), particularly C6 hydrocarbons, have attracted significant attention due to their detrimental effects on human health.1 Benzene, cyclohexane, and n-hexane stand out as particularly problematic compounds, each associated with severe health risks ranging from blood diseases to neurological disorders.Benzene exposure, for instance, has been correlated with serious blood conditions such as anaemia and leukemia,2,3 leading regulatory bodies to continuously reduce acceptable exposure limits below 1 ppm.4 This reduction is particularly crucial given the omnipresence of benzene in urban environments and enclosed spaces, where concentrations can exceed outdoor levels by up to 1.5 times.5 Cyclohexane, while less hazardous than benzene, still poses significant health risks, including cognitive impairment and oxidative stress.6N-Hexane, commonly used as an industrial solvent, is associated with polyneuropathy and maculopathy, disrupting motor and sensory nerve functions through prolonged exposure.7
Beyond health considerations, the separation of benzene and cyclohexane presents an important challenge in industrial processes due to their azeotropic mixture and nearly identical boiling points (80.1 °C for benzene and 80.7 °C for cyclohexane).8 This issue is particularly significant given that 85% of cyclohexane is produced through the catalytic hydrogenation of benzene.8,9
Addressing these challenges requires a multifaceted approach that combines efficient VOC detection and removal from air. Conventional adsorbents like activated carbons and zeolites often struggle with low selectivity and adsorption capacity, particularly in humid environments, limiting their efficacy under real-world conditions.10–12 This has led to intensive research into alternative adsorbents such as metal–organic frameworks (MOFs) appearing as promising candidates to overcome the above limitations.
MOFs, a class of coordination polymers, offer unique advantages due to their structural diversity and tuneable properties.13 These materials, formed through the self-assembly of metal clusters and organic linkers, generally possess high pore volumes and specific surface areas.14 Their key strength lies in the ability to tailor adsorption properties through judicious selection of metals15,16 and ligands,17,18 allowing for remarkable control over the selectivity and adsorption capacity.
Previous studies have demonstrated the potential of MOFs in VOC capture.19 For instance, [Pd(2-pymo)2]n has shown preferential adsorption of benzene over n-hexane, cyclohexane, and toluene, while its isostructural counterpart [Cu(2-pymo)2]n exhibited greater affinity for n-hexane.20 This finding highlights the crucial role of metal centers in determining material selectivity towards specific molecules. Furthermore, investigations of isoreticular MOFs (IRMOFs) have shown that integrating pyrene cores into the structure building ligands (e.g. in IRMOF-14) significantly increases adsorption efficiency for both cyclohexane and benzene, thus highlighting the importance of ligand aromaticity in enhancing host–guest interactions.21
Ligand modification has also proven effective in enhancing MOF performance.22–24 For example, UiO-66-(CF3)2, with perfluorinated groups on its terephthalate ligands, exhibits higher hydrophobicity and stronger interactions with VOCs (e.g. acetic acid,22 ethane23) compared to its non-fluorinated counterpart, making it particularly promising for VOC capture in humid conditions. Additionally, precise control over ligand length allows for tuning of pore size, facilitating either molecular sieving25 or enhanced host–guest interactions through confinement effects.22
A particularly remarkable aspect of MOFs is their potential for stimuli-responsive behavior,26 as demonstrated by structures like MIL-53's27 and zeolitic imidazolate frameworks (ZIFs).28 ZIFs, a subclass of MOFs featuring tetrahedral coordination of metal ions with imidazolate-type linkers, have attracted significant attention due to their structural similarity with zeolites and their unique physico-chemical properties.28,29 Notably, the hydrophobic22,24,30–32 and fairly moisture-resistant29,33,34 ZIF-8_CH3, composed of zinc cations and 2-methylimidazolate ligands, is well-known for its “swing effect” or “gate-opening” phenomenon,35,36 whereby linkers reorient to expand pore apertures upon stimulation.37,38
While this flexibility can be advantageous for certain applications, it can also hinder molecular discrimination by impeding the sieving effect.39 Consequently, strategies to reduce structural flexibility and enhance selectivity have become a focus of many research studied. Methods such as defibrillation under electric fields,40 rapid thermal treatment,41 and post-synthetic functionalization42 have been explored to increase structure stiffening.
Indeed, the flexibility of MOFs is profoundly influenced by their structural characteristics. For instance, replacing zinc with cobalt in ZIF-8_CH3 results in smaller pore apertures due to shorter Co–N bonds, leading to a less flexible framework.43 This approach has been successfully employed to improve CO2/CH4 separation44 in mixed Zn/Co-ZIF-8_CH3 systems.45 Furthermore, functionalizing the imidazolate linkers in ZIF-8 significantly affects its structural and adsorption properties. Research by Chaplais et al.46 demonstrated that the structural flexibility of ZIF materials decreases as follows: ZIF-8_CH3 > ZIF-8_Cl ≫ ZIF-8_Br due to several factors. They suggested that ZIF-8_CH3 and ZIF-8_Cl enable organic linker rotation, allowing the framework to accommodate additional adsorbates more efficiently, whereas the larger bromine atom in ZIF-8_Br creates steric hindrance, thus making the structure more rigid. Additionally, the larger bromine atom reduces the pore size compared to ZIF-8_CH3 and ZIF-8_Cl, thereby limiting the ability of the material to undergo reordering. ZIF-8_CH3 and ZIF-8_Cl can exhibit transition from a cubic-like to a tetragonal-like phase during adsorption, further enhancing their flexibility. However, this transition is inhibited in ZIF-8_Br due to its smaller pores, resulting in increased rigidity. The bulkier bromine atom also restricts linker movement, preventing the structural transitions that are necessary for higher adsorbate uptake, which is not the case with the smaller methyl and chlorine substituents.
Among the numerous ZIFs reported in literature, ZIF-8_Br appears to be one of the rare cases of ZIFs exhibiting intrinsic structural rigidity, making it particularly promising for selective adsorption and separation processes relying on molecular sieving mechanisms.
Our investigation aims to explore the relationship between the structural features of ZIFs and their performance in capturing and separating three structurally similar harmful C6 hydrocarbons: n-hexane (a flexible linear alkane), cyclohexane (a semi-flexible cyclic alkane), and benzene (a rigid planar aromatic).
To elucidate interactions involved, a comprehensive study was conducted comparing the adsorption behaviour of ZIF-8_Br with that of ZIF-8_CH3, aiming to shed a better light to the impact of structural flexibility and stimuli-responsive behaviour on adsorption phenomena. Our experimental approach was complemented by computational investigations involving Grand Canonical Monte Carlo simulations to better understand the underlying adsorption mechanisms.
The obtained results highlight how the structural features of ZIF-8_Br enhance its selectivity, providing valuable insights into the role of structure stiffness in MOF adsorption performance. This study not only enriches the understanding of adsorption processes in flexible MOFs but also paves the way for the rational design of tailored adsorbents for specific environmental and industrial applications. By elucidating the structure–property relationships governing adsorption behaviour, this work contributes to the development of more efficient and selective materials for VOC capture and separation aiming to address critical challenges in air quality management and industrial processes.
Experimental section
Materials and methods
All reagents were procured from commercial suppliers and used as received, without any further purification. The structure and purity of the synthesized materials were verified through several analytical techniques, including Powder X-Ray Diffraction (PXRD), nitrogen adsorption–desorption measurements, thermogravimetric analysis (TGA), and Fourier-Transform Infrared (FTIR) spectroscopy.PXRD patterns of the crystalline samples were recorded using a PANalytical X'Pert Pro diffractometer, with Ni-filtered Cu-Kα radiation set at 45 kV and 25 mA, scanning over a 2θ range of 5 to 50 degrees. Nitrogen adsorption–desorption isotherms were measured at 77 K using a Micromeritics ASAP 2020 apparatus. Samples were degassed for 12 hours at 200 °C under vacuum before measurement. Thermogravimetric analyses were carried out on a TA Instruments SDT 2960 under dry air, with a constant heating rate of 5 °C per minute from 25 to 800 °C. FTIR spectra were obtained in reflection mode, covering the range of 600 to 4000 cm−1 with a resolution of 4 cm−1, using a Nicolet Nexus FT-IR instrument. Vapor adsorption isotherms were measured using a Quartz Crystal Microbalance (QCM) from OpenQCM, equipped with 5 MHz quartz crystals from Novaetech Srl.
Synthesis of ZIF-8_CH3
Synthesis of ZIF-8_CH3 followed a reported procedure.47 Typically, a solution comprising Zn(NO3)2·6H2O (9.87 mmol) in 200 mL of methanol was added to a solution of 2-methylimidazole (79.04 mmol) in 200 mL of methanol with stirring at room temperature. After 1 hour, the solution underwent centrifugation at 9000 rpm for 30 minutes, and the resulting solid was then dispersed in ethanol to eliminate excess reactants. This washing process was repeated three times.Synthesis of ZIF-8_Br
ZIF-8_Br was synthesized by mixing a solution of Zn(CH3COO)2·2H2O (0.5 mmol) in DMF (5 mL) with a solution of methanol (5 mL) containing 2-bromoimidazole (1 mmol) at room temperature. The mixture was stirred for 1 hour at room temperature, followed by centrifugation at 9000 rpm for 30 minutes. The resulting solid was dispersed in ethanol, and this purification step was repeated three times to remove any unreacted species.VOC sorption measurements with QCM
For each MOF material, vapor phase adsorption isotherms were obtained using a QCM setup fitted with quartz crystals (5 MHz). These crystals were coated with the studied MOFs by depositing a suspension containing 2 mg of the MOF powder in 1 mL of methanol. Subsequently, the coated quartz crystals were subjected to solvent exchange activation by immersion in dichloromethane overnight.Before measurement, the recovered quartz samples were removed from dichloromethane and placed in the QCM module, where they were heated to 40 °C under a flux of nitrogen for a minimum of 1 hour to achieve equilibrium. The mass of deposited MOF was determined by the difference in frequencies measured by the QCM before and after deposition at 20 °C, applying the Sauerbrey equation.48
The partial pressure was systematically varied from 0 to 0.9 p/p0 for all three VOCs (n-hexane, cyclohexane, and benzene). During the data collection process, the quartz holder was maintained at 20 °C to ensure the acquisition of the mass uptake isotherm for the sorbed VOC within the MOF.
Computational methods
Computational simulations were conducted to understand the adsorption phenomena of the alcohol-ZIF systems. Initially, the structures of ZIF-8_CH3 (CCDC deposit number: 739168) and ZIF-8_Br (CCDC deposit number: 1860456) were cleaned to remove any residual solvent molecules. Their structural characteristics, including pore-limiting diameter (PLD) and largest cavity diameter (LCD), were determined using Zeo++ software (Table S4†).Partial charges (Mulliken) on MOF structures were determined using periodic density functional theory (DFT) via the CASTEP simulation package.49 This method was selected to accurately reflect the periodic characteristics of MOFs. The Hamiltonian operator was computed with the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional and the Tkatchenko–Scheffler dispersion correction. The molecular wavefunction was described using “on-the-fly” pseudopotentials and a plane wave basis set with a cutoff energy of 570 eV, achieving convergence to within 5.10−7 eV per atom. The Broyden–Fletcher–Goldfarb–Shanno (BFGS) algorithm was employed to explore the potential energy surface for energy minima.
The ZIF structures, with Mulliken charges determined by previous periodic DFT calculations, were prepared as 2 × 2 × 2 supercells, each with dimensions at least twice the van der Waals cutoff value (14 Å). Atoms were modelled treating each as a single Lennard-Jones (L-J) interaction site. Interactions were characterized using the Universal Force Field (UFF)50 and Dreiding force field,51 with host–guest interactions modelled by L-J potentials. Lorentz–Berthelot mixing rules were applied to determine the L-J interaction parameters. To accurately handle long-range electrostatic interactions, the Ewald summation method was used, ensuring a relative accuracy of 10−6 for electrostatic forces and energies. This combined approach allowed the model to effectively capture both short-range van der Waals interactions and long-range electrostatic effects between the host and guest species.
The molecular models of n-hexane, benzene, and cyclohexane were constructed using specific force field parameters adapted for each compound. N-Hexane52,53 was modelled using the TraPPE-UA (united atoms) force field, where each CHx group was represented as a neutral sphere, ensuring flexibility of the carbon chain. Benzene,54,55 on the other hand, was represented using the TraPPE-EH (explicit hydrogen) force field, which explicitly shows hydrogen atoms and incorporates charged pseudo-atoms, reflecting its rigid aromatic structure. In contrast to n-hexane, benzene was modelled as a rigid molecule due to its stable aromatic ring structure. Cyclohexane was simulated as a ring composed of CH2 beads, arranged in a chair conformation based on the configuration detailed by Muñoz-Muñoz et al.56 Although cyclohexane can theoretically adopt multiple conformations at room temperature (chair, half-chair, boat, twisted-boat I, and twisted-boat II), recent studies indicate that the chair conformation predominates significantly,56,57 with only 0.01% of the molecule existing in twisted-boat forms in the vapor phase at 298 K. Therefore, for GCMC simulations, adopting the rigid chair conformation at 293 K is considered an accurate and practical approach. The non-bonded dispersive interactions among molecules were described using the Lennard-Jones (L-J) potential, with force field parameters for n-hexane, benzene, and cyclohexane obtained from literature (Table S2†). Detailed structural models of benzene and cyclohexane, including bond lengths, angles, and positions of pseudo-atoms (C_benz, H_benz, CH2) are provided (Table S3†) for comprehensive understanding and reproducibility in simulations.
Energy minimization techniques were applied to locate the adsorbate precisely within the ZIF structures by searching for the lowest energy potential of the host–guest system, thereby determining the most stable configuration. This simulation was conducted under the NVT ensemble, where the number of particles (N), volume (V), and temperature (T) were kept constant, providing a controlled setting to examine the interactions and energetics of the system accurately. For these simulations, the initialization and production cycles were set to 1000 and 1000![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, respectively, and carried out at a temperature of 293 K.
000, respectively, and carried out at a temperature of 293 K.
The Ideal Adsorbed Solution Theory (IAST) was employed to evaluate material performance based on single-component adsorption isotherms. These isotherms were fitted using the dual-site Langmuir–Freundlich model implemented in the IAST++ software. Selectivity (S) was calculated using the following equation:
| SA/B = qA/yA × yB/qB |
Results and discussion
Adsorption of VOCs
After the complete characterization of the synthesized ZIFs to ensure their correct preparation and purity (ESI†), their adsorption isotherms were recorded at 293 K using three C6 hydrocarbon vapors: n-hexane, cyclohexane, and benzene to evaluate their adsorption behavior. The selected C6 molecules differ in their structure and physico-chemical properties (Table 1) and were chosen to highlight the influence of hydrocarbon type, their linear/cyclic nature, flexibility, kinetic diameter and polarity on the adsorption performance.| Adsorbate | Type | Linearity | Flexibility | Kinetic diameter (Å) | Dielectric constant (F m−1) at 20 °C |
|---|---|---|---|---|---|
| n-Hexane | Alkane | Linear | Flexible | 4.3 | 1.88 |
| Cyclohexane | Alkane | Cyclic | Semi-flexible | 6.0 | 2.02 |
| Benzene | Aromatic | Cyclic | Rigid | 5.8 | 2.27 |
The single-component adsorption isotherms for ZIF-8_CH3 and ZIF-8_Br with C6 hydrocarbon vapors (n-hexane, cyclohexane, and benzene) provide a comprehensive understanding of how structural and physico-chemical properties of adsorbates influence adsorption capacities in the studied MOF structures (Fig. 1). For a more relevant comparison between both structures, the adsorption isotherms are expressed as the number of adsorbed molecules per unit cell rather than the number of moles of adsorbates per gram of adsorbent. Notably, the adsorption experiments revealed no permanent deformation of the flexible ZIF structures, preserving their structural integrity and flexibility.
 | ||
| Fig. 1 Hydrocarbon adsorption isotherms at 293 K for a) ZIF-8_CH3 and b) ZIF-8_Br. | ||
In ZIF-8_CH3, benzene exhibits the highest adsorption capacity (9.5 molecules per unit cell at 0.5 p/p0), followed by n-hexane (8.0 molecules per unit cell) and cyclohexane (1.0 molecule per unit cell). The superior uptake of benzene can be attributed to favorable π–π stacking interactions with the aromatic imidazolate linkers, surpassing the van der Waals forces which govern alkane adsorption. The linear structure of n-hexane facilitates efficient packing within the framework, while the bulky cyclic structure of cyclohexane lead to significant steric hindrance, limiting its access to the pores. It is to note that despite a higher uptake, the adsorption of benzene presents a delay compared with n-hexane which is strongly adsorbed at very low partial pressures.
In contrast, ZIF-8_Br displays significantly a different adsorption profile. Benzene presents the highest uptake (6.5 molecules per unit cell), albeit lower than in ZIF-8_CH3, likely due to a combination of π–Br interactions and enhanced dispersion forces with the polarizable bromine atoms. Surprisingly, cyclohexane shows improved adsorption (5.0 molecules per unit cell) compared to ZIF-8_CH3, suggesting favorable interactions between its semi-flexible structure and the bromine substituents. N-Hexane exhibits drastically reduced uptake (0.5 molecules per unit cell), indicating that linear alkane adsorption is strongly dependent on structure rigidity. Interestingly, for all studied VOCs, the adsorption isotherms obtained with ZIF-8_Br, are shifted to higher partial pressures compared with ZIF-8_CH3, demonstrating the impact of structure rigidity on the adsorbate diffusion and their accommodation in ZIF cavities.
The increased rigidity of ZIF-8_Br, induced by bromine functionalization, plays a crucial role in influencing these adsorption differences. Indeed, the rigidity restricts the conformational flexibility of adsorbates, particularly affecting the ability of the linear n-hexane to adapt to the pore structure. Conversely, the polarizable nature of bromine enhances dispersion interactions, benefiting the adsorption of aromatic and cyclic molecules.
The interdependence between framework rigidity, pore accessibility, and influence of functional groups represents a crucial aspect to investigate the MOF adsorption behavior. For instance, the larger atomic radius of bromine (compared to methyl groups) slightly reduces the effective pore size, significantly impacting adsorbate accommodation. Comparing the isotherms provides insights into the gate-opening phenomenon for both ZIFs and its effect on adsorbate access to the porous structure. It has been shown that, unlike non-aromatic compounds, benzene can penetrate the pores of both structures, indicating that π–π interactions play a more significant role in triggering gate-opening. Hence, despite its relatively large kinetic diameter, benzene can induce pore opening in both flexible and rigid ZIFs. In contrast, non-aromatic hydrocarbons cause a lesser degree of pore opening, as evidenced by the smaller magnitude of this effect.
However, the rigid molecular structure of benzene makes its diffusion into the ZIF porous structure more challenging, requiring higher pressures to achieve full pore entry. In contrast, non-aromatic adsorbates, such as n-hexane and cyclohexane, behave differently. Van der Waals forces have a weaker influence on gate-opening compared to π–π interactions. Consequently, the kinetic diameter of the hydrocarbons has a more pronounced impact on their ability to access ZIF pores. For example, in ZIF-8_CH3, only the smaller, flexible hydrocarbon (n-hexane with a 4.3 Å diameter) can enter the pores, while the larger cyclohexane (6.0 Å diameter) is excluded. The flexibility of n-hexane and its favorable interactions with pore walls result in a steep isotherm slope. In ZIF-8_Br, the more rigid structure significantly reduces pore opening, resulting in a more pronounced sieving effect. Although the high flexibility and low polarity of n-hexane are insufficient to force pore entry via gate-opening, even at higher pressures, the higher rigidity and polarity of cyclohexane enable better accommodation at relatively high pressures, albeit in smaller quantities compared to benzene. This highlights the combined influence of π–π interactions, kinetic diameter, and adsorbate polarity in rigid ZIFs as critical factors for triggering the gate-opening phenomenon.
In addition to these findings, further in-depth investigations would be needed to fully explore the gate-opening mechanisms using advanced in situ characterization techniques such as infrared spectroscopy, inelastic neutron scattering, extended X-ray absorption fine structure (EXAFS), and high-energy resolution fluorescence detection technique to the near edge structure of the X-ray absorption spectra (HERFD-XANES), as documented in the literature.58,59
Computational VOC adsorption study
To better understand the adsorption phenomena, the presence density was simulated to identify the location of each adsorbate within the ZIF structures (Fig. 2). | ||
| Fig. 2 Illustration of a) general ZIF-8_Br structure and b and c) the 4 and 6-membered ring pore windows (4MR and 6MR). Zinc cation polyhedral are colored to delimit and highlight the presence of two types of pore windows (red for 4MR and orange for 6MR). | ||
The 2D density maps (Fig. 3a) exhibit similar patterns for all investigated ZIF types and adsorbates. They indicate that C6 molecules, at a concentration of one molecule per unit cell, are exclusively localized within the β cages of the ZIF structures. Interestingly, even the flexible n-hexane preferentially resides within these cages rather than accommodating the window spaces. This behavior contrasts with that observed in [Pd(2-pymo)2]n, exhibiting also a sodalite-type structure, which allows n-hexane to pass through the 6MR hydrophobic windows.20
 | ||
| Fig. 3 a) 2D representation of the benzene presence density in ZIF-8_Br. b) Illustration of 2 × 2 × 2 supercell of ZIF-8_Br structure and c) 3D representation of the benzene presence density in ZIF-8_Br. | ||
In the case of ZIF-8_(CH3/Br), the preferential confinement of adsorbates within the β cages may be attributed to enhanced steric hindrance effects resulting from the small pore window size of this structure and the maximization of number of host–guest interactions. Both 2D and 3D representations (Fig. 3a–c) demonstrate that adsorbate molecules are mainly localized along the ZIF framework walls, rather than occupying the central regions of the cavities. This spatial distribution strongly suggests the presence of specific host–guest interactions between the adsorbate molecules and the ZIF cavities.
The adsorption configurations of benzene, cyclohexane, and n-hexane within ZIF-8_CH3 and ZIF-8_Br structures were elucidated through energy-minimized Grand Canonical Monte Carlo (GCMC) simulations, showing distinct host–guest interactions influenced by ligand functionalization (Fig. 4 and 5).
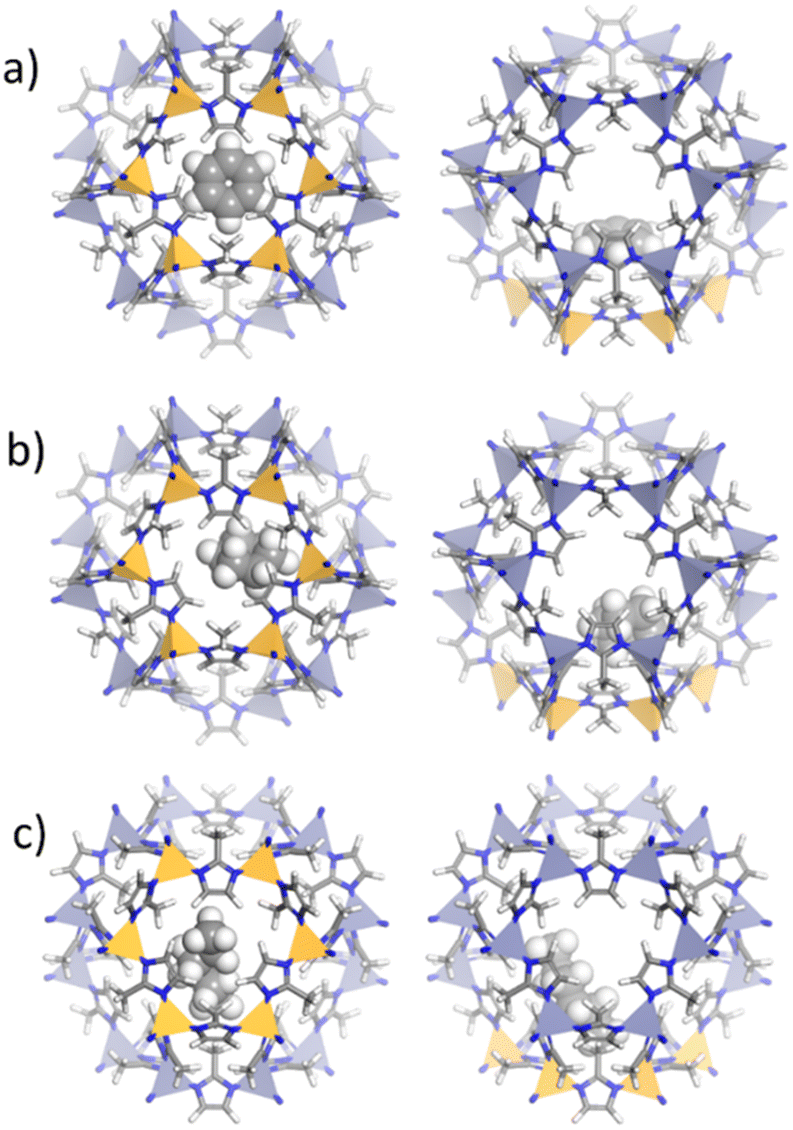 | ||
| Fig. 4 Location of a) benzene, b) cyclohexane and c) n-hexane molecules within ZIF-8_CH3 structure using energy minimization approach through GCMC calculation. Orange polyhedra represent the zinc cations delimiting the 6MR windows in ZIF-8_CH3 cavities. | ||
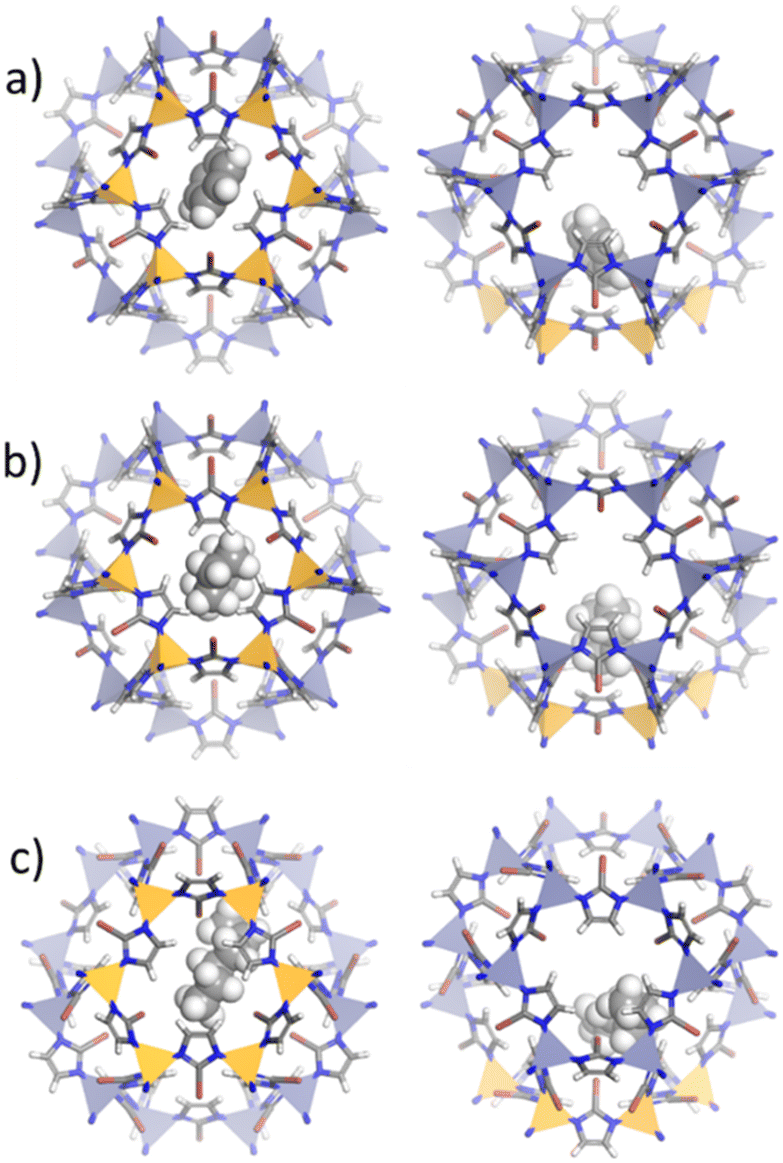 | ||
| Fig. 5 Location of a) benzene, b) cyclohexane and c) n-hexane molecules within ZIF-8_Br structure using energy minimization approach through GCMC calculation. Orange polyhedra represent the zinc cations delimiting the 6MR windows in ZIF-8_Br cavities. | ||
In ZIF-8_CH3, benzene molecules exhibit a pronounced affinity to the 6MR windows, positioning themselves in parallel to the imidazolate linkers. This orientation suggests strong π–π interactions between the aromatic guest and the electron-rich environment of the ligand, a phenomenon well-documented in many MOF systems.60 Cyclohexane, adopting its characteristic chair conformation, interacts with the methyl-functionalized linkers predominantly through van der Waals forces due to its nonpolar nature. The linear n-hexane aligns along the cavity walls, maximizing weak but extensive surface interactions with the hydrophobic framework.30–32
In contrast, ZIF-8_Br induces notable changes in guest orientations. For instance, the benzene molecule adopts a quasi-perpendicular inclination relative to the 6MR windows, likely due to altered electronic interactions induced by the electronegative bromine atoms.61 This modification in structure polarizability potentially enhances π–π interactions. Cyclohexane occupies a more central position within the 6MR windows, while n-hexane appears slightly more constrained, reflecting the influence of the bulkier bromine substituents on the pore environment.
Separation of VOCs
To investigate the separation efficiency of ZIF-8_CH3 and ZIF-8_Br, the IAST method was employed to estimate the selectivity between selected VOC pairs: n-hexane/cyclohexane, n-hexane/benzene, and benzene/cyclohexane at 293 K (Fig. 6). The IAST selectivity analysis of ZIF-8_CH3 and ZIF-8_Br shows significant differences in their separation performance for the considered binary equimolar mixtures over a pressure range of 0.004 to 0.08 bar.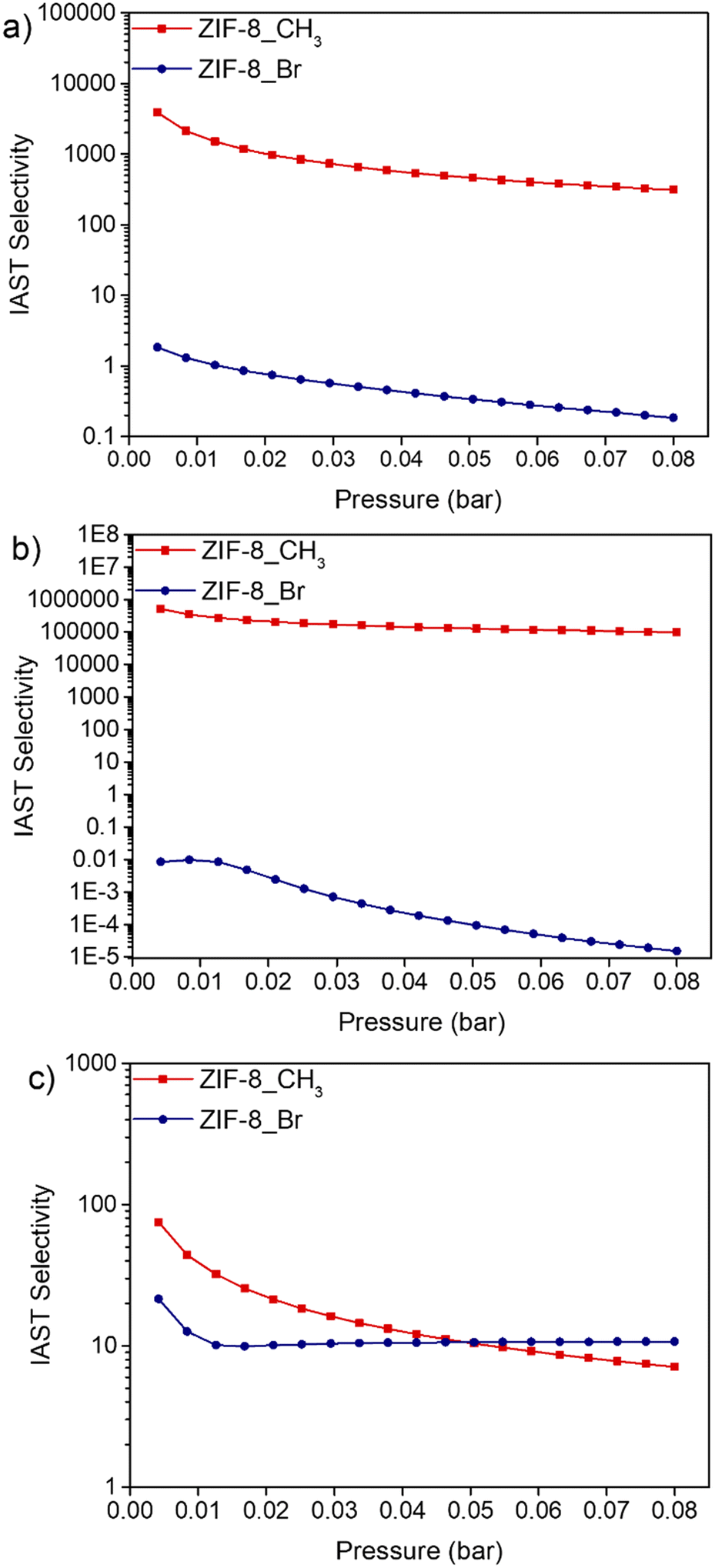 | ||
| Fig. 6 IAST selectivities of VOC pairs at 293 K for ZIF-8_CH3 and ZIF-8_Br including equimolar mixtures of a) n-hexane/cyclohexane, b) n-hexane/benzene and c) benzene/cyclohexane. | ||
For n-hexane/cyclohexane separation, ZIF-8_CH3 exhibits remarkably high initial selectivity (3863 at 0.004 bar), decreasing to 311 at 0.08 bar. This preference for n-hexane can be attributed to its linear structure, allowing more efficient packing and stronger van der Waals interactions within methyl-functionalized pores as discussed elsewhere.62 Conversely, ZIF-8_Br shows minimal selectivity (1.83 to 0.19), likely due to steric effects and increased structural stiffness.
The n-hexane/benzene separation shows even more pronounced differences. ZIF-8_CH3 demonstrates exceptional initial selectivity (512722 at 0.004 bar), decreasing to 99127 at 0.08 bar. This extreme preference for n-hexane is likely due to the hydrophobic nature of methyl groups and steric hindrance effect. ZIF-8_Br, however, exhibits reverse selectivity (0.008 to 0.000015), strongly favoring benzene. This reversal can be attributed to enhanced π–π interactions and potential halogen bonding effects, as reported by Yagi et al.63 for halogen-functionalized MOFs.
For benzene/cyclohexane separation, both structures favor benzene, but at different extent. ZIF-8_CH3 shows higher initial selectivity (75 at 0.004 bar) compared to ZIF-8_Br (21), likely due to enhanced π–π interactions. The selectivity decreases with pressure and is more pronounced in ZIF-8_CH3, suggesting that the gate-opening phenomenon strongly participates to the separation process64,65 while its influence is much more reduced for ZIF-8_Br due to its intrinsic stiffness.
Conclusions
This study highlights the influence of structural flexibility and functionalization in zeolitic imidazolate frameworks on the selective adsorption and separation of C6 hydrocarbons. Through a comparative analysis of ZIF-8_Br and ZIF-8_CH3, we explored how the structural stiffness and dynamic behaviour govern adsorption/separation properties. ZIF-8_Br, exhibiting strong structural rigidity, preferentially adsorbs benzene and cyclohexane over n-hexane, driven by limited conformational freedom and enhanced dispersion interactions. Conversely, the flexibility of ZIF-8_CH3 structure allows n-hexane preferential adsorption while excluding cyclohexane. Moreover, this ZIF analogue also shows increased benzene uptake due to more favourable host–guest interactions and efficient molecular packing within its porous structure. Computational simulations support these findings, elucidating different adsorption mechanisms depending on the structural stiffness and chemical environment in MOF cavities. Moreover, the application of Ideal Adsorbed Solution Theory enabled to shed light to the role of the MOF structure features on separation performance. For instance, ZIF-8_Br shows limited selectivity for n-hexane/cyclohexane and inverse selectivity for n-hexane/benzene. These insights into the interdependence between rigidity, functionalization, and adsorption behaviour, provide valuable information to support rational design of MOFs with tailored properties for efficient VOC capture and separation. In conclusion, this work provides valuable insights into adsorption mechanisms in flexible structures, enhancing the fundamental understanding necessary for developing optimized MOFs. While the findings primarily contribute to scientific knowledge, they may also inspire further research into potential applications in industrial separation processes and air quality management.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization, methodology and validation: KD. Synthesis and characterization of ZIFs, adsorption and computational investigation, and data analysis: KD. Writing – original draft: KD. Writing – review and editing: KD, MD and AJ. Supervision: MD and AJ. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the ANR SensMOFAir project (#ANR-20-CE04-0012).Notes and references
- R. Montero-Montoya, R. López-Vargas and O. Arellano-Aguilar, Ann. Glob. Health, 2018, 84, 225–238 CrossRef.
- M. Belingheri, S. Fustinoni, G. De Vito, A. Porro and M. A. Riva, Med. Lav., 2019, 110, 234–240 Search PubMed.
- O. E. Ifeanyi, Open Access Blood Research & Transfusion Journal, 2018, 2, 1–5 Search PubMed.
- J. Roma-Torres, J. P. Teixeira, S. Silva, B. Laffon, L. M. Cunha, J. Méndez and O. Mayan, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2006, 604, 19–27 CrossRef.
- V. Cocheo, P. Sacco, C. Boaretto, E. De Saeger, P. Perez Ballesta, H. Skov, E. Goelen, N. Gonzalez and A. Baeza Caracena, Nature, 2000, 404, 141–142 CrossRef PubMed.
- T. Campos-Ordonez and O. Gonzalez-Perez, Front. Pharmacol., 2016, 6, 291 Search PubMed.
- N. K. Jørgensen and K.-H. Cohr, Scand. J. Work, Environ. Health, 1981, 7, 157–168 CrossRef PubMed.
- H. Yao, Y. Wang, M. Quan, M. U. Farooq, L. Yang and W. Jiang, Angew. Chem., Int. Ed., 2020, 59, 19945–19950 CrossRef PubMed.
- M. Ayuso, A. Cañada-Barcala, M. Larriba, P. Navarro, N. Delgado-Mellado, J. García and F. Rodríguez, Sep. Purif. Technol., 2020, 240, 116583 CrossRef.
- X. Shen, R. Ou, Y. Lu, A. Yuan, J. Liu, J. Gu, X. Hu, Z. Yang and F. Yang, Environ. Int., 2020, 143, 105774 CrossRef PubMed.
- T. J. Bandosz, in Adsorption by Carbons, Elsevier, 2008, pp. 533–564 Search PubMed.
- B. K. Marcus and W. E. Cormier, Chem. Eng. Prog., 1999, 95, 47–52 Search PubMed.
- W. Lu, Z. Wei, Z. Y. Gu, T. F. Liu, J. Park, J. Park, J. Tian, M. Zhang, Q. Zhang, T. Gentle, M. Bosch and H. C. Zhou, Chem. Soc. Rev., 2014, 43, 5561–5593 RSC.
- X. Zhang, Z. Chen, X. Liu, S. L. Hanna, X. Wang, R. Taheri-Ledari, A. Maleki, P. Li and O. K. Farha, Chem. Soc. Rev., 2020, 49, 7406–7427 RSC.
- K. A. Forrest, T. Pham, S. K. Elsaidi, M. H. Mohamed, P. K. Thallapally, M. J. Zaworotko and B. Space, Cryst. Growth Des., 2019, 19, 3732–3743 CrossRef.
- B. Van De Voorde, M. Hezinová, J. Lannoeye, A. Vandekerkhove, B. Marszalek, B. Gil, I. Beurroies, P. Nachtigall and D. De Vos, Phys. Chem. Chem. Phys., 2015, 17, 10759–10766 RSC.
- R. Banerjee, A. Phan, B. Wang, C. Knobler, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Science, 2008, 319, 939–943 CrossRef PubMed.
- N. M. Padial, E. Quartapelle Procopio, C. Montoro, E. López, J. E. Oltra, V. Colombo, A. Maspero, N. Masciocchi, S. Galli, I. Senkovska, S. Kaskel, E. Barea and J. A. R. Navarro, Angew. Chem., Int. Ed., 2013, 52, 8290–8294 CrossRef PubMed.
- K. Dedecker, E. Dumas, B. Lavédrine, N. Steunou and C. Serre, in Metal-Organic Frameworks (MOFs) for Environmental Applications, Elsevier, 2019, pp. 141–178 Search PubMed.
- K. Dedecker, M. Drobek, V. Rouessac and A. Julbe, ACS Appl. Mater. Interfaces, 2023, 15, 6831–6838 CrossRef PubMed.
- K. Dedecker, M. Drobek and A. Julbe, J. Phys. Chem. B, 2023, 127, 11091–11099 CrossRef PubMed.
- K. Dedecker, R. S. Pillai, F. Nouar, J. Pires, N. Steunou, E. Dumas, G. Maurin, C. Serre and M. L. Pinto, ACS Appl. Mater. Interfaces, 2018, 10, 13886–13894 CrossRef PubMed.
- J. Pires, J. Fernandes, K. Dedecker, J. R. B. Gomes, G. Pérez-Sánchez, F. Nouar, C. Serre and M. L. Pinto, ACS Appl. Mater. Interfaces, 2019, 11, 27410–27421 CrossRef PubMed.
- S. Dasgupta, S. Biswas, K. Dedecker, E. Dumas, N. Menguy, B. Berini, B. Lavedrine, C. Serre, C. Boissière and N. Steunou, ACS Appl. Mater. Interfaces, 2023, 15, 6069–6078 CrossRef PubMed.
- B. Liang, X. Zhang, Y. Xie, R. B. Lin, R. Krishna, H. Cui, Z. Li, Y. Shi, H. Wu, W. Zhou and B. Chen, J. Am. Chem. Soc., 2020, 142, 17795–17801 CrossRef PubMed.
- P. Iacomi and G. Maurin, ACS Appl. Mater. Interfaces, 2021, 13, 50602–50642 CrossRef PubMed.
- F. Millange and R. I. Walton, Isr. J. Chem., 2018, 58, 1019–1035 CrossRef.
- A. Phan, C. J. Doonan, F. J. Uribe-Romo, C. B. Knobler, M. Okeeffe and O. M. Yaghi, Acc. Chem. Res., 2010, 43, 58–67 CrossRef PubMed.
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef PubMed.
- A. U. Ortiz, A. P. Freitas, A. Boutin, A. H. Fuchs and F.-X. Coudert, Phys. Chem. Chem. Phys., 2014, 16, 9940–9949 RSC.
- K. Zhang, R. P. Lively, C. Zhang, R. R. Chance, W. J. Koros, D. S. Sholl and S. Nair, J. Phys. Chem. Lett., 2013, 4, 3618–3622 CrossRef.
- H. Zhang and R. Q. Snurr, J. Phys. Chem. C, 2017, 121, 24000–24010 CrossRef.
- H. Zhang, M. Zhao, Y. Yang and Y. S. Lin, Microporous Mesoporous Mater., 2019, 288, 109568 CrossRef.
- D. Liu, X. Ma, H. Xi and Y. S. Lin, J. Membr. Sci., 2014, 451, 85–93 CrossRef.
- F. Coudert, ChemPhysChem, 2017, 18, 2732–2738 CrossRef.
- D. Fairen-Jimenez, S. A. Moggach, M. T. Wharmby, P. A. Wright, S. Parsons and T. Düren, J. Am. Chem. Soc., 2011, 133, 8900–8902 CrossRef PubMed.
- S. Aguado, G. Bergeret, M. Pera-Titus, V. Moizan, C. Nieto-Draghi, N. Bats and D. Farrusseng, New J. Chem., 2011, 35, 546–550 RSC.
- A. Arami-Niya, G. Birkett, Z. Zhu and T. E. Rufford, J. Mater. Chem. A, 2017, 5, 21389–21399 RSC.
- C. Zhang, R. P. Lively, K. Zhang, J. R. Johnson, O. Karvan and W. J. Koros, J. Phys. Chem. Lett., 2012, 3, 2130–2134 CrossRef PubMed.
- A. Knebel, B. Geppert, K. Volgmann, D. I. Kolokolov, A. G. Stepanov, J. Twiefel, P. Heitjans, D. Volkmer and J. Caro, Science, 2017, 358, 347–351 CrossRef.
- D. J. Babu, G. He, J. Hao, M. T. Vahdat, P. A. Schouwink, M. Mensi and K. V. Agrawal, Adv. Mater., 2019, 31, 1900855 Search PubMed.
- A. Huang, N. Wang, C. Kong and J. Caro, Angew. Chem., Int. Ed., 2012, 51, 10551–10555 CrossRef.
- P. Krokidas, S. Moncho, E. N. Brothers, M. Castier and I. G. Economou, Phys. Chem. Chem. Phys., 2018, 20, 4879–4892 RSC.
- M. Loloei, S. Kaliaguine and D. Rodrigue, Sep. Purif. Technol., 2022, 296, 121391 CrossRef.
- J. Mor, R. B. Nelliyil and S. K. Sharma, Langmuir, 2023, 39, 10056–10065 CrossRef CAS PubMed.
- G. Chaplais, G. Fraux, J.-L. Paillaud, C. Marichal, H. Nouali, A. H. Fuchs, F.-X. Coudert and J. Patarin, J. Phys. Chem. C, 2018, 122, 26945–26955 Search PubMed.
- A. Demessence, C. Boissière, D. Grosso, P. Horcajada, C. Serre, G. Férey, G. J. A. A. Soler-Illia and C. Sanchez, J. Mater. Chem., 2010, 20, 7676 RSC.
- G. Sauerbrey, Z. Phys., 1959, 155, 206–222 CrossRef CAS.
- P. Kratzer and J. Neugebauer, Front. Chem., 2019, 7, 1–18 CrossRef PubMed.
- A. K. Rappé, C. J. Casewit, K. S. Colwell, W. A. Goddard and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef.
- S. L. Mayo, B. D. Olafson and W. A. Goddard, J. Phys. Chem., 1990, 94, 8897–8909 CrossRef CAS.
- M. G. Martin and J. I. Siepmann, J. Phys. Chem. B, 1998, 102, 2569–2577 CrossRef CAS.
- S. J. Keasler, S. M. Charan, C. D. Wick, I. G. Economou and J. I. Siepmann, J. Phys. Chem. B, 2012, 116, 11234–11246 CrossRef CAS PubMed.
- N. Rai and J. I. Siepmann, J. Phys. Chem. B, 2007, 111, 10790–10799 CrossRef CAS PubMed.
- N. Rai and J. I. Siepmann, J. Phys. Chem. B, 2013, 117, 273–288 CrossRef CAS.
- Y. M. Muñoz-Muñoz, G. Guevara-Carrion, M. Llano-Restrepo and J. Vrabec, Fluid Phase Equilib., 2015, 404, 150–160 CrossRef.
- A. Sławek, K. Grzybowska, J. M. Vicent-Luna, W. Makowski and S. Calero, ChemPhysChem, 2018, 19, 3364–3371 CrossRef PubMed.
- M. E. Casco, Y. Q. Cheng, L. L. Daemen, D. Fairen-Jimenez, E. V. Ramos-Fernández, A. J. Ramirez-Cuesta and J. Silvestre-Albero, Chem. Commun., 2016, 52, 3639–3642 RSC.
- R. Boada, J. Chaboy, S. Hayama, L. L. Keenan, A. A. Freeman, M. Amboage and S. Díaz-Moreno, J. Phys. Chem. C, 2022, 126, 5935–5943 CrossRef CAS.
- H. Li, C. Lin, R. Ma and Y. Chen, Chemosphere, 2023, 337, 139377 CrossRef CAS.
- J. Edzards, H.-D. Saßnick, A. G. Buzanich, A. M. Valencia, F. Emmerling, S. Beyer and C. Cocchi, J. Phys. Chem. C, 2023, 127, 21456–21464 CrossRef CAS.
- A. Sławek, K. Roztocki, D. Majda, S. Jaskaniec, T. J. H. Vlugt and W. Makowski, Microporous Mesoporous Mater., 2021, 312, 110730 CrossRef.
- R. Yagi and T. Ueda, Phys. Chem. Chem. Phys., 2023, 25, 20585–20596 RSC.
- N. Nijem, H. Wu, P. Canepa, A. Marti, K. J. Balkus, T. Thonhauser, J. Li and Y. J. Chabal, J. Am. Chem. Soc., 2012, 134, 15201–15204 CrossRef CAS PubMed.
- M. T. Luebbers, T. Wu, L. Shen and R. I. Masel, Langmuir, 2010, 26, 15625–15633 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional material characterizations and computational details. See DOI: https://doi.org/10.1039/d4lf00388h |
| This journal is © The Royal Society of Chemistry 2025 |